
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanomaterial-enhanced electrochemical biosensors for rifampicin monitoring in serum: towards precision tuberculosis therapy
Rohith
Shetty†
a,
Sudhaunsh
Deshpande†
 a,
Anu Mary
Joy
a,
Ajith Mohan
Arjun
a,
Qianming
Xu
ab,
Alison
Holmes
a and
Sanjiv
Sharma
*a
a,
Anu Mary
Joy
a,
Ajith Mohan
Arjun
a,
Qianming
Xu
ab,
Alison
Holmes
a and
Sanjiv
Sharma
*a
aDavid Price Evans Global Health and Infectious Diseases Group, Pharmacology & Therapeutics, Institute of Systems, Molecular and Integrative Biology (ISMIB), University of Liverpool, 6 West Derby Street, Liverpool L7 8TX, UK. E-mail: sanjiv.sharma@liverpool.ac.uk
bDepartment of Biotechnology and Life Science, Tokyo University of Agriculture and Technology, 2-24-16, Naka-cho, Koganei, Tokyo 184-8588, Japan
First published on 6th November 2025
Abstract
Tuberculosis (TB) treatment is hampered by the pharmacokinetic variability of the cornerstone drug, rifampicin (RIF). This can lead to sub-therapeutic dosing, treatment failure, and the subsequent emergence of drug resistance. Therapeutic drug monitoring (TDM) is essential but is often inaccessible in high-burden, resource-limited settings due to its reliance on slow, expensive, and lab-based techniques like HPLC, while point-of-care systems offer a rapid and low-cost alternative. To address this critical gap, we have developed a low-cost, rapid, and scalable electrochemical biosensor for point-of-care RIF monitoring. The sensor platform integrates a highly selective molecularly imprinted polymer (MIP) with a highly porous gold (HPG) nanomaterial on a disposable printed circuit board (PCB) electrode, costing approximately £0.09 per unit. The HPG layer significantly enhances the electroactive surface area and provides exceptional resistance to biofouling, a critical feature for clinical utility. This allows the sensor to operate directly in complex biological matrices, demonstrating robust performance in undiluted human serum. The sensor achieves a clinically relevant detection range of 8–24 μg mL−1 with a limit of detection (LOD) of 0.848 μg mL−1 and a limit of quantification (LOQ) of 1.31 μg mL−1. This work presents a significant step towards democratizing TDM, offering a practical tool to personalize TB therapy and combat drug resistance at the frontline of patient care.
Introduction
Tuberculosis (TB) is an infectious disease caused by the bacterium Mycobacterium tuberculosis, which primarily infects the lungs but can spread to other organs if untreated. Mycobacterium tuberculosis can be transmitted to the lungs by breathing the airborne droplet nuclei created when an infected person coughs, sneezes, or talks.1 Once inhaled, Mycobacterium tuberculosis survives and replicates within alveolar macrophages. Mycobacterium tuberculosis can escape host immune defences by preventing phagosome–lysosome fusion as well as manipulating host cell apoptosis.2 The immune system holds the infection in a latent stage, but if the immune system wanes, the infection can be reactivated and cause active disease.Tuberculosis (TB), caused by Mycobacterium tuberculosis, remains a preventable and curable yet urgent public-health threat shaped by complex, overlapping, and trans-national epidemiology. In 2021, there were an estimated 10.6 million new cases and 1.3 million deaths, making TB the world's leading infectious cause of mortality; most new cases were concentrated across 30 high-burden countries, particularly in South-East Asia and Africa.3 Multidrug-resistant TB (MDR-TB), driven by mutations that render first-line of defense ineffective, is growing; only ∼40% of people with drug-resistant disease were treated as reported in ref. 2 and 3. Resistance is fueled by inappropriate drug use, erroneous prescribing, poor-quality medicines, or premature treatment interruption and, critically, by suboptimal drug exposure that fails to exert adequate selective pressure, enabling resistant Mycobacterium tuberculosis to survive and expand.4,5 Accurate drug monitoring is therefore not just patient management but a public-health intervention to break the resistance cycle.
Rifampicin (RIF) is the mainstay of TB therapy, dramatically improving treatment success since its introduction in the late 1960s and to shorten treatment regimens from ∼24 months to about 6 months.4,6 With only three truly novel TB drugs approved in the last 50 years (bedaquiline, 2012; delamanid, 2014; pretomanid, 2019), it is critical that RIF is preserved.7–9 The clinical dosages of RIF for children under the ages of 11 months and 17 years are 5–10 mg kg−1 and 10 mg kg−1 (600 mg maximum per dose) twice daily, respectively.10 However, for adults the dose is much higher at 600–1200 mg daily divided into two or four doses.10 The clinical role of RIF is further complicated by its highly variable pharmacokinetics with patients receiving the same dose typically showing wide differences in serum and lesion concentrations stemming primarily from absorption issues, genetic variability (i.e., SLCO1B1 polymorphisms) and powerful cytochrome P450 enzyme induction resulting in major drug–drug interactions.4,11,12 Inadequate exposure due to either under-dosing or poor absorption is associated with poorer clinical outcomes, worse bacteriological outcomes, delayed response and development of resistance. Adverse effects of low serum RIF concentrations have also been described for rpoB mutations.4,5
Therapeutic drug monitoring (TDM), measuring blood concentrations to guide dose adjustments to a therapeutic window, offers a practical route to personalize therapy. Exposure metrics such as the area under the curve (AUC)/minimum inhibitory concentration (MIC) are more predictive of outcomes than prescribed doses, and certain groups (drug-resistant TB, HIV co-infection, and renal/hepatic impairment) may benefit the most.4,13 TDM can enable timely, evidence-based adjustments, potentially preventing resistance, accelerating response, and reducing costs.4
However, widespread TDM is constrained by reliance on high-performance liquid chromatography (HPLC) or liquid chromatography mass spectrometry/mass spectrometry (LC-MS/MS) which is accurate and sensitive, but these are expensive and infrastructure-intensive, and slow workflows from sampling to result can take hours to weeks making them ill-suited to point-of-care use in high-burden settings.4,14 This gap underscores the urgent need for rapid, affordable, and user-friendly alternatives for RIF monitoring.4
Electrochemical biosensors are a very exciting new form of technology that could help solve these problems and provide a path towards real-time, point-of-care measurement of drugs in complex biological fluids like blood or serum.15,16 They are compact, easy to use, and work by converting a specific biological recognition event into a quantifiable electrical response. An electrochemical biosensor will typically consist of a biological recognition event and an electrochemical transducer.15 Biosensors have several advantages like high sensitivity/selectivity, low-cost, rapid analysis time, miniaturization and portability, low-cost and ease of use.15,17 There are a few limitations that prevent biosensors from achieving general clinical adoption. The most common two limitations of biosensors are biofouling, which is the non-specific adsorption of biological materials onto the sensor surface and selectivity, which is the ability to detect RIF in the presence of molecules structurally like RIF and in complex sample matrices.16,18
The primary biorecognition element of this biosensor is a molecularly imprinted polymer (MIP) that is designed for the selective binding of RIF. Although aptamers exhibit high affinity and sequence tunability, their use in TDM is more demanding. Optimization is challenging and surface immobilization adds complexity; moreover, ribonucleic acid (RNA) aptamers are susceptible to nuclease degradation in serum.19,20 In contrast, MIPs are highly chemically stable, stable at high temperatures, reusable due to multiplexing regeneration cycles, do not break down due to enzymatic degradation, and fit the portable point-of-care diagnostics perspective in resource-constrained settings.21–23 We replace costly screen-printed electrodes (SPEs), typically priced at £8 per unit, with custom-designed printed circuit boards (PCBs), which are highly scalable and cost as little as £0.09 per unit. The PCB electrode platform was chosen for its low cost, reproducibility, and scalability in manufacturing electrodes with specific geometries, enabling easy integration into compact diagnostic systems. However, to overcome the electrochemical inactivity of the standard ENIG finish, we introduce a silver barrier layer followed by electroplated gold, ensuring stable electrochemical performance. It is also compact and can be modified easily.24–26 The electrode surface was modified with highly porous gold (HPG) to address the challenge of biofouling, as its high surface-area-to-volume ratio and excellent conductivity enhance the electroactive surface area while reducing non-specific protein adsorption, thereby maintaining accessibility for smaller analytes such as RIF.
The MIP layer was electropolymerized directly onto the HPG-modified PCB electrode, using phenol red as the functional monomer and pyrrole as a co-monomer, in the presence of RIF as the template. Pyrrole was selected for its known properties as a conductive polymer, as well as easy electropolymerisation, and it can also produce polar interactions with the naphthoquinone region of RIF and can form hydrogen bond interactions with the carbons from both hydroxyl and carbonyl groups.27 The electropolymerisation of phenol red adds another redox-active dye polymer that can build a film at a low oxidation potential (∼0.08 V), and this considerably reduces interference from redox processes involving ascorbic acid and paracetamol. Electropolymerisation was conducted in a mixed acetonitrile–PBS solvent mixture that would allow rapid template-monomer complexation while adhering to a physiological pH.
The binding mechanism between RIF and the functional monomers is governed by a synergistic combination of hydrogen bonding between the pyrrole NH group and RIF's carbonyl and hydroxyl groups, along with electrostatic interactions between phenol red's sulfonate groups and the protonatable amine groups of RIF, as shown in Fig. 1. Docking simulations showed affinities ranging from −6.39 to −0.43 kcal mol−1, with most values clustering between −6.3 and −5.0 kcal mol−1. The strongest pose (−6.39 kcal mol−1) and an average of −5.14 kcal mol−1 indicate moderate but stable interactions of RIF with phenol red and pyrrole, while higher values suggest weaker or non-specific binding. Following polymerization, the template is removed, revealing recognition cavities that are complementary in shape, size, and chemical functionality to RIF, ensuring high specificity even in the presence of other structurally similar anti-TB drugs.28 The integration of an antifouling HPG-modified PCB electrode with a chemically specific MIP bioreceptor provides a robust sensing interface, combining the stability of inorganic nanostructures, the molecular selectivity of engineered receptors, and the scalability of PCB technology—critical attributes for developing effective point-of-care TDM systems for tuberculosis (Table 1).
 | ||
| Fig. 1 Schematic illustration of the fabrication process of molecularly imprinted polymers. A RIF-functional monomer cocktail is prepared, where the non-covalent bonds are formed. The functional monomer is then crosslinked to form a rigid structure. The template molecule (rifampicin) is then removed. | ||
| Title | Technique | Dynamic range (reported) | Sample matrix | Ref. |
|---|---|---|---|---|
| An ultrasensitive electrochemical sensing platform based on silver nanoparticle-anchored 3D reduced graphene oxide for rifampicin detection | DPV/modified electrode (Ag NPs/3D rGO) | 0.01–45 μM (linear) | Human blood, pharmaceutical samples, aquatic products | 29 |
| A highly sensitive and ecofriendly assay platform for the simultaneous electrochemical determination of rifampicin and isoniazid | DPV on Ni(OH)2@rGO-modified SPCEs | RIF: 1.0 × 10−9–2.0×10−7 M and 2.0 × 10−7–5.0 × 10−5 M (two linear ranges); LOD ≈ 3.0 ×10−9 M | Human serum and pharmaceutical formulations | 30 |
| An electrochemical sensor based on the composite of molybdenum carbides and MWCNT-modified electrodes for ultrasensitive detection of rifampicin | Voltammetric sensor (MWCNT-Mo2C composite on a GCE) | (Reported as ultrasensitive; calibration reported in the paper—LOD at low nM μM−1 levels depending on the technique) | Pharmaceutical formulations/model solutions | 31 |
| Electrochemical determination of rifampicin based on its oxidation using multi-walled carbon nanotube-modified glassy carbon electrodes | CV, DPV, and SWV on MWCNT-modified GCEs | 0.04–10 μM (linear); LOD (DPV) ≈ 7.51 nM | Pharmaceutical dosage forms (tablets), buffer | 32 |
| Rifampicin: electrochemical effect on the blood component by cyclic voltammetry using a nano-sensor | Cyclic voltammetry on nanoparticle-modified electrodes | Reported sensitive electrochemical responses in the blood matrix; LOD/linear range given in the article (see the paper) | Blood (investigates electrochemical oxidation in blood) | 33 |
| A simple UPLC-MS/MS assay of rifampin in a small volume of human plasma | UPLC-MS/MS (MRM) | 0.025–10 μg mL−1 (quantifiable) | Human plasma (0.02 mL sample) | 34 |
| Quantification of rifampicin in human plasma and cerebrospinal fluid by a highly sensitive and rapid LC-MS/MS method | LC-MS/MS (MRM) | 25–6400 ng mL−1 (i.e., 0.025–6.4 μg mL−1) | Human plasma and cerebrospinal fluid (CSF) | 35 |
| Spectrophotometric determination of rifampicin through chelate formation and charge transfer complexation | Visible/charge-transfer spectrophotometry (various reagents) | For different reagents: 5–140 μg mL−1, 2–45 μg mL−1 (or 5–120 μg mL−1), 15–200 μg mL−1; alternate method 10–240 μg mL−1 | Capsules, human serum, and urine | 36 |
| Sub-minute determination of rifampicin and isoniazid in fixed-dose combination tablets by capillary zone electrophoresis with UV detection | Capillary zone electrophoresis (CZE) with UV | LOD for RIF 0.34 mg L−1; LOQ 1.13 mg L−1 (method validated for tablets) | Fixed-dose combination tablets (pharmaceutical formulations) | 37 |
Methods and materials
Reagents
RIF (1GM) was purchased from Sigma-Aldrich (China). Potassium hexacyanoferrate(II) trihydrate (K4[Fe(CN)6]·3H2O, P9387), potassium hexacyanoferrate(III) (K3[Fe(CN)6], 244023), and phosphate-buffered saline (PBS) tablets (P4417) were obtained from Sigma-Aldrich (Dorset, UK). Potassium chloride (KCl, 10735874), gold(III) chloride (AuCl3), ammonium chloride (NH4Cl), and HEPES(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) were purchased from Sigma-Aldrich, and acetonitrile (ACN), pyrrole (Py) (functional monomer) and phenol red sodium salt (PhR) (functional monomer) were procured from Thermo Fisher Scientific (Loughborough, UK). All aqueous solutions were prepared using deionized (DI) water with a resistivity of 15.6 MΩ cm. Human serum (H4522) from human male AB plasma, US origin, from Sigma-Aldrich was sourced as a reagent and stored at −20 °C until use.Electrode fabrication
A printed circuit board (PCB) electrode chip consists of four integrated three-electrode systems. The central circular element serves as the reference electrode (RE), while the surrounding horseshoe-shaped structure functions as the counter electrode (CE). Four outer circular segments act as working electrodes (WEs), enabling simultaneous or comparative electrochemical measurements, and serve as the substrate used in this study. Prior to electrode modification, the PCBs were thoroughly cleaned to eliminate any manufacturing residues or surface contaminants. After immersion in 70% (v/v) ethanol for 15 min, the PCBs were rinsed extensively with deionized (DI) water. After rinsing, the chips were dried under a gentle stream of nitrogen gas to ensure complete removal of residual moisture.The reference (RE) and counter electrodes (CE) were modified by electroplating with a commercially available silver-brush plating solution (Spa Plating, UK). The electroplating was performed by configuring a galvanic cell with a platinum (Pt/Ti type) rod as the counter electrode. Silver was electroplated onto the RE and CE by performing multistep potentiometry (MSP) under vigorous stirring by applying 15 cycles of −0.5 mA for 30 s followed by 0.1 mA for 5 s. The PCBs were then rinsed and dried under a nitrogen stream.
The working electrodes were electroplated by setting a galvanic cell with a Pt/Ti type rod as the CE and the PCB WEs as the WE. Spa plating's gold brush plating solution was used as the electrolyte. 25 cycles of MSP were performed at −1.0 mA for 30 s followed by 0.2 mA for 5 s under vigorous stirring. The electrodes were then rinsed and dried under a nitrogen stream.
Subsequently, the silver-plated reference electrode was converted to an Ag/AgCl pseudo-reference by incubating 20 μL of 3 M KCl solution onto the CE and RE for 30 s, followed by aspiration, rinsing with DI water, and drying under a nitrogen stream. 1.5 μL of 100 mM ferric chloride (FeCl3) solution was carefully added onto the RE only for 30 s. The electrode was then rinsed thoroughly with DI water and dried once more under a gentle nitrogen stream.
