
Investigating the role of amine in InP nanocrystal synthesis: destabilizing cluster intermediates by Z-type ligand displacement†
Dylan C.
Gary
,
Alessio
Petrone
 ,
Xiaosong
Li
and
Brandi M.
Cossairt
*
,
Xiaosong
Li
and
Brandi M.
Cossairt
*
Department of Chemistry, University of Washington, Box 351700, Seattle, WA 98195-1700, USA. E-mail: cossairt@chem.washington.edu
First published on 23rd November 2016
Abstract
The reaction of primary amines with In37P20(O2CR)51 is found to remove In(O2CR)3 subunits from In37P20(O2CR)51. This loss of Z-type ligands coincides with structural rearrangement to alleviate core strain and passivate phosphorus atoms. This result consolidates conflicting claims that primary amines both promote and retard precursor conversion rates for InP nanocrystals.
Primary amine is a common additive in many nanocrystal syntheses and has been assigned a variety of roles including ligand, shape control agent, proton donor, percursor conversion inhibitor, and growth activator.1–4 Specifically in the synthesis of indium phosphide quantum dots (InP QDs) from indium carboxylates and silylphosphines, amine has been proposed to play several distinct, and often conflicting roles.5–7 Xie et al. have suggested that amines act as activating agents for indium carboxylates by increasing their reactivity towards silylphosphines.5 This hypothesis is consistent with the observation that primary amines lowered the temperature necessary to synthesize InP QDs from silylphosphines and indium carboxylates by about 100 °C. This claim was later refuted by Allen et al. who discovered that amine retards In–P bond formation from silylphosphines and indium carboxylates by monitoring the precursor conversion reactions using NMR spectroscopy.6 Based on these observations, a reaction mechanism in which amine competes with phosphine for coordination to In prior to In–P bond formation via an SN2 transition state was proposed.
Xie et al. also demonstrated that InP QDs ranging in size from 2 to 8 nm could be synthesized by altering the concentration of amine and free carboxylic acid employed in these syntheses. Protière et al. observed in their syntheses that addition of primary amine yielded in situ water which would oxidize the surface of the InP QDs, forming an In2O3 shell and effectively halting further growth.7 These apparently contrary observations may be resolved by considering the difference in temperature at which these two studies were carried out. At the lower temperatures reported by Xie et al. (190 °C rather than 270 °C), the condensation of amine and free carboxylic acid would take place at a slower rate than precursor conversion and growth of InP based upon reported reaction times for the direct amidation of carboxylic acids with primary amines.8
Given that amines have been shown to impede precursor conversion and halt nanocrystal growth at elevated temperatures due to the formation of an In2O3 shell, another explanation was needed to account for the observation that the addition of primary amines lowers the temperature necessary to synthesize high quality InP over a large range of sizes. Our group offered an explanation consistent with these observations.9 We repeated the synthesis of InP reported by Xie et al. at lower temperatures (110–160 °C) in the absence of primary amine. Under these reaction conditions, we obtained a magic-size cluster of InP instead of the larger dynamic structures reported by Xie and coworkers. Addition of primary amine to a solution of these clusters held at 160 °C was shown by UV-Vis absorbance spectroscopy to rapidly induce growth of InP through a continuum of sizes. Therefore, amine can both inhibit precursor conversion in a non-rate limiting step, as well as decrease the temperature necessary for the rate-limiting nucleation of InP nanocrystals from clusters since these are temporally distinct events in a two-step nucleation mechanism. Recently, we have been able to determine the identity of the magic-size cluster in this synthesis to be In37P20(O2CR)51 by single crystal X-ray diffraction.10 This result has prompted us to further investigate the reaction of primary amine with In37P20(O2CR)51 in order to elucidate its role in destabilizing this cluster intermediate.
Previously, we were able to determine the reaction product of In37P20(O2CR)51 with one equivalent of H2O in the solid state by exposing single crystals of In37P20(O2CCH2C6H5)51 to moist air for short periods of time. The resultant crystals yielded a new diffraction pattern consistent with a structure in which a single carboxylate ligand had shifted binding modes from bidentate to monodentate and H2O had datively bound to the available coordination site (Fig. 1). This observation suggests that In37P20(O2CR)51 may have site selective reactivity towards Lewis bases, and that the indium centre to which water was bound (In16 and its symmetry equivalent In27) is the most likely candidate for initial binding site of primary amines.
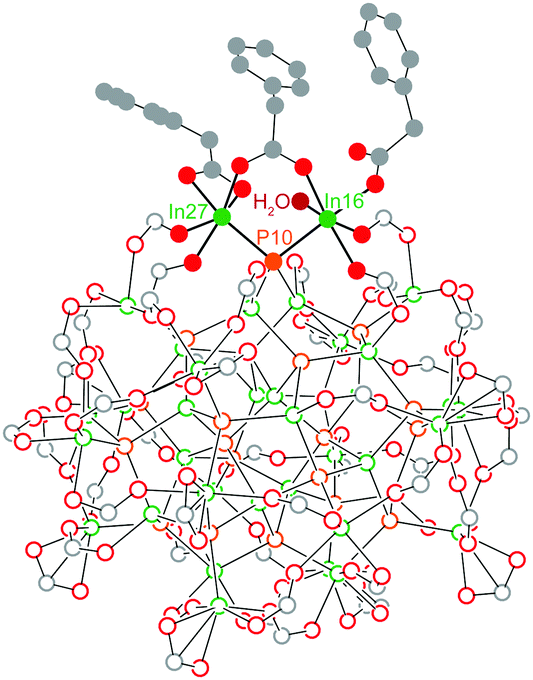 | ||
| Fig. 1 Truncated structure of In37P20(O2CCH2C6H5)51·H2O. Prior to hydration, both In atoms (In16 and In27) bound to P10 exhibited the same first coordination sphere and were related by pseudo C2 symmetry. Post hydration, one phenylacetate ligand on In16 adopted a monodentate binding mode to accommodate H2O. | ||
Our first insight into the reaction scheme between primary amines and In37P20(O2CR)51 was obtained by adding 108 equivalents of benzylamine to In37P20(O2CCH2C6H5)51. From this solution, crystals of In(O2CH2C5H6)3(H2NCH2C6H5)3 were obtained (Fig. S1, ESI†). In this structure, indium exhibits an octahedral geometry in which each of the three datively bound benzylamine ligands are trans to a phenylacetate ligand. The liberation of In(O2CR)3 in an amine-bound form from the surface of In37P20(O2CR)51 is consistent with recent literature illustrating L-type promoted Z-type ligand exchange as a viable strategy for removing anionic carboxylate from QD surfaces using neutral amine.11
In light of this discovery, we examined this reaction further by performing a titration of In37P20(O2CCH2C6H5)51 with 4-fluorobenzylamine (H2NCH2C6H4F). We chose H2NCH2C6H4F in order to specifically probe the fate of the amine additive using 19F{1H} NMR spectroscopy. From the 31P{1H} NMR data, we see evidence for a stoichiometric reaction upon addition of 1 to 3 equivalents of H2NCH2C6H4F with complete conversion to a single new product that preserves the pseudo C2 symmetry as evidenced by shifts in all the major resonances associated with In37P20(O2CCH2C6H5)51 (Fig. 2A). Based on our assignment of the 31P{1H} NMR spectrum for In37P20(O2CCH2C6H5)51 (Fig. S2–S6 and Tables S1–S3, ESI†), we observed that upon addition of the first three equivalents of H2NCH2C6H4F the largest shift in the spectrum is for the phosphorus atom P10 (from −185 ppm to −173 ppm). This interpretation is consistent with our previous assignment of the most acidic site on the cluster residing on In16, which is bound to P10.10 Between the addition of six and twelve equivalents of H2NCH2C6H4F, we observe that all major resonances have shifted and broadened in the 31P{1H} NMR spectrum. This is likely due to the removal of In(O2CH2C6H5)3 in the form of In(O2CCH2C6H5)3(H2NCH2C6H5)3 with a concurrent structural rearrangement.
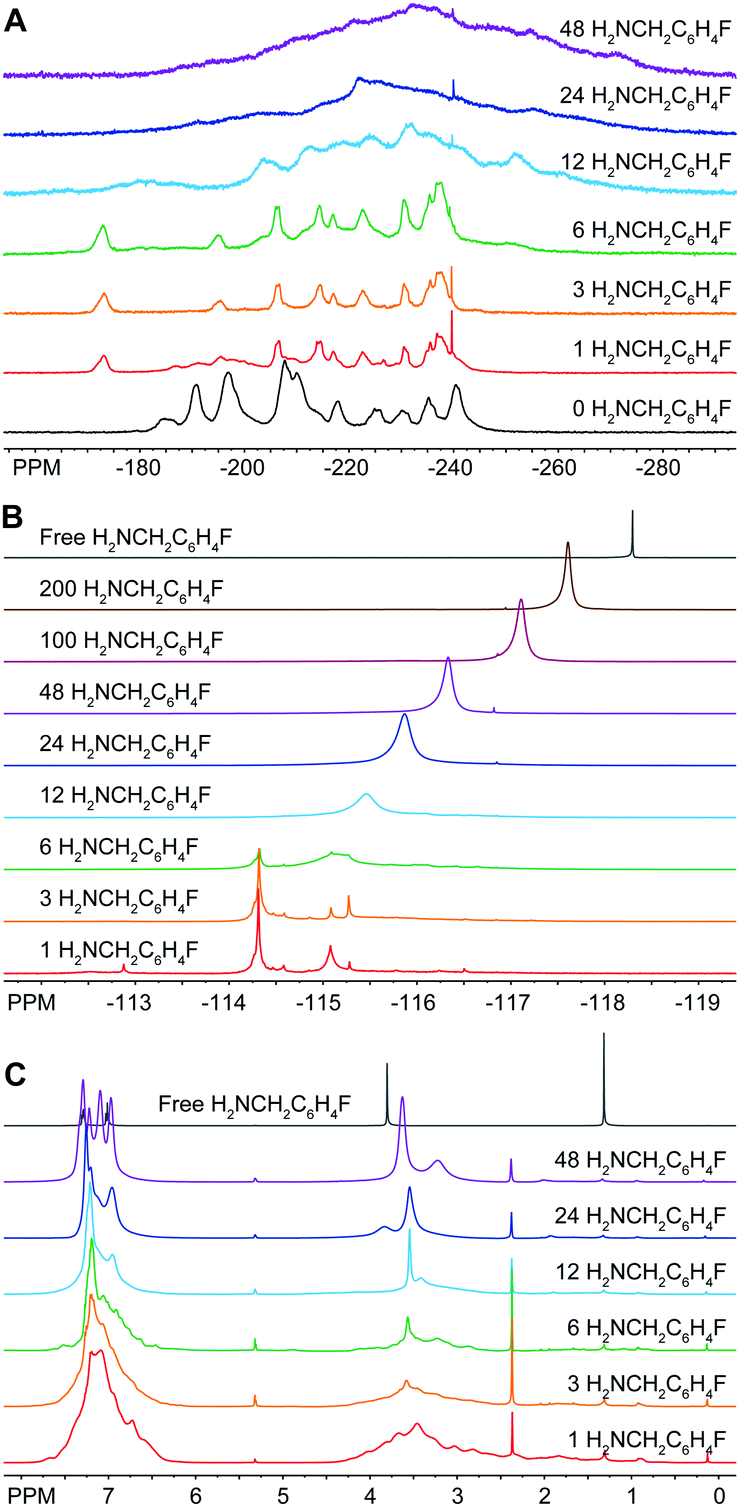 | ||
| Fig. 2 (A) 31P{1H} (202.3 MHz), (B) 19F{1H} (469 MHz), and (C) 1H (500 MHz) NMR spectra of In37P20(O2CCH2C6H5)51 after stoichiometric additions of H2NCH2C6H4F to a solution in CD2Cl2. | ||
19F{1H} NMR spectra were taken of the same solution of In37P20(O2CCH2C6H5)51 with 17 equivalents of α,α,α-trifluorotoluene (C6H5CF3) as an internal standard (Fig. 2B). All of the 19F nuclei are accounted for throughout these additions within experimental error according to the relative integration of H2NCH2C6H4F to C6H5CF3. Upon addition of a single equivalent of H2NCH2C6H4F to In37P20(O2CCH2C6H5)51, three major resonances were observed in the 19F{1H} spectrum at −114.3 ppm, −115.1 ppm, and −115.3 ppm. We attribute these peaks to H2NCH2C6H4F tightly binding to the surface of the In37P20(O2CCH2C6H5)51. This initial reaction went to completion in terms of the consumption of H2NCH2C6H4F since we observed no free H2NCH2C6H4F (singlet at −118.3 ppm). Up to the addition of 6 equivalents of H2NCH2C6H4F, the 19F{1H} spectra can be explained in terms of H2NCH2C6H4F exhibiting three predominant coordination modes to the surface of In37P20(O2CCH2C6H5)51. Given that each of the 31P nuclei in the 31P{1H} NMR spectra could be accounted for with a single structure, but we observed multiple peaks in the 19F{1H} spectra from the addition of 1 to 3 equivalents of H2NCH2C6H4F, there may be multiple modes by which H2NCH2C6H4F can bind to the surface of In37P20(O2CCH2C6H5)51 while preserving a constant phosphide sublattice.
After 12 equivalents of H2NCH2C6H4F were added to the solution, all of the peaks assigned to the initial binding modes of H2NCH2C6H4F were lost and a broad singlet grew in around −115.5 ppm. We interpret this to amine having liberated surface bound indium from In37P20(O2CCH2C6H5)51 in the form of In(O2CCH2C6H5)3(H2NCH2C6H4F)3. This is consistent with our interpretation of the 31P{1H} NMR spectra. Upon further additions of H2NCH2C6H4F, the broad peak attributed to liberated In(O2CCH2C6H5)3(H2NCH2C6H4F)3 continued to grow in, sharpen, and shift upfield toward free H2NCH2C6H4F. Since we observed a single broad peak in the 19F NMR spectra after this point, we attributed this resonance to H2NCH2C6H4F being in equilibrium between datively binding to indium at the surface of the cluster or to In(O2CCH2C6H5)3(H2NCH2C6H4F)3 and freely diffusing in solution.
The 1H NMR spectra of the titration of In37P20(O2CCH2C6H5)51 with H2NCH2C6H4F exhibited line sharpening over the course of the titration in both the alkyl and aromatic regions of the spectrum (Fig. 2C). We have previously shown that the 1H NMR spectrum of In37P20(O2CCH2C6H5)51 exhibits a large number of peaks from about 2.5 to 4.0 ppm.10 We assigned these peaks to distinct methylene protons on the ligand set due to hindered rotation about the C–C bond of the carbonyl and methylene carbons based on the 1H–1H COSY spectrum. Upon addition of 12 equivalents of H2NCH2C6H4F, we observed that the array of peaks spanning from about 2.5 to 4.0 ppm for the methylene protons of In37P20(O2CCH2C6H5)51 were lost and two broad peaks grew in around 3.5 and 3.4 ppm. This suggests that upon removal of In(O2CCH2C6H5)3 from In37P20(O2CCH2C6H5)51, the resultant cluster is no longer so densely ligated as to prevent rotation about the C–C bond of the carbonyl carbon and methylene carbon of the carboxylate ligands. Once again, the point in the titration assigned to the removal of In(O2CCH2C6H5)3 is consistent with the previous assignments for the 19F{1H} and 31P{1H} spectra.
UV-Vis spectra were also acquired from a titration of In37P20(O2CCH2C6H5)51 with H2NCH2C6H4F carried out under inert atmosphere (Fig. 3). In contrast to the 31P{1H}, 19F{1H}, and 1H NMR spectra, the UV-Vis absorbance spectra of the titration exhibited a continuous shift in which the absorption maximum at 386 nm was lost after a single equivalent of H2NCH2C6H4F had been added, and a new broad peak at 404 nm gradually grew in up to the addition of 96 equivalents of H2NCH2C6H4F. No further shift in the absorbance maximum at 404 nm was observed in the UV-Vis spectra after the addition of 6 equivalents of H2NCH2C6H4F, even though removal of In(O2CCH2C6H5)3(H2NCH2C6H4F)3 is inferred from the NMR at this point in the titration. Analysis of difference spectra (Fig. S7, ESI†) for each sequential data set shows that the major changes in electronic structure are complete following the addition of 12 equivalents of H2NCH2C6H4F, consistent with the structural rearrangement and stabilization of the structure after liberation of In(O2CR)3.
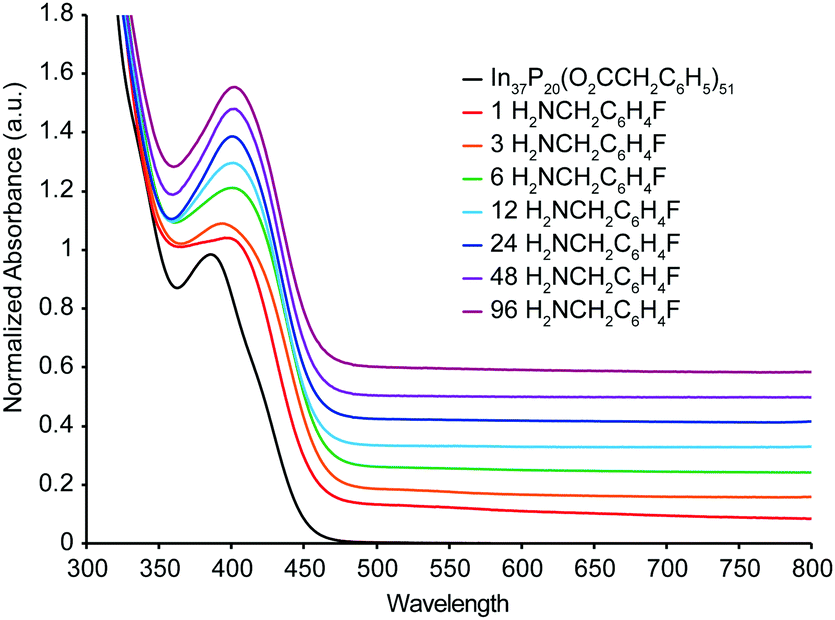 | ||
| Fig. 3 UV-Vis spectra of stoichiometric additions of H2NCH2C6H4F to a solution of In37P20(O2CCH2C6H5)51 carried out under inert atmosphere. Spectra are normalized to the lowest energy peak maximum and offset vertically for clarity. | ||
Previously, we had predicted that introduction of an indium open coordination site on In37P20(O2CCH2C6H5)51 would be readily identified by UV-Vis spectroscopy.10 This hypothesis was based on DFT calculations performed on the structure of In37P20(O2CCH2C6H5)51·H2O and a theoretical structure where H2O was omitted but the displaced carboxylate ligand remained monodentate, leaving an open coordination site on In16. In order to ascertain whether under-coordinated phosphorus atoms (which must be introduced when breaking the In–P bond to liberate In(O2CR)3) would have a similar effect, we performed DFT calculations. These calculations show that the UV-Vis spectrum is strongly affected when an open coordination site is present by removing In16 with three carboxylate ligands from In37P20(O2CCH2C6H5)51 (Fig. S8, ESI†). We then mimicked the structural rearrangement by performing geometry optimizations on structures where the phenylacetate moieties were replaced by acetate, In36P20(O2CCH3)48. In this case, by allowing the system to relax when In16 was removed along with three carboxylate ligands, a significant change in the electronic properties is still observed (Fig. S9 and S10, ESI†). These calculations suggest that under-coordinated phosphorus atoms would also be readily identified by UV-Vis spectroscopy.
Since we observe no clear evidence for a new species in the UV-Vis spectra between the addition of 6 and 12 equivalents where we propose the loss of In(O2CCH2C6H5)3 from analysis of the NMR spectra, we hypothesize that upon removal of In(O2CCH2C6H5)3 from In37P20(O2CCH2C6H5)51 a structural rearrangement occurs in order to both alleviate core strain and fully passivate all phosphorus atoms. Powder X-ray diffraction data is consistent with such a structural rearrangement with a shift in the major reflections to larger angles, potentially indicating a release of compressive strain in the crystallites (Fig. S11, ESI†). Such correlations between surface chemistry and lattice strain have been previously proposed for CdSe and CdS quantum belts.12 If this hypothesis is valid, it serves to highlight the complementary information available from NMR and UV-Vis spectroscopy for In37P20(O2CCH2C6H5)51.
We hypothesize that the number of atoms removed from the surface of each cluster is two. At least 6 equivalents of amine are necessary to remove two indium atoms in the form of In(O2CH2C5H6)3(H2NCH2C6H4F)3, and 12 equivalents are necessary to remove 4 indium atoms. Liberation of In(O2CH2C5H6)3(H2NCH2C6H4F)3 was implicated between the addition 6 and 12 equivalents of H2NCH2C6H4F. Taken together, this restricts the number of In(O2CH2C5H6)3(H2NCH2C6H4F)3 molecules liberated to at most 4. Adding free carboxylic acid to amine treated clusters suggested that a fraction of the added amine remains datively bound to the surface of the cluster after removal of In(O2CH2C5H6)3(H2NCH2C6H4F)3, which would not leave enough amine to account for the removal of four indium atoms (Fig. S12, ESI†). In16 is the most likely candidate for removal and is chemically indistinguishable from In27 prior to addition of amine. If amine is capable of removing In16, it is likely that In27 would also be removed upon further addition of amine. The removal of three indium atoms from the cluster surface would leave an insufficient number available to fully passivate 20 phosphorus atoms in a tetrahedral environment, and therefore at most two indium atoms are proposed to be removed prior to rearrangement.
In37P20(O2CCH2C6H5)51 has a metal to pnictogen ratio that is similar to the metal to chalcogen ratio of Cd35Se20, a known magic-size cluster of CdSe.13 The structure of Cd35Se20 is a zincblende, metal-rich tetrahedron in which each Se is tetrahedrally coordinated to four Cd atoms. Given that 35 In atoms are necessary to passivate an In-rich, zincblende regular tetrahedron of InP with 20 P atoms and we observe no evidence for dangling bonds in the final product by UV-Vis spectroscopy, we propose a reaction scheme between In37P20(O2CR)51 and H2NR′ in which two equivalents of In(O2CR)3(H2NCR′)3 are removed from In37P20(O2CR)51 to yield a regular tetrahedron (or a structure that is symmetrically intermediate between the C2 and Td limits) with the formula In35P20(O2CR)45 (Scheme 1). Given the broadness of the solution 31P{1H} NMR data and the broadening we observe in the powder X-ray diffraction data, it seems likely that the structure might be dynamic with multiple species present as an equilibrium mixture.
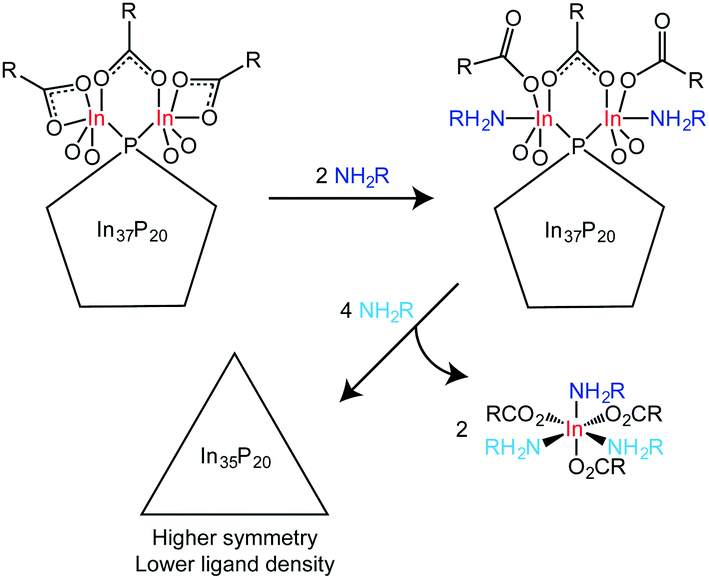 | ||
| Scheme 1 Proposed reaction scheme between In37P20(O2CR)51 and primary amine. | ||
In conclusion, we have explored the reaction of In37P20(O2CR)51 with primary amines via31P{1H}, 19F{1H}, and 1H NMR and UV-Vis spectroscopy. We have determined that addition of up to three equivalents of primary amine results in strong binding of amine to specific indium centres. Removal of fewer than four indium atoms is inferred from the abrupt changes observed in the NMR spectra between 6 and 12 equivalents of amine and the isolation of In(O2CCH2C6H5)3 as a tris(amine) complex from the reaction mixture. Evolution of the inorganic core structure was observed by the 31P{1H} NMR spectroscopy following displacement of In3+. A decrease in ligand density as implicated by the loss of hindered rotation about the methylene and carbonyl carbon atoms in the ligand set was also observed by 1H NMR spectroscopy. These changes were not reflected in the UV-Vis spectroscopy which may be more sensitive to the surface chemistry, especially the presence of under-coordinated indium or phosphorus atoms. The irregular structure observed for the magic-size cluster of InP in the absence of L-type ligands may be due to the preference to fully passivate surface bound indium atoms while simultaneously meeting the demands of charge-balance leading to a dense, interconnected ligand network that distorts the preferred bulk geometry of InP. This would also explain the quasi spherical shape of In37P20(O2CR)51 since this geometry would minimize surface area and thereby maximize the opportunity for bridging binding modes to fully passivate surface bound indium atoms.
This work was supported by the National Science Foundation under grant numbers CHE-1552164 (BMC and DCG – synthesis and characterization), CHE-1565520 and DMR-1408617 (AP, XL – theory). This work was facilitated through the use of advanced computational, storage, and networking infrastructure provided by the Hyak supercomputer system at UW, funded by the Student Technology Fee. We thank Jennifer Stein and Max Friedfeld for the help in acquiring the PXRD data.
References
- R. García-Rodríguez and H. Liu, J. Am. Chem. Soc., 2014, 136, 1968–1975 CrossRef PubMed.
- H. You, X. Liu, H. Liu and J. Fang, CrystEngComm, 2016, 18, 3934–3941 RSC.
- K. Yu, X. Liu, Q. Y. Chen, H. Yang, M. Yang, X. Wang, X. Wang, H. Cao, D. M. Whitfield, C. Hu and Y. Tao, Angew. Chem., Int. Ed., 2014, 53, 6898–6904 CrossRef CAS PubMed.
- K. Kim, D. Yoo, H. Choi, S. Tamang, J.-H. Ko, S. Kim, Y.-H. Kim and S. Jeong, Angew. Chem., Int. Ed., 2016, 55, 3714–3718 CrossRef CAS PubMed.
- R. Xie, D. Battaglia and X. Peng, J. Am. Chem. Soc., 2007, 129, 15432–15433 CrossRef CAS PubMed.
- P. M. Allen, B. J. Walker and M. G. Bawendi, Angew. Chem., Int. Ed., 2010, 49, 760–762 CrossRef CAS PubMed.
- M. Protière and P. Reiss, Chem. Commun., 2007, 2417–2419 RSC.
- L. J. Gooßen, D. M. Ohlmann and P. P. Lange, Synthesis, 2008, 160–164 Search PubMed.
- D. C. Gary, M. W. Terban, S. J. L. Billinge and B. M. Cossairt, Chem. Mater., 2015, 27, 1432–1441 CrossRef CAS.
- D. C. Gary, S. E. Flowers, W. Kaminsky, A. Petrone, X. Li and B. M. Cossairt, J. Am. Chem. Soc., 2016, 138, 1510–1513 CrossRef CAS PubMed.
- N. C. Anderson, M. P. Hendricks, J. J. Choi and J. S. Owen, J. Am. Chem. Soc., 2013, 135, 18536–18548 CrossRef CAS PubMed.
- Y. Zhou, F. Wang and W. E. Buhro, J. Am. Chem. Soc., 2015, 137, 15198–15208 CrossRef CAS PubMed.
- A. N. Beecher, X. Yang, J. H. Palmer, A. L. LaGrassa, P. Juhas, S. J. L. Billinge and J. S. Owen, J. Am. Chem. Soc., 2014, 136, 10645–10653 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental details and additional experimental and computational data and analysis. CCDC 1507652. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc07952k |
| This journal is © The Royal Society of Chemistry 2017 |