
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThinking outside the library: cluster synthesis of diverse molecules on a single robotic platform
Franck
Le Vaillant
 *a,
Luidgi
Gromat†
b,
Clémentine
Pescheteau†
a,
Nicolas
Ducrot
b,
Aurélien
Demilly
b,
Jean-Christophe
Meillon
a,
Nicolas
Do Huu
b and
Quentin
Perron
b
*a,
Luidgi
Gromat†
b,
Clémentine
Pescheteau†
a,
Nicolas
Ducrot
b,
Aurélien
Demilly
b,
Jean-Christophe
Meillon
a,
Nicolas
Do Huu
b and
Quentin
Perron
b
aIktos Robotics Laboratory, 25-27 Avenue du Québec, 91140 Villebon-sur-Yvette, France. E-mail: franck.levaillant@iktos.com
bIktos, 65 rue de Prony, 75017 Paris, France
First published on 28th November 2025
Abstract
The development of a general autonomous platform for organic synthesis that enables faster, flexible and efficient delivery of target molecules is an attractive strategy for many fields such as drug discovery and materials science. Traditionally, automated parallel synthesis relies on the synthesis of libraries of various sizes, sharing the same transformations and name reactions with defined reaction conditions. Herein, we report on the development of our platform and a paradigm shift in high throughput robotic synthesis: from mono-reaction type libraries to multi-reaction type clusters. This fundamentally distinct approach differs from the current strategies by clustering reactions based on their reaction conditions, defined as ranges of acceptable temperature and reaction time by expert chemists. As a result, many different reactions can be merged into a cluster. An algorithm has been developed to help chemists organize the workload into the minimum number of clusters, taking into account the physical and chemical constraints of the platform. We applied this strategy to efficiently organize the synthesis of 135 molecules, using 27 different name reactions in only 6 clusters and 3 synthetic campaigns.
1 Introduction
Autonomous laboratories are often seen as the laboratory of the future.1,2 Such environments are complex and expensive by design and require both robotics3 and artificial intelligence4 with a high degree of integration in order to allow automatic execution, data capture and decision, leading to the completion of additional steps in a defined workflow.5–8 Although much progress has been made in the field of lab automation as well as in the field of chemistry digitalization,9 a fully autonomous lab has yet to be implemented and run on challenging tasks at high-capacity. One of the current bottlenecks is the translation of ideas to real materials ready for testing.10Indeed, research in chemistry relies heavily on navigation through a chemical space, whether it is to find biologically active molecules11 or novel materials12 having specific properties. To accomplish this goal, modern tools such as in silico models have been developed to help chemists navigate larger and larger chemical spaces in a relevant manner.13 However, the acquisition of real-life data points is mandatory to continue driving the research and the next iteration of the classical Design Make Test Analyze cycle (DMTA). Running multiple projects in parallel gives rise to siloed library syntheses, with each project working independently under its own constraints of building block availability, resulting in an overall lack of efficiency. Furthermore, incremental modifications often lead to very similar compounds, prepared via the decoration of a main scaffold using the same transformations, applied with the same reaction conditions for each well. While automated combinatorial chemistry reveals limitations regarding chemistry outcomes and the impact in the exploration of chemical spaces, many strategies already exist to efficiently prepare lists of molecules of various sizes and structural diversity. Among those, diversity-oriented synthesis is aimed at generating high-structural diversity within a library, accelerating the discovery of hits.14 Such a strategy is often embedded in generative AI-tools.15 The counterpart is the challenge associated with the synthesis of a list of chemical compounds that are less similar, thus requiring extensive manual lab work.16
Nowadays, many automated platforms exist, providing robust solutions for high-throughput experimentation (HTE, Fig. 1A)17 and parallel synthesis (Fig. 1B).18,19 Flow chemistry alternatives have also been developed with significant results in rapid reaction optimization20–22 and linear organic synthesis.23–26 However, currently, high-throughput synthesis (HTS) is typically associated with low molecular diversity. Indeed, these two technologies (HTE and parallel synthesis) rely on performing similar types of chemistry experiments in the same campaign with adjusted parameters, allowing optimization of reaction conditions for one transformation in the case of HTE, or access to target libraries with parallel synthesis.
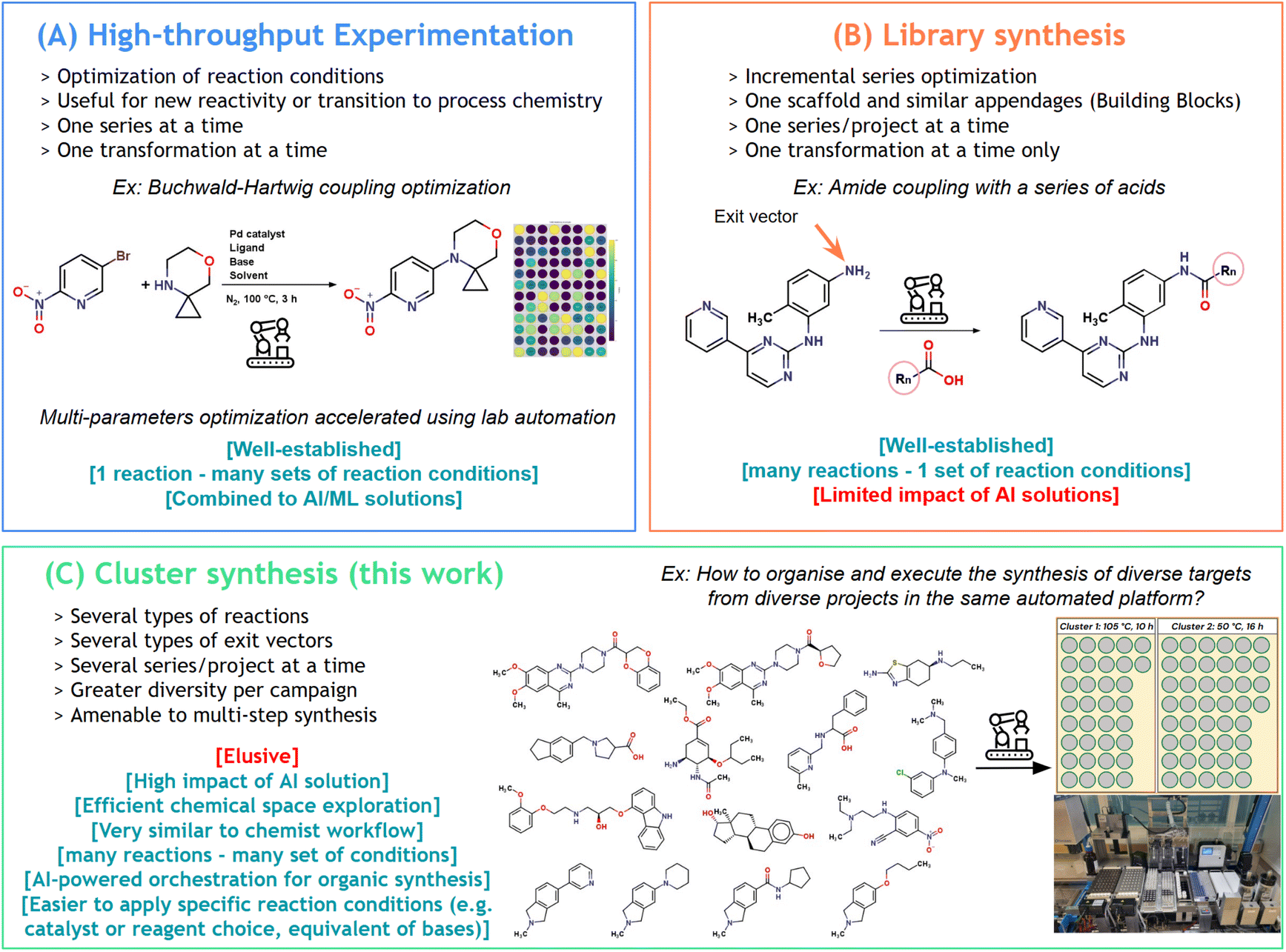 | ||
| Fig. 1 Applications of robotic platforms in organic synthesis: (A) high-throughput experimentation for reaction optimization. (B) Conventional approach: siloed library synthesis for each project; (C) this work: merging and clustering approach for the synthesis of diverse molecules on a single platform. | ||
On the other hand, the automated synthesis of highly diverse molecules, originating from a diversity-oriented strategy or even from different projects, and using a vast variety of reactions simultaneously, is largely unexplored and remains a great challenge (Fig. 1C). Indeed, such a new automated organic synthesis paradigm requires criteria to batch reactions together:
• a queue list of molecules to make;
• a queue list of reactions to perform;
• an interface to make decisions;
• a scheduler that merges reactions together in a cluster based on similar reaction time and temperature;
• a versatile platform with enough positions for solids, solvents and solutions;
• a robust traceability tool, with mapping and barcodes to avoid mistakes;
• a general program that can adapt to any size of campaign and any type of chemistry within the physical limits of the synthesis platform.
To this end, we developed our own Iktos robotic platform (Section 2), based on proprietary existing tools (Subsection 2.1), which have been integrated with newly developed tools (Subsections 2.2 and 2.3). Finally, we will showcase its utilization, focusing on the workload organization for the synthesis of 135 diverse molecules (Section 3).
2 Iktos robotic platform
2.1 Proprietary existing tools as a pillar for building an autonomous platform
Herein, we present an AI-driven platform built to tackle this novel approach to synthetic chemistry, one that can generate molecules, propose a retrosynthetic access, schedule the workload, structure the data and pilot a robot to execute automated organic synthesis (Fig. 2). This balance of human creativity and AI-driven, constraint-aware design ensures that the robot is fully used. Our vision is actually in good agreement with the approach developed by the MicroCycle at Novartis.27 Our platform is specialized in drug discovery, where we operate DMTA cycles to advance medicinal chemistry projects (Fig. 2A); however, it may be generalized to other domains relying on organic synthesis. In our previous work,28 we presented our vision on how DMTA cycles can be handled differently, with an advanced integration of our AI tools for drug design and retrosynthesis, and the use of a robotic platform to accelerate the synthesis part of libraries of molecules. In this work, we report on the implementation of scheduler layers and chemical templates to combine different types of reactions within a single synthetic campaign (Fig. 2B). Indeed, the ultimate goal of our platform is to expedite DMTA cycles, with a smooth data flow throughout the cycle. The requirements are depicted in the next paragraphs and in Fig. 2C, where the silos drug design (1), retrosynthesis (2), orchestration (3) and laboratory execution (4) need to be integrated based on existing tools (1 and 2) or newly developed ones (3 and 4).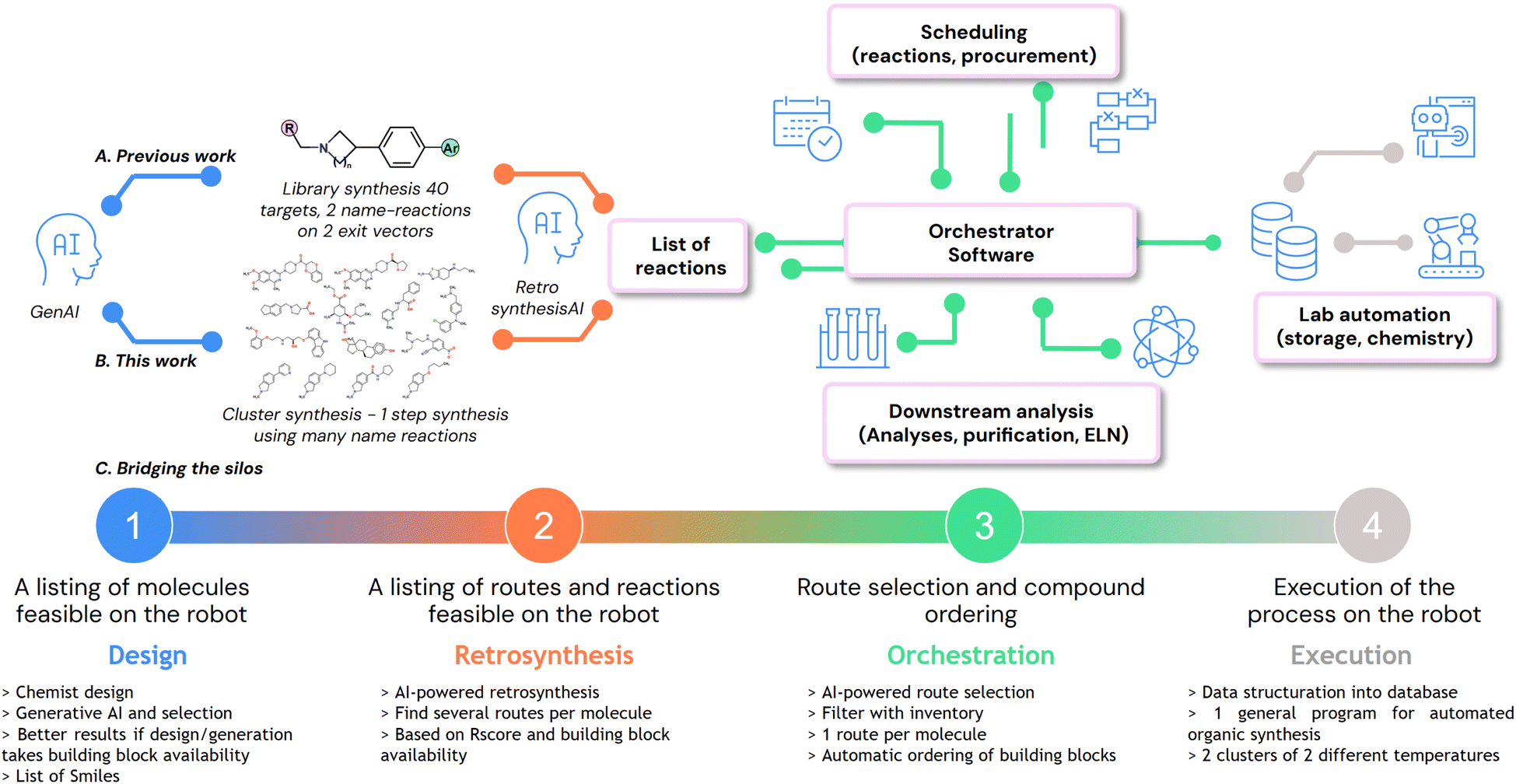 | ||
| Fig. 2 A) Our previous work: generation using GenAI, 40 molecules selected based on 1 scaffold with 2 exit vectors run as library synthesis. (B) This work: generation of a list of unrelated molecules run as cluster synthesis. (C) Overall data workflow: the journey of a molecule from virtual to synthesis using integrated silos. | ||
The available tools were adapted to our specific needs: grippers for vial manipulation, a barcode scanner for inventorization, solid and liquid dispensers, ionizers, heatable and shakable racks of substrates, two independent reactors with cooling and heating systems, an SPE filtration set-up, and MTP racks for collecting final compounds. With the hardware in hand, the goal was then to design a data model compatible with the use of a general program for launching campaigns of automated organic synthesis. We identified 3 different levels of data: campaign parameters, batch parameters, and reaction parameters. For the sake of clarity, all the necessary information to run chemical reactions is now divided into 11 CSV files (campaign, batch, substance, reaction, substrate, powder, solution, solvent, product, config, and preload) (SI). These data correspond to variables in our program. For each action such as powder dispense or solution dispense, we built specific fully parameterized macros. Once operated, these macros can search data in the CSV files, create new dynamic zones where they are actionable, iterate multiple times, and perform tool actions. By utilizing these key programming tools (variables, macros and dynamic zones) in the robot software (AutoSuite), we managed to build a robust program that encompasses all cases of campaigns we may need to set up on our platform, from 1 to 96 reactions. Without such a program, every new campaign would require spending time to prepare complex sequences of actions and parameters, with a great risk of error, potentially leading to failure of the campaigns and hardware incidents. With a general organic synthesis program in hand, we turned our attention to the development of a tool that generates reliably and automatically structured data. Indeed, regarding orchestration (Fig. 2C(3)), extensive development was required on our side to create a functional platform able to connect the four silos, by structuring the data and assisting chemists in making decisions.
2.2 Development and integration of new tools
In order to optimize the synthesis flow of the platform, we aim to group various reactions in the same reactor to run them in parallel: a new synthesis paradigm that we call cluster synthesis. Reactions can share the same reactor if they have similar reaction conditions. Therefore, we implemented two layers of scheduling, aimed at maximizing the number of reactions inside each synthesis campaign. The first layer is called strategic scheduling (detailed in Subsubsection 2.2.1) for route selection. It is followed by the validation of expert chemists and the assignment of reaction conditions (i.e. templates, see Subsubsection 2.2.2), which unlocks the provisioning of chemicals through a procurement system (see Subsubsection 2.2.3). The second layer is the tactical scheduling, which is run on a daily basis to schedule the cluster synthesis (see Subsubsection 2.2.4).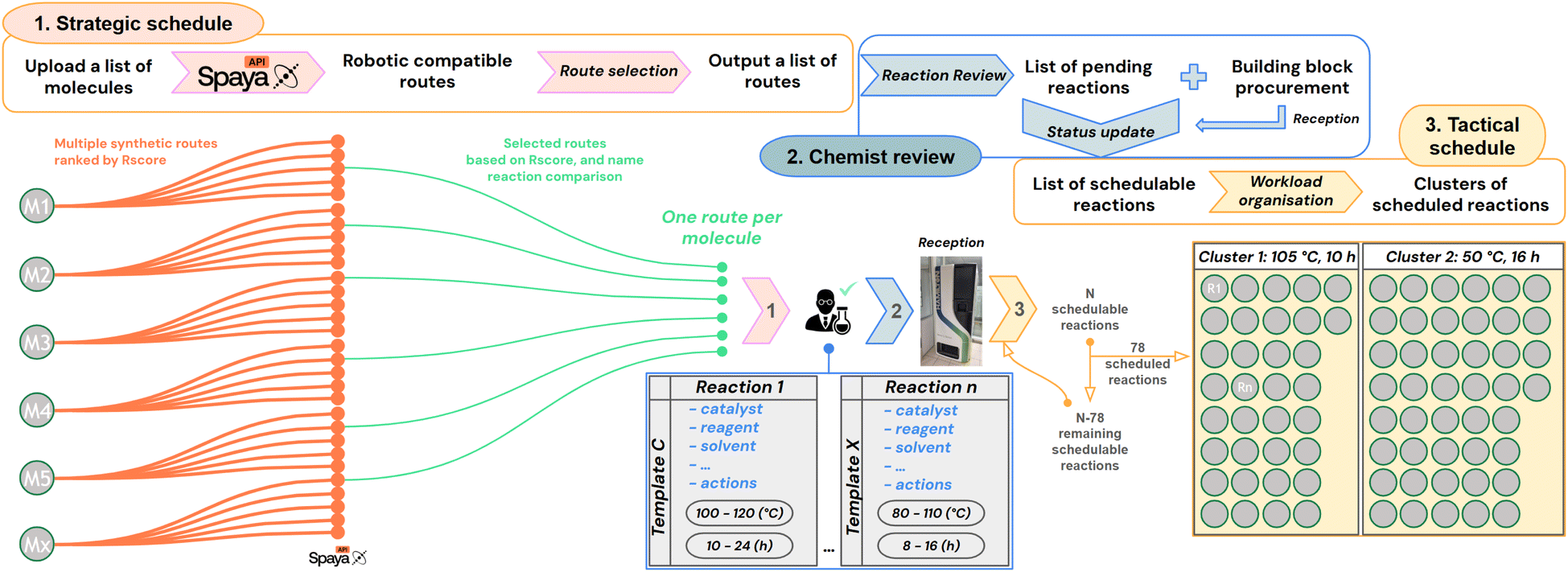 | ||
| Fig. 3 Top: overview of the workflow to translate digital molecular ideas into physically executable reactions. (1) Strategic scheduling: for each input molecule, multiple synthetic routes are generated via the Spaya API under robotic constraints. One route per molecule is selected based on feasibility (Rscore), building block availability, and name reaction comparison. In the output, the Pistachio name reactions are given. (2) Chemist review: Pistachio name reactions are automatically translated into internal lab name reactions, allowing chemists to assign one of the associated reaction templates. Each template encompasses the necessary chemicals and operational conditions, with temperature and time ranges. Building blocks are either sourced from the internal inventory or requested via an integrated procurement system. (3) A scheduling layer evaluates which reactions can be run based on the current inventory and overlapping template conditions. Reactions are clustered into executable batches by finding intersections in condition intervals (e.g., temperature and time), enabling efficient use of robotic reactors through parallel synthesis across diverse projects and chemotypes. Bottom: exemplification of clustered reactions organized for parallel execution. | ||
Therefore, we decided to create our own lab name reactions and associated them with one or several Pistachio name reactions. To these lab name reactions, we then designed and associated sets of conditions we used to call “reaction templates” (see Fig. 3(2)). We defined the required chemicals (reagents, additives, catalysts, ligands, and solvents) from a list of chemicals validated on the platform. We selected their stoichiometry, their position in the robot (solution, powder or solvent), and also the expected final reaction concentration. Several optional robot actions were registered in the template, such as rinsing, quenching, dilution and final SPE filtration steps.
