
Core-ligand modulation alters core–shell coordination to produce stable supercooled phase-change materials for long-term heat storage and release
Yuchen
Pan
,
Tianle
Cheng
,
Rui
Hu
,
Xusheng
Zhang
,
Zeyu
Niu
,
Zeyan
Dong
,
Dong
He
,
Siyuan
Tang
 and
Zhubing
He
*
and
Zhubing
He
*
Department of Materials Science and Engineering, Shenzhen Key Laboratory of Full Spectral Solar Electricity Generation (FSSEG), Southern University of Science and Technology (SUSTech), No. 1088, Xueyuan Rd., Shenzhen, 518055, Guangdong, China. E-mail: hezb@sustech.edu.cn
First published on 11th November 2025
Abstract
Hierarchical core–shell coordinated metal–organic networks came out recently as promising supercooled phase-change materials (SPCMs) that achieve both stable energy storage and controllable release for long-term energy storage and release. However, these new materials need further development, especially regarding the supercooling stability of the high-enthalpy composition. Here, we report a Mn-acetamidomethanol@erythritol (MA@Er) core–shell SPCM with high erythritol load achieved through core ligand modulation, simultaneously achieving both high supercooling stability and controllable crystallization. As revealed, the hydroxylic group of acetamidomethanol interacts with the erythritols in the core–shell, leading to a thermodynamic energy barrier over 5 times higher than the currently reported counterparts, resulting in only 7.8% loss of enthalpy after 1000 successive thermal charge–discharge cycles. MA@Er, with an enthalpy of 161 kJ kg−1, can release its latent heat to attain a temperature peak of 71.7 °C shortly after being triggered by a small shear stress of 8 Pa. Moreover, its thermal energy utilization efficiency reaches 86.3% when consuming low-grade waste heat. MA@Er core–shell SPCMs demonstrate great potential for practical utilization of low-grade thermal energy in the long-term mode.
1 Introduction
Global decarbonization urgently requires efficient heat storage solutions to manage low-grade thermal energy, which includes over 60% of global thermal management of industrial, transport and residential sectors.1,2 Regarding the spatial and time matching requirements of heat supply and demand,3 supercooled phase-change materials (SPCMs) provide reversible latent heat exchange with a time delay that can address these issues.Current SPCMs face various technical challenges. Hydrated salts such as sodium acetate trihydrate, sodium thiosulfate pentahydrate, and calcium chloride hexahydrate possess high volumetric energy storage density, but their irreversible phase precipitation and unstable supercooling behavior severely limit their practical application.4,5 In comparison, sugar alcohols demonstrate good potential owing to their high latent enthalpy and high supercooling degrees while exhibiting distinct crystallization behaviors. The dense hydrogen-bond network causes high viscosity of xylitol and sorbitol, which possess high supercooling stability, but the release of heat by crystallization is difficult due to the overlarge activation energy barriers.6,7 In contrast, erythritol is prone to crystallize spontaneously, although it has a high supercooling degree owing to its lower density of hydrogen-bond networks.8–10 To achieve controllable crystallization for latent heat release, efforts have focused on erythritol in the past decade.11–14 Such polymers as polyvinyl alcohol and polymeric absorbent resin, were employed to suppress the erythritol aggregation for easy crystallization through separating them by hydrogen-bond networks, but cause the difficult triggerability of crystallization.12,13 Alkali hydroxides were used to modulate the hydrogen-bond interactions to enhance the nucleation barrier; however, they suffer from poor triggering efficiency due to the additional energy required to initiate crystallization.11 Other additives, such as carrageenan, have been used to increase the material's viscosity, thereby retarding crystallization, and a large shear force or energy input is required to trigger their crystallization.14 In reality, the balance between supercooling stability and triggerability seems to be an insurmountable challenge for small-molecule-based supercooled phase-change materials in their application for long-term heat storage and release.
Recently, our group reported a new material system based on an innovative core@shell coordination structure of manganese-methylurea@erythritol supercooled liquid for controllable heat storage and release, successfully addressing the intrinsically contradictory issue of supercooling stability and triggerability of purely organic molecules.15 Depending on the synergistic effect of both metal–organic coordination and organic–organic hydrogen bonds, the new material can exhibit both high thermodynamic stability and triggerability. This was an opening work and still needed further development. One direction is to enhance the supercooling stability, especially for materials with high erythritol loads, because higher loads enable higher energy storage capacity. The ligand in the core is the critical point that determines the supercooling stability for the material system.
To address the above issues, we employed acetamidomethanol as the core ligand to successfully synthesize Mn-acetamidomethanol@erythritol (MA@Er) core–shell SPCMs with high erythritol loads and significantly enhanced supercooling stability. Compared with the easily spontaneous crystallization of manganese-methylurea@erythritol, MA@Er demonstrates significantly enhanced supercooling stability at the same erythritol load. Its nucleation energy barrier is over 5 times higher than that of the methylurea-based material. Furthermore, MA@Er exhibits an outstanding cycling durability, retaining 92.2% of its initial enthalpy after 1000 successive charge–discharge cycles. Based on comprehensive characterizations, the remarkable enhancement in supercooling stability can be attributed to the core–shell interactions of the hydroxylic group of the acetamidomethanol with erythritols, more than the effect of the amine group of methylurea. In addition to the systematic endothermal and exothermal tests, the thermodynamics and kinetics of the MA@Er materials were also well explored.
2 Results and discussion
2.1 Structure and properties of the Mn-acetamidomethanol (MA) core
Fig. 1ashows the chemical structure and electrostatic potential of acetamidomethanol, where the carbonyl oxygen exhibits electronegativity due to its lone pair of electrons. One mole of manganese chloride was dissolved in four moles of molten acetamidomethanol, yielding a glassy solid upon natural cooling, which is denoted as MA (Fig. 1b). MA appears brown, corresponding to its transmission spectrum shown in Fig. 1c. This can be attributed to its distinctive absorption bands at 420 and 660 nm, which are ascribed to the 6A1g(S) → 4T1g(4G) and 6A1g(S) → 4Eg(4D) transitions.16 Thermogravimetric analysis (TGA) revealed that the decomposition temperature of MA increased from 130 °C (for pure acetamidomethanol) to 155 °C (Fig. 1d). The X-ray diffraction (XRD) pattern of MA shows that it is amorphous at room temperature (Fig. 1e), although both acetamidomethanol and manganese chloride have transition temperatures (Tg) of −2 °C (Fig. 1f). The cooling curve (black line) further proves that its glass state is stable even below −60 °C. The melting point (Tm) of crystalline MA was determined to be 45 °C, as shown in the red DSC curve in Fig. 1f. The Tg/Tm ratio was 0.85, while that of Mn-methylurea was 0.75,15 significantly higher than the 2/3 threshold of the Kauzmann criterion, indicating its high glass-forming ability (GFA) and strong resistance to crystallization.17,18 The viscosity-temperature behavior was fitted using the Vogel–Fulcher–Tammann (VFT) equation, showing a good consistency (Fig. 1g).19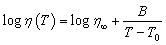 | (1) |
 | ||
| Fig. 1 Property and structure characterization of the MA glass. (a) Acetamidomethanol chemical structure and electrostatic potential diagram. (b) Schematic of the synthesis of MA. (c) UV-vis transmittance spectra of MA. (d) TGA profile of MA. (e) XRD patterns of MnCl2, acetamidomethanol, and MA. (f) Differential scanning calorimetry (DSC) traces for crystalline MA (red line for heating and blue line for cooling) and glassy MA (gray line for heating) at 10 K min−1. (g) Angell plot showing the fragility of MA alongside SiO2. The solid lines represent a fit curve to the measured temperature-dependent viscosity. Reproduced with permission,15 copyright 2025, Wiley. (h) Fourier transform infrared (FTIR) spectra of acetamidomethanol and MA at ambient temperature. (i) Density functional theory (DFT) calculation results for the MA spatial structure. | ||
FTIR analysis revealed a red shift in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration peak (from 1651 to 1632 cm−1) of acetamidomethanol after coordination (Fig. 1h), attributed to the weakening of the CO bond through σ coordination with Mn. The C–O peak (1016 cm−1) exhibited a blue shift, resulting from coordination-induced hydrogen bond rearrangement.22,23 Based on the above understanding, the coordination structure of MA was revealed by DFT-PBE calculations (Fig. 1i). Mn2+ ion adopts an octahedral configuration coordinated by four acetamidomethanol molecules and two Cl− ions.24,25 The relatively weak Mn–Cl bonds can be readily replaced by the incoming strong-field ligands. These asymmetrical interaction in MA allows the larger moving degree of freedom increases the structure disorder and finally raises the GFA of MA.26
O stretching vibration peak (from 1651 to 1632 cm−1) of acetamidomethanol after coordination (Fig. 1h), attributed to the weakening of the CO bond through σ coordination with Mn. The C–O peak (1016 cm−1) exhibited a blue shift, resulting from coordination-induced hydrogen bond rearrangement.22,23 Based on the above understanding, the coordination structure of MA was revealed by DFT-PBE calculations (Fig. 1i). Mn2+ ion adopts an octahedral configuration coordinated by four acetamidomethanol molecules and two Cl− ions.24,25 The relatively weak Mn–Cl bonds can be readily replaced by the incoming strong-field ligands. These asymmetrical interaction in MA allows the larger moving degree of freedom increases the structure disorder and finally raises the GFA of MA.26
2.2 Structure analysis of MA@Er SPCM
Although it possesses a high latent enthalpy of 330 kJ kg−1 and a large supercooling degree, erythritol undergoes spontaneous crystallization due to its weak hydrogen bonding.10 In the core–shell structure of Mn@Er SPCMs, the elongated Mn–Cl bonds in the MA glass core provide two dynamic sites to coordinate two erythritol molecules, while the excess erythritol molecules surround the MA core to form a shell through hydrogen bonding interactions.15 The color changes from brown for the MA core to shallow pink for MA@Er (Fig. S1).In the manganese-methylurea@erythritol materials system, an obvious cold crystallization occurred in the DSC cycle of MME3 sample, announcing it is potentially unstable for the composition with Mn:erythritol molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 although its latent heat enthalpy is as high as 208.5 kJ kg−1.15 To ensure high enthalpy through a high erythritol load, we synthesized MA@Er SPCMs with Mn to erythritol molar ratios of 1:8 (MA@Er1), 1:10 (MA@Er2), 1:12 (MA@Er3), 1:14 (MA@Er4), and 1:16 (MA@Er5). As summarized in Table 1, we compared the enthalpy and supercooling stability of MA@Er SPCMs based on the same molar ratio as the MME series in our former work.15 Because the erythritol weight percentage (wt%) of MA@Er is lower than its MME counterpart due to the larger molecular mass of acetamidomethanol, their corresponding weight-based enthalpy is slightly lower. However, the supercooling stability of the MA@Er series is greatly superior to that of MME. Taking the molar ratio of 1:16 as an example, MA@Er5 will not crystallize for one day, while MME4 starts crystallizing after one hour, as evidenced by the evolution pictures recorded in Fig. S2. After one hour, several crystalline flakes appeared in MME4 while MA@Er5 remained clean. After one day, MME4 was almost fully crystallized, while MA@Er5 had some crystallized flakes inside. That enhancement can be attributed to the stronger interaction between acetamidomethanol and erythritol in the outer layer than methylurea. Among these MA@Er SPCMs from MA@Er1 to MA@Er5, MA@Er1 with that ratio of 1:8 and remained stable after 120 days of storage without any obvious change is expected to own considerably 160 kJ kg−1 and was selected to analyze structural characterization (Fig. S3).
:12 although its latent heat enthalpy is as high as 208.5 kJ kg−1.15 To ensure high enthalpy through a high erythritol load, we synthesized MA@Er SPCMs with Mn to erythritol molar ratios of 1:8 (MA@Er1), 1:10 (MA@Er2), 1:12 (MA@Er3), 1:14 (MA@Er4), and 1:16 (MA@Er5). As summarized in Table 1, we compared the enthalpy and supercooling stability of MA@Er SPCMs based on the same molar ratio as the MME series in our former work.15 Because the erythritol weight percentage (wt%) of MA@Er is lower than its MME counterpart due to the larger molecular mass of acetamidomethanol, their corresponding weight-based enthalpy is slightly lower. However, the supercooling stability of the MA@Er series is greatly superior to that of MME. Taking the molar ratio of 1:16 as an example, MA@Er5 will not crystallize for one day, while MME4 starts crystallizing after one hour, as evidenced by the evolution pictures recorded in Fig. S2. After one hour, several crystalline flakes appeared in MME4 while MA@Er5 remained clean. After one day, MME4 was almost fully crystallized, while MA@Er5 had some crystallized flakes inside. That enhancement can be attributed to the stronger interaction between acetamidomethanol and erythritol in the outer layer than methylurea. Among these MA@Er SPCMs from MA@Er1 to MA@Er5, MA@Er1 with that ratio of 1:8 and remained stable after 120 days of storage without any obvious change is expected to own considerably 160 kJ kg−1 and was selected to analyze structural characterization (Fig. S3).
| Molar ratio of core:erythritol | Material | Weight percentage of erythritol (wt%) | ΔH (J g−1) | Supercooling stability |
|
|---|---|---|---|---|---|
| 1:8 |
MA@Er1 | 66.95 | 161 | 120 day | 47.5 |
| MME2 | 69.83 | 163.1 | 60 day | 8.6 | |
| 1:12 |
MA@Er3 | 75.24 | 197 | 7 day | 36.6 |
| MME3 | 77.64 | 208.5 | 3 day | 7.3 | |
| 1:16 |
MA@Er5 | 80.21 | 210 | 1 day | 25.2 |
| MME4 | 82.23 | 215.7 | 1 hour | 5.4 |
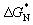
Once triggered mechanically, MA@Er1 transforms from amorphous to crystalline (c-MA@Er1) and from transparent to opaque in color, as shown in Fig. 2a, which was attributed to light scattering caused by erythritol crystals (Fig. 2b). Temperature-dependent in situ XRD results (Fig. 2c) confirm that only erythritol crystallization occurs in the above transition.27 Although pure erythritol typically crystallizes near 33 °C due to its weak hydrogen bond network (Fig. S4), no crystallization exotherm was observed during the cooling of MA@Er1, confirming its supercooled liquid behavior at room temperature. This result is attributed to molecular static displacement or orientation distortion, which shifts the Tg of MA@Er1 from −2 °C for the MA core to −28 °C (Fig. 2d).28 Furthermore, the viscosity was found to be reduced to 1440 Pa s (Fig. S5), resulting in a greatly improved triggerability of MA@Er1. Thus, MA@Er1 exhibited remarkable supercooling stability and triggerability, and has an enthalpy as high as 161 J g−1.
 | ||
| Fig. 2 Structure analysis of MA@Er1. (a) Photographs of MA@Er1 and c-MA@Er1 captured within a glass container (20 cm × 20 cm × 1 cm). (b) XRD patterns of MA@Er1, c-MA@Er1, and erythritol. (c) Temperature-dependent in situ XRD analysis of MA@Er1 (induced by a seed crystal). (d) DSC traces for c-MA@Er1 with a heating and cooling rate of 10 K min−1. (e) Fourier transform infrared spectra of MA@Er1 and c-MA@Er1 at ambient temperature. (f) Experimental 1H nuclear magnetic resonance (NMR) spectra of erythritol, MA@Er1 and MA (d6-dimethyl sulfoxide (DMSO), 400 MHz). | ||
FTIR and nuclear magnetic resonance (NMR) analyses were used to explore the chemical interactions in MA@Er1 and c-MA@Er1. In the transition from the crystalline to the supercooled liquid state, the O–H stretching vibration (3230 cm−1) of erythritol was progressively replaced by the N–H and O–H vibration of acetamidomethanol (Fig. 2e and S6). Concurrently, the C–O vibration (1078 cm−1 and 1051 cm−1) of erythritol exhibits a red shift, indicating its departure from the close-packed state and the formation of a coordinate bond with Mn2+.29 This shift also causes a peak shift in the C–H stretching vibration (2900 cm−1 and 430 cm−1) of erythritol. The red shift and broadening of the N–H and O–H peaks in MA@Er1 suggest the presence of hydrogen bonds with the erythritol O–H groups. The peak shift and broadening of active hydrogen in the 1H NMR spectrum (Fig. 2f) further confirm the formation of a hydrogen-bond network among acetamidomethanol, erythritol, and Cl−.30,31 Notably, the larger peak shift of the hydroxyl group in the 1H NMR spectrum confirms the formation of stronger core–shell interactions between erythritol and the hydroxyl group of acetamidomethanol, indicating that the MA@Er SPCMs formed a denser hydrogen-bond network.
2.3 Thermodynamics and kinetics of the phase-transition
As the loads of erythritol increase, samples MA@Er2–MA@Er5 show similar appearances and chemical structures to MA@Er1 (Fig. S7 and S8). In the crystalline state, the lattice filling rate from c-MA@Er1 to c-MA@Er5 increased progressively with erythritol loads (Fig. S9). The enthalpies for MA@Er1-MA@Er5 in the first DSC cycle were 161, 183, 197, 205 J g−1 and 210 J g−1, respectively (Fig. 3a), which agree well with the phase-change enthalpy of the erythritol loads (Table S1) by subtracting the bond energy difference (EMn–O − EMn–Cl). As expected, no exothermic peaks were observed during cooling, confirming that the thermal energy remained stored in the supercooled liquid. | ||
| Fig. 3 Thermodynamics and kinetics of the MA@Er SPCMs crystallization. (a) The first DSC cycle for MA@Er SPCMs with a heating and cooling rate of 10 K min−1. (b) The second DSC cycle for MA@Er3 and MME3 with heating and cooling rates of 10 K min−1. Reproduced with permission,15 copyright 2025, Wiley. (c) The second DSC cycle for MA@Er SPCMs with heating and cooling rates of 10 K min−1. (d) The threshold shear time for shear-induced crystallization in the SPCMs at 30 °C. (d) Temperature-dependent viscosity measurements of the MA@Er SPCMs. (f) Nucleation energy barrier of MA@Er SPCMs and MME3. Reproduced with permission,15 copyright 2025, Wiley. | ||
In contrast to MME3,15 which exhibited cold crystallization peaks during the second DSC cycle,15 MA@Er3 with the same erythritol load maintained a stable supercooled liquid state without crystallization (Fig. 3b), demonstrating the excellent supercooling stability. Additionally, as the erythritol loads increase, the glass transition temperature of the MA@Er SPCMs gradually decreased, leading to a corresponding reduction in the GFA (Fig. 3c). This indicates that the supercooling stability of the MA@Er SPCMs decreased with higher erythritol loads, while its triggerability was enhanced. To evaluate the triggerability, MA@Er SPCMs were placed between a titanium rotor and a base plate with a rough surface at 30 °C (Fig. S10), which provides nucleation sites, significantly lowering the nucleation barrier.32 Under these conditions, the shear-triggering time thresholds for MA@Er1-MA@Er5 were 640, 265, 115, 30 and 27 seconds, respectively (Fig. 3d),33,34 demonstrating that the MA@Er SPCMs exhibit controllable trigger performance while exhibiting higher supercooling stability than MME. Stronger core–shell interactions form a more dense hydrogen-bond network, resulting in higher viscosity for the MA@Er SPCMs compared to the MME system.15 As shown in Fig. 3e, MA@Er3 achieves a viscosity of 1151 Pa s at room temperature, significantly enhancing its supercooling stability, since higher viscosity implies slower relaxation kinetics. This is also reflected in the crystal growth process, as recorded by optical microscopy (Fig. S11). The viscosity decreases with increasing erythritol load, and MA@Er demonstrates a pattern consistent with the supercooling stability sequence derived from the above triggerability analyses. The temperature dependence of viscosity conformed to the VFT equation (Fig. S12). Extrapolating the fitted curve to a viscosity of 1012 Pa s predicted Tg values of −28, −29, −30, −31 and −33 °C for MA@Er1 to MA@Er5, respectively, agreeing well with the DSC results shown in Fig. 3c.
To further evaluate the enhanced supercooling stability in the MA@Er SPCMs, we conducted an in-depth study of their crystallization thermodynamics. According to the classical nucleation theory, the minimum nucleation free energy (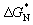 ) can be expressed as:
) can be expressed as:
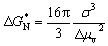 | (2) |
Based on the results of the calculations, Fig. 3f and Table 1 present the nucleation energy barriers for the MA@Er SPCMs. The crystallization energy barriers for each MA@Er SPCM are 47.5 × 10−20, 42.0 × 10−2, 36.6 × 10−20, 31.2 × 10−20 and 25.2 × 10−20 J, respectively. These are approximately 5 times that of the MME counterparts with methylurea as the core ligand. This significant enhancement in energy barrier is attributed to the hydroxyl group of acetylaminomethanol forming stronger core–shell interactions with erythritols, which results in a denser hydrogen-bond network. The nucleation barrier exhibited a decreasing trend with increasing erythritol loading, consistent with the observed variation in supercooling stability across the MA@Er SPCMs.
The crystallization of MA@Er SPCMs can be triggered by a small shear stress of 8 Pa.15,35 This stress disrupts the hydrogen bond network between the erythritol shell and the MA core, thereby initiating a localized crystallization process. The released energy further breaks Mn–O coordination bonds, initiating chain crystallization and releasing the remaining heat.
2.4 Thermal energy storage-release performance
A schematic of the thermal charge–discharge cycling course is shown in Fig. 4a. Crystalline c-MA@Er absorbs waste heat and melts. By natural cooling, the melt MA@Er enters the supercooled liquid state by the release of its sensible heat (Qs). The supercooled MA@Er retains the residual thermal energy stably. Upon application of a low-cost mechanical trigger, the reserved latent heat will be released as QL. In c-MA@Er1, the metal coordination core leads to a significant improvement in the thermal conductivity for erythritol from 0.433 to 0.598 W (m−1 K) (Fig. S13). During thermal charging experiments (Fig. 4b), 200 g of c-MA@Er1 absorbs 71.6 kJ of heat and fully melts within 2 h. Then, the sample cooled naturally to room temperature through the release of 29.6 kJ as Qs. The calculations of heat absorbed and released are described in detail in the SI.36 The cooling curve showed no exothermic peak, indicating a successful transition to the supercooled state and stable latent heat storage. Compared with related SPCMs reported recently, the MA@Er series shows an outstanding triggerability with much lower energy cost (Table S2). A small shear stress of 8 Pa was used to trigger MA@Er1, heating up to 71.7 °C within 693 seconds, and releasing 32.2 kJ as QL (Fig. 4c), which is consistent with the enthalpy of 161 kJ kg−1 determined by its DSC test (Fig. 3a). The released latent heat comprises 45% of the total heat absorbed. Even without rigorous thermal isolation, only 9.8 kJ of heat was dissipated to the environment across the process, and the whole thermal energy utilization efficiency reached 86.3%, which is among the top levels of thermal management systems reported.37,38 The latent heat release course of MA@Er1 was also recorded with an infrared camera (Fig. 4d). Moreover, the MA@Er series can be solar-charged and convert solar energy (Fig. S14), demonstrating its great application potential for future carbon-zero technologies. | ||
| Fig. 4 Thermal energy storage-release of MA@Er. (a) Schematic of MA@Er molecules' arrangement in a thermal charge–discharge cycle with the energy variation. (b) Temperature evolution curve of 200 g MA@Er1 during waste-heat recycling and sensible-heat release processes. (c) Temperature evolution curve of 200 g MA@Er1 during latent heat release process. (d) Time-dependent infrared images of MA@Er1 in the heat-release process triggered by mechanical stirring. (e) Temperature evolution curves in a latent heat release process for 150 g, 100 g and 50 g MA@Er1. (f) The enthalpy loss derived from the DSC curve variation of MA@Er1 across 1000 thermal charge–discharge cycles. | ||
The mass-dependent latent heat release results, ranging from 50 to 200 g, demonstrated that the temperature peak was fixed at 71 °C (Fig. 4e). That agrees well with the Specific Heat Formula (eqn (3)) that their temperature peak is only determined by the latent heat enthalpy (161 kJ kg−1) of MA@Er1.
| Q = cmΔT | (3) |
3 Conclusion
In summary, we successfully synthesized Mn-acetamidomethanol@erythritol (MA@Er) core–shell SPCMs with both high supercooling stability and a facile trigger, for low-grade heat harvesting and release, relying on core ligand engineering. Based on comprehensive characterizations and mechanistic calculations, the thermodynamic energy barriers of MA@Er SPCMs were determined to be 30–50 × 10−20 J, over 5 times higher than the reported methylurea systems, accounting for the high supercooling stability. These results can be attributed to the higher GFA of the MA core and more extensive core–shell interactions between acetamidomethanol and erythritol. The high thermodynamic energy barrier results in a remarkable cyclability, with 92.2% of the enthalpy retained after 1000 cycles. On the other hand, a small shear stress of only 8 Pa can trigger the crystallization of the MA@Er series within hundreds of seconds, releasing the latent heat and attaining a peak temperature of 71.7 °C. The thermal energy utilization efficiency reaches 86.3% when consuming low-grade waste heat. Our MA@Er SPCMs will promote the development of SPCMs for low-grade thermal energy recycling and utilization, especially for long-term storage applications.Author contributions
Z. H. and Y. P. conceived the idea and designed the experiments. Z. H. directed the project. Y. P. synthesized the MA@Er series materials and carried out most of the experiments and characterizations. Y. P. conducted UV-Vis-NIR, DSC, NMR, and FTIR measurements. Z. N. and Z. D. performed SEM measurements. D. H. and S. T. contributed to additional characterizations. Z. H., Y. P., T. C. and R. H. analyzed all the data. Y. P. and T. C. wrote the manuscript. Z. H. revised the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
Original data are available from the corresponding authors upon reasonable request. The data supporting this article have been included as part of the ESI.Acknowledgements
We thank the Department of Core Research Facilities, Materials Science and Engineering and the Center of Computational Science and Engineering of SUSTech for their support with the characterizations and calculations in this work. This work was supported by the Shenzhen Key Laboratory Project (No. ZDSYS201602261933302), the Shenzhen Science and Technology Innovation Committee (No. SGDX20220530111203019) and SUSTech funds (No. G03050K002 and Y01506027).References
- X. Wang, Y.-T. Huang, C. Liu, K. Mu, K. H. Li, S. Wang, Y. Yang, L. Wang, C.-H. Su and S.-P. Feng, Nat. Commun., 2019, 10, 4151 CrossRef CAS
.
- W. Aftab, A. Usman, J. Shi, K. Yuan, M. Qin and R. Zou, Energy Environ. Sci., 2021, 14, 4268–4291 RSC
- C. Liu, Q. Cheng, B. Li, X. Liu and Z. Rao, Renew. Sustain. Energy Rev., 2023, 188, 113805 CrossRef CAS
- W. Hua, X. Lv, X. Zhang, Z. Ji and J. Zhu, J. Energy Storage, 2023, 67, 107378 CrossRef
- M. Dannemand, J. M. Schultz, J. B. Johansen and S. Furbo, Appl. Therm. Eng., 2015, 91, 671–678 CrossRef CAS
- M. Delgado, M. Navarro, A. Lázaro, S. A. E. Boyer and E. Peuvrel-Disdier, Sol. Energy Mater. Sol. Cells, 2021, 220, 110840 CrossRef CAS
- C. G. Fan, G. F. Yuan, Y. Wang, Y. Zhang and Z. F. Wang, Sol. Energy, 2022, 234, 231–239 CrossRef CAS
- S. Shen, S. Tan, S. Wu, C. Guo, J. Liang, Q. Yang, G. Xu and J. Deng, Energy Convers. Manage., 2018, 157, 41–48 CrossRef CAS
- X.-F. Shao, S. Yang, H.-Y. Shi, L.-W. Fan and Y.-P. Yuan, J. Energy Storage, 2023, 64, 107190 CrossRef
- S. Yang, X.-F. Shao, H.-Y. Shi, J.-H. Luo and L.-W. Fan, Sol. Energy Mater. Sol. Cells, 2022, 236, 111538 CrossRef CAS
- X. Li, J. Zhang, Y. Liu, Y. Xu, K. Cui, Z. Yao, B. Fu, C. Song, W. Shang, P. Tao and T. Deng, Chem.–Eng. J., 2023, 452, 139328 CrossRef CAS
- C. Liu, T. Xiao, J. Zhao, Q. Liu, W. Sun, C. Guo, H. M. Ali, X. Chen, Z. Rao and Y. Gu, Renew. Sustain. Energy Rev., 2023, 188, 113814 CrossRef CAS
- M. Pan, D. Wang, L. Wang, N. Dong, H. Xie and W. Yu, Chem.–Eng. J., 2024, 499, 156058 CrossRef CAS
- S. Yang, H.-Y. Shi, J. Liu, Y.-Y. Lai, Ö. Bayer and L.-W. Fan, Nat. Commun., 2024, 15, 4948 CrossRef CAS
- X. Zhang, Z. Du, D. He, Z. Chen, Z. Li, G. Chen, Q. Tan, H. Gao, Z. Niu, S. Ma, T. Cheng, B. Nie, Y. Ding and Z. He, Adv. Mater., 2025, 37, 2412528 CrossRef CAS
- I. Konidakis, C.-P. E. Varsamis, E. I. Kamitsos, D. Möncke and D. Ehrt, J. Phys. Chem. C, 2010, 114, 9125–9138 CrossRef CAS
- T. Ling, P. F. Da, X. L. Zheng, B. H. Ge, Z. P. Hu, M. Y. Wu, X. W. Du, W. B. Hu, M. Jaroniec and S. Z. Qiao, Sci. Adv., 2018, 4, 8 Search PubMed
- K. Ito, C. T. Moynihan and C. A. Angell, Nature, 1999, 398, 492–495 CrossRef CAS
- Q. Zheng and J. C. Mauro, J. Am. Ceram. Soc., 2016, 100, 6–25 CrossRef
- T. D. Bennett, J.-C. Tan, Y. Yue, E. Baxter, C. Ducati, N. J. Terrill, H. H. M. Yeung, Z. Zhou, W. Chen, S. Henke, A. K. Cheetham and G. N. Greaves, Nat. Commun., 2015, 6, 8079 CrossRef CAS PubMed
- J. Orava, A. L. Greer, B. Gholipour, D. W. Hewak and C. E. Smith, Nat. Mater., 2012, 11, 279–283 CrossRef CAS
- J. Kozuch, K. Ataka and J. Heberle, Nat. Rev. Methods Primers, 2023, 3, 70 CrossRef CAS
- P. Hobza and Z. Havlas, Chem. Rev., 2000, 100, 4253–4264 CrossRef CAS
- X.-J. Hou, P. He, H. Li and X. Wang, J. Phys. Chem. C, 2013, 117, 2824–2834 CrossRef CAS
- O. Rojas, G. Mirzoyan, Z. Adamyan, V. V. Papoyan, G. Amatuni and N. Ananikian, Sci. Rep., 2025, 15, 11758 CrossRef CAS PubMed
- Z. Lin, J. J. Richardson, J. Zhou and F. Caruso, Nat. Rev. Chem., 2023, 7, 273–286 CrossRef CAS PubMed
- C. Ceccarelli, G. A. Jeffrey and R. K. McMullan, Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem., 1980, 36, 3079–3083 CrossRef
- V. Nozari, C. Calahoo, L. Longley, T. D. Bennett and L. Wondraczek, J. Chem. Phys., 2020, 153, 204501 CrossRef CAS PubMed
- J. Xue, X. Hua, W. Li, L. Yang, Y. Xu, G. Zhao, G. Zhang, C. Li, K. Liu, J. e. Chen and J. Wu, Carbohydr. Res., 2012, 361, 12–18 CrossRef CAS PubMed
- B. Bernet and A. Vasella, Helv. Chim. Acta, 2000, 83, 995–1021 CrossRef CAS
- M. T. Huggins, T. Kesharwani, J. Buttrick and C. Nicholson, J. Chem. Educ., 2020, 97, 1425–1429 CrossRef CAS
- G. C. Sosso, J. Chen, S. J. Cox, M. Fitzner, P. Pedevilla, A. Zen and A. Michaelides, Chem. Rev., 2016, 116, 7078–7116 CrossRef CAS PubMed
- X.-F. Shao, C.-L. Chen, Y.-J. Yang, X.-K. Ku and L.-W. Fan, Sol. Energy Mater. Sol. Cells, 2019, 195, 142–154 CrossRef CAS
- M. Navarro, Z. Gracia, J. Asín, A. Lázaro, M. Martí and M. Delgado, Sol. Energy Mater. Sol. Cells, 2025, 292, 113803 CrossRef CAS
- J. J. Duffy, A. J. Hill and S. H. Murphy, Appl. Rheol., 2015, 25, 8–13 Search PubMed
-
J. H. Lienhard, A Heat Transfer Textbook: Fifth Edition, Dover Publications, 2019 Search PubMed
- R. A. Kishore, C. Booten, M. V. A. Bianchi, J. Vidal and R. Jackson, Energy Build., 2022, 260, 111937 CrossRef
- X. Chen, H. Gao, Z. Tang, W. Dong, A. Li and G. Wang, Energy Environ. Sci., 2020, 13, 4498–4535 RSC
- B. Androsits, J. Therm. Anal. Calorim., 2000, 61, 331–333 CrossRef
| This journal is © The Royal Society of Chemistry 2026 |