
Dynamic covalent single chain nanoparticles based on hetero Diels–Alder chemistry†
Nils
Wedler-Jasinski
ab,
Thorsten
Lueckerath
ab,
Hatice
Mutlu
 ab,
Anja S.
Goldmann
abe,
Andreas
Walther
*c,
Martina H.
Stenzel
*d and
Christopher
Barner-Kowollik
*abe
ab,
Anja S.
Goldmann
abe,
Andreas
Walther
*c,
Martina H.
Stenzel
*d and
Christopher
Barner-Kowollik
*abe
aPreparative Macromolecular Chemistry, Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstr. 18, 76128 Karlsruhe, Germany. E-mail: christopher.barner-kowollik@kit.edu
bInstitut für Biologische Grenzflächen (IBG), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
cInstitute for Macromolecular Chemistry, Albert-Ludwigs-University of Freiburg, Stefan-Meier-Str. 31, 79104 Freiburg, Germany. E-mail: andreas.walther@makro.uni-freiburg.de
dCentre for Advanced Macromolecular Design (CAMD), The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: m.stenzel@unsw.edu.au
eSchool of Chemistry, Physics and Mechanical Engineering, Queensland University of Technology (QUT), 2 George Street, QLD 4000, Brisbane, Australia. E-mail: christopher.barnerkowollik@qut.edu.au
First published on 15th November 2016
Abstract
We introduce the fully reversible folding of single chain nanoparticles (SCNPs) based on covalent hetero Diels–Alder (HDA) chemistry. A cyclopentadiene (Cp)-protected cyanodithioester (CDTE) monomer is designed and copolymerized with methyl methacrylate (MMA) via RAFT polymerization. The polymer chains are folded and subsequently unfolded by exploiting the reversible nature of the HDA reaction.
Single Chain Nanoparticles (SCNPs) are formed by the intramolecular collapse of a single polymer chain via cross-linking to a denser coil and are typically prepared in highly dilute solutions, exploiting the preference of intramolecular over intermolecular cross-linking under these conditions.1 The major long term aim of SCNPs is to mimic natural proteins in their structure and function with fully synthetic polymers, eventually overcoming their fragility and high costs of isolation. The SCNP approach to folded macromolecules suits a broad range of monomers and chemistries, is applicable on relatively large scale and can potentially operate with high molecular weight chains.2–6 The exceptional level of structural control observed in proteins (primary to quaternary structure) is not yet achieved by SCNPs, however, recent all-atom simulations indicate that relatively few folding/cross-linking points can force a chain into a fixed tertiary structure.7
Chain collapses can be classified with regard to the thermodynamic/kinetic stability of the cross-linking points (covalent, dynamic covalent or non-covalent). The collapse can proceed hetero- or homo-functional (depending on the number of different types of involved moieties) or cross-linker-mediated. If several folding mechanisms proceed independently of each other, collapses are termed orthogonal. Furthermore, two general synthetic approaches can be distinguished: in the repeating unit approach, the folding points are randomly distributed along the polymer, whereas selective point folding proceeds between folding points placed in defined positions in the backbone.2,3,8
Non-covalent and dynamic covalent SCNPs potentially respond structurally to external stimuli, due to their reversible cross-linking chemistry. Such systems have been recently reviewed.2,9 Complementary to non-covalent systems that are, in general, prone to rather weak stimuli (e.g. pressure, concentration, solvent) dynamic covalent bonds are broken when specific external stimuli are applied, due to their more robust nature. The most prominent examples for dynamic covalent SCNPs are based on disulfides (redox stimulus),10–13 hydrazones (exchange + pH stimulus),14,15 enamines (exchange + pH stimulus)9,16,17 and coumarins (photo stimulus).18,19
Since dynamic covalent and non-covalent SCNPs are dense polymer coils with precisely arranged functional groups that respond structurally to one or more chosen external trigger(s), important applications of dynamic SCNPs lie in sensing,20 nanomedicine9 and catalysis.21–23
(Hetero) Diels–Alder (HDA) reactions typically exhibit good yields and tolerance towards other functional groups under ambient conditions. The most important feature in the context of modern polymer chemistry is, however, the often dynamic (thermo-)reversible nature of adduct formation, which can be employed in reversible polymers/networks and self-healing materials.24,25 Thiocarbonyls possess lower LUMO energies than their oxygen analogues, making them less stable in the presence of nucleophiles yet, on the other hand, enable milder HDA conditions.
Dithioesters are relatively stable thiocarbonyls and, if substituted with electron donating groups, can act as RAFT agents. Substituted with electron withdrawing groups (e.g. CN), they readily react with a broad spectrum of dienes but, on the other hand, have to be protected with Cp to prevent dimerization in the presence of nucleophiles.26 The protective Cp group can be subsequently expelled from the cyanodithioester (CDTE) by heating in the presence of dienes that form more stable adducts (e.g. sorbic alcohol).27
Herein, we report a dynamic covalent, cross-linker-mediated chain collapse (SCNP formation) followed by complete re-opening of the SCNP coil, combining for the first time the reversible nature of the HDA reaction of CDTE with Cp and sorbic alcohol (SorbOH) in SCNPs. Thereby, we critically add to the existing dynamic covalent chemistries for SCNP formation and establish a new combined stimulus (diene + temperature) for reversible SCNPs formation.
We introduce a new Cp-protected CDTE-monomer (3), which is copolymerized with methyl methacrylate (MMA) in a RAFT process (P1A-C) and in a conventional free radical copolymerization (P4) in different monomer ratios to obtain SCNP precursors with different molecular weights (P4 > P1A > P1B ≈ P1C). These open chain precursors (P1A-C, P4) are heated in dilute solution with a sorbic bi-linker (4) resulting in SCNPs (P2A-C, P5). One SCNP (P2A) is transformed back into an open chain state (P3) by substitution of the sorbic bi-linker (4) with SorbOH, exploiting the reversible nature of HDA chemistry. The chain collapse and opening are monitored by size exclusion chromatography (SEC), 1H-NMR and Dynamic Light Scattering (DLS).
The CDTE-SorbOH-couple employed for the SCNP formation represents the first example of fully reversible SCNPs on the basis of HDA ligation (Fig. 1).
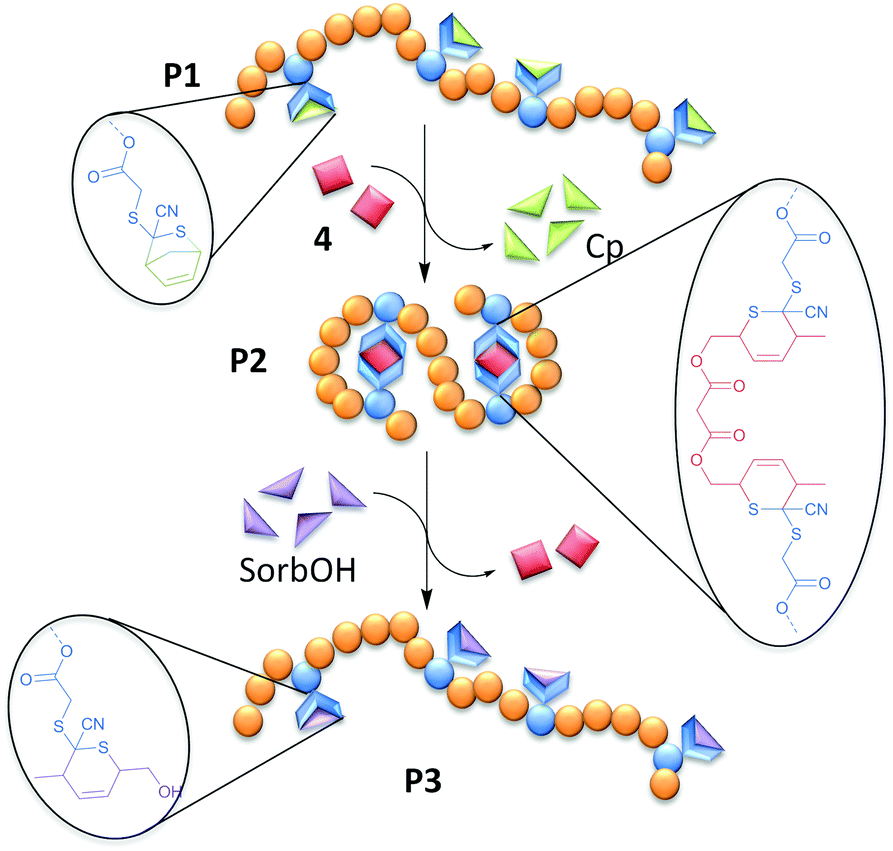 | ||
| Fig. 1 Overview of the dynamic covalent folding and unfolding of SCNPs based on HDA chemistry. | ||
To fabricate reversible SCNPs (Scheme 1), the protected CDTE monomer (3) was synthesized starting with the esterification of (hydroxyethyl)methacrylate (HEMA) with bromoacetyl bromide, resulting in the bromide-functional HEMA 1 (yield: 44%). Intermediate 1 was subjected to a nucleophilic substitution with sodium carbonocyanidodithioate (2), which was synthesized according to Oehlenschlaeger et al.,27 followed by in situ trapping of the free CDTE with freshly distilled Cp in one pot (yield: 42%). Cp-protected CDTE monomer (3) was copolymerized with methyl methacrylate (MMA) in a RAFT process controlled by 2-cyano-2-propyl benzodithioate (CPBD). Comonomer ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 and 1:9 (3:MMA) were selected for the polymerization, and the same ratios have been found in the isolated precursors (P1A-C) verified by 1H-NMR analysis. Two molecular weights have been prepared with a ratio of 1:5 (13.8 kDa for P1A, 9.8 kDa for P1C) and one with 1:9 (10.0 kDa for P1B). In addition, a long precursor (47.8 kDa) with a ratio of 1:5 was synthesized via conventional free radical copolymerization (P4). The sorbic bi-linker (4) was synthesized as cross-linking agent for SCNP formation in one step via esterification (Scheme 1) (yield: 48%).
:5 and 1:9 (3:MMA) were selected for the polymerization, and the same ratios have been found in the isolated precursors (P1A-C) verified by 1H-NMR analysis. Two molecular weights have been prepared with a ratio of 1:5 (13.8 kDa for P1A, 9.8 kDa for P1C) and one with 1:9 (10.0 kDa for P1B). In addition, a long precursor (47.8 kDa) with a ratio of 1:5 was synthesized via conventional free radical copolymerization (P4). The sorbic bi-linker (4) was synthesized as cross-linking agent for SCNP formation in one step via esterification (Scheme 1) (yield: 48%).
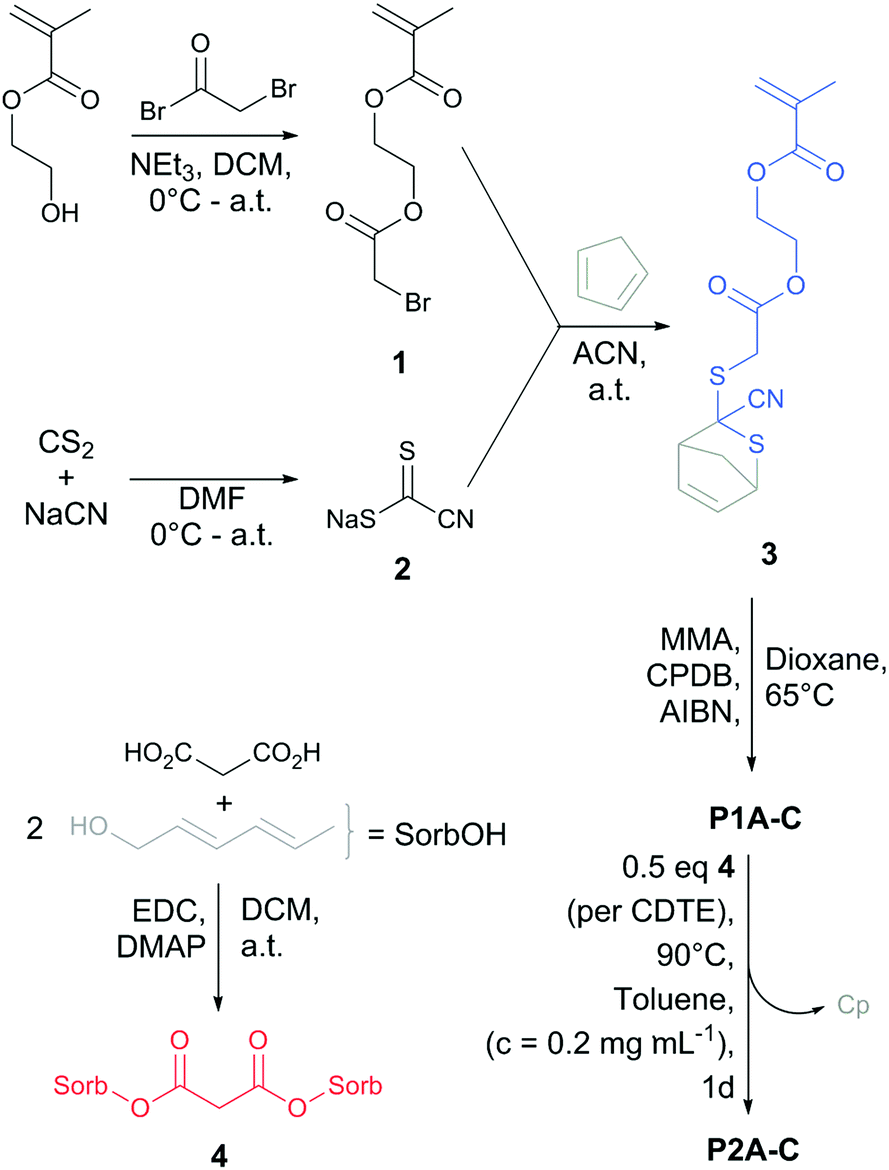 | ||
| Scheme 1 Synthetic route for the preparation of the CDTE monomer 3, sorbic bi-linker 4 and their roles in dynamic covalent SCNP formation (P2A-C). | ||
For the SCNP folding, the linear precursors (P1A-C, P4) were dissolved in toluene (cpolymer = 0.2 mg mL−1) and heated with 0.5 eq. (per CDTE) of the sorbic bi-linker (4) in dilute solution, allowing the sorbic bi-linker (4) to substitute Cp from the protected CDTEs in the polymer backbone of P1A-C. The formation of the more stable HDA adduct of CDTE with 4 resulted in cross-linking between pairs of CDTE in the polymer backbone, forcing the chains in a collapsed state (P2A-C). SCNP P5 was synthesized in an analogue procedure employing P4 as linear precursor.
Applying the reversible nature of HDA chemistry to SCNPs, the sorbic bi-linker (4) was expelled from the CDTEs in the folded chain state of P2A by subsequent reaction with 5 eq. SorbOH in toluene (cpolymer = 0.2 mg mL−1) at 90 °C for 2 d towards an unfolded chain state (P3).
The SEC data of the chain collapses of the linear precursors (P1A-C, P4) to the corresponding SCNPs (P2A-C, P5) and the unfolding of P2A to P3 are summarized in Table 1. Detailed comparative chromatograms of the sequence P1A to P3 are depicted in Fig. 2b, the sequence of the precursors P1B,C is provided in Fig. 3 and the folding of P4 towards P5 is shown in Fig. S9 (ESI†). Further, the kinetics of folding and unfolding of the sequence P1A–P3 are provided in Fig. S6 (ESI†).
| Collapse | MMA:CDTE |
Precursora (kDA, Đ) | Producta (kDa, Đ) | ΔMP,abs (kDa) | ΔMP,rel (%) |
|---|---|---|---|---|---|
| a Size Exclusion Chromatography (SEC) data: MP and Đ. b Synthesized via conventional free radical polymerization. c Unfolding: substitution of bi-linker-CDTE adduct with SorbOH-CDTE adduct. | |||||
| P1A to P2A | 5:1 |
13.8, 1.3 | 11.7, 1.2 | −2.1 | −15 |
| P1B to P2B | 9:1 |
10.0, 1.3 | 9.0, 1.2 | −1.0 | −10 |
| P1C to P2C | 5:1 |
9.8, 1.3 | 8.3, 1.2 | −1.5 | −15 |
| P4 to P5b | 5:1 |
47.8, 1.6 | 38.5, 1.5 | −9.3 | −19 |
| P2A to P3c | 5:1 |
11.7, 1.2 | 14.0, 1.2 | +2.3 | |
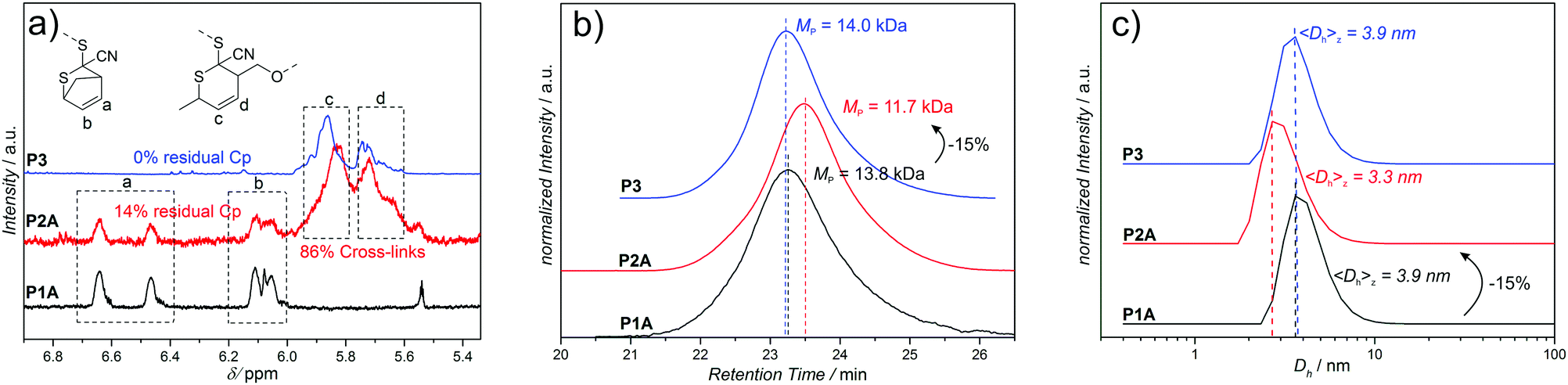 | ||
| Fig. 2 Folding and un-folding sequence of P1A to P3 monitored by (a) 1H-NMR in CDCl3 (6.8–5.4 ppm), (b) SEC in DMAc and (c) DLS in DMSO. Residual Cp-protection of P2A is calculated from the relative integrals of protons a, b, c and d. | ||
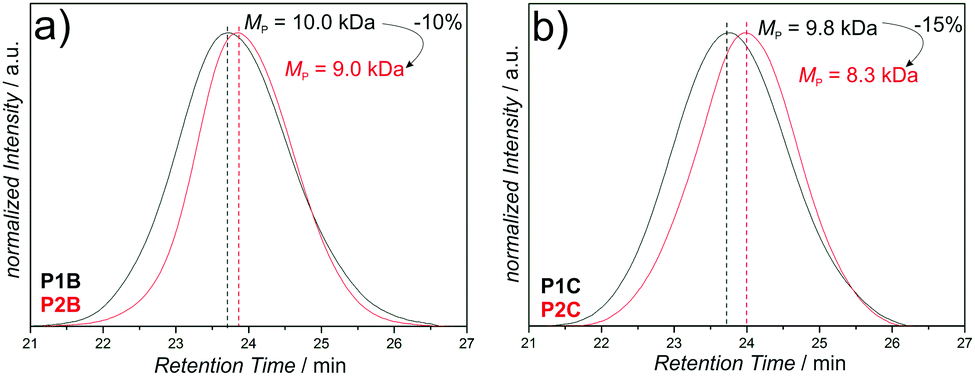 | ||
| Fig. 3 SEC analysis of the folding of P1B (a) and P1C (b). | ||
The SCNPs (P2A-C, P5) show all expected characteristics of single chain folding regarding the influence of cross-linking density and chain length effects on the folding behavior, as well as decrease in dispersity (Table 1): higher amounts of CDTE in the polymer backbone at constant molecular weight lead to larger absolute collapses, as evidenced by P2B (MMA:CDTE = 9:1, −1.0 kDa) and P2C (MMA:CDTE = 5:1, −1.5 kDa) compared with their linear precursors. Longer precursor chains exhibiting similar cross-linker ratios show larger absolute and relative collapses than their shorter analogues, as indicated by P2A (−2.1 kDa, −15%), P2C (−1.5 kDa, −15%) and P5 (−9.3 kDa, −19%). Moreover, all SCNPs independently of the molecular weight or MMA:CDTE ratio show a slight decrease in dispersity during the folding process.
To demonstrate the reversibility of this approach towards dynamic covalent SCNPs, P2A is opened employing the retro-HDA reaction with SorbOH, substituting the cross-linker from the CDTE moieties and introducing OH-functionalities resulting in an open chain (P3). Detailed 1H-NMR, SEC and DLS data of the folding and opening sequence of P1A to P3, reveal the smooth folding and opening of the polymer chain (Fig. 2).
SEC analysis (Fig. 2b) evidences that the hydrodynamic volume of P2A reverts back to the value of the initial precursor before SCNP formation (P1A) during unfolding towards P3. The apparent (peak) molecular weights indicate that the folding of the linear precursor P1A (MP = 13.8 kDa) to the SCNP P2A (MP = 11.7 kDa), corresponding to an apparent decrease in molecular weight of −2.1 kDa (15%), is reversible, since the unfolded SCNP P3 (MP = 14.0 kDa) shows a comparable value to precursor P1A.
1H-NMR analysis (Fig. 2a) of each chain state verifies the presence of the magnetic resonance signals of Cp-protected CDTE protons a (6.4–6.7 ppm) and b (5.9–6.2 ppm) for the open chain precursor P1A, indicating that the protecting group is preserved during polymerization. Comparison of the integral values a and b of the remaining Cp-protected CDTE and the protons c and d (5.5–6.0 ppm), arising from the adduct with 4, reveals that in the SCNP P2A the majority (86%) of CDTE has formed cross-links. Residual Cp-protection (14%) most probably originates from the chain collapse which shields/prevents these remaining Cp-protected CDTE groups inside the SCNP from reaction with 4 (other kinetic effects are excluded, since folding kinetics show no further shift after 1 d). After substitution of bi-linker (4) with neat SorbOH, the magnetic resonances signals arising from protons c and d remain unchanged, due to the identical structure of the adducts. The residual Cp-protection of P2A, however, disappears completely, because of the lack of shielding effects in the open chain state of P3. Parallel and congruent DLS measurements (Fig. 2c) confirm the reversible reduction of chain volume observed in SEC analysis (Fig. 2b): during the folding of P1A (3.9 nm) to the corresponding SCNP P2A (3.3 nm), the hydrodynamic diameter decreases by 0.6 nm (15%) supporting the values obtained by SEC analysis. The unfolding of SCNP P2A to P3 (3.9 nm) results in a shift back towards the value of the initial precursor polymer P1A, further confirming the SEC data. Moreover, the SEC folding kinetics of P1A to P3 (Fig. S6, ESI†) show that folding is faster (1 d) than unfolding (2 d) under the given conditions, indicating that the cross-linkable CDTE groups are indeed harder to access in a collapsed chain than in an open chain (steric hindrance). Thereby, the kinetic data also indicate shielding effects, which were observed in 1H-NMR (Fig. 2a). Additional experiments attempting to fold P3 back to the state of P2A, employing bi-linker 4 under similar reaction conditions as in the transformation of P1A to P2A, were unsuccessful. In the first (and successful) folding step towards P2, the Cp-CDTE adduct is substituted with the more stable Sorb-CDTE adduct, serving as driving force for the collapse. During the unfolding of P2A to P3, bi-linker 4 is substituted from the CDTEs by SorbOH unfolding the chain, while none of the CDTE adducts is favored. In case of the unsuccessful folding of P3 back to P2A (employing bi-linker 4), we hypothesize that the missing driving force is responsible for the lack of collapse.
In summary, we combine dynamic HDA chemistry based on CDTE, Cp and sorbic alcohol resulting in reversible chain folding based on dynamic covalent bonds in a cross-linker-mediated repeating unit approach. A new Cp-protected CDTE methacrylate (3) is synthesized for the first time in three steps and copolymerized with MMA in a RAFT process and conventional free radical polymerization resulting in a library of precursor polymers (P1A-C, P4) that are subject to single chain collapses with a new sorbic bi-linker (4). The successful collapses (P2A-C, P5) are studied systematically regarding chain length and comonomer ratios and one SCNP was transferred back to an open chain state (P3) exploiting the reversible nature of HDA chemistry, monitored by SEC, DLS and 1H-NMR analysis.
C. B.-K. and A. W. acknowledge financial support from the Volkswagen Stiftung in the ‘Integration of Molecular Components in Functional Macroscopic Systems’ Program. C. B.-K. additionally acknowledges continued funding by the Karlsruhe Institute of Technology (KIT) in the context of the Helmholtz BioInterfaces in Technology and Medicine (BIFTM) and Science and Technology of Nanosystems (STN) programs as well as from the Queensland University of Technology (QUT). Additional support in the context of the SFB 1176 (project A1) is gratefully acknowledged by C. B.-K. and M. H. S. Dr Pavel Levkin is acknowledged for providing access to the DLS instrument.
References
- V. W. Kuhn and H. Majer, Die Makromol. Chem., 1956, 18, 239–253 CrossRef.
- S. Mavila, O. Eivgi, I. Berkovich and N. G. Lemcoff, Chem. Rev., 2016, 116, 878–961 CrossRef CAS PubMed.
- O. Altintas and C. Barner-Kowollik, Macromol. Rapid Commun., 2016, 37, 29–46 CrossRef CAS PubMed.
- M. Gonzalez-Burgos, A. Latorre-Sanchez and J. A. Pomposo, Chem. Soc. Rev., 2015, 44, 6122–6142 RSC.
- C. K. Lyon, A. Prasher, A. M. Hanlon, B. T. Tuten, C. A. Tooley, P. G. Frank and E. B. Berda, Polym. Chem., 2015, 6, 181–197 RSC.
- E. J. Foster, E. B. Berda and E. W. Meijer, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 118–126 CrossRef CAS.
- D. Danilov, C. Barner-Kowollik and W. Wenzel, Chem. Commun., 2015, 51, 6002–6005 RSC.
- O. Altintas and C. Barner-Kowollik, Macromol. Rapid Commun., 2012, 33, 958–971 CrossRef CAS PubMed.
- A. Sanchez-Sanchez and J. A. Pomposo, Part. Part. Syst. Charact., 2014, 31, 11–23 CrossRef CAS.
- B. T. Tuten, D. Chao, C. K. Lyon and E. B. Berda, Polym. Chem., 2012, 3, 3068–3071 RSC.
- C. Song, L. Li, L. Dai and S. Thayumanavan, Polym. Chem., 2015, 6, 4828–4834 RSC.
- O. Shishkan, M. Zamfir, M. A. Gauthier, H. G. Borner and J.-F. Lutz, Chem. Commun., 2014, 50, 1570–1572 RSC.
- R. Braslau, F. Rivera Iii and C. Tansakul, React. Funct. Polym., 2013, 73, 624–633 CrossRef CAS.
- D. E. Whitaker, C. S. Mahon and D. A. Fulton, Angew. Chem., Int. Ed., 2013, 52, 956–959 CrossRef CAS PubMed.
- B. S. Murray and D. A. Fulton, Macromolecules, 2011, 44, 7242–7252 CrossRef CAS.
- L. Buruaga and J. A. Pomposo, Polymers, 2011, 3, 1673–1683 CrossRef CAS.
- A. Sanchez-Sanchez, D. A. Fulton and J. A. Pomposo, Chem. Commun., 2014, 50, 1871–1874 RSC.
- J. He, L. Tremblay, S. Lacelle and Y. Zhao, Soft Matter, 2011, 7, 2380–2386 RSC.
- W. Fan, X. Tong, Q. Yan, S. Fu and Y. Zhao, Chem. Commun., 2014, 50, 13492–13494 RSC.
- M. A. J. Gillissen, I. K. Voets, E. W. Meijer and A. R. A. Palmans, Polym. Chem., 2012, 3, 3166–3174 RSC.
- M. Artar, T. Terashima, M. Sawamoto, E. W. Meijer and A. R. A. Palmans, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 12–20 CrossRef CAS.
- S. Basasoro, M. Gonzalez-Burgos, A. J. Moreno, F. L. Verso, A. Arbe, J. Colmenero and J. A. Pomposo, Macromol. Rapid Commun., 2016, 37, 1060–1065 CrossRef CAS PubMed.
- E. Huerta, P. J. M. Stals, E. W. Meijer and A. R. A. Palmans, Angew. Chem., Int. Ed., 2013, 52, 2906–2910 CrossRef CAS PubMed.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- Z. Xu, Y. Zhao, X. Wang and T. Lin, Chem. Commun., 2013, 49, 6755–6757 RSC.
- H. E. Simmons, D. C. Blomstrom and R. D. Vest, J. Am. Chem. Soc., 1962, 84, 4756–4771 CrossRef CAS.
- K. K. Oehlenschlaeger, J. O. Mueller, J. Brandt, S. Hilf, A. Lederer, M. Wilhelm, R. Graf, M. L. Coote, F. G. Schmidt and C. Barner-Kowollik, Adv. Mater., 2014, 26, 3561–3566 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, and mass spectra. See DOI: 10.1039/c6cc07427h |
| This journal is © The Royal Society of Chemistry 2017 |