Highly porous gold surface modification
The working electrodes were modified with highly porous gold (HPG) to enhance the electrochemically active surface area (ECSA) and electron transfer kinetics. This was achieved using a two-step electrochemical procedure in a solution of 20 mM AuCl3 and 2.5 M NH4Cl, mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Initially, a soft gold layer was deposited by performing ten cycles of cyclic voltammetry (CV) from +0.8 V (vs. Ag/AgCl) to +0.3 V (vs. Ag/AgCl) at a scan rate of 0.1 V s−1. The soft gold layer was then electroporated by performing chronoamperometry at −1.2 V (vs. Ag/AgCl) for 60 seconds.25,38–40 At this potential, the evolution of ammonia gas acts as a dynamic template, producing a porous nanostructure within the gold layer.
:1 ratio. Initially, a soft gold layer was deposited by performing ten cycles of cyclic voltammetry (CV) from +0.8 V (vs. Ag/AgCl) to +0.3 V (vs. Ag/AgCl) at a scan rate of 0.1 V s−1. The soft gold layer was then electroporated by performing chronoamperometry at −1.2 V (vs. Ag/AgCl) for 60 seconds.25,38–40 At this potential, the evolution of ammonia gas acts as a dynamic template, producing a porous nanostructure within the gold layer.
Molecularly imprinted polymers (MIPs)
The RIF-imprinted pPhR-pPy polymer was synthesized on a HPG-modified working electrode (WE). A functional monomer analyte cocktail was prepared as follows: a 40 mM stock solution of the functional monomer, pyrrole (Py), was prepared in 100 mM HEPES buffer (pH 6). Separately, a 5 mM stock solution of the co-monomer, phenol red (PhR), was also prepared in 100 mM HEPES and deoxygenated by bubbling nitrogen gas through the solution for 15 min. In a microcentrifuge tube, 100 μL of the Py solution and 100 μL of the 5 mM PhR solution were combined with 250 μL of a 7.2 mg mL−1 RIF solution (prepared in acetonitrile). To this mixture, 50 μL of 100 mM phosphate-buffered saline (PBS, pH 7.4) was added. The resulting solution was briefly vortexed and incubated at room temperature for one hour to facilitate the formation of non-covalent complexes between the monomers and the template molecule.For electropolymerization, a 100 μL aliquot of the functional monomer analyte cocktail solution was carefully dropped onto a PCB electrode surface within a potentiostatic cell, using an external Pt/Ti type counter electrode. To promote the electrostatic attraction of the zwitterionic RIF template onto the working electrode, a pre-conditioning potential of 0.2 V (vs. Ag/AgCl) was applied for 30 s, which allows the electrode potential to reach a steady baseline, minimizing drift during measurement. Subsequently, the cross-linked polymer film was formed by performing 40 cycles of cyclic voltammetry (CV) between 0.3 V and +0.9 V (vs. Ag/AgCl) at a scan rate of 0.1 V s−1.
To remove the RIF template, the sensor was incubated in methanol for 10 min, followed by washing with DI water, and drying under a nitrogen stream.
Electrochemical characterization
Each step of the electrode fabrication and surface modification process was monitored by performing 5 cycles of CV from −0.2 V (vs. Ag/AgCl) to 0.5 V (vs. Ag/AgCl) at a scan rate of 0.1 V s−1 in 5 mM [Fe(CN)6]3−/4− solution with 0.1 M KCl. The synthesis of MIPs was monitored by performing differential pulse voltammetry (DPV) from −0.2 V (vs. Ag/AgCl) to 0.5 V (vs. Ag/AgCl) with an increment of 0.02 V, an amplitude of 0.05 V, a pulse width of 0.1 s and a pulse period of 0.5 s in 10 mM PBS.The biofouling resistance of HPG was assessed by performing CV on freshly prepared HPG from −0.2 V (vs. Ag/AgCl) to 0.5 V (vs. Ag/AgCl) at a scan rate of 0.1 V s−1 in 5 mM [Fe(CN)6]3−/4− solution with 0.1 M KCl. The electrodes were incubated in 100 μL human serum for 30 min. The human serum was then rinsed off and CV was performed under the conditions mentioned above. Planar gold electrodes were used as a control for this study.
Surface characterization
Scanning electron microscopy (SEM) was performed on planar gold, HPG and MIP electrodes using a ZEISS Gemini 2. The samples were prepared by rinsing with DI water and drying under a nitrogen stream, and sputter-coated with chromium at 7 × 106 mBar for 1 min.Sensor performance
For the detection of RIF, dose–response studies were conducted in PBS buffer and in spiked human serum. A 100 μL sample was pipetted onto the surface of the four-electrode system. A baseline signal was first recorded using DPV. The sample was then incubated on the sensor surface for 30 min at room temperature to allow for analyte binding. After incubation, a second DPV measurement was recorded. The sensor response in signal loss was calculated using eqn (1). | (1) |
All experiments were performed in compliance with institutional and national guidelines. Human serum samples used in this study were commercially procured standard reagents (non-identifiable pooled serum) and therefore did not require ethical approval or informed consent.
Results and discussion
Electrochemical characterization
The fabrication of the biosensor was monitored using CV at each step-in an [Fe(CN)6]3−/4− redox probe solution before polymerization and in 10 mM PBS after modification with the polymer. The bare planar gold electrode exhibited reversible redox behavior with peak-to-peak separation denoting normal electron transfer kinetics with a peak current of 91.1 μA. Upon modification with a HPG layer, a substantial enhancement in the redox peak currents and a reduction in peak-to-peak separation were observed with a peak current of 110.3 μA. The peak current for the HPG-modified electrode was 10.58 μA larger than the peak current observed for the planar gold electrode, representing a 1.16-fold increase. This improvement in both oxidation and reduction currents is attributed to the significantly larger electrochemically active surface area provided by the nanostructured morphology of the HPG, which increases the number of active sites and facilitates more efficient electron transfer. The reduced peak-to-peak separation of approximately 0.085 V (85 mV) suggests faster electron transfer kinetics, contributing to enhanced electrochemical performance. These improvements make the HPG-modified gold electrode especially effective for biosensor applications that require high sensitivity and rapid detection.Upon modification with a HPG layer, enhancement in the redox peak currents and a reduction in peak-to-peak separation was observed. HPG exhibits the largest peak currents and the greatest separation in curves, which is indicative of improved electron transfer kinetics. The porous structure increases the number of active sites, thereby increasing the double-layer capacitance and resulting in an increased current density for the same potential sweep. This increase has been attributed to the very high electroactive surface area due to the nanostructure morphology of the HPG, which allows for more efficient electron transfer across the surface of the electrode and the redox probe. The biofouling study shows that exposure to undiluted human serum markedly reduced the redox response on planar Au where the peak reduced from ∼91 μA to ∼59 μA. In contrast, HPG retained most of its signal from ∼110 μA to ∼99 μA with the voltametric profile largely preserved as shown in Fig. 2B. After electropolymerizing a MIP film on the HPG surface, there was a reduction in peak currents as expected. This reduction in signal/fluctuation was expected and demonstrates that a polymer layer has been deposited and therefore insulates the electrode surface.
 | ||
| Fig. 2 A) Cyclic voltammograms comparing the older dual working electrode design (planar gold) with the newer quad-working planar and HPG. B) Cyclic voltammograms comparing the biofouling resistance of HPG with that of planar gold. | ||
After electropolymerizing a MIP film on the HPG surface, a reduction in peak currents is expected due to the presence of two different redox probes. This blocks the diffusion of the redox species to the electrode, thereby lowering the current response, as reported in previous studies by Amouzadeh Tabrizi et al.43 and Siciliano et al.44
An important step to develop a working MIP sensor is the removal of the template molecule to yield specific binding cavities that are free and accessible. The polymer film containing the RIF template molecule is a significant barrier to the redox probe prior to completing the template removal process, effectively decreasing the electrochemical signal output. After initiating the template removal from the incubated polymer film with methanol for the extraction of the RIF template, a marked increase in peak currents is observed. The recovery of the electrochemical signal indicates that the template molecules are fully removed as shown in Fig. S1, as this frees the polymer matrix and creates recognition sites that increase the surface area. This observation was supported by DPV measurements, which also showed a distinct enhancement in the peak current after template removal, confirming improved probe accessibility to the electrode surface. The DPV results confirm the successful creation of the “molecular memory” effect fundamental to the sensor's selectivity.
Surface characterization
The morphological changes at each stage of the electrode modification were visualized using scanning electron microscopy (SEM), as shown in the figure below. The bare planar gold electrode (Fig. 3A) presents a relatively smooth and uniform surface, characteristic of a standard electrode finish. In stark contrast, the surface modified with highly porous gold (HPG) (Fig. 3B) reveals a complex, three-dimensional nanostructure with extensive porosity. This sponge-like morphology is directly responsible for the significantly increased electrochemically active surface area observed in the CV experiments. Following electropolymerization, the planar gold electrode is coated with the MIP film (Fig. 3C), resulting in a more conformal surface that partially fills the underlying pores while retaining significant texture. This visual evidence in Fig. 3D confirms the successful deposition of a stable polymer film onto the HPG scaffold, creating an integrated sensing interface crucial for both signal amplification and selective molecular recognition. The inherent porosity of the HPG is key to its anti-biofouling properties, as it resists the adsorption of large biomolecules from serum while allowing small analytes like RIF to access the recognition sites. | ||
| Fig. 3 Surface characterization: scanning electron micrographs (SEM). A) SEM image of planar gold. B) SEM image of a highly porous gold electrode. C) SEM images of the molecularly imprinted polymer (MIP) on planar gold electrodes. D) SEM image of the molecularly imprinted polymer on HPG electrodes. | ||
Electrochemical detection of RIF
The analytical performance of the fabricated MIP sensor for RIF detection was evaluated using DPV in both phosphate-buffered saline (PBS) (Fig. S2) and human serum. This dual testing approach allowed assessment of the sensor performance in an ideal buffer system and in a complex biological matrix. Fig. 4B illustrates the sensor's response to increasing concentrations of RIF, from 0 μg mL−1 to 32 μg mL−1, in PBS and in spiked human serum. In the absence of RIF (0 μg mL−1), the DPV curves displayed a well-defined peak current corresponding to the redox activity of the pPhR/pPy-based MIP film. Upon introduction of RIF and subsequent incubation, a concentration-dependent decrease in the DPV peak current was observed. This decline is attributed to the rebinding of RIF molecules to the complementary cavities within the MIP film, progressively blocking the electrode surface and restricting electron transfer of the redox probe. The systematic reduction in peak currents with increasing RIF concentrations confirms both the sensor's specificity towards the target analyte and the quantifiable nature of the rebinding process. | ||
| Fig. 4 Analytical performance and specificity of the MIP-based RIF sensor. A) Differential pulse voltammetry (DPV) curves showing a concentration-dependent decrease in peak currents with increasing concentrations of RIF in human serum. B) Calibration curves illustrating the sensor's response (% signal loss) to RIF in PBS versus spiked human serum, fitted with a non-linear Hill equation model. The highlighted green area indicates the therapeutic range for RIF (0–32 μg mL−1). C) Sensor stability analysis showing a consistent dose–response across a physiological pH range of 6.5 to 8.5. D) Specificity test demonstrating that the sensor's response to RIF is unaffected by the presence of the potential interferent, vancomycin, in spiked human serum. However, an interference from paracetamol was observed, saturating the sensor at 24 μg mL−1. Such an interference can be dealt with the integration of machine learning (ML) models that can potentially attenuate the effects of interference. A multiple-input single-output (MISO) model can potentially improve the sensor's performance in complex matrices by using multiple features to predict the concentration more accurately. | ||
Sensor calibration and performance in PBS and human serum
To quantify the connection between the sensor response and RIF concentration, a calibration curve of the percentage signal loss based on the RIF concentration in spiked human serum samples was produced Fig. 4B signal loss is defined as the relative variation in the DPV peak current before and after RIF incubation. The biosensor exhibited an apparent dose-dependent reduction in the current across a clinically relevant concentration range of 8–24 μg mL−1 with measurable responses between 0 μg mL−1 and 32 μg mL−1.The calibration data were analyzed with a non-linear Hill equation model, resulting in reliable coefficients of determination (PBS: R2 = 0.987; human serum: R2 = 0.975). These robust R2 values represent a high degree of correlation between the RIF concentration and percentage signal loss, providing confidence in the reliability of the fit and the capacity of the sensor for quantification. The dynamic range of the biosensor (0–32 μg mL−1) was well within the therapeutic plasma window for RIF; 8–24 μg mL−1, thereby confirming clinical relevance of the biosensor for therapeutic drug monitoring (TDM). The comparative response of the sensor in both PBS and serum solutions, the complexities of biological matrices aside, demonstrates its reliability and practicality for use with direct patient samples.
The sensor demonstrated a limit of detection (LOD) of 0.848 μg mL−1 and a limit of quantification (LOQ) of 2.54 μg mL−1 for the linear range of 0–25 μg mL−1. The LOD and LOQ were calculated using the standard IUPAC eqn (2) and (3)
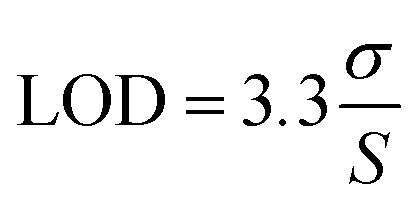 | (2) |
 | (3) |
Sensor specificity and stability
To validate the sensor's robustness for clinical applications, its performance was assessed under varying pH conditions and in the presence of potential chemical interferents (N = 4).Fig. 4C demonstrates the sensor's remarkable stability across a physiologically relevant pH range from 6.5 to 8.5. The dose–response curves at each pH value are highly consistent, indicating that minor fluctuations in the pH of biological samples will not compromise the accuracy of the RIF measurement. More importantly, the sensor's selectivity was confirmed through an interference study, with the results presented in Fig. 4D. The sensor's response to increasing concentrations of RIF was measured in human serum spiked with potentially interfering compounds, paracetamol (15 μg mL−1) and vancomycin (17.5 μg mL−1). The resulting dose–response curves for the spiked samples were nearly superimposable on the curve obtained from unadulterated human serum. This lack of significant deviation confirms that the sensor does not cross-react with these common medications and specifically binds to RIF. This high degree of selectivity is crucial for a diagnostic tool, ensuring reliable and accurate measurements in complex patient samples where multiple drugs may be present.
Advancements in the current work and alignment with AMR goals
This work represents a significant leap forward from our previous sensor iterations, marked by key improvements in design, fabrication, and performance. The transition from a dual-working electrode (2-WE) to a quad-working electrode (4-WE) platform is a major advancement, enabling higher data throughput and the potential for internal calibration or multiplexed detection on a single, low-cost chip. Furthermore, the fabrication protocol has been substantially refined; the optimized electrodeposition of HPG has resulted in a nanostructure with a larger electrochemically active surface area and, consequently, improved electron transfer kinetics compared to our earlier planar gold designs. This enhancement is critical for achieving the high sensitivity needed for clinical applications.This technology directly supports the strategic goals of the UK's 20-year vision and the EU's One Health Action Plan to combat antimicrobial resistance (AMR). A primary driver of drug resistance is sub-optimal antibiotic exposure, which allows resistant strains to survive and multiply. By enabling rapid, affordable, point-of-care therapeutic drug monitoring (TDM), our sensor provides a direct mechanism for antibiotic stewardship. It allows clinicians to personalize RIF dosing, ensuring concentrations remain within the therapeutic window to maximize efficacy and minimize the risk of resistance development. This moves precision medicine from a centralized lab to the frontlines of global health, a core objective in the fight against AMR.
Future work
Looking ahead, while the sensor currently demonstrates excellent selectivity against common drugs like paracetamol and vancomycin, we aim to build an even more robust “smart” sensor system. Our next phase of development will involve the integration of machine learning (ML) regressors. We plan to train an ML model on electrochemical data from samples containing known concentrations of both RIF and various interferents. This model will learn to recognize and deconstruct the subtle voltammetric signatures of each compound, allowing it to computationally compensate for any minor signal interference from paracetamol or other co-administered drugs. The implementation of this ML layer will enhance the sensor's accuracy in complex polypharmacy scenarios, ensuring its reliability for deployment in real-world clinical settings without the need for extensive sample cleanup.Conclusion
In response to the urgent global health need for accessible TB care, this research successfully delivers a pioneering electrochemical biosensor capable of quantifying RIF with high precision directly in human serum. Our key innovation lies in the synergistic integration of three core components:1) a molecularly imprinted polymer (MIP) for specific molecular recognition,
2) a highly porous gold (HPG) nanostructure for signal amplification and outstanding anti-biofouling performance, and
3) a mass-producible printed circuit board (PCB) that reduces the per-unit cost to just £0.09.
This platform directly overcomes the primary obstacles to widespread TDM by eliminating the need for expensive equipment and complex sample pre-treatment. The sensor's ability to operate effectively in undiluted human serum is a critical differentiator, proving its robustness and readiness for real-world clinical applications. By achieving a clinically relevant detection range of 8–24 μg mL−1 and a low limit of detection (0.848 μg mL−1), this device provides the analytical performance required to guide personalized dosing regimens. This work bridges the gap between laboratory-grade precision and point-of-care practicality, presenting a disruptive tool poised to enhance treatment efficacy, reduce the emergence of drug resistance, and advance the personalized management of TB in the settings that need it most.
Author contributions
Rohith Shetty: data curation, formal analysis (equal), investigation (equal), and writing – original draft preparation. Sudhaunsh Deshpande: conceptualization, dormal analysis (equal), investigation (equal), methodology, project administration, software, visualization, and writing – review & editing (lead). Anu Mary Joy: methodology and resources. Arjun Ajith Mohan: writing – review & editing. Qianming Xu: methodology and resources. Alison Holmes: supervision and resources. Sanjiv Sharma: supervision, validation, and review & editing.Conflicts of interest
The authors declare no conflicts of interest.Data availability
The data supporting the findings of this study are available within the article and its supplementary information (SI). Additional raw data, including electrochemical datasets and microscopy images, are available from the corresponding author upon reasonable request.Supplementary information is available. See DOI: https://doi.org/10.1039/d5sd00165j.
Acknowledgements
The authors acknowledge that the SEM images were taken using a ZEISS Gemini 2 at the University of Liverpool Scanning Electron Microscopy Shared Research Facility (SEM SRF).References
- M. Pai, M. A. Behr, D. Dowdy, K. Dheda, M. Divangahi, C. C. Boehme, A. Ginsberg, S. Swaminathan, M. Spigelman, H. Getahun, D. Menzies and M. Raviglione, Nat. Rev. Dis. Primers., 2016, 2, 16076 CrossRef.
- C. J. Cambier, S. Falkow and L. Ramakrishnan, Cell, 2014, 159, 1497–1509 CrossRef CAS PubMed.
- Global Tuberculosis Programme, 2022, https://www.who.int/teams/global-tuberculosis-programme/data.
- A. Alsultan and C. A. Peloquin, Drugs, 2014, 74, 839–854 Search PubMed.
- Y. Zhang and W.-W. Yew, Int. J. Tuberc. Lung Dis., 2015, 19, 1276–1289 Search PubMed.
- P. Sensi, Clin. Infect. Dis., 1983, 5, S402–S406 CrossRef CAS PubMed.
- M. T. Gler, V. Skripconoka, E. Sanchez-Garavito, H. Xiao, J. L. Cabrera-Rivero, D. E. Vargas-Vasquez, M. Gao, M. Awad, S.-K. Park, T. S. Shim, G. Y. Suh, M. Danilovits, H. Ogata, A. Kurve, J. Chang, K. Suzuki, T. Tupasi, W.-J. Koh, B. Seaworth, L. J. Geiter and C. D. Wells, N. Engl. J. Med., 2012, 366, 2151–2160 CrossRef CAS PubMed.
- A. H. Diacon, A. Pym, M. P. Grobusch, J. M. De Los Rios, E. Gotuzzo, I. Vasilyeva, V. Leimane, K. Andries, N. Bakare, T. De Marez, M. Haxaire-Theeuwes, N. Lounis, P. Meyvisch, E. De Paepe, R. P. G. Van Heeswijk and B. Dannemann, N. Engl. J. Med., 2014, 371, 723–732 CrossRef.
- F. Conradie, A. H. Diacon, N. Ngubane, P. Howell, D. Everitt, A. M. Crook, C. M. Mendel, E. Egizi, J. Moreira, J. Timm, T. D. McHugh, G. H. Wills, A. Bateson, R. Hunt, C. Van Niekerk, M. Li, M. Olugbosi and M. Spigelman, N. Engl. J. Med., 2020, 382, 893–902 CrossRef CAS.
- National institute for Health and Care Excellance.
- E. Chigutsa, M. E. Visser, E. C. Swart, P. Denti, S. Pushpakom, D. Egan, N. H. G. Holford, P. J. Smith, G. Maartens, A. Owen and H. McIlleron, Antimicrob. Agents Chemother., 2011, 55, 4122–4127 CrossRef CAS PubMed.
- M. Niemi, J. T. Backman, M. F. Fromm, P. J. Neuvonen and K. T. Kivistö, Clin. Pharmacokinet., 2003, 42, 819–850 CrossRef CAS PubMed.
- C. A. Peloquin, Drugs, 2002, 62, 2169–2183 CrossRef CAS PubMed.
