DOI:
10.1039/D6SC00233A
(Review Article)
Chem. Sci., 2026,
17, 9370-9397
Electrochemical functionalization of pyridines
Received
9th January 2026
, Accepted 7th April 2026
First published on 8th April 2026
Abstract
Pyridines are important nitrogen-containing heteroaromatics with wide applications in pharmaceuticals, agrochemicals, ligands, and materials science. However, their selective C–H functionalization remains challenging due to low nucleophilicity, regioselectivity issues, and catalyst deactivation via nitrogen coordination. Traditional methods such as electrophilic substitution, directed metalation, transition-metal catalysis, and N-oxide transformations have enabled significant advances in this field, but often require harsh conditions or pre-functionalized substrates. In contrast, electrochemical synthesis has emerged as a sustainable and versatile alternative, employing electricity as a clean redox agent to enable direct C–H, C–C, and C–heteroatom bond formation under mild, tunable conditions. This review provides a focused overview of how electro-organic synthesis has addressed key challenges in pyridine functionalization associated with traditional methods, tracing its historical development and highlighting recent advances, with particular emphasis on anodic oxidation, cathodic reduction, paired electrolysis, and mediator or metal-catalyzed strategies, thereby offering readers an in-depth understanding of the advancements in this field and its significance in organic synthesis. Key mechanistic insights and representative applications are highlighted to demonstrate the growing potential of electrochemical strategies for scalable, late-stage pyridine diversification. We hope that this review will highlight the salient features of electrochemical strategies for pyridine functionalization in synthetic applications and stimulate the development of new, more advanced methodologies.
 Satabdi Bera | Satabdi Bera received her MSc degree from Jadavpur University in 2017. Then, she joined the Indian Institute of Technology Kharagpur as a PhD student in 2018 under the supervision of Dr Rajarshi Samanta. Recently, she joined as a research associate at the Indian Institute of Science, Bangalore, with Dr Durga Prasad Hari. Her research interests focus on the development of new synthetic methods by utilising visible light and electricity. |
 Tushar Singha | Tushar Singha earned his BSc in Chemistry from the University of Kalyani in 2018 and his MSc from IIT Madras in 2020. During his MSc, he worked on copper-catalysed C–S bond formation under the guidance of Prof. G. Sekar. He is currently pursuing his PhD at the Indian Institute of Science, Bangalore, with Dr Durga Prasad Hari. His research focuses on ring-strain release and the development of photo- and electrochemical strategies in organic synthesis. |
 Nakul Abhay Bapat | Nakul Abhay Bapat obtained a B·S (Research) degree from IISc in 2024, during which he explored photo- and electrochemical strain release transformations in the group of Dr Durga Prasad Hari. He is currently pursuing a PhD at EPFL, Switzerland, under Dr Jeremy Luterbacher. His research interests are biomass conversion and lignin degradation. |
 Durga Prasad Hari | Durga Prasad Hari earned his MSc degree from IIT Madras in 2010. He then moved to the University of Regensburg in Germany to pursue his PhD under the supervision of Prof. Burkhard Koenig. After completing his doctoral studies in 2014, he became a postdoctoral fellow in the laboratory of Prof. Jerome Waser at EPFL in Switzerland. In 2018, he joined the Aggarwal group at the University of Bristol as a Marie Curie research fellow. Since March 2021, he has held an assistant professor position at the Indian Institute of Science in Bangalore. His research interests focus on discovering new reactivity of strained molecules and carbenes through photo- and electrochemical strategies. |
1. Introduction
Pyridines are among the essential nitrogen-containing heteroaromatic scaffolds widely found in pharmaceuticals,1 agrochemicals,2 ligands,3 and functional materials4 due to their electron-deficient aromatic ring, basic nitrogen atom, and excellent metal-coordination properties. Their prevalence in biologically active compounds1 and catalytic systems3 has driven considerable interest in methods that allow direct and selective functionalization of the pyridine ring. Classical strategies, including electrophilic substitution,5 directed metalation with strong bases,6,7 transition-metal catalyzed cross-coupling8,9 or C–H activation,10–12 photoredox catalysis,13 and transformations via pyridine N-oxides,14 have enabled valuable transformations but often rely on harsh conditions, stoichiometric reagents, or pre-functionalized substrates, limiting their scalability and sustainability.
In recent years, electrochemical functionalization has emerged as a powerful and environmentally benign alternative to stoichiometric oxidants and reductants, employing electric current as a clean redox reagent to generate reactive species directly at the electrode surface under mild and tunable conditions.15 This strategy enables selective C–H functionalization of readily oxidizable electron-rich arenes, allowing C–C and C–heteroatom bond formation.16,17 However, the efficient C–H or C–X bond modification of electron-deficient pyridines remains significantly more challenging. Owing to the operational simplicity of electro-organic synthesis, broad functional-group tolerance, and precise control over redox events through constant-current or constant-potential electrolysis, electrochemical methods have emerged as a powerful platform for the late-stage diversification of pyridine-containing molecules in both academic and industrial settings (Scheme 1A).
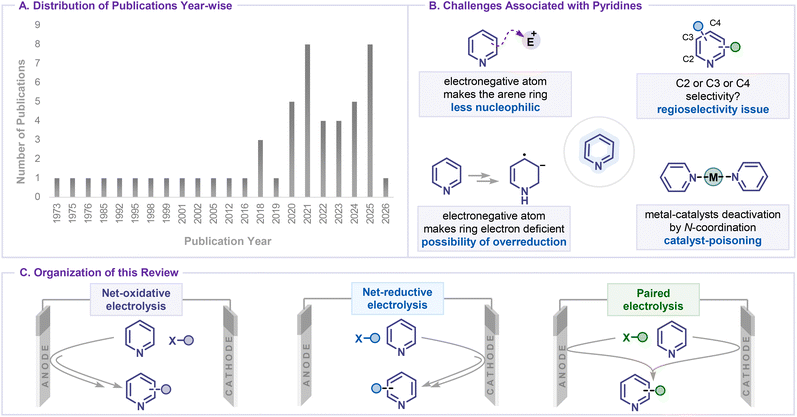 |
| | Scheme 1 (A) Distribution of publications year-wise. (B) Challenges associated with pyridine functionalization. (C) Organization of the review. | |
Although several reviews have discussed the electrochemical functionalization of N-heterocycles in general, only a limited number of pyridine examples are included. While these reviews provide valuable insights into broader electrosynthetic strategies, they primarily focus on other (hetero)arenes (e.g., phenols, indoles, pyrazoles, and imidazoles) and on specific pyridine transformations, such as C2 functionalization via activated pyridinium species and the functionalization of cyano-substituted pyridines.18 However, numerous recent reports have expanded the scope to enable regioselective functionalization at C2, C4, and C3/C5 positions, highlighting the evolving reactivity patterns of pyridine substrates that are not comprehensively captured in existing reviews. Due to their electron-deficient nature and the presence of a coordinating nitrogen atom, pyridines present unique challenges, including site-selective functionalization (C2 vs. C4 vs. C3/C5), susceptibility to overreduction, and catalyst deactivation through strong N-coordination (Scheme 1B). In electrochemical systems, these factors are closely linked to reduction potentials and pyridine activation modes, which critically influence reaction pathways and method selection. Therefore, a dedicated pyridine-focused perspective enables a more systematic understanding of these features and provides practical guidance for electrochemical functionalization of pyridines.
Building on this perspective, this review surveys recent advances in electrochemical methods for pyridine functionalization, organized according to the underlying electrochemical logic. Specifically, the discussed strategies are classified into three broad categories: net-anodic oxidation, net-cathodic reduction, and paired electrolysis, each exploiting the unique redox properties of pyridine and its derivatives. We discuss both direct electrolysis, where the substrate itself undergoes electron transfer at the electrode surface, and mediator-assisted processes, in which redox-active species facilitate selective transformations. The merging of photocatalysis and transition-metal catalysis with electrochemical approaches is also discussed, wherein the synergy of transition metals with electrochemical conditions is shown to enable challenging C–H, C–C, and C–heteroatom bond formations. Key reaction parameters, including the choice of electrolyte, electrode material, solvent, and the application of either constant current or constant voltage, are analyzed, as these factors drastically influence reaction efficiency, selectivity, and functional group tolerance. Furthermore, selected examples of functionalized pyridine derivatives are presented, illustrating the versatility and synthetic utility of electrochemical approaches for the late-stage diversification of this important heterocyclic scaffold. Finally, the mechanistic aspects of representative transformations are explored to provide insight into electron-transfer pathways, the formation of radical or cationic intermediates, and regioselectivity.
This review is organized to first highlight the early examples of electrochemical pyridine functionalization, followed by sections based on the key electrochemical processes: (i) net oxidation, (ii) net reduction, and (iii) paired electrolysis (Scheme 1C). Within each process-oriented section, transformations are further categorized by the type of bond formed, including C–C, C–N, C–O, C–B, C–Si, C–S, and annulation reactions. At the end of each major electrochemical process, the choice of reaction parameters, such as electrode materials, electrolyte and electrochemical control mode, etc., is summarised to inform rational reaction design for pyridine activation and regioselectivity control. Finally, the current challenges and future directions in the field are discussed. We hope this review will shed light on the key features of electrochemical pyridine functionalization for synthetic applications and inspire the development of more advanced methodologies.
2. Early advancements
The first example of electrochemical pyridine functionalization was reported by Inoue and his coworkers in 1973 (Scheme 2a).19 They demonstrated the electrochemical fluorination of pyridines, which predominantly resulted in perfluorinated derivatives. Subsequently, in 1975, Haszeldine and his co-workers extended this approach to alkyl-substituted pyridines, achieving fluorination that primarily produced mixtures of perfluoro-(N-fluoropiperidine) derivatives under constant voltage electrolysis (Scheme 2b).20 A significant milestone was achieved by Ballinger and Teare, who reported the first electrochemical monofluorination of pyridine, affording 2-fluoropyridine in 22% yield.21 Performing the electrolysis at a constant voltage of 2.5 V using platinum electrodes was key for the successful monofluorination of pyridine with Me4NF·2HF serving as both the fluorine source and the supporting electrolyte. The reaction efficiency was highly sensitive to the applied potential and electrolyte concentration, as higher voltages led to pyridine decomposition (Scheme 2c). These studies collectively laid the groundwork for electrochemical fluorination of pyridines, highlighting the critical influence of reaction parameters on selectivity and product distribution.
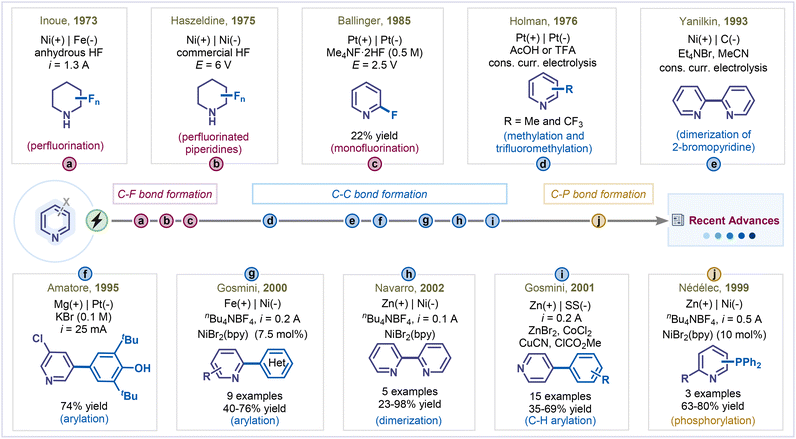 |
| | Scheme 2 Early examples of electrochemical functionalization of pyridines. | |
Building upon these early fluorination efforts, researchers soon began exploring alkylation strategies to expand the functional diversity of pyridine frameworks. In this context, in 1976, Holman and co-workers reported the electrochemical methylation and trifluoromethylation of pyridine by electrolyzing aqueous pyridine solutions containing acetic acid or trifluoroacetic acid, thereby introducing a sustainable pathway for alkyl-functionalized pyridines (Scheme 2d).22
The electrochemical coupling of pyridines has evolved remarkably over the past decades, progressing from early homocoupling studies to highly selective cross-coupling methodologies. The first electrochemical homocoupling of pyridines was reported in 1993 by Yanilkin and co-workers using a Ni-catalyst and 2,2′-bipyridine as the ligand (Scheme 2e).23 In this pioneering study, electrochemically generated Ni(0) undergoes oxidative addition with 2-bromopyridine to produce Ni(II)–Py species. This intermediate is then reduced at the cathode to a Ni(I)–pyridyl species, which undergoes another cycle of oxidative addition followed by reductive elimination, ultimately furnishing the dimerized product. Building upon this foundation, in 1995, Amatore and co-workers reported the first electrochemical cross-coupling of pyridines, utilizing dihalopyridines and 2,6-di-tert-butylphenol as the arylating reagent to achieve C–C bond formation under electrochemical conditions (Scheme 2f).24 This study opened a new avenue for expanding the reactivity landscape of pyridines beyond homocoupling. The field advanced further in 2000, when the electrochemical cross-coupling of pyridines with aryl halides was achieved, tolerating a wide variety of pyridine, aryl, and heteroaryl halides to deliver the corresponding arylated pyridines in good to excellent yields (Scheme 2g).25 Continuing this progress, Navarro and co-workers further modified the homocoupling protocol by employing NiBr2(bpy) as a catalyst and zinc as a sacrificial anode in DMF or acetonitrile, successfully synthesizing 2,2′-bipyridine and dimethyl-2,2′-bipyridines from 2-bromopyridine and 2-bromomethylpyridines, respectively (Scheme 2h).26 Further optimization of nickel catalysts and ligand systems enabled the controlled arylation of polyhalogenated pyridines with improved turnover numbers and selectivity.27 Later, the same group significantly enhanced homocoupling efficiency by introducing an iron anode, which increased yields to 98% by preventing nickel catalyst passivation through iron-ion-mediated trapping of free bipyridine generated during sacrificial anode oxidation.28 Collectively, these advances delineate a clear trajectory from the foundational Ni-catalyzed homocoupling to modern, efficient, and selective cross-coupling methodologies, underscoring the growing maturity and synthetic potential of electrochemical strategies in pyridine chemistry.
Expanding the scope of electrochemical pyridine functionalization beyond halide coupling, Gosmini and co-workers achieved a landmark advancement by reporting the first electrochemical C–H arylation of pyridines, utilizing in situ-generated organozinc reagents as arylating partners in the presence of a copper catalyst (Scheme 2i).29 The organozinc intermediate coupled with pyridinium salts, which is generated in situ from pyridines and methyl chloroformate. The resulting 1,4-dihydropyridine intermediates were then oxidized to furnish 4-phenylpyridines in moderate to high yields. This study not only demonstrated the feasibility of direct C–H arylation under electrochemical control but also highlighted the synthetic versatility of organozinc intermediates in pyridine derivatization.
Nédélec and co-workers developed a C–P bond-forming strategy by employing chlorodiphenylphosphines as phosphorylating agents in the presence of a nickel catalyst, which opened an avenue for C–heteroatom bond formation in pyridine functionalization (Scheme 2j).30
3. Functionalization via net oxidation
Electrochemical net-oxidative strategies have emerged as powerful and sustainable approaches for the formation of C–C and C–heteroatom bonds. These methodologies operate through sequential single-electron transfer (SET) oxidation processes, enabling substrate activation without the need for stoichiometric chemical oxidants. By precisely controlling anodic potentials, electrochemical oxidation facilitates radical–polar crossover pathways and grants access to valuable molecular architectures that are often difficult to obtain using conventional oxidative methods. Among various net oxidative approaches, electrochemical C–H bond functionalization has gained prominence, as it enables regioselective activation and site-selective modification of heteroaromatic systems such as pyridines, thereby providing an efficient route to complex, functionally diverse compounds.
3.1 C–H alkylation
Within oxidative C–H functionalization, C–H alkylation of pyridines represents a significant advancement. In this strategy, alkyl radicals generated either directly at the anode or through mediator-assisted oxidation add to pyridinium intermediates to furnish dihydropyridine derivatives. A subsequent anodic oxidation step restores aromaticity, thereby providing the corresponding alkylated pyridines. In 2020, an electrophotocatalytic Minisci-type functionalization of pyridines was demonstrated by Xu and co-workers using a decarboxylative alkylation strategy (Scheme 3).31 Two distinct optimized conditions were developed for C–H alkylation and C–H carbamoylation, with CeCl3·7H2O and 4CzIPN being employed as photocatalysts, respectively. The alkylation of 2,6-disubstituted pyridine was well tolerated to furnish the product 1; however, C4-substituted pyridines were found to yield a mixture of mono and bis-alkylated products (2 and 3). Notably, pyridine-containing drug molecules such as diflufenican were also compatible, delivering the desired alkylated product 4 in 31% yield. Conversely, the carbamoylation reaction exhibited remarkable selectivity at the C2-position of pyridines and pyridine-containing drug-like molecules, affording mono-substituted products (5–7). Mechanistically, the alkylation reaction was initiated via anodic oxidation of CeIII to CeIV, which coordinated with carboxylic acid to form intermediate I. The alkyl radical III was then proposed to be generated via a photoinduced ligand-to-metal charge transfer (LMCT) in the Ce(IV) complex I, followed by decarboxylation. After the addition of radical III to the activated pyridinium salt, the resulting radical intermediate IV was suggested to lose a proton to generate intermediate V and subsequently undergo oxidation mediated by Ce(IV) to furnish the alkylated product. However, for carbamoylation, the radical I was generated via a single-electron transfer (SET) oxidation of oxamate by the excited-state 4CzIPN. The radical II generated after the Minisci-type addition to pyridinium salt was first reduced by the photocatalyst to give intermediate III, while regenerating the photocatalyst. Subsequent oxidation of IV at the anode to yield the carbamoylated product. Finally, the cathodic reaction was taken care of by the reduction of protons to produce hydrogen gas.
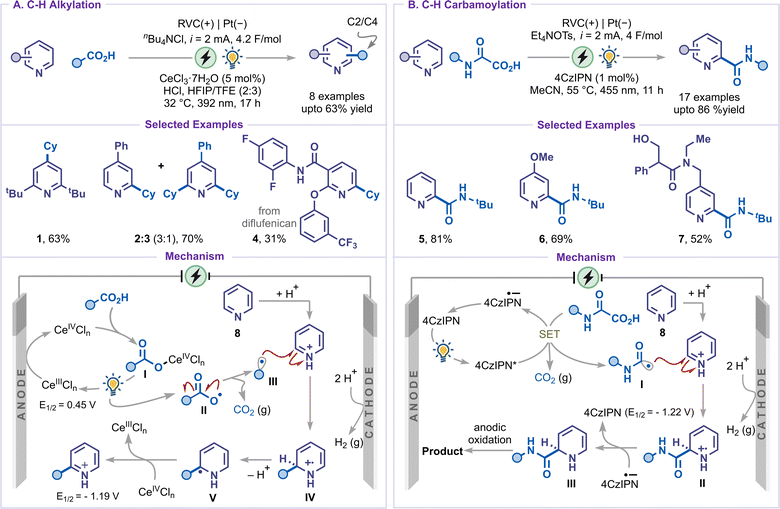 |
| | Scheme 3 (A) C–H alkylation. (B) C–H carbamoylation. | |
3.2 C–H arylation
Heterobiaryls represent valuable structural motifs found in numerous biologically relevant molecules and advanced functional materials.32 Consequently, the development of efficient and sustainable strategies for their synthesis has attracted considerable attention. In line with recent advances in electrochemical methodologies, the direct oxidative C–H arylation of pyridines has been successfully demonstrated in the synthesis of heterobiaryls.
Ackermann and co-workers reported an electrochemical manganese-catalysed strategy for the oxidative C–H arylation of pyridines bearing weakly coordinating amide directing groups, employing Grignard reagents as aryl sources (Scheme 4).33 This approach circumvented the need for stoichiometric oxidants and zinc salts, which are typically required in iron-catalyzed systems to prevent Grignard homocoupling. The electrolysis was performed under a constant current of 3 mA with MnCl2 as the catalyst, neocuproine as the ligand, and TMEDA as an additive in THF solvent. The versatility of this electrocatalytic protocol was demonstrated through its broad substrate scope, accommodating various substituted pyridines and a range of arylating agents, including heteroaryl counterparts (products 9–11). Moreover, the strategy was successfully extended to the methylation of pyridine (product 12), underscoring its synthetic utility and generality. Mechanistic investigations, supported by cyclic voltammetry, scanning electron microscopy coupled with energy-dispersive X-ray spectroscopy (SEM–EDS), and DFT calculations, revealed that C–H activation proceeds through a high-spin ligand-to-ligand hydrogen transfer (LLHT) pathway facilitated by single-electron transfer at the anode. The reaction was initiated through the formation of the active Mn(II) intermediate I in the presence of ligands and the arylating reagent (ArMgBr). This catalytically active Mn(II) species subsequently underwent C–H bond activation of pyridine to form the cyclometalated intermediate IIvia the LLHT process, followed by transmetalation to generate intermediate III. Subsequent anodic oxidation produces intermediate IV, which then undergoes reductive elimination to furnish the arylated pyridine. The resulting Mn(I) species was reoxidized at the anode to regenerate the Mn(II) catalyst, demonstrating that manganese electrocatalysis could effectively activate the C–H bond of pyridines with weakly coordinating amides while suppressing overoxidation and homocoupling.
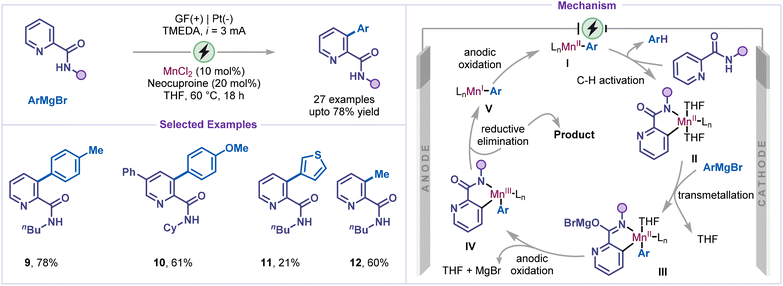 |
| | Scheme 4 Electrochemical Mn-catalyzed C–H bond arylation using aryl Grignard reagents. | |
3.3 C–H borylation
Xu and co-workers recently developed a method for direct C–H borylation of pyridines by merging electrochemistry and photocatalysis, using a borane–trimethylamine (13) complex as the borylating agent (Scheme 5).34 The main objective of this transformation was to enable efficient generation of boryl radicals under mild conditions, without the need for stoichiometric chemical oxidants. This was achieved using the photocatalyst anthraquinone-2-sulfonate (AQS) via hydrogen-atom transfer. This strategy proved effective for C4-substituted pyridines, 2,6-disubstituted pyridines, and 2,3,4-trisubstituted pyridines, all of which underwent smooth conversion to the products (14–16). Upon photoexcitation, the excited anthraquinone sulfonate (AQS) abstracted a hydrogen atom from the borane–trimethylamine complex (13), generating a boryl radical I along with a semiquinone-type radical species (AQS-H˙). The boryl radical I subsequently underwent Minisci-type addition to the pyridinium salt II to produce a radical cation intermediate III, which underwent deprotonation and further oxidation to yield the borylated product 17. The AQS-H˙ species underwent anodic oxidation followed by deprotonation, regenerating the ground-state AQS and thereby completing the catalytic cycle.
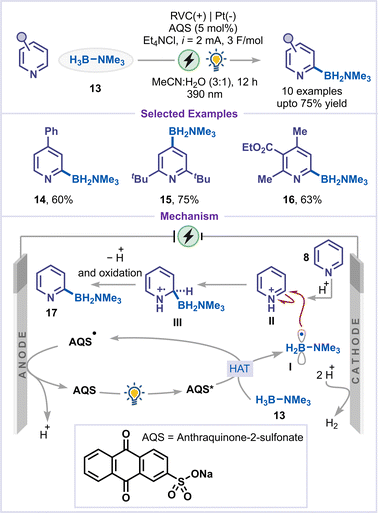 |
| | Scheme 5 C–H borylation. | |
3.4 C–H amination
An electrochemical direct amination of pyridines has also been developed under mild and sustainable conditions. In 2018, Mei and co-workers reported a copper-catalyzed electrochemical C–H amination of pyridines equipped with a directing group (Scheme 6).35 The use of nBu4NI as a redox mediator was key to the success of this protocol. Constant-current electrolysis was carried out in an undivided cell employing Cu(OTf)2 as the catalyst and KOPiv as the base. The substrates bearing picolinamide (NHCO-2-Py) moiety served as an effective bidentate directing group, facilitating coordination of both the amide and ortho-pyridyl N atoms to the copper center and enabling site-selective C–H amination under electrochemical conditions. A wide range of pyridine derivatives was compatible with this method, tolerating both electron-donating (19 and 20) and electron-withdrawing (21) substituents, with selective amination at the C2 position. Mechanistic studies, including kinetic isotope effect measurements, cyclic voltammetry, and radical inhibition experiments, suggested that a high-valent Cu(III) species is involved as a key intermediate. The reaction was believed to be initiated by the coordination of the Cu(II) catalyst with the amine nucleophile 18 and substrate 22, leading to the formation of a copper(II) complex I. This complex was subsequently oxidized by an iodine radical to afford the Cu(III) intermediate II which was proposed to undergo a ligand-to-metal charge transfer (LMCT) process to give radical cation III. The intramolecular amine transfer to radical cation III, followed by a second LMCT event, yielded intermediate IV. The aminated product 19 was then released, and a Cu(I) species was formed. Finally, iodine radical-mediated anodic oxidation of Cu(I) was proposed to regenerate the active Cu(II) catalyst, thereby completing the catalytic cycle.
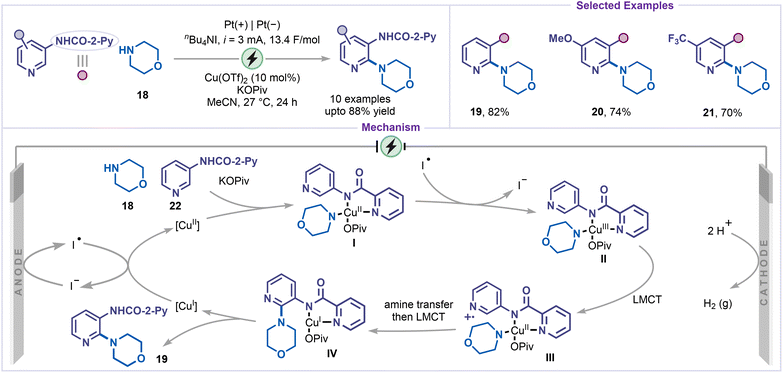 |
| | Scheme 6 C–H amination. | |
Very recently, Malapit and co-workers reported a directing-group-free electrochemical C2-amination of pyridines using selectfluor II (23) as the aminating reagent (Scheme 7).36 Under optimized electrochemical conditions, the desired aminated products (24–26) were obtained in good to excellent yields. The transformation proceeded through a two-step sequence: (i) electrochemical amination to generate DABCOnium salts, followed by (ii) neutralization with KCN to afford the corresponding aminated or piperazine derivatives. Mechanistically, the reaction begins with anodic oxidation of reduced selectfluor II (23), forming the radical dication I, which subsequently adds to the pyridine substrate via a charge-transfer complex II, generating the intermediate III. Further anodic oxidation and proton elimination yield the DABCOnium salt IV. Finally, nucleophilic substitution (SN2) of this intermediate with KCN furnishes the piperazine derivative 27. The high C2-selectivity is attributed to the strong electrophilicity and steric demand of the nitrogen-centered radical dication I, favoring regioselective addition at the C2-position of the pyridine ring.
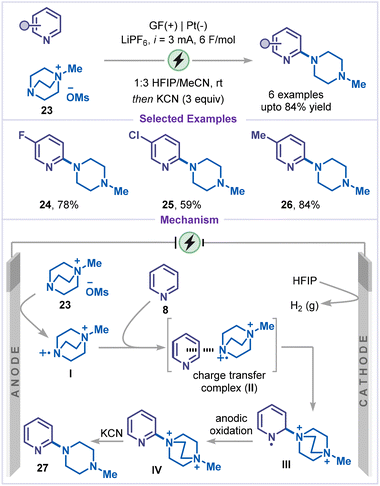 |
| | Scheme 7 C–H amination using reduced selectfluor II. | |
3.5 C–H sulfonylation
Another noteworthy transformation is the electrochemical sulfonylation of pyridines, which enables direct introduction of sulfonyl groups onto the pyridine core and broadens the repertoire of sulfur-containing derivatives.37 In 2024, Yi and co-workers reported a meta-selective electrochemical sulfonylation of pyridines proceeding via a dearomatized oxazino–pyridine species (Scheme 8).38 Electrolysis of the oxazino–pyridine and sulfinate salt was carried out in an undivided cell with a graphite felt (GF) anode and Pt cathode under constant-current conditions using nBu4NBF4 as the supporting electrolyte. Subsequent acidic treatment furnished the desired meta-sulfonylated pyridines. This method proved broadly applicable to a wide range of sulfonyl groups, including (hetero)aryl sulfinates and alkyl sulfinates (products 28–30). The protocol was also successfully extended to late-stage functionalization of drug-like molecules containing pyridine motifs, such as vismodegib (product 31). Mechanistic investigations indicated that preferential anodic oxidation of the sulfinate over the oxazino–pyridine 32 was crucial for generating the sulfonyl radical. The sulfonyl radical I was selectively added at the β-nitrogen site of the oxazino–pyridine 32, yielding a key intermediate II that undergoes further anodic oxidation to generate intermediate III. Subsequent deprotonation and acid-mediated rearomatization afforded the meta-sulfonylated product.
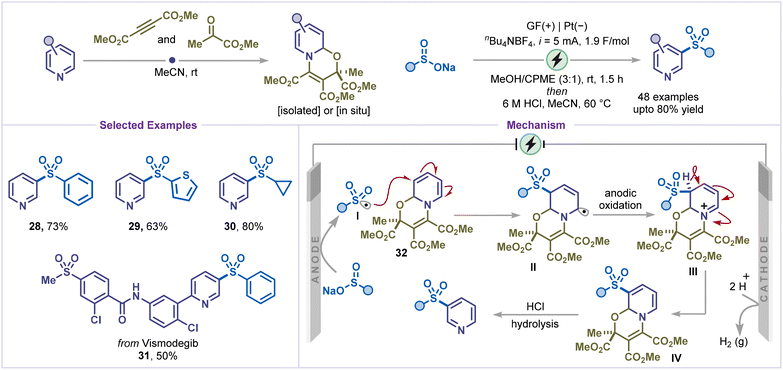 |
| | Scheme 8 C–H sulfonylation. | |
3.6 C–H silylation
Direct C–H silylation represents a powerful strategy in modern organic synthesis, as the resulting silylated products serve as versatile intermediates for further synthetic elaboration.39 In 2023, Wang and co-workers reported an elegant example of site-selective silylation of pyridines under mild electrophotochemical conditions, employing 9,10-phenanthrenequinone (PQ) as both a photocatalyst and a hydrogen-atom-transfer (HAT) agent (Scheme 9).40 The protocol exhibits broad substrate tolerance, accommodating variously monosubstituted pyridines (34 and 35). Silylated products (36 and 37) were efficiently obtained from 2,6- and 2,4-disubstituted pyridines. Mechanistically, the electrophotocatalytic C–H silylation begins with photoexcitation of PQ to PQ*, followed by HAT from tBuMe2SiH to generate the silyl radical I (tBuMe2Si˙) and PQH˙. The silyl radical I is added to the pyridinium salt II, forming a key intermediate III, which subsequently transfers a hydrogen (HAT) atom to PQH˙, followed by deprotonation to yield the silylated pyridine 38. Finally, PQ is regenerated by anodic oxidation of PQH2.
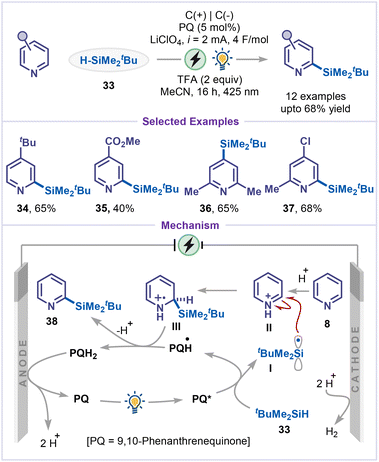 |
| | Scheme 9
Ortho-C–H silylation. | |
An elegant strategy for the meta-C–H silylation of pyridines was recently disclosed by Xu and co-workers (Scheme 10).41 The transformation proceeded without a photocatalyst or external oxidants, relying instead on N-hydroxyphthalimide (NHPI) as a hydrogen-atom-transfer (HAT) catalyst. Constant current electrolysis in acetonitrile at room temperature efficiently generated silyl radicals in situ, which selectively engaged with the pyridine ring. Notably, the presence of an electron-withdrawing substituent on the pyridine ring was found to enhance meta-selectivity, dictating the role of electronic effects in controlling regioselective C–H bond functionalization. While 2,6-dicyanopyridine furnished both mono- and disilylated products (39 and 40), 2,4,6-trisubstituted pyridine exclusively delivered the monosilylated product 41, reflecting the impact of steric effects on second silylation. In addition, the use of bulkier silanes, such as triisopropylsilane was compatible with the reaction, affording the corresponding meta-silylated product 42 in 55% yield. A plausible reaction pathway involves single-electron oxidation of NHPI, followed by proton elimination to generate the N-oxyl radical I, which abstracts a hydrogen atom from silane, generating silyl radical II. The silyl radical II then preferentially adds to the meta position of intermediate III, affording intermediate IV. Finally, N-oxyl radical I abstract a hydrogen atom from intermediate IV, regenerating NHPI and delivering the meta-silylated product 39. The observed meta-selectivity is attributed to a combination of electronic and stabilization effects. Specifically, the strong electron-withdrawing inductive effect of cyano or ester substituents enhances the electrophilic character of the meta position relative to the ortho and para positions.
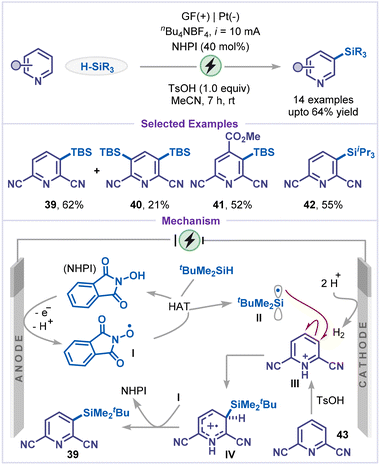 |
| | Scheme 10
Meta-C–H silylation. | |
3.7 C–H halogenation
Halopyridines are highly important intermediates in organic synthesis and medicinal chemistry due to their versatile reactivity and broad applications. They also serve as key building blocks in the synthesis of pharmaceuticals, functional materials, and agrochemicals, enabling precise modification of the pyridine scaffold to tune biological activity or material properties.10,42 Therefore, the electrochemical halogenation of pyridines opened another avenue for their efficient synthesis. In 2016, Gryaznova and co-workers reported a selective electrochemical fluorination of pyridines and ethylpyridines mediated by transition metal catalysts (Scheme 11).43 The regioselectivity of the transformation was found to depend strongly on the metal catalyst employed: nickel catalysts favored fluorination at the C3-position, whereas silver and cobalt salts preferentially promoted C2-fluorination. The methodology was demonstrated on two representative substrates, affording the corresponding fluorinated products (44–47) in good yields. The observed catalyst-dependent regioselectivity likely arises from distinct oxidative fluorination pathways, the mechanistic details of which remain to be fully elucidated.
 |
| | Scheme 11 Transition-metal catalyzed C–H fluorination. | |
In 2021, Chen and co-workers reported an electrochemical meta-C–H bromination of pyridines via an Umpolung strategy (Scheme 12).44 Selective meta-functionalization was achieved by introducing an electron-donating group at the ortho position to enhance nucleophilicity at the meta site, and electrophilic bromine was generated in situ from tetrabutylammonium bromide (48). A broad substrate scope was displayed by the optimized bromination protocol, with both electron-donating (Me) and halogen (F) substituents being tolerated to deliver the products (49 and 50). When the cyclohexylamino (NHCy) group was used, the corresponding brominated pyridine 51 was obtained in 60% yield. Remarkably, preinstallation of methoxy groups at the 2- and 6-positions of the pyridine led to a highly efficient electrolysis, delivering product 52 in an isolated yield of 95%. CV studies indicated that bromine radical is generated via oxidation of bromide, while the 2-aminopyridine alone was found to be electrochemically inactive. Based on these findings and prior reports, a plausible mechanism was proposed in which the bromide ion was anodically oxidized to Br-radical I, which then forms molecular bromine and undergoes electrophilic addition on the amino-pyridine to yield intermediate II. Subsequent deprotonation furnished the meta-brominated product 54.
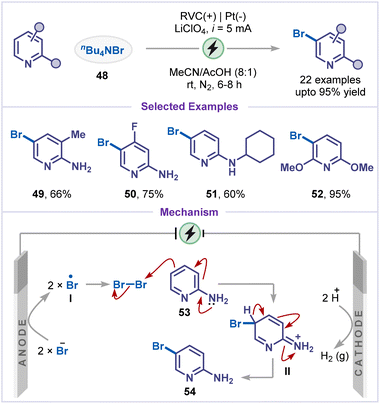 |
| | Scheme 12 C–H bromination. | |
3.8 Annulation of pyridine derivatives
Indolizines are found in biologically active natural products and pharmaceuticals.45 They exhibit a broad spectrum of activities, including anti-inflammatory, antimicrobial, anti-tubercular, and anticancer properties45b,46 and have recently attracted attention for applications in dye-sensitized solar cells (DSSCs) and organic light-emitting devices (OLEDs).47 In 2022, Sharma and co-workers developed a greener strategy for indolizine synthesis via an electrochemical cascade C–H functionalization and annulation (Scheme 13).48 Treatment of pyridines with 2-methylquinazolinone derivatives and chalcone derivatives in an undivided electrochemical cell under constant current (10 mA) efficiently furnished the corresponding annulated indolizine 55. Notably, the replacement of chalcones with isoxazole-containing styryl-alkene also afforded indolizine 56 in good yield. Based on experimental evidence and prior literature, the mechanism was believed to be initiated by anodic oxidation of iodide to generate an iodine radical, which reacted with quinazolinone 57 to form an intermediate Ivia HAT, followed by recombination of the heterobenzyl radical with the iodine radical. Pyridine then reacted with I to form intermediate II, which underwent deprotonation followed by an intermolecular cycloaddition with chalcone yield intermediate III. Finally, anodic oxidation of intermediate III restored aromaticity, thereby forming the heterocyclic indolizine product 58.
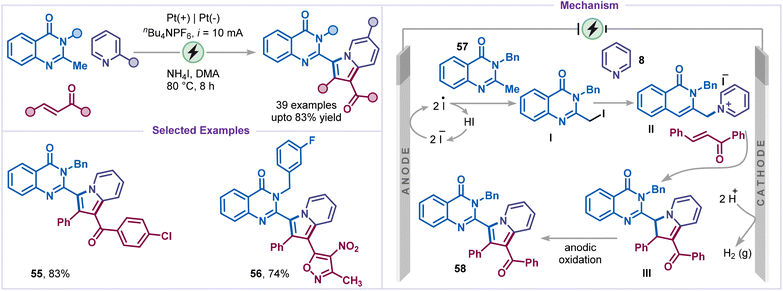 |
| | Scheme 13 Synthesis of indolizines via C–H functionalization of pyridine. | |
Very recently, electrochemical annulation of pyridines was reported by Terent'ev and co-workers for the synthesis of imidazo[1,5-a]pyridines using NH4SCN as a cyanating agent (Scheme 14).49 A wide range of pyridine-2-carboxaldehydes and benzylamines, including chloro-substituted and heteroaryl benzylamines, was compatible and afforded the desired products (59–63) in good to moderate yields. Control experiments suggested that cyanide species were generated prior to core formation, with no involvement of free thiocyanogen. Mechanistically, anodic oxidation of thiocyanate in the presence of water produces the cyanide anion, which attacks the imine intermediate I formed from pyridine-2-carboxaldehyde 64 and benzylamine 65, generating intermediate II. Subsequent oxidation of intermediate II with DMSO or Shono-type anodic-oxidation pathway, furnishes intermediate III, which isomerizes to IV. Finally, intramolecular cyclization of intermediate IV, followed by oxidation and deprotonation, yields the final product, 1-cyano-imidazo[1,5-a]pyridine 59.
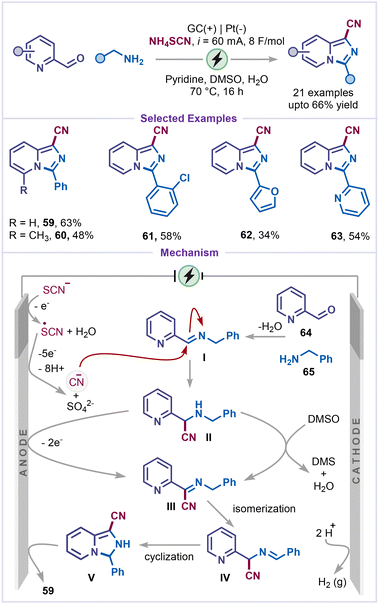 |
| | Scheme 14 Oxidative annulation of pyridine. | |
3.9 Dearomative functionalization
The ‘escape from flatland’ concept has been applied to pyridines to transform their planar aromatic cores into three-dimensional (3D) dehydropiperidine scaffolds with increased molecular complexity.50 Replacing a pyridine core in pharmaceuticals with an sp3-enriched piperidine counterpart can improve solubility and bioavailability.51 Motivated by this strategy, Ye and co-workers have very recently developed a regio- and diastereoselective dearomative multifunctionalization of pyridines using TMS-halide, TMSCN and alcohol under constant current electrolysis (Scheme 15).52 The method exhibits a broad substrate scope across various pyridine derivatives and alcohol nucleophiles; chloro- and bromo-silanes can be employed to facilitate dearomatization with moderate to excellent diastereoselectivity (products 66–68). The mechanism of this transformation begins with the activation of the pyridine 8 by cyanogen bromide, which is generated electrochemically in situ from TMSBr and TMSCN. The activated pyridinium intermediate I is then trapped by methanol to form the dearomative intermediate II. Subsequently, an anodically generated bromine radical adds to the nucleophilic enamine double bond in a regio- and diastereoselective manner, affording the radical intermediate III. Further anodic oxidation of radical III leads to the formation of the iminium ion IV, which is subsequently trapped by a nucleophile to afford the final product. The high diastereoselectivity is supported by DFT calculations, which indicate that the addition of the bromine radical to the enamine from the sterically less hindered face is energetically favored.
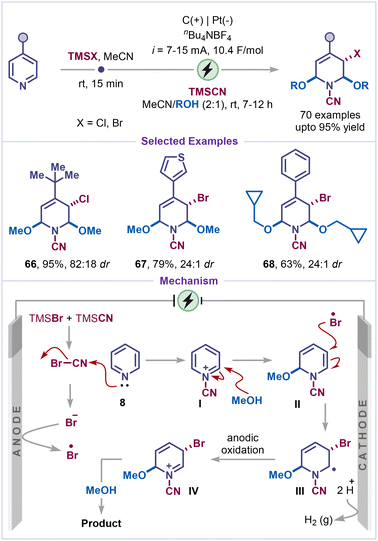 |
| | Scheme 15 Dearomative stereoselective multifunctionalization. | |
Collectively, net-oxidative electrochemical strategies for C–H bond functionalization of pyridines offer an attractive and sustainable alternative to conventional methods. In most reported systems, acidic additives are deliberately chosen to generate more electron-deficient pyridinium species, which enhance regioselective radical addition by increasing the electrophilicity of specific positions on the pyridine ring. Polar aprotic solvents (such as MeCN) are typically selected to stabilize charged intermediates and to buffer proton release during the rearomatization step. From a practical standpoint, robust electrode materials such as reticulated vitreous carbon (RVC), platinum, carbon, and graphite felt (GF) are preferred due to their stability under oxidative conditions and corrosion resistance; at the cathode, these electrodes also enable efficient proton reduction, thereby maintaining charge balance throughout the electrochemical process.
4. Functionalization via net reduction
In contrast to the extensively studied net oxidative pathways, net reductive electrochemical methods provide an equally versatile and sustainable platform for pyridine functionalization. These approaches proceed via distinct mechanistic pathways that involve radical or anionic intermediates generated by cathodic reduction. Therefore, C–X (X = H, Cl, Br, CN) bonds functionalization of pyridines can also be achieved efficiently via cathodic reduction, generating reactive intermediates that facilitate efficient C–C and C–heteroatom bond formation with high regioselectivity and broad functional group tolerance.
4.1 C–H deuteration
Due to the pronounced kinetic isotope effect, deuterated compounds are of great significance in synthetic chemistry, materials science, and pharmaceutical research.53 Although C–H/C–D exchange provides a straightforward route for deuterium introduction,54 traditional methods, such as transition-metal-catalysed exchange55 or deuterodehalogenation56 typically require noble metals, strong acids/bases, or organometallic reagents, and often suffer from limited regioselectivity, low incorporation efficiency, and a restricted substrate scope. In this context, Xiang and co-workers recently reported an electrocatalytic γ-selective deuteration of pyridines using D2O as the deuterium source (Scheme 16).57 A broad range of substituted pyridines bearing aryl and heteroaryl groups afforded the desired deuterated products (69–71) in excellent yields. The practicality of this strategy was further demonstrated through the late-stage deuteration of a pyridine-containing estradiol derivative 71. Mechanistically, the cathodic reduction of 2-phenyl pyridine 72 produced the pyridyl radical anion I, which underwent nucleophilic attack to D2O at the preferred C4-position to form intermediate II. Subsequent oxidation by molecular iodine or molecular oxygen afforded the deuterated pyridine 69. Molecular iodine is generated in situ from iodide at the anode.
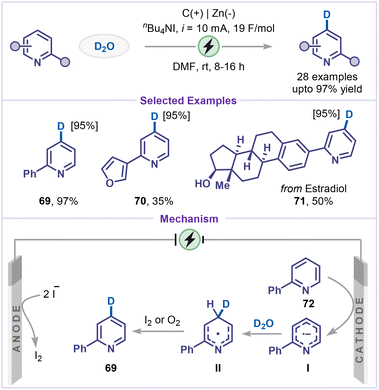 |
| | Scheme 16 C–H deuteration. | |
4.2 C–H carboxylation
Direct carboxylation of organic molecules using CO2 has emerged as a powerful strategy for the incorporation of carboxylic acid functionalities.58 In the context of pyridines, such transformations provide straightforward access to picolinic acid and nicotinic acid derivatives, which serve as valuable building blocks in pharmaceuticals and drug discovery.59 The first study in this area was reported by Zare and coworkers, who demonstrated that a nickel complex could efficiently catalyze the electrochemical reduction of CO2, generating a carboxylate radical anion as a key reactive intermediate.60 This species not only facilitated the indirect electroreduction of pyridines but also coupled directly with pyridyl radical anions, leading to the formation of the corresponding carboxylated pyridine derivatives. However, this reaction is limited to mechanistic studies; its synthetic potential is not explored.
In 2022, Qiu and co-workers made a significant breakthrough by reporting the first site-selective C4-carboxylation of pyridines utilizing CO2 as an inexpensive and sustainable carbon source (Scheme 17).61 This protocol is a metal-free, base-free and allows selective C–H carboxylation of electron-deficient pyridines, affording the products (73–75) with excellent chemo- and regioselectivity. Notably, 2,6-difluoropyridine was converted to 2,6-difluoronicotinic acid (73) in 76% yield rather than to 2,6-difluoroisonicotinic acid, which was likely influenced by the electron-withdrawing effects of the fluorine atoms. Similarly, 2,6-diphenylpyridine was transformed into 2,6-diphenylisonicotinic acid (74) under the same conditions. The reaction was initiated by the cathodic reduction of 2-phenyl pyridine 72, leading to the formation of a pyridyl radical anion I, which was subsequently added to CO2 to generate intermediate II. The high regioselectivity observed was attributed to the intrinsic electronic properties of the substrate. Intermediate II was then oxidized by iodine to produce III, accompanied by the loss of a proton. Finally, protonation afforded the carboxylic acid product 76. Meanwhile, Et4NI was oxidized at the anode, obviating the need for a sacrificial anode.
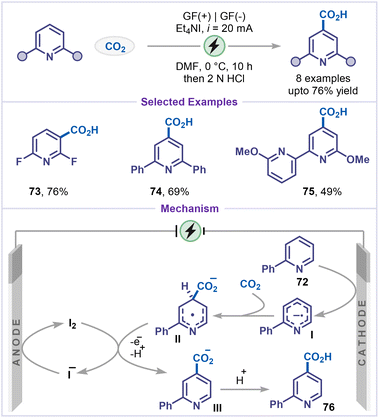 |
| | Scheme 17 Electro reductive C–H carboxylation. | |
Concurrently, Lin, Yu and co-workers developed an electrochemical approach that offers complementary site selectivity, in which regioselectivity is governed entirely by the cell configuration: C5-carboxylation is favored in divided cells, whereas C4-carboxylation predominates in undivided cells (Scheme 18).62 This strategy was successfully applied to a broad range of pyridines (77–80), bipyridine (81), and furan-substituted pyridine (82), demonstrating its wide applicability. For C5-carboxylation, the reaction is proposed to initiate via single-electron reduction of 2-phenyl pyridine 72, generating radical anion I. DFT calculations indicated that the highest electron density of I is located at C5, which undergoes nucleophilic addition to CO2 to form intermediate II. Subsequently, cathodic reduction produces dianion III, which is then oxidatively rearomatized by O2 and methylated in the presence of methyl iodide to furnish the corresponding methyl ester derivative 83. However, the addition of CO2 to intermediate I is reversible and not energetically favorable (endergonic). This reversibility allows the product ratio to depend on the next irreversible step according to the Curtin–Hammett principle. Additionally, DFT calculations showed that if CO2 were to add at the C4 position instead, the resulting intermediate would have a weaker C–H bond at that site (by approximately 3.8 kcal mol−1), thereby facilitating a hydrogen-atom transfer (HAT) process. So, if a hydrogen-atom acceptor were present, the reaction could be pushed toward C4-carboxylation instead of C5. Under undivided-cell conditions, iodine generated anodically from tetrabutylammonium iodide (nBu4NI) functioned as an efficient hydrogen-atom acceptor, engaging in either a direct H-atom transfer (HAT) mechanism or a proton-coupled electron-transfer (PCET) process with intermediate IV to produce intermediate V. Subsequent treatment with methyl iodide furnished the isonicotinic acid ester 84 as the final product. Despite its high C4-selectivity, the method showed reduced current efficiency and slower reaction rates due to continuous charge consumption in regenerating I2 instead of driving the desired transformation.
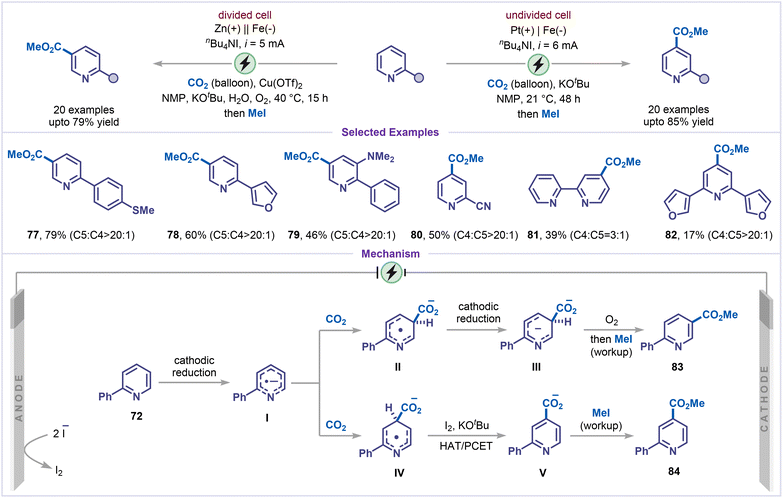 |
| | Scheme 18 Site-selective carboxylation of pyridines. | |
4.3 C–H silylation
Silylated pyridines are valuable synthetic intermediates, and they can also be efficiently accessed through cathodic reduction of pyridines. Recently, Zhang, Hao and co-workers reported an electrochemical C4-silylation of pyridines via a reductive dearomatization strategy in the presence of chlorosilanes (Scheme 19).63 This methodology was demonstrated to be broadly applicable, affording good yields for a wide range of pyridines bearing aryl, heteroaryl and estrone substituents (85–87). However, for a bulky silyl group, C5-silylated pyridine 88 was obtained as the major product. The reaction commenced with the formation of a pyridyl radical anion Ivia single electron-reduction, wherein the nitrogen atom, bearing the highest electron density on the pyridine ring, attacked the chlorosilane, followed by cathodic reduction produced the N-silylated intermediate II, when smaller chlorosilanes were employed. This intermediate II subsequently reacted with another chlorosilane at the C4 position to afford intermediate III, which, upon hydrolysis and rearomatization by oxidation, yielded the C4-silylated product with excellent regioselectivity. In contrast, when bulky chlorosilanes such as chlorotriisopropylsilane (TIPSCl) were used, steric hindrance disfavored the formation of intermediate VI. Instead, silylation occurred at the C5 position via intermediate IV, in which the most electron-rich carbon of the pyridine ring participated. The resulting intermediate V after hydrolysis and aerobic oxidation furnished the C5-silylated product.
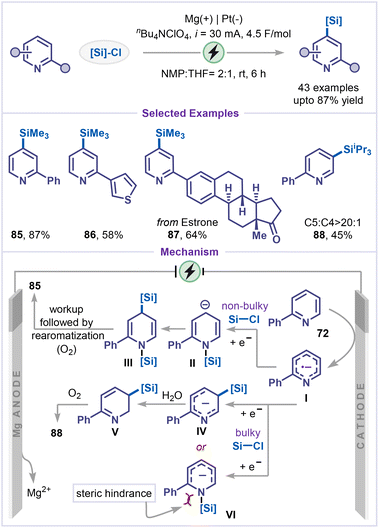 |
| | Scheme 19 Regioselective C–H silylation. | |
4.4 C–H alkylation
Although the alkylation of pyridines has been extensively explored through oxidative electrochemical Minisci-type reactions employing different alkyl radical precursors, these approaches often suffer from C2/C4 regioselectivity issues and overalkylation. Very recently, Zhang, Hao and co-workers furnished a silane-assisted electro reductive C4-alkylation of pyridines (Scheme 20).64 The reaction was compatible with a range of pyridines and alkyl halides, affording the corresponding alkylated pyridines (89–91) in good yields. The reaction was proposed to begin with the formation of a pyridinium intermediate I through the reaction of 2-phenyl pyridine 72 with chlorotrimethylsilane, which was subsequently reduced at the cathode to afford radical intermediate II, while magnesium was oxidized to Mg2+ at the anode. In parallel, cathodic reduction of cyclohexyl bromide 92 generates a cyclohexyl radical III. Radical–radical recombination produced intermediate IV. Subsequent hydrolysis of intermediate IV, followed by aerobic oxidation, furnished the desired C4-alkylated product 93.
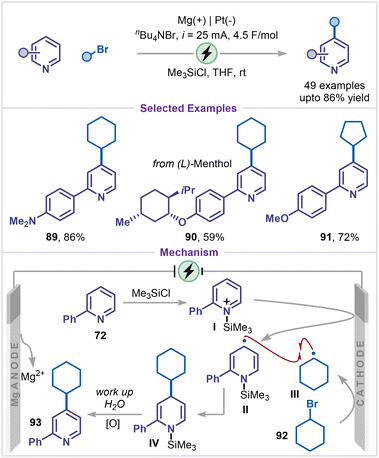 |
| | Scheme 20 C–H bond alkylation. | |
4.5 Decyanative alkylation
Following the electroreductive C–H bond functionalization strategy, electrochemical decyanative functionalization has emerged as another efficient approach for the selective bond functionalization of pyridines. Under electrochemical conditions, cyano-pyridines undergo single-electron reduction to form pyridyl radical anions, which can participate in a variety of bond-forming processes. Subsequent cyanide elimination restores aromaticity.
A pioneering advance was achieved with a nickel-catalyzed electroreductive alkylation of pyridines, employing 4-cyanopyridine and electron-deficient alkenes as coupling partners (Scheme 21).65 A diverse range of enones was explored, affording the corresponding alkylated pyridines (95–97) in moderate to excellent yields. When cinnamamide bearing a chiral auxiliary was employed, the alkylated product 97 was obtained in good yield with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric ratio. Mechanistically, the transformation can proceed through two distinct pathways: in path a, the alkene 98 undergoes cathodic reduction to generate a radical-anion species I, which tautomerizes to the radical-enolate intermediate II. This intermediate then couples with a nickel-bound pyridine complex III at the C4 position, forming intermediate IV. Subsequent reduction of IV produces the carbanion intermediate V, which undergoes cyanide elimination to afford the desired alkylated product 99. In path b, the enolate intermediate II engages in radical coupling with a pyridine radical anion VI, generated in situ from 4-cyanopyridine via cathodic reduction, to form adduct V, which then follows a similar cyanide elimination and protonation to deliver the final product.
:1 diastereomeric ratio. Mechanistically, the transformation can proceed through two distinct pathways: in path a, the alkene 98 undergoes cathodic reduction to generate a radical-anion species I, which tautomerizes to the radical-enolate intermediate II. This intermediate then couples with a nickel-bound pyridine complex III at the C4 position, forming intermediate IV. Subsequent reduction of IV produces the carbanion intermediate V, which undergoes cyanide elimination to afford the desired alkylated product 99. In path b, the enolate intermediate II engages in radical coupling with a pyridine radical anion VI, generated in situ from 4-cyanopyridine via cathodic reduction, to form adduct V, which then follows a similar cyanide elimination and protonation to deliver the final product.
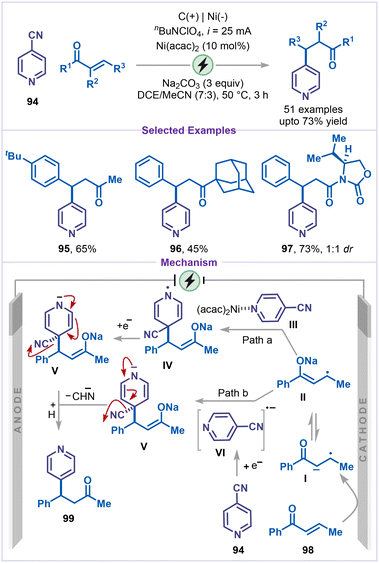 |
| | Scheme 21 Decyanative alkylation using alkene. | |
Rovis and co-workers developed an elegant electrochemical decyanative alkylation of pyridines employing iminium salts as alkylating agents (Scheme 22a).66 This strategy provided a direct and efficient route to pyridine-containing α-tertiary amines via a proton-coupled electron transfer (PCET) pathway, a transformation of high significance owing to the prevalence of such scaffolds in pharmaceuticals and agrochemicals. The method exhibited broad functional-group tolerance, accommodating pyridines substituted with halides and alkynes (100 and 101), and was equally effective with cyclic iminium salts, affording the corresponding cyclic amines (102 and 103). Mechanistically, the reaction proceeds through single-electron cathodic reduction of the iminium salt 104 to generate an α-amino radical I, while cyanopyridine 94 undergoes simultaneous proton-coupled electron transfer (PCET) to yield a pyridyl radical II. The radical–radical coupling between I and II affords intermediate III, which subsequently undergoes deprotonation followed by cyanide elimination to deliver the sterically hindered pyridyl amine 105.
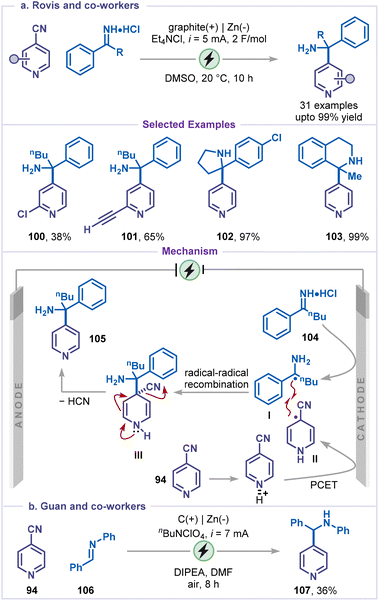 |
| | Scheme 22 Decyanative alkylation using imines. | |
Following this, Guan and co-workers developed an electrochemical protocol for the synthesis of benzylic secondary amines via the coupling of imines with 4-cyanopyridine (Scheme 22b).67 Under the standard reaction conditions, 4-cyanopyridine 94 afforded the corresponding aminated product 107, albeit in modest yield (36%). The reaction was initiated via reduction of both the 4-cyanopyridine 94 and the imine 106via SET to generate the pyridyl radical intermediate and α-amino radical, respectively. Finally, radical–radical cross-coupling followed by cyanide elimination delivered the desired product 107.
Next, electroreductive alkylation of 4-cyanopyridines using aldehydes and ketones as alkylating agents was reported by Yang and co-workers (Scheme 23).68 The reaction exhibited a broad substrate scope, efficiently converting a variety of aliphatic and aromatic aldehydes into the corresponding secondary alcohols (108–110), while ketones also participated to afford tertiary alcohol 111 in good yield. The reaction mechanism involved a ketyl radical intermediate I formed by cathodic reduction of carbonyl compound 112; concurrently, 4-cyanopyridine 94 was reduced to a persistent radical anion II. These radicals underwent radical–radical cross-coupling to form an intermediate III, which then eliminated cyanide to afford alcohol 110. The formation of both transient radical I and persistent radical II was proposed to occur preferentially at the cathode. At the anode, DABCO undergoes sacrificial oxidation, thereby providing the necessary electron balance to drive the cathodic reduction steps.
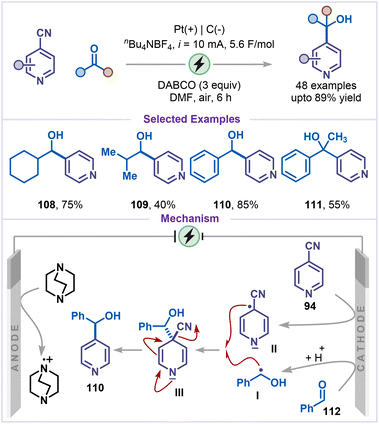 |
| | Scheme 23 Decyanative alkylation using aldehyde/ketones. | |
In a recent advance, Huang and co-workers developed a decyanative alkylation of cyanopyridines using unactivated alkyl bromides (Scheme 24).69 This operationally simple and scalable protocol featured a broad substrate scope including electron-withdrawing, electron-donating groups substituted pyridines, as well as primary, secondary, tertiary and cyclic alkyl bromides (products 113–117), without the need for a sacrificial anode. The transformation proceeded through single-electron reduction of Cs-coordinated cyanopyridines to pyridyl radical anion intermediate I, which subsequently, coupled with alkyl radical intermediate II generated from the corresponding alkyl bromides 118 by reduction at the cathode to give intermediate III. Finally, intermediate III underwent decyanation to furnish product 119.
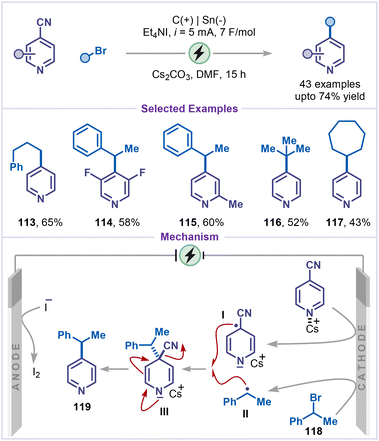 |
| | Scheme 24 Decyanative alkylation using alkyl bromides. | |
Very recently, a complementary decyanative alkylation of 4-cyanopyridines was disclosed by Zeng and co-workers using continuous-flow electrochemical methods (Scheme 25).70 Under flow constant current electrolysis, the combination of alkenes, aryl bromides, and cyanopyridines enabled efficient access to alkylated pyridine frameworks. Aryl bromides bearing electron-withdrawing substituents, such as an ester group, was well tolerated and delivered the corresponding product 120 in 73% yield. Notably, aryl bromides derived from L-menthol also participated effectively, providing the desired product 121 in 62% yield, highlighting the functional-group compatibility of the protocol. A plausible mechanistic pathway was proposed for the observed regioselectivity (Scheme 25). The process was initiated by cathodic reduction of the aryl bromide, followed by debromination to generate an aryl radical I, which subsequently added to the alkene to form a carbon-centred radical intermediate II. In parallel, cyanopyridine underwent cathodic reduction to produce a relatively persistent pyridyl radical anion III. Final radical–radical coupling between these two intermediates, followed by decyanation, furnished the alkylated pyridine product.
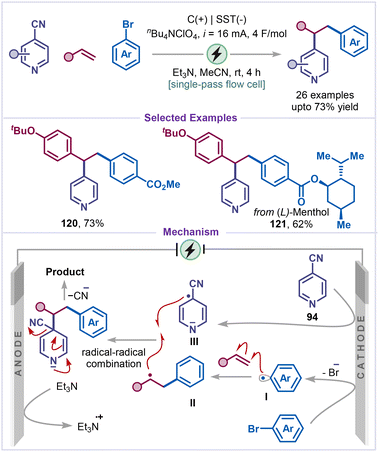 |
| | Scheme 25 Multicomponent decyanative alkylation using alkene. | |
4.6 Decyanative carboxylation
Very recently, Li and co-workers employed a decyanative strategy for the carboxylation of cyanopyridines (Scheme 26).71 A broad range of 4-cyanopyridines bearing various aryl substituents efficiently afforded the corresponding isonicotinic acid derivatives (122–125). The proposed mechanism for this transformation began with the cathodic reduction of 4-cyanopyridine 94, producing a pyridyl radical anion intermediate I that underwent carboxylation with CO2 to generate a cyano-1,4-dihydropyridine-4-carboxylate radical anion II. Further cathodic reduction of II formed the corresponding dianion III, which subsequently undergoes decyanation and protonation to afford the desired isonicotinic acid derivative 126.
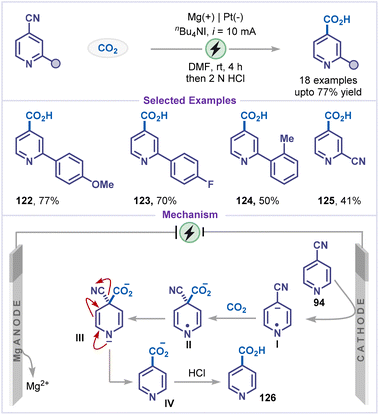 |
| | Scheme 26 Decyanative carboxylation using CO2. | |
4.7 Dehalogenative alkylation
Electrochemical dehalogenative functionalization has become a powerful method, allowing the otherwise inert C–halogen bond in pyridines to be effectively activated under electrochemical conditions, thereby facilitating a wide variety of C–C and C–heteroatom bond-forming reactions.72 In this context, Bio and co-workers reported an electrochemical dehalogenative alkylation of pyridines employing redox-active esters as alkylating reagents, highlighting the potential of electrochemical activation in promoting sustainable and selective pyridine functionalization (Scheme 27).73 Under constant-current electrolysis at 20 mA, this method afforded 3-alkylated pyridines in moderate yields (127 and 128). The reaction proceeds via the following mechanism: cathodic reduction of the redox-active ester results in decarboxylative fragmentation, generating radical II. Simultaneously, Ni(0) underwent oxidative addition with pyridine halides to form intermediate IV. The alkyl radical II was then captured by the nickel complex IV, affording intermediate V. Finally, reductive elimination from intermediate V delivered the C3-alkylated pyridine. The resulting Ni(I) species was subsequently reduced at the cathode, completing the catalytic cycle.
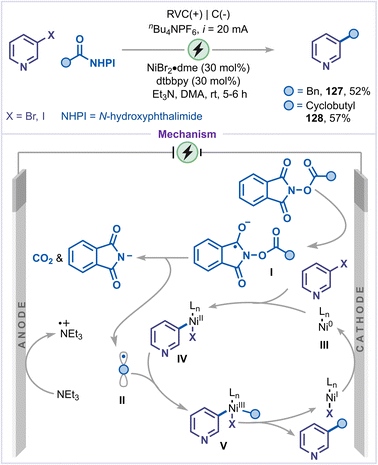 |
| | Scheme 27 Dehalogenative alkylation using redox-active ester. | |
4.8 Dehalogenative deuteration
In 2020, Zhang and co-workers reported an elegant electrochemical dehalogenative deuteration of arenes/pyridines using D2O as the deuterium source (Scheme 28).56a Control experiments with CD3CN/H2O established water as the exclusive hydrogen source. The proposed mechanism involves a cathodic reduction of halopyridine to generate pyridyl radicals, which subsequently couple with deuterium radicals produced via D2O splitting, supported by EPR studies. The suppression of product formation by hydrogen-radical scavenging with tBuOH further corroborated this mechanism, although direct deuterium abstraction from D2O could not be completely excluded. The reaction exhibited broad scope with aryl halides, and a few examples of pyridine substrates were reported in this study (129–132).
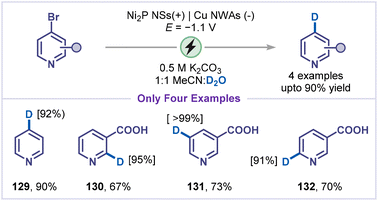 |
| | Scheme 28 Dehalogenative deuteration. | |
4.9 Dehalogenative phosphorylation
In 2020, Wickens and co-workers reported a strategy for phosphorylation of chloropyridine, where a photocatalyst was first activated electrochemically before photoexcitation, overcoming the energy limitations of visible-light photoredox catalysis (Scheme 29).74 This approach enabled radical generation from 2-chloropyridine (133), which are difficult to reduce owing to their high reduction potentials. The reaction is believed to initiate via cathodic reduction of the photocatalyst NpMI, generating a mild reducing radical anion intermediate I, which is then excited by light to form a highly reducing intermediate II. This excited photocatalyst intermediate II then reduced the pyridine chloride 133, producing pyridyl radical intermediate III. Radical III then added to the phosphorous atom of triethyl phosphite 134 to yield intermediate IV. Finally, the radical intermediate IV undergoes β-scission to give phosphorylated pyridine 135.
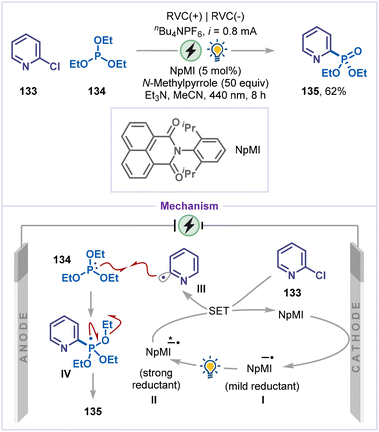 |
| | Scheme 29 Dehalogenative phosphorylation. | |
4.10 Dearomative carboxylation
Meanwell and co-workers developed a mild, highly chemoselective electrochemical Birch carboxylation of pyridines using CO2 as a sustainable C1 source, providing a greener and safer alternative to classical Birch reductions (Scheme 30).75 Electrolysis conducted in DMF using a magnesium sacrificial anode and a platinum cathode enabled selective reduction and C4-carboxylation of pyridines without affecting other aromatic rings, such as bis(trifluoromethyl)-, methylthio-substituted phenyl, and benzofuran moieties, affording the desired products in good yields (products 136–138). A plausible mechanism involves cathodic reduction of the pyridine substrate 139via a Birch-type electrochemical process to form a radical anion intermediate I. Delocalization of the resulting benzylic radical into the aryl substituent stabilizes the dearomatized species. Trapping of the nitrogen-centered anion by CO2 affords intermediate II, which, upon further cathodic reduction, generates a dianionic species III. Capture of a second equivalent of CO2 produces a dicarboxylated intermediate IV, followed by decarboxylation on work-up, furnishes the corresponding 1,4-dihydropyridine product 140.
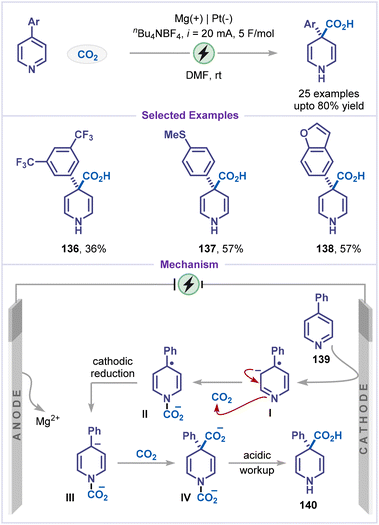 |
| | Scheme 30 Birch-type dearomative carboxylation. | |
From a practical standpoint, net-reductive methods enable diverse C–X (X = H, Cl, Br, CN) functionalization of pyridines by generating pyridyl radical anions via controlled single-electron transfer at the cathode. Polar aprotic solvents such as DMF and MeCN are typically selected because they stabilize charged intermediates, efficiently dissolve salts and CO2, and provide a wide electrochemical window under reductive conditions. Halide-containing supporting electrolytes are often preferred, as they can participate in the overall redox balance and, in some systems, in situ-generated molecular iodine facilitates rearomatization. Carbon-based electrodes paired with oxidizable electron donors such as amines, or sacrificial metal (e.g., magnesium), are commonly employed as anodes, while metal-based electrodes are typically used at the cathode. Furthermore, the choice of electrochemical cell: a divided versus an undivided cell can be used to control regioselective functionalization.
5. Functionalization via paired electrolysis
Paired electrolysis is an electrochemical strategy in which both anodic and cathodic reactions are utilized productively within a single operation. By enabling simultaneous redox events, it maximizes energy efficiency and allows the formation of multiple bonds or products in one step. This approach expands the synthetic scope by integrating complementary oxidation and reduction processes, providing a sustainable and versatile platform for complex molecular transformations.
5.1 C–H deuteration
A direct, efficient, and regioselective C4–H deuteration strategy for pyridines was reported by Qiu and co-workers (Scheme 31).76 Under optimized conditions, a wide range of pyridines and bipyridines were successfully deuterated with excellent yields and high deuterium incorporation (69 and 141). Moreover, the late-stage functionalization of pharmaceutically relevant molecules, including carveol (product 142), further underscored the synthetic utility and practical relevance of this approach. Mechanistic studies supported by cyclic voltammetry suggested the following pathway for deuteration: initially, nBu4NI dissociates to furnish nBu3N (I) and nBuI (II). The nBu3N (I) undergoes anodic oxidation to generate a radical cation III, thereby delivering electrons to the electrochemical cycle. Concurrently, nBuI and pyridine derivative 72 react to give the pyridinium salt IV, which undergoes single-electron reduction at the cathode to form a radical species V. The radical V is further reduced to an anionic intermediate VI. Subsequent reaction of VI with D2O provides a deuterated intermediate VII, which undergoes anodic oxidation to furnish the desired deuterated pyridine 69.
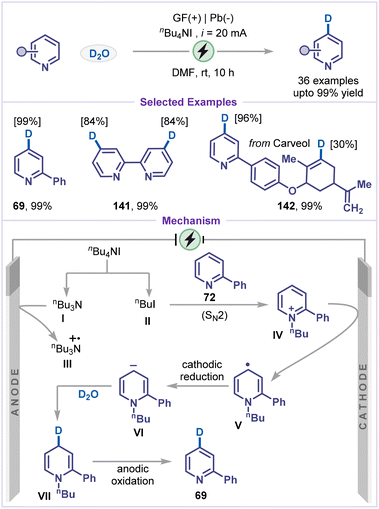 |
| | Scheme 31 C–H bond deuteration via paired electrolysis. | |
5.2 C–H alkylation
Paired electrolysis also enabled C–H alkylation of pyridines under mild conditions. Chen and co-workers reported the first electrochemical deaminative alkylation of pyridines, employing Katritzky salts as alkyl radical precursors (Scheme 32).77 The methodology exhibited a limited scope with respect to pyridine substrates; however, it is applicable to structurally complex pyridine derivatives, affording the corresponding C2- or C4-alkylated products (143–145), albeit in modest yields. A plausible mechanism involved the single-electron reduction of the Katritzky salt 146 at the cathode, leading to the formation of the persistent radical species I, which was subsequently exergonically fragmented to produce the alkyl radical II. The radical then slowly underwent an energy-demanding addition to the pyridinium salt IV, forming intermediate V. Simultaneously, a relatively rapid and exergonic radical–radical combination between I and II generated intermediate III. The reduction of intermediate V was achieved by the excited state photocatalyst [RuII(bpy)3]*, resulting in the formation of intermediate VI and [RuIII(bpy)3]. The oxidized [RuIII(bpy)3] species could oxidize intermediate III, thereby generating the alkyl radical II and completing the photocatalytic cycle. Finally, intermediate VI was oxidized at the anode to furnish the desired product 147.
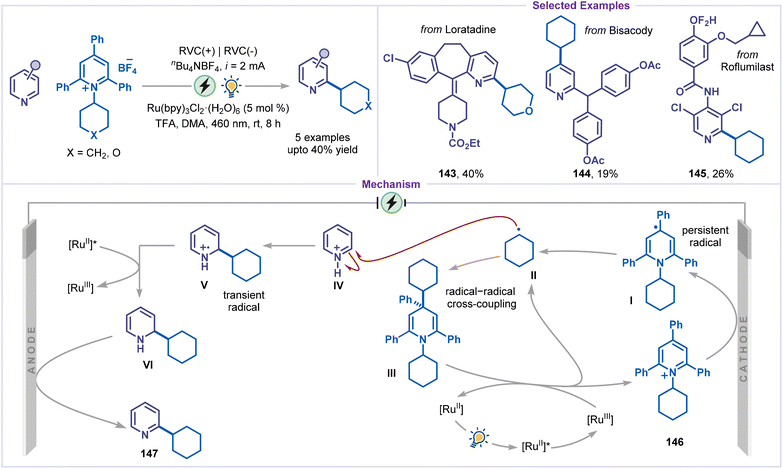 |
| | Scheme 32 Redox-neutral C–H alkylation using Katritzky salts. | |
Cheng and co-workers further utilized a paired electrolysis strategy for developing a protocol for meta-C–H alkylation of 4-cyanopyridines with alkyl halides (Scheme 33).78 Using this strategy, the electrochemical alkylation of pyridines with diverse alkyl bromides, including primary, secondary, and tertiary alkyl bromides, was well tolerated, affording the corresponding products 148–151 in moderate to good yields. The transformation proceeds through the cathodic reduction of 4-cyanopyridine 94, generating the radical anion I, which undergoes meta-selective SN2 alkylation with primary alkyl bromides to afford intermediate II. Subsequent anodic oxidation of intermediate II furnishes the desired product. In contrast, secondary and tertiary bromides undergo single-electron reduction to yield the alkyl radical III, which couples with the radical anion IV, a resonance form of I, to produce the anion V. Finally, anodic oxidation of V delivers the alkylated pyridine. The observed meta-selectivity arises from the highest charge and spin density localised at the meta-position of radical anion I, favouring SN2 substitution with primary alkyl bromides. For secondary and tertiary bromides, the meta-substituted intermediate V is energetically more stable, suggesting that post-transition-state bifurcation may contribute to the origin of selectivity.
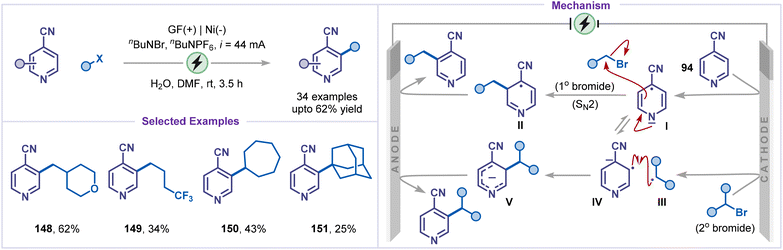 |
| | Scheme 33
Meta-C–H bond alkylation via paired electrolysis. | |
5.3 C–H amination
Malapit and co-workers developed a site-selective electrochemical protocol for the direct amination of aromatic C–H bonds, including pyridines (Scheme 34).79 Under optimized electrochemical conditions, selective functionalization of the meta-C–H bond was observed. Interestingly, substituted pyridine, bipyridine and terpyridine exclusively afforded the corresponding DABCOnium salts 153–155 in moderate to good yields (43–68%). Mechanism of the reaction starts with the single-electron reduction of selectfluor II reagent 152 at the cathode to produce an N-radical dication intermediate I, which engages in a charge-transfer interaction with pyridine and leads to C–N bond formation. This process generated the radical intermediate III, which was then anodically oxidized to give intermediate IV. Finally, deprotonation of IV by a base afforded the pyridine-DABCOnium product 156. Furthermore, it is proposed that triethylamine may function as a sacrificial reductant through anodic oxidation, thereby promoting the efficient reduction of selectfluor II.
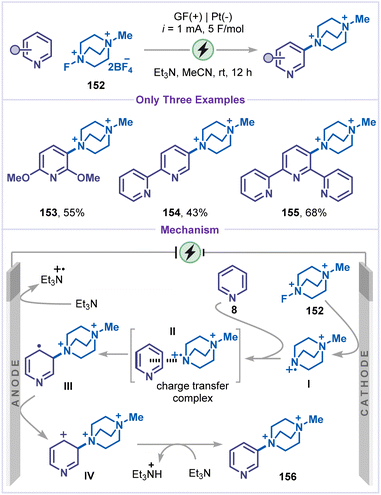 |
| | Scheme 34 C–H amination via paired-electrolysis. | |
5.4 C–H halogenation
Lei and co-workers reported an electrochemical C–H halogenation of 2-aminopyridine 53 by employing a paired electrolysis approach (Scheme 35).80 Using CBr4 (157) as the brominating agent, constant-current electrolysis (12 mA) was performed with a carbon rod as the anode, a nickel plate as the cathode, and nBu4NBF4 as the electrolyte, converting 2-aminopyridine 53 into the corresponding brominated product 54. Mechanistic studies suggest that under electrolysis, CBr4 is reduced at the cathode to generate both bromide ions and tribromomethyl radicals. The bromide ions are subsequently oxidized at the anode to form Br2, which efficiently brominates 2-aminopyridine via electrophilic bromination.
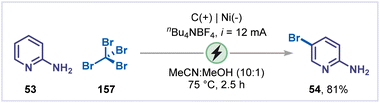 |
| | Scheme 35
Meta-bromination via paired electrolysis. | |
5.5 Dehalogenative alkylation
Ye and co-workers reported an electrochemical aminomethylation of halopyridines catalyzed by nickel (Scheme 36).81 C2 and C4-alkylated pyridines 161 and 162 were obtained from the corresponding bromopyridines 158 and 159. Mechanistically, Ni(II) was first reduced at the cathode via a sequential two-electron transfer to generate Ni(0), which subsequently underwent a rapid Ni(0)/Ni(II) comproportionation to yield Ni(I) species. The oxidative addition of Ni(I) to the bromo-pyridine 158 produced the intermediate I, which was further reduced at the cathode to form intermediate II. Simultaneously, the α-amino carbon radical III was generated via anodic oxidation followed by deprotonation of tertiary-amine 160, which was captured by intermediate II to furnish intermediate IV. Finally, reductive elimination from intermediate IV affords the coupling product 153, thereby regenerating the Ni(I) complex.
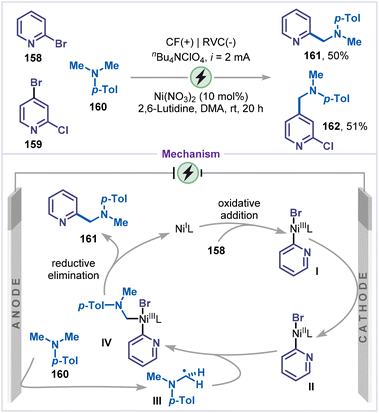 |
| | Scheme 36 Dehalogenative alkylation via paired electrolysis. | |
Given the significance of fluorinated moieties in drug discovery, and their ability to improve pharmacokinetic profiles as well as key physicochemical properties when incorporated into therapeutic or diagnostic molecules, Zhang, Wang and co-workers established a nickel-catalyzed electrochemical fluoroalkylation of iodo-pyridines using sodium difluoromethanesulfinate 165 as the fluoroalkylating reagent (Scheme 37).82 Under mild electrochemical conditions, 3-iodo-pyridine (163) and 2-methoxy-3-iodo-pyridine (164) are converted to fluoroalkylated pyridines 166 and 167 in moderate yields. Based on cyclic voltammetry and electron spin resonance (ESR) spectroscopy, two possible pathways were proposed for this electrochemical fluoroalkylative coupling. Firstly, 3-iodo-pyridine 163 undergoes oxidative addition with Ni(I) to form the intermediate I, which was subsequently reduced at the cathode to generate the Ni(II) species II. Alternatively, Ni(I) was first cathodically reduced to Ni(0), which then underwent oxidative addition with 163 to generate intermediate II. Simultaneously, sodium fluoroalkyl sulfinate 165 was anodically oxidized to produce the fluoroalkyl radical III, which was captured by the Ni(II) species II to form intermediate IV. Finally, reductive elimination from intermediate IV affords the fluoroalkylated product 166 and regenerates Ni(I).
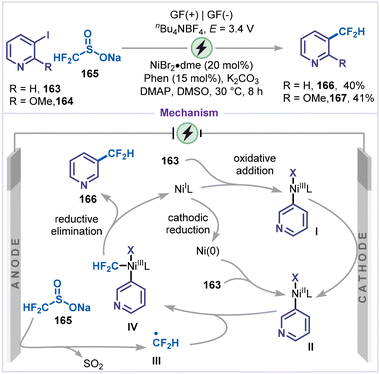 |
| | Scheme 37 Dehalogenative fluoroalkylation via paired electrolysis. | |
5.6 Decyanative arylation
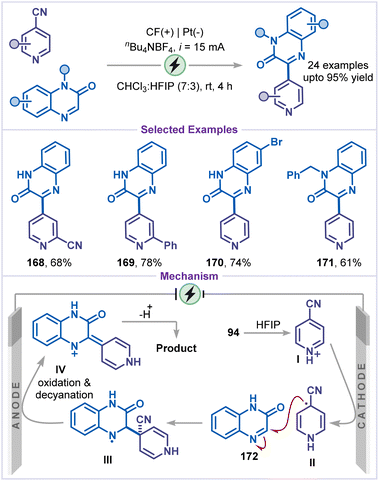
In 2021, Yang and co-workers reported decyanative arylation of pyridines using quinoxalin-2(1H)-ones as a coupling partner (Scheme 38).83 This method demonstrated excellent substrate scope, enabling the synthesis of the desired products bearing various functional groups, including cyano, bromo, and N-benzyl (168–171). The reaction mechanism involves protonation of 4-cyanopyridine (94) with HFIP, followed by cathodic reduction to generate the radical intermediate II. This radical species then adds to substrate 172, forming the nitrogen-centred radical intermediate III, which undergoes anodic oxidation, followed by decyanation to yield the cationic intermediate IV. Finally, intermediate IV undergoes isomerization and deprotonation, affording the desired product.
 |
| | Scheme 38 Decyanative arylation using quinoxalin-2(1H)-ones. | |
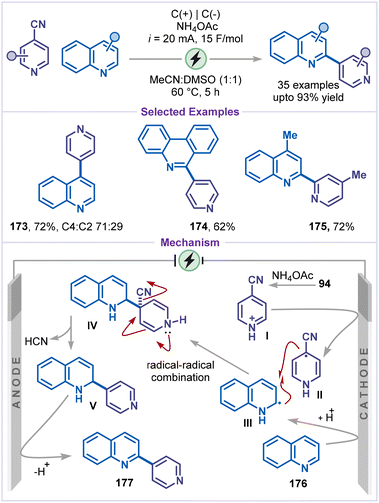
Another strategy for the electrochemical arylation of pyridine was developed by Wen and co-workers by employing quinoline as the arylating agent (Scheme 39).84 Under constant-current electrolysis in an undivided cell, a variety of heterocycles, including quinoline and phenanthridine, were reacted with 4-cyanopyridines to afford 4-arylated pyridines (173–175) in good to excellent yields. Mechanistically, the 4-cyanopyridinium salt I was cathodically reduced to generate the radical intermediate II, while quinoline 176 was reduced to form the radical III. Radicals II and III subsequently underwent a radical–radical coupling to produce intermediate IV, which, upon elimination of HCN, generated intermediate V. Finally, the desired arylated pyridine product 177 was obtained via two-electron anodic oxidation and proton elimination from intermediate V.
 |
| | Scheme 39 Decyanative arylation using quinoline. | |
5.7 Dehalogenative etherification
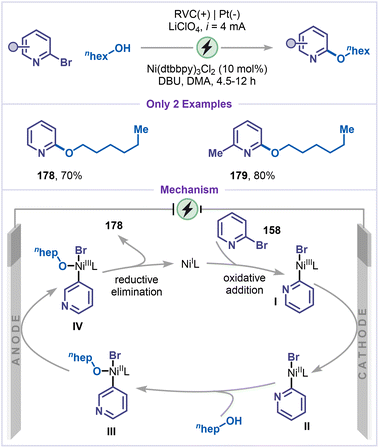
Recent advances in electrochemical etherification have enabled site-selective alkoxylation of arenes, providing a practical and sustainable alternative to conventional ether-forming methods. In this context, Baran and co-workers developed a nickel-catalyzed electrochemical etherification of pyridines and arenes with alcohols as an efficient replacement for classical SNAr strategies (Scheme 40).85 The reaction proceeds under mild conditions, using DBU as the base and nBu4NBr as the electrolyte, affording desired alkoxy-pyridines 178 and 179 in good yields. The mechanism of this C–O cross-coupling is particularly intriguing. The process begins with the cathodic reduction of Ni(II) to generate an active Ni(I) species, which undergoes oxidative addition with 2-bromopyridine (158) to form the Ni(III) complex I. Subsequent reduction of intermediate I generates the Ni(II) species II, which then undergoes ligand exchange with the alcohol to afford intermediate III. Because C–O reductive elimination from the Ni(II)-alkoxy complex III is thermodynamically and kinetically unfavourable at room temperature, and therefore anodic oxidation is employed to generate the Ni(III)-alkoxy species IV. From this higher-valent intermediate IV, facile reductive elimination efficiently affords the C–O coupled product 178.
 |
| | Scheme 40 C–O bond formation from bromopyridines. | |
5.8 Dehalogenative phosphorylation
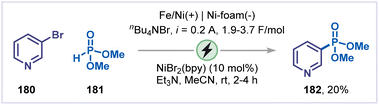
Parallel to the development of C–X bond alkylation and arylation, electrochemical strategies have also been applied to the phosphorylation of pyridines under mild and sustainable conditions. In 2018, Léonel and co-workers reported the first electrochemically assisted Ni-catalyzed phosphorylation of aryl halides (Scheme 41).86 When 3-bromopyridine (180) was reacted with dimethyl phosphonate (181) under constant-current electrolysis (0.2 A) using an iron/nickel rod as the anode and nickel foam as the cathode, the corresponding 3-phosphonylated pyridine 182 was obtained in 20% yield. The reaction was proposed to begin with the cathodic reduction of the Ni(II) pre-catalyst to Ni(0), followed by oxidative addition with the halide. The resulting Ni(II)-aryl complex undergoes single electron reduction to form Ni(I)-aryl complex, followed by ligand exchange with dimethyl phosphonate form a phosphoryl–nickel(I) intermediate. Subsequent anodic oxidation, followed by reductive elimination, furnished the desired product 182, regenerating the Ni(0) catalyst.
 |
| | Scheme 41 Phosphorylation via paired electrolysis. | |
5.9 Annulation reactions
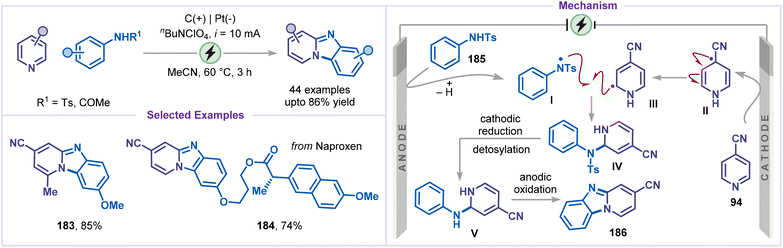
In 2021, Li and co-workers developed an electrochemical intermolecular C–H amination strategy for the coupling of pyridines and anilines, providing an efficient route to benzo[4,5]imidazo[1,2-a]pyridines (Scheme 42).87 The protocol features a broad substrate scope and excellent tolerance of diverse functional groups, enabling streamlined access to a variety of fused heterocyclic frameworks. A variety of substituted pyridines and anilines underwent smooth annulation, affording the desired products in excellent yields (183 and 184). The proposed mechanism begins with anodic oxidation and proton elimination of aniline 185 to generate an nitrogen-centered radical I, and a simultaneous cathodic reduction of the 4-cyano pyridine 94, generating the radical species II. Two radical species, I and II, then recombine to form the intermediate IV, which, on subsequent detosylation via cathodic reduction generates the intermediate V. Finally, anodic oxidation of intermediate V, followed by cyclization, affords the annulated product 186. However, this transformation is limited by its requirement for an electron-withdrawing substituent (e.g., CN or CO2Me) at the C4-position of the pyridine ring, as well as the need for an electron-withdrawing group (e.g., Ts or COMe) on the nitrogen atom of the aniline to ensure successful reaction outcomes.
 |
| | Scheme 42 Synthesis of benzo[4,5]imidazo[1,2-a]pyridines via paired electrolysis. | |
In paired electrolysis strategies, the choice of electrode is particularly critical, as oxidative and reductive events occur concurrently within the same electrochemical system. Carbon-based materials such as carbon, graphite, and reticulated vitreous carbon (RVC) are typically selected as anodes due to their stability under oxidative conditions, whereas nickel, platinum, or metal foams are commonly employed as cathodes owing to their high conductivity and efficient electron-transfer characteristics under reductive conditions. Polar aprotic solvents such as MeCN, HFIP, DMSO, and DMF are frequently chosen because they stabilize radical and ionic intermediates, ensure effective dissolution of electrolytes and substrates, and provide a broad electrochemical window compatible with simultaneous anodic and cathodic processes.
6. Conclusion and outlook
In this review, we have charted the development of electrochemical strategies for pyridine functionalization with a focus on challenges and design principles associated with pyridines, from the earliest pioneering studies to the most recent advances, offering researchers an in-depth perspective on the diverse approaches employed. Beyond these foundational methods, the integration of electrochemistry with photocatalysis and transition-metal catalysis has further enhanced efficiency, selectivity, and sustainability, paving the way for innovative strategies in pyridine derivatization. The synthetic power of these methods is exemplified by their successful application in the late-stage functionalization of complex, pyridine-containing molecules, underscoring their growing relevance to pharmaceutical, agrochemical, and materials research. Such developments not only expand the synthetic utility of electrochemical pyridine functionalization but also promise to unlock entirely new reactivity paradigms. Nevertheless, the studies highlighted in this review reveal that the field of electrochemical pyridine functionalization remains comparatively underdeveloped, with several important areas still requiring systematic exploration.
(1) A significant number of studies summarized in this review utilize pre-functionalized pyridines (e.g., cyano- or halo-substituted derivatives) as starting materials. To further broaden the synthetic versatility and practical applicability of these electrochemical strategies, the direct C–H functionalization of pyridines remains a highly desirable and impactful goal.
(2) Several valuable functional groups, such as trifluoromethyl, thiotrifluoromethyl, nitro and azide have not yet been introduced into pyridine frameworks via electrochemical methodologies. Therefore, developing efficient and general electrochemical protocols for incorporating these functionalities is an important direction for future research.
(3) Despite significant progress in alkylation and arylation cross-coupling reactions of pyridines, alkenylation, alkynylation, and allenylation processes remain unexplored. The development of such transformations would enable greater molecular complexity and structural diversity, thereby expanding the synthetic utility of electrochemical pyridine functionalization.
(4) To date, there are no reported examples of asymmetric electrochemical functionalization of pyridines employing chiral ligands or catalysts, highlighting a significant opportunity for advancement toward enantioselective electrochemical methodologies.
Taken together, these observations highlight the pressing need for continued innovation in catalyst design, reaction development, and mechanistic investigation to fully realize the potential of electrochemical strategies in pyridine functionalization. Overall, we anticipate that bringing together these key historical and recent advances will inspire researchers in both organic synthesis and medicinal chemistry to adopt electrochemical approaches for accessing functionalized pyridines.
Author contributions
S. B. and T. S. prepared schemes and wrote the manuscript. N. B. did a literature survey and helped in preparing the initial draft. D. P. H. wrote and corrected the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Data availability
No new primary research results, software, or code were included, and no data were generated or analyzed as part of this review.
Acknowledgements
Financial support from the Anusandhan National Research Foundation (ANRF), Government of India (File ANRF/ARG/2025/006468/CS) is gratefully acknowledged. D. P. H. thanks IISc Bangalore for the infrastructure. S. B. thanks Council of Scientific & Industrial Research (CSIR), Government of India (File 09/0079(24039)/2025-EMR-I) for the post-doctoral fellowship. T. S. thanks the Ministry of Education, Government of India, for the Prime Minister's Research Fellowship.
References
- A. A. Altaf, A. Shahzad, Z. Gul, N. Rasool, A. Badshah, B. Lal and E. Khan, J. Drug Des. Med. Chem, 2015, 1, 1–11, DOI:10.11648/j.jddmc.20150101.11.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274, DOI:10.1021/jm501100b.
-
S. Pal, in Pyridine: A Useful Ligand in Transition Metal Complexes, ed. P. Pyridine and P. Pandey, IntechOpen, 2018, DOI:10.5772/intechopen.76986.
-
(a) J. K. Kallitsis, M. Geormezi and S. G. Neophytides, Polym. Int., 2009, 58, 1226–1233, DOI:10.1002/pi.2661;
(b) M. N. Zafar, A. H. Atif, M. F. Nazar, S. H. Sumrra, E. S. Gul and R. Paracha, Russ. J. Coord. Chem., 2016, 42, 1–18, DOI:10.1134/S1070328416010097.
-
(a) A. Klapars and E. Vedejs, Chem. Heterocycl. Compd., 2004, 40, 759–766, DOI:10.1023/B:COHC.0000040772.13427.0b;
(b) A. R. Katritzky, E. F. V. Scriven, S. Majumder, R. G. Akhmedova, A. V. Vakulenko, N. G. Akhmedov, R. Murugan and K. A. Abboud, Org. Biomol. Chem., 2005, 3, 538–541, 10.1039/B413285H;
(c) J. N. Levy, J. V. Alegre-Requena, R. Liu, R. S. Paton and A. McNally, J. Am. Chem. Soc., 2020, 142, 11295–11305, DOI:10.1021/jacs.0c04674;
(d) B. T. Boyle, J. N. Levy, L. de Lescure, R. S. Paton and A. McNally, Science, 2022, 378, 773–779, DOI:10.1126/science.add8980;
(e) L. R. Domingo and M. J. Aurell, New J. Chem., 2023, 47, 5193–5202, 10.1039/D2NJ05845F.
- A. Turck, N. Plé, F. Mongin and G. Quéguiner, Tetrahedron, 2001, 57, 4489–4505, DOI:10.1016/S0040-4020(01)00225-3.
- F. Marsais and G. Queguiner, Tetrahedron, 1983, 39, 2009–2021, DOI:10.1016/S0040-4020(01)91919-2.
- M. J. Dias Pires, D. L. Poeira and M. M. B. Marques, Eur. J. Org Chem., 2015, 2015, 7197–7234, DOI:10.1002/ejoc.201500952.
- F. Y. Zhou and L. Jiao, Synlett, 2020, 32, 159–178, DOI:10.1055/s-0040-1706552.
- K. Murakami, S. Yamada, T. Kaneda and K. Itami, Chem. Rev., 2017, 117, 9302–9332, DOI:10.1021/acs.chemrev.7b00021.
- H. Sindhe, M. M. Reddy, K. Rajkumar, A. Kamble, A. Singh, A. Kumar and S. Sharma, Beilstein J. Org. Chem., 2023, 19, 820–863, DOI:10.3762/bjoc.19.62.
- S. M. Manolikakes, N. M. Barl, C. Sämann and P. Knochel, Z. Naturforsch., B:J. Chem. Sci., 2013, 68, 411–422, DOI:10.5560/znb.2013-3061.
- L. Zhang, Z. Q. Wu and L. Jiao, Angew. Chem., Int. Ed., 2020, 59, 2095–2099, DOI:10.1002/anie.201912564.
- I. Habib, K. Singha and M. Hossain, ChemistrySelect, 2023, 8, e202204099, DOI:10.1002/slct.202204099.
-
(a) E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302–308, DOI:10.1021/acscentsci.6b00091;
(b) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319, DOI:10.1021/acs.chemrev.7b00397;
(c) S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018–6041, DOI:10.1002/anie.201712732;
(d) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619, DOI:10.1002/anie.201709766;
(e) Y. Yuan and A. Lei, Nat. Commun., 2020, 11, 802, DOI:10.1038/s41467-020-14322-z.
- S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. Kampf, Jpn. Chem. Rev., 2018, 118, 6706–6765, DOI:10.1021/acs.chemrev.8b00233.
- D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386–12400, 10.1039/D0SC01848A.
-
(a) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619, DOI:10.1002/anie.201711060;
(b) N. Li, R. Sitdikov, A. P. Kale, J. Steverlynck, B. Li and M. Rueping, Beilstein J. Org. Chem., 2024, 20, 2500–2566, DOI:10.3762/bjoc.20.214;
(c) F. A. M. Maria, B. S. Guilherme, B. Eliakin Sato de, B. Timothy John, M. M. Guilherme and T. d. O. Kleber, Chem. Synth., 2025, 5, 41, DOI:10.20517/cs.2024.177;
(d) M. K. Yadav and S. Chowdhury, Org. Biomol. Chem., 2025, 23, 506–545, 10.1039/D4OB01187B.
- Y. Inoue, S. Nagase, K. Kodaira, H. Baba and T. Abe, Bull. Chem. Soc. Jpn., 1973, 46, 2204–2207, DOI:10.1246/bcsj.46.2204.
- V. J. Davis, R. N. Haszeldine and A. E. Tipping, J. Chem. Soc., Perkin Trans. 1, 1975, 1263–1267, 10.1039/P19750001263.
- J. R. Ballinger, F. W. Teare, B. M. Bowen and E. S. Garnett, Electrochim. Acta, 1985, 30, 1075–1077, DOI:10.1016/0013-4686(85)80175-4.
- J. H. P. Utley and R. J. Holman, Electrochim. Acta, 1976, 21, 987–989, DOI:10.1016/0013-4686(76)85075-X.
- Y. G. Budnikova, Y. M. Kargin and V. V. Yanilkin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1992, 41, 1299–1300, DOI:10.1007/BF00864205.
- C. Amatore, C. Combellas, N. E. Lebbar, A. Thiébault and J.-N. Verpeaux, J. Org. Chem., 1995, 60, 18–26, DOI:10.1021/jo00106a009.
- C. Gosmini, S. Lasry, J. Y. Nedelec and J. Perichon, Tetrahedron, 1998, 54, 1289–1298, DOI:10.1016/S0040-4020(97)10225-3.
- K. W. R. de França, M. Navarro, É. Léonel, M. Durandetti and J. Y. Nédélec, J. Org. Chem., 2002, 67, 1838–1842, DOI:10.1021/jo016280y.
- K. W. R. de França, J. de Lira Oliveira, T. Florêncio, A. P. da Silva, M. Navarro, E. Léonel and J. Y. Nédélec, J. Org. Chem., 2005, 70, 10778–10781, DOI:10.1021/jo0517491.
- J. L. Oliveira, M. J. Silva, T. Florêncio, K. Urgin, S. Sengmany, E. Léonel, J. Y. Nédélec and M. Navarro, Tetrahedron, 2012, 68, 2383–2390, DOI:10.1016/j.tet.2012.01.017.
- E. L. Gall, C. Gosmini, J. Y. Nédélec and J. Périchon, Tetrahedron, 2001, 57, 1923–1927, DOI:10.1016/S0040-4020(01)00015-1.
- Y. Budnikova, Y. Kargin, J.-Y. Nédélec and J. Périchon, J. Organomet. Chem., 1999, 575, 63–66, DOI:10.1016/S0022-328X(98)00963-2.
- X.-L. Lai, X.-M. Shu, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2020, 59, 10626–10632, DOI:10.1002/anie.202002900.
-
(a) K. L. Billingsley, K. W. Anderson and S. L. Buchwald, Angew. Chem., Int. Ed., 2006, 45, 3484–3488, DOI:10.1002/anie.200600493;
(b) A. S. Guram, X. Wang, E. E. Bunel, M. M. Faul, R. D. Larsen and M. J. Martinelli, J. Org. Chem., 2007, 72, 5104–5112, DOI:10.1021/jo070341w;
(c) N. Kudo, M. Perseghini and G. C. Fu, Angew. Chem., Int. Ed., 2006, 45, 1282–1284, DOI:10.1002/anie.200503479;
(d) L.-C. Campeau and K. Fagnou, Chem. Soc. Rev., 2007, 36, 1058–1068, 10.1039/B616082D;
(e) U. Kiehne, J. Bunzen, H. Staats and A. Lützen, Synthesis, 2007, 2007, 1061–1069, DOI:10.1055/s-2007-965952;
(f) D. Zhao, J. You and C. Hu, Chem.–Eur. J., 2011, 17, 5466–5492, DOI:10.1002/chem.201003039;
(g) T. Markovic, B. N. Rocke, D. C. Blakemore, V. Mascitti and M. C. Willis, Chem. Sci., 2017, 8, 4437–4442, 10.1039/C7SC00675F.
- L. Massignan, C. Zhu, X. Hou, J. C. A. Oliveira, A. Salamé and L. Ackermann, ACS Catal., 2021, 11, 11639–11649, DOI:10.1021/acscatal.1c02516.
- L. Song, J.-L. Zhuang, P. Xiong and H.-C. Xu, Green Chem., 2025, 27, 10556–10561, 10.1039/D5GC03231H.
- Q.-L. Yang, X.-Y. Wang, J.-Y. Lu, L.-P. Zhang, P. Fang and T.-S. Mei, J. Am. Chem. Soc., 2018, 140, 11487–11494, DOI:10.1021/jacs.8b07380.
- G. Stewart, E. M. Alvarez, C. Rapala, J. H. Sklar, J. A. Kalow and C. A. Malapit, Nat. Synth., 2026, 5, 55–63, DOI:10.1038/s44160-025-00890-9.
- T. Stevens, K. Ekholm, M. Gränse, M. Lindahl, V. Kozma, C. Jungar, T. Ottosson, H. Falk-Håkansson, A. Churg, J. L. Wright, H. Lal and A. Sanfridson, J. Pharmacol. Exp. Ther., 2011, 339, 313–320, DOI:10.1124/jpet.111.182139.
- S. Qin, M. Yang, M. Xu, Z.-H. Peng, J. Cai, S. Wang, H. Gao, Z. Zhou, A. S. K. Hashmi, W. Yi and Z. Zeng, Nat. Commun., 2024, 15, 7428, DOI:10.1038/s41467-024-50644-y.
-
(a) Y. Nakao and T. Hiyama, Chem. Soc. Rev., 2011, 40, 4893–4901, 10.1039/C1CS15122C;
(b) A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388–405, DOI:10.1021/jm3010114;
(c) C. Cheng and J. F. Hartwig, Chem. Rev., 2015, 115, 8946–8975, DOI:10.1021/cr5006414.
- Q. Wan, Z.-W. Hou, X.-R. Zhao, X. Xie and L. Wang, Org. Lett., 2023, 25, 1008–1013, DOI:10.1021/acs.orglett.3c00144.
- Z.-X. Wang, D.-Y. Yang, Z. Zhang, P. Xu, F. Zhou, S. Yang and J. Zhou, J. Org. Chem., 2026, 91, 370–378, DOI:10.1021/acs.joc.5c02473.
-
(a) J. F. Bunnett and R. E. Zahler, Chem. Rev., 1951, 49, 273–412, DOI:10.1021/cr60153a002;
(b)
M. R. Grimmett, Halogenation of Heterocycles: II. Six- and Seven-Membered Rings, in Advances in Heterocyclic Chemistry, A. R. Katritzky, Academic Press, 1993, vol. 58, pp. 271–345, DOI:10.1016/S0065-2725(08)60288-3.
- T. V. Gryaznova, V. V. Khrizanforova, K. V. Kholin, M. N. Khrizanforov and Y. H. Budnikova, Russ. Chem. Bull., 2016, 65, 1798–1804, DOI:10.1007/s11172-016-1513-x.
- Y. Wu, S. Xu, H. Wang, D. Shao, Q. Qi, Y. Lu, L. Ma, J. Zhou, W. Hu, W. Gao and J. Chen, J. Org. Chem., 2021, 86, 16144–16150, DOI:10.1021/acs.joc.1c00923.
-
(a) M. Lebœuf, A. Cavé, A. Ranaivo and H. Moskowitz, Can. J. Chem., 1989, 67, 947–952, DOI:10.1139/v89-145;
(b) L.-L. Gundersen, C. Charnock, A. H. Negussie, F. Rise and S. Teklu, Eur. J. Pharm. Sci., 2007, 30, 26–35, DOI:10.1016/j.ejps.2006.09.006;
(c) J. P. Michael, Nat. Prod. Rep., 2008, 25, 139–165, 10.1039/B612166G.
- K. Bedjeguelal, H. Bienaymé, A. Dumoulin, S. Poigny, P. Schmitt and E. Tam, Bioorg. Med. Chem. Lett., 2006, 16, 3998–4001, DOI:10.1016/j.bmcl.2006.05.014.
-
(a) J. Wan, C.-J. Zheng, M.-K. Fung, X.-K. Liu, C.-S. Lee and X.-H. Zhang, J. Mater. Chem., 2012, 22, 4502–4510, 10.1039/C2JM14904D;
(b) J. H. Delcamp, A. Yella, T. W. Holcombe, M. K. Nazeeruddin and M. Grätzel, Angew. Chem., Int. Ed., 2013, 52, 376–380, DOI:10.1002/anie.201205007;
(c) Y. R. Song, C. W. Lim and T. W. Kim, Luminescence, 2016, 31, 364–371, DOI:10.1002/bio.2968.
- B. K. Malviya, A. K. Jassal, M. Karnatak, V. P. Verma and S. Sharma, J. Org. Chem., 2022, 87, 2898–2911, DOI:10.1021/acs.joc.1c02773.
- S. S. Grishin, O. M. Mulina, V. A. Vil' and A. O. Terent'ev, Org. Chem. Front., 2024, 11, 327–335, 10.1039/D3QO01690K.
-
(a) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756, DOI:10.1021/jm901241e;
(b) Y. Ling, Z.-Y. Hao, D. Liang, C.-L. Zhang, Y.-F. Liu and Y. Wang, Drug Des., Dev. Ther., 2021, 4289–4338, DOI:10.2147/DDDT.S329547.
- Q.-S. Chen, J.-Q. Li and Q.-W. Zhang, Pharm. Fronts, 2023, 05, e1–e14, DOI:10.1055/s-0043-1764218.
- Z. Wang, W.-F. Liang, C.-X. Gong, X. He, J.-H. Li, Y. Lin and K.-Y. Ye, CCS Chem., 2026, 1–19, DOI:10.31635/ccschem.025.202507069.
-
(a) A. Katsnelson, Nat. Med., 2013, 19, 656, DOI:10.1038/nm0613-656;
(b) C. Allais, J. M. Grassot, J. Rodriguez and T. Constantieux, Chem. Rev., 2014, 114, 10829–10868, DOI:10.1021/cr500099b;
(c) A. Mullard, Nat. Rev. Drug Discovery, 2016, 15, 219–221 CrossRef CAS PubMed.
- T. Junk and W. J. Catallo, Chem. Soc. Rev., 1997, 26, 401–406, 10.1039/CS9972600401.
-
(a) E. Alexakis, J. R. Jones and W. J. S. Lockley, Tetrahedron Lett., 2006, 47, 5025–5028, DOI:10.1016/j.tetlet.2006.05.106;
(b) H. Yang, C. Zarate, W. N. Palmer, N. Rivera, D. Hesk and P. J. Chirik, ACS Catal., 2018, 8, 10210–10218, DOI:10.1021/acscatal.8b03717.
-
(a) C. Liu, S. Han, M. Li, X. Chong and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 18527–18531, DOI:10.1002/anie.202009155;
(b) Y. Li, Z. Ye, Y.-M. Lin, Y. Liu, Y. Zhang and L. Gong, Nat. Commun., 2021, 12, 2894, DOI:10.1038/s41467-021-23255-0.
- L. Shi, M. Liu, L. Zheng, Q. Gao, M. Wang, X. Wang and J. Xiang, Org. Lett., 2024, 26, 4318–4322, DOI:10.1021/acs.orglett.4c01296.
-
(a) K. Huang, C. L. Sun and Z. J. Shi, Chem. Soc. Rev., 2011, 40, 2435–2452, 10.1039/C0CS00129E;
(b) J. D. Weaver, A. Recio III, J. A. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846–1913, DOI:10.1021/cr1002744;
(c) S. Wang and C. Xi, Chem. Soc. Rev., 2019, 48, 382–404, 10.1039/C8CS00281A;
(d) J. H. Ye, T. Ju, H. Huang, L. L. Liao and D. G. Yu, Acc. Chem. Res., 2021, 54, 2518–2531, DOI:10.1021/acs.accounts.1c00135;
(e) G.-Q. Sun, L.-L. Liao, C.-K. Ran, J.-H. Ye and D.-G. Yu, Acc. Chem. Res., 2024, 57, 2728–2745, DOI:10.1021/acs.accounts.4c00417.
-
(a) S.-s. Jew, B.-s. Park, D.-y. Lim, M. G. Kim, I. K. Chung, J. H. Kim, C. I. Hong, J.-K. Kim, H.-J. Park, J.-H. Lee and H.-g. Park, Bioorg. Med. Chem. Lett., 2003, 13, 609–612, DOI:10.1016/S0960-894X(02)01041-7;
(b) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537, DOI:10.1002/anie.201102010.
- H. Khoshro, H. R. Zare, A. A. Jafari and A. Gorji, Electrochem. Commun., 2015, 51, 69–71, DOI:10.1016/j.elecom.2014.11.025.
- Z. Zhao, Y. Liu, S. Wang, S. Tang, D. Ma, Z. Zhu, C. Guo and Y. Qiu, Angew. Chem., Int. Ed., 2023, 62, e202214710, DOI:10.1002/anie.202214710.
-
(a) G.-Q. Sun, P. Yu, W. Zhang, W. Zhang, Y. Wang, L.-L. Liao, Z. Zhang, L. Li, Z. Lu, D.-G. Yu and S. Lin, Nature, 2023, 615, 67–72, DOI:10.1038/s41586-022-05667-0;
(b) G. Zhong, Y. Huang and L. He, Chem. Synth., 2023, 3, 19, DOI:10.20517/cs.2023.03;
(c) M. Surke, R. Zhao and L. Ackermann, Sci. China:Chem., 2023, 66, 1549–1550, DOI:10.1007/s11426-023-1585-6.
- Z. Yang, X. Liu, T. Zhang, H. Maekawa, X.-Q. Hao and M.-P. Song, Org. Chem. Front., 2024, 11, 5545–5552, 10.1039/D4QO01066C.
- Z. Yang, X. Liu, Y.-X. Chen, Y. Gao, X. Xiao, H. Maekawa, T. Zhang, X.-Q. Hao and M.-P. Song, Sci. Adv., 2025, 11, eadz3401, DOI:10.1126/sciadv.adz3401.
- S. Zhang, L. Li, X. Li, J. Zhang, K. Xu, G. Li and M. Findlater, Org. Lett., 2020, 22, 3570–3575, DOI:10.1021/acs.orglett.0c01014.
- D. Lehnherr, Y.-h. Lam, M. C. Nicastri, J. Liu, J. A. Newman, E. L. Regalado, D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2020, 142, 468–478, DOI:10.1021/jacs.9b10870.
- W.-M. Zeng, Y.-H. He and Z. Guan, Org. Lett., 2022, 24, 7178–7182, DOI:10.1021/acs.orglett.2c02900.
- X. Zhang, C. Yang, H. Gao, L. Wang, L. Guo and W. Xia, Org. Lett., 2021, 23, 3472–3476, DOI:10.1021/acs.orglett.1c00920.
- W. Deng, X. Li, Z. Li, Y. Wen, Z. Wang, Z. Lin, Y. Li, J. Hu and Y. Huang, Org. Lett., 2023, 25, 9237–9242, DOI:10.1021/acs.orglett.3c03984.
- X. Sun, F. Meng, K. Xu and C. Zeng, Org. Lett., 2025, 27, 12784–12789, DOI:10.1021/acs.orglett.5c04052.
- J.-L. Sui, L. Zeng, M. Hu, F.-J. Sun, P.-Z. Cui and J.-H. Li, Org. Lett., 2025, 27, 11510–11514, DOI:10.1021/acs.orglett.5c03529.
- P. F. Li, G. S. Kou, L. P. Qi and Y. A. Qiu, J. Electrochem., 2024, 30, 2313005, DOI:10.61558/2993-074X.3442.
- H. Li, C. P. Breen, H. Seo, T. F. Jamison, Y.-Q. Fang and M. M. Bio, Org. Lett., 2018, 20, 1338–1341, DOI:10.1021/acs.orglett.8b00070.
- N. G. W. Cowper, C. P. Chernowsky, O. P. Williams and Z. K. Wickens, J. Am. Chem. Soc., 2020, 142, 2093–2099, DOI:10.1021/jacs.9b12328.
- S. Sarkar, Rohit and M. W. Meanwell, Green Chem., 2025, 27, 2900–2906, 10.1039/D4GC05976J.
- Z. Zhao, R. Zhang, Y. Liu, Z. Zhu, Q. Wang and Y. Qiu, Nat. Commun., 2024, 15, 3832, DOI:10.1038/s41467-024-48262-9.
- K. Wang, X. Liu, S. Yang, Y. Tian, M. Zhou, J. Zhou, X. Jia, B. Li, S. Liu and J. Chen, Org. Lett., 2022, 24, 3471–3476, DOI:10.1021/acs.orglett.2c01022.
- J. Pan and X. Cheng, ChemCatChem, 2025, 17, e202402139, DOI:10.1002/cctc.202402139.
- E. M. Alvarez, G. Stewart, M. Ullah, R. Lalisse, O. Gutierrez and C. A. Malapit, J. Am. Chem. Soc., 2024, 146, 3591–3597, DOI:10.1021/jacs.3c11506.
- Z. Zhou, Y. Yuan, Y. Cao, J. Qiao, A. Yao, J. Zhao, W. Zuo, W. Chen and A. Lei, Chin. J. Chem., 2019, 37, 611–615, DOI:10.1002/cjoc.201900091.
- Y. Ma, J. Hong, X. Yao, C. Liu, L. Zhang, Y. Fu, M. Sun, R. Cheng, Z. Li and J. Ye, Org. Lett., 2021, 23, 9387–9392, DOI:10.1021/acs.orglett.1c03500.
- Z. Zou, H. Li, M. Huang, W. Zhang, S. Zhi, Y. Wang and Y. Pan, Org. Lett., 2021, 23, 8252–8256, DOI:10.1021/acs.orglett.1c02997.
- J. Wen, X. Yang, K. Yan, H. Qin, J. Ma, X. Sun, J. Yang and H. Wang, Org. Lett., 2021, 23, 1081–1085, DOI:10.1021/acs.orglett.0c04296.
- C. Niu, J. Yang, K. Yan, J. Xie, W. Jiang, B. Li and J. Wen, iScience, 2022, 25, 104253, DOI:10.1016/j.isci.2022.104253.
- H. J. Zhang, L. Chen, M. S. Oderinde, J. T. Edwards, Y. Kawamata and P. S. Baran, Angew. Chem., Int. Ed., 2021, 60, 20700–20705, DOI:10.1002/anie.202107820.
- S. Sengmany, A. Ollivier, E. Le Gall and E. Léonel, Org. Biomol. Chem., 2018, 16, 4495–4500, 10.1039/C8OB00500A.
- M. J. Luo, X. H. Ouyang, Y. P. Zhu, Y. Li and J. H. Li, Green Chem., 2021, 23, 9024–9029, 10.1039/D1GC02922C.
Footnote |
| † Authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Tushar
Singha†
,
Tushar
Singha†













































