
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirecting robust built-in electric fields via imine linkage orientation in COFs for efficient dehydrogenative organic transformation coupled with H2O2 photosynthesis
Chang
He†
ab,
Weixu
Liu†
b,
Enwei
Zhu
*a,
Yongfa
Zhu
*b and
Chen
Chen
 *b
*b
aKey Laboratory of Preparation and Applications of Environmental Friendly Materials, Jilin Normal University, Changchun, China. E-mail: zhuenwei05@126.com
bEngineering Research Center of Advanced Rare Earth Materials, Department of Chemistry, Tsinghua University, Beijing, China. E-mail: zhuyf@tsinghua.edu.cn; cchen@mail.tsinghua.edu.cn
First published on 12th December 2025
Abstract
The built-in electric field (IEF) can be improved via developing donor–acceptor (D–A) type COF photocatalysts, yet available modulation strategies remain limited and poor. Here, we design a series of D–A COFs with isomeric imine linkages to promote the IEF and redox activity. The orientation of imine linkages on D/A centers can effectively adjust the dipole moment of COFs, which is increased by ∼50%, thereby enhancing the IEF. The charge separation efficiency is also elevated through the enhancement of the IEF, leading to exciton binding energy being reduced by ∼30% and charge-separated state lifetime prolonged 7-fold. Accordingly, the optimal COF exhibits impressive photocatalytic redox activities (5-fold promoted), achieving a remarkable tetrahydroquinoline dehydrogenation rate of 10.02 mmol g−1 h−1, far exceeding reported metal-free photocatalysts and even comparative with metal photocatalysts, along with a H2O2 production rate over 20.01 mmol g−1 h−1. Our findings establish a universal strategy to modulate the IEF, which contributes to designing high-performance COF photocatalysts.
Introduction
To address the energy and environmental crises, directly converting solar energy into chemical energy through photocatalysis is a promising strategy for achieving sustainable development.1–3 However, efficient and stable semiconductor photocatalysts are still lacking.4–6 In recent years, two-dimensional (2D) COFs, featuring porous crystalline structures formed through various types of reversible dynamic covalent bonds linking organic functional units, have attracted much attention.7–9 This resultant architecture in COFs is unique, with π-functional units in-plane periodically linked into 2D polymeric conjugated backbones, and the layer frameworks further noncovalently stacked to form separated columnar π-arrays. Such ordered crystalline structures with channels are ideal semiconductor photocatalysts, particularly in overcoming the limitations of light absorption and active sites found in inorganic semiconductors, which show great potential in photocatalysis.8,10–15 Nevertheless, inefficient photogenerated charge separation severely limits their practical application.16–18 Rational design of 2D COFs via molecular engineering is still a major challenge.Although researchers have explored a diverse array of molecular engineering strategies to enhance the photocatalytic performance of imine-based COFs—such as fine-tuning the dipole moment and creating active sites through the introduction of functional groups,19–21 controlling the interlayer dihedral angle via steric hindrance groups to optimize charge carrier dynamics22–24 and so on, the donor–acceptor (D–A) design strategy has been widely demonstrated as one of the most effective and universal approaches, primarily due to its unparalleled ability to generate a robust IEF that dramatically enhances charge separation efficiency.15–31 2D D–A COFs feature D and A units alternately arranged in-plane and spatially stacked into dual-continuous π-structures with ordered layer arrays.32 This unique alternating D–A conjugation architecture contributes to numerous super-heterojunctions, which are helpful in promoting the IEF to generate long-lived charge-separated states for photocatalysis.33,34 However, strategies for modulating the IEF in 2D D–A COFs are predominantly confined to molecular-level optimization of donor and acceptor units, while the repertoire of available modulation approaches remains substantially constrained. The linkages in 2D D–A COFs play a crucial role in optical absorption and electronic properties, yet the orientation of the D and A groups relative to the COF linkage has been rarely investigated. It can be predicted that the aforementioned orientation is crucial, given that both the D–A system and the linkage are oriented. The proper orientation, as has been recently demonstrated in electrochemiluminescence,35 will effectively superimpose the effects of both components.
Based on the above consideration, a series of 2D D–A COFs featuring spatially separated D/A centers with isomeric imine linkages is rationally designed and successfully synthesized (Scheme 1). The influence of the imine linkage on photocatalytic activity was explored by varying the spatial arrangement of the D/A centers relative to the imine linkage. We reveal that, within the same D–A center frameworks, COFs with the imine-linkage nitrogen adjacent to the A center (DCNA) exhibited better activity on the dehydrogenation oxidation of tetrahydroquinoline and the production of hydrogen peroxide (H2O2) compared to COFs with the imine-linkage nitrogen adjacent to the D center (DNCA). The structure–activity relationship was further investigated using a range of in situ/quasi-in situ tests and DFT calculations. The results indicate that COFs with DCNA orientation exhibit enhanced charge separation efficiency. This can be attributed to a larger overall dipole moment generated from the alignment between the inherent dipole moments of the D–A centers and the polar imine linkage, which is increased by ∼50%, further establishing a more robust IEF with more prolonged charge-separated state lifetimes and lower Eb (reduced by ∼30%). This work highlights the significance of linkage orientation on photocatalytic activity and provides a general strategy for designing high-performance COF photocatalysts.
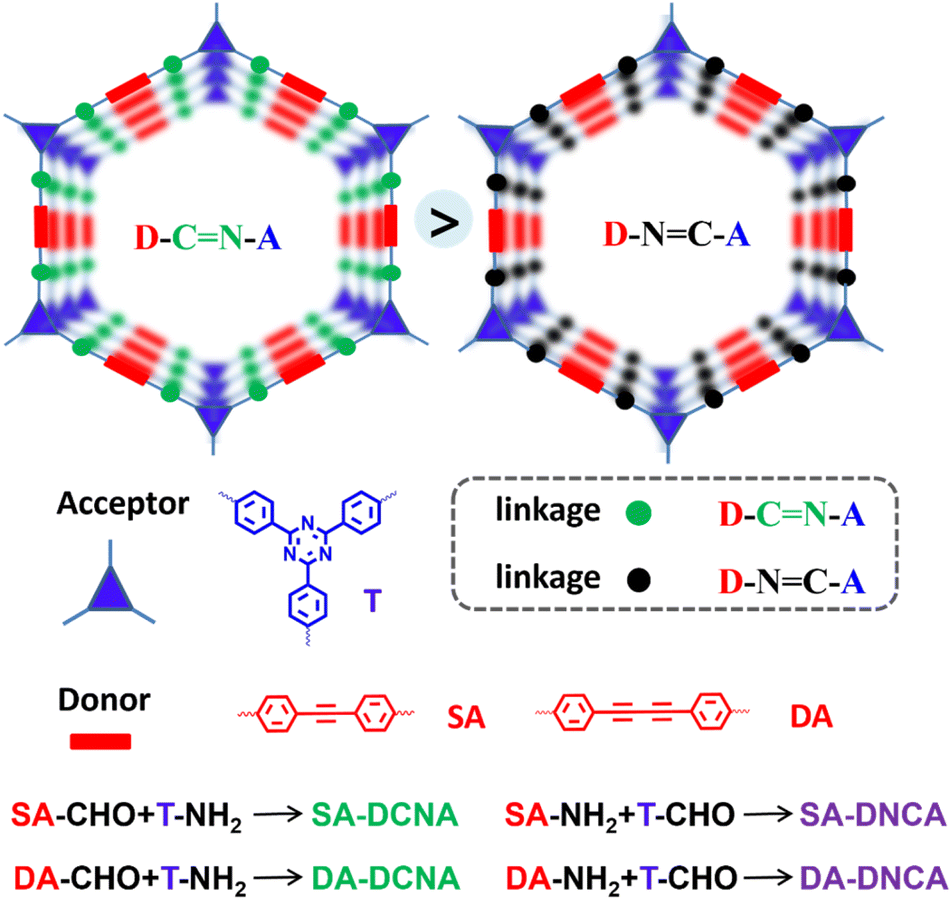 | ||
| Scheme 1 The as-synthesized D–A COFs with isomeric imine linkages. | ||
Results and discussion
Structure confirmation of D–A COFs with isomeric imine linkages
Herein, a series of COFs with the D/A centers spatially arranged opposite to the imine linkage were constructed through an acid-catalyzed Schiff base reaction. Specifically, SA-DCNA and DA-DCNA were synthesized by the condensation of acetylenic aldehyde D units and triazine amine A units to form COFs with the DCNA structure. In contrast, SA-DNCA and DA-DNCA with the DNCA structure were synthesized by the condensation of acetylenic amine D units and triazine aldehyde A units. The structures of SA-DCNA, SA-DNCA, DA-DCNA, and DA-DNCA were confirmed through 13C solid-state nuclear magnetic resonance (13C-ssNMR) spectroscopy, Fourier-transform infrared (FT-IR) spectroscopy, element analysis (EA), thermogravimetric analysis (TGA), powder X-ray diffraction (PXRD), transmission electron microscopy (TEM), BET surface area measurements and structural simulations. The 13C-ssNMR spectra (Fig. 1a and b) show that the carbon atoms of the imine linkage in SA-DCNA and DA-DCNA exhibit broad peaks at 153 ppm, whereas in SA-DNCA and DA-DNCA, they appear at 155 ppm and 160 ppm, indicating the formation of the imine linkage. Meanwhile, the variation in chemical shift is consistent with the change induced by the orientation of the imine linkage. The appearance of a FT-IR signal at ∼1623 cm−1 for –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– and the disappearance of the –CHO stretching signal at ∼1670 cm−1 also confirm the condensation of aldehyde and amine groups of organic units (Fig. S1).36,37 Additionally, the EA results (Table S1) for C, N, and H in COFs are in good agreement with the theoretical predictions, further verifying the successful synthesis of the four COFs.
N– and the disappearance of the –CHO stretching signal at ∼1670 cm−1 also confirm the condensation of aldehyde and amine groups of organic units (Fig. S1).36,37 Additionally, the EA results (Table S1) for C, N, and H in COFs are in good agreement with the theoretical predictions, further verifying the successful synthesis of the four COFs.
 | ||
| Fig. 1 (a and b) 13C ssNMR spectra of SA-DCNA and SA-DNCA, and DA-DCNA and DA-DNCA. (c and d) Experimental and simulated PXRD patterns of SA-DCNA and SA-DNCA, and DA-DCNA and DA-DNCA. (e–h) TEM images of SA-DCNA, SA-DNCA, DA-DCNA, and DA-DNCA. Left inset: enlarged image of the region within the red dotted box. Right inset: corresponding SAED pattern of the region within the red dotted box. | ||
The crystal structures of COFs are determined by PXRD and structural simulations (Fig. S2–S5). Pawley refinement indicates that the differences between the simulated and experimental PXRD patterns are negligible, and all COFs adopt an eclipsed AA-stacking mode. As shown in Fig. 1c, SA-DCNA and SA-DNCA exhibit identical 2θ peaks at 2.1°, 3.4°, 4.2°, and 6.2°, corresponding to the (100), (110), (200), and (210) planes, respectively, due to the same D–A periodic frameworks. Similarly, DA-DCNA and DA-DNCA also present the same 2θ peaks at 1.9°, 3.4°, 4.0° and 5.2°, which are assigned to the (100), (110), (200) and (210) planes, respectively (Fig. 1d). Therefore, the as-synthesized COFs display 2D hexagonal structures with conjugation backbones extended over the x–y plane, as well as forming organized columnar π arrays stacked along the z direction. From the TEM images (Fig. 1e–h, and S6–S9), the periodically ordered channels in the four COFs are observable, further proving the formation of the 2D topology. The porosities of COFs were also evaluated by nitrogen adsorption experiments at 77 K, with the pore diameter ranging from 3.6 nm to 4.1 nm (Fig. S10 and S11). As shown in Fig. S12, scanning electron microscopy (SEM) images reveal a nanocrystalline block morphology. The structural stability of COFs was investigated with TGA (Fig. S13), exhibiting decomposition temperatures above 400 °C under a N2 atmosphere.
The D–A system was confirmed by in situ X-ray absorption near edge structure (XANES) spectra, stimulated and measured diffuse reflectance spectra (DRS), and the DFT calculation. The peak around ∼289.0 eV, associated with the 1s to π* excitation of C atoms in the triazine rings, shows enhanced absorption after irradiation (Fig. S14). Meanwhile, a significant increase is also observed in the N K-edge absorption spectrum at ∼399.0 eV, which is attributed to the 1s to π* excitation of triazine rings (Fig. 2a and b).38 This phenomenon is due to the transfer of photogenerated electrons to the triazine-containing structure. Accordingly, the absorption peak corresponding to the 1s to π* excitation of the C≡C bond at ∼286.0 eV slightly decreases. It reveals that photogenerated electrons tend to concentrate in the triazine-containing acceptor moieties, whereas the holes stay at alkyne-containing donor moieties, thus confirming the formation of the D–A system within COFs. Meanwhile, charge transfer absorption peaks are observed in the DRS spectra (Fig. S15), further demonstrating the internal charge transfer processes.39 The DRS spectra of four COFs were also simulated (Fig. 2c–f), showing that the excitation of S0 → S1 was dominated by charge transfer excitation (CTE) of π–π* from the SA/DA-based unit to the triazine-based unit. Additionally, the HOMO and LUMO distribution of COFs features D–A characteristics (inset of Fig. 2c–f and S16), where the HOMO is distributed mainly on the SA/DA-based unit, and the LUMO is distributed mainly on the triazine-based unit. These findings validate the D–A system of as-prepared COFs, allowing the COFs to generate charge-separated states, where the holes remain on the D centers, while the electrons migrate to the A centers.
 | ||
| Fig. 2 (a and b) In situ irradiated (405 nm) N K-edge XANES spectra of SA-DCNA and SA-DNCA, and DA-DCNA and DA-DNCA. (c–f) Comparison of theoretical and experimental DRS spectra for SA-DCNA, SA-DNCA, DA-DCNA, and DA-DNCA. | ||
Manipulating imine linkage orientation to direct robust IEFs
The distribution of localized charge density can significantly affect the dipole moment of the molecule.27,40–43 Likewise, polarity alterations arising from the orientation of imine linkages may remarkably modulate the overall dipole moment of COFs. Thus, surface electrostatic potential distribution and molecular dipole moment of four COFs were investigated. It was found that SA-DCNA and DA-DCNA can produce larger dipoles than SA-DNCA and DA-DNCA, respectively. As shown in Fig. 3a and c, the dipole moment of SA-DCNA is 1.64 debye, outperforming that of SA-DNCA with 1.01 debye. Similarly, DA-DCNA also exceeds DA-DNCA (1.44 debye > 1.08 debye, Fig. 3b and d). Given that the only difference between SA-DCNA & SA-DNCA and between DA-DCNA & DA-DNCA is the orientation of the imine linkage, we quantitatively determined the influence of the imine linkage orientation on the overall molecular dipole via fragment dipole analysis. Specifically, the dipole moments of the D–A segment and the imine linkage within COFs were calculated. It was found that the dipole of the D–A segment is directed from D to A units, while that of the imine linkage is pointed from the carbon atom to the nitrogen atom, consistent with the classical definition of the dipole moment. According to DFT calculations, the dipole moment of the D–A segment is 0.45 debye for both SA-DCNA and SA-DNCA, and the dipole moment of the imine linkage is 1.15 debye and 1.35 debye, respectively. When the dipole of the D–A segment in SA-DCNA aligns with the dipole direction of the imine linkage, a larger cumulative molecular dipole moment of 1.64 debye is generated. In contrast, the opposite direction in SA-DNCA produces a dipole moment of only 1.01 debye. The modulation of dipoles in DA-DCNA and DA-DNCA by imine linkages is in good agreement with the regulation observed in SA-DCNA and DA-DNCA.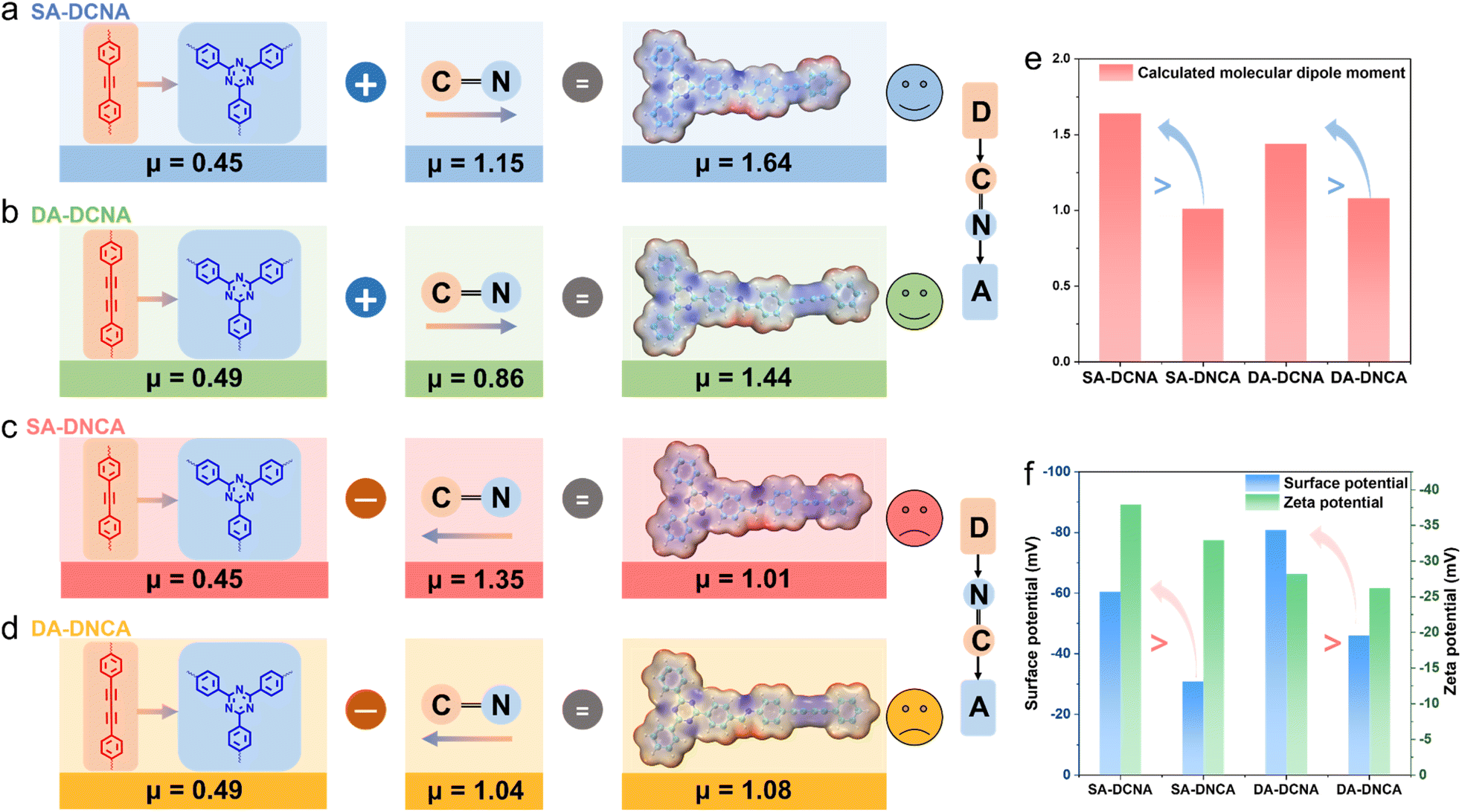 | ||
| Fig. 3 (a–d) Surface electrostatic potential distribution and fragment dipole analysis of SA-DCNA, SA-DNCA, DA-DCNA, and DA-DNCA repeating parts. (e) Comparison of the dipole moment over COFs. (f) Surface potential and zeta potential of COFs. | ||
The above analysis indicates that the orientation of the imine linkage can significantly change the polarity of COFs (Fig. 3e). With the intrinsic orientation of the imine linkage (CN) aligning with the D–A system, the COF would possess a larger overall molecular dipole, which further induces a robust IEF in semiconductors.44 Kelvin Probe Force Microscopy (KPFM) and zeta potential tests were conducted to compare the IEF of the COFs (Fig. 3f and S17). It is reported that higher surface potential and zeta potential suggest a more robust IEF.45–47 We found that the surface potential of SA-DCNA and DA-DCNA is nearly twice as high as that of SA-DNCA and DA-DNCA, with slightly higher zeta potentials. This indicates that the enhancement of molecular polarity caused by the imine linkage orientation could enable DCNA-type COFs to feature a more robust IEF.
More robust IEFs would facilitate carrier separation more,48 which is also confirmed by TAS, TRPL, EPR, XPS and temperature-dependent PL. TAS was performed on four COFs to study their photophysical properties in acetonitrile, as shown in Fig. 4a–d. Under 400 nm excitation, all four COFs showed short-lived stimulated emission (SE) signals at the corresponding wavelengths, accompanied by excited-state absorption (ESA) signals in the range of 550–780 nm. Subsequently, the SE signals decayed rapidly, whereas the ESA signals exhibited different decay rates for four COFs, following the trends of SA-DCNA > SA-DNCA and DA-DCNA > DA-DNCA. The TAS spectra are dominated by the ground state bleaching (GSB) absorption band over a longtime scale. It is well known that the long-time-scale GSB signal was observed not only in 2D COFs,49,50 but also in 2D carbon materials, which is commonly regarded as charge delocalization in π–π stacking units.51–53 Thus, we attribute this GSB to the delocalization of charge carriers within the COFs, indirectly suggesting that the ESA signals in COFs are from charge-separated states. Based on previous studies and the existed D–A systems within these COFs, this ESA signal is ascribed to triazine radical anions.49,50 The spectral broadening features of EAS peaks are likely associated with the extended π-conjugation systems of 2D COFs, which is in good agreement with previous reports.49 By carrying out kinetic fitting at 650 nm for these COFs, two kinetic components were resolved, as shown in Fig. 4e and Table S6. The shorter lifetime component (τ1) is related to the process of the rapid recombination between holes and triazine radical anions generated initially from the separated exciton via the D–A system. The prolonged lifetime component (τ2) is attributed to the slow relaxation and recombination process of holes and triazine radical anions after transport and migration. It was found that both τ1 and τ2 for four COFs exhibit the trends of SA-DCNA > SA-DNCA and DA-DCNA > DA-DNCA. This indicates that SA-DCNA and DA-DCNA manifest relatively slower electron–hole recombination kinetics, resulting in the formation of prolonged charge-separated species and achieving more effective charge separation.
 | ||
| Fig. 4 (a–d) Femtosecond-transient absorption spectroscopy (fs-TAS) 2D plots of SA-DCNA, SA-DNCA, DA-DCNA, and DA-DNCA. (e) Time–absorption profiles of fs-TAS at a selected wavelength (650 nm) and the corresponding fitting curves. (f–i) Evolution-associated spectra (EAS) of the transient species obtained from global analysis of fs-TAS for SA-DNCA, SA-DCNA, DA-DNCA, and DA-DCNA. | ||
A more detailed investigation into the photophysical processes within the COFs was conducted using global fitting, as illustrated in Fig. 4f–i. These fs-TAS datasets were best fit by an A → B → C → D → GS (SA-DCNA and DA-DCNA) and A → B → C → GS (SA-DNCA and DA-DNCA) first-order sequential kinetic model, which could also be validated by the good agreement between the fitting curves and the original fs-TAS data at various wavelengths (Fig. S18).
For the EAS spectra resolved from the COFs, due to the rapid dissociation of excitons into charge-separated (CS) states in D–A type 2D polymers, typically within the instrument response function (IRF) of fs-TAS,49 we attribute the ultrafast ESA1 to the exciton state. It is easily seen that these exciton state spectral features are very similar, especially at <500 nm, which is due to the fact that their major light-absorbing functional groups are identical. For SA-DCNA and DA-DCNA (Fig. 4g and i), the obtained ESA2 and ESA3 resemble each other, both containing characteristic absorption peaks (∼650 nm) of triazine radical anions.49,50 We assigned them respectively to the CT and CS1 states. The CT state represents the unrelaxed hot CS1 state,54–57 which undergoes 5.0 ps and 4.3 ps relaxation to the CS1 state, respectively. The ESA4 state is the long-time-scale GSB signal mentioned above, and we attribute the ESA4 state to the delocalization of triazine radical anions within SA-DCNA and DA-DCNA. Thus the 957.3 ps and 827.7 ps for SA-DCNA and DA-DCNA, respectively, correspond to the processes in which the CS1 state undergoes interlayer charge migration. As for the final evolution, it involves the formation of superoxide radical anions, as well as the recombination or return to the GS of triazine radical anions and holes. Due to the limited time detection range of fs-TAS, accurate time constants for the GSB to GS process cannot be obtained here through kinetic fitting.
For the EAS resolved from SA-DNCA and DA-DNCA (Fig. 4f and h), the results are slightly different. We did not observe CS states and long-lived GSB signals, where the ESA3 showed almost no features. Therefore, the CT states generated from exciton dissociation directly relax to the GS within 1.2 ps and 0.5 ps, respectively. These short-lived and hot CS states, which are not fully dissociated, are unfavorable for photocatalysis and difficult to participate in the redox reaction, further indicating the role of imine linkage orientation in stabilizing and forming long-lived triazine radical anions.
TRPL measurements were employed to further evaluate the excited-state lifetime of COFs in acetonitrile under 440 nm irradiation. As shown in Fig. 5a and Table S7, the PL kinetics for COFs were determined through bi-exponential fitting, which is consistent with the TAS kinetics. The process with a shorter lifetime (∼1 ns) is ascribed to the rapid recombination and radiative luminescence of charge carriers, which do not contribute to the overall photocatalytic process. The process with prolonged lifetime is associated with the slow recombination of charge-separated states, and a larger proportion of the slow process is more beneficial for enhancing photocatalytic activity. It is observed that SA-DCNA and DA-DCNA have a higher proportion of the slow process relative to SA-DNCA and DA-DNCA, giving rise to longer average lifetimes for SA-DCNA and DA-DCNA. This illustrates that SA-DCNA and DA-DCNA have more robust IEFs, resulting in more effective charge separation, which is also confirmed by TDDFT. The electron, hole, and overlap integral distributions with a series of indexes (Fig. S19 and Table S8) indicate that SA-DCNA and DA-DCNA feature less overlap of electrons and holes in excited states compared to those of SA-DNCA and DA-DNCA.
 | ||
| Fig. 5 (a) Time-dependent photoluminescence (TRPL) spectra and the fitted lifetime of COFs. (b) Solid-state electron paramagnetic resonance (EPR) spectra of COF powders. (c–f) Temperature-dependent PL spectra from 140 to 300 K under excitation at 420 nm and the fitted exciton binding energy (Eb) of SA-DCNA, SA-DNCA, DA-DCNA, and DA-DNCA. | ||
Furthermore, EPR and X-ray photoelectron spectroscopy (XPS) reveal that the orientation of the imine linkage in COFs significantly affects the charge separation within the same D–A system, and COFs with the DCNA structure exhibit enhanced charge separation efficiency. As shown in Fig. 5b, under visible light irradiation, all four COFs exhibit a signal at g = 2.004, indicating the presence of unpaired electrons in the conduction band.58 However, there is a notable difference in EPR intensity, with SA-DCNA and DA-DCNA exhibiting higher intensity compared to SA-DNCA and DA-DNCA, respectively. As for XPS, the peak around ∼285.7 eV, corresponding to carbon atoms in triazine rings, exhibits a shift toward lower binding energy after irradiation (Fig. S20), which is due to the transfer of photogenerated electrons to the triazine-containing structure. And the shift magnitude trends for these four COFs remain consistent with those observed in EPR, TAS, and TRPL.
To quantitatively assess the charge separation efficiency of COFs, temperature-dependent PL measurements were performed to calculate Eb, as shown in Fig. 5c–f. In temperature-dependent PL spectra, the fluorescence intensity of exciton transitions diminishes as the temperature increases. This occurs because excitons are more prone to dissipate energy through non-radiative processes at elevated temperatures, such as vibrational relaxation, instead of fluorescence. The PL intensity of four COFs decreases with the rising temperature, indicating that excitons undergo thermally activated non-radiative recombination processes. The Eb values of SA-DCNA, SA-DNCA, DA-DCNA, and DA-DNCA are determined to be 61.7 meV, 118.0 meV, 107.3 meV and 121.0 meV, respectively. This illustrates that the alignment of the imine linkage with the D–A direction in COFs reduces Eb and enhances charge separation, which are consistent with the trend obtained in calculated Eb using TDDFT for the repeating units (Table S8). Therefore, COFs with aligned dipole directions of the D–A segment and imine linkage generate a larger molecular dipole, resulting in a more robust IEF, which reduces Eb and enhances carrier separation.
Enhancement of photocatalytic redox activity via imine linkage orientation
The photocatalytic activity over these COFs needs to be evaluated to confirm the above results. The dehydrogenation reaction of N-heterocycles could be driven by photogenerated holes and activated oxygen species, such as superoxide radicals and singlet oxygen, resulting in high-value products. This sustainable and mild transformation route not only produces valuable organic compounds but also generates H2O2 simultaneously. Hence, the dehydrogenation of tetrahydroquinoline was employed as a model reaction for COFs, which provides a comprehensive assessment of the oxidative and reductive activities of COFs. It is noteworthy that SA-DCNA and DA-DCNA exhibit better activity compared to SA-DNCA and DA-DNCA in different types of solvent (Fig. 6a and S21), which is in accordance with the charge separation analysis. In particular, DA-DCNA with alternated diyne-based D centers exhibited the best photocatalytic activity, achieving a remarkable photocatalytic dehydrogenation rate for tetrahydroquinoline of 10.02 mmol g−1 h−1, along with a hydrogen peroxide production rate of 20.01 mmol g−1 h−1, which represents a top level performance of metal-free catalytic systems, even surpassing many metal photocatalysts (Fig. 6e and Table S10). Comparative experiments as well as quenching experiments (Fig. 6b, c and Table S9) and in situ EPR (Fig. 6d) indicate that ·O2− and holes function as the crucial active species to participate in oxygen and tetrahydroquinoline activation. The energy band structure (Fig. 6f) is determined via UV-vis-NIR DRS (for optical band gap, Fig. S15) and ultraviolet photo-electron spectroscopy (UPS, for Fermi level and valence band positions, Fig. S22), indicating that COFs can thermodynamically activate oxygen to superoxide radicals. Thus, the mechanism for the photocatalytic reaction is as follows (Fig. 6g). The separation of excitons is due to the D–A system after irradiation. Nearby tetrahydroquinoline molecules are rapidly oxidized by holes on the D centers, resulting in the formation of tetrahydroquinoline radical cations. Meanwhile, electrons at the A centers react with dissolved O2 to generate superoxide radicals, participating in the dehydrogenation of tetrahydroquinoline radical cations, simultaneously producing H2O2. | ||
| Fig. 6 (a) Production of quinoline and hydrogen peroxide over COFs. Conditions: 1 atm oxygen, 298 K, 0.1 mmol tetrahydroquinoline, 3 mg catalyst, 5 h, LED light (420 nm). It should be noted that the selectivity is over 99.9% unless otherwise stated. (b and c) Quenching experiments for holes and superoxide radicals. (d) EPR detection for ·O2− under irradiation. (e) Dehydrogenation rate comparison over DA-DCNA and DA-DNCA. (f) Schematic energy band diagrams of COFs. (g) Mechanism diagram of photocatalytic oxidative dehydrogenation coupled with H2O2 photosynthesis over COFs. | ||
Conclusions
In summary, a universal strategy has been developed showing that COFs with a DCNA configuration exhibit a more robust IEF, resulting in a significant enhancement in photocatalytic performance relative to COFs with a DNCA configuration. The underlying cause is that the polar imine linkage orientation induces a dramatic change in overall polarity of COFs. With the intrinsic orientation of the imine linkage aligning with the D–A system, it results in a larger dipole and forms a more robust IEF, significantly improving the carrier separation efficiency of COFs. Consequently, DCNA COFs exhibit more prolonged charge-separated state lifetimes and much lower Eb, obtaining over 5-fold enhancement in photocatalytic oxygen activation compared to DNCA type COFs. This work demonstrates that the linkage orientation has a remarkable impact on the charge separation and provides a general and transformative guidance for designing high-performance COF photocatalysts.Author contributions
C. H., W. L., and E. Z. conceived the project. Y. Z. and C. C. guided the research. C. H., W. L., and E. Z. performed experimental work. W. L. performed the computational studies. C. H., W. L., and E. Z. cowrote the manuscript. All authors participated in data analysis.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc08299d.Acknowledgements
This work was supported by the Beijing Natural Science Foundation (Z250018), National Natural Science Foundation of China (U22B2071, 22136002, and U24A20540), Yunnan Provincial Science and Technology Project at Southwest United Graduate School (202302AO370017), National Key Research and Development Project of China (2020YFA0710304) and Special Fund Project of Jiangsu Province for Scientific and Technological Innovation in Carbon Peaking and Carbon Neutrality (BK20220023). We thank Prof. Wenqing Yao of the Analysis Center, Tsinghua University, for her help with XPS/in situ XPS/UPS. We thank the 1W1A station at the Beijing Synchrotron Radiation Facility for help with PXRD. We thank the Catalysis and Surface Science End station at the BL11U beamline in the National Synchrotron Radiation Laboratory (NSRL) for help with in situ XANES.References
- Z. Teng, Q. Zhang, H. Yang, K. Kato, W. Yang, Y.-R. Lu, S. Liu, C. Wang, A. Yamakata and C. Su, Nat. Catal., 2021, 4, 374–384 Search PubMed.
- J. Kosco, M. Bidwell, H. Cha, T. Martin, C. T. Howells, M. Sachs, D. H. Anjum, S. Gonzalez Lopez, L. Zou and A. Wadsworth, Nat. Mater., 2020, 19, 559–565 CrossRef CAS.
- Y. Zhang, C. Pan, G. Bian, J. Xu, Y. Dong, Y. Zhang, Y. Lou, W. Liu and Y. Zhu, Nat. Energy, 2023, 8, 361–371 CrossRef CAS.
- A. M. Evans, L. R. Parent, N. C. Flanders, R. P. Bisbey, E. Vitaku, M. S. Kirschner, R. D. Schaller, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Science, 2018, 361, 52–57 Search PubMed.
- C. Gropp, T. Ma, N. Hanikel and O. M. Yaghi, Science, 2020, 370, eabd6406 CrossRef CAS.
- X. Liu, D. Huang, C. Lai, G. Zeng, L. Qin, H. Wang, H. Yi, B. Li, S. Liu and M. Zhang, Chem. Soc. Rev., 2019, 48, 5266–5302 Search PubMed.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- M. Wang, M. Ballabio, M. Wang, H.-H. Lin, B. P. Biswal, X. Han, S. Paasch, E. Brunner, P. Liu and M. Chen, J. Am. Chem. Soc., 2019, 141, 16810–16816 Search PubMed.
- R. Chen, Y. Wang, Y. Ma, A. Mal, X.-Y. Gao, L. Gao, L. Qiao, X.-B. Li, L.-Z. Wu and C. Wang, Nat. Commun., 2021, 12, 1354 CrossRef CAS.
- Y. Zhang, J. Zhang, S. Dou, H. Shang, J. Xu, Y. Dong, Y. Zhang, Y. Lou, C. Pan and Y. Zhu, Unveiling the Role of Excited-State Dipole Moment: Governing Non-Sacrificial H2O2 Generation on Porphyrin Photocatalysts, Angew Chem., Int. Ed. Engl., 2025, e202512844, DOI:10.1002/anie.202512844.
- Z. Xue, B. Zhang, Q. Guo, Y. Wang, Q. Li, K. Yang and S. Qiao, Sacrificial-Agent-Triggered Mass Transfer Gating in Covalent Organic Framework for Hydrogen Peroxide Photocatalysis, Adv. Mater., 2025, 37, e10201 CrossRef CAS.
- H. Tan, P. Zhou, Y. Gu, Y. Liu, W. Chen, H. Guo, F. Lin, H. Luo, X. Cao, L. Zeng, M. Luo and S. Guo, Separation-free artificial photosynthesis of concentrated hydrogen peroxide and value-added fuels over Ta atomic sites, Nat. Commun., 2025, 16, 8784 CrossRef PubMed.
- D. Li, C. Li, L. Zhang, H. Li, L. Zhu, D. Yang, Q. Fang, S. Qiu and X. Yao, J. Am. Chem. Soc., 2020, 142, 8104–8108 CrossRef CAS.
- H. Wang, Y. Yang, X. Yuan, W. L. Teo, Y. Wu, L. Tang and Y. Zhao, Mater. Today, 2022, 53, 106–133 Search PubMed.
- C. Kang, K. Yang, Z. Zhang, A. K. Usadi, D. C. Calabro, L. S. Baugh, Y. Wang, J. Jiang, X. Zou and Z. Huang, Nat. Commun., 2022, 13, 1370 CrossRef CAS.
- M. Wang, S. Fu, P. Petkov, Y. Fu, Z. Zhang, Y. Liu, J. Ma, G. Chen, S. M. Gali and L. Gao, Nat. Mater., 2023, 22, 880–887 Search PubMed.
- T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L.-H. Li, Y. Wang, J. Su and J. Li, Science, 2018, 361, 48–52 Search PubMed.
- L. Li, Q. Yun, C. Zhu, G. Sheng, J. Guo, B. Chen, M. Zhao, Z. Zhang, Z. Lai and X. Zhang, J. Am. Chem. Soc., 2022, 144, 6475–6482 CrossRef CAS.
- Y.-Y. Gu, J. Wang, Q. Tang, H. Wei, J. Ning, X. Lan, X. Wang, X. Li, Y. Jia, S. Wang and L. Hao, Insights into Substituent Effects on the Fundamental Photocatalytic Processes of Covalent Organic Frameworks toward H2 Evolution and H2O2 Production Reactions, ACS Catal., 2024, 14, 11262–11272 CrossRef CAS.
- Y. Feng, G. Wang, R. Liu, X. Ye, S. Tao, M. A. Addicoat, Z. Li, Q. Jiang and D. Jiang, Photoresponsive Covalent Organic Frameworks: Visible-Light Controlled Conversion of Porous Structures and Its Impacts, Angew Chem., Int. Ed. Engl., 2024, 63, e202400009 Search PubMed.
- F. Ma, T. Gao, X. Sun, C. Han, Y. Wang, A. Jiang, Y. Zhou, G. Liang, H. Wang, L. Wang, B. Jia, Y. Huang, H. Huang, X. Y. Kong, H. Li, N. Huang, T. Ma and L. Ye, Keto-enol tautomerism as dynamic electron/hole traps promote charge carrier separation for hydrogen peroxide photosynthesis, Nat. Commun., 2025, 16, 7432 CrossRef CAS.
- X. X. Wang, C. R. Zhang, R. X. Bi, Z. H. Peng, A. M. Song, R. Zhang, H. X. He, J. X. Qi, J. W. Gong, C. P. Niu, R. P. Liang and J. D. Qiu, Reducing the Exciton Binding Energy of Covalent Organic Framework Through π-Bridges to Enhance Photocatalysis, Adv. Funct. Mater., 2025, 35, 2421623 CrossRef CAS.
- L. Zou, Z. A. Chen, D. H. Si, S. L. Yang, W. Q. Gao, K. Wang, Y. B. Huang and R. Cao, Boosting CO(2) Photoreduction via Regulating Charge Transfer Ability in a One-Dimensional Covalent Organic Framework, Angew Chem., Int. Ed. Engl., 2023, 62, e202309820 CrossRef CAS.
- K. Yu, P. He, D. Zhang, H. Wu, K. Shu, H. Long, Y. Sun, X. Pei, B. Jiang, K. Cao, Y. Li and L. Ma, Full-Dimensional Control of Covalent Organic Frameworks Through Repulsion-Torsion Effect: Prominent 1D Above 2D and 3D Structures, Angew Chem., Int. Ed. Engl., 2025, 137 Search PubMed.
- Y. Wang, W. Hao, H. Liu, R. Chen, Q. Pan, Z. Li and Y. Zhao, Nat. Commun., 2022, 13, 100 CrossRef CAS.
- W. Li, X. Huang, T. Zeng, Y. A. Liu, W. Hu, H. Yang, Y. B. Zhang and K. Wen, Angew. Chem., Int. Ed., 2021, 60, 1869–1874 CrossRef CAS.
- Z. Chen, J. Wang, M. Hao, Y. Xie, X. Liu, H. Yang, G. I. Waterhouse, X. Wang and S. Ma, Nat. Commun., 2023, 14, 1106 CrossRef CAS.
- Y. Song, A. Li, P. Li, L. He, D. Xu, F. Wu, F. Zhai, Y. Wu, K. Hu and S. Wang, Chem. Mater., 2022, 34, 2771–2778 CrossRef CAS.
- H. Wang, H. Wang, Z. Wang, L. Tang, G. Zeng, P. Xu, M. Chen, T. Xiong, C. Zhou and X. Li, Chem. Soc. Rev., 2020, 49, 4135–4165 RSC.
- R. Xia, X. Zheng, C. Li, X. Yuan, J. Wang, Z. Xie and X. Jing, ACS Nano, 2021, 15, 7638–7648 CrossRef CAS.
- C. Qin, X. Wu, L. Tang, X. Chen, M. Li, Y. Mou, B. Su, S. Wang, C. Feng and J. Liu, Nat. Commun., 2023, 14, 5238 CrossRef CAS.
- X. Zhang, K. Geng, D. Jiang and G. D. Scholes, J. Am. Chem. Soc., 2022, 144, 16423–16432 CrossRef CAS.
- A. M. Evans, A. Giri, V. K. Sangwan, S. Xun, M. Bartnof, C. G. Torres-Castanedo, H. B. Balch, M. S. Rahn, N. P. Bradshaw and E. Vitaku, Nat. Mater., 2021, 20, 1142–1148 CrossRef CAS PubMed.
- Y. Mou, X. Wu, C. Qin, J. Chen, Y. Zhao, L. Jiang, C. Zhang, X. Yuan, E. Huixiang Ang and H. Wang, Angew. Chem., Int. Ed., 2023, 62, e202309480 CrossRef CAS PubMed.
- R. Luo, X. Luo, H. Xu, S. Wan, H. Lv, B. Zou, Y. Wang, T. Liu, C. Wu, Q. Chen, S. Yu, P. Dong, Y. Tian, K. Xi, S. Yuan, X. Wu, H. Ju and J. Lei, J. Am. Chem. Soc., 2024, 146, 16681–16688 Search PubMed.
- J. N. Chang, Q. Li, J. W. Shi, M. Zhang, L. Zhang, S. Li, Y. Chen, S. L. Li and Y. Q. Lan, Angew. Chem., 2023, 135, e202218868 Search PubMed.
- C. Wang, Z. Zhang, Y. Zhu, C. Yang, J. Wu and W. Hu, Adv. Mater., 2022, 34, 2102290 CrossRef CAS.
- H. Cheng, H. Lv, J. Cheng, L. Wang, X. Wu and H. Xu, Adv. Mater., 2022, 34, 2107480 Search PubMed.
- S. Bi, P. Thiruvengadam, S. Wei, W. Zhang, F. Zhang, L. Gao, J. Xu, D. Wu, J.-S. Chen and F. Zhang, J. Am. Chem. Soc., 2020, 142, 11893–11900 CrossRef CAS PubMed.
- K. Wu, X.-Y. Liu, P.-W. Cheng, Y.-L. Huang, J. Zheng, M. Xie, W. Lu and D. Li, J. Am. Chem. Soc., 2023, 145, 18931–18938 CrossRef CAS PubMed.
- H. Ben, G. Yan, H. Liu, C. Ling, Y. Fan and X. Zhang, Adv. Funct. Mater., 2022, 32, 2104519 CrossRef CAS.
- J. P. Jeon, Y. J. Kim, S. H. Joo, H. J. Noh, S. K. Kwak and J. B. Baek, Angew. Chem., 2023, 135, e202217416 CrossRef.
- G. Fu, D. Yang, S. Xu, S. Li, Y. Zhao, H. Yang, D. Wu, P. S. Petkov, Z.-A. Lan and X. Wang, J. Am. Chem. Soc., 2024, 146, 1318–1325 CrossRef CAS PubMed.
- Z. Zhang, Y. Zhu, X. Chen, H. Zhang and J. Wang, Adv. Mater., 2019, 31, 1806626 CrossRef.
- R. Chen, C. Ni, J. Zhu, F. Fan and C. Li, Nat. Protoc., 2024, 1–33 Search PubMed.
- Y. Guo, Q. Zhou, J. Nan, W. Shi, F. Cui and Y. Zhu, Nat. Commun., 2022, 13, 2067 Search PubMed.
- Y. Sheng, W. Li, L. Xu and Y. Zhu, Adv. Mater., 2022, 34, 2102354 CrossRef CAS PubMed.
- R. Chen, S. Pang, H. An, J. Zhu, S. Ye, Y. Gao, F. Fan and C. Li, Nat. Energy, 2018, 3, 655–663 Search PubMed.
- S. Jin, X. Ding, X. Feng, M. Supur, K. Furukawa, S. Takahashi, M. Addicoat, M. E. El-Khouly, T. Nakamura and S. Irle, Angew. Chem., Int. Ed., 2013, 52, 2017–2021 CrossRef CAS PubMed.
- S. Jin, M. Supur, M. Addicoat, K. Furukawa, L. Chen, T. Nakamura, S. Fukuzumi, S. Irle and D. Jiang, J. Am. Chem. Soc., 2015, 137, 7817–7827 CrossRef CAS.
- M. A. Herranz, C. Ehli, S. Campidelli, M. Gutiérrez, G. L. Hug, K. Ohkubo, S. Fukuzumi, M. Prato, N. Martín and D. M. Guldi, J. Am. Chem. Soc., 2008, 130, 66–73 Search PubMed.
- M. Ohtani and S. Fukuzumi, Chem. Commun., 2009, 4997–4999 RSC.
- J. Malig, N. Jux, D. Kiessling, J. J. Cid, P. Vazquez, T. Torres and D. M. Guldi, Angew. Chem., Int. Ed., 2011, 50, 3561–3565 CrossRef CAS PubMed.
- C. Wang, B. Wu, Y. Li, T. Dong, Y. Chai, Y. Zhang and C. Wang, Angew. Chem., Int. Ed., 2023, 62, e202300377 Search PubMed.
- Y. Chai, L. Liu, Y. Xu, X. Liu, C. Wang, Y. Bo, Y. Zhang, Z. Wang, Y. Weng and D. M. Guldi, J. Am. Chem. Soc., 2023, 145, 14190–14195 CrossRef CAS PubMed.
- J. A. Christensen, B. T. Phelan, S. Chaudhuri, A. Acharya, V. S. Batista and M. R. Wasielewski, J. Am. Chem. Soc., 2018, 140, 5290–5299 Search PubMed.
- T. W. Kim, S. Jun, Y. Ha, R. K. Yadav, A. Kumar, C.-Y. Yoo, I. Oh, H.-K. Lim, J. W. Shin and R. Ryoo, Nat. Commun., 2019, 10, 1873 Search PubMed.
- J. Yang, A. Acharjya, M.-Y. Ye, J. Rabeah, S. Li, Z. Kochovski, S. Youk, J. Roeser, J. Grüneberg, C. Penschke, M. Schwarze, T. Wang, Y. Lu, R. van de Krol, M. Oschatz, R. Schomäcker, P. Saalfrank and A. Thomas, Angew. Chem., Int. Ed., 2021, 60, 19797–19803 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |