
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePermanently porous cycloparaphenylene nanohoops via supramolecular engineering
Ashlyn A.
Kamin
 a,
Nathaniel J.
Schuster
a,
Haomiao
Xie
b,
Phuong H.
Le
a,
Kathleen M.
Snook
a,
Sebastian
Krajewski
a,
Werner
Kaminsky
a,
Omar K.
Farha
b,
Ramesh
Jasti
c and
Dianne J.
Xiao
*a
a,
Nathaniel J.
Schuster
a,
Haomiao
Xie
b,
Phuong H.
Le
a,
Kathleen M.
Snook
a,
Sebastian
Krajewski
a,
Werner
Kaminsky
a,
Omar K.
Farha
b,
Ramesh
Jasti
c and
Dianne J.
Xiao
*a
aDepartment of Chemistry, University of Washington, Seattle, Washington 98195, USA. E-mail: djxiao@uw.edu
bDepartment of Chemistry, Northwestern University, Evanston, Illinois 60208, USA
cDepartment of Chemistry and Biochemistry, Materials Science Institute, Knight Campus for Accelerating Scientific Impact, University of Oregon, Eugene, Oregon 97403, USA
First published on 20th November 2025
Abstract
Cycloparaphenylenes, also called carbon nanohoops, are a class of strained macrocycles recognized for their unique size-dependent optoelectronic properties, rich host–guest chemistry, and resemblance to carbon nanotubes. In this work, we establish carbon nanohoops as versatile tectons for permanently porous molecular crystals with well-defined supramolecular architectures and novel functionality. Using fluorinated nanohoops as precursors, we have synthesized seven new difluorodibenzodioxin-, methoxy-, and catechol boron bromide-functionalized derivatives. Structural characterization by single crystal X-ray and electron diffraction reveals porous structures stabilized by diverse noncovalent interactions, including π–π, CH–π, and boron–π interactions. In difluorodibenzodioxin nanohoops, robust intratubular π–π stacking guides the formation of nanotubular arrays with surface areas up to 910 m2 g−1, the highest value reported for cycloparaphenylenes to date. Remarkably, the nanotubular structure persists even in the presence of peripheral functionalization, enabling the formation of surface-decorated nanotubes. In contrast, boron bromide-derivatized nanohoops adopt an unusual 3D pore network that maximizes boron–π interactions. Due to the unique curvature of the carbon nanohoops, dense molecular packing is frustrated and the Lewis acidic boron centers remain partially exposed, leading to enhanced CO2 binding. This work provides a blueprint on how substituted cycloparaphenylenes can be used to achieve permanently porous molecular materials with targeted structure and function.
Introduction
Organic molecules with intrinsic cavities have long been studied as supramolecular hosts to bind and capture specific guests in solution.1–5 More recently, cavity-containing organic macrocycles and cages have gained renewed interest as a means of achieving permanent porosity in the solid state.6–11 Compared to porous frameworks constructed from extended covalent or coordination bonds, porous molecules provide greater solution processability as well as more dynamic and adaptive pore structures. In addition, simple post-synthetic processing techniques can be used to alter materials properties, including co-crystallization12–14 and the formation of different crystal polymorphs.15,16 However, to date, the number of permanently porous organic molecules remains far lower than the number of MOFs and COFs. This can be attributed to challenges in crystal structure engineering and prediction, as molecules tend to pack densely in solid state. Even compounds that appear to contain pores when solvated often rearrange and lose structural integrity upon solvent removal.17,18We have been interested in constructing porous materials from [n]cycloparaphenylenes ([n]CPPs), a class of strained macrocycles composed of n para-linked benzene rings (Fig. 1). Cycloparaphenylenes display a number of attractive properties, including size-dependent optoelectronic properties, good chemical and thermal stability, and well-defined cavities with tunable diameters.19 In solution, their unique concave π-surfaces display strong interactions with complementary guests,20 including C60,21 C70,22 smaller CPP rings,23 and corannulene.24
 | ||
| Fig. 1 Overview of the molecular structure, crystal packing, and gas sorption properties of (a) all-hydrocarbon, (b) fluorinated, (c) difluorodibenzodioxin, and (d) catechol boron bromide cycloparaphenylenes (CPPs). Gray, white, pink, red, green, and brown spheres represent C, H, B, O, F, and Br atoms, respectively. | ||
Despite their rich solution-state host–guest chemistry, remarkably few CPPs have been shown to be porous in the solid-state. All-hydrocarbon CPPs crystals typically adopt dense herringbone packing structures, reducing the accessible void space (Fig. 1a).19 In addition, structural rearrangements often occur upon solvent removal that further reduce porosity. For example, negligible N2 adsorption at 77 K, a common measure of permanent porosity, was reported for the all-hydrocarbon [12]CPP. Higher gas uptake was observed at 195 K, which was attributed to the presence of small pores that become accessible with thermally activated motion.25
To our knowledge, only a single previously reported cycloparaphenylene ring, 12F-[12]CPP, has been shown to adsorb measurable quantities of N2 at 77 K.26,27 In the solid-state, 12F-[12]CPP stacks vertically to form nanotubes, which further assemble in a hexagonal packing arrangement (Fig. 1b).27 This structure is maintained even upon solvent removal. The three perfluorinated aryl rings present in 12F-[12]CPP play key roles in enabling and preserving porosity, as the overall packing structure is stabilized by intratubular C–H⋯F interactions and intertubular arene–perfluoroarene π-stacking.
In this work, we were interested in whether noncovalent interactions beyond C–H⋯F interactions could be used to induce permanent porosity in cycloparaphenylene molecular crystals. Through the synthesis, structural characterization, and gas adsorption characterization of seven new difluorodibenzodioxin-, methoxy-, and catechol boron bromide-substituted cycloparaphenylenes, we show that diverse noncovalent interactions, including π–π, C–H⋯π, and boron–π interactions, can be used to assemble porous architectures with surface areas exceeding 900 m2 g−1 (Fig. 1c and d). In addition, we show that the traditionally inert cycloparaphenylene pore walls can be decorated with Lewis acidic boron centers, leading to enhanced CO2 binding. Together, this work illustrates how the precision synthesis and supramolecular assembly of cycloparaphenylene nanohoops can be exploited to access to new materials that both mimic and expand beyond the porosity of carbon nanotubes.
Results and discussion
Synthetic overview
A summary of the cycloparaphenylene structures and synthetic routes discussed in this work is provided in Fig. 2. The previously reported octa- and dodecafluorinated cycloparaphenylenes 8F-[10]CPP, 8F-[12]CPP, and 12F-[12]CPP were selected as synthetic precursors,26,27 as prior work by us and others has shown that fluorinated CPPs are easily derivatized using nucleophilic aromatic substitution (SNAr).28,29 From these known fluorinated CPPs, new difluorodibenzodioxin-, methoxy-, and catechol boron bromide-derivatized CPPs can be facilely prepared in just 1–2 steps (Fig. 2).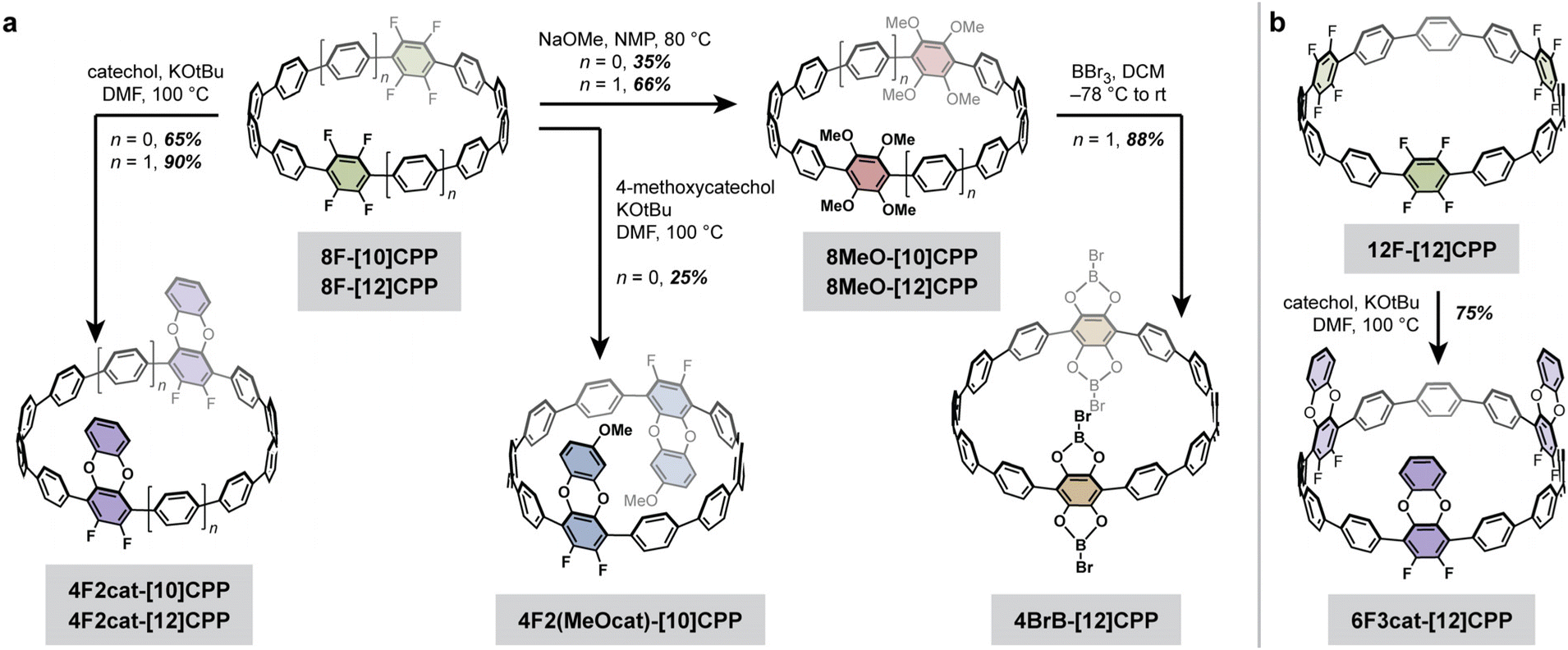 | ||
| Fig. 2 Synthesis of new difluorodibenzodioxin–, methoxy–, and catechol boron bromide-derivatized cycloparaphenylene nanohoops from (a) octafluorinated 8F-[10]CPP and 8F-[12]CPP and (b) dodecafluorinated 12F-[12]CPP. | ||
While the gram scale synthesis of the dodecafluorinated 12F-[12]CPP was previously reported by Jasti and coworkers, the related octafluorinated 8F-[10]CPP and 8F-[12]CPP had only been reported on sub-20 mg scales.26 Therefore, our first goal was to optimize and scale up these synthetic routes (Scheme 1). We found that the curved intermediates 1, 2a, and 2b for the synthesis of 8F-[10]CPP and 8F-[12]CPP could each be prepared on the decagram scale from a simple biphenylene precursor (see the SI for the full synthetic details). These intermediates undergo macrocyclization via palladium-catalyzed cross-coupling to give the triethylsilyl (TES)-protected macrocycles 3a and 3b on a multi-gram scale without any reduction in yield relative to the original reports (44% for 3a, 41% for 3b). Subsequent desilylation followed by reductive aromatization of 3a afforded 8F-[10]CPP in half-gram quantities with 74% yield over two steps. Similarly, desilylation and reductive aromatization of 3b afforded 8F-[12]CPP in multi-hundred-milligram quantities in 35% yield over two steps.
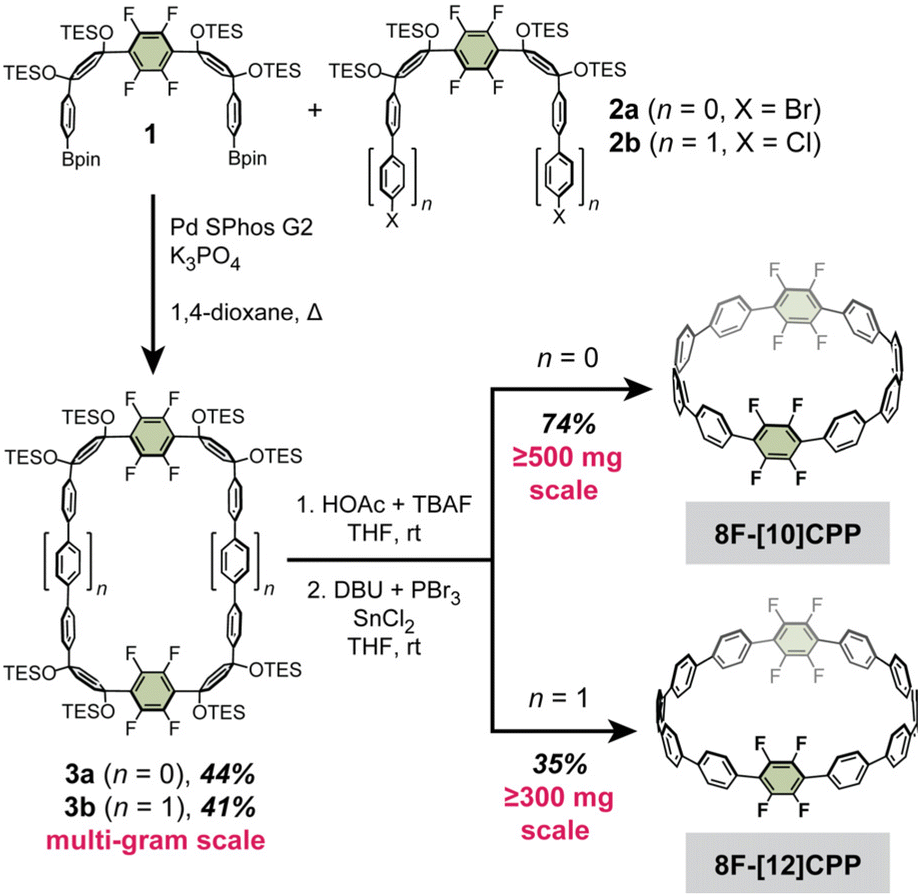 | ||
| Scheme 1 Optimized large-scale syntheses of 8F-[10]CPP and 8F-[12]CPP from the curved precursors 1 and 2a/2b. | ||
Difluorodibenzodioxin cycloparaphenylenes with robust tubular packing
With significant quantities of fluorinated CPPs in hand, we next investigated how functionalization of the CPP ring impacts supramolecular packing. While the substitution of fluorinated CPPs with alkoxide, alkylthiolate, and pyrrolide-based nucleophiles has been previously reported,28,29 we were particularly interested in bidentate aromatic nucleophiles such as catechols. We hypothesized that the planar geometry of the resulting dibenzodioxin linkages could facilitate nanotubular alignment of the CPPs via π–π stacking.We first investigated the benzodioxination of a model tetrafluorinated terphenyl substrate, 4 (Scheme 2). Almost no reaction progress was observed at 80 °C in polar aprotic solvents such as dimethylformamide (DMF) or N-methylpyrrolidone (NMP) when 4 was subjected to excess catechol and tert-butoxide. Increasing the reaction temperature to 120 °C resulted exclusively in the formation of the mono-substituted intermediate, 5 (Scheme 2). Complete substitution to form 6 required reaction temperatures of 240 °C in even higher-boiling solvents such as 1,3-dimethyl-2-imidazolidinone (DMI) or N,N′-dimethylpropyleneurea (DMPU).
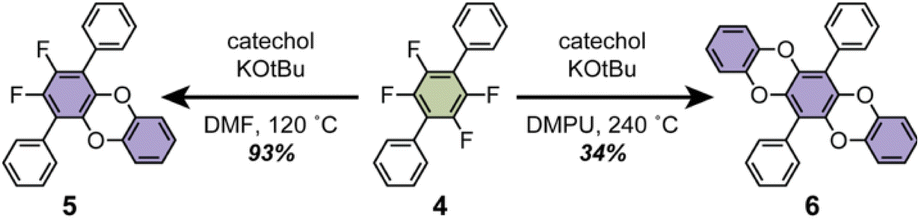 | ||
| Scheme 2 Catecholation of the tetrafluorinated terphenyl 4 to produce monosubstituted 5 and disubstituted 6. | ||
Given the dramatic difference in temperatures required for the partial versus complete substitution of our model terphenyl compound, we hypothesized that one-fold benzodioxination of our fluorinated CPPs could be achieved selectively under relatively mild conditions. Indeed, subjecting 8F-[10]CPP, 8F-[12]CPP, and 12F-[12]CPP to excess catechol and tert-butoxide in DMF at 100 °C led to the selective formation of the desired 4F2cat-[10]CPP, 4F2cat-[12]CPP, and 6F3cat-[12]CPP, respectively (Fig. 3). All three molecules were purified by chromatography-free workups and were fully characterized by high-resolution mass spectrometry and multinuclear NMR. Large single crystals suitable for X-ray diffraction were grown for 4F2cat-[10]CPP and 4F2cat-[12]CPP, whereas thin needle-like crystals suitable for electron diffraction were obtained for 6F3cat-[12]CPP.
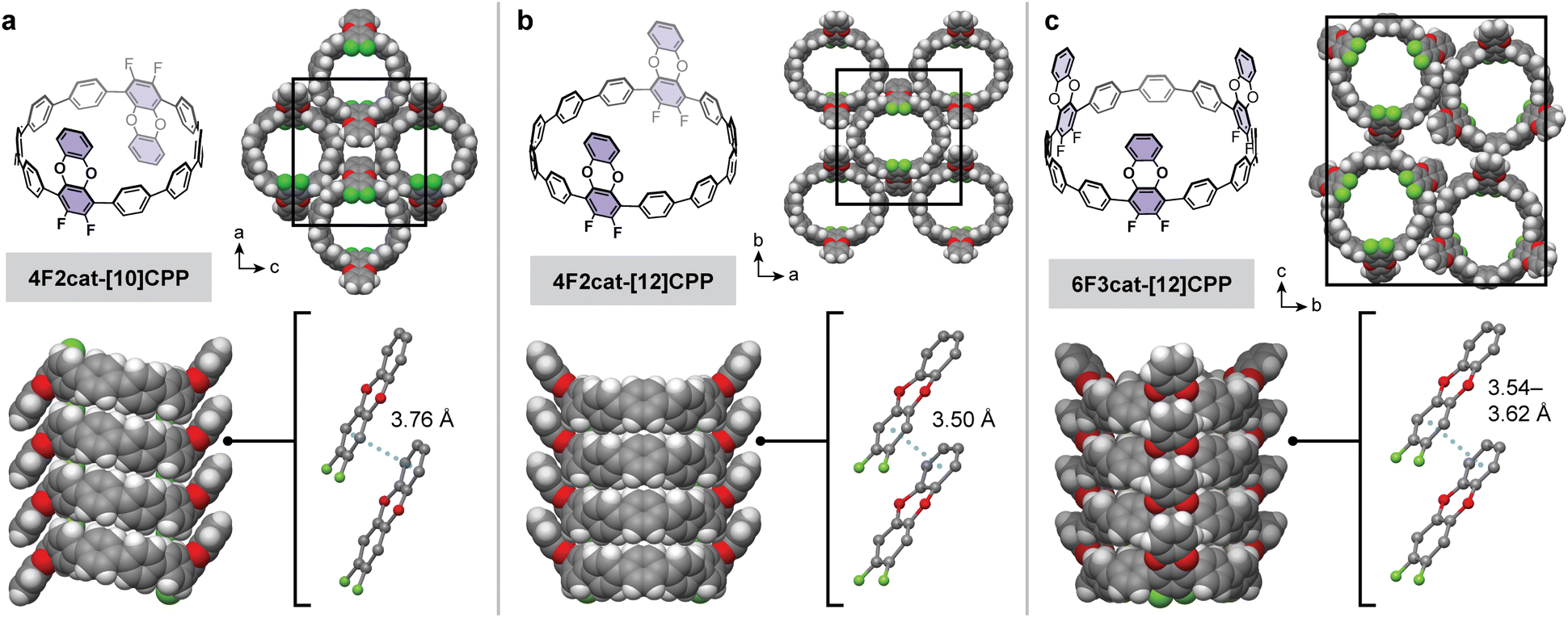 | ||
| Fig. 3 Molecular structure and crystal packing of difluorodibenzodioxin-derivatized CPPs (a) 4F2cat-[10]CPP, (b) 4F2cat-[12]CPP, and (c) 6F3cat-[12]CPP. In all cases, the CPP rings stack vertically to form nanotubular structures that are stabilized by π–π stacking between the difluorodibenzodioxin units. Gray, white, red, and green spheres represent C, H, O, and F atoms, respectively. | ||
The structures of 4F2cat-[10]CPP, 4F2cat-[12]CPP, and 6F3cat-[12]CPP are shown in Fig. 3. Excitingly, all three CPPs stack vertically to form well-defined nanotubes that further order in a rectangular packing arrangement. Within each nanotube, nearly eclipsed π–π stacking is observed between the difluorodibenzodioxin units (Fig. 3), with small horizontal offsets of < 0.65 Å (Table 1). The centroid-to-centroid π-stacking distance varies slightly across the three structures, ranging between 3.50–3.76 Å (Fig. 3). Attraction between the electron-rich and electron-poor π-subcomponents of neighboring difluorodibenzodioxins seems to promote the cofacial geometry.30 In addition to the intratubular π–π stacking, C–H⋯π interactions are also observed between adjacent nanotubes. Most notably, C–H⋯π interactions between the benzodioxin unit and neighboring CPP aromatic rings are present in all structures (Fig. S22). Finally, while electron density was observed inside the pores, we were unable to resolve the packing of the disordered solvent molecules.
While the nanotubular packing of 4F2cat-[10]CPP, 4F2cat-[12]CPP, and 6F3cat-[12]CPP is broadly similar, significant differences in the relative orientation of the difluorodibenzodioxin rings are observed. In 4F2cat-[10]CPP, one difluorodibenzodioxin substituent is oriented upward while the other is oriented downward, giving the molecule a “chair”-like appearance when viewed from the side. In contrast, the difluorodibenzodioxin units in 4F2cat-[12]CPP and 6F3cat-[12]CPP are oriented in the same direction, giving the appearance of a “boat” or bowl-like structure. While only a single conformation is observed in the solid-state, we hypothesize that each CPP rapidly interconverts between the “boat” and “chair” conformations in solution, as only a single set of signals is apparent in our 1H NMR spectra at 298 K.
Given the remarkably similar nanotubular packing found in these three difluorodibenzodioxin-substituted CPPs, we were curious if the nanotubular assembly would persist after peripheral substitution of the benzodioxin ring. Therefore, we reacted excess 4-methoxycatechol with 8F-[10]CPP and tert-butoxide in DMF at 100 °C to produce the methoxydifluorodibenzodioxin-CPP, 4F2(MeOcat)-[10]CPP (Fig. 4). Bulk recrystallization enabled the isolation of a single regiopure product that was confirmed by HRMS and multinuclear NMR (Fig. 4). The single crystals adopted a needle-like habit that was too thin for conventional X-ray diffraction but could be solved by electron diffraction.
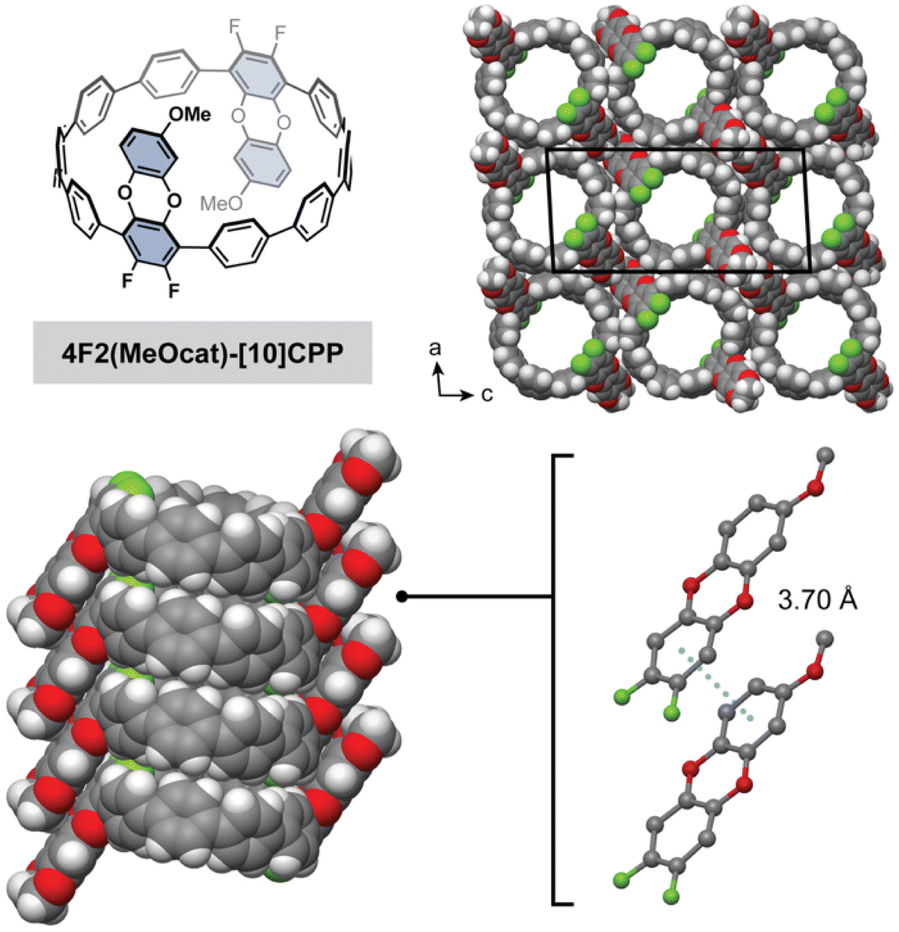 | ||
| Fig. 4 Single crystal electron diffraction structure of 4F2(MeOcat)-[10]CPP highlighting the nanotubular packing and π–π interactions between the difluorodibenzodioxin rings. Gray, white, red, and green spheres represent C, H, O, and F atoms, respectively. | ||
Excitingly, the crystal structure revealed that 4F2(MeOcat)-[10]CPP also successfully adopts a nanotubular structure. The additional steric bulk of the methoxy groups does not hinder π–π stacking between the difluorodibenzodioxin units, and a short centroid-to-centroid distance of 3.70 Å is observed (Fig. 4). Similar to 4F2cat-[10]CPP, the methoxy-substituted difluorodibenzodioxin rings are oriented in opposite directions in a “chair”-like conformation. The powder X-ray diffraction pattern of the as-synthesized crude product, which represents a mixture of regioisomers, also closely matched the nanotubular packing of 4F2cat-[10]CPP and 4F2(MeOcat)-[10]CPP (Fig. S25). Together, this suggests that the nanotubular packing is highly robust, persisting even in the presence of additional steric bulk and structural disorder.
While accessible channels are clearly present in the crystal structures of all four difluorodibenzodioxin-functionalized CPPs, this does not always translate to permanent porosity, as pore collapse upon solvent removal is common in molecular crystals. For example, in the case of fluorinated CPPs, we found that the accessible surface area is highly dependent on the number of C–H⋯F and fluoroarene–arene interactions. Following recrystallization from trichloroethylene and activation at 125 °C, we observed a 77 K N2 Brunauer–Emmett–Teller (BET) surface area of 770 m2 g−1 for 12F-[12]CPP, which is even higher than the previously reported literature value of 608 m2 g−1.26 However, the BET surface area drops precipitously upon removal of a single tetrafluoroarene unit. The surface areas of 8F-[12]CPP and 8F-[10]CPP were significantly lower, at 130 m2 g−1 and 250 m2 g−1, respectively, suggesting structural rearrangements occur upon solvent removal (Table 2).
| Category | Compound name | BET SA (m2 g−1) |
|---|---|---|
| Fluorinated CPPs | 12F-[12]CPP | 770 |
| 8F-[12]CPP | 130 | |
| 8F-[10]CPP | 250 | |
| Difluorodibenzodioxin CPPs | 6F3cat-[12]CPP | 730 |
| 4F2cat-[12]CPP | 910 | |
| 4F2cat-[10]CPP | 400 | |
| 4F2(MeOcat)-[10]CPP | 700 | |
| Catechol boron bromide CPPs | 4BrB-[12]CPP | 180 |
Surprisingly, the packing of the difluorodibenzodioxin-functionalized CPPs appears significantly more robust than their fluorinated precursors (Table 2). The 77 K N2 adsorption isotherms are shown in Fig. 5, and additional desorption data are provided in Fig. S32–38. Remarkably, the BET surface area of 4F2cat-[12]CPP is 910 m2 g−1, a considerable improvement on its fluorinated precursor and the highest surface area measured in this study. The BET surface area of 6F3cat-[12]CPP was found to be 730 m2 g−1, which is close to the value obtained for its dodecafluorinated analogue. Impressively, despite the increased steric bulk, the BET surface area of the methoxy-substituted 4F2(MeOcat)-[10]CPP is nearly identical at 700 m2 g−1. This value also closely matches the expected value of ∼740 m2 g−1 calculated from the crystal structure. The BET surface area of 4F2cat-[10]CPP is slightly lower (400 m2 g−1), though still appreciably higher than its fluorinated precursor. Relatively narrow DFT pore size distributions centered near 10 Å and 11 Å were observed for the [10]CPP and [12]CPP derivatives, respectively (Fig. S40). This is consistent with the crystal structures, which show a diameter of ∼13–14 Å for the [10]CPP rings and ∼15–16 Å for the shorter dimension of the [12]CPP rings. In all cases, powder X-ray diffraction confirms persistence of the nanotubular packing after activation (Fig. S43–48).
 | ||
| Fig. 5 Nitrogen isotherms collected at 77 K for difluorodibenzodioxin CPPs and corresponding BET surface areas. | ||
The maintenance of robust nanotubular packing in our difluorodibenzodioxin CPPs despite changes to the overall ring size, number of difluorodibenzodioxin units, and peripheral substitution pattern is unusual, as subtle changes in molecular structure generally trigger dramatic changes in the crystal packing.10 Going forward, further elaboration of the difluorodibenzodioxin motif may be a promising route towards precise nanotube assemblies with tailored surface functionality.
Catechol boron bromide CPPs with exposed Lewis acidic centers
While the fluorinated and difluorodibenzodioxin-functionalized CPPs described in the previous section display high surface areas, the pore walls are relatively inert. Pores lined with reactive chemical functionalities, such as Lewis acidic centers, are often advantageous for selective gas separations, storage, and catalysis. As a proof of concept, we hypothesized that Lewis acidic boron centers could be installed onto fluorinated CPPs via a straightforward two-step sequence, specifically methoxylation followed by treatment with BBr3 (Fig. 2). This should generate catechol boron bromides, which are less reactive than the parent BBr3 but still retain moderate Lewis acidity.31To test this hypothesis, methoxylation of our octafluorinated CPPs was carried out using the standard SNAr conditions established in our previous work.29 The substitution of 8F-[12]CPP with sodium methoxide in NMP at 80 °C followed by purification via column chromatography afforded pure 8MeO-[12]CPP in 66% yield (Fig. 2a). The methoxylation of 8F-[10]CPP under the same conditions gave a mixture of partially substituted intermediates in addition to the desired fully-substituted product, 8MeO-[10]CPP. We attribute this incomplete conversion to the decreased solubility of the [10]CPP relative to the [12]CPP. However, purification by column chromatography still allowed for the isolation of pure 8MeO-[10]CPP in 35% yield.
Given the greater yield and solubility of the [12]CPP derivative, we focused on 8MeO-[12]CPP for subsequent studies. The catechol boron bromide derivative 4BrB-[12]CPP was isolated in 88% yield upon treatment of a suspension of 8MeO-[12]CPP in dichloromethane (DCM) with BBr3 at −78 °C, followed by slow warming to room temperature and precipitation with anhydrous n-pentane. Small, colorless single crystals suitable for X-ray diffraction were isolated from the crude reaction mixture. The presence of a single boron environment was confirmed by 11B NMR, and single crystal X-ray diffraction confirmed that the methyl groups were removed and replaced with B–Br units.
Unlike the herringbone or nanotubular packing commonly observed in other CPP derivatives, the crystal structure of 4BrB-[12]CPP reveals an unusual interconnected 3D pore network (Fig. 6). Along the ab plane, the CPP rings assemble side-by-side to form a slightly corrugated 2D layer (Fig. 6a). These 2D layers are separated by additional CPP molecules, which are sandwiched between the layers via offset π-stacking between a di(bromodioxaborole)benzene unit and a CPP aromatic ring (centroid-to-centroid distance of 3.56 Å, see Fig. 6c). Within each 2D layer, pairs of di(bromodioxaborole)benzene units assemble in a parallel offset arrangement, with a short boron–carbon distance of 3.29(5) Å (Fig. 6b). Similar boron–π interactions, also known as triel bonds, have been observed both experimentally and computationally.32 For example, the calculated structure of a BCl3–benzene complex displays a boron–carbon distance of 3.15 Å.32
 | ||
| Fig. 6 Overview of the single crystal X-ray structure of 4BrB-[12]CPP, including the (a) overall molecular packing. (b) boron–π interactions, and (c) π–π interactions. Gray, white, pink, red, and brown spheres represent C, H, B, O, and Br atoms, respectively. | ||
Due to the unique radial π-system of cycloparaphenylene rings, all of the Lewis acidic boron centers remain partially exposed in 4BrB-[12]CPP. Close boron–π contacts can only form on the outer face of the CPPs, allowing guests, in principle, to approach boron centers through the concave interior face. This is in stark contrast to the linear terphenyl analogue, which we have also synthesized and crystallized. The linear analogue crystallizes to form infinite 1D stacks that completely block both faces of the Lewis acidic boron centers (Fig. S49).
While a high surface area exceeding 2000 m2 g−1 is expected based on the crystal structure of 4BrB-[12]CPP, partial pore collapse occurs during activation, leading to an experimental BET surface area of 180 m2 g−1 (Table 2). Carbon dioxide was used as a probe molecule to determine whether the boron centers remain accessible despite the moderate surface area (Fig. 7). Compared to the relatively inert aromatic walls, the Lewis acidic boron centers should lead to stronger physisorption of more polarizable gases such as CO2.
 | ||
| Fig. 7 (a) Variable temperature CO2 isotherms of 4BrB-[12]CPP, and (b) isosteric heats of CO2 adsorption (−Qst) for 4BrB-[12]CPP and 4F2cat-[12]CPP. | ||
Variable temperature CO2 isotherms were measured and fit to obtain isosteric heats of CO2 adsorption for both 4BrB-[12]CPP as well as 4F2cat-[12]CPP, which served as a reference compound that does not contain Lewis acidic centers (Fig. 7 and S51). Excitingly, CO2 binding was significantly stronger in 4BrB-[12]CPP, which displayed an initial heat of adsorption (−Qst) of 24(1) kJ mol−1, compared to 17(1) kJ mol−1 for 4F2cat-[12]CPP (Fig. 7b). The value obtained for 4BrB-[12]CPP is similar to metal–organic frameworks containing weak Lewis acids, such as the coordinatively unsaturated Cu2+ and Zn2+ centers in Cu- and Zn-MOF-74, respectively (22 kJ mol−1 for Cu2+, 27 kJ mol−1 for Zn2+).33 Together, the gas sorption properties of 4BrB-[12]CPP highlight how the curvature of CPP rings can be exploited to frustrate molecular packing and maintain the accessibility of reactive functional groups.
Conclusions
Through the synthesis of seven new cycloparaphenylenes functionalized with difluorodibenzodioxin, methoxy, and catechol boron bromide substituents, we have shown how diverse noncovalent interactions can be exploited to obtain permanently porous architectures. In particular, we have identified the difluorodibenzodioxin unit as a new supramolecular synthon for inducing robust cofacial π–π stacking and nanotubular CPP packing in the solid-state. The nanotubular packing not only persists upon solvent removal but is also tolerant of peripheral substitution, enabling future studies on the chemistry and materials properties of surface-functionalized nanotube assemblies. Such systems may find future use in separations applications, for example as synthetic nanopores for selective guest transport across membranes.34 Finally, we have shown that reactive groups such as coordinatively unsaturated boron centers can be installed on cycloparaphenylenes, and that these Lewis acidic moieties lead to enhanced binding of guest molecules such as CO2. Together, these studies establish cycloparaphenylenes as versatile building blocks for porous molecular crystals with well-defined supramolecular architectures and novel functionality.Author contributions
A. A. K. and D. J. X. designed the research and wrote the manuscript; A. A. K. carried out the synthesis and characterization of all species; N. J. S. collected all gas sorption data; H. X. collected and refined the single crystal electron diffraction data; S. K. and W. K. collected and refined the single crystal X-ray diffraction data; P. H. L. and K. M. S. assisted with CPP characterization. All authors interpreted the results and contributed to reviewing and editing the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: additional experimental details and data, including detailed synthetic procedures, experimental methods for diffraction and gas sorption, and supplementary tables and figures. See DOI: https://doi.org/10.1039/d5sc07685d.CCDC 2484714–2484723 contain the supplementary crystallographic data for this paper.35a–j
Acknowledgements
This work is primarily supported by the Arnold and Mabel Beckman Foundation through a Beckman Young Investigator Award. The gas sorption studies were additionally supported by the David and Lucile Packard Foundation through a Packard Fellowship for Science and Engineering. RJ was supported by NSF CHE-2400147. AAK is supported by an NSF graduate research fellowship. The NMR facility at the UW Department of Chemistry is supported by NIH Award Number S10OD030224-01A1. The X-ray facility of the UW Department of Chemistry is supported by NSF Award Number CHE-0840520. This work made use of the IMSERC (RRID:SCR_017874) Crystallography facility at Northwestern University, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-2025633). Purchase and installation of the electron diffractometer (Rigaku ED-Synergy) used to obtain results included in this publication was supported by the Air Force Office of Scientific Research (AFOSR) through the Defense University Research Instrumentation Program Grant FA9550-24-1-0058. We thank Dr Tara Clayton for insightful discussions on the synthesis of fluorinated CPPs.Notes and references
- V. Böhmer, Calixarenes, Macrocycles with (Almost) Unlimited Possibilities, Angew Chem. Int. Ed. Engl., 1995, 34(7), 713–745, DOI:10.1002/anie.199507131.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, The Cucurbit[n]Uril Family, Angew. Chem., Int. Ed., 2005, 44(31), 4844–4870, DOI:10.1002/anie.200460675.
- G. Crini, Review: A History of Cyclodextrins, Chem. Rev., 2014, 114(21), 10940–10975, DOI:10.1021/cr500081p.
- T. Ogoshi, T. Yamagishi and Y. Nakamoto, Pillar-Shaped Macrocyclic Hosts Pillar[n]Arenes: New Key Players for Supramolecular Chemistry, Chem. Rev., 2016, 116(14), 7937–8002, DOI:10.1021/acs.chemrev.5b00765.
- Comprehensive Supramolecular Chemistry II, ed. J. L. Atwood, G. W. Gokel and L. J. Barbour, Elsevier, Amsterdam, 2017 Search PubMed.
- J. Tian, P. K. Thallapally and B. P. McGrail, Porous Organic Molecular Materials, CrystEngComm, 2012, 14(6), 1909–1919, 10.1039/C2CE06457J.
- T. Hasell and A. I. Cooper, Porous Organic Cages: Soluble, Modular and Molecular Pores, Nat. Rev. Mater., 2016, 1(9), 16053, DOI:10.1038/natrevmats.2016.53.
- S. Das, P. Heasman, T. Ben and S. Qiu, Porous Organic Materials: Strategic Design and Structure–Function Correlation, Chem. Rev., 2017, 117(3), 1515–1563, DOI:10.1021/acs.chemrev.6b00439.
- A. I. Cooper, Porous Molecular Solids and Liquids, ACS Cent. Sci., 2017, 3(6), 544–553, DOI:10.1021/acscentsci.7b00146.
- M. A. Little and A. I. Cooper, The Chemistry of Porous Organic Molecular Materials, Adv. Funct. Mater., 2020, 30(41), 1909842, DOI:10.1002/adfm.201909842.
- G. Zhang, W. Lin, F. Huang, J. Sessler and N. M. Khashab, Industrial Separation Challenges: How Does Supramolecular Chemistry Help?, J. Am. Chem. Soc., 2023, 145(35), 19143–19163, DOI:10.1021/jacs.3c06175.
- J. T. A. Jones, T. Hasell, X. Wu, J. Bacsa, K. E. Jelfs, M. Schmidtmann, S. Y. Chong, D. J. Adams, A. Trewin, F. Schiffman, F. Cora, B. Slater, A. Steiner, G. M. Day and A. I. Cooper, Modular and Predictable Assembly of Porous Organic Molecular Crystals, Nature, 2011, 474(7351), 367–371, DOI:10.1038/nature10125.
- T. Hasell, S. Y. Chong, K. E. Jelfs, D. J. Adams and A. I. Cooper, Porous Organic Cage Nanocrystals by Solution Mixing, J. Am. Chem. Soc., 2012, 134(1), 588–598, DOI:10.1021/ja209156v.
- T. Hasell, S. Y. Chong, M. Schmidtmann, D. J. Adams and A. I. Cooper, Porous Organic Alloys, Angew. Chem., Int. Ed., 2012, 51(29), 7154–7157, DOI:10.1002/anie.201202849.
- A. Avellaneda, P. Valente, A. Burgun, J. D. Evans, A. W. Markwell-Heys, D. Rankine, D. J. Nielsen, M. R. Hill, C. J. Sumby and C. J. Doonan, Kinetically Controlled Porosity in a Robust Organic Cage Material, Angew. Chem., Int. Ed., 2013, 52(13), 3746–3749, DOI:10.1002/anie.201209922.
- T. Hasell, J. L. Culshaw, S. Y. Chong, M. Schmidtmann, M. A. Little, K. E. Jelfs, E. O. Pyzer-Knapp, H. Shepherd, D. J. Adams, G. M. Day and A. I. Cooper, Controlling the Crystallization of Porous Organic Cages: Molecular Analogs of Isoreticular Frameworks Using Shape-Specific Directing Solvents, J. Am. Chem. Soc., 2014, 136(4), 1438–1448, DOI:10.1021/ja409594s.
- L. J. Barbour, Crystal Porosity and the Burden of Proof, Chem. Commun., 2006, 11, 1163, 10.1039/b515612m.
- L. R. Nassimbeni, Physicochemical Aspects of Host–Guest Compounds, Acc. Chem. Res., 2003, 36(8), 631–637, DOI:10.1021/ar0201153.
- E. R. Darzi and R. Jasti, The Dynamic, Size-Dependent Properties of [5]–[12]Cycloparaphenylenes, Chem. Soc. Rev., 2015, 44(18), 6401–6410, 10.1039/c5cs00143a.
- Y. Xu and M. Von Delius, The Supramolecular Chemistry of Strained Carbon Nanohoops, Angew. Chem., Int. Ed., 2020, 59(2), 559–573, DOI:10.1002/anie.201906069.
- T. Iwamoto, Y. Watanabe, T. Sadahiro, T. Haino and S. Yamago, Size-Selective Encapsulation of C60 by [10]Cycloparaphenylene: Formation of the Shortest Fullerene-Peapod, Angew. Chem., Int. Ed., 2011, 50(36), 8342–8344, DOI:10.1002/anie.201102302.
- D. Lu, G. Zhuang, H. Wu, S. Wang, S. Yang and P. Du, A Large π-Extended Carbon Nanoring Based on Nanographene Units: Bottom-Up Synthesis, Photophysical Properties, and Selective Complexation with Fullerene C70, Angew. Chem., Int. Ed., 2017, 56(1), 158–162, DOI:10.1002/anie.201608963.
- S. Hashimoto, T. Iwamoto, D. Kurachi, E. Kayahara and S. Yamago, Shortest Double-Walled Carbon Nanotubes Composed of Cycloparaphenylenes, ChemPlusChem, 2017, 82(7), 1015–1020, DOI:10.1002/cplu.201700097.
- T. Matsuno, M. Fujita, K. Fukunaga, S. Sato and H. Isobe, Concyclic CH-π Arrays for Single-Axis Rotations of a Bowl in a Tube, Nat. Commun., 2018, 9(1), 3779, DOI:10.1038/s41467-018-06270-6.
- H. Sakamoto, T. Fujimori, X. Li, K. Kaneko, K. Kan, N. Ozaki, Y. Hijikata, S. Irle and K. Itami, Cycloparaphenylene as a Molecular Porous Carbon Solid with Uniform Pores Exhibiting Adsorption-Induced Softness, Chem. Sci., 2016, 7(7), 4204–4210, 10.1039/c6sc00092d.
- J. M. Van Raden, E. J. Leonhardt, L. N. Zakharov, A. Pérez-Guardiola, A. J. Pérez-Jiménez, C. R. Marshall, C. K. Brozek, J. C. Sancho-García and R. Jasti, Precision Nanotube Mimics via Self-Assembly of Programmed Carbon Nanohoops, J. Org. Chem., 2020, 85(1), 129–141, DOI:10.1021/acs.joc.9b02340.
- E. J. Leonhardt, J. M. Van Raden, D. Miller, L. N. Zakharov, B. Alemán and R. A. Jasti, Bottom-Up Approach to Solution-Processed, Atomically Precise Graphitic Cylinders on Graphite, Nano Lett., 2018, 18(12), 7991–7997, DOI:10.1021/acs.nanolett.8b03979.
- H. Shudo, M. Kuwayama, Y. Segawa, A. Yagi and K. Itami, Half-Substituted Fluorocycloparaphenylenes with High Symmetry: Synthesis, Properties and Derivatization to Densely Substituted Carbon Nanorings, Chem. Commun., 2023, 59(90), 13494–13497, 10.1039/D3CC04887J.
- A. A. Kamin, T. D. Clayton, C. E. Otteson, P. M. Gannon, S. Krajewski, W. Kaminsky, R. Jasti and D. J. Xiao, Synthesis and Metalation of Polycatechol Nanohoops Derived from Fluorocycloparaphenylenes, Chem. Sci., 2023, 14(36), 9724–9732, 10.1039/D3SC03561A.
- C. R. Martinez and B. L. Iverson, Rethinking the Term “Pi-Stacking.”, Chem. Sci., 2012, 3(7), 2191, 10.1039/c2sc20045g.
- R. K. Boeckman and J. C. Potenza, Catechol Boron Halides: Mild and Selective Reagents for Cleavage of Common Protecting Groups, Tetrahedron Lett., 1985, 26(11), 1411–1414, DOI:10.1016/S0040-4039(00)99058-0.
- S. J. Grabowski, Triel Bonds–Interactions without Borders, Similarly as Hydrogen Bonds, Cryst. Growth Des., 2025, 25(12), 4539–4552, DOI:10.1021/acs.cgd.5c00466.
- W. L. Queen, M. R. Hudson, E. D. Bloch, J. A. Mason, M. I. Gonzalez, J. S. Lee, D. Gygi, J. D. Howe, K. Lee, T. A. Darwish, M. James, V. K. Peterson, S. J. Teat, B. Smit, J. B. Neaton, J. R. Long and C. M. Brown, Comprehensive Study of Carbon Dioxide Adsorption in the Metal–Organic Frameworks M2(dobdc) (M = Mg, Mn, Fe, Co, Ni, Cu, Zn), Chem. Sci., 2014, 5(12), 4569–4581, 10.1039/C4SC02064B.
- J. Shen, G. Liu, Y. Han and W. Jin, Artificial Channels for Confined Mass Transport at the Sub-Nanometre Scale, Nat. Rev. Mater., 2021, 6(4), 294–312, DOI:10.1038/s41578-020-00268-7.
- (a) CCDC 2484714: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2pdk2r; (b) CCDC 2484715: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2pdk3s; (c) CCDC 2484716: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2pdk4t; (d) CCDC 2484717: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2pdk5v; (e) CCDC 2484718: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2pdk6w; (f) CCDC 2484719: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2pdk7x; (g) CCDC 2484720: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2pdk8y; (h) CCDC 2484721: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2pdk9z; (i) CCDC 2484722: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2pdkb0; (j) CCDC 2484723: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2pdkc1.
| This journal is © The Royal Society of Chemistry 2026 |