
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExposed high-concentration Ir active sites and metal–support interaction endow Ir/MoO2-Mo2C hybrids with high atom utilization and stability for pH-universal hydrogen evolution reaction
Yuehuan
Zhang
and
Qiang
Yuan
 *
*
State Key Laboratory of Green Pesticide, Center for R&D of Fine Chemicals, College of Chemistry and Chemical Engineering, Guizhou University, Guiyang, Guizhou province 550025, PR China. E-mail: qyuan@gzu.edu.cn
First published on 15th December 2025
Abstract
Constructing active metal–support interfacial structures is crucial for enhancing the activity and stability of electrocatalysts during the hydrogen evolution reaction (HER) across a wide pH range. Herein, ultrasmall mixed Ir species anchored on MoO2–Mo2C hollow nanoflowers assembled from nanosheets were synthesized via high-temperature sintering (600–800 °C). The X-ray absorption fine structure and X-ray photoelectron spectroscopy revealed the metal–support interaction (MSI) in Ir/MoO2-Mo2C-800. In situ attenuated total reflection surface-enhanced infrared absorption spectroscopy and in situ Raman spectroscopy reveal that Ir/MoO2-Mo2C-800 can reconstruct the distribution equilibrium of the hydrogen-bond network of interfacial water to promote water adsorption and dissociation. The highly exposed Ir active sites and MSI enable Ir/MoO2-Mo2C-800 to exhibit extremely low overpotentials of 32 (alkaline), 14 (acidic), and 29 (neutral) mV at 10 mA cm−2, respectively. Moreover, the mass activities of Ir/MoO2-Mo2C-800 (9.23, 60.99, and 8.34 A mg−1PGM) are 2.68-, 28.27-, and 3.93-fold higher than those of commercial Pt/C (3.45, 2.16, and 2.12 A mg−1PGM), respectively. The assembled Ir/MoO2-Mo2C-800 ‖ IrO2 anion-exchange membrane (AEM) water electrolyzer demonstrates stable operation for over 700/220 h at a high current density of 0.2/1 A cm−2 and Ir/MoO2-Mo2C-800 can maintain its initial morphology. This study proposes a feasible strategy to achieve highly efficient and stable pH-universal HER by developing electrocatalysts with high concentration Ir active sites and MSI engineering.
Introduction
Electrocatalytic water splitting for green hydrogen (H2) production offers promising prospects for carbon neutrality and alleviates global energy shortages.1–4 For the hydrogen evolution reaction (HER) at the cathode during water electrolysis, platinum-group metals (PGMs) are the benchmark HER catalysts owing to their high performance and favorable Gibbs free energy of H* adsorption (ΔGH*).5–8 However, practical application of PGMs is highly limited owing to their high cost and scarcity.9–11 Moreover, the HER kinetics of PGMs in neutral or alkaline media are generally one to several orders of magnitude slower than in acidic media.12–14 Therefore, it is crucial to develop an effective strategy for maximum utilization of active platinum-group atoms while maintaining high catalytic activity across a wide pH range.Clusters and single-atom catalysts have attracted extensive attention owing to the advantages they offer such as high atomic utilization, excellent activity, and abundant low-coordination atoms. However, their limited lifespan due to sintering or loss of active metal sites is a considerable limitation.15–17 Moreover, the metal–support interaction (MSI) formed between the support and active metal plays a crucial role in preventing the agglomeration and detachment of metal nanoparticles (NPs) under a high current density.18–21 Therefore, selecting suitable supports to anchor PGM clusters and develop MSI is essential for developing catalysts with superior activity and enhanced stability.22–25 A suitable support system can anchor active metal NPs to expose active sites, modulating their electronic structure and charge transfer.26–29 It optimizes the adsorption/desorption of intermediates and enhances reaction kinetics, thereby improving the intrinsic HER activity of electrocatalysts.30–33 Zhao et al. attributed the exceptional HER activity and stability of Ir–V2O3 under alkaline and acidic conditions to the distinct advantage of direct Ir–V metallic bonding. This bonding configuration enables optimal binding energies with H and OH species.34 Feng et al. anchored Ir NPs on Co-BPDC nanosheets. Density functional theory (DFT) calculations revealed that electron transfer between Ir and Co-BPDC enhances electron transfer kinetics and optimizes the H2O adsorption energy on Ir@Co-BPDC.35 Leung et al. demonstrated that the MSI between NiCo foam and Ir clusters facilitates rapid electron transfer. DFT calculations revealed that the weakened hydrogen adsorption energy and optimized ΔGH* on Ir-nc@m-NiCo contribute to the enhanced HER performance.36 Despite notable progress in constructing MSI, developing ultrasmall cluster catalysts with metallic bonding to enhance activity and stability for practical applications remains a formidable challenge.
Herein, we present an effective strategy to enhance the HER activity and stability of the catalyst for all pH by loading ultrasmall mixed Ir species onto a hollow MoO2–Mo2C support, thereby developing MSI. Specifically, the Ir/MoO2-Mo2C-800 catalyst requires overpotentials of only 32, 14, and 29 mV to achieve a current density of 10 mA cm−2 in alkaline, acidic, and neutral solutions, respectively, outperforming commercial Pt/C and Ir/C catalysts. Notably, the anion exchange membrane water electrolyzer (AEMWE) with Ir/MoO2-Mo2C-800 as a cathode demonstrates stable operation for over 700 h at 200 mA cm−2. Detailed structural characterization reveals that the formation of MSI effectively induces the electronic distribution of the active Ir sites. In situ attenuated total reflection surface-enhanced infrared absorption spectroscopy and in situ Raman spectroscopy demonstrate that the Ir/MoO2-Mo2C-800 catalyst optimizes the dynamic equilibrium of the hydrogen-bond network distribution at the catalyst/electrolyte interface. This optimization remarkably enhances water molecule adsorption and subsequent dissociation processes, resulting in enhanced HER activity. This study presents an electrocatalyst with MSI engineering, abundant low-coordination atoms, and a high concentration of active Ir sites for efficient and stable pH-universal HER.
Results and discussion
The Mo2C substrate was synthesized via ammonia-catalyzed self-polymerization. Its transmission electron microscopy (TEM) images reveal a hollow nanoflower architecture assembled from nanosheets (Fig. S1a and b). This hollow nanoflower structure, with preferentially exposed edges, enhances electrocatalytic activity by promoting mass transfer and reaction kinetics.37 The characteristic Mo2C peaks (PDF#35-0787) observed in the corresponding X-ray diffraction (XRD) pattern confirm its successful synthesis (Fig. S1c). The Ir3+ was subsequently reduced to Ir clusters via high-temperature calcination (800 °C) and anchored onto the MoO2–Mo2C substrate with a hollow nanoflower architecture to form Ir/MoO2-Mo2C-800.The morphology of Ir/MoO2-Mo2C-800 was characterized using scanning electron microscopy (SEM), TEM, and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). Ir/MoO2-Mo2C-800 retains the inherent three-dimensional structure of Mo2C (Fig. 1a), ensuring the uniform distribution of Ir clusters throughout the hollow nanoflowers with an average diameter of 1.41 nm (Fig. 1b–e). The XRD peaks of Ir/MoO2-Mo2C-800 in Fig. 1f match with those of Ir (PDF#06-0598), Mo2C (PDF#35-0787), and MoO2 (PDF#73-1807), confirming the formation of these three phases in Ir/MoO2-Mo2C-800. Additionally, the characteristic peak intensity of Mo2C is lower than that of MoO2, which is attributed to the oxidation and conversion of a significant amount of Mo2C into MoO2 during the high-temperature sintering process. In addition, the Ir clusters in the control samples of Ir/MoO2-Mo2C-700 and Ir/MoO2-Mo2C-600 have sizes of 1.58 and 1.49 nm, respectively, and their XRD patterns exhibit a similar phase composition to that of Ir/MoO2-Mo2C-800 (Fig. S2 and S3). HAADF-STEM images and the corresponding energy-dispersive spectroscopy (EDS) elemental mapping demonstrate the uniform distribution of Ir, Mo, C, N, and O in Ir/MoO2-Mo2C-800 (Fig. 1g). Inductively coupled plasma optical emission spectrometry (ICP-OES) measurements indicate Ir loadings of 14.92 wt%, 14.77 wt%, and 14.62 wt% for Ir/MoO2-Mo2C-800, Ir/MoO2-Mo2C-700, and Ir/MoO2-Mo2C-600, respectively (Table S1).
 | ||
| Fig. 1 (a) Low and high magnification SEM images, (b and c) TEM images, (d) AC-HAADF-STEM image, (e) particle size statistics of Ir clusters, (f) XRD pattern, and (g) HAADF-STEM and elemental mapping images of Ir/MoO2-Mo2C-800. | ||
As shown in Fig. 2a–d, the atomic-resolution HAADF-STEM images of Ir/MoO2-Mo2C-800 indicate that the Ir metal species are primarily present as amorphous clusters, along with a small number of single atoms, sub-nanometer clusters, and crystalline clusters. These provide abundant low-coordination atoms and a high concentration of active sites, thereby enhancing the catalytic kinetics.38–41Fig. 2e shows lattice spacings of 0.191 and 0.241 nm, which correspond to the (200) planes of MoO2 and (200) Ir, respectively, demonstrating the formation of an Ir and MoO2 interface. The lattice fringe spacing measured in Fig. 2f is 0.115 nm, corresponding to the (311) plane of Ir, and the fast Fourier transform (FFT) pattern in the inset jointly confirms the existence of interfaces between Ir and MoO2–Mo2C. Fig. 2g illustrates clear interfaces among Ir nanoclusters (marked by a red circle), Mo2C (orange circle), and MoO2 (blue circle). The corresponding inverse fast Fourier transform images (Fig. 1h–j) reveal lattice spacings of 0.222, 0.228, and 0.213 nm, which match the (111) planes of face-centered cubic (fcc) Ir, (101) Mo2C, and (210) MoO2, respectively, further confirming the existence of Ir and MoO2–Mo2C interfaces.42 These results imply the presence of MSI between Ir and MoO2–Mo2C.
 | ||
| Fig. 2 (a and c) Atomically resolved HAADF-STEM images of Ir/MoO2-Mo2C-800. (b and d) Enlarged AC-HAADF-STEM images (top) and the corresponding FFT images (bottom) for the inset of (a and c). The red circles, blue circles, orange rectangles, and green rectangles correspond to the Ir single atoms, Ir sub-nanoclusters, amorphous Ir clusters, and crystalline Ir clusters, respectively. (e–g) HAADF-STEM images of Ir/MoO2-Mo2C-800 (inset: the corresponding FFT images). (h–j) Inverse FFT patterns of the red elliptical region, orange elliptical region, and blue elliptical regions shown in (g), respectively. | ||
X-ray photoelectron spectroscopy (XPS) measurements were taken to investigate the electronic structure and chemical state of the Ir/MoO2-Mo2C-800 catalyst. The full XPS spectrum of Ir/MoO2-Mo2C-800 reveals the presence of Ir, Mo, C, N, and O signals, which is consistent with the EDS results (Fig. S4). The peaks observed at approximately 60.85 and 64.01 eV can be attributed to the Ir 4f7/2 and Ir 4f5/2 of Ir NPs, while the peaks at 61.40 and 64.49 eV indicate the presence of Ir–O (Fig. 3a).43 As shown in Fig. 3b, the binding energy peaks at 231.99 and 235.12 eV correspond to Mo4+ 3d5/2 and Mo4+ 3d3/2, respectively, in Ir/MoO2-Mo2C-800. The two characteristic binding energy peaks at 232.71 and 235.89 eV are attributed to Mo6+ 3d5/2 and Mo6+ 3d3/2, respectively.44 Compared with Mo2C, the Mo4+ 3d5/2 peak in Ir/MoO2-Mo2C-800 demonstrates a negative binding energy shift of approximately 0.38 eV, confirming the interaction between Ir and MoO2–Mo2C. The O 1s spectrum of Ir/MoO2-Mo2C-800 exhibits three peaks at 530.57, 531.84, and 533.14 eV, corresponding to metal–O, surficial/interfacial O, and C–O, respectively (Fig. 3c).45 The C 1s spectrum of Ir/MoO2-Mo2C-800 shows three deconvoluted peaks at 284.45, 285.90, and 288.35 eV, which correspond to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–N, and C–O bonds, respectively (Fig. S5a).46 The N 1s spectrum of Ir/MoO2-Mo2C-800 can be fitted into four components with binding energies of 396.6, 398.4, 399.9, and 401.75 eV, attributed to Mo 3p, pyridinic N, pyrrolic N, and graphitic N, respectively (Fig. S5b).47 In summary, electron redistribution can influence the electrocatalytic performance for hydrogen production by optimizing the adsorption/desorption energy between intermediates and active sites.48
C, C–N, and C–O bonds, respectively (Fig. S5a).46 The N 1s spectrum of Ir/MoO2-Mo2C-800 can be fitted into four components with binding energies of 396.6, 398.4, 399.9, and 401.75 eV, attributed to Mo 3p, pyridinic N, pyrrolic N, and graphitic N, respectively (Fig. S5b).47 In summary, electron redistribution can influence the electrocatalytic performance for hydrogen production by optimizing the adsorption/desorption energy between intermediates and active sites.48
 | ||
| Fig. 3 XPS spectra of (a) Ir 4f, (b) Mo 3d, and (c) O 1s for Ir/MoO2-Mo2C-600, Ir/MoO2-Mo2C-700, Ir/MoO2-Mo2C-800, and Mo2C. (d) Ir L3-edge XANES spectra and (e and f) corresponding FT-EXAFS curves of Ir/MoO2-Mo2C-800, Ir foil, and IrO2. EXAFS wavelet transform plots of (g) Ir/MoO2-Mo2C-800, (h) IrO2, and (i) Ir foil. | ||
X-ray absorption spectroscopy (XAS) measurements were taken to further examine the valence state and coordination environment of Ir in Ir/MoO2-Mo2C-800. Fig. 3d shows the Ir L3-edge XANES spectrum of Ir/MoO2-Mo2C-800, where the white line peak intensity is higher than that of the Ir foil but lower than that of IrO2, demonstrating that the Ir valence state in Ir/MoO2-Mo2C-800 lies between that of metallic Ir and IrO2. The atomic coordination environment of Ir/MoO2-Mo2C-800 was further confirmed by extended X-ray absorption fine structure (EXAFS) spectroscopy. The EXAFS spectrum of Ir/MoO2-Mo2C-800 exhibits peaks at 1.65 and 2.59 Å, which correspond to the Ir–O and Ir–Mo/Ir bonds, respectively (Fig. 3e and f). The EXAFS fitting curve in Ir k-space for Ir/MoO2-Mo2C-800 and the corresponding fitting parameters reveal coordination numbers of ∼2.4 for Ir–O and 1.1 for Ir–Mo bonds (Fig. S6 and Table S2), characteristic of a typical low-coordinated Ir cluster structure.27 Wavelet transform analysis further confirms the formation of Ir–Mo metallic bonds in Ir/MoO2-Mo2C-800 (Fig. 3g–i). In summary, XAS combined with HAADF-STEM images and XPS analysis demonstrates the successful anchoring of 1.41 nm Ir clusters on MoO2–Mo2C, confirming the MSI between Ir and Mo species. Moreover, the low coordination number and MSI between Ir and MoO2–Mo2C can regulate the local electronic structure of active sites, optimizing the adsorption behavior of catalytic sites toward reaction intermediates, thereby enhancing catalytic reaction kinetics.49,50
The HER activities of Ir/MoO2-Mo2C-800, Ir/MoO2-Mo2C-700, Ir/MoO2-Mo2C-600, commercial Pt/C, and Ir/C were first evaluated in a 1.0 M KOH solution. Linear sweep voltammetry (LSV) curves reveal that Ir/MoO2-Mo2C-800 exhibits the best HER performance. Specifically, Ir/MoO2-Mo2C-800 requires only 32 mV overpotential at 10 mA cm−2, outperforming Ir/MoO2-Mo2C-700 (59 mV), Ir/MoO2-Mo2C-600 (83 mV), Pt/C (36 mV), and Ir/C (51 mV) (Fig. 4a and b). Ir/MoO2-Mo2C-800 exhibits a Tafel slope of 35.76 mV dec−1, which is lower than those of Ir/MoO2-Mo2C-700 (98.28 mV dec−1), Ir/MoO2-Mo2C-600 (128.76 mV dec−1), Pt/C (55.51 mV dec−1), and Ir/C (92.87 mV dec−1) (Fig. 4c). This indicates that Ir/MoO2-Mo2C-800 follows a rapid Tafel mechanism, demonstrating faster hydrogen generation kinetics compared with commercial Pt/C and Ir/C.51,52 At overpotentials of 100 and 150 mV, the mass activity of the Ir/MoO2-Mo2C-800 catalyst reached 9.23 and 16.94 A mg−1PGM, respectively, which are 2.67 and 2.61 times higher than those of Pt/C (3.45 and 6.48 A mg−1PGM) (Fig. 4d). Owing to its enhanced catalytic activity, Ir/MoO2-Mo2C-800 demonstrates notable potential as an electrocatalyst for hydrogen production. Subsequently, we evaluated the electrochemical active surface area (ECSA) of the synthesized catalysts by measuring the double-layer capacitance (Cdl) using cyclic voltammetry (CV) curves at different scan rates.53 As shown in Fig. 4e and S7, Ir/MoO2-Mo2C-800 exhibits a Cdl value of 2.93 mF cm−2, which is higher than that of other control samples. This indicates that the MSI between Ir nanoclusters and MoO2–Mo2C generates additional active sites, thereby increasing the accessible active surface area.54 The Nyquist plot shown in Fig. 4f reveals that Ir/MoO2-Mo2C-800 possesses the lowest charge transfer resistance compared with other catalysts, indicating faster reaction kinetics and enhanced charge transfer capability.55 This is likely attributed to the accelerated electron transfer during the HER process, facilitated by the interaction between the highly conductive MoO2–Mo2C substrate and the active sites of Ir nanoclusters. At overpotentials of 50 and 100 mV, Ir/MoO2-Mo2C-800 demonstrated high turnover frequencies (TOFs) of 3.66 and 8.58 s−1, respectively, which are considerably higher than those of Pt/C (1.47 and 3.48 s−1) (Fig. 4g). Stability is another crucial parameter for evaluating catalyst performance. Therefore, we compared the LSV curves of Ir/MoO2-Mo2C-800 and commercial Pt/C before and after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 CV cycles. As shown in Fig. 4h and Table S3, Ir/MoO2-Mo2C-800 shows a smaller negative shift in its LSV curve compared with that in Pt/C, indicating superior stability. This confirms the high atomic utilization and excellent catalytic activity of Ir/MoO2-Mo2C-800 for the HER under alkaline conditions.
000 CV cycles. As shown in Fig. 4h and Table S3, Ir/MoO2-Mo2C-800 shows a smaller negative shift in its LSV curve compared with that in Pt/C, indicating superior stability. This confirms the high atomic utilization and excellent catalytic activity of Ir/MoO2-Mo2C-800 for the HER under alkaline conditions.
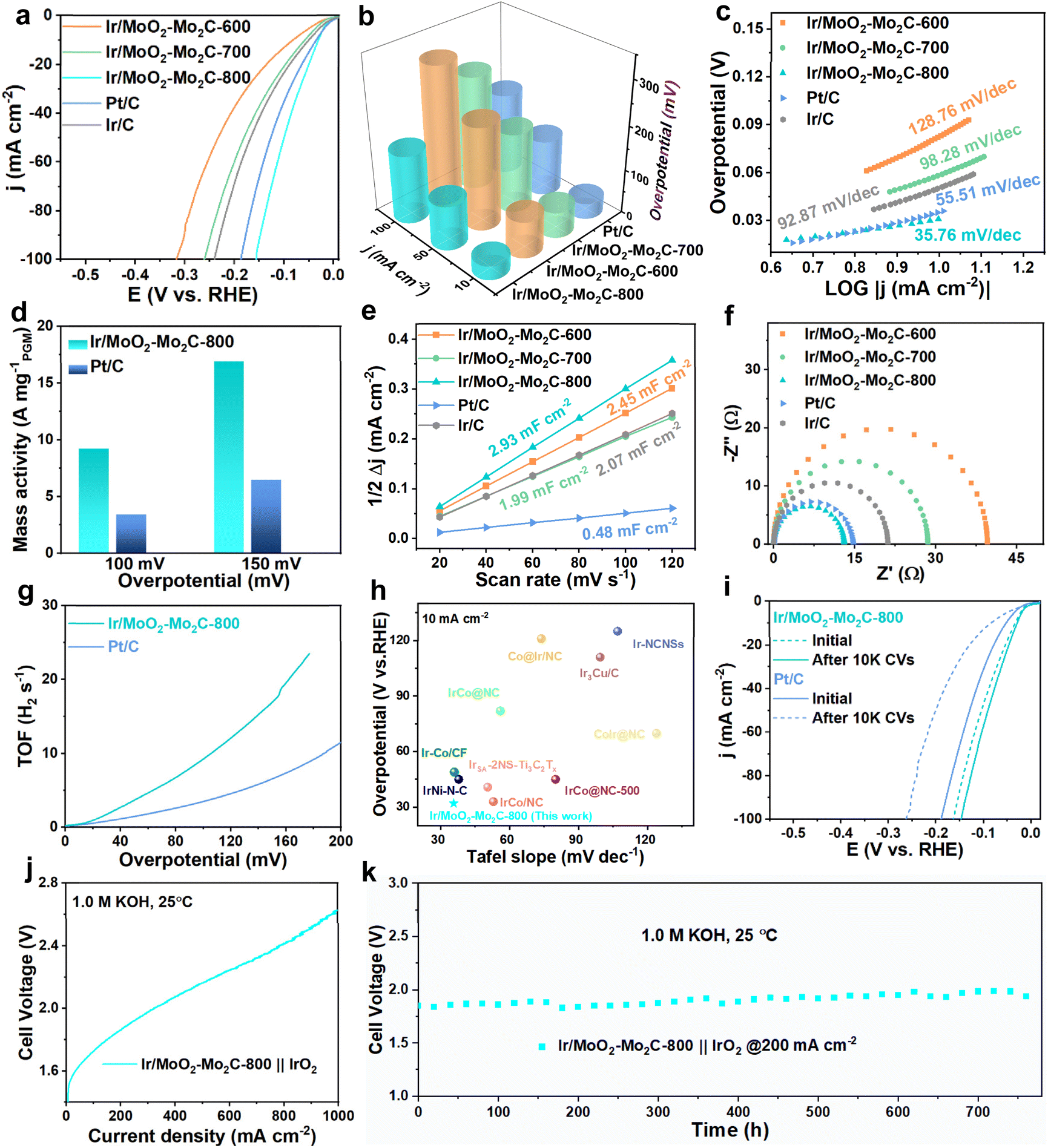 | ||
| Fig. 4 (a) HER LSV curves for Ir/MoO2-Mo2C-800 and other contrast catalysts in 1.0 M KOH. (b) Comparison of the overpotentials of the prepared catalysts. (c) Tafel plots. (d) Mass activity. (e) Corresponding electrochemical double-layer capacitances. (f) EIS spectra. (g) TOF values of Ir/MoO2-Mo2C-800 and Pt/C. (h) Comparison of the overpotential and Tafel slope of Ir/MoO2-Mo2C-800 and recently reported catalysts in 1.0 M KOH. (i) HER polarization curves of Ir/MoO2-Mo2C-800 and Pt/C before and after 10000 CV cycles. (j) Polarization curves of Ir/MoO2-Mo2C-800 at the AEMWE. (k) Stability tests of the AEMWE at 200 mA cm−2. | ||
Owing to its exceptional performance in a two-electrode system, we further evaluated its practical application potential in an anion exchange membrane (AEM) water electrolyzer. The AEM system employed Ir/MoO2-Mo2C-800 as the cathode catalyst and IrO2 as the anode catalyst, with carbon paper and nickel foam as gas diffusion layers. The corresponding polarization curve measured at room temperature showed that only 1.86 V was required to reach a current density of 200 mA cm−2 (Fig. 4j). Long-term stability tests demonstrated that Ir/MoO2-Mo2C-800 could maintain stable water electrolysis for over 700 h at 200 mA cm−2 (Fig. 4k). Additionally, this AEMWE requires only 2.62 V to achieve 1 A cm−2 while maintaining stability for over 220 h (Fig. S8), proving its remarkable stability even at high current densities. Post-stability characterization using TEM and EDS (Fig. S9 and S10) reveals that Ir/MoO2-Mo2C-800 largely retains its original morphology, with elements remaining uniformly distributed on the catalyst surface.
Given the excellent HER performance of Ir/MoO2-Mo2C-800 under alkaline conditions, we further investigated its HER activity in acidic (0.5 M H2SO4) and neutral (1.0 M PBS) media. As expected, Ir/MoO2-Mo2C-800 demonstrated outstanding HER performance under both the conditions. As shown in Fig. 5a and d, the Ir/MoO2-Mo2C-800 required only 14 and 29 mV to achieve 10 mA cm−2 in acidic and neutral media, respectively, outperforming commercial Pt/C, Ir/C, and other control samples. The Tafel slopes of Ir/MoO2-Mo2C-800, derived from LSV curves (Fig. 5b and e), are 16.57 mV dec−1 (acidic) and 34.21 mV dec−1 (neutral), which are considerably lower than those of Ir/C (41.17 and 177.08 mV dec−1) and commercial Pt/C (22.83 and 79.01 mV dec−1). This indicates a faster Volmer-Tafel mechanism and superior reaction kinetics in both the media.56,57 Electrochemical impedance spectroscopy (EIS) was performed to further understand the enhanced HER activity. As shown in Fig. S11, Ir/MoO2-Mo2C-800 exhibited lower charge transfer resistance (Rct) values than Pt/C and Ir/C in 0.5 M H2SO4 and 1.0 M PBS solutions. This indicates that the interfacial charge redistribution caused by Ir–Mo bond formation enables ultrafast electron transfer. The mass activities, normalized to noble metal content (Fig. S12), reached 60.99 A mg−1PGM at 30 mV (acidic) and 8.34 A mg−1PGM at 100 mV (neutral), notably exceeding those of Pt/C (2.16 and 2.12 A mg−1PGM). These results demonstrate the superior noble metal utilization efficiency of Ir/MoO2-Mo2C-800. Moreover, Ir/MoO2-Mo2C-800 exhibited the highest Cdl and TOF values in alkaline and neutral media (Fig. S13–S15), indicating its largest active surface area and intrinsic activity. In addition to excellent catalytic activity, stability is another crucial factor that determines practical applicability. As shown in Fig. 5c and f, Ir/MoO2-Mo2C-800 demonstrates a smaller shift in LSV curves after 10000 cycles compared with Pt/C, confirming its outstanding long-term stability. Collectively, these experimental results demonstrate that Ir/MoO2-Mo2C-800 exhibits superior HER catalytic activity across the entire pH range, outperforming most previously reported Ir-based catalysts (Fig. 4g, Table S4 and S5). Furthermore, its exceptional stability under pH-universal HER conditions is attributed to the Ir–Mo chemical bonding that effectively prevents catalyst agglomeration.
 | ||
| Fig. 5 (a and b) HER polarization curves and (c and d) Tafel plots of the as-synthesized samples in 0.5 M H2SO4 and in 1.0 M PBS. (e and f) Polarization curves for Ir/MoO2-Mo2C-800 before and after CV potential cycles in 0.5 M H2SO4 and in 1.0 M PBS. (g) Performance comparison between the Ir/MoO2-Mo2C-800 and reported advanced HER catalysts in all-pH hydrogen evolution. | ||
To further investigate the catalytic process of Ir/MoO2-Mo2C-800 in the HER, we examined the possible adsorption sites and evolution of reaction intermediates using in situ attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) and in situ Raman spectroscopy.58 As shown in Fig. 6a and b, the ATR-SEIRAS of Ir/MoO2-Mo2C-800 exhibit peaks at 1642 (1639) cm−1 and a broad band spanning 2900–3700 cm−1, which are attributed to the bending vibration mode (δH–O–H) and stretching vibration mode (νOH) of interfacial water molecules, respectively.59 As the HER potential increases, the intensities of the H–O–H bending and OH stretching peaks of Ir/MoO2-Mo2C-800 gradually strengthen, confirming that its surface facilitates water molecule adsorption and dissociation more effectively.60 Additionally, the signals at 2076 cm−1 (alkaline) and 2103 cm−1 (acidic) can be attributed to the interaction between H* intermediates and Ir (νIr–H*).61 Their intensities gradually increase with more negative applied potentials, indicating significantly enhanced hydrogen adsorption capability.62In situ Raman analysis was employed to reveal the crucial role of interfacial water molecule changes during the catalytic HER process. As shown in Fig. 6c, during the alkaline HER process, a broad peak in the range of 3000–3700 cm−1 can be observed, corresponding to the O–H vibration of water.63,64 This peak can be deconvoluted into three distinct bands, assigned to tetrahedrally coordinated water (ν1), triply coordinated water (ν2), and interfacial water (ν3).65 Among them, ν1 and ν2 represent active water molecules involved in the HER process, while ν3 corresponds to inactive water. Therefore, changes in ν3 can be used to identify surface variations on Ir/MoO2-Mo2C-800. As shown in Fig. 6d and Fig. S16a, as the potential shifts from −1.0 to −1.35 V, the proportion of ν3 decreases from 21.83% to 16.22%. This indicates that the presence of inactive water species diminishes as the potential increases, disrupting the hydrogen-bonded water network within the electric double layer, thereby facilitating subsequent water adsorption and dissociation processes.8,14 During the acidic HER process, the peak at 1052 cm−1 is attributed to the stretching vibration mode of SO42− (Fig. 6e).66 In addition, within the range of 2950–3810 cm−1, the O–H stretching vibration of interfacial water can be fitted into three distinct Gaussian peaks: four-hydrogen-bonded water (4-HB·H2O, ν1), two-hydrogen-bonded water (2-HB·H2O, ν2), and weakly hydrogen-bonded hydronium ions (H·H2O, ν3) (Fig. 6f and S16b).67,68 As the applied potential increases negatively from −1.00 V to −1.04 V, the percentage of ν3 decreases from 22.64% to 18.25%, indicating favorable proton transfer kinetics in the HER for Ir/MoO2-Mo2C-800.69 Overall, the Ir–Mo interfacial effect in Ir/MoO2-Mo2C-800 promotes intermediate generation, thereby accelerating HER kinetics across a wide pH range (Fig. 6g).
 | ||
| Fig. 6 In situ electrochemical ATR-SEIRAS profiles of Ir/MoO2-Mo2C-800 measured in (a) alkaline and (b) acidic solutions. (c) In situ Raman spectra of Ir/MoO2-Mo2C-800 in 1.0 M KOH. (d) The normalized intensities of the three peaks of interfacial water on Ir/MoO2-Mo2C-800 with potentials from −1.0 V to −1.35 V. (e) In situ Raman spectra of Ir/MoO2-Mo2C-800 in 0.5 M H2SO4. (f) The normalized intensities of the three peaks of interfacial water on Ir/MoO2-Mo2C-800 with potentials from −1.0 V to −1.04 V. (g) Proposed H2 evolution mechanism on Ir/MoO2-Mo2C-800 in alkaline and acidic media. | ||
Conclusion
In summary, we successfully constructed MSI by integrating ultrasmall Ir species with nanosheet-assembled hollow nanoflower-like MoO2–Mo2C, endowing the catalyst with outstanding activity and stability across the entire pH range. Comprehensive XAS and XPS spectroscopic analyses demonstrated that the development of MSI between Ir and MoO2–Mo2C effectively modulated the electronic structure of the active Ir centers. Operando ATR-SEIRAS and in situ Raman spectroscopy revealed that the Ir/MoO2-Mo2C-800 catalyst effectively disrupts the hydrogen-bond network of water molecules within the electric double layer, thereby promoting water adsorption and dissociation. Owing to the high-concentration Ir sites and MSI, in alkaline, acidic, and neutral solutions, the Ir/MoO2-Mo2C-800 catalyst demonstrates low overpotentials of 32, 14, and 29 mV at 10 mA cm−2 and high mass activities of 9.23, 60.99, and 8.34 A mg−1PGM, respectively. Furthermore, an AEMWE employed Ir/MoO2-Mo2C-800 as the cathode electrocatalyst and achieved stable operation for over 700/220 h at 0.2/1 A cm−2 while maintaining its initial morphology. This study introduced an effective strategy to boost the efficiency of Ir atom utilization and the stability of Ir-based HER electrocatalysts.Author contributions
The manuscript was written through contributions of all authors.Conflicts of interest
The authors declare no competing financial interest.Data availability
All relevant data are included in the manuscript and its supplementary information (SI). Supplementary information: experimental section, TEM images, XRD patterns, CV curves, Nyquist plots, and tables. See DOI: https://doi.org/10.1039/d5sc08863a.Acknowledgements
This work was supported by the Guizhou Province Department of Education (2021312), National Natural Science Foundation of China (22465009), and Science Foundation for Postgraduate Students of Guizhou Province (2024YJSKYJJ024).Notes and references
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K. C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- Q. Sha, S. Wang, L. Yan, Y. Feng, Z. Zhang, S. Li, X. Guo, T. Li, H. Li, Z. Zhuang, D. Zhou, B. Liu and X. Sun, Nature, 2025, 639, 360–367 CrossRef CAS PubMed.
- A. A. Feidenhans’l, Y. N. Regmi, C. Wei, D. Xia, J. Kibsgaard and L. A. King, Chem. Rev., 2024, 124, 5617–5667 CrossRef PubMed.
- A. H. Shah, Z. Zhang, Z. Huang, S. Wang, G. Zhong, C. Wan, A. N. Alexandrova, Y. Huang and X. Duan, Nat. Catal., 2022, 5, 923–933 CrossRef CAS.
- M. Xu, Y. Kang, L. Wang, Y. Zhang, G. Jiang, Y. Cai, Y. Tu, Q. Zhao, J. Chi, W. Song, H. Yu, J. Hu, W. Liu, R. Huang, L. Yu, J. Lu, X. Bao and D. Deng, Joule, 2025, 9, 101968 CrossRef CAS.
- Y. Chen, Y. Liu, L. Li, T. Sakthivel, Z. Guo and Z. Dai, Adv. Funct. Mater., 2024, 34, 2401452 CrossRef CAS.
- X. Deng, K. Kusada, T. Yamamoto, T. Toriyama, Y. Murakami, J.-C. Tseng, H. Yamada, O. Sakata, H. Ashitani, S. Kawaguchi, Y. Kubota and H. Kitagawa, Angew. Chem., Int. Ed., 2025, 64, e202414786 CrossRef CAS PubMed.
- H. Ling, Q. Yuan, T. Sheng and X. Wang, J. Colloid Interface Sci., 2025, 685, 371–381 CrossRef CAS PubMed.
- D. Guo, X.-X. Xue, M. Jiao, J. Liu, T. Wu, X. Ma, D. Lu, R. Zhang, S. Zhang, G. Shao and Z. Zhou, Chem. Sci., 2024, 15, 16281–16290 RSC.
- Z. Yuehuan and Q. Yuan, Chem. Commun., 2024, 60, 7188–7191 RSC.
- J.-H. Baek, S. H. Kweon, H.-J. Noh, D. H. Kweon, J.-M. Seo, S. J. Lee, S. K. Kwak and J.-B. Baek, Adv. Sci., 2025, 12, 2414012 CrossRef CAS PubMed.
- Z. Kou, Y. Liu, W. Cui, B. Yang, Z. Li, R. D. Rodriguez, Q. Zhang, C. L. Dong, X. Sang, L. Lei, T. Zhang and Y. Hou, Energy Environ. Sci., 2024, 17, 1540–1548 RSC.
- I. Ledezma-Yanez, W. D. Z. Wallace, P. Sebastián-Pascual, V. Climent, J. M. Feliu and M. T. M. Koper, Nat. Energy, 2017, 2, 17031 CrossRef CAS.
- Y. Feng, W. Zhu, J. Xu, D. Zhang, Q. Ma, L. Zhao, L. Lin, Q. Su, Y. Wang, Q. Liu, Y. Wei, X. Li, J. Huang, Y. Ye, J. Zhao and B. Wu, ACS Nano, 2025, 19, 7948–7961 CrossRef CAS PubMed.
- S. Kumari, T. Masubuchi, H. S. White, A. Alexandrova, S. L. Anderson and P. Sautet, J. Am. Chem. Soc., 2023, 145, 5834–5845 CrossRef CAS PubMed.
- H.-Q. Cai, Q.-H. Yang, L.-L. Chen, R.-L. Gao, M.-H. Liao, J.-S. Xing, W.-B. Zhou, N. Pu, J. Gu and Y.-C. Huang, Rare Met., 2025, 44, 4701–4711 CrossRef CAS.
- D. Liu, L. Xu, S. Li, A. Xu, Y. Sun, T. Liu, M. Liu, H. Wang, X. Liu, T. Yao and T. Ding, Nano Res., 2024, 17, 6993–7000 CrossRef CAS.
- Z. Gao, A. Li, X. Liu, M. Peng, S. Yu, M. Wang, Y. Ge, C. Li, T. Wang, Z. Wang, W. Zhou and D. Ma, Nature, 2025, 638, 690–696 CrossRef CAS PubMed.
- J. Zhao, R. Urrego-Ortiz, N. Liao, F. Calle-Vallejo and J. Luo, Nat. Commun., 2024, 15, 6391 CrossRef CAS PubMed.
- N. Nie, D. Zhang, Z. Wang, W. Yu, S. Ge, J. Xiong, Y. Gu, B. Yang, J. Lai and L. Wang, Appl. Catal., B, 2022, 318, 121808 CrossRef CAS.
- T.-T. Zhou, K.-Y. Dong, Z. Zheng and Q. Yuan, Rare Met., 2025, 44, 3119–3129 CrossRef CAS.
- J. Liu, L. Chen and X. Liu, ACS Catal., 2024, 14, 1987–2002 CrossRef CAS.
- E. D. Goodman, A. S. Asundi, A. S. Hoffman, K. C. Bustillo, J. F. Stebbins, S. R. Bare, S. F. Bent and M. Cargnello, Adv. Mater., 2021, 33, 2104533 CrossRef CAS PubMed.
- X. Wu, Z. Wang, D. Zhang, Y. Qin, M. Wang, Y. Han, T. Zhan, B. Yang, S. Li, J. Lai and L. Wang, Nat. Commun., 2021, 12, 4018 CrossRef CAS PubMed.
- T. Lu, T. Li, D. Shi, J. Sun, H. Pang, L. Xu, J. Yang and Y. Tang, SmartMat, 2021, 2, 591–602 CrossRef CAS.
- C. Ye, J. Shan, C. Zhu, W. Xu, L. Song, Y. Zhu, Y. Zheng and S.-Z. Qiao, Adv. Energy Mater., 2023, 13, 2302190 CrossRef CAS.
- B. Zhang, J. Wang, G. Liu, C. M. Weiss, D. Liu, Y. Chen, L. Xia, P. Zhou, M. Gao, Y. Liu, J. Chen, Y. Yan, M. Shao, H. Pan and W. Sun, Nat. Catal., 2024, 7, 441–451 CrossRef CAS.
- J. Chen, Z. Hu, Y. Ou, Q. Zhang, X. Qi, L. Gu and T. Liang, J. Mater. Sci. Technol., 2022, 120, 129–138 CrossRef CAS.
- S. Harsha, R. K. Sharma, M. Dierner, C. Baeumer, I. Makhotkin, G. Mul, P. Ghigna, E. Spiecker, J. Will and M. Altomare, Adv. Funct. Mater., 2024, 34, 2403628 CrossRef CAS.
- A. Mosallanezhad, C. Wei, P. Ahmadian Koudakan, Y. Fang, S. Niu, Z. Bian, B. Liu, T. Huang, H. Pan and G. Wang, Appl. Catal., B, 2022, 315, 121534 CrossRef CAS.
- J. Li, Y. Tan, M. Zhang, W. Gou, S. Zhang, Y. Ma, J. Hu and Y. Qu, ACS Energy Lett., 2022, 7, 1330–1337 CrossRef CAS.
- J. Yuan, J. Feng, G. Li, Y. Song, N. Yu, X. Chen, G. He, Y. Tan and M. Ni, Chem. Eng. J., 2025, 514, 163260 CrossRef CAS.
- W. Zhou, Y. Huang, H. Cai, T. Wang, H. Li, C. Zhang, L. Zhao, L. Chen, M. Liao, Z. Tang, K. Chen, J. Gu, W. Gao, Z. Fan and Z. Wen, Nano-Micro Lett., 2025, 17, 296 CrossRef CAS PubMed.
- Y. Zheng, W. Geng, S. Xiao, T. Ma, C. Cheng, Y. Liao, Z. Zeng, S. Li and C. Zhao, Angew. Chem., Int. Ed., 2024, 63, e202406427 CrossRef CAS PubMed.
- R. Li, X. Chen, H. Zhang, Y. Wang, Y. Lv, H. Jiang, B. Guo and X. Feng, Inorg. Chem., 2024, 63, 2282–2288 CrossRef CAS PubMed.
- J. Yuan, J. Zhou, Z. Peng, G. Li, Y. Hou and M. K. H. Leung, J. Mater. Chem. A, 2024, 12, 2383–2390 RSC.
- Y. Xie, S. Xu, A. C. Meng, B. Zheng, Z. Chen, J. M. Tour and J. Lin, Energy Environ. Sci., 2024, 17, 8670–8682 RSC.
- Y. Guo, M. Wang, Q. Zhu, D. Xiao and D. Ma, Nat. Catal., 2022, 5, 766–776 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- R. Chen, S. Chen, L. Wang and D. Wang, Adv. Mater., 2024, 36, 2304713 CrossRef CAS PubMed.
- G. Luo, M. Song, Q. Zhang, L. An, T. Shen, S. Wang, H. Hu, X. Huang and D. Wang, Nano-Micro Lett., 2024, 16, 241 CrossRef PubMed.
- L. Hou, C. Li, H. Jang, M. G. Kim, J. Z. Jiang, J. Cho, S. Liu and X. Liu, Adv. Mater., 2024, 36, 2410039 CrossRef CAS PubMed.
- J. Yang, Y. Shen, Y. Sun, J. Xian, Y. Long and G. Li, Angew. Chem., Int. Ed., 2023, 62, e202302220 CrossRef CAS PubMed.
- L. Peng, M. Liao, X. Zheng, Y. Nie, L. Zhang, M. Wang, R. Xiang, J. Wang, L. Li and Z. Wei, Chem. Sci., 2020, 11, 2487–2493 RSC.
- X. Zhu, M. Fang, B. Yang, M. Zhan, S. Ke, F. Yang, X. Wu, Y. Liu, Z. Huang and X. Min, J. Mater. Chem. A, 2024, 12, 4108–4122 RSC.
- Y. Li, S. Zhang, C. He, H. Yao, C. Guo, W. Wang and Y. Hu, ACS Catal., 2025, 15, 2315–2327 CrossRef CAS.
- P. Kuang, Y. Wang, B. Zhu, F. Xia, C.-W. Tung, J. Wu, H. M. Chen and J. Yu, Adv. Mater., 2021, 33, 2008599 CrossRef CAS PubMed.
- Y. Feng, Z. Li, S. Li, M. Yang, R. Ma and J. Wang, J. Energy Chem., 2022, 66, 493–501 CrossRef CAS.
- X. Liu, Y. Deng, L. Zheng, M. R. Kesama, C. Tang and Y. Zhu, ACS Catal., 2022, 12, 5517–5526 CrossRef CAS.
- S.-W. Yu, S. Kwon, Y. Chen, Z. Xie, X. Lu, K. He, S. Hwang, J. G. Chen, W. A. Goddard III and S. Zhang, Adv. Funct. Mater., 2024, 34, 2402966 CrossRef CAS.
- L.-M. Cao, L.-H. Yu, H.-B. Huang, C.-J. Gao, X. Huang, X.-F. Zhang, X.-H. Zhang, Z.-Y. Du and C.-T. He, Adv. Funct. Mater., 2024, 34, 2411111 CrossRef CAS.
- J. Chen, G. Fu, Y. Tian, X. Li, M. Luo, X. Wei, T. Zhang, T. Gao, C. Chen, S. Chaemchuen, X. Xu, X. Sun, T. Bu, F. Verpoort, J. Wang and Z. Kou, Interdiscip. Mater., 2024, 3, 595–606 CAS.
- M. Duan, T. Shu, J. Li, D. Zhang, L. Y. Gan, K. X. Yao and Q. Yuan, Nano Res., 2023, 16, 8836–8844 CrossRef CAS.
- Z. Wang, B. Xiao, Z. Lin, Y. Xu, Y. Lin, F. Meng, Q. Zhang, L. Gu, B. Fang, S. Guo and W. Zhong, Angew. Chem., Int. Ed., 2021, 60, 23388–23393 CrossRef CAS PubMed.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K.-Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- K.-G. Qu, Z.-F. Chen, L.-H. Wang, H.-B. Li, S.-Y. Zeng, R. Li, L.-J. Meng, H.-Y. Chen and Q.-X. Yao, Rare Met., 2025, 44, 2094–2102 CrossRef CAS.
- Q. Ma and S. Mu, Interdiscip. Mater., 2023, 2, 53–90 Search PubMed.
- T. Parker, Y. Zhang, K. Shevchuk, T. Zhang, V. Khokhar, Y.-H. Kim, G. Kadagishvili, D. Bugallo, M. Tanwar, B. Davis, J. Kim, Z. Fakhraai, Y.-J. Hu, D.-e. Jiang, D. V. Talapin and Y. Gogotsi, ACS Nano, 2025, 19, 22228–22239 CrossRef CAS PubMed.
- Z.-T. Yan, S. Tao, J. Wang, X.-L. Lu and T.-B. Lu, Adv. Mater., 2024, 36, 2411942 CrossRef CAS PubMed.
- W. Wu, K. Zhang, S. Wei, Y. Wang, Z. Zhang, P. Guo and G. Liu, Chem. Eng. J., 2024, 496, 154030 CrossRef CAS.
- T. Senna, N. Ikemiya and M. Ito, J. Electroanal. Chem., 2001, 511, 115–121 CrossRef CAS.
- Y. Zheng, Z. Xing, S. Xiao, D. Ye, Y. Kong, S. Zhang, T. Ma, C. Cheng, S. Li and C. Zhao, Adv. Mater., 2025, 37, 2508994 CrossRef CAS PubMed.
- Y. Zhu, M. Klingenhof, C. Gao, T. Koketsu, G. Weiser, Y. Pi, S. Liu, L. Sui, J. Hou, J. Li, H. Jiang, L. Xu, W.-H. Huang, C.-W. Pao, M. Yang, Z. Hu, P. Strasser and J. Ma, Nat. Commun., 2024, 15, 1447 CrossRef CAS PubMed.
- R. Li, H. Zhao, L. Wang, Q. Zhou, X. Yang, L. Jiang, X. Luo, J. Yu, J. Wei and S. Mu, Chem. Sci., 2025, 16, 4383–4391 RSC.
- J. Zhao, J. Wang, J. Yao, L. Li, D. Chen, G. Li and G. Zhang, Angew. Chem., Int. Ed., 2025, 64, e202505031 CrossRef CAS PubMed.
- H. Chen, Z. Gao, S. Ren, R.-T. Gao, L. Wu and L. Wang, Adv. Energy Mater., 2025, 15, 2403067 CrossRef CAS.
- Y.-H. Wang, S. Zheng, W.-M. Yang, R.-Y. Zhou, Q.-F. He, P. Radjenovic, J.-C. Dong, S. Li, J. Zheng, Z.-L. Yang, G. Attard, F. Pan, Z.-Q. Tian and J.-F. Li, Nature, 2021, 600, 81–85 CrossRef CAS PubMed.
- K. Zhu, H. Yang, G. Guo, Y. Wang, W. Tan, F. Ma, H. Zhang and S. Peng, ACS Catal., 2025, 15, 9563–9573 CrossRef CAS.
- M.-R. Qu, Y.-X. Cheng, S.-H. Feng, J. Xu, J.-K. Yao, W.-S. Yan, S. Zhu, L. Cao, R. Wu and S.-H. Yu, Energy Environ. Sci., 2025, 18, 5985–5997 RSC.
| This journal is © The Royal Society of Chemistry 2026 |