
Band engineering and hybridization of competing arsenene allotropes: a computational study†
 ab
and
Yue
Chen
*ab
ab
and
Yue
Chen
*ab
Abstract
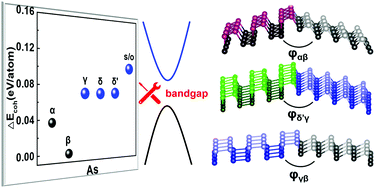
Arsenene is an emerging two-dimensional material similar to its group-V isologue phosphorene. Based on first-principles calculations and finite-temperature ab initio molecular dynamics (AIMD), we investigated the dynamical stability and electronic properties of three competing phases of two-dimensional (2D) arsenic, namely γ-As, the newly discovered δ′-As, and s/o-As. Phonon calculations and AIMD results confirm their dynamical stabilities. It is also found that the band structures of these allotropes can be effectively engineered by changing the number of layers or in-layer strain, realizing direct–indirect bandgap and metal–insulator transitions. The highly tunable electronic structures and the broad range bandgaps evidence potential applications in nanoelectronics and optoelectronics. Moreover, δ′-As is predicted to transform to a higher symmetry δ-As phase when the number of layers is sufficiently large. An intriguing phenomenon unveiled is the potential hybridization of different allotropes at an energy cost lower than 0.02 eV Å−1. This becomes particularly valuable for assembling heterostructures with different well-defined regions in one contiguous arsenene layer.
- This article is part of the themed collection: 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

