
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoselective additions to alkenylphosphonium salts for the synthesis of P-stereogenic compounds
Xiao-Bing
Chen
 ,
Damián
Padín†
* and
Ben L.
Feringa
*
,
Damián
Padín†
* and
Ben L.
Feringa
*
Stratingh Institute for Chemistry, University of Groningen, Groningen, 9747AG, The Netherlands. E-mail: b.l.feringa@rug.nl
First published on 20th February 2026
Abstract
The stereoselective functionalisation of alkenyl P(V) compounds via conjugate additions represents an attractive approach to synthesise chiral organophosphorus compounds. However, asymmetric conjugate additions to alkenyl P(V) compounds are scarce and, in the presence of P-stereogenic centers, diastereoinduction is often low. Here, we report the use of BINOL-based alkenylphosphonium salts for the generation of two non-consecutive P- and C-stereogenic centers via addition of C-nucleophiles in a single operation. These alkenylphosphonium salts behave as activated alkenylphosphonamidate surrogates with increased reactivity and stereocontrol. This methodogy allows the versatile preparation of enantioenriched organophosphorus building-blocks in high yield and stereoselectivity.
Introduction
The stereoselective synthesis of P-stereogenic building blocks has become a major goal in organic synthesis due to their increasingly widespread use in ligand-design for asymmetric catalysis,1–3 medicinal chemistry and agrochemistry.4,5 Yet, routes to access chiral organophosphorus compounds are limited and less explored than C-stereogenic compounds. Recently, notable advancements in the field have been reported by using chiral auxiliaries,6–15 resolution of enantiomers, formation of diastereomeric complexes16 or catalytic asymmetric synthesis.17–20 Importantly, approaches that render multiple orthogonal functionalisation sites at P-chiral building blocks have gained particular attention, as they allow a straightforward and modular access to a myriad of chiral enantioenriched compounds from a relatively small number of precursors.8,18,21–25Chiral alkenyl P(V) compounds (e.g. alkenylphosphonates, alkenyl phosphine oxides or alkenylphosphonamidates, among others) would represent a suitable platform for preparing P-stereogenic compounds. In principle, the combination of a P-stereocenter and a mildly electrophilic olefin would allow the functionalisation at the β-carbon via conjugate addition and, potentially, the formation of a new C-stereocenter.26 Not surprisingly, this motif has attracted considerable attention in recent years and several methods have been devised by our group27 and others using asymmetric hydrophosphonylation of alkynes,28–35 Csp2-P(III)/P(V) cross-coupling reactions27,36,37 or desymmetrisation reactions.38–42
However, stereoselective functionalisations of alkenyl P(V) compounds via conjugate additions are not trivial, and current methodologies suffer from certain limitations. First, our group43 and others44–49 have developed catalytic asymmetric conjugate additions to alkenyl P(V) compounds, however, the introduction of P-stereocenters was not explored using this approach (Fig. 1A). Second, alkenyl P(V) compounds are challenging Michael acceptors that often require the presence of activating groups (Fig. 1B).50–53 Third, conjugate additions to alkenyl P(V) compounds bearing a P-stereogenic centre show, in most cases, poor diastereoselectivity (from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 5:1 d.r.), as a result of an inefficient transfer of chiral information from the P-stereocenter to the β position in the substrate (Fig. 1C).28–30,33–35,42,54,55
:1 to 5:1 d.r.), as a result of an inefficient transfer of chiral information from the P-stereocenter to the β position in the substrate (Fig. 1C).28–30,33–35,42,54,55
 | ||
| Fig. 1 State-of-the-art and challenges in stereoselective conjugate additions using (chiral) alkenyl P(V) compounds as electrophiles. | ||
In our previous report on Ni(0)-catalysed alkenylation of BINOL-based phosphoramidites towards chiral-at-P alkenylphosphonamidates, we identified alkenylphosphonium salts as key intermediates.27 These intermediates proved to be reasonably stable under ambient conditions and, upon hydrolysis under acidic conditions, provided versatile P-stereogenic alkenylphosphonamidates with high optical purity. While we were unsuccessful to selectively functionalise the β position of the final alkenylphosphonamidates, preliminary studies suggested that the alkenylphosphonium intermediates could actually be more reactive and selective towards formal conjugate addition processes. In the literature, simple alkenylphosphonium salts, such as triphenylvinylphosphonium bromide, have been widely explored as electrophiles with a variety of nucleophiles.56–60 However, to the best of our knowledge, no stereoselective additions have been reported, so far. Moreover, none of the reported methodologies allow the access to P-stereogenic compounds in a single operation. Addressing this challenge, we report here the use of readily available BINOL-based alkenylphosphonium salts as activated alkenyl P(V) precursors in stereoselective additions with a wide range of C-nucleophiles (Fig. 1D). Remarkably, the final phosphonamidates obtained by this methodology feature two non-consecutive stereocenters, i.e., a P-stereocenter and a C-stereocenter, controlled by a highly efficient axial-to-point chirality transfer promoted by the BINOL moiety. In addition, these chiral organophosphorus compounds present several functionalisation sites suitable for further derivatization downstream.
Results and discussion
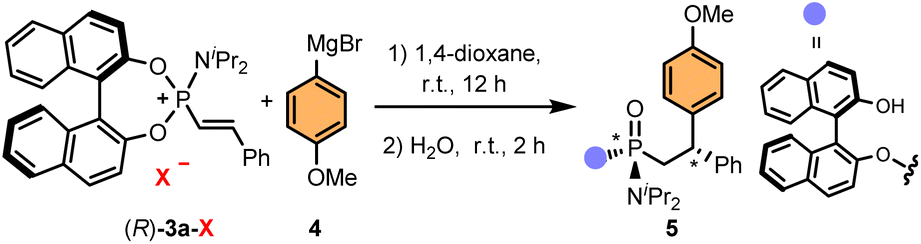
Chiral alkenylphosphonium salts, such as (R)-3a, could be easily accessed in multi-gram scale and nearly quantitative yields from readily available BINOL-based phosphoramidites and β-bromostyrenes using a variation of our previously reported Ni(0)-catalysed C–P coupling reaction (Fig. 2).27 These phosphonium salts proved to be reasonably bench-stable, although partial hydrolysis in the presence of moisture or during purification by column chromatography was occasionally observed. Interestingly, although the crude reaction mixture after evaporation of the solvent provided a reasonably pure phosphonium salt, Ni(II) contamination, which rendered the crude product a characteristic bright green colour, proved to be detrimental in subsequent manipulations of the product. Nonetheless, a simple recrystallization of the crude enabled the removal of any Ni(II) impurities and provided the phosphonium salts as off-white solids. | ||
| Fig. 2 Practical preparation of chiral alkenylphosphonium salt (R)-3a through a Ni(0)-catalysed C–P coupling reaction. | ||
For the development of an asymmetric addition to alkenylphosphonium salts, we chose phosphonium salt (R)-3a and a commercially available Grignard reagent, 4-methoxyphenylmagnesium bromide, as model substrates (Table 1). Encouraged by our previous experience on catalytic asymmetric 1,4-additions using organometallic reagents,61–67 we envisioned that the addition of CuI/CuII salts should promote the addition reaction. Indeed, preliminary experiments using different copper salts as catalysts in THF at room temperature provided nearly quantitative conversions of (R)-3a, which, upon hydrolysis under acidic conditions, led to adduct 5 as a mixture of diastereoisomers (Table 1, entry 1). Importantly, while the addition of the Grignard reagent generates the C-stereocenter (referred to condition 1 in Table 1), the hydrolysis step generates the P-stereocenter (condition 2). Given the presence of the enantiopure BINOL auxiliary, four diastereoisomers can be observed in the 31P-NMR of the crude mixture (see SI for NMR monitoring of the reaction). Interestingly, this two-step, one-pot procedure using copper salts as catalysts led us to observe the predominant formation of one of the possible diastereoisomers, albeit in moderate stereoselectivity. Subsequent control experiments allowed us to discover that the use of a catalyst was, in fact, not necessary (entry 2). In an attempt to improve the diastereoselectivity, the reaction was conducted at low temperatures (entries 3 and 4), however, poor conversions and moderate stereoselectivities were observed. After extensive optimisation studies (see SI for a detailed optimisation table), we established that the pH of the hydrolysis step had a critical role in controlling the stereoselection. While in our previous Pd-catalysed arylation of phosphoramidites12 the addition of Cs2CO3 proved crucial for obtaining high yields and stereoselectivities, and in the Ni-catalysed alkenylation,27 addition of aqueous HCl led to the highest diastereoselectivities, in the current study, addition of neutral water produced better results than Cs2CO3 or HCl (compare entries 2, 5 and 6). Curiously, a remarkable diastereodivergency was observed when performing the hydrolysis step in water or in the presence of Cs2CO3 (entry 2 vs. entries 5 and 6). Finally, further screening of the reaction conditions allowed us to identify 1,4-dioxane as the optimal solvent for both, the Grignard addition and the hydrolysis steps (entry 7). Under these conditions, adduct (R,Rp,S)-5 was obtained in high yield (87%) and excellent diastereoselectivity (88:0:4:8 d.r.). Notably, the diastereomeric purity of the final product could be often improved after purification of the crude compound by simple column chromatography. Thus, in practice, only two, out of four possible stereoisomers, were isolated after purification (vide infra).
|
|
|||
|---|---|---|---|
| Entry | Conditions | Conversionb (%) | d.r.b |
| a Reactions performed with (R)-3a (0.1 mmol), 4-methoxyphenylmagnesium bromide (1.5 equiv.), under the indicated conditions. For the full optimisation table, see the SI. b Determined by 31P-NMR and 1H-NMR (CDCl3) of the crude reaction mixture. c Isolated yield in brackets. | |||
| 1 | (1) CuI/CuII salts, THF, r.t. | 70–100 | 15:6:14:65–14:1:10:75 |
| (2) HClaq | |||
| 2 | (1) THF, r.t. | 84 | 11:2:10:77 |
| (2) HClaq | |||
| 3 | (1) THF,-78 °C | 35 | 21:3:10:66 |
| (2) HClaq | |||
| 4 | (1) THF, 0 °C | 84 | 11:2:10:76 |
| (2) HClaq | |||
| 5 | (1) THF, r.t. | 92 | 81:11:1:7 |
| (2) H2O | |||
| 6 | (1) THF, r.t. | 90 | 63:7:3:27 |
| (2) Cs2CO3 (2 equiv.) | |||
| 7 | (1) 1,4-Dioxane, r.t. | 98 (87)c |
88![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :0:4:8 :0:4:8 |
| (2) H 2 O | |||

With the optimised conditions in hand, we evaluated the generality of the methodology and its limitations. First, we analysed the effect of the phosphonium salt counterion on the yield and stereoselectivity (Table 2). Curiously, while phosphonium bromide (R)-3a-Br and iodide (R)-3a-I provided the desired phosphonamidate 5 in good yield and stereoselectivity, triflate (R)-3a-OTf led to a diminished yield, albeit improved stereoselectivity, and chloride (R)-3a-Cl only produced trace amounts of (R,Rp,S)-5.
|
|
||
|---|---|---|
| X | Yieldb | d.r.c |
| a Conditions: (R)-3a-X (0.1 mmol), 4-methoxyphenylmagnesium bromide (1.5 equiv.) in 1,4-dioxane at room temperature for 12 h, then H2O was added and stirred for 2 h. b Isolated yield. c Diastereomeric ratio (d.r.) determined by 31P-NMR and 1H-NMR (CDCl3) of the crude reaction mixture after hydrolysis with H2O. | ||
| Cl | Trace | — |
| Br | 87 | 88:0:4:8 |
| I | 90 | 85:8:1:6 |
| OTf | 40 | 94:0:0:6 |
We next examined the use of other commercially available organometallic reagents as nucleophiles (Scheme 1). The reaction tolerated well the use of a wide range of organomagnesium, organolithium, organocuprate and organozinc C-nucleophiles, which allowed the installation of alkyl (6–10, 16), vinyl (11) and aryl groups (5, 12–15) in good overall yields with moderate to excellent diastereoselectivities (up to >20:1 d.r.). Notably, arene-based nucleophiles proved to be excellent coupling partners for the synthesis of functionalised chiral diarylmethanes (5, 12–15). Curiously, while these Csp3- and Csp2-based nucleophiles cleanly reacted with alkenylphosphonium (R)-3a, 1-propynylmagnesium bromide, as a prototypic Csp-based nucleophile, failed to provide any identifiable product. A similar fate was observed with (1,3-dioxolan-2-ylmethyl)magnesium bromide.
 | ||
| Scheme 1 Scope of organometallic reagents. General conditions: (R)-3a (0.1 mmol), organometallic reagent (2.0 equiv.) in 1,4-dioxane (0.1 M) at room temperature for 18 h, then, H2O (1 mL) was added and stirred for 6 h. Diastereomeric ratio determined by 31P NMR (CDCl3). For the sake of clarity, the functionalities highlighted in orange indicate the moiety installed with the organometallic reagent. | ||
The scope of electrophiles was subsequently explored using different alkenylphosphonium salts and Grignard reagents, which are readily available and often provide better diastereoselectivities and yields (Scheme 2).68 The addition reaction took place with a wide variety of alkenylphosphonium salts bearing different substitution patterns, including electron-donating groups (13, 17–21, 24, 25) as well as electron-poor substrates (22) at different positions. The reaction also tolerated well the introduction of one (26–29) or even two heteroaromatic rings (30). Remarkably, the resulting functionalised phosphonamidates were obtained in high overall yields (43–92%) and excellent stereoselectivities (>20:1 d.r. in most cases).
 | ||
| Scheme 2 Scope of alkenylphosphonium salts. General conditions: (R)-3 (0.1 mmol), organometallic reagent (2.0 equiv.) in 1,4 dioxane (0.1 mL) at room temperature for 18 h, then, H2O (1 mL) was added and stirred for 6 h. Diastereomeric ratio determined by 31P NMR (CDCl3). For the sake of clarity, the functionalities highlighted in orange indicate the moiety installed with the organometallic reagent. a Compounds 13 and 13′ are epimers. (see SI for more details). b The (S,Sp,R)-5′ was prepared following the same procedure starting from (S)-1b (see SI for more details). | ||
The simplicity of this formal conjugate addition/hydrolysis approach to generate non-consecutive P- and C-stereocenters, together with the presence of multiple functionalisation sites in the final phosphonamidates led us to explore the scalability of the reaction and develop some synthetic applications. Gratifyingly, the reaction could be easily scaled up to without modifying the standard reaction conditions and compound (R,Rp,S)-5 could be isolated in gram scale (1.77 g) and in excellent yield (82%) and diastereomeric purity (95:5 d.r., Fig. 3a). First, we demonstrated that the amine moiety of phosphonamidate (R,Rp,S)-5 could be replaced by a methoxy group under acidic conditions,27,69 furnishing chiral phosphonate 31 in an unoptimized 28% yield and moderate diastereospecificity (88:12 d.r., Fig. 3b). Interestingly, upon protection of the BINOL moiety as silylether, the chiral auxiliary could be chemoselectively replaced with different arene motifs by addition of an aryl Grignard reagent. The stereospecificity in these nucleophilic substitutions was substrate-dependent, obtaining compound 33 with significant erosion of the diastereomeric purity and 34 in nearly perfect diastereospecificity (Fig. 3c).
 | ||
| Fig. 3 Gram-scale synthesis of 5 and synthetic manipulations of phosphonamidates. | ||
The unusually high level of remote stereocontrol exerted by the BINOL auxiliary on the β-carbon led us to study in more detail the structure of the alkenylphosphonium salt. DFT-based modelling of alkenylphosphonium salt (R)-3a-Br (see SI for details) revealed a key synergistic role between the amine and the BINOL substituents at the phosphorus centre (Fig. 4a). The axially chiral BINOL group appears to induce a rotation of the P–N (iPr)2 bond to minimize the steric repulsion between these two groups. As a consequence, the bulky iso-propyl groups of the amine fragment block one of the diastereotopic faces of the olefin, ultimately leaving only the pro-S face available for nucleophilic attack by the organometallic reagent (Fig. 4b).
 | ||
| Fig. 4 Stereochemical rationale. | ||
Conclusions
In summary, stereoselective additions to chiral alkenyl-PV compounds have usually proved challenging to accomplish due to their low reactivity of and poor diastereoselectivity. Here, we have demonstrated that the combination of an alkenylphosphonium salt, acting as a more electrophilic alkenylphosphonamidate surrogate, and a BINOL moiety as chiral auxiliary, enabled the smooth and highly stereoselective nucleophilic addition of a wide range of C-nucleophiles to the β-carbon. This allowed us to access to functionalised P-stereogenic building-blocks featuring two non-consecutive stereocenters.Author contributions
Conceptualization: X.-B. C., D. P. and B. L. investigation and methodology: X.-B. C. and D. P. writing – original draft, review and editing: X.-B. C., D. P. and B. L. supervision: D. P. and B. L. funding acquisition: B. L.Conflicts of interest
There are no conflicts to declare.Data availability
CCDC 2526188 contains the supplementary crystallographic data for this paper.70The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental procedures, characterisation of new compounds, computational details, crystallographic data, and extended discussion of the scope of the reaction. See DOI: https://doi.org/10.1039/d5sc09946c.
Acknowledgements
This work was supported by the Netherlands Organization for Scientific Research (B. L. F.), the Royal Netherlands Academy of Arts and Sciences (B. L. F.), the Dutch Ministry of Education, Culture, and Science (Gravitation Program 024.001.035 to B. L. F.), the European Research Council (Advanced Investigator Grant 694345 to B. L. F.). The COFUND project oLife has received funding from the European Union's Horizon 2020 research and innovation programme under Grant Agreement No. 847675 (oLife postdoctoral fellowship). We gratefully acknowledge the support from the China Scholarship Council (CSC PhD Fellowship No. 202108330058 to X. C.). The authors are also grateful to J. L. Sneep for collecting high-resolution mass spectrometry data for all newly reported compounds. We sincerely thank Lotte Stindt for testing the single crystals of our compounds and for her valuable academic discussions.Notes and references
- W. S. Knowles, Acc. Chem. Res., 1983, 16, 106–112 CrossRef CAS.
- G. Xu, C. H. Senanayake and W. Tang, Acc. Chem. Res., 2019, 52, 1101–1112 CrossRef CAS PubMed.
- T. Imamoto, Chem. Rev., 2024, 124, 8657–8739 CrossRef CAS PubMed.
- U. Pradere, E. C. Garnier-Amblard, S. J. Coats, F. Amblard and R. F. Schinazi, Chem. Rev., 2014, 114, 9154–9218 CrossRef CAS PubMed.
- J. B. Rodriguez and C. Gallo-Rodriguez, ChemMedChem, 2019, 14, 190–216 CrossRef CAS PubMed.
- Z. S. Han, N. Goyal, M. A. Herbage, J. D. Sieber, B. Qu, Y. Xu, Z. Li, J. T. Reeves, J.-N. Desrosiers, S. Ma, N. Grinberg, H. Lee, H. P. R. Mangunuru, Y. Zhang, D. Krishnamurthy, B. Z. Lu, J. J. Song, G. Wang and C. H. Senanayake, J. Am. Chem. Soc., 2013, 135, 2474–2477 CrossRef CAS PubMed.
- D. Xu, N. Rivas-Bascón, N. M. Padial, K. W. Knouse, B. Zheng, J. C. Vantourout, M. A. Schmidt, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2020, 142, 5785–5792 CrossRef CAS PubMed.
- Y. Zhang, P. Zhao, S. Sun, Q. Wu, E. Shi and J. Xiao, Commun. Chem., 2023, 6, 133 CrossRef CAS PubMed.
- T. Murai, Chem. Lett., 2023, 52, 703–714 CrossRef CAS.
- K. W. Knouse, J. N. deGruyter, M. A. Schmidt, B. Zheng, J. C. Vantourout, C. Kingston, S. E. Mercer, I. M. McDonald, R. E. Olson, Y. Zhu, C. Hang, J. Zhu, C. Yuan, Q. Wang, P. Park, M. D. Eastgate and P. S. Baran, Science, 2018, 361, 1234–1238 CrossRef CAS PubMed.
- C. Endo, Y. Inoue, T. Maruyama, M. Minoura and T. Murai, Synthesis, 2022, 55, 934–944 Search PubMed.
- A. Mondal, N. O. Thiel, R. Dorel and B. L. Feringa, Nat. Catal., 2022, 5, 10–19 CrossRef CAS.
- K. Kuwabara, Y. Maekawa, M. Minoura, T. Maruyama and T. Murai, J. Org. Chem., 2020, 85, 14446–14455 CrossRef CAS PubMed.
- K. V. Rajendran, K. V. Nikitin and D. G. Gilheany, J. Am. Chem. Soc., 2015, 137, 9375–9381 CrossRef CAS PubMed.
- E. Bergin, C. T. O'Connor, S. B. Robinson, E. M. McGarrigle, C. P. O'Mahony and D. G. Gilheany, J. Am. Chem. Soc., 2007, 129, 9566–9567 CrossRef CAS PubMed.
- F. A. Kortmann, M.-C. Chang, E. Otten, E. P. A. Couzijn, M. Lutz and A. J. Minnaard, Chem. Sci., 2014, 5, 1322–1327 RSC.
- M. Formica, T. Rogova, H. Shi, N. Sahara, B. Ferko, A. J. M. Farley, K. E. Christensen, F. Duarte, K. Yamazaki and D. J. Dixon, Nat. Chem., 2023, 15, 714–721 CrossRef CAS PubMed.
- M. Formica, B. Ferko, T. Marsh, T. A. Davidson, K. Yamazaki and D. J. Dixon, Angew. Chem., Int. Ed., 2024, 63, e202400673 Search PubMed.
- G. J. Lovinger, M. H. Sak and E. N. Jacobsen, Nature, 2024, 632, 1052–1059 Search PubMed.
- K. C. Forbes and E. N. Jacobsen, Science, 2022, 376, 1230–1236 Search PubMed.
- K. W. Knouse, D. T. Flood, J. C. Vantourout, M. A. Schmidt, I. M. McDonald, M. D. Eastgate, P. S. Baran and A. C. S. Cent, Sci, 2021, 7, 1473–1485 CAS.
- G.-L. Zheng, Y. Zhang, J.-M. Zhang, L. Chen, X.-S. Xue and Z.-T. He, J. Am. Chem. Soc., 2025, 147, 13566–13576 CrossRef CAS PubMed.
- K. Ding, J.-J. Xue, J.-X. Zou, Q.-W. Zhang and B. Su, Angew. Chem., Int. Ed., 2026, 65, e22534 CrossRef CAS PubMed.
- L. Zhang, M. Wang, Y. Luo, Z. Zhang, D. Liu and W. Zhang, J. Am. Chem. Soc., 2025, 147, 47712–47721 CrossRef CAS PubMed.
- B. S. Daniels, B. G. Blackburn, S. J. Scribner and V. M. Dong, J. Am. Chem. Soc., 2025, 147, 21339–21346 CrossRef CAS PubMed.
- S. R. Harutyunyan, T. den Hartog, K. Geurts, A. J. Minnaard and B. L. Feringa, Chem. Rev., 2008, 108, 2824–2852 CrossRef CAS PubMed.
- X.-B. Chen, D. Padín, C. N. Stindt and B. L. Feringa, Angew. Chem., Int. Ed., 2023, 62, e202307450 Search PubMed.
- J. Kang, K. Ding, S.-M. Ren, W.-J. Yang and B. Su, Angew. Chem., Int. Ed., 2025, 64, e202415314 Search PubMed.
- W.-H. Wang, Y. Wu, P.-J. Qi and Q.-W. Zhang, ACS Catal., 2023, 13, 6994–7001 CrossRef CAS.
- Y.-Q. Zhang, X.-Y. Han, Y. Wu, P.-J. Qi, Q. Zhang and Q.-W. Zhang, Chem. Sci., 2022, 13, 4095–4102 RSC.
- X. Xie, S. Li, Q. Chen, H. Guo, J. Yang and J. Zhang, Org. Chem. Front., 2022, 9, 1589–1592 RSC.
- D. Ji, J. Jing, Y. Wang, Z. Qi, F. Wang, X. Zhang, Y. Wang and X. Li, Chem, 2022, 8, 3346–3362 CAS.
- X.-T. Liu, Y. Wu and Q.-W. Zhang, Synlett, 2021, 33, 301–306 Search PubMed.
- X.-T. Liu, X.-Y. Han, Y. Wu, Y.-Y. Sun, L. Gao, Z. Huang and Q.-W. Zhang, J. Am. Chem. Soc., 2021, 143, 11309–11316 Search PubMed.
- Q. Dai, L. Liu, Y. Qian, W. Li and J. Zhang, Angew. Chem., Int. Ed., 2020, 59, 20645–20650 CrossRef CAS PubMed.
- C. Wang, Q. Yang, Y.-H. Dai, J. Xiong, Y. Zheng and W.-L. Duan, Angew. Chem., Int. Ed., 2023, 62, e202313112 CrossRef CAS PubMed.
- A. Modi, C. Gosmini and A. Auffrant, Chem.–Asian J., 2025, 20, e202401780 CrossRef CAS PubMed.
- For a recent review see: K. Ding and B. Su, Eur. J. Org Chem., 2024, 27, e202301160 CrossRef CAS.
- J. S. Harvey, S. J. Malcolmson, K. S. Dunne, S. J. Meek, A. L. Thompson, R. R. Schrock, A. H. Hoveyda and V. Gouverneur, Angew. Chem., Int. Ed., 2009, 48, 762–766 Search PubMed.
- H. Fernández-Pérez and A. Vidal-Ferran, Org. Lett., 2019, 21, 7019–7023 Search PubMed.
- Y. Zhang, F. Zhang, L. Chen, J. Xu, X. Liu and X. Feng, ACS Catal., 2019, 9, 4834–4840 Search PubMed.
- Z. Wang and T. Hayashi, Angew. Chem., Int. Ed., 2018, 57, 1702–1706 Search PubMed.
- V. Hornillos, C. Vila, E. Otten and B. L. Feringa, Angew. Chem., Int. Ed., 2015, 54, 7867–7871 CrossRef CAS PubMed.
- S. C. Cullen and T. Rovis, Org. Lett., 2008, 10, 3141–3144 CrossRef CAS PubMed.
- J. Kim, H. Lee and J. Yun, Tetrahedron, 2019, 75, 4250–4254 Search PubMed.
- L. Ge and S. R. Harutyunyan, Chem. Sci., 2022, 13, 1307–1312 RSC.
- W.-J. Yue, J.-Z. Xiao, S. Zhang and L. Yin, Angew. Chem., Int. Ed., 2020, 59, 7057–7062 CrossRef CAS PubMed.
- D. Hong, W. Mao, E. Irran and M. Oestreich, Synthesis, 2022, 54, 2049–2056 CrossRef CAS.
- H. Li, L. Cheng, G. Li, T. Xu, S. Zhang and F. Zeng, Org. Lett., 2023, 25, 488–493 CrossRef CAS PubMed.
- P. E. McDermott, M. P. Ó. Fearraigh, A. M. Horan and E. M. McGarrigle, Org. Biomol. Chem., 2023, 21, 1027–1032 RSC.
- S. Sulzer-Mossé, A. Alexakis, J. Mareda, G. Bollot, G. Bernardinelli and Y. Filinchuk, Chem.–Eur. J., 2009, 15, 3204–3220 CrossRef PubMed.
- T. Janecki, J. Kędzia and T. Wąsek, Synthesis, 2009, 1227–1254 Search PubMed.
- F. Vaghi, P. Soppelsa, K. Wurst, G. Licini and M. Orlandi, Org. Lett., 2025, 27, 6654–6658 CrossRef CAS PubMed.
- W.-H. Wang, Y. Wu, H.-T. Wang, P.-J. Qi, W.-N. Lan and Q.-W. Zhang, Nat. Synth., 2022, 1, 738–747 CrossRef CAS.
- S.-M. Ren, W.-J. Yang, K. Ding, J. Kang, J.-X. Zou and B. Su, Org. Lett., 2025, 27, 6082–6087 CrossRef PubMed.
- Y. Shen and J. Yao, J. Org. Chem., 1996, 61, 8659–8661 CrossRef CAS.
- D. Filippini and M. Silvi, Nat. Chem., 2022, 14, 66–70 CrossRef CAS PubMed.
- A. Kuźnik, R. Mazurkiewicz and B. Fryczkowska, Beilstein J. Org. Chem., 2017, 13, 2710–2738 CrossRef PubMed.
- M. Yoshida, M. Sawamura and Y. Masuda, ChemCatChem, 2022, 14, e202200744 CrossRef CAS.
- D. Ikeshita, Y. Masuda, N. Ishida and M. Murakami, Org. Lett., 2022, 24, 2504–2508 CrossRef CAS PubMed.
- B. L. Feringa, Acc. Chem. Res., 2000, 33, 346–353 CrossRef CAS PubMed.
- F. López, S. R. Harutyunyan, A. J. Minnaard and B. L. Feringa, J. Am. Chem. Soc., 2004, 126, 12784–12785 CrossRef PubMed.
- C. Vila, V. Hornillos, M. Fañanás-Mastral and B. L. Feringa, Chem. Commun., 2013, 49, 5933–5935 Search PubMed.
- A. K. Schoonen, M. Á. Fernández-Ibáñez, M. Fañanás-Mastral, J. F. Teichert and B. L. Feringa, Org. Biomol. Chem., 2014, 12, 36–41 RSC.
- T. den Hartog, Y. Huang, M. Fañanás-Mastral, A. Meuwese, A. Rudolph, M. Pérez, A. J. Minnaard and B. L. Feringa, ACS Catal., 2015, 5, 560–574 CrossRef.
- K. P. McGrath, A. K. Hubbell, Y. Zhou, D. P. Santos, S. Torker, F. Romiti and A. H. Hoveyda, Adv. Synth. Catal., 2020, 362, 370–375 Search PubMed.
- S. F. Pizzolato, P. Štacko, J. C. M. Kistemaker, T. van Leeuwen and B. L. Feringa, Nat. Catal., 2020, 3, 488–496 CrossRef CAS.
- For an Expanded Study of the Scope of Electrophilic Partners See Section 10 in SI..
- Z.-K. Xie, Z.-Y. Pan, Y.-H. Cui, H.-J. Jiang, M.-L. Shen, C.-Z. Yao, Q. Li and J. Yu, ACS Catal., 2025, 15, 15894–15901 CrossRef CAS.
- CCDC 2526188: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qspy5.
Footnote |
| † Present address: Department of Chemistry, University of Cambridge, Cambridge CB2 1EW, UK, E-mail: E-mail: dp721@cam.ac.uk |
| This journal is © The Royal Society of Chemistry 2026 |