
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible photon energy storage by [2+2] cycloaddition of Pd-oxazolones
Qianfeng
Qiu
a,
Junichi
Usuba
b,
Wai Lean
Koay
c,
Vincent J. O.
Conrad
 d,
Nathan M.-W.
Wu
a,
Shao-Liang
Zheng
e,
Vinh Xuan
Truong
c and
Grace G. D.
Han
*ad
d,
Nathan M.-W.
Wu
a,
Shao-Liang
Zheng
e,
Vinh Xuan
Truong
c and
Grace G. D.
Han
*ad
aDepartment of Chemistry, Brandeis University, Waltham, MA, USA. E-mail: grace_han@ucsb.edu
bResearch Center for Net Zero Carbon Society, Institute of Innovation for Future Society, Nagoya University, Nagoya 464-8603, Japan
cInstitute of Sustainability for Chemicals, Energy and Environment (ISCE2), Agency for Science, Technology and Research (A*STAR), 1 Pesek Road, Jurong Island, Singapore 627833, Republic of Singapore
dDepartment of Chemistry and Biochemistry, University of California, Santa Barbara, CA, USA
eDepartment of Chemistry and Chemical Biology, Harvard University, Cambridge, MA, USA
First published on 11th December 2025
Abstract
We report a new class of ortho-palladated oxazolone complexes that capture and release solar energy through visible-light-triggered [2 + 2] photocycloaddition reactions. The free oxazolone ligands weakly absorb near 400 nm and undergo slow and incomplete intermolecular dimerization, yielding multiple stereoisomers of cycloadducts. Upon ortho-palladation, the resulting Pd(II) complexes exhibit strong charge-transfer absorption bands in the visible range (430–570 nm) and adopt a clamshell geometry that preorganizes the reactive alkenes in close spatial proximity. This unique coordination environment facilitates fast, near-quantitative, and reversible [2 + 2] photocycloaddition in both solution and amorphous solid state under the irradiation of green light or standard sunlight. The cycloadducts formed are stable, isolable solids with long thermal half-lives (up to 504 days at 298 K), enabling long-term energy storage. The combination of visible-light responsiveness, robust reversibility, and solid-state reactivity demonstrates the potential of transition metal complexes as a promising molecular platform for molecular solar thermal energy storage applications.
Introduction
Molecular solar thermal (MOST) energy storage systems harness reversible photo-induced reactions to capture and store photon energy in metastable chemical bonds, subsequently releasing it as heat upon external stimulation. Historically, MOST compounds have largely revolved around established photoswitches like azo(hetero)arenes,1–4 hydrazones,5 dihydroazulenes,6,7 fulvalene diruthenium complexes,8,9 and norbornadienes.10–15 These systems typically rely on E–Z isomerization1–5 or intramolecular cycloadditions10–15 to achieve substantial enthalpic differences between their thermodynamically stable and metastable states.Despite significant research dedicated to optimizing light absorption ranges, photoisomerization quantum yields, thermal half-lives, and energy storage densities in these photoswitches,3,6,11,14,16 energy storage via intermolecular photochemical reactions has remained comparatively underexplored. Intermolecular [2 + 2] photocycloadditions, in particular, offer a unique avenue to access highly strained cycloadducts, thereby elevating their energy state. Recent progress in solid-state photochemistry has greatly expanded the scope of reactions in the field,17–31 leading to novel materials with tunable optical and physical properties.20,27,29 Particularly, advancements in topologically-controlled, intermolecular cycloadditions17,18,21,22,24–26,31 have hinted at the feasibility of solid-state MOST systems based on crystalline [2 + 2] and [4 + 4] photodimerizations, further demonstrated by compounds like styrylpyryliums,32 anthracene derivatives,33 and diazetidines.34 However, these systems face a critical limitation: the shallow light penetration depth caused by surface reflection in solids. While such cycloadditions can proceed in solution,35–37 they typically suffer from competing side reactions and the formation of multiple stereoisomeric cycloadducts in modest yields. Moreover, conventional [2 + 2] and [4 + 4] cycloadditions generally require ultraviolet (UV, <400 nm) activation, further constraining their practical utility.
Oxazolone-based metal complexes present a promising solution to these challenges. While some free oxazolone derivatives with λmax ∼ 380 nm have been reported to undergo high-yield dimerization (75% to 100%),38 relatively high concentrations and long reaction times are needed (Fig. 1a). In an optimal flow microreactor, 200 mM THF-d8 solutions undergo complete conversion in 30 minutes, while batch reactions in 6 mM CD2Cl2 require 72 hours. Stereoselectivity for the most thermodynamically stable product, i.e., the epsilon isomer, ranges from 38% to 88%, among a mixture of up to 4 isomers, partly due to Z–E isomerization of oxazolone. However, the regioselective ortho-palladation of the oxazolones via C–H activation dramatically transforms their properties.39 These resulting Pd(II) complexes display significant bathochromic shifts (λmax ∼460 nm),40 which enables efficient visible-light-triggered [2 + 2] photocycloaddition (Fig. 1b). The formation of a clamshell structure with carboxylate bridges organizes the oxazolones to undergo efficient [2 + 2] cycloaddition with high stereoselectivity.
 | ||
| Fig. 1 (a) Chemical structures of oxazolones 1 and 2 and their partial [2 + 2] cycloaddition in solutions. (b) Chemical structures of Pd-oxazolone complexes and their complete and reversible [2 + 2] cycloaddition. (c) The energy diagram of visible-light-triggered (at 505 nm) photon energy storage via [2 + 2] cycloaddition of Pd-oxazolones and energy release upon thermally triggered reversion of their cycloadducts. ΔH‡: enthalpy of thermal activation for cycloreversion; ΔHstorage: energy storage density of metastable state. | ||
Historically, studies on these complexes have primarily emphasized their synthetic utility,39,41,42 most notably the stereoselective photocycloaddition enabling access to functionalized 1,3-diaminotruxillic acids.39,42 Investigations into the stability of various cycloadducts have shown that those incorporating electron-donating substituents or heteroaryl groups often undergo partial or complete cycloreversion in the dark.39,43 While such behavior was originally regarded as a limitation to the synthetic scope of these reactions, it in fact provides strong motivation to exploit the reversibility of the process for MOST energy storage and release. To the best of our knowledge, however, no prior studies have evaluated palladated cycloadducts as potential molecular energy storage units.
Herein, we introduce ortho-palladated oxazolones that undergo reversible inter-ligand [2 + 2] photocycloaddition in both solution and solid states. The palladation not only enables visible-light absorption (430–570 nm) but also enforces a geometry conducive to controlled and reversible dimerization. This work represents the first demonstration of solar thermal energy storage in Pd complexes and provides a critical molecular design framework for the future development of sunlight-driven, solution-free, and scalable MOST systems.
Results and discussion
We synthesized the oxazolone ligands (1 and 2) and their corresponding Pd(II) complexes (1-Pd and 2-Pd) using established methods, achieving satisfactory yields (details in the experimental section, Fig. S1). This synthetic protocol is straightforward, readily scalable, and potentially sustainable, as the oxazolone ligands can be prepared from bio-sourced compounds such as vanillin and hippuric acid. The 2-(acetyloxy)ethoxy group (R) was additionally incorporated in ligand 2 and 2-Pd with the specific aim of tuning its solid-state phase and reactivity (vide infra). The chemical structures of all ligands and complexes were confirmed through nuclear magnetic resonance (NMR) spectroscopy (Fig. S2–S9) and high-resolution mass spectrometry (HR-MS) (Fig. S10 and S11).Upon 405 nm irradiation in solutions (4.5–5.4 mM, ∼2 mg mL−1), ligands 1 and 2 underwent slow [2 + 2] cycloaddition to generate multiple stereoisomers, reaching conversions of 75% (1-CBs) and 62% (2-CBs), respectively, after 3 days of continuous irradiation (Fig. S12 and S13). Both 1-CBs and 2-CBs could near-quantitatively revert to the respective ligands upon heating up to 200 °C over 1 hour (Fig. S12 and S13), but no detectable heat release was observed, indicating a negligible energy difference between the ligands and their cycloadducts. Consequently, further structural identification of stereoisomers and thermal kinetics characterization of the ligand CBs were not pursued.
In contrast, the ortho-palladated oxazolones, 1-Pd and 2-Pd (1.5–1.7 mM, ∼2 mg mL−1), demonstrate drastically improved inter-ligand [2 + 2] cycloaddition under the irradiation at 505 nm within 3 hours, leading to the near-quantitative formation of their metastable cycloadduct, 1-Pd-CB and 2-Pd-CB, respectively, as a single product (Fig. 1b and S14–S15). The bidentate coordination of Pd(II) rigidly fixes the distance between the two reactive oxazolone ligands. This preorganization is instrumental in facilitating the [2 + 2] cycloaddition reaction, dramatically enhancing reactivity compared to the bimolecular reaction among free ligands in solution.38 This photochemical transformation is the core mechanism by which photon energy is captured and stored within the strained chemical bonds of the newly formed cyclobutane rings. The stored energy, quantifiable as the enthalpy difference (ΔHstorage) between the thermodynamically stable Pd-oxazolone complexes and their high-energy cyclobutane adducts, can then be released as heat through a thermally triggered reversion (Fig. 1c and S16–S17). We precisely measured ΔHstorage by differential scanning calorimetry (DSC), providing a direct assessment of the energy storage capacity (vide infra).
Our molecular design was carefully constructed to enable visible light responsiveness. First, the strategic ortho-palladation via C–H activation plays a crucial role in this case. The dπ–pπ conjugation induces a red-shift in absorption along with an increase in molar absorptivity of Pd complexes.40 In addition, we placed electron-donating methoxy and 2-(acetyloxy)ethoxy groups onto the phenyl rings. To evaluate the influence of the electron-donating groups on the photophysical properties of Pd complexes, we performed structural optimizations and vertical excitation energy calculations on 1-Pd, along with reference compounds (1-Pd′ and 1-Pd″) in which the methoxy groups were partially or completely replaced by hydrogen atoms (Fig. 2a, S18–S19 and Tables S1–S3). In the methoxy-functionalized structures, the pronounced elevation of the HOMO and HOMO − 1 level reduces the energy for the S0–S1 and S0–S2 transitions. Vertical excitation energy calculations clearly show that 1-Pd with eight methoxy substituents presents the lowest-energy S0–S2 transition (HOMO − 1 → LUMO excitation) compared to other compounds with four or no methoxy groups. These computational results indicate that the introduction of electron-donating groups extends orbital delocalization and induces a red-shift in the absorption wavelength.
Such molecular designs give rise to striking differences in the absorption profiles of the ligands and their Pd complexes. Ligands 1 and 2 display primary absorption bands in the 400–450 nm range (Fig. 2b and S20a), whereas the corresponding Pd complexes (Fig. 2c and S20b) exhibit a red-shifted profile with strong absorption around 500 nm and an onset near 570 nm. The photophysical responses are also markedly different: in dilute solutions (2.0 × 10−5 M), ligands 1 and 2 show little or no spectral change upon irradiation at 430 nm over 1 hour, indicating the negligible yield of dimerization (Fig. 2b and S20a). This result suggests that the concentration of ligands plays a critical role in governing the rate of intermolecular cycloaddition. In contrast, the dilute solutions of 1-Pd and 2-Pd (2 × 10−5 M) undergo rapid [2 + 2] cycloaddition under 2 min upon exposure to 505 nm light (Fig. 2c and S20b), displaying negative photochromism characterized by the near-complete loss of absorption peaks at 500 nm and 400 nm, and the emergence of a blue-shifted band around 360 nm, consistent with cyclobutane formation. At the photostationary state (PSS), the conversion to the cycloadducts was nearly quantitative, reaching 97% for 1-Pd and 95% for 2-Pd, characterized by NMR (Fig. S14 and S15).
The practical utility of these complexes was further highlighted by their near-quantitative (97–98%) conversion upon direct sunlight irradiation over 6 hours at higher concentration (1.5–1.7 mM, ∼2 mg mL−1) (NMR characterization shown in Fig. S14 and S15), demonstrating their potential for direct solar energy harvesting. Furthermore, we determined substantial photochemical reaction quantum yields (QYs) of 25.8 ± 2.4% for 1-Pd and 29.9 ± 0.5% for 2-Pd (Fig. S21 and S22). These QYs are at the higher end of those reported for molecular systems in which two photochromes are covalently linked to promote intramolecular [2 + 2] or [4 + 4] cycloadditions (Fig. 2d). Notably, 1-Pd and 2-Pd are the only molecular systems that undergo such cycloadditions under visible light irradiation (>500 nm), whereas all other systems strictly require UV (300–400 nm) irradiation. The strong visible-light (505 nm) absorption, negative photochromism, near-quantitative PSS ratios, and high QYs (Table 1) underscore the high efficiency of Pd-oxazolone system for solar energy harvesting.
 | ||
| Fig. 2 (a) Energy diagram and TD-DFT vertical excitation energies of 1-Pd, 1-Pd′, and 1-Pd″ calculated at the (TD-)B3LYP functionals with 6-31G** (for H, C, N, O, and F) and Lanl2DZ (for Pd atoms) basis sets. Solution-state UV-vis absorption spectra of (b) 2 and (c) 2-Pd in DCM (2 × 10−5 M), upon light irradiation. 2-Pd-CB reverts to 2-Pd upon heating, restoring the initial absorption spectrum. (d) Quantum yield (QY) of intramolecular photo-cycloaddition for covalently-linked systems: bis-thiomaleimides (TMI, green, λmax = 354),35ortho-dianthryl benzenes (ortho-9-ABz, purple, avg. λmax = 372),44 bis(18-crown-6) stilbene-alkanediammonium ions (SB-ADA, light blue, avg. λmax = 332),36 bis(9-anthryl) alkanes (9-AA, orange, λmax = 395),45 styrylbenzoquinoline (SBQ, dark blue, λmax = 323)37 and Pd-linked oxazolones (1- and 2-Pd, red, avg. λmax = 472 nm). | ||
| Cycloaddition | Cycloreversion | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| λ max (nm) | PSS505 (% CB) | PSSSun (% CB) | Φ 505 | ΔH‡ (kJ mol−1) | ΔS‡ (J mol−1 K−1) | ΔG‡ (kJ mol−1) | ΔHstorage (kJ mol−1) | t 1/2 (days) | |
| a λ max: wavelength of maximum absorption; PSS505 and PSSSun: photostationary state of cycloaddition (Pd to Pd-CB) under 505 nm and standard solar irradiation; Φ505: quantum yield of cycloaddition under 505 nm light irradiation; ΔH‡, ΔS‡, ΔG‡: enthalpy, entropy and Gibbs free energy values of thermal activation for cycloreversion, respectively; ΔHstorage: energy released as heat during cycloreversion; t1/2: thermal half-life of metastable state. | |||||||||
| 1-Pd | 474 | 97 | 98 | 0.26 | 98.1 | −29.1 | 106.1 | 34.2 | 276 |
| 2-Pd | 471 | 95 | 97 | 0.30 | 98.1 | −34.2 | 107.4 | 35.6 | 504 |
In order to understand their potential energy storage in solid state, we systematically examined the structure and phase behavior of the Pd complexes, revealing a pronounced difference between 1-Pd and 2-Pd. For crystalline 1-Pd, we performed single-crystal X-ray diffraction analysis (Fig. 3a, S23–S24, and Tables S4–S5). The crystal structure of 1-Pd reveals a head-to-tail stacking arrangement between two oxazolone ligands within the complex, largely driven by the favorable π interactions. The distances between reactive C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds were measured to be 3.24 and 3.33 Å (Fig. 3b), which falls within the accepted range required for [2 + 2] cycloaddition per Schmidt's principle.
C bonds were measured to be 3.24 and 3.33 Å (Fig. 3b), which falls within the accepted range required for [2 + 2] cycloaddition per Schmidt's principle.
 | ||
| Fig. 3 (a) Refined single-crystal structure of 1-Pd and (b) the zoomed-in top view and side view, showing head-to-tail stacking of two adjacent oxazolone ligands and the distance and dihedral angle between potentially reactive CC bonds. (c) Hirshfeld surface analysis of the crystal structure (1-Pd) showing the intermolecular interactions (C–H⋯O) between methoxy and adjacent methyl groups. | ||
However, 1-Pd did not exhibit any photo-reactivity in crystalline state despite the proximity between the neighboring reactive alkenes (Fig. S25). In the crystal structure, we noticed a significant dihedral angle (40.6°) between the two CC bonds (C8–C7–C28–C27), which may prevent the necessary orbital overlap required for efficient photocycloaddition in crystals (Fig. 3b). In addition, Hirshfeld surface analysis suggests a dense packing of 1-Pd complexes and insufficient void space in the crystal (Fig. 3c), which could limit the solid-state reactivity of the compound. This finding highlights that in crystalline-state photoreactions, both close molecular proximity and precise geometric alignment of reacting alkenes are essential.
Surprisingly, solid-state 2-Pd underwent successful [2 + 2] photocycloaddition under the irradiation of 505 nm and sunlight, despite the minor difference in the chemical structure from 1-Pd (Fig. S26). In powder X-ray diffraction (PXRD) analysis, 2-Pd exhibited amorphous features, and its cycloadducts also remained amorphous (Fig. 4a). We were unable to obtain any single crystals of 2-Pd or generate a microcrystalline form, so no crystalline reference exists for a quantitative phase purity analysis. In contrast, 1-Pd displayed sharp diffraction peaks that shifted but remained well-defined upon cycloaddition. The PXRD pattern obtained from the single-crystal structure of 1-Pd closely matched the powder PXRD data (Fig. 4a). Differential scanning calorimetry further corroborated the phase distinction: 1-Pd showed a clear melting transition at 175 °C (Fig. S27), consistent with crystallinity, whereas 2-Pd remained amorphous across the temperature range studied (−90 to 200 °C), with no observable melting or crystallization transitions upon heating or cooling (Fig. S28). Thermogravimetric analysis (Fig. S29) confirmed the high thermal stability of both complexes under 200 °C, regardless of their solid-state phase.
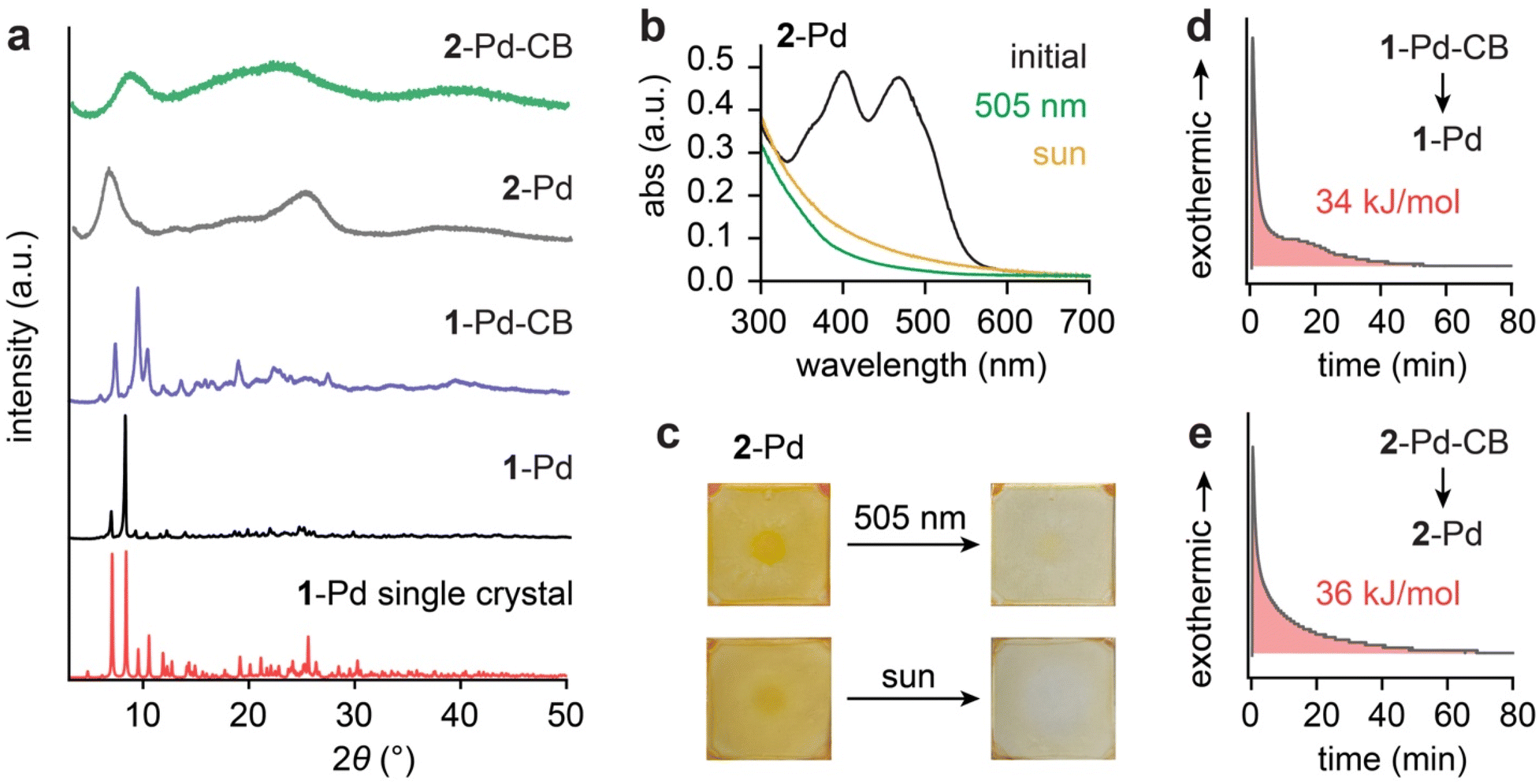 | ||
| Fig. 4 (a) Powder XRD patterns of 1-Pd (black), 1-Pd-CB (purple), 2-Pd (grey), and 2-Pd-CB (green) along with the simulated XRD pattern of 1-Pd (red) from single-crystal diffraction. (b) Solid-state UV-vis absorption spectra of 2-Pd (black) and 2-Pd-CB (green and yellow) thin films after 40 hours of irradiation. (c) Optical images of thin film samples after 40 hours of irradiation. DSC isothermal plots illustrating the cycloreversion of (d) 1-Pd-CB to 1-Pd at 110 °C and (e) 2-Pd-CB to 2-Pd at 80 °C. | ||
We hypothesize that the presence of the flexible 2-(acetyloxy)ethoxy group in 2-Pd disrupts the orderly molecular packing, thereby preventing the formation of a crystalline lattice. We examined the solid-state cycloaddition of amorphous 2-Pd in thin films under the irradiation of simulated sunlight (AM 1.5, 1 Sun) and 505 nm LED over 40 hours (Fig. 4b and c). Disappearance of 2-Pd's spectral features was observed in the solid-state UV-vis spectra, indicating the successful conversion to 2-Pd-CB. We confirmed that the observed spectral changes arose exclusively from the cycloaddition by preparing thicker sandwich films of 2-Pd (Fig. S26), which showed no signs of degradation by 1H NMR analysis and exhibited partial conversion to 2-Pd-CB (28%) upon solar irradiation. These results, together with the lack of reactivity observed for solid 1-Pd (Fig. S25), strongly suggest that in the crystalline state, precise geometric alignment of the alkenes is essential for photocycloaddition. In contrast, these stringent requirements are relaxed in solution or amorphous states due to the conformational flexibility available in such media. Specifically, we hypothesize that the weaker non-covalent interactions among the disordered molecules in the amorphous solid provide the molecular flexibility needed for intramolecular rotation in 2-Pd, allowing the alkene groups to transiently adopt the parallel alignment and close distance required for the photocycloaddition. These weak interactions also permit the substantial changes in molecular shape and volume that accompany formation of the cyclobutane photoproduct.
The quantitative measurement of energy storage density (ΔHstorage) was performed via isothermal DSC, which monitored the exothermic heat release during the cycloreversion of CB. The isothermal DSC condition was chosen to isolate the exothermic signal from the cycloreversion, avoiding the overlap with the subsequent endothermic phase transition of the regenerated Pd complexes that is observed in conventional DSC scans over a wide temperature range (Fig. S27 and S28). The reversion of 1-Pd-CB to 1-Pd released about 34 kJ mol−1 (29 J g−1) of energy (Fig. 3d) and 2-Pd-CB to 2-Pd approximately 36 kJ mol−1 (27 J g−1) (Fig. 3e). We note that these energy densities are on the lower side when compared to the reported [2 + 2] MOST systems, such as norbornadienes (89 kJ mol−1 or 399 J g−1),14 diazetidines (146 kJ mol−1 or 318 J g−1)34 and styrylpyriliums (42 kJ mol−1 or 51 J g−1).32 This difference can be largely attributed to the nature of the energy storage mechanism in the Pd-oxazolone complexes. Unlike systems that undergo photoinduced dearomatization, such as 2-phenylbenzoxazoles forming diazetidines via [2 + 2] cycloaddition, or norbornadienes converting to quadricyclanes with severely strained fused rings, energy storage in our Pd complexes arises primarily from the ring strain of the cyclobutane photoproduct. As a result, the stored energy is moderate, comparable to that of styrylpyrylium systems, which also generate cyclobutanes. While not reaching high energy densities, the strong visible light absorption at 505 nm, rapid and near-quantitative isomerization, (photo)chemical robustness, and facile cycloreversion and heat release are unique advantages the Pd-oxazolone system offers. We note that introducing a sterically bulky substituent at the benzylic position may increase ring strain in the cyclobutane, thereby further enhancing the ΔHstorage value. In addition, incorporating lighter metal centers such as Ni in place of Pd can reduce the overall molecular weight of the system, which is expected to improve the gravimetric energy density.
Crucial for practical applications, high thermal stability of the metastable state (Pd-CB) is desired for long-term energy storage without losses over time until triggered. We characterized the thermal half-lives of 1-Pd-CB and 2-Pd-CB at 298 K, determining them to be exceptionally long: 276 days and 504 days, respectively (Table 1 and Fig. S30–S31). The notable difference in half-life, with 2-Pd-CB exhibiting superior stability, is primarily attributed to a higher activation entropy associated with its reversion back to 2-Pd, suggesting a more kinetically hindered thermal decay pathway for the 2-(acetyloxy)ethoxy functionalized complex.
Conclusions
We developed a sunlight responsive MOST system based on ortho-palladated oxazolone complexes that undergo reversible [2 + 2] photocycloaddition near-quantitatively in solution and in amorphous solid states. These complexes exhibit substantial quantum yields and long thermal half-lives, allowing for efficient and long-term solar energy storage. Importantly, we confirm the importance of the conformational freedom of the [2 + 2] cycloaddition system in amorphous solids, which enabled the reactions that are otherwise unfavorable in crystalline state. We believe the molecular design strategy and phase engineering demonstrated here will inspire new efforts in developing sunlight-driven, amorphous-state solar thermal materials with practical energy storage and release capabilities.Author contributions
Q. Q. performed TGA, DSC, solution UV-vis spectroscopy, PSS and quantum yield measurements, and PXRD characterization. J. U. carried out DFT calculations and Hirshfeld surface analysis. W. L. K. synthesized, purified, and structurally characterized the ligands and Pd complexes. V. J. O. C.. conducted thin-film spectroscopy and revised figures. N. M.-W. W. provided insights into absorption properties of compounds. S.-L. Z. collected and analyzed single-crystal X-ray diffraction data. V. X. T. and G. G. D. H. conceived the project and supervised the research. All authors discussed the results and contributed to manuscript preparation.Conflicts of interest
The authors declare no conflicts of interest.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc07833d.CCDC 2493202 (1-Pd) contains the supplementary crystallographic data for this paper.46
Acknowledgements
This material is based upon work supported by the Air Force Office of Scientific Research under award number FA9550-22-1-0254. G.G.D.H. acknowledges the NSF CAREER award (DMR-2142887), 2025 Marion Milligan Mason Award for Women in the Chemical Sciences (AAAS), Alfred P. Sloan Foundation (FG-2022-18328), and the Camille and Henry Dreyfus Foundation (TC-23-028). We acknowledge NIH Shared Instrumentation grant S10OD034395 for purchase of an NMR spectrometer. We thank the support to the X-ray core facility from the Major Research Instrumentation (MRI) Program of the National Science Foundation (NSF) under Award Numbers 2216066. Q. Q was partly supported by Brandeis MRSEC (DMR-2011846), and N.M.-W.W. was supported by RCSA Collaborative Innovation Award (SA-SM3-2024-057b). V.X.T. acknowledges funding support from the A*STAR Industry Alignment Fund—Pre-Positioning Programme (IAF-PP: H23J2a0130) for the development of new polymer crosslinking technologies. PXRD patterns were obtained on a powder X-ray diffractometer funded by the AFOSR DURIP award (FA9550-23-1-0072).References
- C. Averdunk, M. Shamsabadi, K. Moth-Poulsen and H. A. Wegner, Structure–property relationship of p-alkoxyazobenzenes as molecular solar thermal phase change material energy storage systems (MOST-PCM), J. Mater. Chem. C, 2025, 13, 13337–13346 RSC.
- A. Gonzalez, M. Odaybat, M. Le, J. L. Greenfield, A. J. P. White, X. Li, M. J. Fuchter and G. G. D. Han, Photocontrolled Energy Storage in Azobispyrazoles with Exceptionally Large Light Penetration Depths, J. Am. Chem. Soc., 2022, 144, 19430–19436 Search PubMed.
- Q. Qiu, Q. Qi, J. Usuba, K. Lee, I. Aprahamian and G. G. D. Han, Visible light activated energy storage in solid-state Azo-BF2 switches, Chem. Sci., 2023, 14, 11359–11364 Search PubMed.
- D. Schatz, C. Averdunk, R. Fritzius and H. A. Wegner, An Azobenzene-Based Liquid Molecular Solar Thermal (MOST) Storage System–Energy Carrier and Solvent, Small, 2025, 21, 2502938 Search PubMed.
- Q. Qiu, S. Yang, M. A. Gerkman, H. Fu, I. Aprahamian and G. G. D. Han, Photon Energy Storage in Strained Cyclic Hydrazones: Emerging Molecular Solar Thermal Energy Storage Compounds, J. Am. Chem. Soc., 2022, 144, 12627–12631 CrossRef CAS PubMed.
- M. B. Nielsen, N. Ree, K. Mikkelsen and M. Cacciarini, Tuning the dihydroazulene - vinylheptafulvene couple for storage of solar energy, Russ. Chem. Rev., 2020, 89, 573–586 CrossRef CAS.
- Z. Wang, J. Udmark, K. Börjesson, R. Rodrigues, A. Roffey, M. Abrahamsson, M. B. Nielsen and K. Moth-Poulsen, Evaluating Dihydroazulene/Vinylheptafulvene Photoswitches for Solar Energy Storage Applications, ChemSusChem, 2017, 10, 3049–3055 CrossRef CAS.
- Y. Kanai, V. Srinivasan, S. K. Meier, K. P. C. Vollhardt and J. C. Grossman, Mechanism of Thermal Reversal of the (Fulvalene)tetracarbonyldiruthenium Photoisomerization: Toward Molecular Solar–Thermal Energy Storage, Angew. Chem., Int. Ed., 2010, 49, 8926–8929 Search PubMed.
- A. Lennartson, A. Lundin, K. Börjesson, V. Gray and K. Moth-Poulsen, Tuning the photochemical properties of the fulvalene-tetracarbonyl-diruthenium system, Dalton Trans., 2016, 45, 8740–8744 RSC.
- A. S. Aslam, M. Shamsabadi, R. J. Salthouse, J. Andréasson and K. Moth-Poulsen, Norbornadiene Quadricyclane as Multimode Photoswitches: Synergistic Light and Protonation-Controlled Heat Release, ChemSusChem, 2025, 18, e202501005 CrossRef CAS.
- E. Franz, N. Oberhof, D. Krappmann, N. Baggi, Z. Hussain, K. Moth-Poulsen, H. Hölzel, A. Hirsch, A. Dreuw, O. Brummel and J. Libuda, Tuning Electrocatalytic Energy Release in Norbornadiene Based Molecular Solar Thermal Systems Through Substituent Effects, Chem.–Eur. J., 2025, 31, e02294 CrossRef CAS PubMed.
- K. Jorner, A. Dreos, R. Emanuelsson, O. El Bakouri, I. F. Galván, K. Börjesson, F. Feixas, R. Lindh, B. Zietz, K. Moth-Poulsen and H. Ottosson, Unraveling factors leading to efficient norbornadiene–quadricyclane molecular solar-thermal energy storage systems, J. Mater. Chem. A, 2017, 5, 12369–12378 RSC.
- L. Magson, D. Maggiolo, A. S. Kalagasidis, S. Henninger, G. Munz, M. Knäbbeler-Buß, H. Hölzel, K. Moth-Poulsen, I. Funes-Ardoiz and D. Sampedro, Sustainable Heat Generation in Flow from a Molecular Solar Thermal Energy Storage System, Adv. Energy Sustainability Res., 2025, 6, 2400230 CrossRef CAS.
- J. Orrego-Hernández, A. Dreos and K. Moth-Poulsen, Engineering of Norbornadiene/Quadricyclane Photoswitches for Molecular Solar Thermal Energy Storage Applications, Acc. Chem. Res., 2020, 53, 1478–1487 CrossRef PubMed.
- Z. Wang, A. Roffey, R. Losantos, A. Lennartson, M. Jevric, A. U. Petersen, M. Quant, A. Dreos, X. Wen, D. Sampedro, K. Börjesson and K. Moth-Poulsen, Macroscopic heat release in a molecular solar thermal energy storage system, Energy Environ. Sci., 2019, 12, 187–193 RSC.
- R. R. Weber, C. N. Stindt, A. M. J. Van Der Harten and B. L. Feringa, Push-Pull Bis-Norbornadienes for Solar Thermal Energy Storage, Chem.–Eur. J., 2024, 30, e202400482 CrossRef CAS PubMed.
- M. Barba Hernández, M. G. Vasquez-Ríos, H. Höpfl and L. R. MacGillivray, Crystal Engineering of Multiple Supramolecular Heterosynthons in Cocrystals of Boronic Acids: Structures, Photoreactivities, and Catalysis, Cryst. Growth Des., 2025, 25, 38–52 CrossRef.
- G. Bolla, Q. Chen, G. Gallo, I.-H. Park, K. C. Kwon, X. Wu, Q.-H. Xu, K. P. Loh, S. Chen, R. E. Dinnebier, W. Ji and J. J. Vittal, Multifunctional Properties of a Zn(II) Coordination Complex, Crys. Growth Des., 2021, 21, 3401–3408 CrossRef CAS.
- J. Haimerl, G. C. Thaggard, B. K. P. Maldeni Kankanamalage, R. Bühler, J. Lim, K. C. Park, J. Warnan, R. A. Fischer and N. B. Shustova, Shifting Gears: Photochromic Metal–Organic Frameworks with Stimulus-Adaptable Performance, J. Am. Chem. Soc., 2025, 147, 19918–19930 CrossRef CAS.
- X. Hou, C. Ke, C. J. Bruns, P. R. McGonigal, R. B. Pettman and J. F. Stoddart, Tunable solid-state fluorescent materials for supramolecular encryption, Nat. Commun., 2015, 6, 6884 CrossRef PubMed.
- N. Juneja, G. C. George and K. M. Hutchins, Simultaneous Cycloadditions in the Solid State via Supramolecular Assembly, Angew. Chem., Int. Ed., 2025, 64, e202415567 Search PubMed.
- D. Kitagawa and S. Kobatake, in Advances in Organic Crystal Chemistry, ed. S. Kobatake and H. Uekusa, Springer Nature, Singapore, 2025, pp. 27–40 Search PubMed.
- K. Kuntze, J. Viljakka, M. Virkki, C.-Y. Huang, S. Hecht and A. Priimagi, Red-light photoswitching of indigos in polymer thin films, Chem. Sci., 2023, 14, 2482–2488 RSC.
- A. Lal and K. M. Sureshan, Isomer-dependent reactivity in the solid state: topochemical [4 + 4] vs. [4 + 2] cycloaddition reactions, Chem. Sci., 2025, 16, 13496–13502 Search PubMed.
- W. Li, T. J. Gately, D. Kitagawa, R. O. Al-Kaysi and C. J. Bardeen, Photochemical Reaction Front Propagation and Control in Molecular Crystals, J. Am. Chem. Soc., 2024, 146, 32757–32765 CrossRef CAS.
- L. Ma, G. C. George III, S. P. Kelley and K. M. Hutchins, Programmable Solid-State [2 + 2] Photocycloadditions of Dienes Directed by Structural Control and Wavelength Selection, J. Am. Chem. Soc., 2025, 147, 18249–18256 CrossRef CAS PubMed.
- P. H. Nguyen, A. M. Scheuermann, A. Nikolaev, M. L. Chabinyc, C. M. Bates and J. Read de Alaniz, Reversible Modulation of Conductivity in Azobenzene Polyelectrolytes Using Light, ACS Appl. Polym. Mater., 2023, 5, 4698–4703 CrossRef CAS.
- K. C. Park, J. Lim, G. C. Thaggard, B. K. P. Maldeni Kankanamalage, I. Lehman-Andino, Y. Liu, J. M. Burrell, C. R. Martin, A. T. Ta, A. B. Greytak, J. W. Amoroso, D. D. Diprete, M. D. Smith, S. R. Phillpot and N. B. Shustova, Stimuli-responsive photoswitch–actinide binding: a match made in MOFs, Chem. Sci., 2025, 16, 14115–14126 RSC.
- K. Wang, J. Y. Park, Akriti and L. Dou, Two-dimensional halide perovskite quantum-well emitters: A critical review, EcoMat, 2021, 3, e12104 CrossRef CAS.
- N. M.-W. Wu, H.-L. Wong and V. W.-W. Yam, Photochromic benzo[b]phosphole oxide with excellent thermal irreversibility and fatigue resistance in the thin film solid state via direct attachment of dithienyl units to the weakly aromatic heterocycle, Chem. Sci., 2017, 8, 1309–1315 Search PubMed.
- Y. Ye, D. Wu, Y. Sun, D. Wang, Y. Wang, N. Wang, H. Hao, L. Li, P. Naumov and C. Xie, A solid-solution approach for controllable photomechanical crystalline materials, Nat. Commun., 2025, 16, 6647 Search PubMed.
- S. Cho, J. Usuba, S. Chakraborty, X. Li and G. G. D. Han, Solid-state photon energy storage via reversible [2+2] cycloaddition of donor-acceptor styrylpyrylium system, Chem, 2023, 9, 3159–3171 CAS.
- S. Chakraborty, H. P. Q. Nguyen, J. Usuba, J. Y. Choi, Z. Sun, C. Raju, G. Sigelmann, Q. Qiu, S. Cho, S. M. Tenney, K. E. Shulenberger, K. Schmidt-Rohr, J. Park and G. G. D. Han, Self-activated energy release cascade from anthracene-based solid-state molecular solar thermal energy storage systems, Chem, 2024, 10, 3309–3322 CAS.
- H. P. Q. Nguyen, A. Mukherjee, J. Usuba, J. Wan and G. G. D. Han, Large and long-term photon energy storage in diazetidines via [2+2] photocycloaddition, Chem. Sci., 2024, 15, 18846–18854 RSC.
- R. Malde, M. A. Parkes, M. Staniforth, J. M. Woolley, V. G. Stavros, V. Chudasama, H. H. Fielding and J. R. Baker, Intramolecular thiomaleimide [2 + 2] photocycloadditions: stereoselective control for disulfide stapling and observation of excited state intermediates by transient absorption spectroscopy, Chem. Sci., 2022, 13, 2909–2918 Search PubMed.
- E. N. Ushakov, T. P. Martyanov, A. I. Vedernikov, O. V. Pikalov, A. A. Efremova, L. G. Kuz’mina, J. A. K. Howard, M. V. Alfimov and S. P. Gromov, Self-assembly through hydrogen bonding and photochemical properties of supramolecular complexes of bis(18-crown-6)stilbene with alkanediammonium ions, J. Photochem. Photobiol., A, 2017, 340, 80–87 CrossRef CAS.
- M. F. Budyka, T. N. Gavrishova, V. M. Li and S. A. Tovstun, Styrylbenzoquinoline dyads as a new type of fluorescing photochromes operating via [2 + 2] photocycloaddition mechanism: Optimization of the structure, Spectrochim. Acta, Part A, 2024, 320, 124666 CrossRef CAS.
- A. García-Montero, A. M. Rodriguez, A. Juan, A. H. Velders, A. Denisi, G. Jiménez-Osés, E. Gómez-Bengoa, C. Cativiela, M. V. Gómez and E. P. Urriolabeitia, Metal-Free [2 + 2]-Photocycloaddition of (Z)-4-Aryliden-5(4H)-Oxazolones as Straightforward Synthesis of 1,3-Diaminotruxillic Acid Precursors: Synthetic Scope and Mechanistic Studies, ACS Sustainable Chem. Eng., 2017, 5, 8370–8381 CrossRef.
- E. Serrano, A. Juan, A. García-Montero, T. Soler, F. Jiménez-Márquez, C. Cativiela, M. V. Gomez and E. P. Urriolabeitia, Stereoselective Synthesis of 1,3-Diaminotruxillic Acid Derivatives: An Advantageous Combination of C–H-ortho-Palladation and On-Flow [2+2]-Photocycloaddition in Microreactors, Chem.–Eur. J., 2016, 22, 144–152 CrossRef CAS PubMed.
- Y. B. Dudkina, K. V. Kholin, T. V. Gryaznova, D. R. Islamov, O. N. Kataeva, I. K. Rizvanov, A. I. Levitskaya, O. D. Fominykh, M. Y. Balakina, O. G. Sinyashin and Y. H. Budnikova, Redox trends in cyclometalated palladium(II) complexes, Dalton Trans., 2017, 46, 165–177 RSC.
- D. Dalmau, O. Crespo, J. M. Matxain and E. P. Urriolabeitia, Fluorescence Amplification of Unsaturated Oxazolones Using Palladium: Photophysical and Computational Studies, Inorg. Chem., 2023, 62, 9792–9806 CrossRef CAS.
- C. Carrera, A. Denisi, C. Cativiela and E. P. Urriolabeitia, Functionalized 1,3-Diaminotruxillic Acids by Pd-Mediated C–H Activation and [2+2]-Photocycloaddition of 5(4H)-Oxazolones, Eur. J. Inorg. Chem., 2019, 2019, 3481–3489 CrossRef CAS.
- D. Roiban, E. Serrano, T. Soler, I. Grosu, C. Cativiela and E. P. Urriolabeitia, Unexpected [2 + 2] C–C bond coupling due to photocycloaddition on orthopalladated (Z)-2-aryl-4-arylidene-5(4H)-oxazolones, J. Chem. Soc., 2009, D, 4681–4683 Search PubMed.
- N. Baggi, L. M. Muhammad, Z. Liasi, J. L. Elholm, P. Baronas, E. Molins, K. V. Mikkelsen and K. Moth-Poulsen, Exploring ortho-dianthrylbenzenes for molecular solar thermal energy storage, J. Mater. Chem. A, 2024, 12, 26457–26464 Search PubMed.
- W. R. Bergmark, G. Jones II, T. E. Reinhardt and A. M. Halpern, Photoisomerization of bis(9-anthryl)methane and other linked anthracenes. The role of excimers and biradicals in photodimerization, J. Am. Chem. Soc., 1978, 100, 6665–6673 Search PubMed.
- Q. Qiu, CCDC 2493202: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2ppcwn.
| This journal is © The Royal Society of Chemistry 2026 |