Importantly, we defined a temperature range and a reaction time range where we believe the template will be valid. For example, for amide bond formation via peptidic coupling, one template used in the following study was set up between 25 and 35 °C for 6 to 18 h. These ranges allow the creation of clusters based on their intersection with other ones, and running different types of reactions in the same campaign (see Subsubsection 2.2.4). Overall, for the sake of synthesis flexibility, we can associate to each name reaction several templates having different reaction conditions.
For each molecule designed, the strategic schedule returns one route with the associated name reaction of each step. Similarly to Coley et al.,23 we consider CASP as a recommendation problem, where the proposed routes should be validated. In this perspective, we put in place a reaction review panel, which allows chemists to choose which reactions should be continued towards synthesis, and easily discard reactions they consider not appropriate in the given context. At this stage, chemists will review each reaction, verify that the name reaction extraction was correct, update it if necessary, and select one of the available templates. Selecting a template will automatically fill in all the necessary parameters required to run a reaction: the only things the chemist needs to choose will be the limiting reactant, the stoichiometry and the reaction scale. If further changes are expected, it is possible to choose another template or to edit an existing one. So far we have developed 130 templates, associated with 68 lab name reactions that encompassed 127 Pistachio name reactions, representing 36.5% of the patented literature (SI). If the proposed route involves multi-step synthesis, chemists will review all reactions individually and the validation of each step is necessary to validate the route.
Indeed, all building blocks required for a reaction are pre-weighed, by chemists or an external provider, into 2 mL barcoded vials and registered into the Ilaka inventory before the campaign set-up. Finally, we seamlessly integrated a smart robotic storage system (Hamilton Verso Q20) by connecting it to Ilaka via API. This straightforward communication allows an automatic reception and update of substrates, as well as a fast and errorless recovery of a list of vials for a specific campaign.
Moreover, depending on the slot capacities, some slots can be shared for multiple reactions: if two reactions share the same solvent, this solvent can be stored in only one slot if the combined volume is small enough, allowing for extended capabilities rather than limiting the number of reactions to the number of solvent slots of the robot. This can also be done for powders and solutions. This parameter, taken into account in the optimization problem, is referred to as a bin-packing problem.38
In multi-step synthesis, another molecule type is involved in Ilaka, naturally called “intermediate”. The status of the intermediate stays as “pending” until the completion of its synthesis, when it will then be marked as “in stock”. When available in the inventory, the next synthetic step will be updated from “pending” to “schedulable” and will therefore be directly considered in the next tactical schedule. The tactical schedule then unleashes its powerful ability to (re)program very quickly the optimized synthesis planning of the day based on stock availability.
2.3 Integration in Ilaka GUI
With a complex workflow and multiple novel tools in hand, we turned our efforts to the development of a user-friendly interface. Indeed, user interface design in laboratory automation systems represents a critical bottleneck in pharmaceutical research, where sophisticated AI-driven synthesis processes remain underutilized due to poor human–computer interaction paradigms.39,40 While automated laboratory orchestration technologies have matured significantly,41,42 their adoption is severely hampered by interface complexity that fails to align with researchers’ workflow requirements. This design-user mismatch creates cognitive overhead that diminishes the effectiveness of otherwise powerful automation capabilities, ultimately limiting research productivity and system adoption rates.To address these challenges, we developed Ilaka, our user-centered web application for laboratory orchestration supervision (SI). Our approach prioritizes researcher workflows while leveraging advanced technical architecture: an intuitive front-end utilizing GraphQL for optimized data exchange,46 coupled with a distributed microservices back-end implementing event sourcing patterns. The resulting platform enables researchers to maintain their domain expertise and decision-making authority while fully exploiting AI and automation capabilities, effectively bridging the gap between technological sophistication and practical usability in laboratory settings.
| Trust mechanism | Implementation | User benefit |
|---|---|---|
| Decision point transparency | Clear explanation of logic | Confidence in automated decisions |
| Calculation transparency | Visible auto-calculations for reaction parameters, equivalent ratios, amount of substance, mass and volume | User verify and modify chemical calculations |
| Comprehensive reporting | Visualize and export any data | Users can export detailed analysis |
| Workflow phase visibility | Step-by-step activities and status tracking | Process understanding |
| Cross-context operations | Unified state transitions | Seamless experience |
Overall, we assembled and integrated both software and hardware into a functional, responsive and resilient automated discovery platform that can handle many use cases occurring in a chemistry laboratory. From a list of targets and a list of routes in hand, our chemists can populate our orchestration software to organize the work as schedules and prepare the corresponding campaigns. This digitization process already accelerates the Make step of the DMTA cycle. The intuitive utilization of our Chemspeed robotic platform is an additional way to accelerate this step. From our orchestrator software, machine-readable data are obtained and used as input for the Chemspeed platform. In our previous study, we exemplified the parallel synthesis of a small library of 40 compounds, made in 2 steps (Suzuki coupling and reductive amination).28 In the next section, we will apply it in the context of the synthesis of a large queue of various molecules.
3 Experiments, results and discussion
3.1 Generation of a list of molecules
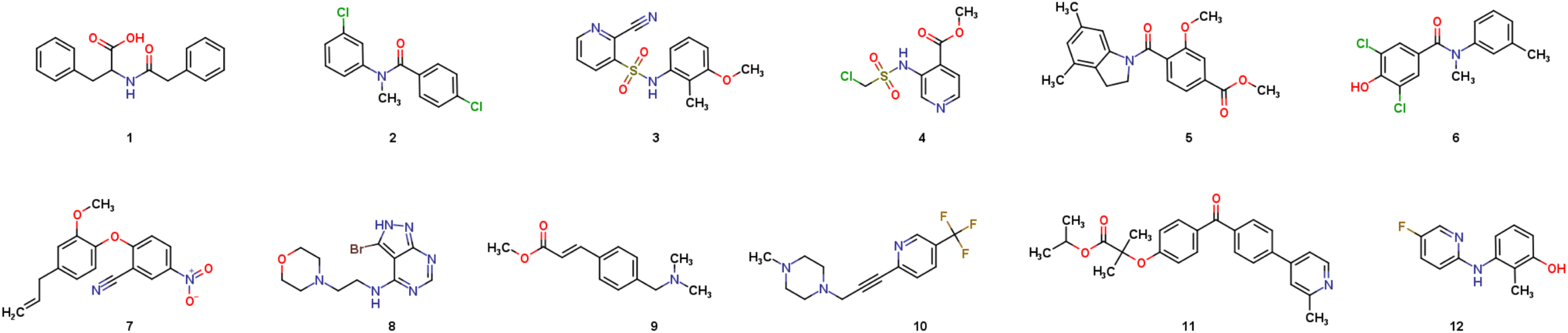
Our study started with the generation of the chemical space accessible in one step based on available chemicals in our inventory and the name reactions validated on our platform. At this stage, we considered 68 name reactions, and selected about 350 compounds as starting materials. A one step forward-generation using our growing optimizer29 was performed, leading to a chemical space of over 4000 molecules (SI). A retrosynthesis search was then performed, requiring only few hours, and the output was additionally filtered to ensure having routes compatible with the building blocks available in our inventory. The output was a list of reaction SMILES, the name reaction associated, and the building-blocks required. To limit the number of experiments performed on the robot while testing it on challenging conditions (by maximizing diversity), we then applied further filters, such as limiting the maximum number of reactions per name reaction (25/NR), limiting the maximum of 5 reactions per substrate, and one reaction maximum for compounds in a batch of 50 mg. Finally, since diversity of chemical reactivity was important for this work in order to assess the clustering approach, we tried to get the maximum number of name reactions possible in this list. This filtration process led to 335 molecules in a dataset having good quality synthesizability. Among these 335 molecules, which can be prepared in one step, we finally performed a manual selection to get a final list of 135 molecules along with 135 reactions to perform in this project.A projection of the synthetically accessible chemical space is illustrated in Fig. 4B, with the 3616 molecules represented in blue; and the final selection of 135 targets in orange. We were pleased to see that these selected targets allowed covering a wide chemical space. Furthermore, based on the distribution displayed in Fig. 4B, 27 lab name reactions were used, thus highlighting the wide diversity of chemical reactions. In summary, 135 diverse molecules have been selected by our process (scheduler + expert chemists), ready to be synthesized in one-step routes.
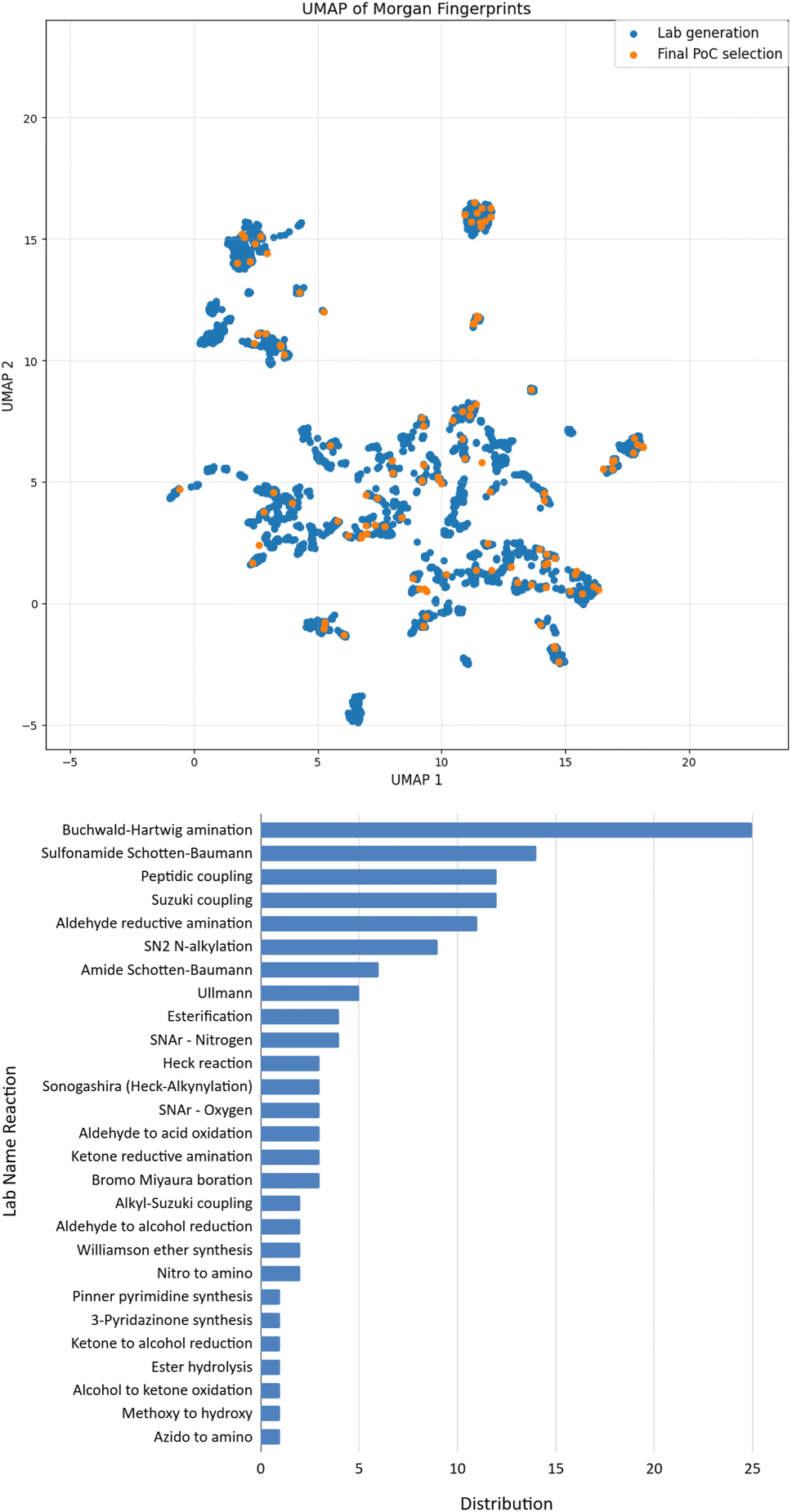 | ||
| Fig. 4 Top: the chemical space 2D representation shows the coverage of the 135 molecules (in blue) selected among the 3616 synthetically accessible generated molecules (in grey). Such a representation has been obtained using UMAP based on the pairwise distances between the Morgan fingerprints of the molecules (radius 2, 2048 bits); Bottom: distribution of 27 name reactions used for the synthesis of the 135 molecules. | ||
3.2 Reaction review to assign chemical templates
With these 135 validated targets and disconnections in hand, we aimed at preparing the necessary data for real-life synthesis. At this stage, we were able to review each reaction in Ilaka, verify that the name reaction extraction was correct, update it if necessary (e.g. typically to make a distinction between SNAr and Buchwald–Hartwig amination), and select one of the available templates. For each reaction, we defined the limiting reactant, the stoichiometry, and the scale of the reaction was fixed at 0.10 mmol. The reaction review process for the 135 reactions took us roughly 2 hours, meaning an average of one minute per reaction.Then, the following step is the procurement of the reactants. Since all compounds were already in our inventory, we did not need to buy them. However, it was necessary to reformat them in the 2 mL barcoded vials, and assign a barcode to each building block. Loading this information in our software unlocked the building block status from incoming to in stock and the reactions status from pending to schedulable.
3.3 Workload organization based on chemical templates and robotic constraints
The tactical scheduling version used in this study produced variable results, due both to the stochastic nature of the run and to its limitation of handling only one campaign at a time. Therefore, when running the tactical scheduling for the 135 reactions, several iterations were processed in order to have a combination of schedules that allowed running all reactions in only three campaigns. The chosen iteration of the tactical scheduling suggested a first campaign with 95 scheduled reactions in 2 clusters of 48 and 47 reactions. These clusters contain both 10 lab name reactions, associated with 23 and 20 Pistachio name reactions respectively. The proposed schedule was 90 °C for 10 h and 25–35 °C for 14–18 h. In the case of the 2nd cluster, we decided to run the reactions at 35 °C for 16 h. After approval of this first schedule, a second schedule was attempted on the remaining 40 reactions, and 29 reactions were scheduled in 2 batches of 15 and 14 reactions each at 105 and 70 °C, respectively. Finally, a third schedule was obtained from the last 11 reactions, with 2 final batches containing 7 and 4 reactions.Overall, for 135 reactions, 6 clusters were obtained with a good distribution of temperature (10, 35, 40, 70, 90, and 105 °C), and they could be run in only 3 campaigns according to our robotic constraints (see result output Fig. 5).
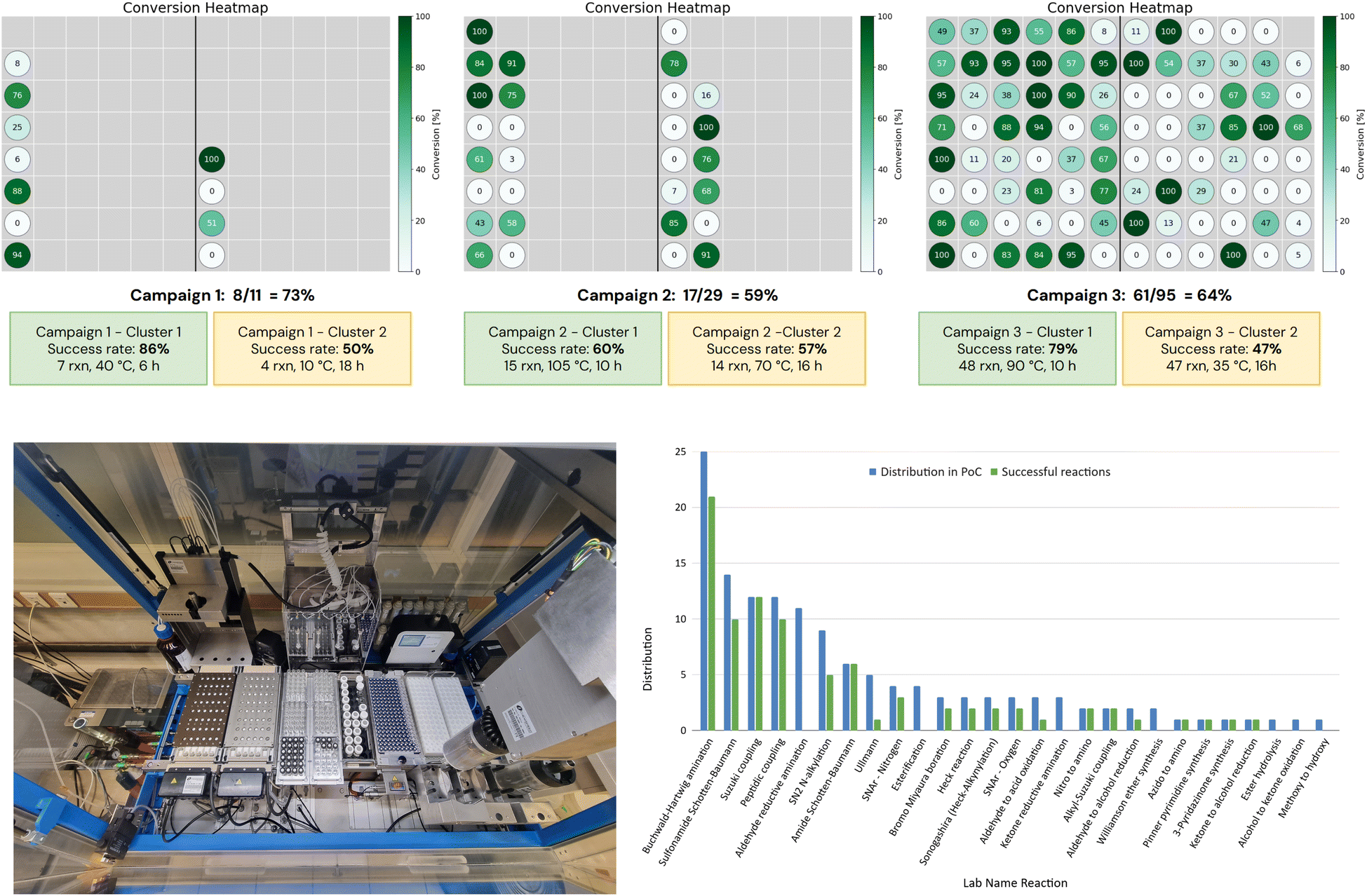 | ||
| Fig. 5 Top: conversion heatmap by campaigns and clusters; bottom left: robotic set-up for performing Campaign 3 of 95 reactions; bottom right: success rates by name reactions. | ||
3.4 Synthesis campaign set-up
Once the reactions are scheduled, our interface Ilaka gives information to the chemist to prepare the campaign and facilitate the transition from virtual to reality. It includes the total mass of each powder, the volumes of each solvent needed, a tool for automatic calculations to easily prepare stock solutions in barcoded vials and a mapping of each rack. Finally, all data are structured in a machine readable format and can be exported in CSV files or pushed in a database connected to the Chemspeed automated platform.After loading the platform with all necessary components using SPE-cartridges, and both non-barcoded vials (solvent) and barcoded vials (building blocks, stock solutions, powders, and crude collection), the robot can start executing the program. As mentioned in Subsubsection 2.1.3, our workflow is general and can adapt to any number of reactions included in the campaign: in other terms, we will use only one program to set up the next three campaigns of this study (11, 29 and 95 reactions), with only the required data being different.
The program begins with inventory tasks, prompting the user to confirm the positions of non-barcoded vials. The robot then scans all barcoded vials and powder containers to verify the presence of expected materials and record the exact location of each item—since vials can be placed randomly to reduce human error. Once the robot has validated the inventory and mapped all vial positions, the chemistry workflow can begin. The first step is substrate dissolution: appropriate solvents are dispensed into the 2 mL vials, then heated at 35 to 40 °C and shaken for 15 minutes. Substrates are then transferred into the reactors, followed by stock solutions, powders, and any remaining solvent if required. Each reactor – capable of handling up to 48 reactions – is heated and stirred according to the campaign parameters (temperature, agitation, and duration). Post-reaction operations are predefined in the execution files (CSV), including dilution of the reaction mixture, quenching of reactive intermediates or excess reagents, and filtration.
3.5 Synthesis results
In terms of synthetic output, all reaction mixtures were analyzed using acidic and/or basic HPLC methods. The success threshold for a reaction was set at 5% of the total UV area in the trace of the best detection method (acidic or basic). This level was considered sufficient to allow purification of a minimum amount of compound, since in our drug discovery approach only 1 mg is needed for testing, with active compounds being re-synthesized as required. Then, a conversion rate was estimated for each reaction based on LC-MS analysis interpretation. Resulting heatmaps, with detailed success rates per campaign and per cluster, are depicted in Fig. 5 (top).Overall, 86 expected products have been detected after 3 campaigns, representing 64% of the 135 reactions. This is a good success rate, considering that no optimization was performed. Excellent reactivity outcome was observed for classical reactions (see Fig. 5 bottom right), such as palladium catalyzed couplings (Suzuki and alkyl-Suzuki couplings at 100%, Buchwald–Hartwig amination at 84%, Boration, and Heck and Sonogashira couplings at 67%), amide bond formation (amide Schotten Baumann at 100% and peptidic coupling at 83%), sulfonamide synthesis (71%). On the other hand, reactions based on C–O bond formation proved unsatisfactory with only SNAr being above average (SNAr – oxygen at 67%, Ullmann condensation at 20%, and Williamson ether synthesis and esterification at 0%). Although these statistics on a limited number of reactions should be taken with care, they still provide first insights into the strategy of running many chemical reactions simultaneously. Furthermore, 2 name reactions (namely aldehyde and ketone reductive aminations) led to 10% of the global failure. These failures could be potentially attributed to one or several of these issues: the choice of the template, the quality of a reagent (e.g. NaBH(OAc)3) or solvent, the length of the set-up for an automated process on a very large campaign, or substrate-specific sensitivity of a reaction. Since the 0% success rate for reductive aminations was clearly not in line with our statistics on these name reactions (typically 65–80% success rate per campaign), we decided to run again the 14 reactions in a new campaign by changing the template and the batch of the reducing agent. The alternative template included a larger amount of the reducing agent (NaBH(OAc)3) and the use of DCM as the solvent (instead of DMA used previously). With the clustering parameters being similar, this additional campaign was run at 35 °C for 16 h, like in Campaign 3 – Cluster 2. Overall, it would have led to the same six clusters obtained for the 135 reactions. We were pleased to observe the formation of 11 products out of the 14 reactions (SI) in LC-MS analysis, representing 79% of success rate, a result more in line with our current statistics on reductive aminations. Taking into account these additional successes among the total of 135 reactions, we could increase the global success rate from 64% to 72%.
3.6 Purification of samples
Out of the 97 successful reactions, 12 were selected for purification, primarily for time and cost considerations, as purification was outsourced. The goal was to choose a representative subset of compounds, covering structural diversity, a wide range of name reactions, and varying levels of %UV area observed in LC-MS analysis (from 6% to 100%) (see Table 2). This selection aimed at assessing the practical difficulty of purification by comparing HPLC-based UV detection % with the actual isolated yields.| Molecule | Reaction ID | Lab name reaction | % Area (UV), LC-MS method | Net weight (mg) | Purity (%) | Isolated yield (%) | Yield/UV detection |
|---|---|---|---|---|---|---|---|
| 1 | IKT-R-26 | Amide Schotten–Baumann | 6%, acidic | 1.1 | 93 | 4 | 0.67 |
| 2 | IKT-R-81 | Amide Schotten–Baumann | 76%, acidic | 13.7 | 98 | 48 | 0.63 |
| 3 | IKT-R-42 | Sulfonamide Schotten–Baumann | 67%, basic | 0.4 | 0 | 0 | 0 |
| 4 | IKT-R-39 | Sulfonamide Schotten–Baumann | 21%, basic | 1.8 | 95 | 6 | 0.33 |
| 5 | IKT-R-31 | Peptidic coupling | 52%, acidic | 10.8 | 100 | 32 | 0.62 |
| 6 | IKT-R-46 | Peptidic coupling | 30%, basic | 7.9 | 97 | 25 | 0.83 |
| 7 | IKT-R-36 | SNAr – oxygen | 78%, acidic | 5.6 | 97 | 18 | 0.23 |
| 8 | IKT-R-130 | SNAr – nitrogen | 93%, acidic | 10 | 90 | 28 | 0.30 |
| 9 | IKT-R-47 | Heck reaction | 100%, acidic | 0.6 | 3 | 0 | 0 |
| 10 | IKT-R-10 | Sonogashira (Heck-alkynylation) | 100%, acidic | 3.1 | 90 | 10 | 0.10 |
| 11 | IKT-R-134 | Suzuki coupling | 83%, acidic | 14.5 | 100 | 35 | 0.42 |
| 12 | IKT-R-33 | Buchwald–Hartwig amination | 87%, acidic | 4.1 | 100 | 19 | 0.22 |
|
|||||||
Our external partner succeeded in the purification of 10 compounds, i.e. 83% of success rate, with isolated masses from 1.1 to 14.5 mg, final purities above 90%, and corresponding yields from 4 to 48%. Surprisingly, we noted that some compounds that are well detected by UV could be difficult to purify, with the examples of reactions IKT-R-42, -47 and -10. They showed respectively 67, 100 and 100% of UV detection of compounds 3, 9 and 10, but below 10% of yield, due to low recovered quantity and very low purity. Degradation or issues during the automated outsourced purification process could explain these disappointing results. Indeed, the time between sending the crude materials and receiving back the purified products was about 3 weeks, which encourages us to internalize the analysis and purification process on our platform.
More positively, some compounds that appeared at first glance difficult to purify, either due to low % in UV (IKT-R-26, molecule 1) or to closeness with other peaks (IKT-R-46, molecule 6), finally showed relatively good recovery, highlighted by a comparison rate Yield/UV detection of 0.67 and 0.83, respectively. In the case of reaction IKT-R-26, even if the yield of 4% was low, we considered the purification successful since we obtained more than 1 mg of isolated compound 1, with 93% of purity, enough to perform a future biological assay. Other purification results were quite satisfactory, with acceptable recovered quantities and Yield/UV detection rates between 0.22 and 0.63.
Therefore, based on these results, we could conclude that the probability of purification success was not directly linked to the initial UV detection %, and cannot be easily predicted. Indeed, we assume that the limited number of purifications performed in this project could not allow generalization of the calculated rates. However, we were able to confirm that performing the full process of one-step robotic synthesis, analysis, and purification was possible to obtain a large diversity of pure molecules, in sufficient quantities for further biological testing in the context of a medicinal chemistry project.
3.7 Discussion
In the context of our study, we decided to schedule all the available reactions. The goal was to determine the set of temperatures and times for both reactors of 48 slots and to maximize the number of reactions compatible with this setup. Thus, the planning of the 135 reactions was possible in 6 clusters resulting in only 3 synthesis campaigns on our platform. However, we could note that this version of our algorithm, which relied on optimizing the filling of the reactors for each campaign on a daily basis, tends to provide a variability of 3 to 4 campaigns, with very unbalanced sizes of campaigns. As a result, the time of preparation for the chemists and robotic set-up will be largely affected by the number of reactions, making it potentially unpractical for very large campaigns, and inefficient for small ones. Moreover, some additional parameters like the numbers of solvents, substrates, solutions, and powders to be used are not optimized in such a schedule, but just verified when running the filling of the available positions.To address such limitations, we later upgraded our tactical scheduling algorithm to represent all constraints natively in a linear optimization problem (see Subsection 5.1) (SI). The optimization problem incorporates all robotic hardware constraints, including the limited number of slots for each compound type (solvents, substrates, solutions, and powders) and reactor capacity limitations. We define a clustering problem where each cluster represents a reactor, and pairs of clusters form execution groups (potential synthesis campaign). This approach offers several advantages: first, it provides complete visibility into the entire reaction queue through a single tactical request, enabling chemists to predict the total number of groups required and choose a group based on their available time. Second, each approved group of clusters becomes a campaign ready for robotic execution.
To validate this improved algorithm, we retrospectively applied it to the 135 reactions from our previous synthesis campaign, resulting in 6 well-balanced clusters with more evenly distributed group sizes compared to the original approach. The comparison between the two versions is shown in Table 3. All reactions could be still performed within 3 groups, containing respectively 71, 52 and 12 reactions (note that the reaction numbers can vary depending on the runs of the tactical scheduling, because there could be multiple optimal solutions to the problem as discussed in Subsection 5.1), ensuring a more practical set-up for chemists when possible. We could also note that temperature and time distributions are similar to the first ones, and that name reactions diversity in each group seems more balanced. Overall, a more optimized solution is found by this new version of our scheduling.
 )/version 2 after optimization (
)/version 2 after optimization ( )
)
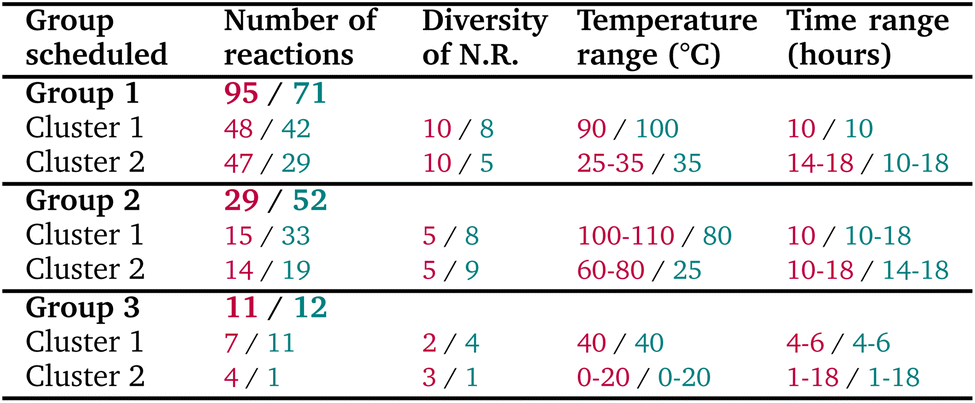
|
Retrospectively, the key takeaways from this project are (1) the safe and reproducible data flow and (2) the significant time saved through the use of our automated workflow. For each campaign, the total preparation and execution time was tracked (SI). As expected, the process becomes increasingly efficient as the number of reactions increases. While little time was saved in the 11-reaction campaign, substantial time saving was observed for the 29- and 95-reaction campaigns, freeing the chemist to focus on other tasks. In addition, the use of barcodes and standardized, reproducible procedures help ensure an error-free reaction setup.
We compared the estimated time to plan and execute the synthesis of our 135-reaction set using a manual process or an automated workflow, either via a library mode or via our clustering approach (SI). Thus, our workflow proved to be at least 2 to 4 times faster than manual or automated library approaches, while offering greater reproducibility and higher-quality data. However, a manual setup allows more tailored adjustment of reaction conditions for each individual transformation, potentially leading to higher success rates. Our current approach is designed to accelerate the execution of large and diverse sets of syntheses, prioritizing throughput over yield optimization.
Interestingly, since Ilaka tracks all events and stores the data of our platform, it would be possible to flag any unexpectedly poor outcome at the campaign or name reaction level. This could be performed simply by comparing the performance of a campaign (campaign success rate) or of a name reaction (name reaction success rate) and compare it directly to historical data. With the implementation of a trigger (e.g. <10% of success rate or < “historical success rate”/2), a notification could directly draw the attention of the chemist to investigate more deeply the reasons behind the failure. In the long term approach towards a more autonomous laboratory, it could be envisioned to automatically trigger a new campaign with the next most probable template. This could have been applied to handle the issue encountered with the reductive amination.
An additional advantage of the robotic platform is its scalability: if higher throughput is required, the system can be expanded by adding more robots. This modularity enables a straightforward path to increase capacity without fundamentally changing the workflow, making it well-suited for projects of varying size and ambition. In a context where new targets are continuously added to the pipeline, the advantages of a robust and fully automated workflow become even more significant. To further improve performance, several enhancements are under development, such as enabling reaction duplicates under varying conditions and dynamically optimizing or refining synthetic templates based on usage history. These improvements aim to increase the overall success rate of high-diversity, clustered synthesis campaigns.
4 Conclusion and outlook
Our robotic platform, launched three years ago, has rapidly evolved through the stepwise integration of key components—including our chemical template system, AI-based scheduling tool, and procurement services, all seamlessly orchestrated by our central software Ilaka. This platform represents a paradigm shift in chemical synthesis, enabling high-throughput execution by merging pending reactions into a queue and clustering them based on compatible temperature and time parameters. Additional robotic constraints are also considered to ensure that the generated schedules are executable in practice, making the platform not only efficient but fully operational in real-world settings.This scheduling algorithm is embedded within a proprietary software suite that offers an integrated environment to manage retrosynthesis, procurement, inventory, compound storage, and robotic execution. We believe this new synthesis paradigm has the potential to accelerate discovery across all domains where organic chemistry is a key enabler of innovation, while delivering structured and reproducible data. Within our internal research pipeline, it already supports the efficient development of drug candidates,47 handling multiple targets and projects in parallel on a single automated platform.
The synthesis workflow we have developed for our Chemspeed system is highly adaptable, compatible with a wide variety of chemical reactions and scalable for campaign sizes ranging from 1 to 96 reactions. Current limitations include the inability to perform staged additions or pre-activation steps (e.g., sequential additions with intermediate stirring) due to the nature of our clustering and reagent distribution strategies. However, we are actively exploring solutions to overcome these challenges. A new study that mimics real world situations (multi-step syntheses, delay of chemical delivery etc.) is under consideration in our laboratory.
Looking ahead, our focus for the next two years is to further enhance the platform by developing and integrating automated analysis and purification capabilities48–50 into Ilaka, making the full process more intelligent, schedulable, and efficient. We are also progressing toward the integration of a bio-assay platform to enable increasingly autonomous DMTA cycles.
Finally, we record every decision made by chemists within Ilaka—including template selection and reaction choices—with the long-term vision of building AI models capable of selecting the most appropriate synthetic template based on the context. This data-driven approach paves the way toward our ultimate goal: the creation of a fully autonomous laboratory.
5 Methods
5.1 Tactical schedule
In this section, we want to cluster reactions as effectively as possible, which means minimizing the number of synthesis campaigns. Each cluster of reactions is identified by its campaign cp and its reactor j, with j ∈ {0; 1}.To do so, we defined an integer linear programming problem that takes a list of Nr reactions as input, and outputs the assignment of each reaction to a cluster (cp, j), with each reaction ri having ranges of accepted temperatures Ti and durations Di.
Let C = {cp∣p ∈ {1, …, Nr}} be the set of available campaigns. We have that
| ∀ p ∈ {1, …, Nr}, cp = {ri∣xip = 1} |
The objective is to minimize the number of campaigns used:
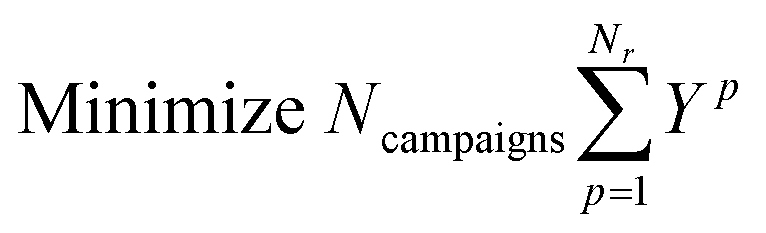
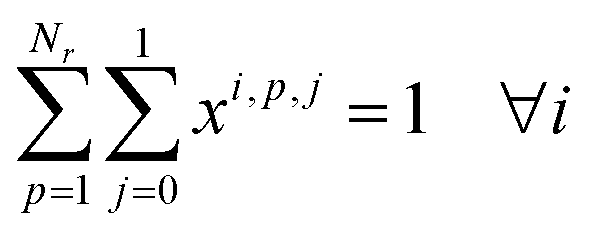
| xi,p,j ≤ yp,j ∀i,p,j |
2. Cluster capacity: the number of reactions assigned to a cluster must be smaller than the capacity of the reactor.
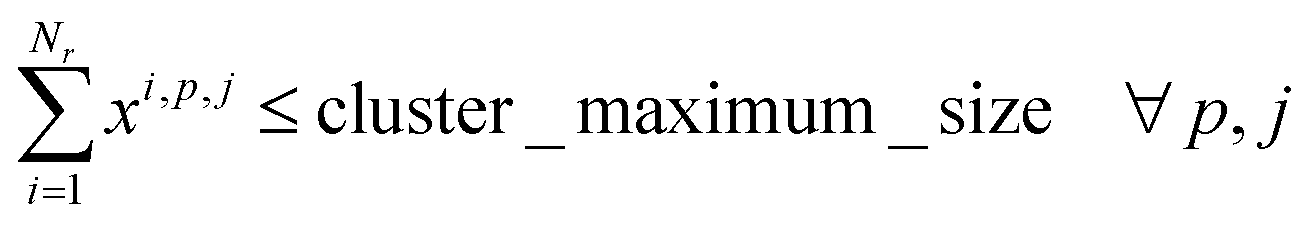
3. Temperature and duration assignment:
To deal with temperature and duration constraints, we let ztempp,j,T be the binary variable indicating if temperature T is assigned to cluster (cp, j) and zdurationp,j,D the binary variable indicating if duration D is assigned to cluster (cp, j).
Each used cluster (containing at least one reaction) has exactly one temperature and duration time assigned.
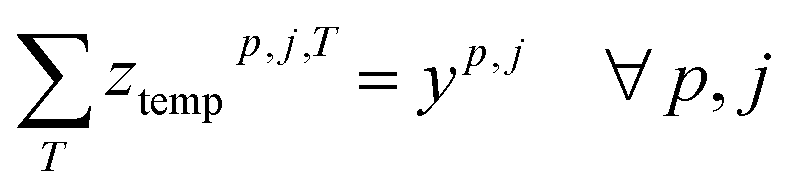
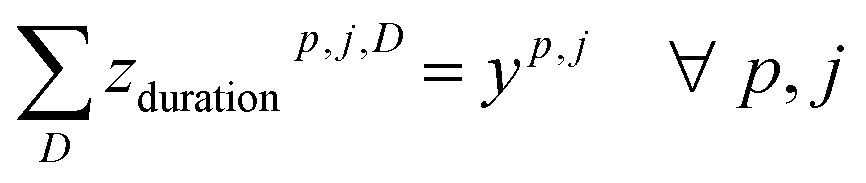
4. Reaction compatibility: if a reaction i is in cluster (cp, j), the assigned temperature and duration of the cluster are contained in the templates of reaction ri
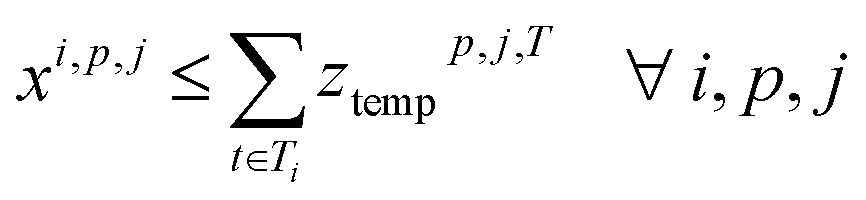
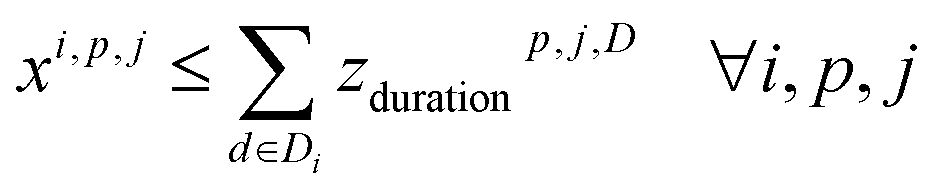
5. Campaign usage: a campaign is used if at least one of its clusters is used.
| Yp ≥ yp,0 ∀ p |
| Yp ≥ yp,1 ∀ p |
6. Capacity constraints and bin-packing38 formulation:
To ensure feasibility with respect to robotic hardware limitations, we impose capacity constraints per campaign. For each campaign p, the total number of substrates used by all reactions assigned to that campaign must not exceed the available slots:
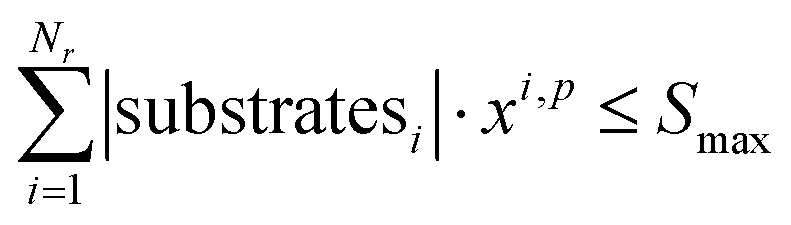
For solvents, solutions, and powders, the allocation is modeled as a bin-packing problem for each unique chemical identifier (SMILES). Each usage of a SMILES by each reaction in a campaign must be distributed among a finite number of slots (or bins), Bmax, each with a fixed capacity Vmax. Let wi,p,sm,b be a binary variable indicating whether the usage of SMILES sm by reaction ri in campaign cp is assigned to bin b, and binUsedp,sm,b indicate whether bin b is used for SMILES sm. We also define ui,sm as the quantity (mL or mg) of a SMILES sm required by reaction ri, where δ is a safety margin:
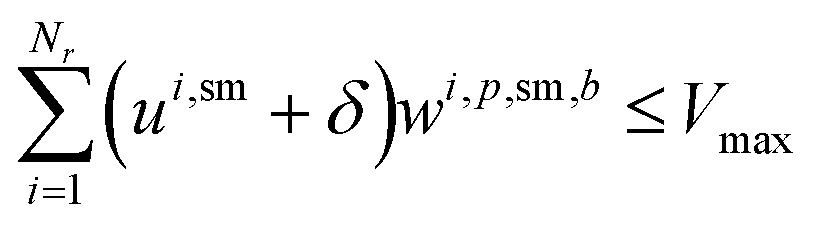
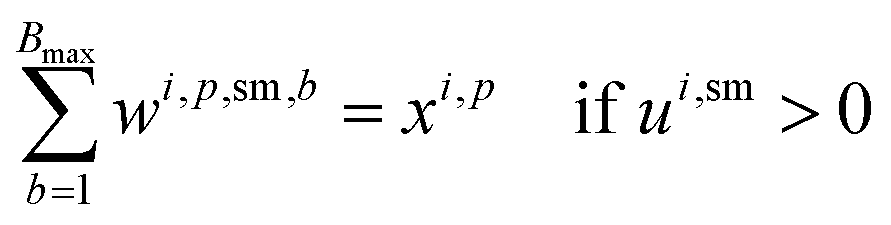
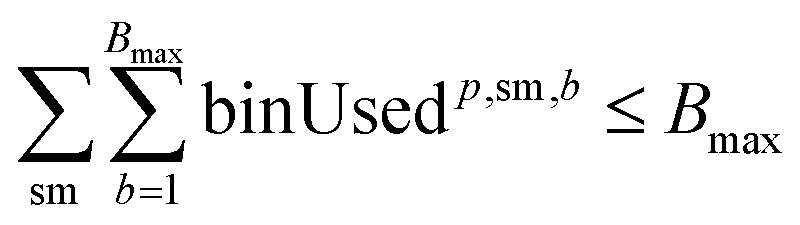
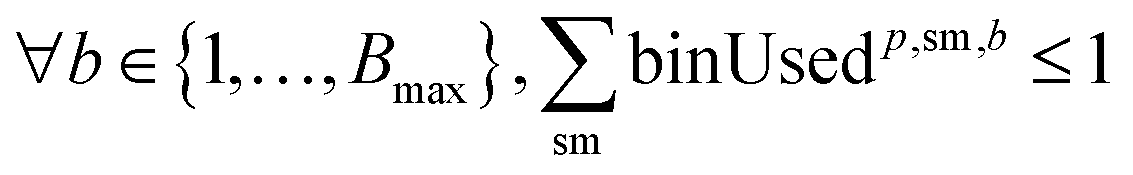
This bin-packing approach ensures that, for each chemical, the total amount required by all reactions assigned to a campaign can be feasibly distributed among the available slots, respecting both per-slot and total slot limits.
This integer linear programming has been implemented using Pulp library,51 and solving the scheduling problem described in Section 3 took approximately 1 minute. It is worth mentioning that this problem can yield multiple clustering solutions, all of which achieve the optimal value of the objective function.
5.2 Chemical synthesis
The complete experimental part, including general information for the generation and retrosynthesis workflow, methods for chemical synthesis, the layout of our Chemspeed platform, the model of CSV instructions, synthetic procedures used for each reaction, analysis result tables and HPLC chromatograms can be found in the SI.Author contributions
Q. P. and N. D. H. conceived the Iktos Robotics Laboratory, and F. L. V. conceived and coordinated the application study herein reported. N. D. designed the strategic and tactical scheduler algorithms and conducted the chemical space generation in this study. L. G. conceived the Ilaka platform and designed the Autosuite automated workflow with F. L. V. and C. P. F. L. V. and C. P. developed the templates of chemical reactions and conducted the reaction reviews in Ilaka with J.-C. M. A. D. and J.-C. M. designed the procurement system, integrated by L. G. in Ilaka. C. P. and F. L. V. conducted the campaign organization, performed the chemical experiments using the robot and interpreted the analysis results. F. L. V., N. D., L. G. and C. P. wrote the manuscript, and Q. P. participated in its revision and proofreading. All authors reviewed and approved the final manuscript.Conflicts of interest
All authors are employees or former employees of Iktos. This work was conducted as part of their professional activities at Iktos. No competitive interests are declared.Data availability
A repository including an open-source version of the code related to the tactical schedule and the necessary data to use the program is available on GitHub at https://github.com/iktos/tactical-scheduling.The data supporting this article have been included as part of the supplementary information (SI).
Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc07668d.
Acknowledgements
We thank BPI France for funding under the project reference IKS-Robotics-AMI. This work was also co-funded by the European Union (EIC Accelerator Grant). We would like to thank our partners Oncodesign for recording the LC-MS analysis of crude materials and Edelris for the HPLC-purification of the selected samples. We thank François Pacquet, from BetterChem, for insightful discussions on the utilization and integration of the Chemspeed platform. We would like to express our sincere gratitude to Maxime Laugeois for fruitful discussions prior to this application study and to everyone at Iktos who contributed to this project in any capacity. Their expertise and support have been indispensable. We are deeply grateful to the engineering team for granting us access to the computing tools and servers that enabled the execution of schedulers and smooth connectivity between databases necessary to run our experiments. Lastly, we would like to extend our sincere thanks to Maxime Laugeois, Stefani Gamboa, Hamza Tajmouati, Noémie Bergues and Clarisse Descamps for proofreading this manuscript. Their insightful comments and suggestions have greatly improved the clarity and quality of our work.Notes and references
- B. Burger, P. M. Maffettone, V. V. Gusev, C. M. Aitchison, Y. Bai, X. Wang, X. Li, B. M. Alston, B. Li and R. Clowes, et al. , Nature, 2020, 583, 237–241 CrossRef PubMed.
- J. Li, C. Ding, D. Liu, L. Chen and J. Jiang, Digital Discovery, 2025, 4, 1672–1684 RSC.
- G. Fang, D.-Z. Lin and K. Liao, Chin. J. Chem., 2023, 41, 1075–1079 CrossRef.
- Y. Zhao, Y. Zhao, J. Wang and Z. Wang, Ind. Eng. Chem. Fundam., 2025, 64, 4637–4668 CrossRef.
- F. Strieth-Kalthoff, H. Hao, V. Rathore, J. Derasp, T. Gaudin, N. H. Angello, M. Seifrid, E. Trushina, M. Guy and J. Liu, et al. , Science, 2024, 384, eadk9227 CrossRef PubMed.
- J.-M. Lu, J.-Z. Pan, Y.-M. Mo and Q. Fang, Artif. Intell. Chem., 2024, 100057 CrossRef.
- T. Dai, S. Vijayakrishnan, F. T. Szczypiński, J.-F. Ayme, E. Simaei, T. Fellowes, R. Clowes, L. Kotopanov, C. E. Shields and Z. Zhou, et al. , Nature, 2024, 1–8 Search PubMed.
- IBM, IBM RXN, https://rxn.res.ibm.com/rxn/robo-rxn/welcome, 20241, accessed: 2024 Search PubMed.
- W. Gao, P. Raghavan and C. W. Coley, Nat. Commun., 2022, 13, 1075 CrossRef PubMed.
- C. A. Nicolaou, C. Humblet, H. Hu, E. M. Martin, F. C. Dorsey, T. M. Castle, K. I. Burton, H. Hu, J. Hendle and M. J. Hickey, et al. , ACS Med. Chem. Lett., 2019, 10, 278–286 CrossRef PubMed.
- P. G. Polishchuk, T. I. Madzhidov and A. Varnek, J. Comput.-Aided Mol. Des., 2013, 27, 675–679 CrossRef PubMed.
- C. Y. Cheng, J. E. Campbell and G. M. Day, Chem. Sci., 2020, 11, 4922–4933 RSC.
- S. Sosnin, Drug Discovery Today, 2025, 104392 CrossRef PubMed.
- S. L. Schreiber, Science, 2000, 287, 1964–1969 CrossRef CAS PubMed.
- E. Lenci and A. Trabocchi, Eur. J. Org Chem., 2022, 2022, e202200575 CrossRef CAS.
- M. D. Burke and S. L. Schreiber, Angew. Chem., Int. Ed., 2004, 43, 46–58 CrossRef PubMed.
- N. Gesmundo, K. Dykstra, J. L. Douthwaite, Y.-T. Kao, R. Zhao, B. Mahjour, R. Ferguson, S. Dreher, B. Sauvagnat and J. Saurí, et al. , Nat. Synth., 2023, 2, 1082–1091 CrossRef CAS.
- T. Jiang, S. Bordi, A. E. McMillan, K.-Y. Chen, F. Saito, P. L. Nichols, B. M. Wanner and J. W. Bode, Chem. Sci., 2021, 12, 6977–6982 RSC.
- T. Jiang, G. Coin, S. Bordi, P. L. Nichols, J. W. Bode and B. M. Wanner, Eur. J. Org Chem., 2025, 28, e202401258 CrossRef CAS.
- A. Slattery, Z. Wen, P. Tenblad, J. Sanjosé-Orduna, D. Pintossi, T. den Hartog and T. Noël, Science, 2024, 383, eadj1817 CrossRef CAS PubMed.
- C. Mateos, M. J. Nieves-Remacha and J. A. Rincón, React. Chem. Eng., 2019, 4, 1536–1544 RSC.
- G.-N. Ahn, B. M. Sharma, S. Lahore, S.-J. Yim, S. Vidyacharan and D.-P. Kim, Commun. Chem., 2021, 4, 53 CrossRef CAS PubMed.
- C. W. Coley, D. A. Thomas III, J. A. Lummiss, J. N. Jaworski, C. P. Breen, V. Schultz, T. Hart, J. S. Fishman, L. Rogers and H. Gao, et al. , Science, 2019, 365, eaax1566 CrossRef CAS.
- S. Steiner, J. Wolf, S. Glatzel, A. Andreou, J. M. Granda, G. Keenan, T. Hinkley, G. Aragon-Camarasa, P. J. Kitson and D. Angelone, et al. , Science, 2019, 363, eaav2211 CrossRef CAS.
- E. Rial-Rodríguez, J. D. Williams, D. Cantillo, T. Fuchß, A. Sommer, H.-M. Eggenweiler, C. O. Kappe and G. Laudadio, Angew. Chem., Int. Ed., 2024, 63, e202412045 CrossRef.
- J. S. Manzano, W. Hou, S. S. Zalesskiy, P. Frei, H. Wang, P. J. Kitson and L. Cronin, Nat. Chem., 2022, 14, 1311–1318 CrossRef CAS.
- C. E. Brocklehurst, E. Altmann, C. Bon, H. Davis, D. Dunstan, P. Ertl, C. Ginsburg-Moraff, J. Grob, D. J. Gosling and G. Lapointe, et al. , J. Med. Chem., 2024, 67, 2118–2128 CrossRef CAS.
- M. Medcalf, V. Jain, S. Gamboa, B. Atwood, M. Lhuillier-Akakpo, V. Cachoux, F. Le Vaillant, J.-C. Meillon, C. Pescheteau and Q. Perron, ChemRxiv, 2024, Version2, preprint, DOI:10.26434/chemrxiv-2024-0z7g6-v2.
- C. Descamps, V. Bouttier, J. Sanz García, M. Lhuillier-Akakpo, Q. Perron and H. Tajmouati, Briefings Bioinf., 2025, 26, bbaf482 CrossRef CAS.
- R. Arora, N. Brosse, C. Descamps, N. Devaux, N. Do Huu, P. Gendreau, Y. Gaston-Mathé, M. Parrot, Q. Perron and H. Tajmouati, Computational Drug Discovery: Methods and Applications, Wiley Online Library, 2024, vol. 12, pp. 275–298 Search PubMed.
- M. H. Segler, M. Preuss and M. P. Waller, Nature, 2018, 555, 604–610 CrossRef CAS PubMed.
- N. M. Software, Pistachio, https://www.nextmovesoftware.com/pistachio.html, 2024, accessed: 2024 Search PubMed.
- E. J. Corey and W. T. Wipke, Science, 1969, 166, 178–192 CrossRef CAS.
- M. H. Todd, Chem. Soc. Rev., 2005, 34, 247–266 RSC.
- M. Parrot, H. Tajmouati, V. B. R. da Silva, B. R. Atwood, R. Fourcade, Y. Gaston-Mathé, N. Do Huu and Q. Perron, J. Cheminf., 2023, 15, 83 Search PubMed.
- M. Vaškevičius, J. Kapočiūtė-Dzikienė, A. Vaškevičius and L. Šlepikas, PeerJ Comput. Sci., 2023, 9, e1511 CrossRef PubMed.
- Q. Ai, F. Meng, R. Wang, J. C. Klein, A. G. Godfrey and C. W. Coley, Digital Discovery, 2025, 4, 486–499 RSC.
- M. R. Garey and D. S. Johnson, Analysis and design of algorithms in combinatorial optimization, Springer, 1981, pp. 147–172 Search PubMed.
- B. Lutnick, A. J. Ramon, B. Ginley, C. Csiszer, A. Kim, I. Flament, P. F. Damasceno, J. Cornibe, C. Parmar and K. Standish, et al. , J. Pathol. Inf, 2023, 14, 100337 CrossRef.
- O. Kapustina, P. Burmakina, N. Gubina, N. Serov and V. Vinogradov, Artif. Intell. Chem., 2024, 2, 100072 CrossRef.
- J.-C. Cousty, T. Cavagna, A. Schmidt, E. Mariano, K. Villat, F. de Nanteuil and P. Miéville, Digital Discovery, 2024, 3, 2434–2447 RSC.
- W. Zhang, L. Hao, V. Lai, R. Corkery, J. Jessiman, J. Zhang, J. Liu, Y. Sato, M. Politi and M. E. Reish, et al. , Nat. Commun., 2025, 16, 5182 CrossRef.
- L. M. Roch, F. Häse, C. Kreisbeck, T. Tamayo-Mendoza, L. P. E. Yunker, J. E. Hein and A. Aspuru-Guzik, PLoS One, 2020, 15, e0229862 CrossRef.
- M. Sim, M. G. Vakili, F. Strieth-Kalthoff, H. Hao, R. J. Hickman, S. Miret, S. Pablo-García and A. Aspuru-Guzik, Matter, 2024, 7, 2959–2977 CrossRef.
- M. Seifrid, F. Strieth-Kalthoff, M. Haddadnia, T. C. Wu, E. Alca, L. Bodo, S. Arellano-Rubach, N. Yoshikawa, M. Skreta and R. Keunen, et al. , Digital Discovery, 2024, 3, 1319–1326 RSC.
- A. Lawi, B. L. Panggabean and T. Yoshida, Computers, 2021, 10, 138 CrossRef.
- A. M. Jordan, A. Miccoli, A. Charnock, A. Denis, E. Fordyce, C. Franklin, E. Gheyouche, I. Henderson, C. Housseman and M. Laugeois, et al. , Cancer Res., 2025, 85, 4191 CrossRef.
- C. Ginsburg-Moraff, J. Grob, K. Chin, G. Eastman, S. Wildhaber, M. Bayliss, H. M. Mues, M. Palmieri, J. Poirier and M. Reck, et al. , SLAS Technol., 2022, 27, 350–360 CrossRef.
- K. Chen, J. L. Dores-Sousa, A. Fontana, C. Grosanu, H. M. McAllister, G. Bai, K. Bartkowiak, S. Cañellas, D. Corens and A. De Groot, et al. , J. Chromatogr. A, 2025, 1742, 465648 CrossRef.
- A. Baranczak, N. P. Tu, J. Marjanovic, P. A. Searle, A. Vasudevan and S. W. Djuric, ACS Med. Chem. Lett., 2017, 8, 461–465 CrossRef.
- S. Mitchell, M. O'Sullivan and I. Dunning, PuLP : A Linear Programming Toolkit for Python, 2011 Search PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |