
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic asymmetric P(III)-additions to salicylaldehydes enable divergent stereoselective dearomatizations of phenols
Aidan J.
Clarkson†
 ,
Kimberly A.
Alley†
,
Bryn K.
Werley
,
Jacob G.
Robins
,
Shubin
Liu
and
Jeffrey S.
Johnson
*
,
Kimberly A.
Alley†
,
Bryn K.
Werley
,
Jacob G.
Robins
,
Shubin
Liu
and
Jeffrey S.
Johnson
*
Department of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599-3290, USA. E-mail: jsj@unc.edu
First published on 15th December 2025
Abstract
Oxidative dearomatizations are useful tools in complex molecule synthesis and the development of enantioselective variants offers substantial opportunities for the field. In this article, we describe a platform for a formal enantioselective dearomatization that employs chiral nonracemic α-hydroxyphosphonates derived from salicylaldehydes to direct diastereoselective functionalization of the phenolic core. As a requisite first step, we developed a new bis(oxazoline)Cu(II)-catalyzed hydrophosphonylation (Pudovik reaction) of salicylaldehydes, allowing access to enantioenriched material in one step. The derived α-hydroxyphosphonates participated in complementary stereoselective dearomatization sequences. A tandem phosphono-Adler–Becker oxidation/[4 + 2]-cycloaddition furnished bicyclo[2.2.2]-octanes bearing phosphonoepoxyketone functionalities in excellent regio- and diastereoselectivity for both steps. Alternatively, oxidation with phenyliodine(III) diacetate (PIDA) provided access to either 2,5- or 2,4-cyclohexadienones, with the latter reacting in a complementary [4 + 2]-cycloaddition. Experimental and computational studies of the two paths reveal distinct modes of stereochemical relay from the α-hydroxyphosphonate adducts to the derived dearomatized Diels–Alder products. The removable α-hydroxyphosphonate and phosphonoepoxide functionalities enable downstream synthetic modifications to the stereochemically defined cycloadducts.
Introduction
Enantioselective phenol dearomatization is a desirable tool in small molecule synthesis, providing access to the latent functionality within the nucleus of readily available hydroxyarene feedstocks.1 This enabling paradigm reveals useful reactive nonaromatic building blocks (Scheme 1A), but it can carry limitations associated with electronic constraints of the substrate and catalyst (i), inherent characteristics of the reaction mechanism (ii),2 and the challenges associated with overcoming aromatic stabilization energy.3 Current methods for enantioselective phenol dearomatization1,4 invoke the use of reagents such as hypervalent iodine,5–8 enzymes,9 transition metals,10,11 or chiral phase transfer catalysts,12 with varying degrees of success and efficiency. Among these examples, naphthol dearomatizations are comparatively more common, although highly enantioselective catalytic phenol spirolactonizations6,7 serve to demonstrate the functional group tolerance of chiral hypervalent iodine oxidations and the promise of the approach. Advances in enantioselective dearomative fluorination reactions of both naphthols and phenols reveal the continual progression of this field.13–15 These examples simultaneously allude to the extant challenges in developing complementary enantioselective oxidative dearomatization reactions with the same degree of selectivity and generality. | ||
| Scheme 1 Proposed reaction platform for enantioselective dearomatization of salicylaldehydes. | ||
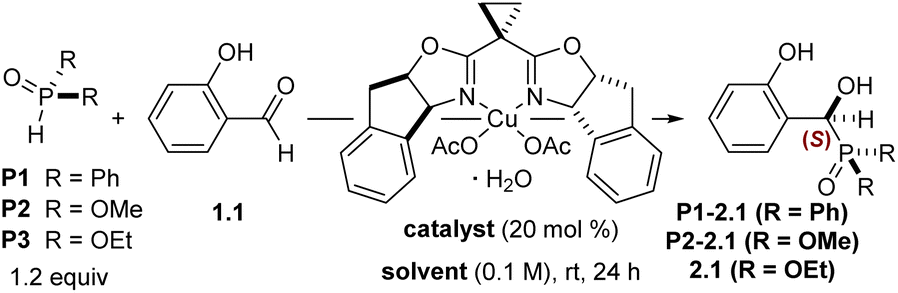
Within the scope of readily available phenols, salicylaldehydes might possess a unique opportunity for stereodifferentiating dearomatization reactions.16 We hypothesized that heretofore unknown enantioselective aldehyde additions could be leveraged as a source of absolute stereochemical information to be relayed through a diastereoselective dearomatization/cycloaddition sequence to access value-added complex cyclohexanes (Scheme 1B). A key facet of this approach is the decoupling of the asymmetric catalysis (I → II, step 1) and dearomatization events (II → III/IV, step 2), which we anticipated would confer greater synthetic flexibility by permitting variable oxidation conditions to access distinct non-aromatic products (III and IV). Strategically, the source of chirality would ideally serve as a versatile functional handle for downstream transformations and thwart unwanted reactivity at the carbonyl – protecting it – until later in the sequence.
With these design criteria in mind, the use of α-hydroxyphosphonates was considered.17–28 Their stereocontrolled synthesis is usually accomplished through the addition of a tri- or pentavalent phosphorus compound to an aldehyde (i.e., the Pudovik reaction),29 which has been shown to be reversible under alkaline conditions.21,25,30,31 We sought to employ chiral nonracemic α-hydroxyphosphonates (II) derived from salicylaldehydes (I) as a point of entry to value-added complex cyclohexanes (V and VI) via oxidative dearomatizations of salicyl alcohol derivatives. Furthermore, we hoped to expand the possible product diversity by employing either a dearomative epoxidation (Adler–Becker oxidation) or an oxidation with phenyliodine(III) diacetate (PIDA).32,33 While the latter has been demonstrated enantioselectively,6,7 there are no known examples of an enantioselective variant of the former.2 This point is further emphasized by a recent report that demonstrates both the challenges and potential utility of the Adler–Becker reaction in complex molecule synthesis.34 We envisioned that this integrated strategy might enable access to enriched dearomatization products through either oxidation pathway, relying instead on the ability of the nascent stereogenic hydroxyphosphonate to induce diastereoselectivity in the dearomatization/cycloaddition cascade. Literature precedent13,35–39 suggested the possibility of applying related tandem oxidative dearomatization/Diels–Alder reactions to trap the resulting dienones (Scheme 1B, step 3); however, this work would have several key distinctions. First, this method would provide access to cycloadducts in enantioenriched fashion, which has not been achieved previously using Adler–Becker oxidation chemistry. Second, we anticipated that slow or reversible diene dimerization might allow for a greater scope of employable dienophiles relative to the prior art. Lastly, the phosphonate functionality would serve as a functional handle for enhanced synthetic utility.
Several potential reactivity and stereochemical challenges emerged upon consideration this experimental plan. First, there are scant reports for the synthesis of racemic salicyl α-hydroxyphosphonates40,41 and no extant methods for accessing enantioenriched variants, a circumstance that stands in contrast to the plethora of reported reactions for other benzaldehydes.42–49 Enantioselective nucleophilic additions of other nucleophiles to salicylaldehydes are known,50–55 but they are usually illustrated as single examples within a scope of benzaldehydes. One reason for this rarity may be the acidic nature of the phenolic proton, which results in deactivation of the electrophile upon deprotonation by a basic catalyst. Second, the phosphonate group's impact on the efficiency and selectivity of the oxidative dearomatization and subsequent dienone functionalization was an open question. Adler–Becker reactions in which the aryl-benzylic C–C bonds experience free rotation are rare and exhibit variable stereocontrol.56–58 Lastly, we suspected that the presence of the phosphonate might affect the well-established propensity of cyclohexadienones to dimerize by [4 + 2]-cycloaddition.1,11,59–62
Herein, we describe an integrated approach to stereoselective dearomatization reliant on the development of a new enantioselective Cu(II)-catalyzed addition of P(III) nucleophiles to salicylaldehydes (Scheme 1C). The derived α-hydroxyphosphonates can be processed by distinct oxidative dearomatization paths to give dienone intermediates that participate in diastereoselective cycloadditions operating under divergent stereocontrol mechanisms; consequently, Pudovik adducts are convertible to chiral building blocks with enantiomeric carbocyclic core structures and complementary peripheral functionality based on the oxidation path selected.
