
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh energy density quasi-solid-state lithium batteries using in situ polymerized gel electrolytes
Zehui
Fan
a,
Yingxin
Liu
b,
Lanhua
Ma
a,
Hang
Liu
a,
Qinghao
Chen
a,
Yi
Wang
c,
Ewa
Zygadło-Monikowska
 d and
Yunhua
Xu
*a
d and
Yunhua
Xu
*a
aSchool of Materials Science and Engineering, State Key Laboratory of Advanced Materials for Intelligent Sensing, National Industry-Education Platform for Energy Storage, Tianjin University, Tianjin 300072, China. E-mail: yunhua.xu@tju.edi.cn
bSchool of Chemical Engineering and Technology, Tianjin Key Laboratory of Advanced Carbon and Electro-chemical Energy Storage, Tianjin University, Tianjin 300072, China
cCollage of Materials and Science Engineering, Guiyang University, Guiyang, Guizhou 550005, China
dFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland
First published on 8th April 2026
Abstract
Commercial lithium-ion batteries that use liquid electrolytes suffer from limitations in energy density and face significant safety concerns. The adoption of quasi-solid-state electrolytes, particularly when paired with high-voltage cathodes and high-capacity anodes, offers a promising approach to address these issues. Among these, in situ polymerized gel electrolytes have garnered considerable attention due to their excellent interfacial contact with electrodes and facile fabrication process. However, constructing in situ polymerized quasi-solid-state lithium batteries that simultaneously achieve high energy density and enhanced safety remains a significant challenge. Key obstacles include the strong oxidation of high-voltage cathodes, poor interfacial stability with high-capacity anodes, and the inherent safety risks associated with high-energy-density lithium batteries. Therefore, there is an urgent need to develop innovative solutions to overcome these challenges. This review provides a comprehensive summary of recent progress in this field. In addition, future perspectives on the development of high-energy-density, safe lithium batteries prepared via in situ polymerization are discussed.
1. Introduction
With the continuous growth of global energy demand, lithium batteries have become the dominant electrochemical energy storage technology due to their high energy density, long cycle life, low self-discharge rate, and lightweight characteristics. They are widely deployed in portable electronic devices, electric vehicles, and grid-scale energy storage systems.1,2 Beyond their technological significance, lithium batteries play a pivotal role in enabling the global low-carbon transition and mitigating climate change by reducing carbon emissions. Continuous advancements in lithium battery technology directly influence global carbon neutrality goals by accelerating the large-scale integration of renewable energy sources and the electrification of transportation.3,4Despite these advancements, commercial lithium batteries are increasingly constrained by insufficient performance and safety limitations. In particular, the energy density of conventional lithium batteries is approaching its theoretical ceiling, primarily due to the limited specific capacity of electrode materials and relatively low cut-off voltages.5–7 As the demand for higher energy density intensifies, especially for electric and unmanned aerial vehicles, the development of next-generation electrochemical energy storage systems has become an urgent priority.8,9 However, increasing energy density often comes at the expense of safety. Conventional organic liquid electrolytes are volatile and highly flammable, making them susceptible to leakage, thermal runaway, and catastrophic failure. These intrinsic safety risks severely hinder the deployment of lithium batteries in high-energy-density and safety-critical applications.10–13
To mitigate these issues, polymer electrolytes have emerged as promising alternatives to traditional liquid electrolytes.14–16 Owing to their superior mechanical strength, improved electrochemical stability, and reduced risk of leakage, polymer electrolytes offer a compelling pathway toward safer lithium batteries. Nevertheless, solid polymer electrolytes still suffer from inherent limitations.17,18 Their relatively low ionic conductivity (<1 mS cm−1) significantly restricts charge and discharge kinetics, rendering them unsuitable for high-energy-density or high-power applications. Moreover, the solid-state nature of polymer electrolytes often results in poor interfacial contact with electrode materials, leading to high interfacial resistance and degraded cycling stability under practical operating conditions.19
To address these issues, polymer gel electrolytes, also referred to as quasi-solid-state electrolytes, have been developed.20 By synergistically combining the advantages of solid and liquid electrolytes, polymer gel electrolytes incorporate plasticizers to markedly enhance ionic conductivity, while retaining improved mechanical integrity and safety.21 This balance between ionic transport and mechanical stability makes polymer gel electrolytes particularly attractive for advanced lithium battery systems.
Among the various polymer gel electrolyte systems, in situ polymerized gel electrolytes have emerged as a particularly promising strategy to address persistent interfacial challenges.22–24In situ polymerization enables the electrolyte to form directly within the assembled battery, ensuring intimate and conformal contact with electrodes. As a result, quasi-solid-state lithium batteries employing in situ polymerized gel electrolytes typically exhibit reduced interfacial impedance and more efficient Li+ transport across electrolyte/electrode interfaces. The homogeneous distribution of lithium salts and functional components within the polymer matrix further facilitates continuous and uniform Li+ transport pathways, thereby enhancing electrochemical performance. Additionally, the quasi-solid-state architecture minimizes electrolyte leakage and mechanical failure under stress, substantially improving safety even at elevated energy densities. Importantly, the simplicity and scalability of the in situ polymerization process offer clear advantages in manufacturing efficiency and cost reduction. In situ polymerized quasi-solid-state lithium batteries are widely regarded as promising candidates for next-generation high-performance energy storage systems.25–27
Despite these advantages, realizing in situ polymerized quasi-solid-state lithium batteries that simultaneously achieve high energy density and robust safety remains a formidable challenge (Fig. 1).28–30 To increase energy density, high-voltage, high-capacity cathode materials should be adopted, such as high-nickel layered oxides (NCM and NCA) and lithium cobalt oxide (LCO).31–33 However, their high cut-off voltages and strong catalytic activity inevitably accelerate electrolyte decomposition, leading to rapid interfacial degradation and capacity fading.34–37 On the anode side, replacing graphite with high-capacity alternatives such as lithium metal or silicon-based anodes can dramatically enhance energy density, but introduces severe challenges including uncontrolled side reactions, dendrite growth, and large volume changes.38–40 Furthermore, although in situ polymerized gel electrolytes significantly improve safety by reducing leakage and enhancing mechanical robustness, the long-term safety and reliability of high-energy-density quasi-solid-state lithium batteries based on this strategy remain insufficiently validated.41–44 Intensive research efforts have focused on the rational design and optimization of monomers, plasticizers, lithium salts, initiators, and polymerization mechanisms to address these interrelated challenges and unlock the full potential of in situ polymerized quasi-solid-state lithium batteries.45
 | ||
| Fig. 1 Schematic diagram showing the challenges for achieving in situ polymerized quasi-solid-state lithium batteries with high energy density and enhanced safety. | ||
This review provides a comprehensive overview of recent progress in quasi-solid-state lithium batteries fabricated via in situ polymerization. Strategies aimed at overcoming the critical challenges associated with achieving both high energy density and enhanced safety are systematically discussed, with particular emphasis on improving electrode/electrolyte interfacial stability, electrochemical performance, and battery safety. Finally, the remaining challenges and future research directions for high-energy-density and safe lithium batteries based on in situ polymerization are discussed to provide guidance for future developments in this rapidly evolving field.
2. In situ polymerized gel electrolytes
To fabricate quasi-solid-state lithium batteries via in situ polymerization, a homogeneous precursor solution containing monomers, plasticizers, lithium salts, and initiators is first prepared. After injection into the cells, quasi-solid-state electrolytes are subsequently formed through in situ polymerization reactions (Fig. 2).46 In contrast to polymer gel electrolytes prepared ex situ, the high fluidity of the precursor solution prior to polymerization enables intimate and conformal contact with electrode surfaces, thereby facilitating efficient Li+ transport across the electrolyte/electrode interface. Consequently, lithium batteries using in situ polymerized gel electrolytes typically exhibit significantly reduced interfacial impedance.22 | ||
| Fig. 2 Schematic illustration of quasi-solid-state lithium batteries fabricated via in situ polymerization. | ||
Benefiting from their high ionic conductivity, exceptional interfacial compatibility, and substantially reduced leakage risk, in situ polymerized gel electrolytes are regarded as promising candidates for the commercialization of high-performance quasi-solid-state lithium batteries.47,48 Notably, the electrochemical properties of the electrolytes are strongly governed by the chemical structure of the polymer matrix. A wide range of in situ polymerized gel electrolytes based on different monomers, including cyclic ethers, acrylates and carbonates, have been developed in recent years (Table 1).
| Monomer | Molecular formula | Monomer structure | Polymerization mechanism | Ref. |
|---|---|---|---|---|
| 1,3-Dioxolane (DOL) | C3H6O2 |

|
Ring-opening | 53 |
| 1,3,5-Trioxane (TXE) | C3H6O3 |
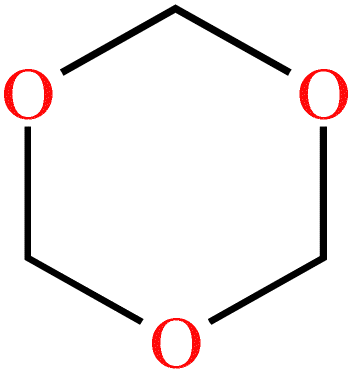
|
Ring-opening | 54 |
| Vinylene carbonate (VC) | C3H2O3 |

|
Free radical | 55 |
| Vinyl ethylene carbonate (VEC) | C5H6O3 |

|
Free radical | 56 |
| Methyl methacrylate (MMA) | C5H8O2 |

|
Free radical | 57 |
| Butyl acrylate (BA) | C7H12O2 |

|
Free radical | 58 |
Cyclic ethers are regarded as ideal monomers for in situ polymerized gel electrolytes due to their low viscosity and excellent reduction stability.49 These characteristics confer good compatibility with lithium metal anodes. Furthermore, cyclic ethers can undergo ring-opening polymerization at room temperature through a simple and efficient reaction pathway.50 This behavior arises from the presence of oxygen atoms with a lone pair of electrons, which are highly susceptible to cation attack, thereby initiating ring-opening polymerization. Consequently, protonic and Lewis acids are commonly employed as initiators for cyclic ether polymerization. The resulting C–O–C bonds in the cyclic ether-based polymer matrix can coordinate effectively with Li+, promoting ion dissociation and enhancing ionic conductivity.51 Despite these advantages, cyclic ether monomers face notable challenges in high-energy-density lithium batteries. Their limited oxidation stability leads to rapid electrolyte degradation when paired with high-voltage cathodes.52 In addition, the inherently uncontrollable nature of ring-opening polymerization complicates uniform precursor infiltration in pouch cells, resulting in incomplete wetting and non-uniform polymerization throughout the electrolyte layer.
In contrast, acrylate and carbonate monomers, ester-based compounds containing unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, form polymer gel electrolytes through free radical polymerization.59,60 This process is initiated by radical generation from thermal decomposition or ultraviolet irradiation of initiators, which subsequently attack the π-electron of the CC bonds, leading to chain propagation and eventual termination. Among these approaches, thermally initiated polymerization is particularly attractive due to its low cost and controllability with existing battery manufacturing processes. Polymer gel electrolytes derived from acrylate and carbonate monomers typically exhibit a high degree of polymerization and minimal residual monomer content. Furthermore, they demonstrate superior oxidation stability compared to cyclic ether-based electrolytes, making them more suitable for high-voltage cathodes.61,62 However, the CO bonds in the ester-based polymer matrix exhibit weaker coordination with Li+ than C–O–C bonds in the cyclic ether-based polymer matrix, resulting in limited ionic mobility.63 This insufficient Li+ transport capability significantly constrains the electrochemical performance of quasi-solid-state lithium batteries, particularly under high-rate and high-energy-density conditions.
C bonds, form polymer gel electrolytes through free radical polymerization.59,60 This process is initiated by radical generation from thermal decomposition or ultraviolet irradiation of initiators, which subsequently attack the π-electron of the CC bonds, leading to chain propagation and eventual termination. Among these approaches, thermally initiated polymerization is particularly attractive due to its low cost and controllability with existing battery manufacturing processes. Polymer gel electrolytes derived from acrylate and carbonate monomers typically exhibit a high degree of polymerization and minimal residual monomer content. Furthermore, they demonstrate superior oxidation stability compared to cyclic ether-based electrolytes, making them more suitable for high-voltage cathodes.61,62 However, the CO bonds in the ester-based polymer matrix exhibit weaker coordination with Li+ than C–O–C bonds in the cyclic ether-based polymer matrix, resulting in limited ionic mobility.63 This insufficient Li+ transport capability significantly constrains the electrochemical performance of quasi-solid-state lithium batteries, particularly under high-rate and high-energy-density conditions.
As a result, neither cyclic ethers nor ester monomers alone can fully satisfy the stringent requirements of high-energy-density, high-safety quasi-solid-state lithium batteries. To overcome these intrinsic trade-offs, hybrid strategies, such as combining multiple monomers, introducing crosslinkers, and optimizing plasticizer composition, are employed.64 Furthermore, advanced functional regulation of in situ polymerized gel electrolytes, including molecular-level structural design and interfacial chemistry engineering, remains essential to simultaneously enhance ionic conductivity, electrochemical stability, and safety.
3. Gel electrolyte and interphase design towards high-voltage cathodes
To meet the growing demand for enhanced battery performance across a range of applications, improving the energy density of lithium batteries has become a critical focus of technological development. Consequently, high-voltage cathodes have emerged as promising candidates for achieving elevated energy density, owing to their higher operating voltage and increased specific capacity (Fig. 3a).65 However, despite their potential, the practical application of high-voltage cathodes presents significant challenges.67 The high voltage, coupled with the strong catalytic activity of transition metals within the cathodes, leads to severe side reactions between the electrolytes and cathodes, resulting in the formation of an unstable cathode electrolyte interphase (CEI).68 Furthermore, the CEI is highly susceptible to disruption during cycling due to the volume changes in high-voltage cathodes. This disruption renders the CEI incapable of suppressing continuous electrolyte decomposition. Consequently, severe sides reactions are triggered, leading to gas generation, transition-metal ion dissolution, irreversible phase transition, and structure degradation of the cathodes, ultimately resulting in rapid performance deterioration.65 | ||
| Fig. 3 (a) Reversible capacities and working voltage windows of intercalation cathode materials. The gravimetric energy density is calculated based on cathode materials. Reproduced with permission.65 Copyright 2021, Royal Society of Chemistry. (b) Schematic open-circuit energy diagram of an electrolyte. Reproduced with permission.66 Copyright 2010, American Chemical Society. | ||
To address the aforementioned issues, various strategies have been developed in recent years. Although these approaches involve diverse mechanisms aimed at enhancing oxidation stability of polymer gel electrolytes, they can be classified into two major categories: (1) increasing the intrinsic oxidative stability of polymer gel electrolytes through modulation of the molecular structures of the polymer matrix and the solvents; (2) facilitating the formation of a high-quality CEI on the surfaces of high-voltage cathodes. In the following section, we discuss the research progress in enhancing the stability of in situ polymerized gel electrolytes towards high-voltage cathodes.
3.1 Polymer gel design
Polymer gel electrolytes fabricated via in situ polymerization from different monomers exhibit distinct anodic stability due to variations in their highest occupied molecular orbital (HOMO) energy levels. Acrylates and carbonates, which have low HOMO energy levels, are commonly used as antioxidant monomers in the preparation of in situ polymerized gel electrolytes, particularly when paired with high-voltage cathodes (Fig. 3b).66 Despite their advantages, electrolytes based on carbonates or acrylates undergo rapid performance degradation at high voltage due to the strong catalytic activity of the cathodes. Therefore, enhancing the intrinsic oxidative stability of polymer gel electrolytes has become a critical focus of research.Due to the exceptional high electronegativity and strong electron-withdrawing capability of fluorine (F) atoms, F-containing functional groups have garnered considerable attention for the const ruction of high-voltage in situ polymerized gel electrolytes.71,72 Zhou et al.73 reported an in situ polymerized strategy to design and synthesize an all-fluorinated polymer electrolyte (AFPE) using a fluorinated acrylate monomer, thereby effectively extending the ESW to 4.89 V. Consequently, AFPE enables Li‖LCO cells to exhibit excellent cycling stability, exhibiting 92% capacity retention after 500 cycles at a cut-off voltage of 4.5 V.
In addition to fluorinated monomers, fluorinated crosslinkers have also been introduced into in situ polymerized gel electrolytes. Tang et al. used vinyl ethylene carbonate (VEC) as a monomer and 2,2,3,3,4,4,5,5-octafluoro-1,6-hexanediol diacrylate (OFHDODA) as a fluorinated crosslinker to enhance oxidation resistance of electrolytes (P(IL-OFHDODA-VEC)), achieving a wide ESW of up to 5.08 V (Fig. 4a and b).74 The Li‖NCM523 (LiNi0.5Co0.2Mn0.3O2) cells using OFHDODA-based electrolytes at a cut-off voltage of 4.5 V deliver a high discharge specific capacity and retain approximately 90% capacity after 200 cycles. Notably, even in polyether systems that inherently exhibit poor oxidation stability, incorporation of fluorinated functional groups can significantly enhance oxidation stability.77 Li et al. selected 1,3-dioxolane (DOL) as a monomer and 2,2′-(2,2,3,3,4,4,5,5-octafluorohexane-1,6-diyl)bis(oxirane) (OFHDBO) as a polyfluorinated crosslinker to prepare a polymer gel electrolyte (PDOL-OFHDBO), which exhibits an ESW exceeding 5.6 V.78 As a result, PDOL-OFHDBO enables Li‖NCM811 cells with a high cathode loading of 3.8 mAh cm−2 to retain a high capacity retention of 89.1% over 100 cycles at a cut-off voltage of 4.5 V. Remarkably, pouch cells using PDOL-OFHDBO achieve a high energy density of 401.8 Wh kg−1.
 | ||
| Fig. 4 (a) Schematic illustration of the preparation of P(IL-OFHDODA-VEC). (b) Linear sweep voltammetry curves of Li|P(IL-OFHDODA-VEC)|carbon cells. Reproduced with permission.74 Copyright 2023, Springer Nature. (c) Schematic illustration of the surface side reactions and interphase enhancement by HVQE. (d) Gas generation of CO2 analyzed by in situ DEMS. Reproduced with permission.75 Copyright 2025, Elsevier. (e). Schematic illustration of the FEOP design. (f) Cycling performance of the Li‖NCM811 cells using different electrolytes. Reproduced with permission.76 Copyright 2025, Wiley-VCH. | ||
Besides widening the ESW, the introduction of fluorinated functional groups can also reduce the surface reactivity of high-voltage cathodes and facilitate the formation of F-rich electrode/electrolyte interphases. Fan et al. reported a high-voltage quasi-solid-state electrolyte (HVQE) via the in situ polymerization of 2,2,2-trifluoroethyl acrylate (TFEA).75 TFEA exhibits a hydrogen transfer energy of −0.74 eV, which is lower than that of other components in HVQE. Consequently, HVQE effectively mitigates the solvent oxidative decomposition, gas generation, and irreversible phase transitions at NCM811 cathodes (Fig. 4c and d). Benefiting from these characteristics, Li‖NCM811 cells using HVQE exhibit a high capacity retention of 84% after 500 cycles at a cut-off voltage of 4.5 V. Remarkably, lithium metal pouch cells using HVQE deliver a high energy density of 416 Wh kg−1 along with an exceptional safety performance. Zhu et al. used 3,3,3-tfifluoropropene oxide (TFPO) as a fluorinated monomer to in situ fabricate fluorinated gel polymer electrolyte (FGPE), in which the electron-withdrawing –CF3 group not only enhances high-voltage stability but also promotes the formation of a LiF-rich electrolyte/electrode interphase (Fig. 4e).79 More recently, Zhu et al. further reported a main-chain fluorinated and ethylene oxide polymer electrolyte (FEOP) using bis(2′,3′-epoxypropyl)perfluorobutane (BEPFB) as a fluorinated crosslinker.76 FEOP exhibits outstanding high-voltage stability, enabling NCM811 cells to deliver an impressive cycle life of 2000 cycles at 4.5 V (Fig. 4f). Moreover, lithium metal pouch cells with FEOP deliver an energy density of 405.3 Wh kg−1 and retain stable cycling over 70 cycles at a higher voltage of 4.7 V.
In addition to F-containing functional groups, cyano (–CN) groups are also widely introduced into electrolytes to enhance their oxidative stability.82 CN-containing functional groups not only lower the HOMO energy level of monomers but also promote the formation of Li3N-containing interphases, thereby improving cycling stability and rate performance. Furthermore, cyano groups incorporated into the polymer matrix can mitigate undesirable side reactions at anodes caused by free nitrile solvents, particularly for lithium metal and graphite anodes. –CN groups, with strong polarity, can coordinate with Li+, thereby improving the ionic conductivity. The in situ polymerized gel electrolytes containing cyano groups exhibit good compatibility with high-voltage cathodes and enable enhanced battery performance.83 Zeng et al. reported a gel polymer electrolyte design using acrylonitrile (AN) and OFHDODA as monomers, along with polyethylene glycol diacrylate (PEGDA) as the crosslinker.84 The electrolytes with both cyano and F-containing groups exhibit an extended ESW of 5.1 V. In addition, the high electron transport capacity of AN promotes the generation of Li3N, while OFHDODA enhances electron transfer kinetics and facilitates C–F bond cleavage to generate LiF. The in situ co-growth of Li3N and LiF-rich interphases enables dendrite-free lithium deposition. As a result, these electrolytes paired with LCO cathodes deliver a capacity retention exceeding 82.8% after 200 cycles.
Beyond electron-withdrawing capabilities, incorporating cations into the polymer matrix can also effectively enhance intrinsic anodic stability.85 Liu et al. propose a host–guest recognition polymer gel electrolyte strategy by integrating metal–organic frameworks (MOFs) into the polymer matrix through in situ polymerization.86 In this design, MOFs containing abundant non-redox-active Ti centers serve as a host platform for electrolytes, not only acting as highly effective accelerators for Li+ transport but also imparting improved mechanical strength and high-voltage endurance. As a result, the electrolytes enable LiNi0.9Co0.05Mn0.05O2‖graphite full cells to achieve a good cycling stability, with 71.4% capacity retention after 250 cycles at a cut-off voltage of 4.3 V. Hou et al. reported a Zn2+ ion-bridged polyether electrolyte (Zn-IBPE) with an extended ESW of exceeding 5 V (Fig. 5a).80 This electrolyte endows 4.5 V Li‖LCO cells with excellent cycling stability, retaining 92% of the initial capacity after 280 cycles (Fig. 5b and c). Notably, SiO-graphite‖NCM811 and Li‖LiNi0.9Co0.05Mn0.05O2 pouch cells using Zn-IBPE achieve high energy densities of 303 Wh kg−1 and 452 Wh kg−1, respectively. Furthermore, a 4 Ah graphite‖NCM811 pouch cell with Zn-IBPE demonstrates superior safety, exhibiting no combustion or smoke during a nail-penetration test.
 | ||
| Fig. 5 (a) Schematic illustration of lithium deposition in the conventional polyether electrolyte and Zn-IBPE. (b) Cycling performance and (c) voltage profiles of the Li‖LCO cells using Zn-IBPE. Reproduced with permission.80 Copyright 2025, Springer Nature. (d) Reaction mechanisms of the in situ polymerization of DOL and DOX. (e) LSV curves of the electrolytes. (f) Electrochemical floating test of the electrolytes using an NCM111 cathode. Reproduced with permission.81 Copyright 2023, Royal Society of Chemistry. | ||
In addition, regulation of the alkyl chain structure of the polymer matrix also contributes to enhanced oxidation stability. Liu et al. reported in situ polymerized gel electrolytes by tuning the monomer from five-membered cyclic DOL to six-membered cyclic 1,3-dioxane (DOX) (Fig. 5d).81 Density function theory (DFT) calculations reveal that the polymer gel electrolytes derived from DOX (P-DOX) exhibit a lower HOMO energy level than those from DOL (Fig. 5e), indicating improved oxidation stability. Linear sweep voltammetry (LSV) measurements further confirm that the P-DOX features an extended EWS to 4.7 V (Fig. 5f), consistent with the DFT results. Therefore, P-DOX effectively stabilizes NCM811 cathodes at a high voltage of 4.5 V, enabling stable cycling in Li‖NCM811 cells.
Fluorinated solvents, which are widely employed in both polymer gel and liquid electrolytes, offer notable advantages.92 They not only improve the anodic stability and flame-retardant properties of electrolytes, but also increase the coordination number of anions in the primary solvation sheath of Li+, thereby facilitating the formation of inorganic-rich interphases.93 This behavior is attributed to the weak coordination between fluorinated solvents and Li+, arising from the extremely high electronegativity of fluorine atoms. Nevertheless, solvents with a high degree of fluorination significantly reduce the solubility of lithium salts, which consequently diminishes the ionic conductivity of electrolytes.94 This limitation poses a substantial challenge to the practical application of in situ polymerized gel electrolytes. Therefore, achieving a balance among oxidation stability, interphase formation, and ionic conductivity is crucial for the effective utilization of fluorinated solvents.
Sulfone-based solvents, known for their excellent anodic stability, are frequently used in liquid electrolytes.95 However, their high viscosity can result in reduced ionic conductivity when incorporated into polymer gel electrolytes.96 This limits their practical use in high-performance energy storage devices. Further development is needed to enhance the ionic conductivity of these solvents without compromising their high-voltage stability.
In contrast to fluorinated and sulfone-based solvents, nitrile-based solvents offer good solubility for lithium salts and low viscosity, making them favorable for achieving high ionic conductivity in polymer gel electrolytes.97 However, these solvents tend to undergo severe side reactions at lithium metal anodes, which compromises their stability over extended cycling.98 The challenge lies in preventing these side reactions while retaining the benefits of high ionic conductivity.
To overcome these limitations, recent studies have explored non-fluorinated molecules that enhance oxidative stability without the disadvantages of fluorine-based solvents.99,100 These new solvents offer a promising avenue for designing in situ polymerized gel electrolytes that combine high ionic conductivity with satisfactory oxidative stability, thus addressing the critical challenges posed by traditional solvents.
3.2 Interphase engineering on cathodes
To mitigate parasitic reactions at the interface between electrolytes and cathodes under high-voltage conditions, constructing a high-quality CEI film on the cathode surface is one of the most prevalent approaches.101–103 An ideal CEI is expected to serve as a uniform and stable charge transfer barrier, suppressing side reactions between electrolytes and high-voltage cathodes.104,105 However, high voltage exacerbates electrolyte decomposition, leading to the thickening and even breakage of the CEI film. These effects can further decrease kinetics and even induce structural degradation of cathodes, ultimately resulting in rapid performance decline.106Due to the electron deficiency of B atoms, B-containing additives can complex with anions and promote the formation of a stable CEI.114,115 For instance, lithium difluoro(oxalate)borate (LiDFOB), a widely utilized B-containing sacrificial salt in liquid electrolytes, preferentially decomposes at high voltage, resulting in the formation of a robust borate-rich interphase and thereby enhancing the high-voltage performance.87,116 Park et al. reported a fluorinated and plastic-crystal-embedded elastomeric electrolyte (F-PCEE), fabricated via the in situ polymerization of hexafluorobutyl acrylate (HFBA) and PEGDA.117 LiDFOB was incorporated into F-PCEE, which not only effectively suppresses corrosion of the aluminum (Al) current collector but also promotes the formation of an inorganic-rich CEI. Consequently, the Li‖NCM811 cells using F-PCEE exhibit high capacity retention at a high cut-off voltage of 4.5 V.
Furthermore, these additives can behave differently in in situ polymerized gel electrolytes compared with liquid electrolytes. Sun et al. reported that LiDFOB not only acts as a sacrificial additive to promote the formation of a high-quality CEI but also serves as an initiator to facilitate the ring-opening polymerization of DOL (Fig. 6a).118 The introduction of this dual-functional additive mitigates the adverse effects associated with introducing separate initiators into polymer gel electrolytes. The polymer gel electrolytes containing LiDFOB (PDOL-FB) enable the formation of a CEI with a higher content of F-containing species than those derived from polymer gel electrolytes without LiDFOB (PDOL-AT) (Fig. 6b and c). In addition, transmission electron microscopy (TEM) analysis shows that a more uniform and stable CEI is formed in PDOL-FB (Fig. 6d–g). Benefiting from these characteristics, PDOL-FB exhibits an extended ESW of 5 V, enabling remarkable cycling performance in Li‖LCO cells. Wu et al. reported a eutectic solution using 1,2,3-trioxane (TXE) and succinonitrile (SN) for the preparation of in situ polymerized gel electrolytes.120 In this work, LiDFOB was also utilized both as a CEI-forming additive and a polymerization initiator, avoiding the introduction of impurities. The resulting electrolyte exhibits a wide ESW of 4.5 V, enabling excellent cycling stability in Li‖LCO cells.
 | ||
| Fig. 6 (a) Schematic illustration showing the dual functionality of LiDFOB in the PDOL electrolytes. Atomic profiles of F 1s in (b) the PDOL-AT and (c) PDOL-FB electrolytes after different sputtering times. TEM images of CEI formed in (d and e) PDOL-AT and (f and g) PDOL-FB after (d and f) 3 cycles and (e and g) 50 cycles. Reproduced with permission.118 Copyright 2025, American Chemical Society. (h) HOMO and LUMO energy levels of different components in TH@DOL. (i) P 2p, (j) F 1s, and (k) N 1s XPS spectra of NCM811 cathodes after 300 cycles. Reproduced with permission.119 Copyright 2025, Elsevier. | ||
Sulfone-based additives with high HOMO energy levels are also used to construct a high-quality CEI in in situ polymerized gel electrolytes.121 Lv et al. fabricated a polymer gel electrolyte via in situ polymerization of PEGDA, incorporating tetramethylene sulfone (TMS) and bis(2,2,2-trifluoromethyl) carbonate (TFEC) as additives (denoted as in situ-TF).122 DFT calculations reveal that TMS exhibits a high HOMO energy level of −7.110 eV, enabling preferential oxidative decomposition to form a stable CEI on the cathode surface, thereby enhancing high-voltage performance. Moreover, TMS is expected to facilitate the dissolution of lithium bis(trifluoromethanesulphonyl)imide (LiTFSI) within the polymer matrix, effectively increasing the ionic conductivity to 0.62 mS cm−1. Consequently, the Li‖NCM cells using in situ-TF exhibit excellent cycling stability, achieving a capacity retention of 86.8% after 1000 cycles.
Cyclotriphosphazene-based compounds have attracted widespread research interest as flame-retardant additives for lithium batteries.133 This interest arises from their high phosphorus and nitrogen content, along with highly tunable chemical structures, which provide a unique combination of flame-retardant properties and electrochemical stability. Furthermore, cyclotriphosphazene compounds facilitate the formation of robust electrode/electrolyte interphases, thereby ensuring long-term cycling stability. Liu et al. prepared a fluorine-free ether-based electrolyte by incorporating a phosphazene (TH) additive, denoted as TH@DOL.119 Owing to its bulky steric hindrance and strong electron-withdrawing properties, TH effectively mitigates oxidation of PDOL-based electrolytes at high-voltages. In addition, TH can adsorb PF6−, suppressing HF generation while simultaneously regulating the HOMO energy level of PF6−, thereby promoting the formation of a stable CEI (Fig. 6h). X-ray photoelectron spectroscopy (XPS) spectra reveal that the CEI formed in TH@DOL is enriched with P, F and N elements (Fig. 6i–k), significantly enhancing the oxidation stability of the electrolyte. Consequently, the Li‖NCM811 cells using TH@DOL retain 76.21% of their capacity after 300 cycles at a high cut-off voltage of 4.5 V, demonstrating superior cycling stability. Furthermore, TH@DOL exhibits excellent flame retardance at elevated temperatures due to the high concentration of flame-retardant elements in TH.
Silicon-based additives containing Si–O bonds not only contribute to the formation of polymeric CEI films but also mitigate parasitic reactions by scavenging HF and PF5 species.134 Ma et al. designed a bilayer heterostructure gel polymer electrolyte (BGPE), fabricated using functional boron-containing monomers to construct a three-dimensional (3D) crosslinked network, with tris(trimethylsilyl) phosphite (TMSPi) as an additive.135 Owing to its higher HOMO energy level, TMSPi undergoes preferential oxidative decomposition on the cathode surface, contributing to the formation of a stable and homogeneous CEI film. In addition, TMSPi effectively removes corrosive species such as HF, alleviating damage to the CEI, stabilizing the cathode structure, and suppressing the dissolution of transition metals. Consequently, the Li‖NCM523 cells using TMSPi exhibit stable cycling performance along with excellent rate performance.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 650 cylindrical lithium-based batteries to operate stably, with both high energy density and enhanced safety.
650 cylindrical lithium-based batteries to operate stably, with both high energy density and enhanced safety.
In addition to the introduction of diluent, the fluorinated polymer matrix can also act as a pseudo-diluent to construct an anion-incorporating Li+ solvation structure. As mentioned in Section 3.1.1, PDOL-OFHDBO not only exhibits an extended ESW but also promotes the formation of an anion-rich solvation structure, which is attributed to the introduction of fluorinated OFHDBO.78 Lu et al. reported an in situ polymerized gel electrolyte that employs fluorinated backbones (TF) as pseudo-diluents in high-concentration ether-based liquid electrolytes (HCE), denoted as TF + HCE (Fig. 7a).123 The weakened solvation capability in the TF + HCE system facilitates the formation of an anion-incorporating solvation structure (Fig. 7b), thereby enabling the formation of a robust and inorganic-rich CEI. TF + HCE enables the Li‖NCM811 full cells to achieve stable cycling performance at 4.4 V and lithium metal pouch cells to deliver a high energy density of 465.63 Wh kg−1. Impressively, Huang et al. designed an in-built fluoropolyether-based quasi-solid-state polymer electrolyte (PTF-PE-SPE) composed of strongly solvating polyether segments and weakly solvating fluorohydrocarbon pendants, thereby creating an anion-rich solvation structure (Fig. 7c).124 The configuration generates a LiF-rich outer layer that provides a high ionic conductivity pathway to facilitate oxygen redox activation, along with an inner layer featuring Mn–F bonds that mitigate oxygen overoxidation on the surface of lithium-rich manganese-based layered oxide (LRMO) (Fig. 7d–f). PTF-PE-SPE enables long-term stability over 500 cycles in Li‖LRMO coin cells at a high cut-off voltage of 4.7 V. Remarkably, the Li‖LRMO pouch cells using PTF-PE-SPE demonstrate an ultrahigh energy density of 604 Wh kg−1(1, 027 Wh L−1) and improved safety.
 | ||
| Fig. 7 (a) Schematic illustration of the Li+ solvation structure in different electrolytes. (b) Raman spectra of different electrolytes. Reproduced with permission.123 Copyright 2025, Royal Society of Chemistry. (c) Stabilized LRMO surface oxygen by an anion-derived CEI in fluoropolyether-based polymer electrolytes. (d) TOM-SIMs 3D and 2D tomography of the LRMO cathodes after 100 cycles using PTF-PE-SPE. (e) Mn 2p and (f) O 1s XPS spectra of the cycled LRMO cathodes using PTF-PE-SPE after sputtering. Reproduced with permission.124 Copyright 2025, Springer Nature. | ||
In fact, the anion-rich solvation structures regulated by these methods facilitate the formation of inorganic-rich interphases, including not only a CEI on the cathode surface but also a solid electrolyte interphase (SEI) on the anode surface. These robust and inorganic-rich SEIs effectively enhance the interfacial stability of electrolytes towards anodes, which will be discussed in subsequent sections.
4. Gel electrolyte and interphase design towards high-capacity anodes
To overcome the capacity limitation of traditional graphite anodes (372 mAh g−1) and meet the development demands of high-energy-density electrochemical devices, lithium metal anodes142,143 and silicon-based anodes,144,145 which possess ultrahigh theoretical specific capacities, have emerged as promising alternatives. However, their practical applications remain constrained by several critical issues. (1) The high reactivity of lithium metal anodes induces severe parasitic reactions with electrolytes, resulting in poor lithium plating/stripping behavior and the formation of lithium dendrites. (2) Silicon-based anodes undergo volumetric expansion exceeding 300% during charge and discharge cycles, leading to electrode structural fracture and continuous growth of the SEI layer. These challenges directly compromise the cycling stability and safety of the devices. Therefore, the rational design of in situ polymerized gel electrolytes is essential to optimize their compatibility with these two high-capacity anodes.4.1 Gel electrolytes for lithium metal anodes
Due to the lowest redox potential (−3.04 V vs. the standard hydrogen electrode) and high specific capacity (3860 mAh g−1) of lithium metal anodes, lithium metal batteries (LMBs) can achieve an energy density exceeding 400 Wh kg−1 and are considered one of the most promising next-generation energy storage technologies.146,147 However, the high reactivity of lithium metal anodes leads to low plating/stripping coulombic efficiency (CE), unstable SEI formation, extensive ‘dead lithium’ accumulation, and dendrite growth.148–150 These issues not only result in poor cycling stability but also pose significant safety hazards. Solid-state electrolytes are widely considered an ideal solution to suppress lithium dendrite growth and improve safety in LMBs.151–153 Nevertheless, their practical application is severely hindered by poor interfacial contact and large interfacial impedance. In contrast, in situ polymerized gel electrolytes are widely recognized as an effective strategy to alleviate these challenges,154,155 which is attributed to their superior interfacial contact with electrodes, high ionic conductivity, and intrinsic quasi-solid-state nature. Nonetheless, the inherently poor mechanical strength of polymer gel electrolytes, combined with the highly reactive nature of lithium metal anodes, still inevitably leads to compromised interfacial stability and compatibility.156,157By systematically summarizing recent studies on quasi-solid-state LMBs fabricated via in situ polymerization (Table 2), it is evident that the rapid capacity degradation of LMBs under practical conditions is primarily attributed to the insufficient compatibility with lithium metal anodes, ultimately resulting in battery failure.160,161 This issue arises from the extremely thin lithium metal anodes employed in practical LMB configurations. Poor lithium plating/stripping behavior accelerates the depletion of lithium metal over a limited number of cycles, resulting in a rapid capacity decay.162 It is noteworthy that PTF-PE-SPE prepared by Huang et al.124 and GMFN by Peng et al.163 both enable lithium metal pouch cells to achieve energy densities exceeding 600 Wh kg−1. Remarkably, pouch cells using GMFN, with an energy density of 604.2 Wh kg−1, exhibit stable cycling performance, retaining 92.83% of their energy after 100 cycles. This is attributed to GMFN's ability to confine unstable free carbonate solvents, thereby suppressing parasitic electrolyte reactions. Furthermore, the formulated bicontinuous gradient polymer layer (BGPL) on the lithium anode surface not only enhances the reaction kinetics but also protects lithium from detrimental moisture, oxygen, and aggressive electrolyte components. Benefiting from the synergistic effects of GMFN and the modified lithium metal anode, superior lithium plating/stripping behaviour is achieved, significantly contributing to the excellent cycling lifespan of the high-energy-density pouch cells. Therefore, regulating in situ polymerized gel electrolytes to mitigate anode/electrolyte side reactions, suppress lithium dendrite growth, and enhance lithium plating/stripping performance is of critical importance.
| Electrolytes | Cathode-anode/cut-off voltage (V) | Rate/cycle number/capacity retention/mass loading (mg cm−2) | Li‖Li cycling performance | Rate/cycle number/capacity retention/mass loading (mg cm−2) | Ref. |
|---|---|---|---|---|---|
| PDOL | NCM90-Li/4.3 | 0.2C/280/78%/2.5 | 0.2 mA cm−2/0.2 mAh cm−2/2000 h | 0.1C/10/98.3%/∼30 | 125 |
| P(VC + DAC + MBA) | NCM811-Li/4.5 | 1C/500/82.2%/3–3.5 | 1 mA cm−2/1 mAh cm−2/5600 h | 0.2C/100/88.9%/10 | 126 |
| P(DEVP + MMA + ADC) | LCO-Li/4.3 | 0.5C/230/81.2%/2–3 | 0.2 mA cm−2/0.2 mAh cm−2/900 h | 0.1C/38/80.8%/∼12 | 127 |
| PETEA + OV-POSS | LCO-Li/4.4 | 1C/270/81%/2 | 0.2 mA cm−2/0.2 mAh cm−2/1000 h | 0.5C/120/100%/5.4 | 128 |
| P(TFPO-PEE) | NCM622-Li/4.3 | 0.5C/1000/78%/2 | 0.5 mA cm−2/mAh cm−2/2000 h | 0.2C/100/95%/21 | 79 |
| PTFMA + FEP | NCM811-Li/4.3 | 1C/300/92%/1.6 | 0.5 mA cm−2/0.5 mAh cm−2/1500 h | 0.1C/50/100%/7 | 129 |
| P(VEC + PEGDA) | LCO-Li/4.45 | 0.2C/400/67.5%/1.5 | 0.1 mA cm−2/0.1 mAh cm−2/1500 h | 0.1C/90/85.4%/4.6 | 130 |
| PVHF-HFP/HAP | NCM811-Li/4.3 | 1C/1000/86.8%/1–2 | 0.3 mA cm−2/0.3 mAh cm−2/1500 h | 0.5C/400/82.8%/5 | 122 |
| P(TFEA + PEGDA) | |||||
| P(AEEO) | NCM85-Li/4.3 | 1C/500/80%/2 | 0.5 mA cm−2/0.5 mAh cm−2/3000 h | 0.2C/200/91.8%/15 | 131 |
| IWSWN-SPE | NCM622-Li/4.3 | 0.5C/300/73.5%/3 | 0.2 mA cm−2/0.2 mAh cm−2/2600 h | 0.02C/100/75%/15.8 | 132 |
In quasi-solid-state lithium metal batteries fabricated via in situ polymerization, the intimate and conformal electrolytes/electrode interface ensures rapid Li+ migration, thereby promoting uniform lithium deposition on the lithium anode surface. However, the volume expansion of lithium metal anodes during repeated charge and discharge cycles significantly degrades the interfacial integrity. To address this challenge, sufficient mechanical strength is required to preserve continuous contact between electrolytes and electrodes. In addition, fast Li+ transport within polymer gel electrolytes is crucial to promote homogeneous lithium deposition during extended cycling. To synergistically enhance the mechanical strength and Li+ conductivity of in situ polymerized gel electrolytes, numerous strategies have been proposed, including plasticizer optimization, construction of crosslinked networks, and incorporation of inorganic fillers.
To enhance the Li+ conductivity of in situ polymerized gel electrolytes, substantial amounts of plasticizers are typically introduced, which inevitably compromise their mechanical strength.131 To balance high Li+ conductivity and robust mechanical properties, Wang et al. reported a competitive coordination strategy by incorporating highly polar SN into a poly(methyl methacrylate-co-methacrylamide) (PMAm) matrix (Fig. 8a).158 The strong affinity of SN to Li+ displaces Li+ from polymer hydrogen-bonding sites, thereby preserving the structural integrity of the polymer while enhancing mechanical strength (Fig. 8b and c). In addition, molecular aggregation induced by hydrogen bonding between polymer chains compresses the polymer matrix, creating fast Li+ diffusion pathways. The PMAm-SN polymer gel electrolyte exhibits excellent toughness (24.8 MJ m−3), high Li+ conductivity (2.8 mS cm−1), and a high Li+ transference number of 0.76. Consequently, PMAm-SN enables stable lithium plating/stripping over 1000 h at 0.3 mA cm−2 and 0.3 mAh cm−2. Furthermore, the Li‖NCM811 pouch cells using PMAm-SN achieve an enhanced cycling lifespan and outstanding safety with an energy density of 456 Wh kg−1. Lv et al. incorporated trace amounts of sulfone (SL) into polyacrylic-based electrolytes (in situ-SL2), forming dynamic ion transport channels.164 The intrinsic flexibility of SL facilitates rapid conformational changes and dynamic coordination with Li+, reducing the energy barriers for Li+ elastic hopping and thus enhancing both Li+ conductivity and Li+ transference. In situ-SL2 effectively suppresses lithium dendrite growth and enables rapid and uniform lithium deposition.
 | ||
| Fig. 8 (a) Schematic illustration of the lithium ion solvation structure and its migration through the collective solvent motion or by hopping between solvent molecules. (b) Uniaxial tensile stress–strain curves of a free-standing PMAm membrane. (c) Photographs of PMAm-SN during uniaxial stretching. Reproduced with permission.158 Copyright 2025, Wiley-VCH. (d) Comparison of the Li stripping/plating in liquid electrolyte (top) and MOF-(Co–Ti) (bottom). Reproduced with permission.159 Copyright 2025, Royal Society of Chemistry. | ||
Constructing 3D crosslinked polymer networks can provide continuous ion-conduction channels, which effectively enhance the ionic conductivity while ensuring mechanical integrity.165,166 Du et al. developed a phosphorus-enriched quasi-solid-state electrolyte (QSPE) via in situ copolymerization of nano-phosphorus methyl methacrylate (BPn-MMA) and phenylphosphine dienyl ester (PDE).167 Benefiting from the 3D crosslinked network, QSPE exhibits superior mechanical strength, achieving an elastic modulus of 7.65 MPa and an extraordinary elongation at break of 652%. QSEP mechanically suppresses lithium dendrite growth, enabling stable cycling performance in Li‖Li symmetric cells while retaining a low overpotential of 84 mV after 1000 cycles.
In addition, the incorporation of inorganic fillers can also enhance the Li+ transport kinetics in in situ polymerized gel electrolytes. Liu et al. fabricated a Ti–Co-based bimetallic MOF host membrane with abundant catalytic sites (MOF-(Co–Ti)).159In situ polymerized gel electrolytes were prepared by incorporating MOF-(Co–Ti) into PDOL. Featuring uniformly distributed Co-rich sites within its porous framework, this MOF leverages spontaneous redox processes to effectively enhance interfacial charge-transfer kinetics, modulate the local coordination environment of Li+, and promote Li+ transport (Fig. 8d). These electrolytes improve Li+ utilization and create additional diffusion pathways, thereby enhancing ion flux and diffusion. Furthermore, the introduction of MOF-(Co–Ti) effectively improves the mechanical strength of in situ polymerized gel electrolytes. Consequently, the Li‖Li symmetric cells using these electrolytes exhibit an impressive cycling life of 1000 h at a current density of 1 mA cm−2.
4.2 Gel electrolytes for silicon-based anodes
Silicon anodes have garnered significant attention due to their ultra-high theoretical capacity (3579 mAh g−1) and low redox potential (−0.3 V vs. Li/Li+).168,169 Over the past three decades, this exceptional capacity has positioned Si anodes as promising candidates for high-energy-density lithium batteries.170–172 However, the substantial volume change (∼300%) during the lithiation/delithiation cycles induces severe pulverization of Si particles, leading to degradation of coulombic efficiency, poor cycling performance and significant safety hazards.173 Polymer gel electrolytes have attracted considerable attention because their soft polymer matrix can partially accommodate the expansion stress of Si anodes. Nevertheless, the dramatic volume variation still causes repeated destruction and reconstruction of the interphases on the Si anode surface, significantly enhancing the porosity of the interfacial regions between the electrolyte and Si nodes. Consequently, the deteriorated interfacial contact between electrolyte and Si anodes results in severe interfacial degradation and rapid capacity decay.In recent years, in situ polymerized gel electrolytes have attracted increasing attention in Si-based lithium batteries, as they effectively mitigate the volume expansion of Si anodes and suppress the surface side reaction, thereby enhancing the cycling stability of Si anodes.174,175 This improvement is primarily attributed to the enhanced mechanical strength and elasticity of polymer gel electrolytes. Cheng et al. utilized PDOL electrolytes to stabilize micro-sized silicon (SiMPs) anodes.176 The conformal nature of the in situ polymerized gel electrolytes effectively suppresses the pulverization of Si microparticles, thereby alleviating capacity decay (Fig. 9a). Scanning electron microscopy (SEM) images reveal that the cycled Si anodes in PDOL electrolytes retain a more intact surface morphology and exhibit a lower expansion ratio compared to those in liquid electrolytes, confirming that the conformality of polymer gel electrolytes mitigates Si anode pulverization (Fig. 9b–g). The Si anodes demonstrated enhanced cycling performance, retaining a reversible capacity of 1837.1 mAh g−1 after 100 cycles at 500 mA g−1. Bai et al. reported a quasi-solid-state electrolyte (PVCM-GPE) to enhance the mechanical integrity of Si anodes, which was fabricated via the in situ polymerization of vinylene carbonate (VC).177 Young's modulus measurements show that the stress distribution becomes more homogeneous in PVCM-GPE than in liquid electrolyte (Fig. 9h). The synergistic combination of the designed Si anode structure and the mechanically enforced PVCM-GPE enables effective stress dissipation without compromising Li+ diffusion. PVCM-GPE not only enables excellent cycling and rate performance, but also chelates transition-metal cations to stabilize interfaces. By coupling conformal polymer gel electrolyte encapsulation with Si anodes and NCM811 cathodes, a 2.7 Ah pouch cell exhibits a high capacity retention of 88% over 2000 cycles (Fig. 9i). In addition, pouch cells using PVCM-GPE achieve excellent rate capability and wide-temperature operability (Fig. 9j), along with a maximum energy density of 355 Wh kg−1 (Fig. 9k). Yu et al. utilized quasi-solid-state polymer electrolytes (QSPEs) in combination with silicon monoxide (SiOx) anodes to address the poor cycling stability of Si-based anodes.178 QSPE was prepared via in situ cationic ring-opening polymerization using TXE as the monomer. The resulting QSPEs show a high elastic modulus of 42 MPa, thereby effectively suppressing the volume expansion of the SiOx anodes during cycling. The SiOx anodes demonstrate improved cycling stability, retaining 81.9% of their capacity after 200 cycles.
 | ||
| Fig. 9 (a) Structure evolution of the SiMP anodes in liquid and polymer electrolytes. SEM images of SiMP anodes with (b–d) CLE and (e–g) PDOL electrolytes. Reproduced with permission.176 Copyright 2024, Wiley-VCH. (h) Stress distribution modeling for LE and PVCM-GPE at different lithiation states, along with the stress distribution of a single composite particle at a deeply lithiated state. (i) Cycling performance of different pouch cells. (j) Rate performance and capacity retention at different temperatures of the pouch cells using GPE. (k) Gravimetric/volumetric energy densities of PVCM-GPE-based pouch cells using different anodes. Reproduced with permission.177 Copyright 2025, Springer Nature. | ||
In situ polymerized gel electrolytes with self-healing properties can effectively alleviate cracks formed on Si anodes during cycling.179 Liu et al. reported a self-healing quasi-solid-state electrolyte (SHQSE) fabricated by in situ polymerization.180 SHQSE employed hydroxyethyl acrylate as a molecular bridge to integrate acrylate and polyurethane through disulfide and complementary hydrogen bonding interactions. These multiple dynamic bonding mechanisms enable a high ionic conductivity of 0.72 mS cm−1 while simultaneously imparting strong self-healing capability. Additionally, the self-healing polymer (SHP) is utilized as an ion-conductive agent within silicon/carbon (Si/C) anodes, providing continuous Li+ transport pathways and preserving electrode structural integrity. Consequently, the 2 Ah NCM811‖Si/C@SHP pouch cells using SHQSE demonstrate superior cycling stability and enhanced safety performance.
4.3 Interphase engineering of anodes
In quasi-solid-state lithium batteries employing lithium metal and Si-based anodes, the substantial volume changes during charge–discharge cycling progressively deteriorate interfacial contact, leading to increased interfacial impedance between the electrodes and the in situ polymer gel electrolyte.181,182 In addition to these issues, the interphases formed on the surfaces of these anodes also become increasingly uneven and thickened over cycling, severely compromising interfacial compatibility between the negative electrodes and the electrolytes.183,184 Therefore, the instability of interphases derived from polymer gel electrolytes remains a critical bottleneck, limiting the battery performance and impeding their broader adoption.185–187 As the challenges associated with interphases between high-voltage cathodes and in situ polymerized gel electrolytes have been discussed above, this section primarily focuses on interfacial challenges between in situ polymerized gel electrolytes and high-capacity anodes.To achieve enhanced high-voltage stability, plasticizers with lower HOMO energy levels, such as carbonates, are commonly incorporated into in situ polymerized gel electrolytes. Nevertheless, these components typically exhibit lower lowest unoccupied molecular orbital (LUMO) energy levels, making them susceptible to reductive decomposition. This leads to the formation of an unstable, organic-rich SEI on the surfaces of high-capacity anodes, causing repeated SEI fracture and reformation, severe parasitic reactions, and rapid capacity degradation. To obtain robust and uniform SEI films on the anode surface, numerous strategies have been developed, including the construction of anion-dominant solvation structures and additive engineering.
The molecular structural design of monomers can effectively regulate the solvation structure of in situ polymerized gel electrolytes. As discussed above, fluorinated functional groups, which exhibit weak interaction with Li+, can induce anion-involved solvation structures, thereby facilitating the formation of uniform and stable SEI films. Both PTF-PE-SPE fabricated by Huang et al.124 and TF + HCE fabricated by Lu et al.123 exhibit stable lithium stripping/plating behavior. Beyond weak interactions, Zhou et al. reported a dual-shielding solvent strategy in an in situ polymerized gel electrolyte (PHTGPE), using 1,1,1,3,3,3-hexafluoroisopropyl acrylate as a high dipole-moment monomer to displace solvents adsorbed on the lithium metal anode (Fig. 10a).188 Strong dipole–dipole interactions between high dipole-moment monomers and solvents inhibit solvent coordination with Li+, resulting in an anion-involved solvation structure. This forms a stable and uniform SEI rich in LiF and Li2S (Fig. 10b and c), achieving stable cycling for 5000 h at 0.25 mA cm−2 and 0.25 mAh cm−2 in Li‖Li coin cells.
 | ||
| Fig. 10 (a) SEI properties and Li+ transport in PHTGPE. (b) 3D rendered time-of-flight secondary ion mass spectrometry (TOF-SIMS) images and (c) corresponding analysis of the SEI layers in the Li‖Li cells with PHTGPE. Reproduced with permission.188 Copyright 2025, American Chemical Society. (d) Raman spectra of HB-GPE, MB-GPE and LB-GPE. (e) Coordination oxygen number profile of Li+ with solvent and TFSI− in different electrolytes. (f) Ionic conductivities of different electrolytes. Reproduced with permission.189 Copyright 2025, Royal Society of Chemistry. (g) TOF-SIMS depth profiles of the cycled Li anode in F-Gel. (h) Long term cyclability of Li‖NCM811 pouch cells using F-Gel at −20 °C. Reproduced with permission.190 Copyright 2025, Royal Society of Chemistry. | ||
Regulating plasticizers can also enhance the coordination number of anions with Li+. For example, incorporating hydrofluoroethane into in situ polymerized gel electrolytes as a diluent can significantly enhance the number of coordinated anions in the Li+ solvation sheath, promoting the formation of an inorganic-rich SEI. Such electrolytes, characterized by a localized high-concentration solvation structure, demonstrate superior stability against lithium metal anodes. Similarly, replacing conventional solvents with fluorinated solvents can promote anion participation in the Li+ solvation sheath. However, the weak interactions between fluorinated solvents and Li+ reduce lithium salt solubility, limiting Li+ transport in the electrolyte. Achieving an optimal balance between weak solvation modulation and Li+ transport is therefore critical.
Zhang et al. fabricated three in situ polymerized gel electrolytes with high, moderate, and low Li+-solvent binding strengths using fluorinated solvents to identify the optimal system.189 The in situ polymerized gel electrolytes with moderate Li+-solvent binding (denoted as MB-GPE), incorporating fluorinated ethylene carbonate (FEC) and methyl 2,2,2-trifluoroethyl carbonate (FEMC), not only form an anion-involved solvation structure but also retain sufficient lithium salt solubility (Fig. 10d and e). MB-GPE exhibits a high ionic conductivity of 1.95 mS cm−1 and facilitates the formation of a robust LiF-rich SEI on the lithium metal anode surface (Fig. 10f). The Li‖Li symmetrical cells with MB-GPE can cycle stably for over 3200 h at 0.5 mA cm−2 and 0.5 mAh cm−2.
Wei et al. reported similar findings. They fabricated in situ polymerized gel electrolytes containing a carboxylic ester solvent with an ethoxy side-chain difluoro-substitution (–OCH2CF2H).190 The gel electrolyte is denoted as F-Gel. This electrolyte promotes rapid Li+ migration and desolvation at low temperatures while facilitating the formation of a LiF-rich interphase (Fig. 10g), thereby suppressing lithium dendrite growth and parasitic reactions on the anode surface. The Li‖NCM811 pouch cells using F-Gel achieve a high energy density of 450 Wh kg−1 and demonstrate enhanced cycling stability, retaining 83.4% of their capacity after 75 cycles at −20 °C (Fig. 10h).
As a representative fluorinated solvent, FEC is widely used as an additive or co-solvent to construct robust and stable SEI layers.198,199 Owing to its lower LUMO energy level compared with non-fluorinated solvents, FEC is preferentially reduced during initial cycling, resulting in the formation of a LiF-rich SEI on the lithium metal anode surface.200 Furthermore, the strong electron-withdrawing effect of the F atoms lowers the HOMO energy level of FEC, thereby enhancing the oxidative stability of the electrolytes.201 Therefore, FEC is commonly regarded as an effective sacrificial additive in in situ polymerized gel electrolytes.202,203 However, the FEC decomposition can generate corrosive HF species under high-voltage conditions, which may deteriorate the electrode–electrolyte interface and compromise long-term cycling stability.204,205 To address this issue, Wang et al. reported a hybrid in situ polymerized gel polymer electrolyte based on pure FEC and a pentafluorophenyl methacrylate (PFPMA) monomer, denoted as HGPE.206 This system promotes the formation of a mixed interphase containing LiDFOB− derived species, effectively suppressing the continuous decomposition of FEC (Fig. 11a). HGPE exhibits stable lithium plating/stripping behavior and enables Li‖NCM811 full cells to achieve a high capacity retention of 83.4% after 300 cycles at a high cut-off voltage of 4.5 V (Fig. 11b). Beyond FEC, fluorinated linear carbonates such as TEFC also possess relatively low LUMO energy levels (Fig. 11c), resulting in their preferential reduction on the lithium metal anode surface. This reduction pathway induces the formation of an SEI enriched with LiF and –CF3-containing species (Fig. 11d).207 Similarly, Lv et al. fabricated in situ-TF using TFEC as an additive to regulate the SEI component and improve lithium metal anode stability.122
 | ||
| Fig. 11 (a) Schematic structure of Li‖NCM811 cells with HGPE. (b) Galvanostatic cycling performance of Li‖Li symmetric cells using LCE and HGPE. Reproduced with permission.206 Copyright 2025, Wiley-VCH. (c) HOMO and LUMO energy levels of different components in in situ-TF. (d) 3D TOF-SIMS rendered images of the SEIs in in situ-T and in situ-TF. Reproduced with permission.122 Copyright 2024, American Chemical Society. (e) Schematic illustration showing that an N-rich SEI effectively stabilizes the lithium metal anode. (f) Galvanostatic cycling performance of Li‖Li symmetric cells using different electrolytes. Reproduced with permission.208 Copyright 2023, Royal Society of Chemistry. | ||
Nitrogen-containing additives, particularly LiNO3, are also widely utilized in LMBs to promote the formation of Li3N-rich SEIs, which significantly enhance lithium plating/stripping reversibility. Zhang et al. incorporated LiNO3 and FEC into poly(vinyl carbonate)-based in situ polymerized gel electrolytes, forming a Li3N–LiF reinforced SEI that effectively suppresses lithium dendrite growth.209 Despite its favorable SEI-forming ability, LiNO3 exhibits strong interactions with Lewis acid, which can hinder the polymerization of cyclic ethers by reducing the polymerization rate. To overcome this limitation, Wang et al. used triethylene glycol dinitrate (TEGDN) as an additive to replace LiNO3. Similar to LiNO3, TEGDN facilitates the formation of a dense, N-rich SEI on the surface of the lithium metal anode, suppressing parasitic reactions (Fig. 11e).208 Importantly, unlike LiNO3, TEGDN does not interfere with the polymerization of DOL, resulting in a high performance polymer electrolyte. The resulting electrolyte exhibits a high ionic conductivity of 2.87 mS cm−1 and stable lithium plating/stripping behavior, allowing Li‖Li symmetric cells to undergo stable cycling for 550 h at 1 mA cm−2 and 1 mAh cm−2 (Fig. 11f). Additionally, the polymerization-retarding effect of NO3− has been intentionally explored to control cyclic ether polymerization. Wang et al. introduced LiNO3 into DOL to construct a high-concentration polymer electrolyte (HCPE), slowing the polymerization reaction and enabling dissolution of 6 M LiFSI.210 This approach induces an anion-involved solvation structure, resulting in a mechanically robust SEI rich in LiF and Li3N with fast Li+ transport. Si-based anodes paired with HCPE deliver a high capacity of 1765 mAh g−1 and retain 2000 mAh g−1 after 100 cycles at 2C. Similarly, Liu et al. used the partial inhibitory effect of LiNO3 to control DOL polymerization, forming a stable amorphous polymer gel electrolytes with a high Li+ conductivity of 5.10 mS cm−1.211 Furthermore, LiNO3 promotes the formation of an inorganic-dominated and stable SEI, enabling excellent fast-charging cycling performance in graphite‖Li cells. Boron-containing additives have been investigated for constructing high-quality SEI layers. BGPE fabricated by Ma et al. used tri(pentafluorophenyl)borane (TPFPB) as an additive, promoting the formation of a chemically robust SEI enriched in B–O and LiF species.135 The B–O components significantly enhance interfacial stability, while the LiF-rich domains help suppress lithium dendrite growth, enabling highly stable lithium plating/stripping and improved cycling stability. Similarly, Li et al. incorporated tris(hexafluoroisopropyl) borate (THB) into in situ polymerized gel electrolytes, denoted as THB-PDOL.212 THB serves a dual role. It acts as a Lewis acid catalyst to initiate the ring-opening polymerization of DOL and simultaneously functions as an SEI-forming additive. This synergistic effect facilitates the formation of a uniform and stable SEI enriched with F- and B-containing species. The Li‖Li symmetric cells using THB-PDOL demonstrate long-term cycling stability for 600 h at a capacity of 0.5 mAh cm−2.
Due to their superior reductive stability, cyclic ether-based polymer gel electrolytes have attracted increasing attention, motivating the development of multifunctional additives that integrate polymerization with interfacial stabilization. Zhao et al. designed nonflammable quasi-solid-state electrolytes (NQSEs) through an in situ polymerization method.214 In this system, 1,3,2-dioxathiolan-2,2,oxide (DTD), a S-containing additive, simultaneously acts as a polymerization initiator and an SEI-forming agent. DFT calculations reveal that the LUMO energy levels of Li+-DTD complexes and their coordination structures with BF4− are lower than those of competing solvated species, leading to their preferential reduction at the surface of the lithium metal anode. This process induces the formation of a compact and uniform SEI enriched in F- and S-containing species, effectively protecting the lithium metal anode and enabling stable lithium plating/stripping in Li‖Li symmetric cells.
In addition to sulfur-based additives, metal-salt-type multifunctional additives were explored. Zheng et al. reported tin trifluoromethanesulfonate (Sn(OTf)2) as a dual-function additive in DOL-based electrolytes.215 Sn(OTf)2 not only promotes the in situ polymerization of DOL but also undergoes interfacial reduction to generate a Li–Sn alloy layer on the lithium metal anode surface. This alloy interphase enhances interfacial charger transfer kinetics and increases the uniformity and density of lithium deposition, thereby substantially improving the cycling stability of lithium metal anodes.
5. Safety
The development of high-energy-density lithium batteries is fundamentally constrained by safety concerns, particularly under extreme conditions such as thermal abuse (e.g., elevated temperature), mechanical abuse (e.g., nail penetration), and electrical abuse (e.g., short circuit). These abuse conditions can induce a rapid increase in internal battery temperature, triggering a cascade of exothermic reactions and gas generation that ultimately lead to thermal runaway. The thermal runaway process typically involves multiple sequential steps. It is initiated by the decomposition or mechanical failure of interphases on the electrode surfaces, followed by continuous parasitic reactions between the electrodes and electrolytes. As the temperature further increases, the polymer separator melts, causing direct contact between the cathode and anode and inducing internal shorting. The resulting heat accumulation accelerates exothermic reactions, eventually leading to combustion or explosion of the battery. Therefore, developing electrolytes with high thermal stability is critically important for mitigating thermal runaway and improving the intrinsic safety of lithium batteries.Quasi-solid-state electrolytes fabricated via in situ polymerization have attracted significant interest owing to their non-flowing nature and superior mechanical strength. However, the presence of a substantial fraction of liquid components within in situ polymerized gel electrolytes still poses potential safety risks. To address these challenges and enhance the safety of quasi-solid-state lithium batteries, extensive efforts have been devoted to electrolyte design strategies in recent years.
5.1 Flame-retardant properties of polymer gel electrolytes
Conventional polyolefin separators typically melt at temperatures around 130 °C, leading to direct contact between the cathode and anode and triggering internal short circuits. This process accelerates heat accumulation and significantly increases the risk of thermal runaway. Therefore, improving the flame-retardant properties of in situ polymerized gel electrolytes is an effective approach to suppress the thermal runaway by maintaining physical separation between electrodes.216 To this end, two primary strategies have been adopted: (1) incorporating flame-retardant plasticizers into polymer gel electrolyte; (2) introducing flame-retardant functional groups into the polymer matrix through molecular-level design.Phosphate-based solvents, known for their excellent flame-retardant properties, are commonly utilized as plasticizers in in situ polymerized gel electrolytes. Yang et al. reported a polymer gel electrolyte comprising FEC and triethyl phosphate (TEP) as the plasticizer (GPE-TEP).217 With a TEP content of 33 vol%, the electrolyte exhibited remarkable flame-retardant behavior, remaining non-ignitable after a 2 s ignition attempt in a self-extinguishing test (SET). Thermogravimetric analysis (TGA) revealed negligible weight loss below 100 °C, indicating effective confinement of plasticizers within the polymer matrix. GPE-TEP demonstrates excellent flame retardancy and thermal stability, contributing to enhanced safety. Tang et al. reported an in situ polymerized gel electrolyte by incorporating TEP into PDOL, which exhibits both superior flame-retardant properties and high-voltage stability, providing an effective method for fabricating quasi-solid-state lithium batteries with high energy density and high safety.218 Similarly, trimethyl phosphate (TMP) has been widely employed as a flame-retardant solvent in polymer gel electrolytes.219 PTF-PE-SPE fabricated by Huang et al. contains 30 wt% TMP as a flame-retardant plasticizer and exhibits intrinsic nonflammability.124 Notably, PTF-PE-SPE demonstrated outstanding safety performance in Ah-level pouch cells. Fully charged Li‖LRMO pouch cells (charged to 4.7 V) using PTF-PE-SPE did not ignite during nail penetration tests. Extended volume-accelerating rate calorimeter (EV-ARC) measurements revealed significantly higher heat-release onset temperature (Tonset) and thermal runaway temperature (Ttr) compared with liquid electrolytes. Furthermore, the cell voltage remained above 3.6 V throughout the EV-ARC test, indicating the absence of severe internal shorting prior to thermal runaway. These results demonstrate that incorporating flame-retardant plasticizers into in situ polymerized gel electrolytes can substantially enhance battery safety, even under extreme abuse conditions.
Despite their good flame retardancy, phosphate-based solvents often exhibit poor compatibility with lithium metal anodes, resulting in severe parasitic reactions and rapid capacity fading. An alternative strategy involves chemically grafting flame-retardant functional groups, particularly phosphorus-containing functional groups, into the polymer matrix. This approach enhances flame retardancy and thermal stability while minimizing direct contact between flame-retardant species and the lithium metal anode. Li et al. developed a flame-retardant solid polymer electrolyte (FRSPE) via in situ copolymerization of methacrylate (MMA), allyl diglycol carbonate (ADC), and diethyl vinyl phosphonate (DEVP) (Fig. 12a).127 In this design, phosphorus-containing groups are covalently bonded to the polymer matrix, effectively suppressing parasitic reactions with the lithium metal anode. As a result, FRSPE exhibits excellent flame retardancy and thermal stability while simultaneously delivering improved electrochemical performance (Fig. 12b).
 | ||
| Fig. 12 (a) Schematic comparison of phosphate-free FRGPE and phosphate-bonded FRSPE. (b) Ignition tests of FRGPE and FRSPE. Reproduced with permission.127 Copyright 2024, Wiley-VCH. (c) Schematic illustration and (d) photographs of ignition tests of the ADN-based polymer gel electrolyte. Reproduced with permission.213 Copyright 2025, Wiley-VCH. | ||
In addition, polymer matrices incorporating high-bond-energy functional groups, such as F- and –CN-containing functional groups, have also demonstrated enhanced flame-retardant properties. For example, HVQE fabricated by Fan et al.75 and GMFN developed by Peng et al.163 showed that F-containing polymer matrices not only exhibit excellent flame retardancy but also enable high-energy-density pouch cells to retain thermal stability during nail penetration tests, without smoke or flame generation. Furthermore, Dai et al. reported a quasi-solid-state electrolyte via in situ polymerization of a butyl acrylate (BA) monomer in adiponitrile (ADN)-based electrolyte.213 Owing to the presence of –CN triple bonds, this electrolyte remained nonflammable even under prolonged flame exposure, demonstrating outstanding flame-retardant performance (Fig. 12c and d).
5.2 Thermal-responsive polymer gel electrolytes
Even with highly flame-retardant polymer gel electrolytes, safety risks can still arise under severe abuse conditions. Therefore, thermal-responsive polymer gel electrolytes that actively regulate ion transport at elevated temperatures have emerged as an advanced safety strategy.220,221 The Cui group developed an in situ polymerized gel electrolyte that exhibits thermally induced interfacial ion-blocking capability to address these safety concerns.222 This electrolyte is synthesized via copolymerization of VC and 2-ethyl-hexanoic acid vinyl ester (EAVE), resulting in a low polymerization conversion. Upon heating, residual VC monomers (∼50 mol% remaining) undergo secondary polymerization, forming a significantly thickened ion-blocking layer on the lithium metal anode surface. This process sharply increases cell impedance, thereby enabling an effective thermal shutdown behavior. More recently, the Cui group further reported a smart polymer gel electrolyte fabricated through in situ polymerization of cyanoethylurea-containing methacrylate and isocyanate-based methacrylate monomers in carbonate-based electrolytes, denoted as PCIE.223 When the temperature exceeds 120 °C, PCIE undergoes further cross-linking via nucleophilic addition reactions between urea and isocyanate groups (Fig. 13a). This thermally triggered cross-linking blocks ion transport and suppresses crosstalk induced by gas evolution, thereby significantly enhancing battery safety performance. Accelerating rate calorimetry measurements indicate that cells using PCIE exhibit lower Tonset and Ttr compared with those using liquid electrolytes, along with a reduced self-heating rate during the thermal runaway (Fig. 13b). These results underscore the effectiveness of PCIE in mitigating thermal hazards. | ||
| Fig. 13 (a) Schematic illustration of the in situ formed smart gel polymer electrolyte. (b) Time–temperature and self-heating rate (SHR) curves during ARC measurements of 1 Ah pouch cells (100% SOC) (inset shows the self-heating rate curve). Reproduced with permission.223 Copyright 2025, Springer Nature. | ||
6. Summary and perspective
This review provides a comprehensive overview of recent progress in in situ polymerized gel electrolytes for lithium batteries. Compared with conventional polymer gel electrolytes, the precursor solutions used in in situ polymerization can fully infiltrate and wet the electrode materials, ensuring intimate electrode/electrolyte contact and significantly reducing interfacial impedance. Benefiting from polymer matrix design and the incorporation of suitable plasticizers, in situ polymerized gel electrolytes generally exhibit high ionic conductivity. Therefore, quasi-solid-state lithium batteries fabricated via in situ polymerization have attracted substantial attention and are widely regarded as promising candidates for next-generation electrochemical energy storage devices. In addition, the relatively simple fabrication process associated with in situ strategies offers the potential to reduce manufacturing complexity and costs.Despite these advancements, the realization of high-energy-density and safe in situ polymerized quasi-solid-state lithium metal batteries still faces several critical challenges. These include oxidative decomposition of electrolytes at high-voltage cathode interfaces, severe parasitic reactions at electrolyte/high-capacity anode interfaces, and safety risks associated with high-energy-density lithium pouch cells. Considerable research efforts have been devoted to addressing these issues in recent years. Accordingly, this review systematically summarizes the recent advances in overcoming the aforementioned challenges and presents perspectives on future research directions.
For the fabrication of in situ polymerized quasi-solid-state lithium batteries, thermally initiated ester monomers containing unsaturated CC bonds are more suitable than cyclic ether-based monomers. This preference arises from the requirement that precursor solutions must fully infiltrate and wet the electrodes prior to polymerization. Unlike the uncontrollable ring-opening polymerization of cyclic ethers, thermally initiated free radical polymerization allows precise regulation of polymerization kinetics through temperature control. Therefore, in situ polymerized gel electrolytes derived from these monomers show great promise for constructing high-energy-density lithium pouch cells.
The adoption of high-voltage cathodes is essential for increasing the energy density of in situ polymerized quasi-solid-state lithium pouch cells. However, elevated cut-off voltage combined with the strong catalytic activity of transition metal ions can accelerate electrolyte oxidation at the cathode interface. To mitigate these effects, improving the intrinsic oxidant stability of electrolytes and constructing robust CEIs should be jointly considered during electrolyte design. Moreover, although increasing the cut-off voltage can further enhance energy density, it often comprises cycling stability and safety. Therefore, identifying an optimal cut-off voltage that balances energy density, durability, and safety remains a critical consideration.
Compatibility between in situ polymerized gel electrolytes and high-capacity anodes largely determines the cycling lifespan of lithium batteries, particularly in LMBs. Under practical operating conditions, rapid capacity decay is primarily caused by poor interfacial stability between the electrolyte and high-capacity anodes, ultimately leading to cell failure. Therefore, ensuring excellent electrolyte/anode compatibility should be a top priority in electrolyte design to achieve long-term cycling stability in high-energy-density lithium batteries.
In situ polymerized gel electrolytes with flame retardancy or thermal-responsive properties can substantially enhance the intrinsic safety of quasi-solid-state lithium batteries. Nevertheless, interfacial stability between electrodes and electrolytes remains equally crucial. Stable interfaces not only enhance electrochemical performance but also suppress heat generation associated with SEI degradation, thereby reducing the risk of thermal runaway. It should be emphasized that in situ polymerized quasi-solid-state LMBs still face significant safety challenges, which warrant continued and in-depth investigation.
Room-temperature ionic conductivity is another important parameter governing the performance of in situ polymerized gel electrolytes. While recent studies have predominantly focused on interfacial engineering, insufficient attention has been paid to ionic conductivity in some cases, resulting in suboptimal performance under practical conditions. Constructing a crosslinked network using cyclic ether-based or acrylate-based monomers can provide a rapid ion-conduction channel, which is a promising strategy to improve the ionic conductivity. Especially for quasi-solid-state batteries employing high-loading electrodes and lean electrolyte configurations, achieving high ionic conductivity is crucial for stable operation and should receive more emphasis in future research.
From a manufacturing perspective, the fabrication process of in situ polymerized quasi-solid-state lithium batteries is largely compatible with existing production lines for liquid-electrolyte-based lithium batteries, minimizing additional costs associated with process modifications. However, the nonuniformity of in situ polymerization gel electrolytes remains a major obstacle to commercialization. Spraying monomers and initiators onto the separator or regulating the in situ polymerization is regarded as a promising strategy to promote the uniform formation of quasi-solid-state electrolytes, thereby improving battery consistency for industrial production.
In summary, in situ polymerized gel electrolytes offer significant opportunities for the development of high-energy-density and safe quasi-solid-state lithium batteries, while also presenting substantial challenges. Through a comprehensive synthesis of recent studies, this review highlights effective strategies for addressing these challenges and outlines future research directions. With continued advances in in situ polymerization chemistry and increasing research focus, in situ polymerized quasi-solid-state lithium batteries are expected to make meaningful progress toward practical and commercial applications.
Author contributions
Z. H. F. and Y. H. X. conceived the idea and defined the structure of the review. All authors contributed to the writing, reviewing, and editing of the manuscript. Y. H. X. supervised the overall project.Conflicts of interest
The authors declare no conflict of interest.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no.: 22579126).References
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS.
- A. Innocenti, S. Beringer and S. Passerini, Nat. Rev. Mater., 2024, 9, 347–357 CrossRef CAS.
- J. B. Goodenough, Energy Environ. Sci., 2014, 7, 14–18 RSC.
- F. Duffner, N. Kronemeyer, J. Tübke, J. Leker, M. Winter and R. Schmuch, Nat. Energy, 2021, 6, 123–134 CrossRef CAS.
- K. Kang, S. M. Ying, J. Breger, C. P. Grey and G. Ceder, Science, 2006, 311, 977–980 CrossRef CAS PubMed.
- B. K. O'Regan, Nat. Energy, 2025, 10, 1181–1182 CrossRef.
- G. Harper, R. Sommerville, E. Kendrick, L. Driscoll, P. Slater, R. Stolkin, A. Walton, P. Christensen, O. Heidrich, S. Lambert, A. Abbott, K. Ryder, L. Gaines and P. Anderson, Nature, 2019, 575, 75–86 CrossRef CAS PubMed.
- V. Viswanathan, A. H. Epstein, Y. M. Chiang, E. Takeuchi, M. Bradley, J. Langford and M. Winter, Nature, 2022, 601, 519–525 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- X. B. Cheng, R. Zhang, C. Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- S. Chen, J. Zheng, L. Yu, X. Ren, M. H. Engelhard, C. Niu, H. Lee, W. Xu, J. Xiao, J. Liu and J.-G. Zhang, Joule, 2018, 2, 1548–1558 CrossRef CAS.
- M. Li, R. P. Hicks, Z. Chen, C. Luo, J. Guo, C. Wang and Y. Xu, Chem. Rev., 2023, 123, 1712–1773 CrossRef CAS.
- P. Lennartz, B. A. Paren, A. Herzog-Arbeitman, X. C. Chen, J. A. Johnson, M. Winter, Y. Shao-Horn and G. Brunklaus, Joule, 2023, 7, 1471–1495 CrossRef CAS.
- Q. Zhou, J. Ma, S. Dong, X. Li and G. Cui, Adv. Mater., 2019, 31, e1902029 CrossRef PubMed.
- G. Xi, M. Xiao, S. Wang, D. Han, Y. Li and Y. Meng, Adv. Funct. Mater., 2021, 31, 2007598 CrossRef CAS.
- S. Huo, L. Sheng, W. Xue, L. Wang, H. Xu, H. Zhang and X. He, InfoMat, 2023, 5, e12394 CrossRef CAS.
- F. Chen, X. Wang, M. Armand and M. Forsyth, Nat. Mater., 2022, 21, 1175–1182 CrossRef CAS PubMed.
- H. Wang, L. Sheng, G. Yasin, L. Wang, H. Xu and X. He, Energy Storage Mater., 2020, 33, 188–215 CrossRef.
- Z. Huang, Z. Wang, X. Chen, L. Yang, T. Huang, X. He, W. Huang, J. Chen, X. Yang, L. Chen, B. Liao, X. Ouyang, J. Liu, X. Ren, F. Pan, Q. Zhang and J. Hu, Adv. Mater., 2025, 37, e04186 CrossRef CAS PubMed.
- X. Zhou, Y. Zhou, L. Yu, L. Qi, K. S. Oh, P. Hu, S. Y. Lee and C. Chen, Chem. Soc. Rev., 2024, 53, 5291–5337 RSC.
- Y. G. Cho, C. Hwang, D. S. Cheong, Y. S. Kim and H. K. Song, Adv. Mater., 2019, 31, e1804909 CrossRef PubMed.
- C. Ma, W. Cui, X. Liu, Y. Ding and Y. Wang, InfoMat, 2021, 4, e12232 CrossRef.
- S. Zhang, B. Xie, X. Zhuang, S. Wang, L. Qiao, S. Dong, J. Ma, Q. Zhou, H. Zhang, J. Zhang, J. Ju, G. Xu, Z. Cui and G. Cui, Adv. Funct. Mater., 2024, 34, 2314063 CrossRef CAS.
- G. Xiao, H. Xu, C. Bai, M. Liu and Y.-B. He, Interdiscip. Mater., 2023, 2, 609–634 CAS.
- H. Zhang, X. Xu, W. Fan, J. Zhao and Y. Huo, Chem.–Eur. J., 2024, 30, e202402798 CrossRef CAS PubMed.
- P. Cui, Y. Li, Y. Liu, S. Wang, X. Tang, Y. Ye, H. Su and C. Sun, RSC Adv., 2024, 14, 36152–36160 RSC.
- T. Liu, J. Zhang, W. Han, J. Zhang, G. Ding, S. Dong and G. Cui, J. Electrochem. Soc., 2020, 167, 070527 CrossRef CAS.
- C. Wang, J. Sun, X. Qu, X. Liu, S. Dong and G. Cui, Curr. Opin. Electrochem., 2022, 33, 100962 CrossRef CAS.
- Q. Liu, L. Wang and X. He, Adv. Energy Mater., 2023, 13, 2300798 CrossRef CAS.
- H. Su, Z. Chen, M. Li, P. Bai, Y. Li, X. Ji, Z. Liu, J. Sun, J. Ding, M. Yang, X. Yao, C. Mao and Y. Xu, Adv. Mater., 2023, 35, e2301171 CrossRef PubMed.
- T. Zhou, J. Wang, L. Lv, R. Li, L. Chen, S. Zhang, H. Zhang, B. Ma, J. Huang, B. Wu, L. Chen, T. Deng and X. Fan, Energy Environ. Sci., 2024, 17, 9185–9194 RSC.
- C. Cai, Z. Fan and Y. Xu, Chem. Res. Chin. Univ., 2025, 41, 1620–1627 CrossRef CAS.
- Z. Wang, J. Chen, J. Fu, Z. Li and X. Guo, Energy Mater., 2024, 4, 400050 CAS.
- P. Shi, J. Ma, M. Liu, S. Guo, Y. Huang, S. Wang, L. Zhang, L. Chen, K. Yang, X. Liu, Y. Li, X. An, D. Zhang, X. Cheng, Q. Li, W. Lv, G. Zhong, Y.-B. He and F. Kang, Nat. Nanotechnol., 2023, 18, 602–610 CrossRef CAS PubMed.
- Z. Lin, Y. Li, P. Ding, C. Lin, F. Chen, R. Yu and Y. Xia, Battery Energy, 2025, 4, e20240063 CrossRef CAS.
- C. Z. Zhao, Q. Zhao, X. Liu, J. Zheng, S. Stalin, Q. Zhang and L. A. Archer, Adv. Mater., 2020, 32, e1905629 CrossRef PubMed.
- Z. Ning, G. Li, D. L. R. Melvin, Y. Chen, J. Bu, D. Spencer-Jolly, J. Liu, B. Hu, X. Gao, J. Perera, C. Gong, S. D. Pu, S. Zhang, B. Liu, G. O. Hartley, A. J. Bodey, R. I. Todd, P. S. Grant, D. E. J. Armstrong, T. J. Marrow, C. W. Monroe and P. G. Bruce, Nature, 2023, 618, 287–293 CrossRef CAS PubMed.
- S. Zou, Y. Yang, J. Wang, X. Zhou, X. Wan, M. Zhu and J. Liu, Energy Environ. Sci., 2024, 17, 4426–4460 RSC.
- Z. Y. Wang, C. Z. Zhao, N. Yao, Y. Lu, Z. Q. Xue, X. Y. Huang, P. Xu, W. Z. Huang, Z. X. Wang, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2024, 64, e202414524 CrossRef PubMed.
- B. Yang, T. Li, Y. Pan, L. Yang, K. Li, J. Chen, Z. Yan, A. Hu and J. Long, Energy Mater., 2024, 4, 400061 CAS.
- J. Xiang, Y. Zhang, B. Zhang, L. Yuan, X. Liu, Z. Cheng, Y. Yang, X. Zhang, Z. Li, Y. Shen, J. Jiang and Y. Huang, Energy Environ. Sci., 2021, 14, 3510–3521 RSC.
- X. Ma, Y. Lu, Y. Ou, S. Yan, W. Hou, P. Zhou and K. Liu, Nano Res., 2024, 17, 8754–8771 CrossRef CAS.
- S. J. Yang, H. Yuan, N. Yao, J. K. Hu, X. L. Wang, R. Wen, J. Liu and J. Q. Huang, Adv. Mater., 2024, 36, e2405086 CrossRef PubMed.
- Y. S. Meng, V. Srinivasan and K. Xu, Science, 2022, 378, eabq3750 CrossRef CAS PubMed.
- J. Yu, X. Lin, J. Liu, J. T. T. Yu, M. J. Robson, G. Zhou, H. M. Law, H. Wang, B. Z. Tang and F. Ciucci, Adv. Energy Mater., 2021, 12, 2102932 CrossRef.
- V. Vijayakumar, B. Anothumakkool, S. Kurungot, M. Winter and J. R. Nair, Energy Environ. Sci., 2021, 14, 2708–2788 RSC.
- P. Xu, Z.-Y. Shuang, C.-Z. Zhao, X. Li, L.-Z. Fan, A. Chen, H. Chen, E. Kuzmina, E. Karaseva, V. Kolosnitsyn, X. Zeng, P. Dong, Y. Zhang, M. Wang and Q. Zhang, Sci. China Chem., 2023, 67, 67–86 CrossRef.
- S. Wang, L. Zhang, Q. Zeng, J. Guan, H. Gao, L. Zhang, J. Zhong, W. Y. Lai and Q. Wang, Adv. Energy Mater., 2023, 14, 2302876 CrossRef.
- M. Li, J. Yang, Y. Shi, Z. Chen, P. Bai, H. Su, P. Xiong, M. Cheng, J. Zhao and Y. Xu, Adv. Mater., 2022, 34, e2107226 CrossRef PubMed.
- H. Yang, M. Jing, L. Wang, H. Xu, X. Yan and X. He, Nano-Micro Lett., 2024, 16, 127 CrossRef PubMed.
- H. Yang, B. Zhang, M. Jing, X. Shen, L. Wang, H. Xu, X. Yan and X. He, Adv. Energy Mater., 2022, 12, 2201762 CrossRef CAS.
- Q. Zhao, X. Liu, S. Stalin, K. Khan and L. A. Archer, Nat. Energy, 2019, 4, 365–373 CrossRef CAS.
- Z. Li, R. Yu, S. Weng, Q. Zhang, X. Wang and X. Guo, Nat. Commun., 2023, 14, 482 CrossRef PubMed.
- J. Chai, Z. Liu, J. Ma, J. Wang, X. Liu, H. Liu, J. Zhang, G. Cui and L. Chen, Adv. Sci., 2017, 4, 1600377 CrossRef PubMed.
- Q. Zhou, C. Fu, R. Li, X. Zhang, B. Xie, Y. Gao, G. Yin and P. Zuo, Chem. Eng. J., 2022, 437, 135419 CrossRef CAS.
- Q. Sun, S. Wang, Y. Ma, D. Song, H. Zhang, X. Shi, N. Zhang and L. Zhang, Adv. Mater., 2023, 35, e2300998 CrossRef PubMed.
- W. Zhang, J. Zhang, X. Liu, H. Li, Y. Guo, C. Geng, Y. Tao and Q. H. Yang, Adv. Funct. Mater., 2022, 32, 2201205 CrossRef CAS.
- T. Dong, H. Zhang, R. Hu, P. Mu, Z. Liu, X. Du, C. Lu, G. Lu, W. Liu and G. Cui, Energy Storage Mater., 2022, 50, 525–532 CrossRef.
- W. Min, L. Li, M. Wang, S. Ma, H. Feng, W. Wang, H. Ding, T. Cheng, Z. Li, T. Saito, H. Yang and P. F. Cao, Angew. Chem., Int. Ed., 2025, 64, e202422510 CrossRef CAS PubMed.
- P. Li, S. Wang, J. Hao, X. Wang, S. M. Hao, Y. Lu, H. Li, W. Zhou and Y. Li, Angew. Chem., Int. Ed., 2023, 62, e202309613 CrossRef CAS PubMed.
- L. Ma, J. Zhao, M. Li, H. Su, Y. Li, Y. Liu, H. Liu, E. Zygadlo-Monikowska and Y. Xu, ACS Appl. Mater. Interfaces, 2025, 17, 18206–18216 CrossRef CAS PubMed.
- Y. Zhao, Y. Bai, W. Li, M. An, Y. Bai and G. Chen, Chem. Mater., 2020, 32, 6811–6830 CrossRef CAS.
- J. Holoubek, M. Yu, S. Yu, M. Li, Z. Wu, D. Xia, P. Bhaladhare, M. S. Gonzalez, T. A. Pascal, P. Liu and Z. Chen, ACS Energy Lett., 2020, 5, 1438–1447 CrossRef CAS.
- X. Fan and C. Wang, Chem. Soc. Rev., 2021, 50, 10486–10566 RSC.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- J. Li, J. Chen, X. Xu, J. Shen, Z. Wang, Z. Guo, P. Lin, J. Sun, B. Huang and T. Zhao, Adv. Mater., 2025, 37, e2501006 CrossRef PubMed.
- J. Xiao, N. Adelstein, Y. Bi, W. Bian, J. Cabana, C. L. Cobb, Y. Cui, S. J. Dillon, M. M. Doeff, S. M. Islam, K. Leung, M. Li, F. Lin, J. Liu, H. Luo, A. C. Marschilok, Y. S. Meng, Y. Qi, R. Sahore, K. G. Sprenger, R. C. Tenent, M. F. Toney, W. Tong, L. F. Wan, C. Wang, S. E. Weitzner, B. Wu and Y. Xu, Nat. Energy, 2024, 9, 1463–1473 CrossRef CAS.
- W. Zhang, T. Yang, X. Liao, Y. Song and Y. Zhao, Energy Storage Mater., 2023, 57, 249–259 CrossRef.
- Y. Huang, B. Cao, X. Xu, X. Li, K. Zhou, Z. Geng, Q. Li, X. Yu and H. Li, Adv. Energy Mater., 2024, 14, 2400943 CrossRef CAS.
- Y. Wang, Z. Li, Y. Hou, Z. Hao, Q. Zhang, Y. Ni, Y. Lu, Z. Yan, K. Zhang, Q. Zhao, F. Li and J. Chen, Chem. Soc. Rev., 2023, 52, 2713–2763 RSC.
- Y. Lu, X. Zhang, Y. Wu, H. Cheng and Y. Lu, Ind. Chem. Mater., 2025, 3, 151–177 RSC.
- G. Zhou, J. Yu and F. Ciucci, Energy Storage Mater., 2023, 55, 642–651 CrossRef.
- L. Tang, B. Chen, Z. Zhang, C. Ma, J. Chen, Y. Huang, F. Zhang, Q. Dong, G. Xue, D. Chen, C. Hu, S. Li, Z. Liu, Y. Shen, Q. Chen and L. Chen, Nat. Commun., 2023, 14, 2301 CrossRef CAS PubMed.
- Z. Fan, Y. Liu, H. Su, S. Chi, Q. Dong, Y. Liu, L. Wang, Q.-H. Yang and Y. Xu, Chem. Eng. J., 2026, 527, 171745 CrossRef CAS.
- J. Zhu, P. Bian, G. Sun, J. Zhang, G. Lou, X. Song, R. Zhao, J. Liu, N. Xu, A. Li, X. Wan, Y. Ma, C. Li, H. Zhang and Y. Chen, Angew. Chem., Int. Ed., 2025, 64, e202424685 CrossRef CAS PubMed.
- D. Zhang, Y. Liu, D. Li, S. Li, Q. Xiong, Z. Huang, S. Wang, H. Hong, J. Zhu, H. Lv and C. Zhi, Energy Environ. Sci., 2025, 18, 227–235 RSC.
- S. Li, H. Hong, X. Yang, D. Li, Q. Xiong, D. Zhang, S. Wang, Z. Huang, H. Lv and C. Zhi, Adv. Mater., 2025, 37, e2504333 CrossRef PubMed.
- J. Zhu, R. Zhao, J. Zhang, X. Song, J. Liu, N. Xu, H. Zhang, X. Wan, X. Ji, Y. Ma, C. Li and Y. Chen, Angew. Chem., Int. Ed., 2024, 63, e202400303 CrossRef CAS PubMed.
- T. Hou, D. Wang, B. Jiang, Y. Liu, J. Kong, Y. He, Y. Huang and H. Xu, Nat. Commun., 2025, 16, 962 CrossRef CAS PubMed.
- Y. Liu, H. Zou, Z. Huang, Q. Wen, J. Lai, Y. Zhang, J. Li, K. Ding, J. Wang, Y. Lan and Q. Zheng, Energy Environ. Sci., 2023, 13, 6110–6119 RSC.
- Y. Dai, Z. Hou, X. Yu, Z. Yan, Y. Feng, G. Luo, D. Deng, W. Peng, Z. Wang, H. Guo, H. Duan, X. Ou, J. C. Zheng and J. Wang, ACS Appl. Mater. Interfaces, 2025, 17, 23197–23208 CrossRef CAS PubMed.
- Q. An, Q. Liu, P. Mao, L. Duan, H. Y. Zhu, L. Liu, G. Zhao, Y. Zha, L. Yang, M. Sun, Y. Fan, F. Xie, G. Hu and H. Guo, Angew. Chem., Int. Ed., 2025, 64, e202422539 CrossRef CAS PubMed.
- K. Zeng, Q. Liu, H. Ma, G. Zhao, Q. An, C. Zhang, Y. Yang, M. Sun, Q. Xu, L. Duan and H. Guo, Energy Storage Mater., 2024, 70, 103564 CrossRef.
- F. Pei, Y. Huang, L. Wu, S. Zhou, Q. Kang, W. Lin, Y. Liao, Y. Zhang, K. Huang, Y. Shen, L. Yuan, S. G. Sun, Z. Li and Y. Huang, Adv. Mater., 2024, 36, e2409269 CrossRef PubMed.
- Q. Liu, L. Yang, Z. Mei, Q. An, K. Zeng, W. Huang, S. Wang, Y. Sun and H. Guo, Energy Environ. Sci., 2024, 17, 780–790 RSC.
- Z. Piao, H. R. Ren, G. Lu, K. Jia, J. Tan, X. Wu, Z. Zhuang, Z. Han, C. Li, R. Gao, X. Tao, G. Zhou and H. M. Cheng, Angew. Chem., Int. Ed., 2023, 62, e202300966 CrossRef CAS PubMed.
- H. Xu, W. Deng, L. Shi, J. Long, Y. Zhang, L. Xu and L. Mai, Angew. Chem., Int. Ed., 2024, 63, e202400032 CrossRef CAS PubMed.
- J. Sun, Y. Wang, Y. Li and W. Feng, Sci. China Mater., 2024, 67, 1393–1402 CrossRef CAS.
- B.-H. Zhang, W.-X. Wen, H.-Y. Wang, Y.-L. Hou, J.-Z. Chen and D.-L. Zhao, Chem. Eng. J., 2023, 472, 144990 CrossRef CAS.
- L. Jin, J. Chen, X. Xu, Z. Wang, Z. Guo, P. Lin, J. Sun, B. Huang and T. Zhao, Sci. Adv., 2025, 11, eadz5203 CrossRef PubMed.
- G. Zhang, T. Zhang, Y. Liu, Q. Wang, R. He, P. Li, Y. Cui, Z. Liu, C. Wang, Y. Deng, J. Chang and J. Lu, Angew. Chem., Int. Ed., 2025, 64, e202506056 CrossRef CAS PubMed.
- X. Fan, X. Ji, L. Chen, J. Chen, T. Deng, F. Han, J. Yue, N. Piao, R. Wang, X. Zhou, X. Xiao, L. Chen and C. Wang, Nat. Energy, 2019, 4, 882–890 CrossRef CAS.
- Y. Yang, Z. Fang, Y. Yin, Y. Cao, Y. Wang, X. Dong and Y. Xia, Angew. Chem., Int. Ed., 2022, 61, e202208345 CrossRef CAS PubMed.
- L. Xue, K. Ueno, S.-Y. Lee and C. A. Angell, J. Power Sources, 2014, 262, 123–128 CrossRef CAS.
- C.-C. Su, M. He, P. C. Redfern, L. A. Curtiss, I. A. Shkrob and Z. Zhang, Energy Environ. Sci., 2017, 10, 900–904 RSC.
- N. Yang, Y. Cui, H. Su, J. Peng, Y. Shi, J. Niu and F. Wang, Angew. Chem., Int. Ed., 2023, 62, e202304339 CrossRef CAS PubMed.
- M. Li, Y. Liu, X. Yang, Q. Zhang, Y. Cheng, L. Deng, Q. Zhou, T. Cheng and M. D. Gu, Adv. Mater., 2024, 36, e2404271 CrossRef PubMed.
- J. Zhang, H. Zhang, S. Weng, R. Li, D. Lu, T. Deng, S. Zhang, L. Lv, J. Qi, X. Xiao, L. Fan, S. Geng, F. Wang, L. Chen, M. Noked, X. Wang and X. Fan, Nat. Commun., 2023, 14, 2211 CrossRef CAS PubMed.
- Z. Wang, X. Che, D. Wang, Y. Wang, X. He, Y. Zhu and B. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202404109 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- Y. Hou, Y. Wang, Z. Wei, Z. Wu, D. Li, Q. Li, S. Li, Z. Chen, Y. Wang, G. Liang, K. Wang and C. Zhi, Angew. Chem., Int. Ed., 2025, 64, e202505147 CrossRef CAS PubMed.
- B. Li, J. Zhao, Z. Zhang, C. Zhao, P. Sun, P. Bai, J. Yang, Z. Zhou and Y. Xu, Adv. Funct. Mater., 2019, 29, 1807137 CrossRef.
- W. Xu, Y. Mu, Y. Yin, A. Hu, Y. Li, J. Wang, Q. Liu, J. Long, L. Zeng and S. Chen, Energy Environ. Sci., 2026, 19, 384–396 RSC.
- J. Chen, Y. Lin, Q. Li, H. Ren, L. Zhang, Y. Sun, S. Zhang, X. Shang, W. Zhou, M. Wu and Z. Li, Angew. Chem., Int. Ed., 2024, 63, e202407024 CrossRef CAS PubMed.
- M. S. Choi, S. G. Kang, J. Choi, J. Ko and J. H. Park, Angew. Chem., Int. Ed., 2025, 64, e202424568 CrossRef CAS PubMed.
- E. Markevich, G. Salitra and D. Aurbach, ACS Energy Lett., 2017, 2, 1337–1345 CrossRef CAS.
- T. Schedlbauer, S. Krüger, R. Schmitz, R. W. Schmitz, C. Schreiner, H. J. Gores, S. Passerini and M. Winter, Electrochim. Acta, 2013, 92, 102–107 CrossRef CAS.
- K. Xu, S. Zhang, T. R. Jow, W. Xu and C. A. Angell, Electrochem. Solid-State Lett., 2002, 5, A26–A29 CrossRef CAS.
- S. Wu, Y. Lin, L. Xing, G. Sun, H. Zhou, K. Xu, W. Fan, L. Yu and W. Li, ACS Appl. Mater. Interfaces, 2019, 11, 17940–17951 CrossRef CAS PubMed.
- W. Zhao, B. Zheng, H. Liu, F. Ren, J. Zhu, G. Zheng, S. Chen, R. Liu, X. Yang and Y. Yang, Nano Energy, 2019, 63, 103815 CrossRef CAS.
- N. Xu, H. Zhou, Y. Liao, G. Li, M. Xu and W. Li, Solid State Ionics, 2019, 341, 115049 CrossRef CAS.
- Y.-K. Han, J. Yoo and T. Yim, J. Mater. Chem. A, 2015, 3, 10900–10909 RSC.
- Z. Wang, L. Xing, J. Li, M. Xu and W. Li, J. Power Sources, 2016, 307, 587–592 CrossRef CAS.
- M. Mao, X. Ji, Q. Wang, Z. Lin, M. Li, T. Liu, C. Wang, Y. S. Hu, H. Li, X. Huang, L. Chen and L. Suo, Nat. Commun., 2023, 14, 1082 CrossRef CAS PubMed.
- M. Mao, B. Huang, Q. Li, C. Wang, Y.-B. He and F. Kang, Nano Energy, 2020, 78, 105282 CrossRef CAS.
- J. Park, H. Seong, C. Yuk, D. Lee, Y. Byun, E. Lee, W. Lee and B. J. Kim, Adv. Mater., 2024, 36, e2403191 CrossRef PubMed.
- Z. Sun, J. Yang, Y. Wu, F. Meng, Y. Niu, H. Xu, Y. Zhu, B. Hong, Z. Chen, J. Zhu, Q. He, G. Wu and W. Chen, J. Am. Chem. Soc., 2025, 147, 18064–18073 CrossRef CAS PubMed.
- G. Liu, H. Yu, T. Zhu, H. Jia, X. Dong, W. Liu, J. Sun, X. Gu and S. Zhang, Nano Energy, 2025, 145, 111471 CrossRef CAS.
- H. Wu, B. Tang, X. Du, J. Zhang, X. Yu, Y. Wang, J. Ma, Q. Zhou, J. Zhao, S. Dong, G. Xu, J. Zhang, H. Xu, G. Cui and L. Chen, Adv. Sci., 2020, 7, 2003370 CrossRef CAS PubMed.
- T. Yim, K. S. Kang, J. Mun, S. H. Lim, S.-G. Woo, K. J. Kim, M.-S. Park, W. Cho, J. H. Song, Y.-K. Han, J.-S. Yu and Y.-J. Kim, J. Power Sources, 2016, 302, 431–438 CrossRef CAS.
- Q. Lv, C. Li, Y. Liu, Y. Jing, J. Sun, H. Wang, L. Wang, H. Ren, B. Wu, T. Cheng, D. Wang, H. Liu, S. X. Dou, B. Wang and J. Wang, ACS Nano, 2024, 18, 23253–23264 CrossRef CAS PubMed.
- Y. Lu, Y. Liu, S. Zhang, Y. Wu, H. Cheng and Y. Lu, Energy Environ. Sci., 2025, 18, 9512–9523 RSC.
- X.-Y. Huang, C.-Z. Zhao, W.-J. Kong, N. Yao, Z.-Y. Shuang, P. Xu, S. Sun, Y. Lu, W.-Z. Huang, J.-L. Li, L. Shen, X. Chen, J.-Q. Huang, L. A. Archer and Q. Zhang, Nature, 2025, 646, 343–350 CrossRef CAS PubMed.
- P. Xu, Y. C. Gao, Y. X. Huang, Z. Y. Shuang, W. J. Kong, X. Y. Huang, W. Z. Huang, N. Yao, X. Chen, H. Yuan, C. Z. Zhao, J. Q. Huang and Q. Zhang, Adv. Mater., 2024, 36, e2409489 CrossRef PubMed.
- G. Ye, L. Zhu, Y. Ma, M. He, C. Zheng, K. Shen, X. Hong, Z. Xiao, Y. Jia, P. Gao and Q. Pang, J. Am. Chem. Soc., 2024, 146, 27668–27678 CrossRef CAS PubMed.
- Z. Li, S. Zhu, S. Gao, Y. He, H. Ding, D. Yang, H. Yang and P. F. Cao, Adv. Funct. Mater., 2024, 34, 2409836 CrossRef CAS.
- C. Fu, X. Zhang, H. Huo, J. Zhu, H. Xu, L. Wang, Y. Ma, Y. Gao, G. Yin, P. Zuo and J. Lu, Adv. Funct. Mater., 2024, 34, 2312187 CrossRef CAS.
- S. Liu, W. Tian, J. Shen, Z. Wang, H. Pan, X. Kuang, C. Yang, S. Chen, X. Han, H. Quan and S. Zhu, Nat. Commun., 2025, 16, 2474 CrossRef CAS PubMed.
- S. Ma, D. Zhang, Z. Tang, W. Li, Y. Zhang, Y. Zhang, K. Ji and M. Chen, ACS Appl. Mater. Interfaces, 2024, 16, 20430–20442 CAS.
- P. Li, J. Hao, S. He, Z. Chang, X. Li, R. Wang, W. Ma, J. Wang, Y. Lu, H. Li, L. Zhang and W. Zhou, Nat. Commun., 2025, 16, 3727 CrossRef CAS PubMed.
- J. Chen, C. He, X. Peng, J. Li, X. Xu, Y. Zhou, J. Shen, J. Sun, Y. Li and T. Zhao, Nat. Commun., 2025, 16, 8494 CrossRef CAS PubMed.
- Y. Zou, Z. Ma, G. Liu, Q. Li, D. Yin, X. Shi, Z. Cao, Z. Tian, H. Kim, Y. Guo, C. Sun, L. Cavallo, L. Wang, H. N. Alshareef, Y. K. Sun and J. Ming, Angew. Chem., Int. Ed., 2023, 62, e202216189 CrossRef CAS PubMed.
- H. Lyu, Y. Li, C. J. Jafta, C. A. Bridges, H. M. Meyer, A. Borisevich, M. P. Paranthaman, S. Dai and X.-G. Sun, J. Power Sources, 2019, 412, 527–535 CrossRef CAS.
- L. Ma, J. Tan, Z. Ren, B. Feng, Z. Liu, P. Yi, S. Cao, W. Lu, Y. Liu, C. Ye, M. Ye, H. Fang and J. Shen, Adv. Funct. Mater., 2024, 35, 2414816 CrossRef.
- Y. Jin, P. M. L. Le, P. Gao, Y. Xu, B. Xiao, M. H. Engelhard, X. Cao, T. D. Vo, J. Hu, L. Zhong, B. E. Matthews, R. Yi, C. Wang, X. Li, J. Liu and J.-G. Zhang, Nat. Energy, 2022, 7, 718–725 CrossRef CAS.
- Z. Wu, R. Li, S. Zhang, L. lv, T. Deng, H. Zhang, R. Zhang, J. Liu, S. Ding, L. Fan, L. Chen and X. Fan, Chem, 2023, 9, 650–664 CAS.
- S. Zhang, R. Li, T. Deng, Q. Ma, X. Hong, H. Zhang, R. Zhang, S. Ding, Y. Wu, H. Zhu, M. Li, H. Zhang, D. Lu, B. Ma, L. Lv, Y. Li, L. Chen, Y. Shen, R. Guo and X. Fan, Nat. Energy, 2024, 9, 1285–1296 CrossRef CAS.
- J. Xu, J. Zhang, T. P. Pollard, Q. Li, S. Tan, S. Hou, H. Wan, F. Chen, H. He, E. Hu, K. Xu, X.-Q. Yang, O. Borodin and C. Wang, Nature, 2023, 614, 694–700 CrossRef CAS PubMed.
- Y. Meng, D. Zhou, R. Liu, Y. Tian, Y. Gao, Y. Wang, B. Sun, F. Kang, M. Armand, B. Li, G. Wang and D. Aurbach, Nat. Energy, 2023, 8, 1023–1033 CrossRef CAS.
- X. Song, R. Zhao, J. Zhu, J. Zhang, N. Xu, J. Liu, Y. Liu, H. Zhang, Y. Ma, C. Li and Y. Chen, Natl. Sci. Rev., 2025, 12, nwaf016 CrossRef CAS PubMed.
- H. Kwon, S. Kim, J. Hyun, H. E. Lee, S. S. Kim, Y. Kim, I. J. Kim, K. Shin, S. Kim, C. Park, H. Kim, D. Shin and H.-T. Kim, Nat. Energy, 2025, 10, 1132–1145 CrossRef CAS.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 RSC.
- D. H. S. Tan, Y.-T. Chen, H. Yang, W. Bao, B. Sreenarayanan, J.-M. Doux, W. Li, B. Lu, S.-Y. Ham, B. Sayahpour, J. Scharf, E. A. Wu, G. Deysher, H. E. Han, H. Jin Hah, H. Jeong, J. B. Lee, Z. Chen and Y. S. Meng, Science, 2021, 373, 1494–1499 CrossRef CAS PubMed.
- W. Yan, Z. Mu, Z. Wang, Y. Huang, D. Wu, P. Lu, J. Lu, J. Xu, Y. Wu, T. Ma, M. Yang, X. Zhu, Y. Xia, S. Shi, L. Chen, H. Li and F. Wu, Nat. Energy, 2023, 8, 800–813 CrossRef CAS.
- H. Huang, Y. Hu, Y. Hou, X. Wang, Q. Dong, Z. Zhao, M. Ji, W. Zhang, J. Li, J. Xie, H. Guo, X. Han, X. Ouyang and W. Hu, Nature, 2025, 644, 660–667 CrossRef CAS PubMed.
- H. Ji, J. Xiang, Y. Li, M. Zheng, L. Yuan, Y. Liao, L. Du, Z. Li, Z. Xie, K. Huang, X. Lin, Z. Xie, Y. Shen, M. Chen, T. Li, G. Feng, Y. Sun, L. Qie, H. Li, F. Zhang, R. Guo, X. Feng, W. Chen, X. Ai, J. Lu and Y. Huang, Nature, 2025, 643, 1255–1262 CrossRef CAS PubMed.
- J. Xiao, Q. Li, Y. Bi, M. Cai, B. Dunn, T. Glossmann, J. Liu, T. Osaka, R. Sugiura, B. Wu, J. Yang, J.-G. Zhang and M. S. Whittingham, Nat. Energy, 2020, 5, 561–568 CrossRef CAS.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- G. M. Hobold, J. Lopez, R. Guo, N. Minafra, A. Banerjee, Y. Shirley Meng, Y. Shao-Horn and B. M. Gallant, Nat. Energy, 2021, 6, 951–960 CrossRef CAS.
- L. Chen, T. Gu, J. Mi, Y. Li, K. Yang, J. Ma, X. An, Y. Jiang, D. Zhang, X. Cheng, S. Guo, Z. Han, T. Hou, Y. Cao, M. Liu, W. Lv, Y. B. He and F. Kang, Nat. Commun., 2025, 16, 3517 CrossRef CAS PubMed.
- G. Ye, X. Hong, M. He, J. Song, L. Zhu, C. Zheng, Y. Ma, Y. An, K. Shen, W. Shi, Y. Jia, M. B. Shafqat, P. Gao, D. Xia, F. Chen and Q. Pang, Adv. Mater., 2025, 37, e2417829 CrossRef PubMed.
- H. An, M. Li, Q. Liu, Y. Song, J. Liu, Z. Yu, X. Liu, B. Deng and J. Wang, Nat. Commun., 2024, 15, 9150 CrossRef CAS PubMed.
- J. Zhang, J. Zhu, R. Zhao, J. Liu, X. Song, N. Xu, Y. Liu, H. Zhang, X. Wan, Y. Ma, C. Li and Y. Chen, Energy Environ. Sci., 2024, 17, 7119–7128 RSC.
- K. He, S. H. Cheng, J. Hu, Y. Zhang, H. Yang, Y. Liu, W. Liao, D. Chen, C. Liao, X. Cheng, Z. Lu, J. He, J. Tang, R. K. Y. Li and C. Liu, Angew. Chem., Int. Ed., 2021, 60, 12116–12123 CrossRef CAS PubMed.
- G. Feng, Q. Ma, D. Luo, T. Yang, Y. Nie, Z. Zheng, L. Yang, S. Li, Q. Li, M. Jin, X. Wang and Z. Chen, Angew. Chem., Int. Ed., 2025, 64, e202413306 CrossRef CAS PubMed.
- L. P. Hou, N. Yao, J. Xie, P. Shi, S. Y. Sun, C. B. Jin, C. M. Chen, Q. B. Liu, B. Q. Li, X. Q. Zhang and Q. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202201406 CrossRef CAS PubMed.
- H. Wang, J. Yang, X. Xu, J. Geng, X. Lin, H. Xu and Y. Huang, Adv. Mater., 2025, 37, e04625 CrossRef CAS PubMed.
- Q. Liu, Q. An, K. Zeng, M. Yang, H. Zhu, X. Liang, G. Zhao, M. Sun, Y. Zha, L. Yang, L. Duan, G. Zhao, Y. Sun and H. Guo, Energy Environ. Sci., 2025, 18, 4934–4948 RSC.
- K. Hatzell, W. Chang, W. Bao, M. Cai, T. Glossmann, S. Kalnaus, B. Liaw, Y. S. Meng, R. Mohtadi and Y. Wang, Joule, 2024, 8, 1550–1555 CrossRef CAS.
- Y. Chen, Z. Wang, X. Li, X. Yao, C. Wang, Y. Li, W. Xue, D. Yu, S. Y. Kim, F. Yang, A. Kushima, G. Zhang, H. Huang, N. Wu, Y. W. Mai, J. B. Goodenough and J. Li, Nature, 2020, 578, 251–255 CrossRef CAS PubMed.
- C. Niu, D. Liu, J. A. Lochala, C. S. Anderson, X. Cao, M. E. Gross, W. Xu, J.-G. Zhang, M. S. Whittingham, J. Xiao and J. Liu, Nat. Energy, 2021, 6, 723–732 CrossRef CAS.
- X. Peng, Y. Zhang, Q. Wang, L. Chen, N. Chen, Y. Jiang, Z. Yang, H. Pan, H. Li, T. Zhao and Y. Li, Nat. Commun., 2025, 16, 11695 CrossRef CAS PubMed.
- Q. Lv, L. A. Li, X. Zhang, R. Wang, N. Wen, L. Xue, H. Wang, L. Shen, D. Chen, F. Ciucci and J. Wang, J. Am. Chem. Soc., 2025, 147, 27611–27623 CrossRef CAS PubMed.
- Z. Li, Q. Liu, Y. Deng, M. Zhou, W. Tang, H. Dong, W. Zhao and R. Liu, Mater. Today Energy, 2023, 31, 101198 CrossRef CAS.
- M. Wang, H. Zhang, Y. Li, R. Liu and H. Yang, J. Energy Chem., 2024, 94, 635–645 CrossRef CAS.
- Y. Du, L. Chen, C. Long, Y. Zhou, R. Xu, Z. Li, S. Yin, C. Tao, N. Li, W. Lv and L. Qiao, Energy Storage Mater., 2026, 84, 104751 CrossRef.
- J. He, Y. Deng, J. Han, T. Xu, J. Qi, J. Li, Y. Zhang, Z. Zhao, Q. Li, J. Xiao, J. Zhang, D. Kong, W. Wei, S. Wu and Q. H. Yang, Nat. Commun., 2025, 16, 4858 CrossRef CAS PubMed.
- Z. Liu, Q. Yu, Y. Zhao, R. He, M. Xu, S. Feng, S. Li, L. Zhou and L. Mai, Chem. Soc. Rev., 2019, 48, 285–309 RSC.
- H. Kim, M. Seo, M. H. Park and J. Cho, Angew. Chem., Int. Ed., 2010, 49, 2146–2149 CrossRef CAS PubMed.
- R. Jain, A. S. Lakhnot, K. Bhimani, S. Sharma, V. Mahajani, R. A. Panchal, M. Kamble, F. Han, C. Wang and N. Koratkar, Nat. Rev. Mater., 2022, 7, 736–746 CrossRef CAS.
- Y. Li, K. Yan, H.-W. Lee, Z. Lu, N. Liu and Y. Cui, Nat. Energy, 2016, 1, 15029 CrossRef CAS.
- H. Li, Z. Zhao, M. Sun, N. Kuang, Y. Liu, Y. Guo, Y. Zhang, F. Chen, Q. Zhan, A. Liu, Y. Zhai, Q. He, Y. Yue, Y. Tian, S. Wu and Q. H. Yang, Adv. Mater., 2025, 37, e2503745 CrossRef PubMed.
- H. Wu, G. Yu, L. Pan, N. Liu, M. T. McDowell, Z. Bao and Y. Cui, Nat. Commun., 2013, 4, 1943 CrossRef.
- Y. Zhai, Z. Zhong, N. Kuang, Q. Li, T. Xu, J. He, H. Li, X. Yin, Y. Jia, Q. He, S. Wu and Q. H. Yang, J. Am. Chem. Soc., 2024, 146, 15209–15218 CrossRef CAS PubMed.
- Z. Cheng, W. Chen, Y. Zhang, J. Xiang, D. Tang, H. Ji, J. Li, Y. Huang and L. Yuan, Adv. Funct. Mater., 2024, 34, 2408145 CrossRef CAS.
- M. Bai, X. Tang, M. Zhang, H. Wang, Z. Wang, A. Shao and Y. Ma, Nat. Commun., 2024, 15, 5375 CrossRef CAS PubMed.
- M. Yu, J. Ma, L. Dong, M. Huang, B. Xue, Y. Sun and H. Xiang, ACS Appl. Mater. Interfaces, 2025, 17, 14097–14108 CrossRef CAS PubMed.
- Y. Zhang, Y. Han, Y. Lei, J. Ni, W. Zhou and Q. Xiao, Batter. Supercaps, 2024, 7, e202400030 CrossRef CAS.
- H. Liu, S. Luo, Y. Yang, X. Zhao, G. Huang, X. Jin, T. Zhong, M. Guan, J. Liu and Y. Li, Energy Storage Mater., 2025, 78, 104250 CrossRef.
- K. Zaghib, M. Armand and M. Gauthier, J. Electrochem. Soc., 1998, 145, 3135–3140 CrossRef CAS.
- M. Li, H. Liu, H. Su, Z. Fan, Y. Liu, J. Yang, W. Zhu, Q. Chen and Y. Xu, Chem. Sci., 2026, 17, 5451–5462 RSC.
- X. Cao, X. Ren, L. Zou, M. H. Engelhard, W. Huang, H. Wang, B. E. Matthews, H. Lee, C. Niu, B. W. Arey, Y. Cui, C. Wang, J. Xiao, J. Liu, W. Xu and J.-G. Zhang, Nat. Energy, 2019, 4, 796–805 CrossRef CAS.
- S. Jurng, Z. L. Brown, J. Kim and B. L. Lucht, Energy Environ. Sci., 2018, 11, 2600–2608 RSC.
- Q.-K. Zhang, X.-Q. Zhang, J. Wan, N. Yao, T.-L. Song, J. Xie, L.-P. Hou, M.-Y. Zhou, X. Chen, B.-Q. Li, R. Wen, H.-J. Peng, Q. Zhang and J.-Q. Huang, Nat. Energy, 2023, 8, 725–735 CrossRef CAS.
- B. Jagger and M. Pasta, Joule, 2023, 7, 2228–2244 CrossRef CAS.
- G. M. Hobold, C. Wang, K. Steinberg, Y. Li and B. M. Gallant, Nat. Energy, 2024, 9, 580–591 CrossRef CAS.
- X. Zhou, Y. Chen, F. Zhen, Y. Wu, W. Li, X. Yin, L. Liu, S. Ding and W. Yu, ACS Nano, 2025, 19, 14073–14084 CrossRef CAS PubMed.
- S. Zhang, Z. Li, Y. Z. Zhang, X. Wang, P.-Y. Dong, S. Lei, W. Zeng, J. Wang, X. Liao, X. Chen, D. Li and S. Mu, Energy Environ. Sci., 2025, 18, 3807–3816 RSC.
- Y. Wei, H. Wang, X. Lin, T. Wang, Y. Cui, Y. Huang, J. Yang, T.-H. Liu, Y. Ren, X. Fan, H. Xu and Y. Huang, Energy Environ. Sci., 2025, 18, 786–798 RSC.
- Y. Jie, S. Wang, S. Weng, Y. Liu, M. Yang, C. Tang, X. Li, Z. Zhang, Y. Zhang, Y. Chen, F. Huang, Y. Xu, W. Li, Y. Guo, Z. He, X. Ren, Y. Lu, K. Yang, S. Cao, H. Lin, R. Cao, P. Yan, T. Cheng, X. Wang, S. Jiao and D. Xu, Nat. Energy, 2024, 9, 987–998 CrossRef CAS.
- R. Qiao, Y. Zhao, S. Zhou, H. Zhang, F. Liu, T. Zhou, B. Sun, H. Fan, C. Li, Y. Zhang, F. Liu, X. Ding, J. W. Choi, A. Coskun and J. Song, Chem, 2025, 11, 102306 CAS.
- F. Zhen, H. Liu, Y. Wu, X. Zhou, W. Lin, Y. Chen, Y. Zhou, H. Wang, X. Yin, S. Ding, X. Chen and W. Yu, Energy Environ. Sci., 2025, 18, 4690–4703 RSC.
- H. Su, H. Zhang, Z. Chen, M. Li, J. Zhao, H. Xun, J. Sun and Y. Xu, Chin. Chem. Lett., 2023, 34, 108640 CrossRef CAS.
- M. Zhou, P. Bai, X. Ji, J. Yang, C. Wang and Y. Xu, Adv. Mater., 2021, 33, e2003741 CrossRef PubMed.
- X. Zheng, L. Huang, X. Ye, J. Zhang, F. Min, W. Luo and Y. Huang, Chem, 2021, 7, 2312–2346 CAS.
- L. Sun, G. Lu, Q. Han, H. Li, O. Sheng and C. Jin, Energy Storage Mater., 2025, 82, 104650 CrossRef.
- X. Q. Zhang, X. B. Cheng, X. Chen, C. Yan and Q. Zhang, Adv. Funct. Mater., 2017, 27, 1605989 CrossRef.
- E. Markevich, G. Salitra, Y. Talyosef, U.-H. Kim, H.-H. Ryu, Y.-K. Sun and D. Aurbach, ACS Appl. Energy Mater., 2018, 1, 2600–2607 CrossRef CAS.
- Z. Li, W. Tang, Y. Deng, M. Zhou, X. Wang, R. Liu and C. Wang, J. Mater. Chem. A, 2022, 10, 23047–23057 RSC.
- K. Fridman, R. Sharabi, R. Elazari, G. Gershinsky, E. Markevich, G. Salitra, D. Aurbach, A. Garsuch and J. Lampert, Electrochem. Commun., 2013, 33, 31–34 CrossRef CAS.
- Q. Hao, J. Yan, Y. Gao, F. Chen, X. Chen, Y. Qi and N. Li, ACS Appl. Mater. Interfaces, 2024, 16, 44689–44696 CrossRef CAS PubMed.
- J. Wang, C. Tao, J. Cao, X. Jiao, L. Wang and T. Liu, Chemelectrochem, 2022, 9, e202200957 CrossRef CAS.
- D. Aurbach, E. Markevich and G. Salitra, J. Am. Chem. Soc., 2021, 143, 21161–21176 CrossRef CAS PubMed.
- H. Zhang, Z. Zeng, F. Ma, X. Wang, Y. Wu, M. Liu, R. He, S. Cheng and J. Xie, Adv. Funct. Mater., 2023, 33, 2212000 CrossRef CAS.
- M. Wang, M. Li, J. Wu, Y. Meng, J. Hao, D. Zhou, C. Han and B. Li, Adv. Mater., 2025, 37, e2502076 CrossRef PubMed.
- S. Zhang, S. Li, X. Wang, C. Li, Y. Liu, H. Cheng, S. Mao, Q. Wu, Z. Shen, J. Mao, H. Pan and Y. Lu, Nano Energy, 2023, 114, 108639 CrossRef CAS.
- Z. Wang, Y. Wang, L. Shen, Z. Jin, H. M. Law, A. Wang, W. Wang and F. Ciucci, Energy Environ. Sci., 2023, 16, 4084–4092 RSC.
- X. Zhang, G. Gao, W. Wang, J. Wang, L. Wang and T. Liu, ACS Appl. Mater. Interfaces, 2022, 14, 49811–49819 CrossRef CAS PubMed.
- Y. Wang, T. Li, X. Yang, Q. Yin, S. Wang, H. Zhang and X. Li, Adv. Energy Mater., 2024, 14, 2303189 CrossRef CAS.
- X. Liu, L. Guo, Z. Zhang, J. Wang, H. Lin, G. Li, X. Ou, D. Wang and W. Zheng, Adv. Funct. Mater., 2024, 35, 2408525 CrossRef.
- T. Li, K. Chen, B. Yang, K. Li, B. Li, M. He, L. Yang, A. Hu and J. Long, Chem. Sci., 2024, 15, 12108–12117 RSC.
- C. Dai, F. Liu, Y. Wang, X. Meng, F. Liu, Y. Deng, J. Wang, J. M. Tarascon and M. Shao, Angew. Chem., Int. Ed., 2025, 65, e20476 CrossRef PubMed.
- J. Zhao, M. Li, H. Su, Y. Liu, P. Bai, H. Liu, L. Ma, W. Li, J. Sun and Y. Xu, Small Methods, 2023, 7, e2300228 CrossRef PubMed.
- J. Zheng, C. H. Weidong Zhang, Z. Shen, X. Wang, J. Guo, S. Li, S. Mao and Y. Lu, Mater. Today Energy, 2022, 26, 100984 CrossRef CAS.
- K. Li, A. Hu, R. Xu, W. Xu, B. Yang, T. Li, Y. Li, Z. W. Seh, J. Long and S. Chen, Adv. Energy Mater., 2025, 15, 2501236 CrossRef CAS.
- H. Yang, Y. Chen, W. Tian, S. Yuan, P. Liu, Q. Wang, T. Jin and L. Jiao, Angew. Chem., Int. Ed., 2025, 64, e202506349 CrossRef CAS PubMed.
- W. Tang, T. Zhou, Y. Duan, M. Zhou, Z. Li and R. Liu, Carbon Neutralization, 2024, 3, 386–395 CrossRef CAS.
- X. Zhang, M. Jia, Q. Zhang, N. Zhang, X. Wu, S. Qi and L. Zhang, Chem. Eng. J., 2022, 448, 137743 CrossRef CAS.
- Q. Zhou, S. Dong, Z. Lv, G. Xu, L. Huang, Q. Wang, Z. Cui and G. Cui, Adv. Energy Mater., 2020, 10, 1903441 CrossRef CAS.
- J. Zhang, H. Wu, X. Du, H. Zhang, L. Huang, F. Sun, T. Liu, S. Tian, L. Zhou, S. Hu, Z. Yuan, B. Zhang, J. Zhang and G. Cui, Adv. Energy Mater., 2023, 13, 2202529 CrossRef CAS.
- H. Zhang, L. Huang, H. Xu, X. Zhang, Z. Chen, C. Gao, C. Lu, Z. Liu, M. Jiang and G. Cui, eScience, 2022, 2, 201–208 CrossRef CAS.
- L. Du, G. Xu, C. Sun, Y. H. Zhang, H. Zhang, T. Dong, L. Huang, J. Ma, F. Sun, C. Li, X. Zhuang, S. Zhang, J. Li, B. Xie, J. Wang, J. Zhao, J. Ju, Z. Hu, F. H. Chang, C. Y. Kuo, C. T. Chen, A. Hilger, I. Manke, S. Dong and G. Cui, Nat. Commun., 2025, 16, 2979 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |