
Mechanisms for hydrogen evolution on transition metal phosphide catalysts and a comparison to Pt(111)†

Abstract
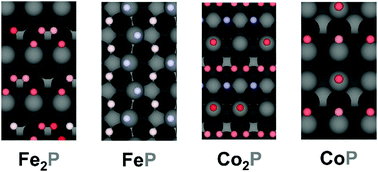
Earth-abundant transition metal phosphides have been demonstrated to be promising alternative catalysts to replace Pt for hydrogen evolution reaction (HER). However, the mechanism for the hydrogen evolution reaction on transition metal phosphides remains unclear. Here, we explore the catalytically active sites and the reaction mechanisms on a variety of model transition metal phosphide surfaces by building cluster expansion models and running Monte Carlo simulations. We demonstrate that the effect of hydrogen coverage, interaction between hydrogen atoms and desorption kinetics all dictate the HER mechanisms and the active sites, and we propose mechanisms that are in good agreement with experimental studies. The present method provides a general and effective way to probe the active sites and study the mechanisms of catalytic reactions, which can facilitate the rational design of highly active electrocatalysts.
- This article is part of the themed collection: 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