Results and discussion
Development of a copper-catalyzed hydrophosphonylation of salicylaldehydes
Our initial efforts began with investigating the asymmetric Pudovik reaction using a bis(oxazoline)Cu(II) (box) complex. Work by Evans and coworkers demonstrated that a (box)Cu(II) complex bearing mildly basic acetate ligands promotes the asymmetric nitroaldol reaction of aldehydes.63 Based on the similar pKa values of dialkyl phosphites and nitroalkanes,64,65 we speculated that a similar system might successfully promote the title reaction; however, salicylaldehyde is absent from the Evans work, making a direct comparison difficult. Direct application of literature conditions from a catalytic hydrophosphonylation of aryl and styrenyl aldehydes48 to our desired substrate (salicylaldehyde 1.1) highlighted the unique challenges posed by salicylaldehydes, affording the desired product in 25% yield (by 1H NMR spectroscopy) and 51![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :49 er. Further experiments revealed that 20 mol% of an in situ prepared inda(box)Cu(II) complex (Table 1) catalyzed nucleophilic addition of diphenylphosphine oxide to salicylaldehyde 1.1 in good yield but poor enantioselectivity (entry 1). Using dimethyl phosphite and switching from THF to MeOH as the solvent produced a marked improvement in enantioselectivity (entry 2). Using diethyl phosphite, the reaction proceeded with moderate selectivity in various solvents (entries 3–6), with methanol providing the α-hydroxyphosphonate 2.1 in 47% NMR yield and 87:13 er (entry 7). The use of diisopropyl phosphite was also evaluated but showed decreased yield with respect to diethyl phosphite (not shown). Several other box, py(box), and inda(box) ligands were screened (see SI for additional optimization details), but no other ligand further improved the observed enantioselectivities. A solvent screen revealed that protic solvents gave markedly higher rates of conversion and selectivities, with isopropanol and ethanol outperforming other branched and straight-chain alcohols. This trend in coordinating solvents is in agreement with other (box)Cu(II)-catalyzed aldehyde additions.66 Lowering the reaction temperature to 0 °C reduced conversion and yield by 10% and had no impact on the enantioselectivity. Increasing the reaction temperature or extending the reaction times also did not produce a significant change to the enantioselectivity. The catalyst loading could be reduced to 5 mol% with no impact on enantioselectivity; however, increased catalyst loadings enabled full conversion of starting material while only requiring 1.5 equivalents of phosphite at 0.5 M (entry 9). Ultimately, isopropanol was used instead of ethanol due to increased conversion and consistency across all substrates. Small amounts of dichloromethane as a cosolvent were found to promote more complete conversion of starting material with negligible effect on enantioselectivity (entries 11 and 12).
:49 er. Further experiments revealed that 20 mol% of an in situ prepared inda(box)Cu(II) complex (Table 1) catalyzed nucleophilic addition of diphenylphosphine oxide to salicylaldehyde 1.1 in good yield but poor enantioselectivity (entry 1). Using dimethyl phosphite and switching from THF to MeOH as the solvent produced a marked improvement in enantioselectivity (entry 2). Using diethyl phosphite, the reaction proceeded with moderate selectivity in various solvents (entries 3–6), with methanol providing the α-hydroxyphosphonate 2.1 in 47% NMR yield and 87:13 er (entry 7). The use of diisopropyl phosphite was also evaluated but showed decreased yield with respect to diethyl phosphite (not shown). Several other box, py(box), and inda(box) ligands were screened (see SI for additional optimization details), but no other ligand further improved the observed enantioselectivities. A solvent screen revealed that protic solvents gave markedly higher rates of conversion and selectivities, with isopropanol and ethanol outperforming other branched and straight-chain alcohols. This trend in coordinating solvents is in agreement with other (box)Cu(II)-catalyzed aldehyde additions.66 Lowering the reaction temperature to 0 °C reduced conversion and yield by 10% and had no impact on the enantioselectivity. Increasing the reaction temperature or extending the reaction times also did not produce a significant change to the enantioselectivity. The catalyst loading could be reduced to 5 mol% with no impact on enantioselectivity; however, increased catalyst loadings enabled full conversion of starting material while only requiring 1.5 equivalents of phosphite at 0.5 M (entry 9). Ultimately, isopropanol was used instead of ethanol due to increased conversion and consistency across all substrates. Small amounts of dichloromethane as a cosolvent were found to promote more complete conversion of starting material with negligible effect on enantioselectivity (entries 11 and 12).
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Phosphite | Yielda (%) | erb |
| a Yields determined by 1H NMR spectroscopic analysis using phenanthrene as an internal standard. b Enantiomeric ratio determined by HPLC analysis. c Reaction performed at 0.5 M using 1.5 equiv. phosphite. d Reaction performed at 0.25 M using 1.5 equiv. phosphite. | ||||
| 1 | THF | P1 | 72 | 65:35 |
| 2 | MeOH | P2 | n.d. | 81:19 |
| 3 | THF | P3 | 63 | 82:18 |
| 4 | MeCN | P3 | 21 | 81:19 |
| 5 | DCM | P3 | 10 | 81:19 |
| 6 | Et2O | P3 | 42 | 81:19 |
| 7 | MeOH | P3 | 47 | 87:13 |
| 8 | EtOH | P3 | 58 | 90:10 |
| 9c | EtOH | P3 | 95 | 88:12 |
| 10d | iPrOH | P3 | 86 | 88:12 |
| 11d | CH2Cl2/iPrOH (1:10) |
P3 | 90 | 90:10 |
| 12 |
CH
2
Cl
2
/
i
PrOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :7) :7)
|
P3 | 97 |
90:10
|
The system tolerated both electron-poor and electron-rich arenes as well as various substitution patterns on the arene, providing α-hydroxyphosphonates in moderate to good yields with enantiomeric ratios equal to or exceeding 87:13, with most products equal to or exceeding 90:10 er (Table 2). The enantiomeric ratio of α-hydroxyphosphonate 2.11 was further upgraded by recrystallization from dichloromethane, affording the desired product as a single observable enantiomer by chiral HPLC. The reactions could be scaled up to at least 5.0 mmol scale without detriment to the yield or enantioselectivity of the transformation. Arenes with substitution at the 6-position proved challenging in this system (2.16–2.18), likely due to steric crowding during phosphite addition.
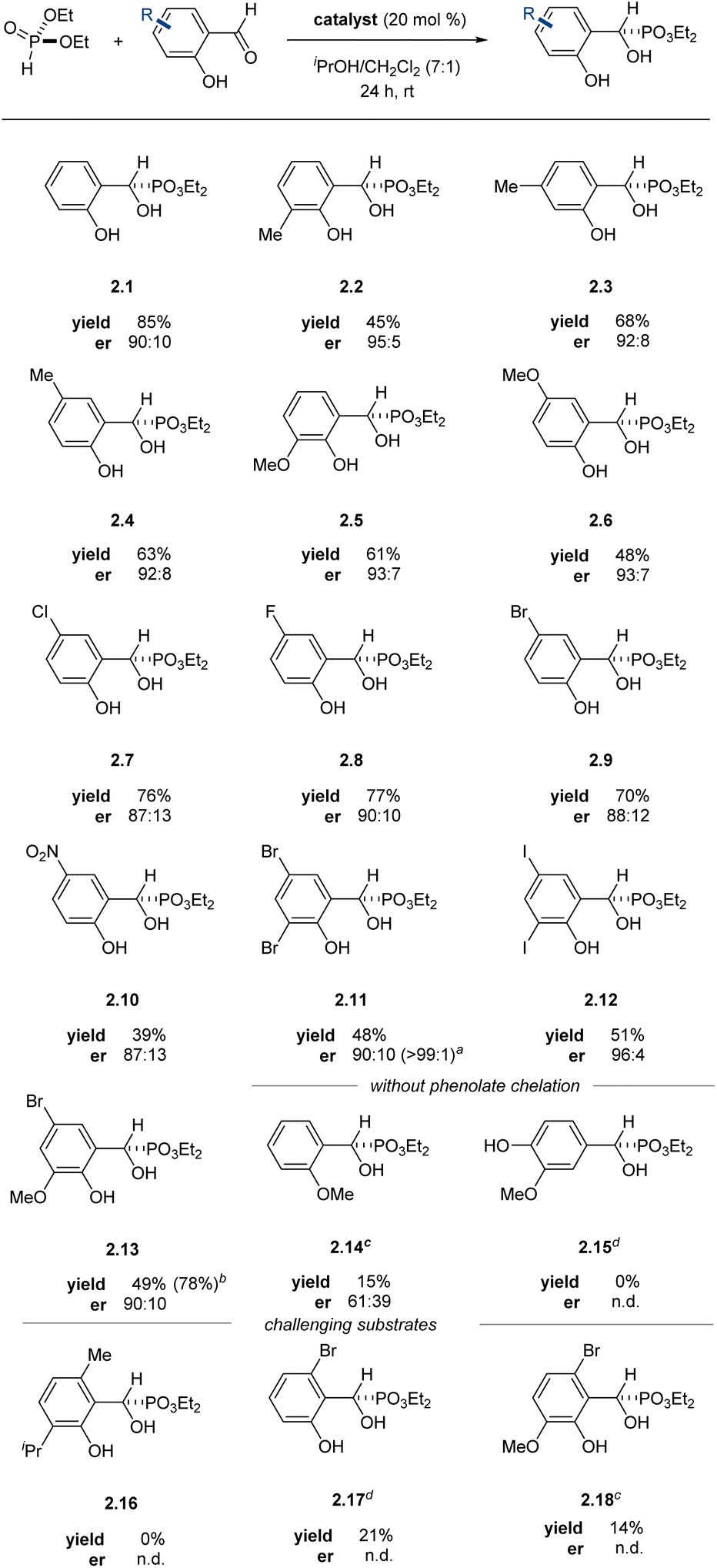
The configuration of the new asymmetric center was established as (S) through an X-ray diffraction study of a derivative (vide infra). This stereochemical outcome constitutes a reversal in aldehyde enantiotopic facial preference relative to prior art,63 indicating a probable change in stereocontrol mechanism. The unique nature of the salicylaldehyde electrophile emerged as the most probable source of this stereochemical reversal. As a point of departure, we formulated a simple mechanism for evaluation. Twofold X-ligand exchange in the (indabox)Cu(OAc)2 complex with one equivalent each of salicylaldehyde and diethyl phosphite was proposed to liberate HOAc (2 equiv.) and create the reactive ternary complex (indabox)Cu-A (Fig. 1). Maximal LUMO-lowering Lewis acid activation of the carbonyl should occur in the ligand plane and optimal HOMO-raising activation of the nucleophilic phosphite should be realized in the apical site, consistent with precedent and established trends with Cu(II).67 When the (indabox)Cu(OAc)2 complex was treated with 3,5-dibromosalicylaldehyde (1.11, 2 equiv.), the crystalline bis(phenolate) octahedral complex (indabox)Cu(1.11)2 was obtained through X-ligand exchange (Fig. 1). As expected, based on Jahn–Teller considerations,63,67 the phenolate ligands lie in the ligand plane while the chelating aldehydes reside in apical sites. Admixture of this complex with diethyl phosphite led to the Pudovik adduct 2.11. The direct outer sphere addition of phosphite to (indabox)Cu(1.11)2 was deemed unlikely: the re-face of the aldehydes is more exposed for nucleophilic attack in this complex, which would lead to the minor (R)-product.
 | ||
| Fig. 1 Simplified mechanistic scheme and a test of ligand exchange. a Yield determined by 1H NMR spectroscopic analysis using mesitylene as an internal standard. | ||
Carbonyl additions via the ternary complexes were evaluated using density functional theory at the level of M062X approximate functional and a compound Pople basis set in chloroform solvent (Fig. 2, see the SI for additional details). Visual inspection suggested differences in steric crowding for phosphite approach in the cyclic diastereomeric transition states, and the impact of these interactions was reflected in the lower calculated barrier for addition to the si-face (ΔΔG‡ = 1.6 kcal mol−1). The analysis is predicated on a pre-equilibrium isomerization away from the more stable geometric isomer into one that is more reactive, intersecting with a more typical model of in-plane coordination of chelating substrates for Cu(II).68 The absolute barrier for the P–C bond forming step is quite low (8.6 kcal mol−1) despite the fact that nucleophilic addition to an anionic (X-type) ligand is obligatory.
 | ||
| Fig. 2 Density functional theory-(DFT) calculated transition states for enantioselectivity-determining step of the of Pudovik reaction. | ||
To examine if the product could be acting as an additional stereodifferentiating ligand, we simulated the reaction at 50% conversion in the presence of freshly prepared catalyst (at the same concentration as our optimal conditions) using enantioenriched 2.1. The reaction proceeds to completion with no effect on stereoselectivity, indicating that the presence of enantioenriched product does not influence the stereoselectivity-determining step. The ortho-phenol proved critical for the title reaction to proceed: poor yield and enantioselectivity was observed with 2-methoxybenzaldehyde and 0% yield with p-vanillin (2.14 and 2.15). These observations further underscore the importance of covalent substrate activation.
Relaying chirality through a tandem dearomatization/cycloaddition sequence
With a developed method to access enantioenriched phenolic α-hydroxyphosphonates, we began investigating the behavior of these products in oxidative dearomatizations. Adaptation of Adler–Becker reactions conditions from a previous report from our lab36 enabled a one-pot oxidative dearomatization/acylnitroso Diels–Alder cycloaddition; however, competitive oxidation of the α-hydroxyphosphonate to the putative corresponding acylphosphonate could not be suppressed. With this initial success, we examined the scope of suitable dienophiles and arenes that could be tolerated in this one-pot transformation. Pudovik adduct 2.2 performed well under optimized reaction conditions, furnishing maleimide adduct 4.1 in 78% yield and 11:1 dr (Table 3). As anticipated by our hypothesis, the stereochemical information imparted by the asymmetric Pudovik reaction was extended in the Adler–Becker oxidation to the adjacent spirocyclic center and the ensuing cycloaddition occurred diastereoselectively. The relative stereochemistry of the cycloaddition was confirmed by X-ray crystallographic analysis of cycloadduct 4.1, with the Adler–Becker oxidation occurring stereoselectively and the subsequent cycloaddition occurring exclusively syn to the epoxide oxygen.69
| a The reaction was performed at 0.03 M. |
|---|

|
Efforts to expand the scope to higher LUMO dienophiles were initially met with a few challenges. In addition to competing with oxidation to the achiral acyl phosphonate, dienone dimerization was competitive with the desired [4 + 2]-cycloaddition. Under standard reaction conditions, dimer 4.7 was isolated in 70% yield in a double diastereoselective cycloaddition. We hypothesized that this dimerization may be reversible at higher temperatures while the desired cycloaddition may provide a thermodynamic sink to sequester the target cycloadducts.70 Indeed, subjecting the spiroepoxydienone dimer to 10 equiv. of dienophile at 85 °C produced the desired Diels–Alder adducts with excellent conversion and high regio- and diastereoselectivities. Oxidation occurred in under 3 h at 0–23 °C, at which point a dienophile could be added and the temperature raised to 85 °C to promote monomerization and the desired [4 + 2]-cycloaddition. Under these modified conditions, both normal and inverse-electron demand Diels–Alder cycloadducts were accessed in good yields and selectivities. Performing the reaction between 0 and 23 °C enabled access to acylnitroso Diels–Alder adduct 4.8 in 49% yield as a single diastereomer and regioisomer. Maximum efficiency in the nitroso Diels–Alder reaction was achieved by dropwise addition of a hydroxycarbamate solution to a chilled mixture of starting material with excess sodium periodate, establishing an excess of diene relative to the active nitroso species, which has a very short lifetime.36,71,72 While alkyl-substituted arenes were well-suited in this platform, unproductive reactivity was observed when using either of the methoxy-substituted Pudovik adducts (2.5 and 2.6), presumably due to formation of the corresponding quinone species (e.g. quinone (±)-5, Scheme 2). Brominated arenes 2.9 and 2.11 also underwent oxidation and cycloaddition; however, bromide displacement during oxidation led to complex reaction mixtures and incomplete conversion. Despite these substrate constraints, this method addresses the major limitations of our initial report,36 providing comparable products in enantioenriched fashion and demonstrating an expansive scope of suitable dienophiles for this dearomative manifold.
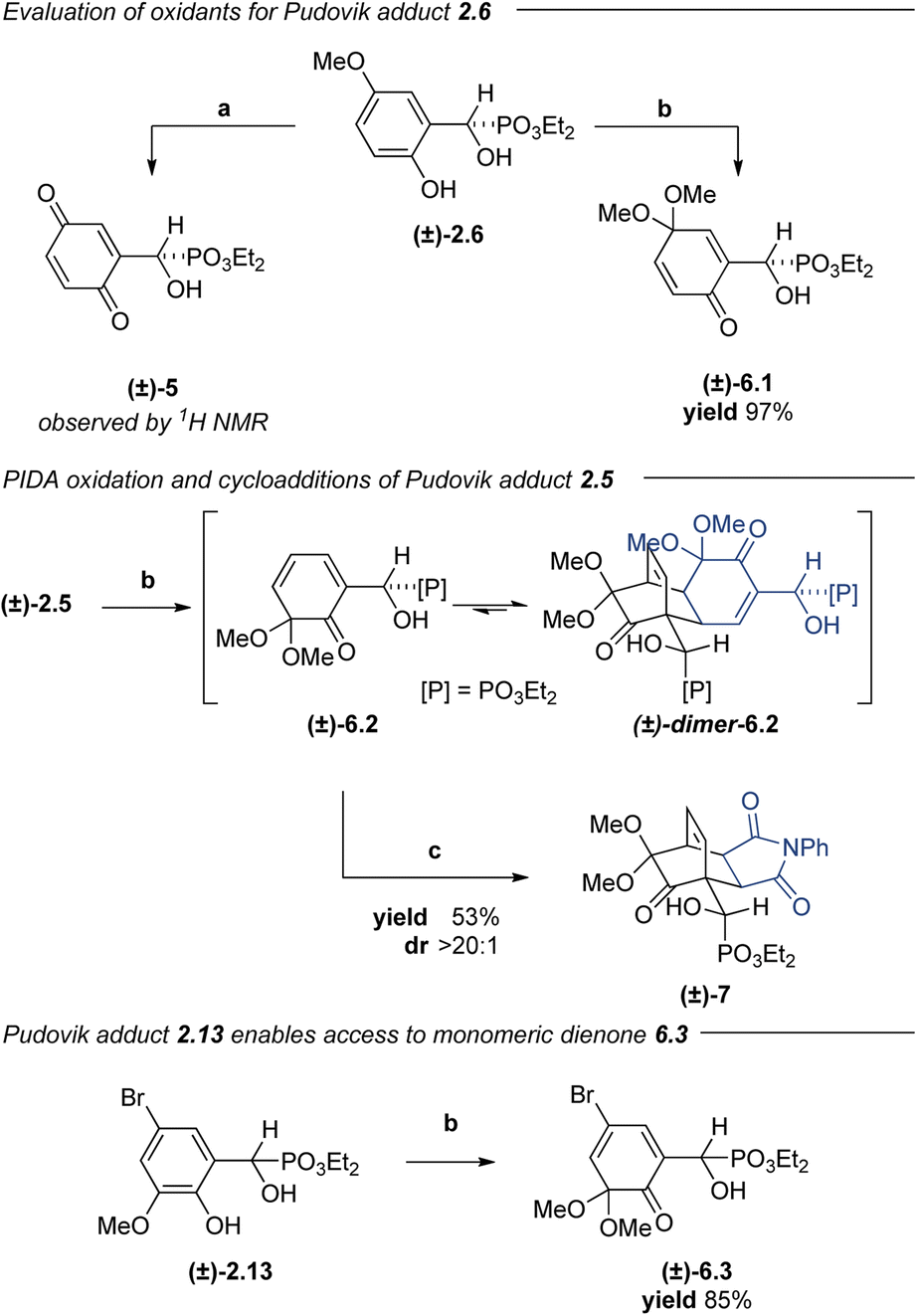 | ||
| Scheme 2 Exploration of PIDA as alternative phenol oxidant. (a) NaIO4, DCE:H2O (2.5:1), TEBACl, 0 to 23 °C, 3 h. (b) PIDA, MeOH, 0 to 23 °C, 20 min. (c) N-Phenylmaleimide, CH2Cl2, 150 °C, 3 h. | ||
In complementary investigations, α-hydroxyphosphonate (±)-2.1 underwent dearomative oxidation by PIDA in methanol to give 2,5-cyclohexadienone (±)-6.1 in 71% yield (not shown). Under similar conditions, α-hydroxyphosphonate 2.6 could also be dearomatized to give dienone (±)-6.1 in 97% yield. Suspecting that the regioselectivity of this oxidation might be heavily influenced by arene substitution patterns,35 we subjected α-hydroxyphosphonate (±)-2.5 to the same conditions to afford dimerized (±)-6.2. We anticipated that dienone (±)-6.2 might behave similarly to the spiroepoxydienones with respect to cycloaddition reactivity. Indeed, heating of dimeric (±)-6.2 at 150 °C in the presence of N-phenylmaleimide provided the desired cycloadduct (±)-7 in 53% yield as a single diastereomer after purification. While this experiment demonstrated the propensity of these substrates to undergo both a reversible dimerization and a diastereoselective [4 + 2]-cycloaddition, we desired a platform that would enable cycloadditions to proceed under milder conditions.
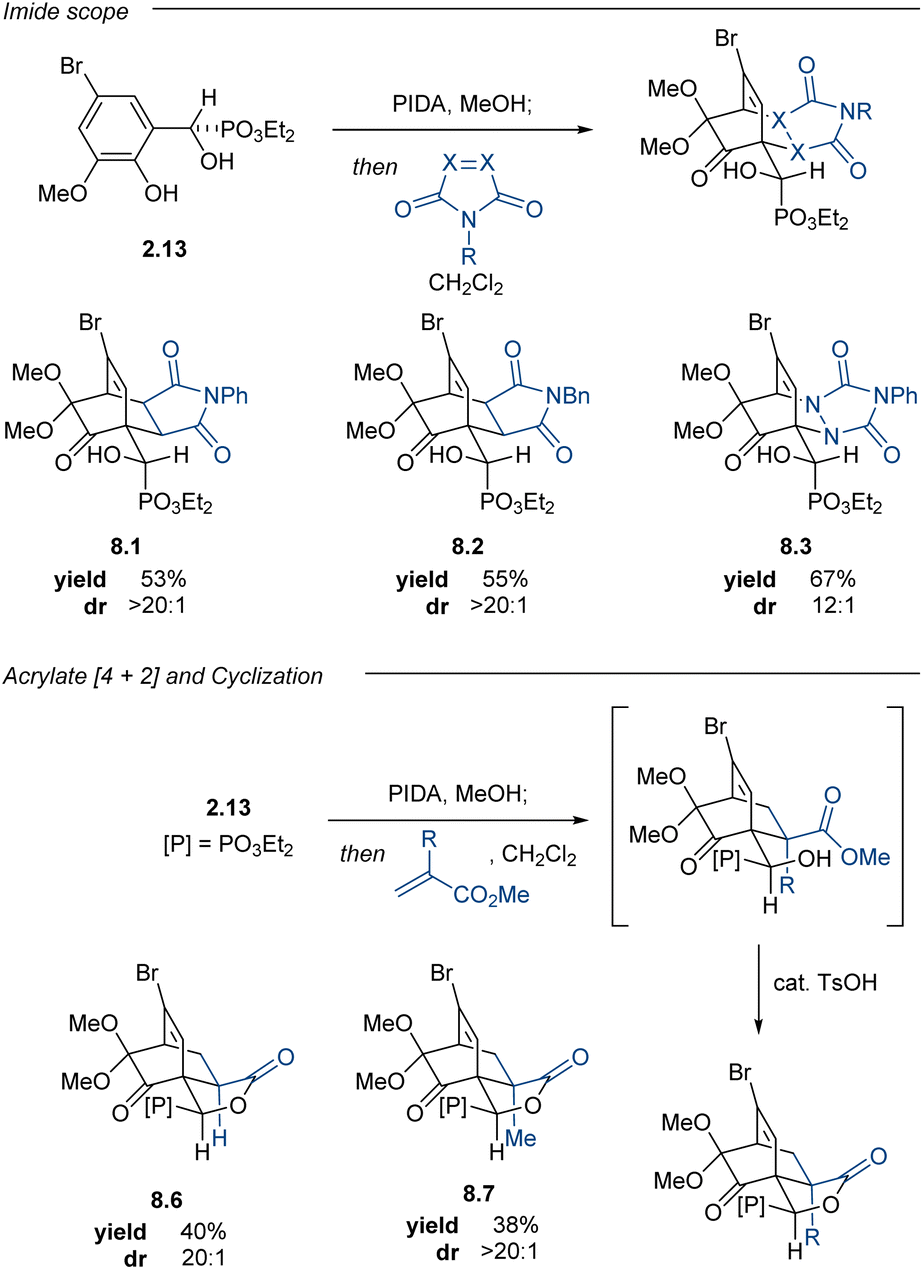
2-Methoxy-4-substituted phenols oxidize to the corresponding 2,4-cyclohexadienones and dimerize much more slowly than the corresponding 4-unsubstituted phenols.73 Aryl α-hydroxyphosphonate (±)-2.13 was subjected to PIDA oxidation conditions to afford ortho-quinone monoketal (±)-6.3 in 85% yield. Dimerization of this compound was slow in the solution phase and could be mitigated by immediate isolation to give monoketal (±)-6.3 as a crystalline solid. Subsequent [4 + 2]-cycloadditions with normal-electron demand dienophiles proceeded well, furnishing cycloadducts (±)-8.1–8.3 in good yields and good to excellent diastereoselectivities (Table 4). Given the promising preliminary data using α-hydroxyphosphonate (±)-6.3 with the maleimide and PTAD dienophiles, we screened a nitroso carbamate dienophile and acrylonitrile ((±)-8.4 and (±)-8.5); however, these cycloadditions proved to be poorly diastereoselective and regioselective, giving complex mixtures that could not be purified further. Cycloadditions with methyl acrylate and methyl methacrylate proceeded smoothly, affording a mixture of the predicted cycloadduct and the corresponding lactonized products (±)-8.6 and (±)-8.7. The addition of catalytic quantities of p-toluenesulfonic acid (TsOH) to the reaction mixture converted the intermediate hydroxy esters to the lactones, which were isolated in 61% and 66% yields, respectively.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Y.
Y.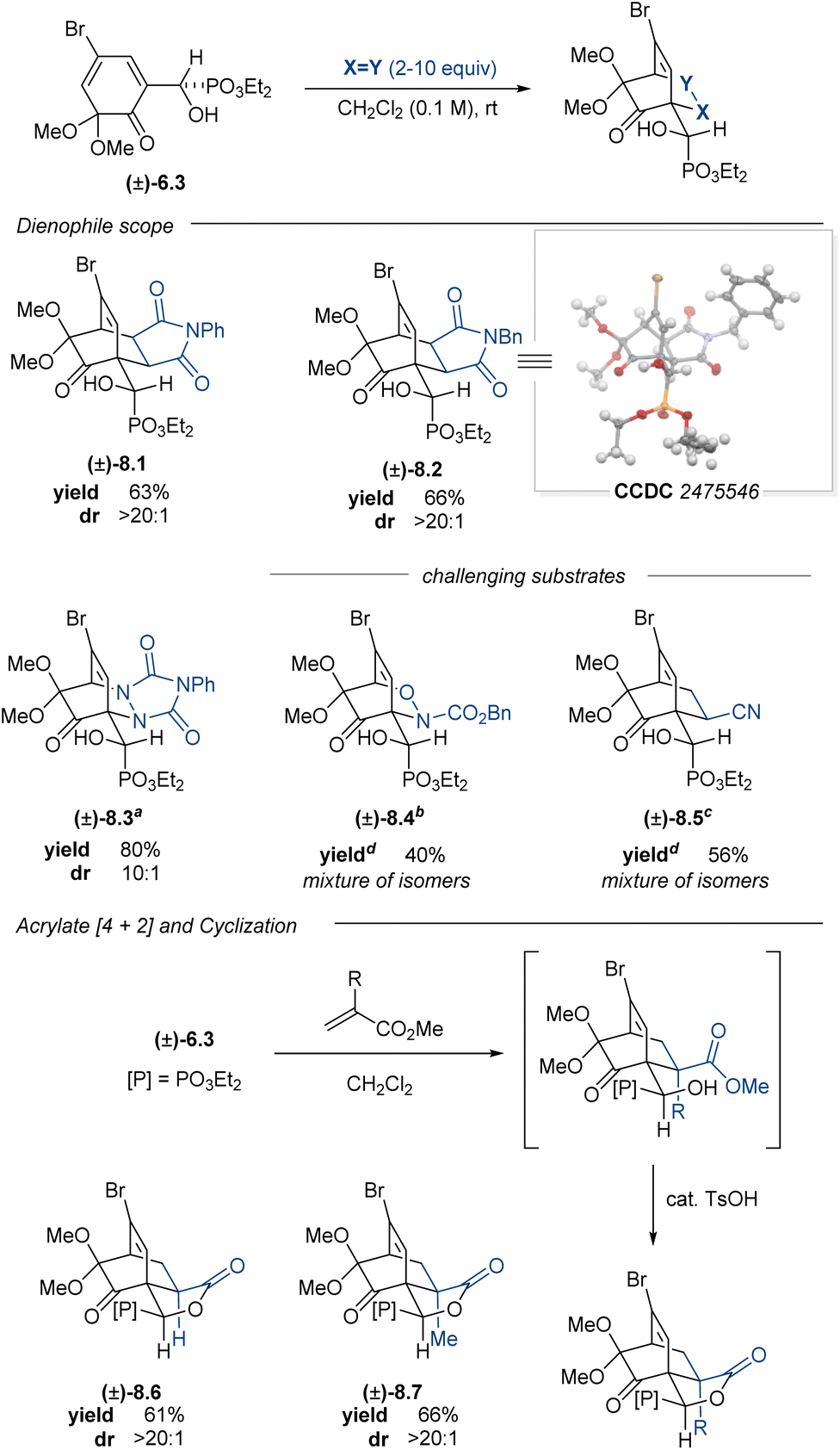
In contrast to quinone monoketal (±)-6.3, the enantioenriched dienone (S)-6.3 did not readily crystallize, even upon purification. The corresponding oil dimerized upon standing at room temperature; however, this challenge was addressed by devising a one-pot oxidative dearomatization/[4 + 2]-cycloaddition sequence. This strategy enabled the synthesis of enantioenriched cycloadducts in good yields and moderate to excellent diastereoselectivities (Table 5). This one-pot process enabled access to enantioenriched cycloadducts 8.1–8.3, all with comparable yields to those reactions performed in two steps with the racemic material. The acrylate cycloadditions also proceeded smoothly in one-pot fashion, providing lactones 8.6 and 8.7 as single diastereomers in synthetically useful yields upon subjecting the crude reaction to catalytic quantities of TsOH. The enantioenrichment of the cycloadduct 8.7 matched that of the starting material, confirming that no racemization occurred during the oxidation or cycloaddition. This PIDA oxidation/[4 + 2]-cycloaddition sequence offers a complementary strategy to access cycloadducts bearing enantiomeric carbocyclic cores relative to the products from the Adler–Becker chemistry.
|
Complementary oxidation pathways reveal distinct mechanisms of stereoinduction
The influence of the stereogenic center established during the enantioselective phosphite/salicylaldehyde addition manifests itself in different ways depending on the chosen oxidant. For the Adler–Becker oxidation, the stereochemical outcome can be understood in stages (Fig. 3a). First, the cyclic I(VII) intermediate II-NaIO4 can lead to diastereomeric products depending on the illustrated gray or blue 90° bond rotation between the arene-benzylic C–C bond during the oxidative cyclization that extrudes the reduced I(V) fragment. The stereochemical relay to the ipso-carbon is highly selective, possibly due to a preference for the trans-phosphono epoxy ketone in the favored path.74,75 In the second stage, the diastereotopic faces of III are sterically differentiated in a manner that precludes reagent approach syn to the phosphonate, thereby completing the stereochemical relay from the ipso-carbon to the asymmetric centers established in the cycloaddition. | ||
| Fig. 3 (a) Steric constraints imposed by the rigid dialkoxyphosphinyl epoxide govern the facial selectivity of the cycloaddition. (b) Hydrogen-bonding between the α-hydroxyphosphonate and the dienophile directs facial selectivity of the cycloaddition. | ||
The diastereofacial preference exhibited by hydroxyphosphonate dienones was ascertained by X-ray crystallographic analysis of cycloadduct (±)-8.2. Computational investigation suggested that hydrogen bonding between the α-hydroxyphosphonate and the incoming dienophile dictated the facial selectivity of the cycloaddition (Fig. 3b, directed model). Substrate pre-organization through internal chelation (Fig. 3b, internal chelate model) does not figure prominently as a stereocontrol feature (ΔΔG‡ = 0.5–3.0 kcal mol−1, see Table S4 for more details). Exocyclic hydroxyl-directed cycloadditions have been reported;76,77 however, this mode of selectivity generally requires a metal chelating agent to strengthen the interaction between the exocyclic alkoxy group and the incoming dienophile to achieve high selectivity.78–80 A previous study from our own laboratories also suggests the hydrogen bonding through an exocyclic hydroxyl group can influence diastereoselectivity; however, the exact consequences of additional substitution appear system-dependent.37
The hydrogen-bonding hypothesis is further corroborated in this work by the decreased selectivity observed with acrylonitrile adduct (±)-8.5, since this dienophile is unlikely to participate in the same degree of hydrogen bonding in the cycloaddition transition state as those dienophiles in the maleimide and acrylate cycloadditions. We hypothesize that the α-hydroxyphosphonate acts as a strong directing group due to enhanced acidity and hydrogen bond donor capability of the hydroxyl proton relative to that of an aliphatic or benzylic alcohol.
Leveraging the α-hydroxyphosphonate as a functional handle
To demonstrate the practicality of the described enantioselective dearomatization strategy, we desired a method that would leverage the α-hydroxyphosphonate as a diversifiable functional group. There exist several literature precedents for both the reductive epoxide opening and phosphonate expulsion tranformations;31,81 however, single-electron reductants including Cp2TiCl and SmI2 proved inadequate for promoting keto epoxide opening.82 Instead, Zn/AcOH proved effective in opening epoxide 4.1 to the corresponding α-hydroxyphosphonate 10 in excellent yield and modest diastereoselectivity.83 While the diastereomers were separable by chromatography, the selectivity proved inconsequential for phosphite removal in the subsequent step. The reported alkaline retro-Pudovik conditions21,31 resulted in elimination of water and formation of a vinyl phosphonate species. To circumvent this competitive elimination, we developed an alternative procedure wherein deprotonation of intermediate 10 with excess LiHMDS and trapping of the α-alkoxyphosphonate with 1.2 equivalents of TMSCl provided the corresponding β-keto aldehyde 11 in 64% yield (Scheme 3), which existed predominantly as the enol tautomer. In contrast, the more sterically congested cycloadduct 8.1 could be oxidized to the corresponding acyl phosphonate 12, which proved unexpectedly recalcitrant to nucleophilic acyl substitution.28 To address that challenge, we developed a Cu(II)-catalyzed esterification that proceeded to afford methyl ester 13 in 51% yield. | ||
| Scheme 3 Phosphite removal through reductive and oxidative paths. (a) Zn/AcOH, MeOH; (b) LiHMDS, TMSCl; then TFA, EtOH; (c) DMP, CH2Cl2; (d) Cu(OTf)2, MeOH. | ||
Conclusions
In summary, we have developed a platform for the enantioselective dearomative functionalization of salicylaldehydes using an integrated asymmetric catalysis/oxidative dearomatization sequence. Stereodefined salicyl α-hydroxyphosphonates were synthesized using a new catalytic asymmetric Pudovik reaction. We anticipate that the reported Cu(II)-catalyzed transformation will inform further development of new enantioselective nucleophilic additions to salicylaldehydes. In the latter half of the described strategy, oxidative dearomatization enabled diversification of the phenolic cores to access distinct classes of enantioenriched cycloadducts with excellent diastereo- and regiocontrol. Lastly, we demonstrated the removal of the phosphonate functionality to access densely functionalized and stereochemically complex polycyclic scaffolds with ample functionality for applications towards complex molecule synthesis.Author contributions
AJC and KAA: conceptualization; formal analysis; methodology; investigation; project administration; writing – original draft and editing. BKW: conceptualization; formal analysis; methodology; investigation; writing – original draft and editing. JGR: conceptualization; formal analysis; methodology. SL: investigation; formal analysis. JSJ: conceptualization; funding acquisition; supervision; project administration; writing – review and editing.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental procedures, characterization data, X-ray diffraction data, computational details of DFT studies. See DOI: https://doi.org/10.1039/d5sc08571c.CCDC 2475547 ((indabox)Cu(1.11)2), 2475545 (4.1) and 2475546 ((±)-8.2) contain the supplementary crystallographic data for this paper.84a–c
Acknowledgements
The project described was supported by Award No. R35 GM118055 from the National Institute of General Medical Sciences. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship Program to B. K. W. under Grant No. DGE-2439854. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation. We thank Dr B. Ehrmann and D. Weatherspoon (UNC Chemistry Mass Spectrometry Core Laboratories) for their assistance with mass spectrometry. We thank Drs M. ter Horst, A. Camp, and E. Roos (UNC Department of Chemistry NMR Core Laboratory) for their assistance with NMR analysis. We thank Dr C.-H. Chen and Dr E. Crawford for their assistance with X-ray crystallography experiments. X-ray diffraction experiments were supported by the National Science Foundation under Grant No. (CHE-2117287).References
- S. P. Roche and J. A. Porco, Angew. Chem., Int. Ed., 2011, 50, 4068–4093 CrossRef CAS PubMed.
- A. A. Rosatella and C. A. M. Afonso, Biomedical and Biopharmaceutical Research, 2024, 21, 1–11 Search PubMed.
- J. L. Franklin, J. Am. Chem. Soc., 1950, 72, 4278–4280 CrossRef CAS.
- Y. You, S.-L. Yu, Q. Li, Y.-P. Zhang, Z.-H. Wang, J.-Q. Zhao, L. Yang and W.-C. Yuan, ACS Catal., 2025, 9503–9543 CrossRef CAS.
- T. Dohi, A. Maruyama, N. Takenaga, K. Senami, Y. Minamitsuji, H. Fujioka, S. B. Caemmerer and Y. Kita, Angew. Chem., Int. Ed., 2008, 47, 3787–3790 CrossRef CAS PubMed.
- M. Uyanik, T. Yasui and K. Ishihara, Angew. Chem., Int. Ed., 2010, 49, 2175–2177 CrossRef CAS PubMed.
- M. Uyanik, T. Yasui and K. Ishihara, Angew. Chem., Int. Ed., 2013, 52, 9215–9218 CrossRef CAS PubMed.
- W. Sun, G. Li, H. Liang and R. Wang, Org. Biomol. Chem., 2016, 14, 2164 RSC.
- S. A. Baker Dockrey, A. L. Lukowski, M. R. Becker and A. R. H. Narayan, Nat. Chem., 2017, 10, 119–125 CrossRef PubMed.
- J. Zhu, N. P. Grigoriadis, J. P. Lee and J. A. Porco, J. Am. Chem. Soc., 2005, 127, 9342–9343 CrossRef CAS PubMed.
- S. Dong, J. Zhu and J. A. Porco, J. Am. Chem. Soc., 2008, 130, 2738–2739 CrossRef CAS PubMed.
- M. F. McLaughlin, E. Massolo, S. Liu and J. S. Johnson, J. Am. Chem. Soc., 2019, 141, 2645–2651 CrossRef CAS PubMed.
- R. J. Phipps and F. D. Toste, J. Am. Chem. Soc., 2013, 135, 1268–1271 CrossRef CAS PubMed.
- H. Egami, T. Rouno, T. Niwa, K. Masuda, K. Yamashita and Y. Hamashima, Angew. Chem., Int. Ed., 2020, 59, 14101–14105 CrossRef CAS PubMed.
- T. Stünkel, K. Siebold, D. Okumatsu, K. Murata, L. Ruyet, C. G. Daniliuc and R. Gilmour, Chem. Sci., 2023, 14, 13574–13580 RSC.
- S. Chakrabarty, E. O. Romero, J. B. Pyser, J. A. Yazarians and A. R. H. Narayan, Acc. Chem. Res., 2021, 54, 1374–1384 CrossRef CAS PubMed.
- T. Hata, A. Hashizume, M. Nakajima and M. Sekine, Tetrahedron Lett., 1978, 19, 363–366 CrossRef.
- R. E. Koenigkramer and H. Zimmer, Tetrahedron Lett., 1980, 21, 1017–1020 CrossRef CAS.
- E. Öhler, E. Zbiral and M. El-Badawi, Tetrahedron Lett., 1983, 24, 5599–5602 CrossRef.
- J. Binder and E. Zbiral, Tetrahedron Lett., 1984, 25, 4213–4216 CrossRef CAS.
- G. Agnel and E. Negishi, J. Am. Chem. Soc., 1991, 113, 7424–7426 CrossRef CAS.
- J. Stawinski and A. Kraszewski, Acc. Chem. Res., 2002, 35, 952–960 CrossRef CAS PubMed.
- M. D. Khidre, Phosphorus, Sulfur Silicon Relat. Elem., 2003, 178, 2147–2158 CrossRef CAS.
- X. Linghu, J. R. Potnick and J. S. Johnson, J. Am. Chem. Soc., 2004, 126, 3070–3071 CrossRef PubMed.
- R. Gancarz, I. Gancarz and U. Walkowiak, Phosphorus, Sulfur Silicon Relat. Elem., 1995, 104, 45–52 CrossRef CAS.
- A. Kraszewski and J. Stawinski, Pure Appl. Chem., 2007, 79, 2217–2227 CrossRef CAS.
- T. Cytlak, M. Skibińska, P. Kaczmarek, M. Kaźmierczak, M. Rapp, M. Kubicki and H. Koroniak, RSC Adv., 2018, 8, 11957–11974 RSC.
- S. Chakrabarty and J. M. Takacs, ACS Catal., 2018, 8, 10530–10536 CrossRef CAS PubMed.
- R. Engel, in Organic Reactions, 2004, pp. 175–248 Search PubMed.
- F. A. Kortmann, M.-C. Chang, E. Otten, E. P. A. Couzijn, M. Lutz and A. J. Minnaard, Chem. Sci., 2014, 5, 1322–1327 RSC.
- B. J. Rowe and C. D. Spilling, J. Org. Chem., 2003, 68, 9502–9505 CrossRef CAS PubMed.
- R. M. Moriarty, C. J. Chany II, J. W. Kosmeder II and J. D. Bois, in Encyclopedia of Reagents for Organic Synthesis, 2006 Search PubMed.
- R. Varala, V. Seema and N. Dubasi, Organics, 2023, 4, 1–40 CrossRef CAS.
- A. A. Rosatella and C. A. M. Afonso, ACS Omega, 2022, 7, 11570–11577 CrossRef CAS PubMed.
- D. Magdziak, S. J. Meek and T. R. R. Pettus, Chem. Rev., 2004, 104, 1383–1430 CrossRef CAS PubMed.
- S. N. Good, R. J. Sharpe and J. S. Johnson, J. Am. Chem. Soc., 2017, 139, 12422–12425 CrossRef CAS PubMed.
- J. G. Robins and J. S. Johnson, Org. Lett., 2022, 24, 559–563 CrossRef CAS PubMed.
- S. Quideau, L. Pouységu and D. Deffieux, Synlett, 2008, 2008, 467–495 CrossRef.
- L. Pouységu, D. Deffieux and S. Quideau, Tetrahedron, 2010, 66, 2235–2261 CrossRef.
- S. A. Beers, E. A. Malloy, W. Wu, M. P. Wachter, U. Gunnia, D. Cavender, C. Harris, J. Davis, R. Brosius, J. L. Pellegrino-Gensey and J. Siekierka, Bioorg. Med. Chem., 1997, 5, 2203–2211 CrossRef CAS PubMed.
- R. M. N. Kalla, H. R. Lee, J. Cao, J. W. Yoo and I. Kim, New J. Chem., 2015, 39, 3916–3922 RSC.
- P. Merino, E. Marqués-López and R. P. Herrera, Adv. Synth. Catal., 2008, 350, 1195–1208 CrossRef CAS.
- J. P. Abell and H. Yamamoto, J. Am. Chem. Soc., 2008, 130, 10521–10523 CrossRef CAS PubMed.
- X. Zhou, X. Liu, X. Yang, D. Shang, J. Xin and X. Feng, Angew. Chem., Int. Ed., 2008, 47, 392–394 CrossRef CAS PubMed.
- D. Uraguchi, T. Ito and T. Ooi, J. Am. Chem. Soc., 2009, 131, 3836–3837 CrossRef CAS PubMed.
- K. Suyama, Y. Sakai, K. Matsumoto, B. Saito and T. Katsuki, Angew. Chem., Int. Ed., 2010, 49, 797–799 CrossRef CAS PubMed.
- D. Zhao and R. Wang, Chem. Soc. Rev., 2012, 41, 2095–2108 RSC.
- T. Deng and C. Cai, RSC Adv., 2014, 4, 27853–27856 RSC.
- J. Guin, Q. Wang, M. Van Gemmeren and B. List, Angew. Chem., Int. Ed., 2015, 54, 355–358 CrossRef CAS PubMed.
- B. R. Matthews, W. R. Jackson, G. S. Jayatilake, C. Wilshire and H. A. Jacobs, Aust. J. Chem., 1988, 41, 1697–1709 CrossRef CAS.
- P. J. Gerrits, J. Marcus, L. Birikaki and A. van der Gen, Tetrahedron:Asymmetry, 2001, 12, 971–974 CrossRef CAS.
- H. Bühler, F. Effenberger, S. Förster, J. Roos and H. Wajant, ChemBioChem, 2003, 4, 211–216 CrossRef PubMed.
- J. D. White and S. Shaw, Org. Lett., 2012, 14, 6270–6273 CrossRef CAS PubMed.
- A. Zhang, L. Yang, N. Yang and Y. Liu, Tetrahedron:Asymmetry, 2014, 25, 289–297 CrossRef CAS.
- S. V. Kumbhar and C. Chen, Catal. Commun., 2021, 148, 106166 CrossRef CAS.
- M. A. Tius and N. K. Reddy, Synth. Commun., 1994, 24, 859–869 CrossRef CAS.
- D. S. Yamashita, V. P. Rocco and S. J. Danishefsky, Tetrahedron Lett., 1991, 32, 6667–6670 CrossRef CAS.
- H. Kageji, N. Mori, H. Takikawa and H. Watanabe, Tetrahedron, 2018, 74, 7074–7081 CrossRef CAS.
- E. Adler, K. Holmberg, H. Miyake, O. F. Nielsen, P. Klæboe and S. Kachi, Acta Chem. Scand., 1971, 25, 2775–2776 CrossRef CAS.
- E. Adler, S. Brasen, H. Miyake, O. F. Nielsen, P. Klæboe and S. Kachi, Acta Chem. Scand., 1971, 25, 2055–2069 CrossRef CAS.
- E. Adler, K. Holmberg, H. Kjøsen, S. Liaaen-Jensen, C. R. Enzell and B. Mannervik, Acta Chem. Scand., Ser. B, 1974, 28, 465–472 CrossRef CAS.
- D. Gabrilidis, C. Kalogiros and L. Hadjiarapoglou, Synlett, 2004, 2004, 2566–2569 CrossRef.
- D. A. Evans, D. Seidel, M. Rueping, H. W. Lam, J. T. Shaw and C. W. Downey, J. Am. Chem. Soc., 2003, 125, 12692–12693 CrossRef CAS PubMed.
- J. N. Li, L. Liu, Y. Fu and Q. X. Guo, Tetrahedron, 2006, 62, 4453–4462 CrossRef CAS.
- A. M. Liquori, A. Dami-ani, J. L. DeCoen, J. Mol Biol, D. Ajo, M. Bossa, A. Damiani, R. Fidenzi, S. Gigli, L. Lanzi, A. Lapic-cirella, J. Theor Biol, W. S. Matthews, J. E. Bares, J. E. Bartmess, F. G. Bordwell, F. J. Cornforth, G. E. Drucker, Z. Margolin, R. J. McCallum, G. J. McCollum and N. R. Vanier, J. Am. Chem. Soc., 2002, 97, 7006–7014 Search PubMed.
- S. K. Ginotra and V. K. Singh, Org. Biomol. Chem., 2007, 5, 3932–3937 RSC.
- B. J. Hathaway and D. E. Billing, Coord. Chem. Rev., 1970, 5, 143–207 CrossRef CAS.
- J. S. Johnson and D. A. Evans, Acc. Chem. Res., 2000, 33, 325–335 CrossRef CAS PubMed.
- V. Singh, Acc. Chem. Res., 1999, 32, 324–333 CrossRef CAS.
- R. Hashimoto, K. Hanaya, T. Sugai and S. Higashibayashi, Bull. Chem. Soc. Jpn., 2022, 95, 663–672 CrossRef CAS.
- G. W. Kirby, Chem. Soc. Rev., 1977, 6, 1–24 RSC.
- A. D. Cohen, B.-B. Zeng, S. B. King and J. P. Toscano, J. Am. Chem. Soc., 2003, 125, 1444–1445 CrossRef CAS PubMed.
- C.-H. Lai, Y.-L. Shen, M.-N. Wang, N. S. Kameswara Rao and C.-C. Liao, J. Org. Chem., 2002, 67, 6493–6502 CrossRef CAS PubMed.
- E. Öhler, M. El-Badawi and E. Zbiral, Chem. Ber., 1985, 118, 4099–4130 CrossRef.
- A. S. Demir, M. Emrullahoglu, E. Pirkin and N. Akca, J. Org. Chem., 2008, 73, 8992–8997 CrossRef CAS PubMed.
- K. N. Houk, S. R. Moses, Y. D. Wu, N. G. Rondan, V. Jager, R. Schohe and F. R. Fronczek, J. Am. Chem. Soc., 1984, 106, 3880–3882 CrossRef CAS.
- H.-C. Tseng, A. K. Gupta, B.-C. Hong and J.-H. Liao, Tetrahedron, 2006, 62, 1425–1432 CrossRef CAS.
- D. E. Ward and M. S. Abaee, Org. Lett., 2000, 2, 3937–3940 CrossRef CAS PubMed.
- L. Barriault, P. J. A. Ang and R. M. A. Lavigne, Org. Lett., 2004, 6, 1317–1319 CrossRef CAS PubMed.
- L. W. K. Moodie and D. S. Larsen, Eur. J. Org Chem., 2014, 2014, 1684–1694 CrossRef CAS.
- G. Agnel and E. Negishi, J. Am. Chem. Soc., 1991, 113, 7424–7426 CrossRef CAS.
- D. B. G. Williams, J. Caddy and K. Blann, Org. Prep. Proced. Int., 2003, 35, 307–360 CrossRef CAS.
- V. K. Singh, P. T. Deota and A. V. Bedekar, J. Chem. Soc., Perkin Trans. 1, 1992, 903–912 RSC.
- (a) CCDC 2475545: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2p3094; (b) CCDC 2475546: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2p30b5; (c) CCDC 2475547: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2p30c6.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |