
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Halogen bonding-induced 1,3-carbohydroxylation of allyl carboxylates via 1,2-cationic acyloxy migration (1,2-CAM)
Sahil
Sharma
 a,
Gaoyuan
Zhao
a,
Loay
Bedda
a,
Arman
Khosravi
a,
Djamaladdin G.
Musaev
b and
Ming-Yu
Ngai
*a
a,
Gaoyuan
Zhao
a,
Loay
Bedda
a,
Arman
Khosravi
a,
Djamaladdin G.
Musaev
b and
Ming-Yu
Ngai
*a
aJames Tarpo Jr. and Margaret Tarpo Department of Chemistry, Purdue University, West Lafayette, Indiana 47907, USA. E-mail: mngai@purdue.edu
bCenter for Scientific Computation, Department of Chemistry, Emory University, Atlanta, GA 30322, USA. E-mail: dmusaev@emory.edu
First published on 24th December 2025
Abstract
Halogen bonding has emerged as a powerful yet underexplored tool for modulating radical reactivity. Here we demonstrate that halogen-bonding interactions between alkyl iodides and water can lower the C–I bond dissociation energy, enabling visible-light-induced photolysis to generate alkyl radicals under mild conditions. Harnessing this activation mode, we achieved a previously unknown 1,3-carbohydroxylation of allyl carboxylates, wherein radical addition is coupled with 1,2-cationic acyloxy migration (CAM) to furnish β-acyloxy alcohols. The transformation exhibits broad structural tolerance, accommodating diverse esters, thioesters, amides, and perfluoroalkyl iodides, and is effective in the late-stage diversification of natural products and drug-derived scaffolds. Mechanistic studies, including isotopic labeling, radical trapping, UV-vis spectroscopy, and DFT calculations, reveal a pathway in which halogen bonding initiates radical alkene addition, followed by rearrangement and carbocation capture. These findings showcase halogen-bonding-assisted photochemistry as a viable platform for radical–cationic cascades, opening new opportunities for reaction development.
Introduction
Halogen bonding (XB), a directional noncovalent interaction between a Lewis base (LB) and an electrophilic region on a halogen atom (σ-hole), has emerged as a powerful activation mode in molecular design and synthesis.1 The σ-hole strength depends strongly on the substituents: electron-withdrawing groups, like fluorines, in CF3I lower the carbon orbital energy, shifting the LUMO coefficient onto iodine and creating a pronounced σ-hole (Fig. 1A).2 Consequently, CF3I forms stronger XB interactions than CH3I, whose σ-hole is negligible. In a typical XB interaction, the lone-pair of the Lewis base (XB acceptor) donates electron density into the antibonding σ*(C–X) orbital of the XB donor, lowering the bond dissociation energy (BDE) of the C–X bond and enabling selective activation under mild conditions.1b Depending on the energy input, the weakened bond can undergo either a two-electron (thermal heterolysis) or a one-electron (photolysis) process, generating cationic or radical intermediates, respectively.3 This simple yet powerful interaction has emerged as a general activation strategy in organic synthesis, enabling molecular recognition, catalysis, and selective bond transformations.3 For example, Ritter and co-workers demonstrated condensed-phase XB adducts of CF3I and C2F5I for perfluoroalkylation under visible light,4 while Chen and colleagues showed that water itself can act as an XB acceptor to iodoperfluoro compounds, triggering radical generation.5 Yamaguchi and Itoh showed that photoirradiation of halogen-bonded complexes between haloarenes or haloalkanes and amines or phenols efficiently generates carbon radicals without metal catalysts or oxidants.6 Collectively, these studies highlight halogen bonding as a versatile platform for C–X bond activation and molecular functionalization. | ||
| Fig. 1 Halogen bonding (XB) in C–X bond activation and acyloxy migration-enabled alkene functionalization. (A) Electronic origin of halogen bonding in alkyl iodides and its effect on C–I bond weakening. (B) Previous work: visible-light-induced phosphine-catalyzed 1,3-carbobromination of allyl carboxylates via 1,2-radical acyloxy migration (1,2-RAM). (C) This work: visible-light-induced halogen-bonding-assisted formal 1,3-carbohydroxylation of allyl carboxylates via 1,2-cationic acyloxy migration (1,2-CAM). | ||
Allyl carboxylates have continued to serve as privileged scaffolds in organic synthesis due to their diverse reactivity.7 They participate in transition-metal-catalyzed nucleophilic (e.g., Tsuji–Trost and decarboxylative allylations)7a–f,j,n,8 and electrophilic substitutions,7g,h,k,9 and undergo anionic acyloxy migrations and rearrangements through two-electron pathways.10 More recently, radical-based strategies have enabled novel 1,2-functionalizations and rearrangements, expanding the synthetic utility of this substrate class beyond traditional sigmatropic and substitution manifolds.11 Despite these advances, formal 1,3-difunctionalization reactions, wherein two distinct groups add across the allyl unit in a 1,3-relationship accompanied by acyloxy migration, remain rare and mechanistically challenging. To address this limitation, our laboratory recently reported a visible-light-induced, phosphine-catalyzed 1,3-carbobromination of allyl carboxylates, which proceeded via a 1,2-radical acyloxy migration (RAM) mechanism (Fig. 1B).12 This transformation exploited the interaction between bisphosphine (dppm) catalysts and bromodifluoroacetates to promote photolytic C–Br bond cleavage under visible light irradiation.
During these investigations, we observed an unexpected divergent pathway: iododifluoroacetates furnished 1,3-carbohydroxylation products even in the absence of a phosphine catalyst. Mechanistic studies indicated that water forms a halogen-bonded complex with the iododifluoroacetate, promoting visible-light-induced C–I bond photolysis. The resulting perfluoroalkyl radical adds regioselectively to allyl carboxylates, followed by a 1,2-cationic acyloxy migration and hydration to afford β-acyloxy alcohols (Fig. 1C). This transformation is significant because it (i) establishes a radical–cationic cascade reaction platform with broad substrate scope, including late-stage modification of bioactive molecules, (ii) expands allyl carboxylate chemistry beyond classical two-electron pathways and radical 1,2-migrations, (iii) demonstrates that water serves as a simple yet powerful halogen-bond acceptor for C–I activation, (iv) enables site-selective synthesis of mono-protected 1,2-diol products, (v) represents the first example of XB-induced 1,3-carbohydroxylation of allyl carboxylates, and (vi) leverages perfluoroalkyl iodides, valuable precursors to fluorinated motifs of pharmaceutical importance,13 under mild, photocatalyst-free conditions.
Results and discussion
Building on these mechanistic insights, we next optimized the reaction conditions to validate the proposed pathway and establish general parameters for this transformation. To this end, 2-methylbut-3-en-2-yl benzoate (1a) and ethyl iododifluoroacetate (2a) were selected as model substrates (Table 1). The optimal conditions were identified as the coupling of 1a with 2a under 100 W blue LED irradiation in 1,2-dichloroethane (0.10 M) at 90 °C for 24 h, in the presence of Na2CO3 (2.5 equiv.) and degassed H2O (10 equiv.), furnishing the desired 1,3-carbohydroxylated product 3a in 99% NMR yield (entry 1). The reaction completely shut down in the absence of Na2CO3 (entry 2), and substitution with other carbonate bases delivered diminished efficiencies (entries 3 and 4), underlining a pronounced counter-cation effect, likely by influencing ion pairing and solvation in DCE.14 A non-carbonate ionic base was less effective (entry 5). Concentration studies revealed that dilution to 0.05 M had little effect on yield, whereas increasing the concentration to 0.20 M diminished product formation (entries 6 and 7). Control experiments confirmed that light, heat, and an oxygen-free environment are essential for achieving high reaction efficiency (entries 8–10).|
|
||
|---|---|---|
| Entry | Deviation from the standard conditions | NMR-yield (%) |
| a See SI for experimental details. Reaction yields were determined by 1H-NMR using CH2Br2 as an internal standard. | ||
| 1 | None | 99 |
| 2 | Without Na2CO3 | <2 |
| 3 | K2CO3 instead of Na2CO3 | 60 |
| 4 | NaHCO3 instead of Na2CO3 | 83 |
| 5 | t BuOK instead of Na2CO3 | <2 |
| 6 | 0.05 M instead of 0.10 M | 99 |
| 7 | 0.20 M instead of 0.10 M | 61 |
| 8 | Without light | <2 |
| 9 | Without heat | 15 |
| 10 | In air | <2 |

With the optimized conditions in hand, we evaluated the substrate scope of the halogen-bonding-induced formal 1,3-carbohydroxylation of allyl carboxylates (Table 2). A broad array of acyclic and cyclic allyl carboxylates underwent efficient 1,3-difunctionalization, furnishing the corresponding β-acyloxy alcohols (3a–3k) in good yield (Table 2A). Symmetrical acyclic allyl carboxylates, such as 1a, furnished the desired product 3a in 76% yield. Importantly, the reaction was readily scalable to the 5 mmol level, affording 0.97 g of 3a in 63% yield. Cyclic systems ranging from five- to twelve-membered rings, including those incorporating a cyclic ether (3c) or an ethylene ketal (3d), were well tolerated, affording products 3b–3f in 55–75% yield. Furthermore, nonsymmetrical allyl carboxylates bearing dialkyl groups (3g), additional ester units (3h), halogens (3i, 3j), and a phthalimide motif (3k) also participated smoothly, highlighting the broad functional group compatibility of the transformation.

The reaction also proved effective with a variety of difluoroester partners (Table 2B). Acyclic esters spanning primary, secondary, and tertiary substitution were all competent, including those bearing a trimethylsilyl group (3l), long-chain substituents containing ester functionalities (3m), and a sterically demanding tert-butyl group (3n), affording the desired products in good yield. Secondary and tertiary cyclic esters within 5–7-membered ring systems, including the bulky adamantyl group (3r), furnished the corresponding β-acyloxy alcohols in 60–68% yield. Notably, thioesters bearing linear alkyl chains, additional ester substituents, or adamantyl were also effective (3s–3u), thereby extending the scope to sulfur-containing substrates.
Encouraged by these results, we next investigated iododifluoroacetamides, which provided the desired β-acyloxy alcohols (3v–3ae) in moderate to excellent yield (Table 2C). Both symmetrical and nonsymmetrical amides, encompassing cyclic and acyclic frameworks, were tolerated. Six-membered cyclic amides incorporating ether (3w), thioether (3x), N-phenyl (3y), or ethylene ketal (3z) functionalities afforded the desired products in 62–85% yield. Acyclic diethyl-substituted derivatives (3aa) as well as N-substituted systems, such as adamantyl (3ab), tert-butyl (3ac), phenylpropyl (3ad), and phenyl (3ae) were also compatible.
We further expanded the scope to iodoalkyl derivatives, which delivered β-acyloxy alcohols (3af–3ak) in good to excellent yield (Table 2D). Perfluoroalkyl iodides, including perfluorohexyl (3af), perfluorooctyl (3ag), perfluoroisopropyl (3ah), and perfluorobenzyl (3ai), were successfully engaged to give products in 51–75% yield. Notably, a fluoroalkyl iodide bearing a phenylsulfonyl group (3aj) exhibited excellent reactivity and afforded the desired product in 91% yield. Even iodoacetonitrile, without fluorine substituents, participated efficiently to afford the product in good yield (3ak).
We next investigated the effect of substituents on the migrating carboxylate group (Table 3A). Aryl esters bearing electron-donating substituents at the para- or meta-positions provided β-acyloxy alcohols (3al–3an) in 62–80% yield. Electron-withdrawing groups, including halogens and trifluoromethyl, were also tolerated across para, meta, and ortho positions (3ao–3as), with yields remaining largely unaffected by substitution pattern. A heteroaryl ester such as a thiophene derivative furnished the desired product (3at) in 57% yield, while an aliphatic ester migrated effectively to give product (3au) in 53% yield.
| a See SI for experimental details. Standard reaction conditions: 1 (0.20 mmol, 1.0 equiv.), 2 (0.70 mmol, 3.5 equiv.), Na2CO3 (0.50 mmol, 2.5 equiv.) and degassed H2O (2.0 mmol, 10 equiv.), DCE (0.10 M), blue LED, 90 °C, 24 h. |
|---|

|
Late-stage modification of bioactive, structurally complex molecules is a powerful strategy for the discovery of new medicinal agents.15 To demonstrate the applicability of the halogen-bonding-induced 1,3-carbohydroxylation of allyl carboxylates to late-stage diversification, a series of natural product- and drug-derived substrates were subjected to the standard conditions (Table 3B). Substrates derived from febuxostat (gout preventive), ibuprofen (analgesic/antipyretic), oxaprozin (anti-arthritic), and probenecid (gout treatment) smoothly underwent 1,3-carbohydroxylation, affording products 3av–3ay in 55–74% yield. An allyl benzoate derived from pentoxifylline (vasodilator) furnished product (3az) in 50% yield. Iododifluoroacetate derivatives of naturally occurring alcohols such as (+)-menthol, (+)-borneol, and cholesterol were also competent, affording products 3aaa–3aac in 74–78% yield. Notably, an amide incorporating the tropane alkaloid scaffold, a motif common in CNS-active agents, participated to provide the corresponding β-acyloxy alcohol (3aad) in 54% yield.
To gain further insight into the reaction mechanism, we performed a series of experimental and computational studies (Fig. 2). Crossover experiments with substrates 1u and 1e yielded exclusively the corresponding non-crossover products 3u and 3e, indicating that the acyloxy migration proceeds via an intramolecular pathway (Fig. 2A). 18O-Labeling provided additional mechanistic evidence: reaction of 18O-labeled substrate 18O-1a (95% 18O incorporation) furnished product 18O-3a with 94% 18O retention, consistent with a five-membered [2,3]-acyloxy shift involving a dioxolium carbocation VII (Fig. 2B). Complementarily, conducting the standard reaction in the presence of degassed H218O afforded 18O-3a-2 with 77% 18O incorporation in the benzoyl carbonyl group, implicating nucleophilic attack of H2O at the benzylic position of carbocation intermediate VII. The slight decrease in 18O incorporation is likely attributable to the hygroscopic nature of Na2CO3, which may introduce unlabeled water into the reaction medium.
 | ||
Fig. 2 Mechanistic studies of halogen-bonding-induced 1,3-carbohydroxylation of allyl carboxylates. (A) Crossover experiment demonstrating intramolecular acyloxy migration. (B) 18O-Labeling experiments supporting formation of a dioxolium cation intermediate. (C) Radical-trapping experiment with TEMPO. (D) Reactivity of a preformed 1,2-iododifluoroalkylated intermediate. (E) Job's plot indicating 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexation between iododifluoro reagent and H2O. (F) UV-vis absorption changes consistent with halogen-bonding interactions. (G) Light on/off and quantum-yield experiments probing radical chain propagation. :1 complexation between iododifluoro reagent and H2O. (F) UV-vis absorption changes consistent with halogen-bonding interactions. (G) Light on/off and quantum-yield experiments probing radical chain propagation. | ||
Further evidence for a radical pathway was obtained by radical-trapping experiments. The addition of TEMPO markedly suppressed product formation, and the corresponding TEMPO-difluoroacetate adduct (4) was detected by 19F NMR and HRMS (Fig. 2C). In addition, a pre-synthesized 1,2-iododifluoroalkylated intermediate (5) could be converted to the desired 1,3-carbohydroxylated product 3a in 41% yield under the standard conditions (Fig. 2D), suggesting that the transformation may proceed via an initial 1,2-iododifluoroalkylation followed by intramolecular substitution reaction to give the dioxolium carbocation VII.
To further probe the presence of halogen bonding, Job's plot analysis revealed a 1:1 complexation between iododifluoroacetate 2a and H2O (Fig. 2E). Consistently, UV-vis absorption spectra of iododifluoroamide 2z in the presence of water exhibited an enhancement in absorption (Fig. 2F), indicative of halogen-bonding interactions between water and the iododifluoro reagent. We propose that this H2O⋯I–RF complex serves as the key precursor to visible-light-induced generation of the difluoroacetate radical under the reaction conditions. Nevertheless, we cannot rule out the possibility that Na2CO3 may also engage in halogen bonding and contribute to halide activation. Furthermore, to assess whether the transformation proceeds via a radical chain process, we conducted light on/off experiments and quantum-yield measurements. As shown in Fig. 2G, the combined yield of the 1,2- and 1,3-substituted products did not increase upon cessation of irradiation, and the quantum yield measurement gave Φ = 0.20. These observations suggest that extended radical chain propagation pathway is unlikely.
DFT calculations revealed that, upon water-assisted photolysis of 2a, the resulting RF radical II can add to allyl carboxylate 1a to form the more stable secondary radical V (Fig. 3). This step is exergonic by ΔG = −13.4 kcal mol−1, with an associated transition state (TS1) barrier of ΔG‡ = 18.1 kcal mol−1. Recombination of V with the iodine radical is barrierless and highly exergonic (ΔG = −35.8 kcal mol−1). In contrast, a radical-chain pathway is disfavored due to the high transition state barrier for halogen atom transfer (TS2, ΔG‡ = 28.1 kcal mol−1), consistent with the light on/off experiments and quantum yield measurements (Fig. 2G). Intermediate VI subsequently undergoes a 1,2-cationic acyloxy migration (1,2-CAM), forming a dioxolium ion intermediate (VII) via a moderate barrier (TS3, ΔG‡ = 23.8 kcal mol−1). The alternative 3-membered TS3′ is significantly less favorable by 13.1 kcal mol−1. The resulting VII then undergoes carbonate-promoted hydrolysis through a six-membered transition-state complex (TS4, ΔG‡ = 13.7 kcal mol−1) to afford intermediate VIII, which is in good agreement with the 18O-labeling experiments (Fig. 2B). Finally, intermediate VIII undergoes NaHCO3-assisted ring opening and proton transfer to yield the desired and kinetically favored 1,3-product (3a) viaTS5 (ΔG‡ = 16.3 kcal mol−1) in an overall thermodynamically favorable step (ΔG = −13.95 kcal mol−1). In contrast, the competing ring-opening pathway leading to the 1,2-product (3a′) is kinetically disfavored, with a higher barrier (TS5′, ΔG‡ = 18.2 kcal mol−1). This computational finding aligns with experimental results, where only the 1,3-product (3a) was observed.
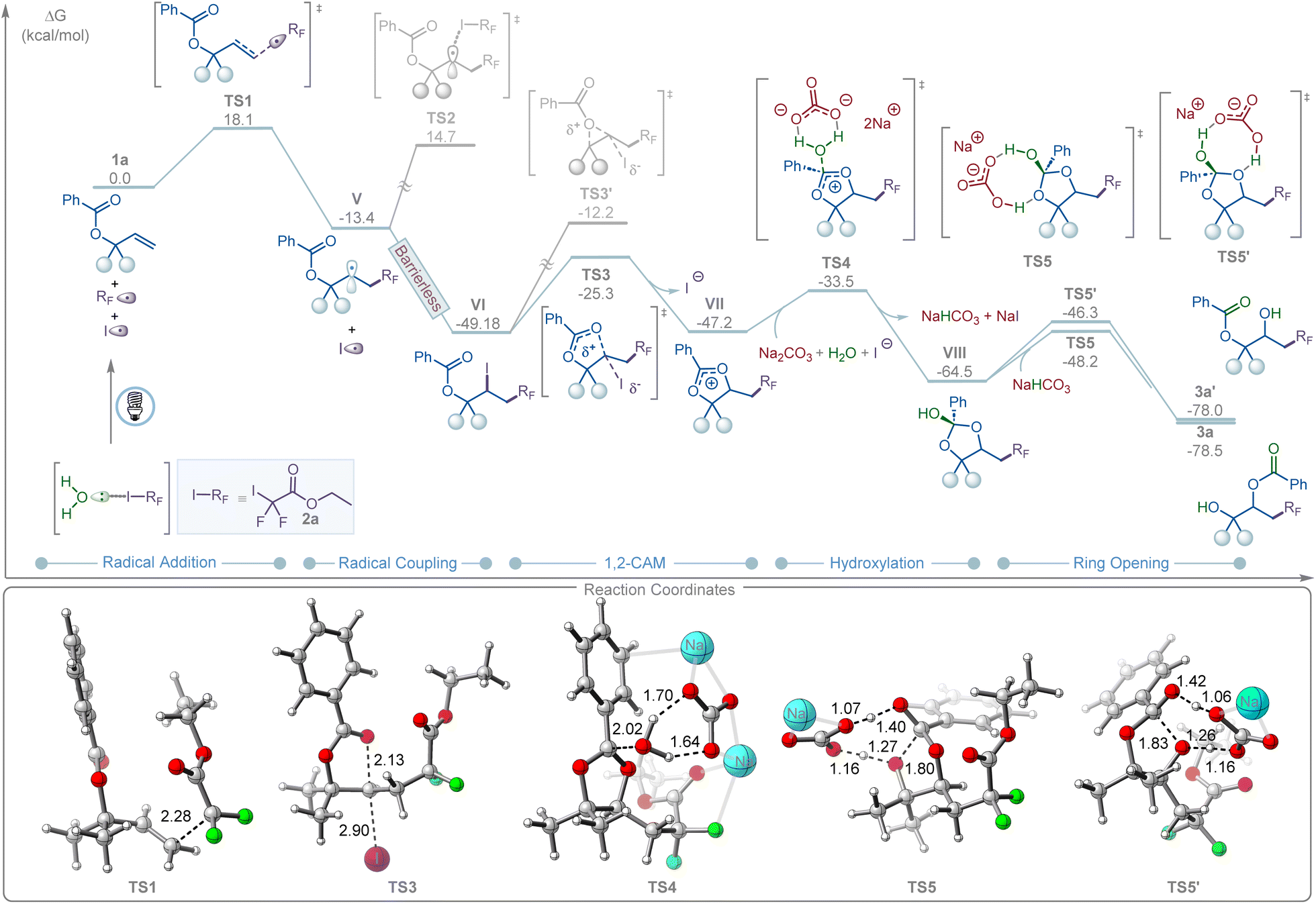 | ||
| Fig. 3 Energy profile of halogen bonding-induced 1,3-carbohydroxylation of allyl carboxylates 1a with iododifluoroester 2avia 1,2-CAM. DFT calculations were performed at the M06/6-311+G(d,p)-SDD/SMD(dichloroethane)//B3LYP-D3(BJ)/6-31 G(d)-SDD/SMD(dichloroethane) level of theory. The 3D representation was prepared by using CYLview.16 | ||
Conclusions
In summary, we have developed the first halogen-bonding-induced, visible-light-mediated formal 1,3-carbohydroxylation of allyl carboxylates via a 1,2-CAM pathway, furnishing β-acyloxy alcohols in good yields with broad functional-group tolerance. Mechanistic and computational studies reveal that water serves not only as the halogen-bond acceptor enabling photocatalyst-free C–I activation, but also as the source of the oxygen atom incorporated into the carbonyl group of the acyl fragment in the acyloxy group. This work establishes a radical–cationic cascade that bridges halogen-bonding activation with ionic rearrangement. We anticipate that the 1,2-CAM reactivity of allyl carboxylates and halogen-bonding-induced radical activation will inspire new strategies for tandem radical–cationic transformations in organic synthesis.Author contributions
S. S., G. Z. and A. K. performed the experiments, synthesized starting materials, developed the substrate scope, and conducted detailed mechanistic studies. L. B. and D. G. M. designed and performed the DFT calculations. G. Z., S. S. and M.-Y. N. conceived the idea and designed the research. S. S., L. B. and M.-Y. N. wrote the manuscript. All the authors commented on the final draft of the manuscript and contributed to the analysis and interpretation of the data.Conflicts of interest
There are no conflicts to declare.Data availability
Additional data are available from the corresponding author upon reasonable request.All experimental procedures, characterization data (1H, 13C, and 19F NMR spectra), and computational details supporting the findings of this study are provided in the supplementary information (SI). Supplementary information: Cartesian coordinates and energies for all computed structures. See DOI: https://doi.org/10.1039/d5sc08514d.
Acknowledgements
We thank the National Institutes of Health (R35-GM119652 to M.-Y. N.) for supporting research described in this manuscript. A. K. gratefully acknowledges support from the Charles H. Viol Memorial Chemistry Fellowship at Purdue University. DFT calculations were carried out at the University of Pittsburgh Center for Research Computing and the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program, supported by NSF award numbers OAC-2117681, OAC-1928147, and OAC-1928224. D. G. M acknowledges the support of the National Science Foundation under the CCI Center for Selective C–H Functionalization (CHE-1700982), and the use of the resources of the Cherry Emerson Center for Scientific Computation at Emory University. We acknowledge constructive discussions within the Catalysis Innovation Consortium, which facilitated this collaborative study.Notes and references
-
(a) T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS
; (b) P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 Search PubMed
-
(a) K. Morokuma, L. Pedersen and M. Karplus, J. Chem. Phys., 1968, 48, 4801–4802 CrossRef CAS
-
(a)
P. Kirsch, Modern fluoroorganic chemistry: synthesis, reactivity, applications, John Wiley & Sons, 2006 Search PubMed
- F. Sladojevich, E. McNeill, J. Börgel, S.-L. Zheng and T. Ritter, Angew. Chem., Int. Ed., 2015, 54, 3712–3716 CrossRef CAS PubMed
- C.-Z. Fang, B.-B. Zhang, B. Li, Z.-X. Wang and X.-Y. Chen, Org. Chem. Front., 2022, 9, 2579–2584 RSC
-
(a) K. Matsuo, E. Yamaguchi and A. Itoh, J. Org. Chem., 2020, 85, 10574–10583 CrossRef CAS
-
(a) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395–422 CrossRef CAS PubMed
-
(a) Y. Cheng, C. Mueck-Lichtenfeld and A. Studer, J. Am. Chem. Soc., 2018, 140, 6221–6225 CrossRef CAS
-
(a) M. Vogt, S. Ceylan and A. Kirschning, Tetrahedron, 2010, 66, 6450–6456 CrossRef CAS
-
(a) R. B. Martin and R. I. Hedrick, J. Am. Chem. Soc., 1962, 84, 106–110 Search PubMed
-
(a) L. Huang, S.-C. Zheng, B. Tan and X.-Y. Liu, Org. Lett., 2015, 17, 1589–1592 CrossRef CAS PubMed
- G. Zhao, S. Lim, D. G. Musaev and M.-Y. Ngai, J. Am. Chem. Soc., 2023, 145, 8275–8284 CrossRef CAS
-
(a) M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed
- N. I. Prakoso, N. V. Nugroho and D. Rubiyanto, AIP Conf. Proc., 2020, 2229, 030001 CrossRef CAS
-
(a) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC
-
C. Y. Legault
CYLview20; Universié de Sherbrooke, 2020; http://www.cylview.org accessed 9/15/2025 Search PubMed
| This journal is © The Royal Society of Chemistry 2026 |