
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA nucleic acid-based electrochemical detection method for post hoc sample analysis
Logan T.
Echeveria
 a,
Sadi
Shahriar
b,
Allison M.
Yorita
a and
Erkin
Seker
*c
a,
Sadi
Shahriar
b,
Allison M.
Yorita
a and
Erkin
Seker
*c
aLawrence Livermore National Laboratory, Livermore, California 94550, USA
bDepartment of Materials Science and Engineering, University of California – Davis, Davis, California 95616, USA
cDepartment of Electrical and Computer Engineering, University of California – Davis, Davis, California 95616, USA. E-mail: eseker@ucdavis.edu
First published on 17th November 2025
Abstract
This work introduces a new electrochemical sensing approach, where the liquid sample containing nucleic acid targets can be blotted onto an electrode that is pre-functionalized with probe DNA. The post-hybridization signal and probe DNA signal (obtained by melting the hybrid) can be successively measured later, making the sensing scheme resilient to probe layer deterioration and circumventing the need to measure probe signal immediately before sample collection, ultimately mitigating the need for electrochemical sensing equipment at the sample collection site.
Introduction
The rapid detection and identification of biomolecules is essential for the diagnosis and management of diseases.1–4 In particular, nucleic acids (DNA and RNA) carry significant diagnostic and therapeutic potential as biomarkers.5 For example, nucleic acids purified from bodily fluids allow for early detection of disorders (e.g., infection,6 cancer7), pre-natal diagnosis,8 prognostic surveillance after trauma,9 and microbiota analysis.10Nucleic acid-based biomarkers often require purification of the sample followed by amplification of the nucleic-acid (e.g., PCR), which are time-intensive, laborious, and expensive. Moreover, these technologies often require technical expertise and specialized equipment, which are often not present in remote locations.
Electrochemical sensors have shown considerable promise for molecular diagnostics with their sensitive detection capabilities in complex biological matrices via enhanced working electrode coating architectures.11,12 Conventional detection schemes for nucleic-acid based electrochemical sensors involve a layer of single-stranded DNA (ssDNA) capture probe that is attached to the working electrode at one end and labeled with a redox reporter at the opposite end (Fig. 1). Upon electrochemical interrogation (e.g., square wave voltammetry [SWV]), the probe layer produces a current,13 which is attenuated upon hybridization with the target DNA or RNA biomarker due to conformational change of the ssDNA.14 The decrease in the current, referred to as percent signal suppression (%SS) and shown in eqn (1), indicates successful hybridization.15 Since the probe and target signals need to be successively acquired for accurately determining %SS, this method is not suitable for sample collection at a site without electrochemical analysis capabilities. In other words, the conventional method relies on a stable and precise interrogation of the probe DNA layer without any target molecule present. In the scenario of sample collection at a remote site, a baseline probe signal needs to be acquired before the chip is shipped out for exposure to the sample (e.g., blood, water). Once the chip is returned to a site with electrochemical analysis capabilities, the post-hybridization current can be acquired. However, since the %SS metric will be calculated based on the previously acquired probe signal, the changes in the signal due to environmental factors/aging during shipment will influence the %SS determination.
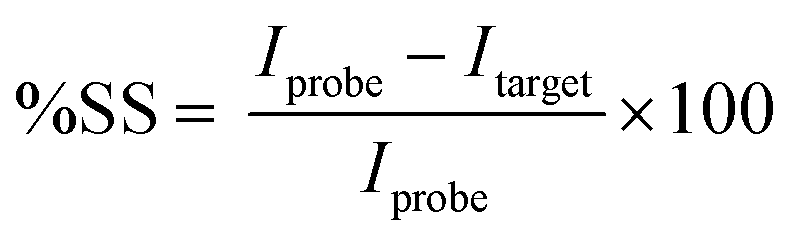 | (1) |
 | (2) |
 | ||
| Fig. 1 Representative electrochemical current reading via square wave voltammetry for (1) probe DNA layer immobilization, (2) hybridization with target DNA – basis for %SS metric, and (3) melting the DNA duplexes in warm DI water – basis for %SE metric. | ||
It was recently shown that DNA-functionalized gold electrodes used as electrochemical biosensors have a shelf life of at least six months if stored properly16 with the caveat that the probe signal still varies, hence necessitating the new method described here. Nevertheless, this finding, combined with the method introduced here, should make it possible to ship out probe-functionalized chips, perform sample collection at a remote location, and return the chips to the analysis site without significantly hindering sensor performance.
Results and discussion
We first assessed the reliability of %SE as a detection metric. Briefly, planar gold (pl-Au) electrodes were fabricated using previously established microfabrication protocols and the electrodes were surface-functionalized with 26-mer single-stranded probe DNA labeled with a methylene blue redox reporter and derivatized with a thiol group attached via C6 spacer for immobilization onto the gold electrodes.12 Mercaptohexanol was used to backfill the bare electrode surfaces, which creates a well-ordered ssDNA monolayer and reduces the capacitive background current.17 The probe sequence (Integrated DNA Technology [IDT]) was: /5ThioMC6-D/ CG TGT TAT AAA ATG TAA TTT GGA ATT /3MeBlN/. The functionalized electrodes were assembled into a custom 3D printed Teflon electrochemical cell with 150 μl sample volume (Fig. S1). The electrochemical signal from the probe layer was acquired in 1× phosphate buffered saline solution (PBS, Thermo Fisher Scientific) with a BioLogic SP-300 potentiostat. The complementary target DNA (IDT) was diluted to 2 μM and 100 nM in 1× PBS and incubated on the electrode for 45 minutes. The electrochemical cell was next rinsed thoroughly with 1× PBS to remove non-specifically-bound 26-mer target DNA followed by SWV interrogation of the hybridized layer. The electrode was then treated with 150 μl of deionized water at 70 °C for 15 minutes to melt the DNA duplex. The cell was then washed with 1× PBS followed by a second SWV measurement of the denatured layer (Fig. S2). The details of the protocol and raw SWV measurements are included in the SI (Fig. S4–S6). All statistical comparisons were performed via Wilcoxon–Mann–Whitney rank sum test (Kaleidagraph), where p < 0.05 was deemed statistically significant.Fig. 2 compares the assessment of DNA concentration in the system, as determined by %SS and %SE. The average %SE value was 8% and 14% less than the corresponding %SS value at 2 μM and 100 nM target DNA concentration, respectively; however, the difference is not statistically significant (p = 0.1 and p = 0.3, respectively for 2 μM and 100 nM), suggesting that the proposed detection metric (%SE) performs as well as the conventional detection metric (%SS).
 | ||
| Fig. 2 Comparison of %SS and %SE metrics for detection of 2 μM and 100 nM target concentration in PBS. | ||
Detection of DNA in clinically-relevant samples via PCR requires purification steps that may not be accessible during sample collection in remote destinations. Similarly, cellular debris and proteins can substantially affect electrochemical sensor performance by adsorbing onto the working electrode.18 The adsorbed layer hinders molecular transport of target molecules to the probe layer and blocks charge transfer between the electrode and redox molecules.19 To mitigate biofouling in electrochemical sensors, various strategies that range from anti-biofouling polymeric coatings20 to electrode nanostructuring21,22 have been explored. One such strategy utilizes size-selective transport phenomenon in nanoporous gold (np-Au) coatings, which enable inherent biofouling-resilient properties.22 Specifically, the porous structure acts as a size exclusion matrix that permits the entry of small fibrillar nucleic acids into the pores while simultaneously blocking larger debris and proteins.
We assessed biofouling resilience of np-Au and pl-Au electrodes using the same electrochemical experiment protocol previously described. The np-Au electrodes were fabricated by co-sputtering 600 nm-thick gold–silver alloy (36% Au and 64% Ag, atomic %) onto a glass coverslips following a 160 nm-thick chromium adhesion layer and an 80 nm-thick gold seed layer (Lesker Labline Sputter System). The coating was dealloyed in 70% nitric acid at 55 °C for 15 minutes to create the np-Au thin films.12,22 The np-Au and pl-Au electrodes were functionalized with probe DNA, assembled into the electrochemical cell, and challenged with 2 μM target DNA in 1× PBS and 10% fetal bovine serum (FBS) in 1× PBS to mimic the complex media present in clinically-relevant samples.22
As shown in Fig. 3, the detection of the target DNA in the complex buffer using pl-Au electrodes resulted in a 48% decrease in average %SE compared to that for detection in PBS (p = 0.2). When np-Au electrodes are used, the complex buffer only caused a 1.2% decrease in %SE compared to that for detection in PBS (p = 0.8). The %SS metrics acquired from the same experiments displayed a similar trend to those with %SE (Fig. S3). These results further validate the utility of %SE metric as a substitute for the conventional %SS and suggest that the np-Au coating with its biofouling resilience is a suitable candidate for electrochemical detection in complex biological media. Moreover, this result suggests that the traditional %SS metric is representative of how the sensor would perform with the new %SE metric, since the foundational biomolecular capture mechanism is the same. To that end, we have previously shown that different probe sequences still produce measurable %SS when exposed to DNA targets with complementary sequences, while non-complementary sequences result in negligible %SS, highlighting the selectivity/specificity of the sensor.22,23 In addition, we have reported that 500 nM 15-mer RNA target in diluted pig blood can be detected using the %SS, demonstrating electrochemical detection in a clinically-relevant matrix.24 Taken together, np-Au electrodes produce measurable %SS in a variety of different scenarios, which is indicative of the sensing performance using the %SE metric.
 | ||
| Fig. 3 Comparison of np-Au and pl-Au electrode performance using %SE metric in detecting 2 μM target in PBS and in 10% FBS in PBS. | ||
An additional requirement for remote sample collection is that the protocol should be simple and require a small amount of sample volume (e.g., finger prick blood collection). To assess np-Au′s performance in collecting liquid biopsy samples via blotting, microliter-sized droplets are placed onto the substrate, which rapidly evaporate in seconds to minutes due to enhanced liquid–air interface caused by imbibition of liquid into the nanostructure.25–27 Droplet evaporation has the potential to enhance detection, especially for low target concentrations, since diffusion-limited transport from bulk solution onto the electrode surface is eliminated.28 As the droplet evaporates, the surrounding buffer disappears and eventually all target molecules converge onto the sensor surface, consequently interacting with the probe DNA. In addition, drying the sample upon hybridization may stabilize DNA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :RNA duplexes protecting them from degradation via nucleases.29
:RNA duplexes protecting them from degradation via nucleases.29
To emulate liquid biopsy sample collection, 0.75 μl droplets of 2 μM target DNA in PBS and 10% FBS were blotted onto the probe DNA-functionalized np-Au and pl-Au electrodes. Once the droplets evaporated (Fig. 4b), the electrodes were assembled into the electrochemical cell, where first the SWV current due to the hybridized probe-target layer was measured through the sequence illustrated in Fig. 1. Subsequently, the electrodes with the hybridized DNA layer were denatured in warm DI water and interrogated with SWV to obtain the %SE.
 | ||
| Fig. 4 The target DNA is introduced onto the electrode surface as a micro-droplet for subsequent evaporation and acquisition of %SE. (a) %SE metric from blotted np-Au electrodes with target DNA in PBS and in 10% FBS in PBS. (b) Optical image of an evaporated droplet. (c) Scanning electron microscope top-view image of the np-Au electrode. | ||
The np-Au electrodes successfully detected target DNA in both PBS and its biofouling FBS-containing counterpart (Fig. 4a) with no statistically-significant difference in performance (p = 0.8). The inter-measurement variability was higher for the blotting-based detection (Fig. 4) compared to direct addition of target DNA into the electrochemical cell (Fig. 2 and 3). We attribute this to the droplet footprint spreading outside the electrochemically-active region defined by our custom Teflon cell. The geometric misalignment results in a portion of the electrode hybridized with DNA not being electrochemically measured, thereby skewing the %SE determination. This problem can be addressed by micropatterning electrodes so that they can be more accurately assembled into the electrochemical cell. We also observed that the magnitude of %SE decreased when comparing the detection in an electrochemical cell (Fig. 3) to the droplet-based technique. The sample volume significantly decreases when using the droplet method, from 150 μl to 0.75 μl. Since both experiments were conducted at 2 μM concentration, the total amount of target DNA in the sample scales with the volume change. Interestingly, the sample volume underwent a 99% reduction yet the %SE only has an average decrease of 66% and 81% for droplet detection in PBS and 10% FBS, respectively. This suggests that the droplet evaporation process is more efficient than a standard diffusion-based electrochemical cell in promoting hybridization of a smaller number of target DNA molecules.
Finally, it is worth emphasizing that the primary goal of this study was to demonstrate the utility of the %SE metric-based sensing approach to enable remote sample collection and subsequent detection at a facility with electrochemical experiment resources. The comparable %SS and %SE metrics, as shown in Fig. 1 and S3, imply that the new sensing approach is applicable to clinically-relevant sample matrices that contain confounding fragments of DNA and RNA molecules.22–24 In the future, the limit of detection and dynamic range of the np-Au-based sensor can be further enhanced by tuning the pore morphology as demonstrated before.14 The key design consideration is that pore size should be large enough to permit unhindered transport of nucleic acids while small enough to block permeation of biofouling species into the deeper probe DNA-functionalized surfaces.
Conclusions
In conclusion, we demonstrated a new electrochemical detection protocol to overcome the necessity of collecting the probe DNA signal prior to sample collection. We expect this method to enable off-site sample collection via blotting similar to at-home paper-based tests, where the electrochemical quantification (for nucleic acids, proteins, or small molecules30) can then be performed at a centralized facility.Author contributions
Logan Echeveria: conceptualization, formal analysis, data curation, investigation, writing – original draft, visualization. Sadi Shahriar: investigation, writing – review & editing. Allison Yorita: methodology, resources, writing – review & editing. Erkin Seker: conceptualization, methodology, validation, resources, writing – review & editing, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI).Supplementary information: [pictorial representation of the experimental protocol, experimental details, peak current obtained from SWV of pl-Au electrode undergoing various treatments, comparison of %SS and %SE for np-Au and pl-Au for PBS and 10% FBS in PBS, raw SWV data that constitute the values shown in Fig. 2–4]. See DOI: https://doi.org/10.1039/d5sd00164a.
Acknowledgements
S. S. and E. S. gratefully acknowledge the support from the National Science Foundation via DMR-2003849, the National Institutes of Healthvia NIBIB R01-EB034279, and from the University of California – Davis via College of Engineering Next Level Research Funds and IGNITION Seed Funding. This work was performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory (LLNL) under the Contract DE-AC52-07NA27344. LLNL release number LLNL-JRNL-2009480. This project also benefited from the resources of the Center for Nano/Micro-Manufacturing (CNM2) facility at University of California, Davis and the Center for Micro- and Nanotechnology at Lawrence Livermore National Laboratory.Notes and references
- H. Lord and S. O. Kelley, J. Mater. Chem., 2009, 19, 3127–3134 RSC.
- J. O. Esteves-Villanueva, H. Trzeciakiewicz and S. Martic, Analyst, 2014, 139, 2823–2831 RSC.
- J. S. Daniels and N. Pourmand, Electroanalysis, 2007, 19, 1239–1257 CrossRef CAS.
- E. Gumus, H. Bingol and E. Zor, J. Pharm. Biomed. Anal., 2023, 225, 115206 CrossRef CAS.
- J. Wang, Anal. Chim. Acta, 2002, 469, 63–71 CrossRef CAS.
- A. Niemz, T. M. Ferguson and D. S. Boyle, Trends Biotechnol., 2011, 29, 240–250 CrossRef CAS PubMed.
- H. Schwarzenbach, D. S. Hoon and K. Pantel, Nat. Rev. Cancer, 2011, 11, 426–437 CrossRef CAS PubMed.
- Y. D. Lo, N. Corbetta, P. F. Chamberlain, V. Rai, I. L. Sargent, C. W. Redman and J. S. Wainscoat, Lancet, 1997, 350, 485–487 CrossRef CAS.
- Y. D. Lo, T. H. Rainer, L. Y. Chan, N. M. Hjelm and R. A. Cocks, Clin. Chem., 2000, 46, 319–323 CrossRef CAS.
- E. E. Ferapontova, Curr. Opin. Electrochem., 2020, 19, 153–161 CrossRef CAS.
- R. Gasparac, B. J. Taft, M. A. Lapierre-Devlin, A. D. Lazareck, J. M. Xu and S. O. Kelley, J. Am. Chem. Soc., 2004, 126, 12270–12271 CrossRef CAS.
- Z. Matharu, P. Daggumati, L. Wang, T. S. Dorofeeva, Z. Li and E. Seker, ACS Appl. Mater. Interfaces, 2017, 9, 12959 CrossRef CAS PubMed.
- K. Kerman, D. Ozkan, P. Kara, B. Meric, J. J. Gooding and M. Ozsoz, Anal. Chim. Acta, 2002, 462, 39–47 CrossRef CAS.
- Y. Xiao, R. Y. Lai and K. W. Plaxco, Nat. Protoc., 2007, 2, 2875–2880 CrossRef CAS PubMed.
- S. O. Kelley, J. K. Barton, N. M. Jackson and M. G. Hill, Bioconjugate Chem., 1997, 8, 31–37 CrossRef CAS.
- J. Chung, A. Billante, C. Flatebo, K. K. Leung, J. Gerson, N. Emmons, T. E. Kippin, L. Sepunaru and K. W. Plaxco, Sens. Diagn., 2024, 3, 1044–1050 RSC.
- F. V. Oberhaus, D. Frense and D. Beckmann, Biosensors, 2020, 10, 45 CrossRef CAS PubMed.
- S. A. Bhakta, E. Evans, T. E. Benavidez and C. D. Garcia, Anal. Chim. Acta, 2015, 872, 7–25 CrossRef CAS.
- A. Barfidokht and J. J. Gooding, Electroanalysis, 2014, 26, 1182–1196 CrossRef CAS.
- S. Saxena, P. Sen, L. Soleymani and T. Hoare, Adv. Sens. Res., 2024, 3, 2300170 CrossRef CAS.
- J. Patel, L. Radhakrishnan, B. Zhao, B. Uppalapati, R. C. Daniels, K. R. Ward and M. M. Collinson, Anal. Chem., 2013, 85, 11610–11618 CrossRef CAS PubMed.
- P. Daggumati, Z. Matharu, L. Wang and E. Seker, Anal. Chem., 2015, 87, 8618 CrossRef CAS PubMed.
- J. Veselinovic, Z. Li, P. Daggumati and E. Seker, Nanomaterials, 2018, 8, 351 CrossRef PubMed.
- J. Veselinovic, M. Alangari, Y. Li, Z. Matharu, J. M. Artés, E. Seker and J. Hihath, Electrochim. Acta, 2019, 313, 116–121 CrossRef CAS.
- T. Gambaryan-Roisman, Curr. Opin. Colloid Interface Sci., 2014, 19, 320–335 CrossRef CAS.
- E. Seker, M. R. Begley, M. L. Reed and M. Utz, Appl. Phys. Lett., 2008, 92, 013128 CrossRef.
- Y. Xue, J. Markmann, H. Duan, J. Weissmüller and P. Huber, Nat. Commun., 2014, 5, 4237 CrossRef CAS.
- Y. Wang, F. Liu, Y. Yang and L.-P. Xu, Mater. Chem. Front., 2021, 5, 5639–5652 RSC.
- I. V. Kornienko, O. Y. Aramova, A. A. Tishchenko, D. V. Rudoy and M. L. Chikindas, Molecules, 2024, 29, 5978 CrossRef CAS.
- V. Kesler, K. Fu, Y. Chen, C. H. Park, M. Eisenstein, B. Murmann and H. T. Soh, Adv. Funct. Mater., 2023, 33, 2208534 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |