
A theoretical benchmark study of the spectroscopic constants of the very heavy rare gas dimers
Abstract
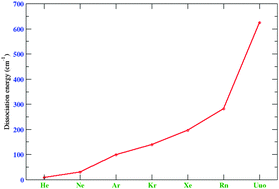
Spectroscopic constants for the homonuclear dimers of the very heavy rare gases radon (Rn) and eka-radon (Uuo) are reported. A computational protocol using the eXact 2-Component molecular-mean field Hamiltonian has been established based on extensive calculations of the xenon dimer. We find that reliable results require CCSD(T) calculations at the extrapolated basis set limit. In this limit counterpoise corrected results are closer to experimentally derived values than uncorrected ones. Furthermore, in an attempt to reduce the computational cost while retaining very high accuracy, we studied the performance of range-separated density functional theory. Although we observe a somewhat more favorable basis set convergence and reduced importance of connected triples by range-separated methods compared to pure wave function theory, in practice we have to employ the same computational protocol for obtaining converged results. At the Dirac–Coulomb level we find an almost fourfold increase of binding energy when going from the radon to the eka-radon dimer, but the inclusion of spin-other orbit interaction reduces the dissociation energy of the heaviest dimer by about 40%.
 Please wait while we load your content...
Please wait while we load your content...

