
Theoretical rationalization for the equilibrium between (μ–η2:η2-peroxido)CuIICuII and bis(μ-oxido)CuIIICuIII complexes: perturbational effects from ligand frameworks†
 b
and
Kazunari
Yoshizawa
*a
b
and
Kazunari
Yoshizawa
*a
Abstract
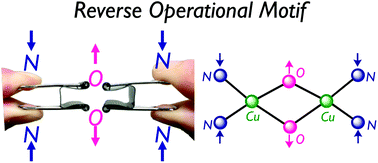
DFT calculations are carried out to investigate the geometric effects of the supporting ligands in the relative energies of the (μ–η2:η2-peroxido)CuIICuII complex 1 and the bis(μ-oxido)CuIIICuIII complex 2. The N3-tridentate ligand bearing acyclic propane diamine framework La preferentially provided 1, whereas the N3-tridentate ligand with cyclic diamine framework such as 1,4-diazacycloheptane Lb gave 2 after the oxygenation of the corresponding CuI complexes as reported previously [S. Itoh, et al., Inorg. Chem., 2014, 53, 8786–8794]. Calculations at the B3LYP*-D3 level of theory can reasonably explain the experimental results in relative energies, structures and harmonic frequencies of 1 and 2. Perturbational effects of the diamine chelates of La and Lb especially on the equilibrium of 1 and 2 are investigated in detail. In the range from 2.30 Å to 3.40 Å of the N–N distance in the diamine moiety, 1 is more stable than 2 by 8.4 kcal mol−1 at the distance of 3.40 Å. Calculated potential energies indicate that the decrease in the N–N distance is associated with a decrease in energy of 2, leading that 2 can be most stabilized at the N–N distance of 2.60 Å. Furthermore, molecular orbitals analyses are performed to explain that the energy gaps between the σ* orbital of the O–O bond and the dx2−y2 orbitals of the CuII ions of 1 get small as the diamine moiety is shrunk, leading to facilitate the O–O bond cleavage from 1 to 2.
 Please wait while we load your content...
Please wait while we load your content...