- J. Kuhlin, M. G. G. Sturkenboom, S. Ghimire, I. Margineanu, S. H. J. Van Den Elsen, N. Simbar, O. W. Akkerman, E. M. Jongedijk, R. A. Koster, J. Bruchfeld, D. J. Touw and J.-W. C. Alffenaar, Clin. Mass Spectrom., 2019, 14, 34–45 CrossRef.
- C. Dincer, R. Bruch, A. Kling, P. S. Dittrich and G. A. Urban, Trends Biotechnol., 2017, 35, 728–742 CrossRef CAS.
- J. S. Daniels and N. Pourmand, Electroanalysis, 2007, 19, 1239–1257 CrossRef CAS PubMed.
- Y. Shao, J. Wang, H. Wu, J. Liu, I. A. Aksay and Y. Lin, Electroanalysis, 2010, 22, 1027–1036 CrossRef CAS.
- F. Cui and H. S. Zhou, Biosens. Bioelectron., 2020, 165, 112349 CrossRef CAS.
- A. G. Ayankojo, J. Reut and V. Syritski, Biosensors, 2024, 14, 71 CrossRef CAS.
- A. Fallah, A. A. Imani Fooladi, S. A. Havaei, M. Mahboobi and H. Sedighian, Biochem. Biophys. Rep., 2024, 40, 101852 CAS.
- Z. Liu, Z. Xu, D. Wang, Y. Yang, Y. Duan, L. Ma, T. Lin and H. Liu, Polymer, 2021, 13, 2657 CAS.
- K. Haupt and K. Mosbach, Chem. Rev., 2000, 100, 2495–2504 CrossRef CAS PubMed.
- R. D. Crapnell, N. C. Dempsey-Hibbert, M. Peeters, A. Tridente and C. E. Banks, Talanta Open, 2020, 2, 100018 CrossRef.
- S. Deshpande, A. M. Arjun, G. Liu, K. Pawlak and S. Sharma, Adv. Healthcare Mater., 2025, e03155 CrossRef.
- A. M. Arjun, S. Deshpande, G. Liu, D. Miura, K. Pawlak, T. Takahiko, M. Higuchi, M. Matsumoto, T. Umemura, K. Tsukakoshi and S. Sharma, ACS Sens., 2025 DOI:10.1021/acssensors.5c01816.
- G. Dutta, A. Regoutz and D. Moschou, in EUROSENSORS 2018, MDPI, 2018, p. 741 Search PubMed.
- Y. Hua, D. Kukkar, R. J. C. Brown and K.-H. Kim, Crit. Rev. Environ. Sci. Technol., 2023, 53, 258–289 CrossRef CAS.
- H. Noor, I. G. David, M. L. Jinga, D. E. Popa, M. Buleandra, E. E. Iorgulescu and A. M. Ciobanu, Sensors, 2023, 23, 976 CrossRef CAS PubMed.
- Q. Zhang, S. Ma, X. Zhuo, C. Wang, H. Wang, Y. Xing, Q. Xue and K. Zhang, Analyst, 2022, 147, 2156–2163 RSC.
- E. Sharafi and S. Sadeghi, New J. Chem., 2023, 47, 500–514 RSC.
- Q. Huang, X. Li, S. Feng, W. Zhuge, F. Liu, J. Peng and S. Mo, Anal. Methods, 2018, 10, 3594–3601 RSC.
- D. Kul, Thai J. Pharm. Sci., 2020, 17, 398–407 CAS.
- M. M. Radhi, A. I. Ibrahim, Y. K. Al-Haidarie, S. A. Al-Asadi and E. A. J. Al-Mulla, Nano Biomed. Eng., 2019, 11, 150–156 CAS.
- T. Sano, T. Ishii, K. Hotta and Y. Mano, ACS Omega, 2023, 8, 36261–36268 CrossRef CAS PubMed.
- A. Srivastava, D. Waterhouse, A. Ardrey and S. A. Ward, J. Pharm. Biomed. Anal., 2012, 70, 523–528 CrossRef CAS PubMed.
- S. Sadeghi and E. Karimi, Chem. Pharm. Bull., 2006, 54, 1107–1112 CrossRef CAS PubMed.
- L. M. Duarte, T. L. Amorim, P. R. Chellini, L. H. C. Adriano and M. A. L. De Oliveira, J. Sep. Sci., 2018, 41, 4533–4543 CrossRef CAS PubMed.
- P. Bollella, Nanomaterials, 2020, 10, 722 CrossRef CAS PubMed.
- P. Bollella, S. Sharma, A. E. G. Cass, F. Tasca and R. Antiochia, Catalysts, 2019, 9, 580 CrossRef CAS.
- S. Deshpande, A. M. Arjun, G. Liu, K. Pawlak and S. Sharma, Adv. Healthcare Mater., 2025, e03155 CrossRef PubMed.
- E. J. Begg, M. L. Barclay and C. J. Kirkpatrick, Br. J. Clin. Pharmacol., 1999, 47, 23–30 CrossRef CAS.
- M. E. Stocker and J. E. Montgomery, Nano Biomed. Eng., 2001, 87, 638–640 CAS.
- M. Amouzadeh Tabrizi, J. P. Fernández-Blázquez, D. M. Medina and P. Acedo, Biosens. Bioelectron., 2022, 196, 113729 CrossRef CAS PubMed.
- G. Siciliano, M. S. Chiriacò, F. Ferrara, A. Turco, L. Velardi, M. A. Signore, M. Esposito, G. Gigli and E. Primiceri, Analyst, 2023, 148, 4447–4455 RSC.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |