
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric synthesis of planar-chiral metacyclophanes via aromatic amination enabled enantioselective desymmetrization
Changyu
Zhou
a,
Jinmiao
Zhou
a,
Lele
Zhang
a,
Wansen
Xie
a,
Huanchao
Gu
a,
Hua
Liu
a and
Xiaoyu
Yang
 *ab
*ab
aSchool of Physical Science and Technology, ShanghaiTech University, Shanghai 201210, China. E-mail: yangxy1@shanghaitech.edu.cn
bState Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, China
First published on 12th February 2026
Abstract
Metacyclophanes are a type of macrocyclic cyclophanes with the ansa chain linked to the aryl rings at the two meta positions, which have been widely found in natural products and bioactive small molecules. However, due to the stringent requirements for maintaining planar chirality and a crowded environment for macrocyclization, the catalytic enantioselective synthesis of planar-chiral metacyclophanes has received significantly less attention compared to their paracyclophane counterparts. Herein, we present an efficient method for enantioselective synthesis of planar-chiral metacyclophanes through an organocatalyzed desymmetrization strategy. By utilizing the chiral phosphoric acid (CPA)-catalyzed asymmetric aromatic amination reaction between arylamines and azodicarboxylates, we successfully broke the mirror symmetry of prochiral m-phenylenediamine-derived metacyclophanes, which yielded various planar-chiral metacyclophanes with good to high enantioselectivities. Notably, when the prochiral metacyclophane substrates feature additional meta-substituents, chiral cyclophanes with both planar chirality and C–N axial chirality could be generated in a single step. The planar-chiral metacyclophane products have demonstrated good configurational stability and the potential for diverse derivatizations, which underscored the value of this method.
Introduction
Macrocycles, including cyclic peptides and oligonucleotides, have garnered substantial research interest in drug discovery due to their functional diversity and stereochemical complexity within a relatively rigid, preorganized structure.1 These characteristics enable them to bind with higher affinity and selectivity to targets that are challenging to engage using their acyclic counterparts. Macrocyclic cyclophanes are a class of macrocycles that feature one or more aromatic rings integrated into their cyclic framework. These macrocyclic cyclophanes can be further categorized into three classes based on the position of the connection between the ansa chain and the aryl rings: paracyclophanes, metacyclophanes, and orthocyclophanes (Fig. 1a).2 Planar chirality is one of the most intriguing characteristics of cyclophanes, arising from the restricted flipping of the aryl ring by the ansa chain and arene substitutions, which is typically found in paracyclophanes and metacyclophanes. However, due to their distinct connecting geometry, the asymmetric synthesis of planar-chiral paracyclophanes has received significantly more attention than planar-chiral metacyclophanes.3 Specifically, the smaller angle (120°) between the exit vectors on the arene of metacyclophanes, compared to the larger angle in paracyclophanes (180°), resulted in increased tendency for ring flipping, thereby necessitating a steric group at the C-2 position to ensure configurational stability. On the other hand, the introduction of a sterically hindered group at the C2-position presents challenges for efficient macrocyclization in metacyclophanes (Fig. 1b). Consequently, although metacyclophane structures have been found in numerous natural products and pharmaceutics (Fig. 1c),4 planar-chiral metacyclophane moieties remain relatively scarce, likely due to the challenges associated with their formation. | ||
| Fig. 1 (a) Three types of cyclophanes. (b) Planar-chiral metacyclophanes. (c) Natural products and pharmaceuticals featuring achiral metacyclophane structures. (d) Asymmetric synthesis of planar-chiral metacyclophanes by Tanaka and co-workers.8 (e) Asymmetric synthesis of planar-chiral metacyclophanes by Li and co-workers.9 (f) Asymmetric synthesis of planar-chiral metacyclophanes by Wang et al.10 and Zhou et al.11 (g) Asymmetric synthesis of planar-chiral metacyclophanes by Li and co-workers.12 (h) This work: asymmetric synthesis of planar-chiral metacyclophanes via aromatic amination-enabled desymmetrization. | ||
Compared to the extensive studies on the catalytic asymmetric synthesis of planar-chiral paracyclophanes utilizing strategies such as asymmetric macrocyclization,5 (dynamic) kinetic resolution6 and others,7 only a limited number of methods have been reported for the asymmetric synthesis of planar-chiral metacyclophanes. In 2007, Tanaka and co-workers reported the first catalytic enantioselective synthesis of planar-chiral metacyclophanes through construction of the arene moiety via Rh-catalyzed intramolecular [2 + 2 + 2] cycloadditions of triynes, albeit with modest yields (Fig. 1d).8 It was not until 2023 that Li and co-workers presented another example of enantioselective synthesis of planar-chiral metacyclophanes featuring trisubstituted pyridines, achieved through macrocyclization via Pd-catalyzed C–O cross-coupling9 (Fig. 1e). Recently, both the Wang group10 and the Zhou group11 disclosed the asymmetric synthesis of planar-chiral indole-derived cyclophanes via organocatalyzed macrocyclizations, in which the ansa chain was connected in a meta-disubstituted manner (Fig. 1f). Interestingly, despite these elegant asymmetric macrocyclization methods, to the best of our knowledge, the application of this strategy to the asymmetric synthesis of planar-chiral metacyclophanes incorporating a substituted phenyl ring remains unexplored. Notably, during the preparation of this manuscript, Li and co-workers reported an alternative tactic for the asymmetric synthesis of planar-chiral metacyclophanes, in which they desymmetrized the prochiral metacyclophanes' arene moiety through a sequential asymmetric Povarov reaction and aromatization12 (Fig. 1g).
With our continuous interest in the asymmetric synthesis of planar-chiral molecules,5d,6b,13 we herein disclose the enantioselective synthesis of planar-chiral metacyclophanes via an organocatalyzed desymmetrization method.14 By utilizing the chiral phosphoric acid15 (CPA)-catalyzed asymmetric aromatic amination reaction with azodicarboxylates,16 we efficiently broke the mirror symmetry of prochiral m-phenylenediamine-derived metacyclophanes, yielding various planar-chiral metacyclophanes with good to high enantioselectivities (Fig. 1h). Notably, with metacyclophane substrates featuring additional meta-substitutions, chiral cyclophanes with both planar chirality and C–N axial chirality17 could be generated with high enantio- and diastereoselectivity.
Results and discussion
We began our study by selecting the prochiral 15-membered metacyclophane 1a as the model substrate (Table 1). Promisingly, the asymmetric aromatic amination of 1a with dibenzyl azodicarboxylate (2a) in toluene, catalyzed by CPA A1 (10 mol%), yielded the desymmetrized planar-chiral metacyclophane 3a in 89% yield with 86![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :14 enantiomeric ratio (er) at 20 °C (entry 1). Subsequently, a range of BINOL- and H8-BINOL-derived CPA catalysts were screened for this reaction (entries 2–11), which indicated that the sterically hindered CPA catalysts exhibited notably poor enantioselectivity control (entry 3 and entry 5). Moreover, with the same substitutions, the BINOL-derived CPA displayed better enantioselectivity compared to the corresponding H8-BINOL-derived catalyst (entries 8 and 9). Encouragingly, the perfluorobiphenyl-substituted BINOL-derived CPA A11 provided the highest yield and enantioselectivity, resulting in the formation of 3a in 99% yield with 89:11 er (entry 11). Utilizing the optimal CPA catalyst A11, we investigated a series of solvents, which suggested that more polar solvents resulted in decreased enantioselectivities (entries 12–15). In contrast, the nonpolar CCl4 emerged as the optimal choice, delivering product 3a with 92:8 er (entry 16). Finally, reducing the reaction temperature was explored to enhance the enantioselectivity (entries 17 and 18). We were pleased to find that conducting the reaction at −20 °C resulted in the formation of chiral metacyclophane 3a in 91% yield with 97:3 er, although a longer reaction time (48 h) was required (entry 18).
:14 enantiomeric ratio (er) at 20 °C (entry 1). Subsequently, a range of BINOL- and H8-BINOL-derived CPA catalysts were screened for this reaction (entries 2–11), which indicated that the sterically hindered CPA catalysts exhibited notably poor enantioselectivity control (entry 3 and entry 5). Moreover, with the same substitutions, the BINOL-derived CPA displayed better enantioselectivity compared to the corresponding H8-BINOL-derived catalyst (entries 8 and 9). Encouragingly, the perfluorobiphenyl-substituted BINOL-derived CPA A11 provided the highest yield and enantioselectivity, resulting in the formation of 3a in 99% yield with 89:11 er (entry 11). Utilizing the optimal CPA catalyst A11, we investigated a series of solvents, which suggested that more polar solvents resulted in decreased enantioselectivities (entries 12–15). In contrast, the nonpolar CCl4 emerged as the optimal choice, delivering product 3a with 92:8 er (entry 16). Finally, reducing the reaction temperature was explored to enhance the enantioselectivity (entries 17 and 18). We were pleased to find that conducting the reaction at −20 °C resulted in the formation of chiral metacyclophane 3a in 91% yield with 97:3 er, although a longer reaction time (48 h) was required (entry 18).
|
|
|||||
|---|---|---|---|---|---|
| Entry | CPA | Sol. | Temp (oC) | Yieldb (%) | erc |
| a Reactions were performed with 1a (0.05 mmol), 2a (0.05 mmol), and the CPA catalyst (0.005 mmol, 10 mol%) in solvent (1.0 mL) at 20 °C for 12 h. b Isolated yield. c The er values were determined by HPLC analysis on a chiral stationary phase. d The reaction time was 48 h. e 3 Å MS (35 mg) was added. | |||||
| 1 | A1 | Tol. | 20 | 89 | 86:14 |
| 2 | A2 | Tol. | 20 | 82 | 83.5:16.5 |
| 3 | A3 | Tol. | 20 | 68 | 64.5:35.5 |
| 4 | A4 | Tol. | 20 | 81 | 80:20 |
| 5 | A5 | Tol. | 20 | 75 | 52.5:47.5 |
| 6 | A6 | Tol. | 20 | 95 | 85:15 |
| 7 | A7 | Tol. | 20 | 94 | 82:18 |
| 8 | A8 | Tol. | 20 | 86 | 84.5:15.5 |
| 9 | A9 | Tol. | 20 | 82 | 74.5:25.5 |
| 10 | A10 | Tol. | 20 | 89 | 87.5:12.5 |
| 11 | A11 | Tol. | 20 | 99 | 89:11 |
| 12 | A11 | DCM | 20 | 90 | 55:45 |
| 13 | A11 | CHCl3 | 20 | 69 | 68.5:31.5 |
| 14 | A11 | Et2O | 20 | 91 | 81.5:18.5 |
| 15 | A11 | EtOAc | 20 | 81 | 62.5:37.5 |
| 16 | A11 | CCl4 | 20 | 91 | 92:8 |
| , 17 | A11 | CCl4 | 0 | 92 | 96:4 |
| , 18 | A11 | CCl4 | −20 | 91 | 97:3 |
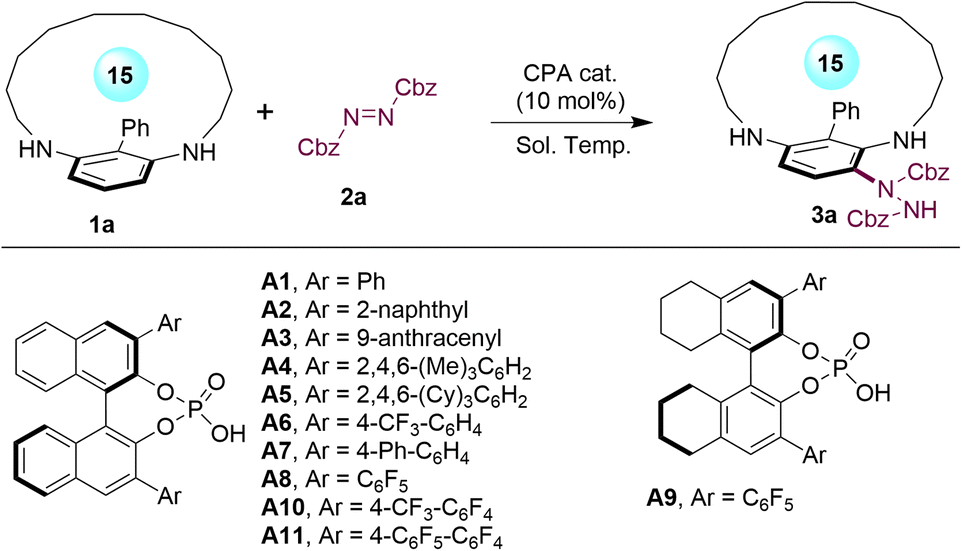
With the optimal conditions established, we embarked on exploring the scope of this method for enantioselective synthesis of planar-chiral metacyclophanes (Scheme 1). Modifications of the azodicarboxylate to another commercially available di-(p-Cl-benzyl) azodicarboxylate yielded the corresponding desymmetrized metacyclophane 3b in 81% yield with 95.5:4.5 er. Moreover, a series of para-substituted phenyl groups were examined at the C-2 position of metacyclophane substrates. Notably, various substituents with varying electronic properties were well tolerated, including the electron-neutral (3c and 3d), electron-donating (3h) and electron-withdrawing (3i) groups, as well as various functional halides (3e–3g), although some exhibited slightly diminished enantioselectivities. Additionally, the meta-substituted phenyl group was also examined, which produced product 3j with a reduced er value, indicating the method's sensitivity toward the C-2 group. Two types of heteroaryl groups were found to be compatible with this method, leading to the formation of planar-chiral metacyclophanes with excellent enantioselectivies of up to 99:1 er (3k–3l). Furthermore, an examination of the ansa chain length of the metacyclophanes was conducted. Both the reduction to a 14-membered macrocycle (3m) and expansion to up to a 17-membered macrocycle (3n–3p) were feasible, producing the planar-chiral metacyclophanes with high enantioselectivities. Notably, replacing the alkyl ansa chain with a polyethylene glycol-type ansa chain did not compromise the high stereoselectivity control of this method (3o). However, extending the macrocycle to an 18-membered structure led to a decrease in the enantioselectivity of the product (3q).
 | ||
| Scheme 1 Scope for asymmetric synthesis of planar-chiral metacyclophanes with C-2 aryl groups. Conditions: reactions were performed with 1 (0.1 mmol), 2 (0.1 mmol), CPA (R)-A11 (0.01 mmol, 10 mol%), and 3 Å MS (70 mg) in CCl4 (2.0 mL) at −20 °C for 48–72 h. Yields refer to isolated yields. The er values were determined by HPLC analysis on a chiral stationary phase. | ||
The modification of the pivotal C-2 groups of the metacyclophanes from (hetero)aryl groups to an alkyl group was investigated (Scheme 2). Given the reduced steric hindrance offered by the methyl group, the substrate's macrocycle was downsized to a 13-membered structure (4a) to ensure configurational stability of the chiral product (Scheme 2a). Applying the previous optimal conditions for the C-2 aryl-substituted substrate on substrate 4a resulted in the corresponding product ent-5a with an unsatisfactory enantioselectivity (11:89 er), underscoring the sensitivity of the C-2 group to the stereoselectivity of this method. A brief optimization of the reaction conditions revealed that the reaction between 4a and azodicarboxylate 2a, catalyzed by a sterically hindered CPA A12 in toluene at −40 °C, furnished product 5a in 86% yield with 95.5:4.5 er (see Table S1 in the SI for details). Surprisingly, despite CPA A11 and A12 sharing the same absolute configuration (R), the products obtained under these two conditions displayed opposite enantiomeric senses, suggesting completely distinct induction models for these two CPA catalysts in this reaction. With these conditions in hand, the scope for the asymmetric synthesis of C-2 alkyl-substituted planar-chiral metacyclophanes was investigated (Scheme 2b). The substitution of the azodicarboxylate with di-(p-Cl-benzyl) azodicarboxylate resulted in 5b with high enantioselectivity, whereas a switch to diethyl azodicarboxylate led to the product 5c with reduced enantioselectivity. Moreover, increasing the size of the macrocycle to a 14-membered structure led to notably decreased enantioselectivity (5d), highlighting the significant differences between the C-2 alkyl-substituted and aryl-substituted metacyclophanes. Remarkably, the prochiral metacyclophane with 2,5-disubstitutions was also feasible with this method, which yielded product 5e, featuring both planar chirality and C–N axial chirality, in 76% yield with 97:3 er and >20:1 dr. The absolute configuration of 5e was determined as (Rp, Sa) through X-ray crystallography analysis. Furthermore, modifications of the C-5 groups were explored. Notably, reducing the sizes of the halide substitutions led to reduced diastereoselectivities (14:1 dr for Cl-substituted 5i and 1.8:1 dr for F-substituted 5g), probably due to the reduced steric hindrance restricting the rotation of the C–N bond. Moreover, the C-5 aryl- and alkyl-substituted metacyclophane substrates also yielded products with both planar and axial chirality (5h and 5i), indicating that steric hindrance, rather than potential hydrogen bonding with the halogen atoms, restricts the rotation of the C–N bond. Notably, while the C-5 aryl-substituted metacyclophane substrate produced product 5h with poor diastereoselectivity, both diastereomers were generated with excellent enantioselectivities (≥99:1 er).
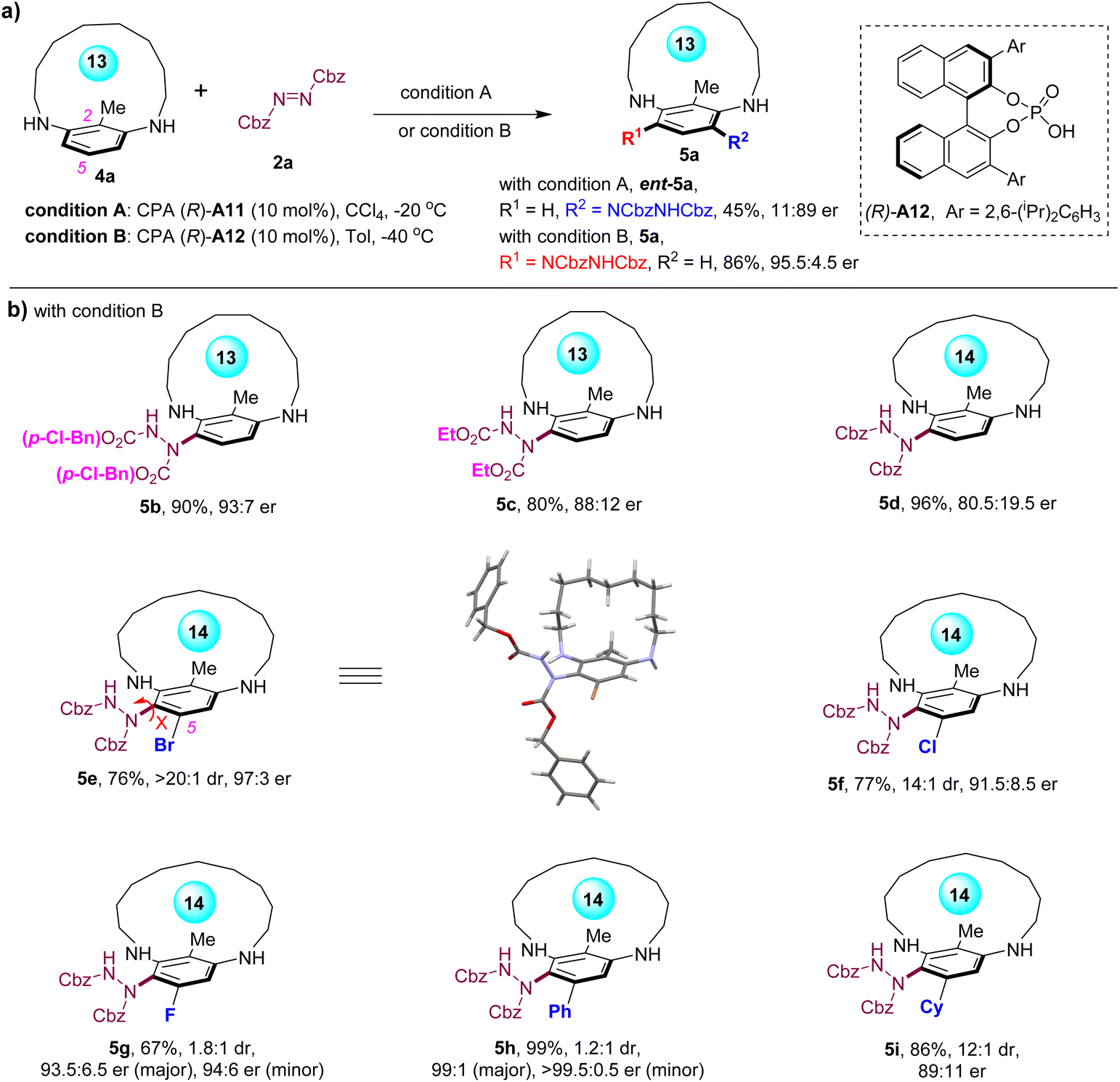 | ||
| Scheme 2 (a) Enantiodivergent asymmetric synthesis of planar-chiral metacyclophane 5a. (b) Scope for asymmetric synthesis of planar-chiral metacyclophanes with C-2 alkyl groups. | ||
The configurational stability of planar-chiral cyclophanes is essential for their further applications; however, the stereochemical properties of metacyclophanes have received less attention compared to those of paracyclophanes. Consequently, a thorough investigation was conducted on the synthesized chiral metacyclophane products (Fig. 2a). Notably, the thermal racemization experiment on the C-2 phenyl-substituted 15-membered metacyclophane 3a revealed its excellent configurational stability, with no notable decrease in enantiopurity observed at 120 °C in diphenyl ether after 5 hours, suggesting an estimated racemization barrier exceeding 37 kcal mol−1. Additionally, the 18-membered metacyclophane 3q also demonstrated good configurational stability, showing no loss of enantiopurity at 100 °C after 5 hours. This result suggested that the relatively modest enantioselectivity of 3q cannot be attributed to its racemization, but rather to the inferior stereoselectivity control of the asymmetric reaction resulting from the enlarged ring size. Moreover, the configurational stabilities of the C-2 methyl-substituted metacyclophanes 5 were investigated. The thermal racemization experiment of the 14-membered metacyclophane 5d suggested a racemization barrier of 29.2 kcal mol−1. In sharp contrast, the 15-membered metacyclophane 5l only exhibited a single peak in HPLC analysis using various chiral stationary phases, indicating its achiral nature. These results clearly underscored the critical role of the size of the C-2 group in the configurational stability of planar-chiral metacyclophanes. Moreover, the stereochemical characteristics of metacyclophane 5e, containing both axial and planar chirality, was studied. Placing 5e in diphenyl ether at 50 °C revealed a notable decrease in the diastereomeric ratio over time, indicating an epimerization half-life of 11.6 hours at 50 °C and an epimerization barrier of 25.9 kcal mol−1. On the other hand, the enantiopurity of 5e could be effectively maintained at this temperature.
 | ||
| Fig. 2 (a) Studies on configurational stability. (b) Control experiments with various diazo reagents. (c) Control experiments on N-substitution. (d) Proposed reaction mechanism. | ||
To elucidate the mechanism of this reaction, several control experiments were conducted using a series of electrophilic diazo reagents (Fig. 2b). In contrast to the high enantioselectivity obtained with the dibenzyl azodicarboxylate 2a, the use of diethyl azodicarboxylate 2c resulted in significantly reduced enantioselectivity, highlighting the critical role of the benzyl groups within azodicarboxylate in controlling enantioselectivity. Additionally, di-tert-butyl azodicarboxylate 2d failed to yield the corresponding amination product, likely due to the high steric hindrance of the N-Boc group. Although the cyclic diazo reagent 2e reacted effectively to give the desymmetrized planar-chiral metacyclophane 3ae, the enantioselectivity was low. Moreover, control experiments on the N-substitutions of the macrocyclic substrate were conducted (Fig. 2c). Methylation of both N-atoms of the substrate (1a′) resulted in no formation of the corresponding aromatic amination product 3a′, highlighting the importance of potential hydrogen bonding interactions. Surprisingly, the racemic mono-N-Me-substituted substrate 1a″ also did not undergo the amination reaction under standard conditions, which may be attributed to the sterically crowded environment around the reactive site caused by the rigid conformation of the substrate.
Building upon previous studies,6b,18 a plausible reaction mechanism is proposed (Fig. 2d). With the dual hydrogen-bonding activation of both the arylamine unit and azodicarboxylate by the CPA catalyst, the para-selective addition of the phenyl ring to azodicarboxylate from the opposite face of the ansa chain afforded the corresponding dearomatized amination intermediate INT-A. This step was believed to be the enantiodetermining step of this reaction, which was followed by a facile rearrangement aromatization to yield the final aminated planar-chiral metacyclophane products. Given the intriguing results where the use of CPA A11 and CPA A12, both with the same (R)-configuration, resulted in the formation of metacyclophane with opposite enantiomeric senses, we propose two potential transition states to rationalize this outcome.19 When employing CPA (R)-A11, the favorable noncovalent interaction between the CH3 group and the fluorinated aryl ring reduced the activation energy of this transition state, leading to the formation of (Sp)-5a. Conversely, with CPA (R)-A12, the analogous transition state revealed significant steric repulsion between the bulky 2,6-disubstituted aryl group and the CH3 group, leading to an energetically unfavorable transition state and consequently promoting the formation of the opposite enantiomer, (Rp)-5a.
To showcase the utilities of this method, the derivatizations of the planar-chiral metacyclophane products were explored (Scheme 3). Electrophilic aromatic bromination of metacyclophane 3a produced the brominated product 6a, which, upon treatment under strong basic conditions, resulted in the dehydrazinylation product 7a, enabling further derivatizations (Scheme 3a). Moreover, the catalytic hydrogenation of 3a provided the primary amine-containing metacyclophane 8a, which displayed limited stability under an air atmosphere owing to its susceptibility to oxidation (Scheme 3b). Consequently, protection of the NH2 group with the Boc group led to the formation of 9a in 85% yield with 95:5 er. In addition, treatment of 8a with 1,1′-carbonyldiimidazole (CDI) yielded the imidazolone-containing metacyclophane 10a. Interestingly, utilizing triphosgene as the condensation reagent resulted in the formation of metacyclophane 11a with an additional N-chlorocarbonyl group in 70% yield with 95:5 er, which could be further employed in coupling reactions. The structure of 11a was unambiguously determined through X-ray crystallography analysis. Furthermore, diazotization of the NH2 group using NaNO2 was followed by a subsequent cyclization to yield the benzotriazole-containing metacyclophane 12a in 68% yield.
 | ||
| Scheme 3 (a) Bromination-based derivatization of planar-chiral metacyclophane 3a. (b) Diverse derivatizations of planar-chiral metacyclophane via cyclization reactions. | ||
Conclusions
In conclusion, we have developed an efficient catalytic enantioselective desymmetrization method for the asymmetric synthesis of planar-chiral metacyclophanes, a class of structurally distinct cyclophanes that have received considerably less attention compared to the paracyclophane counterparts.20 By utilizing the CPA-catalyzed asymmetric aromatic amination reaction with azodicarboxylates, we successfully broke the mirror symmetry of prochiral m-phenylenediamine-derived metacyclophanes, which produced a variety of planar-chiral metacyclophanes with diverse substitutions and ring sizes with good to high enantioselectivities. Moreover, a series of metacyclophanes featuring both planar chirality and C–N axial chirality were generated in a single step when the prochiral metacyclophanes included an additional meta-substitution. Thermal racemization experiments were conducted to investigate the configurational stabilities of these novel metacyclophanes, and various control experiments were performed to elucidate the origin of stereoselectivity. Notably, the synthesized planar-chiral metacyclophanes could undergo various derivatizations to produce diverse planar-chiral products, including those incorporating heterocycles, thereby underscoring the potential of this method.Author contributions
Z. C. performed most of the experimental studies. J. Z., L. Z., and W. X. prepared some of the substrates and catalysts. H. G. solved the X-ray crystal structures. H. L. performed the high-temperature NMR experiments. X. Y. directed the project and wrote the paper with feedback from other authors.Conflicts of interest
There are no conflicts to declare.Data availability
CCDC 2500738 and 2500739 contain the supplementary crystallographic data for this paper.21a,bThe data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc09849a.
Acknowledgements
The authors thank the NSFC (grant no. 22571199 X. Y., 22222107 X. Y. and 22171186 X. Y.) and ShanghaiTech University start-up funding for financial support. Prof. Guangxin Liang is thanked for sharing the optical rotation polarimeter. The authors acknowledge the support from the Analytical Instrumentation Center (# SPST-AIC10112914), SPST, ShanghaiTech University.Notes and references
- (a) E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef CAS PubMed; (b) D. Garcia Jimenez, V. Poongavanam and J. Kihlberg, J. Med. Chem., 2023, 66, 5377–5396 CrossRef CAS PubMed; (c) B. Acharya, D. Saha, D. Armstrong, B. A. Jabali, M. Hanafi, A. Herrera-Rueda, N. R. Lakkaniga and B. Frett, RSC Med. Chem., 2024, 15, 399–415 RSC.
- R. Gleiter and H. Hopf, in Modern Cyclophane Chemistry, WILEY-VCH Verlag GmbH & Co. KGaA, 2004 Search PubMed.
- (a) R. López and C. Palomo, Angew. Chem., Int. Ed., 2022, 61, e202113504 Search PubMed; (b) K. Zhu, L. Yang, Y. Yang, Y. Wu and F. Zhang, Chin. Chem. Lett., 2025, 36, 110678 Search PubMed; (c) L. Yang, Y.-P. Zhang, Y. You, Z.-H. Wang, W.-C. Yuan and J.-Q. Zhao, Chem. Rec., 2025, 25, e202500085 Search PubMed; (d) Y. H. Zhao, D. Zhu and Z. M. Chen, ChemCatChem, 2024, 16, e202401312 Search PubMed; (e) G. Yang and J. Wang, Angew. Chem., Int. Ed., 2024, 16, e202412805 Search PubMed; (f) Z. Dong, J. Li and C. Zhao, Eur. J. Org Chem., 2024, 27, e202400841 Search PubMed.
- (a) J. Zhu, G. Islas-Gonzalez and M. Bois-Choussy, Org. Prep. Proced. Int., 2000, 32, 505–546 CrossRef CAS; (b) A. D. William, A. C. H. Lee, K. C. Goh, S. Blanchard, A. Poulsen, E. L. Teo, H. Nagaraj, C. P. Lee, H. Wang, M. Williams, E. T. Sun, C. Hu, R. Jayaraman, M. K. Pasha, K. Ethirajulu, J. M. Wood and B. W. Dymock, J. Med. Chem., 2012, 55, 169–196 Search PubMed; (c) M. M. Aleman, S. Jindal, N. Leksa, R. Peters and J. Salas, Blood, 2018, 132, 2461 Search PubMed.
- (a) K. Tanaka, T. Hori, T. Osaka, K. Noguchi and M. Hirano, Org. Lett., 2007, 9, 4881–4884 CrossRef CAS PubMed; (b) Q. Ding, Q. Wang, H. He and Q. Cai, Org. Lett., 2017, 19, 1804–1807 Search PubMed; (c) C. Gagnon, É. Godin, C. Minozzi, J. Sosoe, C. Pochet and S. K. Collins, Science, 2020, 367, 917 CrossRef CAS PubMed; (d) S. Yu, G. Shen, F. He and X. Yang, Chem. Commun., 2022, 58, 7293–7296 RSC; (e) G. Yang, Y. He, T. Wang, Z. Li and J. Wang, Angew. Chem., Int. Ed., 2023, 63, e202316739 CrossRef PubMed; (f) J. Wang, M. Wang, Y. Wen, P. Teng, C. Li and C. Zhao, Org. Lett., 2024, 26, 1040–1045 CrossRef CAS PubMed; (g) X. Lv, F. Su, H. Long, F. Lu, Y. Zeng, M. Liao, F. Che, X. Wu and Y. R. Chi, Nat. Commun., 2024, 15, 958 CrossRef CAS PubMed; (h) H. Zhai, K. Lv, J. Li, J. Wang, T. Liu and C. Zhao, J. Am. Chem. Soc., 2024, 146, 29214–29223 Search PubMed; (i) G. Yang, S. Liu, S. Ji, X. Wu and J. Wang, Chem. Sci., 2024, 15, 19599–19603 RSC; (j) J. Wang, K. Lv, Y. Wen, T. Liu and C. Zhao, Nat. Commun., 2025, 16, 3170 Search PubMed.
- (a) K. Kanda, K. Endo and T. Shibata, Org. Lett., 2010, 12, 1980–1983 CrossRef CAS PubMed; (b) D. Wang, Y. B. Shao, Y. Chen, X. S. Xue and X. Yang, Angew. Chem., Int. Ed., 2022, 61, e202201064 CrossRef CAS PubMed; (c) Z. Zhou, K. Kasten, A. P. McKay, D. B. Cordes and A. D. Smith, Angew. Chem., Int. Ed., 2025, 64, e202507126 Search PubMed; (d) S. Huh, E. Linne, L. Estaque, G. Pieters, M. Devereux and O. Baudoin, Angew. Chem., Int. Ed., 2025, 64, e202500653 Search PubMed; (e) D. Zhu, T. Mu, Z. L. Li, H. Y. Luo, R. F. Cao, X. S. Xue and Z. M. Chen, Angew. Chem., Int. Ed., 2024, 63, e202318625 CrossRef CAS PubMed; (f) J. Li, Z. Dong, Y. Chen, Z. Yang, X. Yan, M. Wang, C. Li and C. Zhao, Nat. Commun., 2024, 15, 2338 CrossRef CAS PubMed; (g) J. Li and C. Zhao, ACS Catal., 2023, 13, 14155–14162 Search PubMed; (h) Z. Dong, J. Li, T. Yao and C. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202315603 CrossRef CAS PubMed.
- T. Araki, K. Noguchi and K. Tanaka, Angew. Chem., Int. Ed., 2013, 52, 5617–5621 Search PubMed.
- (a) K. Tanaka, H. Sagae, K. Toyoda and M. Hirano, Tetrahedron, 2008, 64, 831–846 Search PubMed; (b) K. Tanaka, H. Sagae, K. Toyoda, K. Noguchi and M. Hirano, J. Am. Chem. Soc., 2007, 129, 1522–1523 CrossRef CAS PubMed.
- S. Wei, L.-Y. Chen and J. Li, ACS Catal., 2023, 13, 7450–7456 Search PubMed.
- G. Yang, Y. He, T. Wang, Z. Li and J. Wang, Angew. Chem., Int. Ed., 2024, 63, e202316739 CrossRef CAS PubMed.
- H.-H. Chen, J.-T. Jiang, Y.-N. Yang, L.-W. Ye and B. Zhou, Angew. Chem., Int. Ed., 2025, 64, e202505167 Search PubMed.
- Z. Wang, J. Lin, Y.-B. Shao, B. Wang and X. Li, ACS Catal., 2025, 17938–17949 CrossRef CAS.
- S. Yu, H. Bao, D. Zhang and X. Yang, Nat. Commun., 2023, 14, 5239 Search PubMed.
- (a) A. Borissov, T. Q. Davies, S. R. Ellis, T. A. Fleming, M. S. W. Richardson and D. J. Dixon, Chem. Soc. Rev., 2016, 45, 5474–5540 RSC; (b) Y. Xu, T.-Y. Zhai, Z. Xu and L.-W. Ye, Trends Chem., 2022, 4, 191–205 Search PubMed; (c) W. Liu, D. Wang, D. Zhang and X. Yang, Synlett, 2022, 33, 1788–1812 CrossRef CAS.
- (a) T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566–1568 Search PubMed; (b) D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356–5357 Search PubMed; (c) T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS PubMed; (d) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed; (e) W. Liu and X. Yang, Beilstein J. Org. Chem., 2025, 21, 1864–1889 Search PubMed.
- (a) M. Tang and X. Yang, Chin. J. Org. Chem., 2025, 45, 1785–1818 CrossRef CAS; (b) H. Bao, Y. Chen and X. Yang, Angew. Chem., Int. Ed., 2023, 62, e202300481 CrossRef CAS PubMed; (c) M. Tang, J. Zhou, W. Xie, J. Ren, Z. Ye, H. Gu and X. Yang, Chem, 2026, 12, 102694 CrossRef CAS; (d) M. Yuan, W. Xie, S. Yu, T. Liu and X. Yang, Nat. Commun., 2025, 16, 3943 CrossRef CAS PubMed; (e) Q. Jiang, D. Zhang, M. Tang, H. Liu and X. Yang, Sci. China Chem., 2024, 67, 973–980 CrossRef CAS.
- Y.-J. Wu, G. Liao and B.-F. Shi, Green Synth. Catal., 2022, 3, 117–136 Search PubMed.
- D. Zhang, Y.-B. Shao, W. Xie, Y. Chen, W. Liu, H. Bao, F. He, X.-S. Xue and X. Yang, ACS Catal., 2022, 12, 14609–14618 Search PubMed.
- (a) S.-G. Wang, Q. Yin, C.-X. Zhuo and S.-L. You, Angew. Chem., Int. Ed., 2015, 54, 647–650 CrossRef CAS PubMed; (b) A. Changotra, S. Das and R. B. Sunoj, Org. Lett., 2017, 19, 2354–2357 CrossRef CAS PubMed.
- Z. Dong, K. Lv, C. Yuan, L. Wang, H. Wang, J. Li, X.-E. Yan, T. Liu, P. Zheng and C. Zhao, ACS Catal., 2026, 16, 2182–2193 CrossRef CAS.
- (a) CCDC 2500738: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2py6zv; (b) CCDC 2500739: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2py70x.
| This journal is © The Royal Society of Chemistry 2026 |