
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePressure-induced emission enhancement through a synergistic effect between suppression of excimer formation and activation of aggregation-induced emission
Xinqi
Yang†
a,
Yuxiang
Dai†
 b,
Xiaoxiang
Zhang
b,
Daojie
Yang
a,
Ru
Guo
a,
Hai-Le
Yan
c,
Lili
Zhang
d,
Kai
Wang
*e,
Haichao
Liu
*a and
Bing
Yang
*a
b,
Xiaoxiang
Zhang
b,
Daojie
Yang
a,
Ru
Guo
a,
Hai-Le
Yan
c,
Lili
Zhang
d,
Kai
Wang
*e,
Haichao
Liu
*a and
Bing
Yang
*a
aState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun 130012, China. E-mail: hcliu@jlu.edu.cn; yangbing@jlu.edu.cn
bDepartment of Materials Physics and Chemistry, School of Materials Science and Engineering, Northeastern University, Shenyang 110819, China
cKey Laboratory for Anisotropy and Texture of Materials (Ministry of Education), School of Material Science and Engineering, Northeastern University, Shenyang 110819, China
dShanghai Synchrotron Radiation Facility, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201204, China
eSchool of Physics Science and Information Technology, Liaocheng University, Liaocheng 252000, China. E-mail: kaiwang@lcu.edu.cn
First published on 19th November 2025
Abstract
Conventional organic π-systems usually suffer from pressure-induced emission quenching due to enhanced π–π stacking, which leads to the formation of dark-state excimers and impedes their high-pressure applications. Herein, we present a new anthracene derivative, 9-(2,2-di([1,1′-biphenyl]-4-yl)vinyl)anthracene (QP-AN), which exhibits significant pressure-induced emission enhancement (PIEE) at 2.49 GPa. Single-crystal analysis reveals a unique π–π dimer packing motif characterized by an exceptionally small interplanar distance (Dπ–π = 3.280 Å) and a minimal overlap area (Sπ–π = 9.57%). Under high pressure, this motif not only suppresses excimer formation by preventing π-plane slippage but also amplifies the aggregation-induced emission (AIE) effect by restricting the motion of the biphenyl rotors in QP-AN. By comparing QP-AN with a control anthracene derivative that exhibits excimer formation and pressure-induced emission quenching, we propose that the synergy between excimer suppression and AIE activation is key to achieving PIEE. Our findings underscore the critical role of modulating excimer formation in the AIE system for achieving PIEE.
Introduction
Stimuli-responsive organic luminescent materials exhibit targeted optical responses to external stimuli, enabling smart functionalities and serving as fundamental components in next-generation adaptive optoelectronic devices.1–4 Among various stimuli, pressure, as a mechanical input independent of temperature and chemical composition, provides a unique approach to modulating luminescence by compressing intermolecular distances and altering electronic structures.5–10 Under high pressure, organic π-conjugated systems generally exhibit red-shifted and quenched emission, which has become a widely observed phenomenon.11–14 This phenomenon typically arises from pressure-enhanced π–π interactions, which promote the formation of non-emissive excimers (i.e., excited-state dimers).15,16 Within these excimers, compression increases orbital overlap, leading to a reduction in the energy gap and a consequent red-shift in emission. At the same time, nonradiative decay pathways become dominant, resulting in significant emission quenching.17–21 Since this excimer-mediated quenching under pressure severely limits applications that demand efficient emission (e.g., smart sensing,22–27 information encryption,28–30 and optical devices31,32), the development of strategies to mitigate this effect is of paramount importance.In recent years, the emergence of aggregation-induced emission (AIE) materials has inspired new strategies for high-pressure luminescence modulation. AIE motifs, such as tetraphenylethylene, enhance emission by restricting intramolecular motions upon aggregation, thereby suppressing nonradiative decay pathways under ambient conditions.33–37 It has been demonstrated that external hydrostatic pressure can increase molecular rigidity, potentially amplifying AIE effects under high pressure.38–42 In principle, AIE systems are promising candidates for achieving pressure-induced emission enhancement (PIEE). Recent comprehensive reviews on PIEE have extensively summarized key design strategies and underlying mechanisms.43–45 However, in AIE molecules incorporating large π-conjugated units, pressure-enhanced π–π interactions could dominate, promoting excimer formation.40,46 This excimer formation, diverting excited-state energy into nonradiative pathways, negates the benefits of AIE, ultimately leading to emission quenching. Therefore, coordinating excimer suppression with AIE activation is crucial for overcoming high-pressure emission quenching. Moreover, direct observation and controlled regulation of excimer emission under high pressure remain limited and experimentally challenging.
Given that excimer formation typically originates from pre-associated dimers within molecular aggregates,47–50 systems integrating dimeric packing with AIE motifs represent ideal models to investigate their interplay under high pressure. In our previous study, we investigated 9-(2,2-diphenylvinyl)anthracene (DP-AN),51 a molecule featuring a double bond as a bridge, linking anthracene to two phenyl rings. This molecule exhibits AIE properties due to the introduction of two phenyl rotors, while the molecular steric hindrance simultaneously promotes the formation of discrete antiparallel π–π dimer stacking, resulting in excimer emission from the anthracene units in the crystal. However, the strong π–π interaction (π–π overlap area Sπ–π = 54.13% and interplanar π–π distance Dπ–π = 3.635 Å for the anthracene plane) leads to typical excimer-dominated emission quenching from 1 atm to 3.11 GPa, where excimer emission overwhelms the AIE contribution. Consequently, a key objective of our investigation was to definitively establish the causal relationship between excimer formation and high-pressure emission quenching, while simultaneously identifying and incorporating novel “decisive factors” for emission enhancement.
To address the aforementioned challenges, we designed a new molecule, 9-(2,2-di([1,1′-biphenyl]-4-yl)vinyl)anthracene (QP-AN), by extending the side groups from phenyl to biphenyl. This modification not only introduces additional steric hindrance to weaken π–π stacking but also provides AIE activation sites. Crystal structure analysis reveals a “geometrically frustrated” dimer in QP-AN, characterized by an unusually short Dπ–π = 3.280 Å but a minimal Sπ–π = 9.57%, which effectively suppresses excimer formation. By employing in situ high-pressure modulation, the synergistic effect between excimer formation suppression and AIE activation effectively inhibits nonradiative decay pathways, enabling emission enhancement. These findings not only advance our understanding of the intricate interplay between AIE and excimer formation in pressure-induced luminescence but also provides an effective strategy for designing organic materials with superior high-pressure luminescence performance.
Results and discussion
In our previous work, we reported the construction of discrete π–π stacking dimers in the crystals of DP-AN (i.e., the DP-AN-Y crystals). The crystals exhibit typical excimer emission (λem = 540 nm) at ambient pressure (Fig. S1). Although DP-AN displays AIE characteristics at ambient pressure due to restricted phenyl rotor motion, in situ high-pressure photoluminescence (PL) measurements reveal a significant decrease in emission intensity from 0 to 3.11 GPa (Fig. S2 and S3). This behaviour is consistent with the typical pressure-induced emission quenching observed in excimer systems.52–55 In other words, the AIE effect fails to effectively energize excimer emission once dimer formation occurs. This observation highlights a key limitation of AIE–excimer systems under pressure: intramolecular motion restriction alone is inadequate to counteract the inherent quenching tendency of excimer states.To further probe the role of excimer formation in pressure-induced emission quenching, we designed a new derivative, QP-AN, derived from the molecular structure of DP-AN through extension of the side group from phenyl to biphenyl (Fig. 1a). This modification introduces an additional phenyl ring as an AIE-active rotor while simultaneously increasing steric hindrance on one side of the anthracene core. The design strategy was intended to enhance AIE behaviour while modulate π–π interactions within anthracene dimers in the crystal. QP-AN was synthesized via a three-step procedure (Scheme S1), and its molecular structure was fully characterized by 1H NMR, 13C NMR, and mass spectrometry (MS), successfully confirming target synthesis. Single crystals were grown via solvent diffusion, and their structure and packing modes were determined by single-crystal X-ray diffraction (SCXRD).
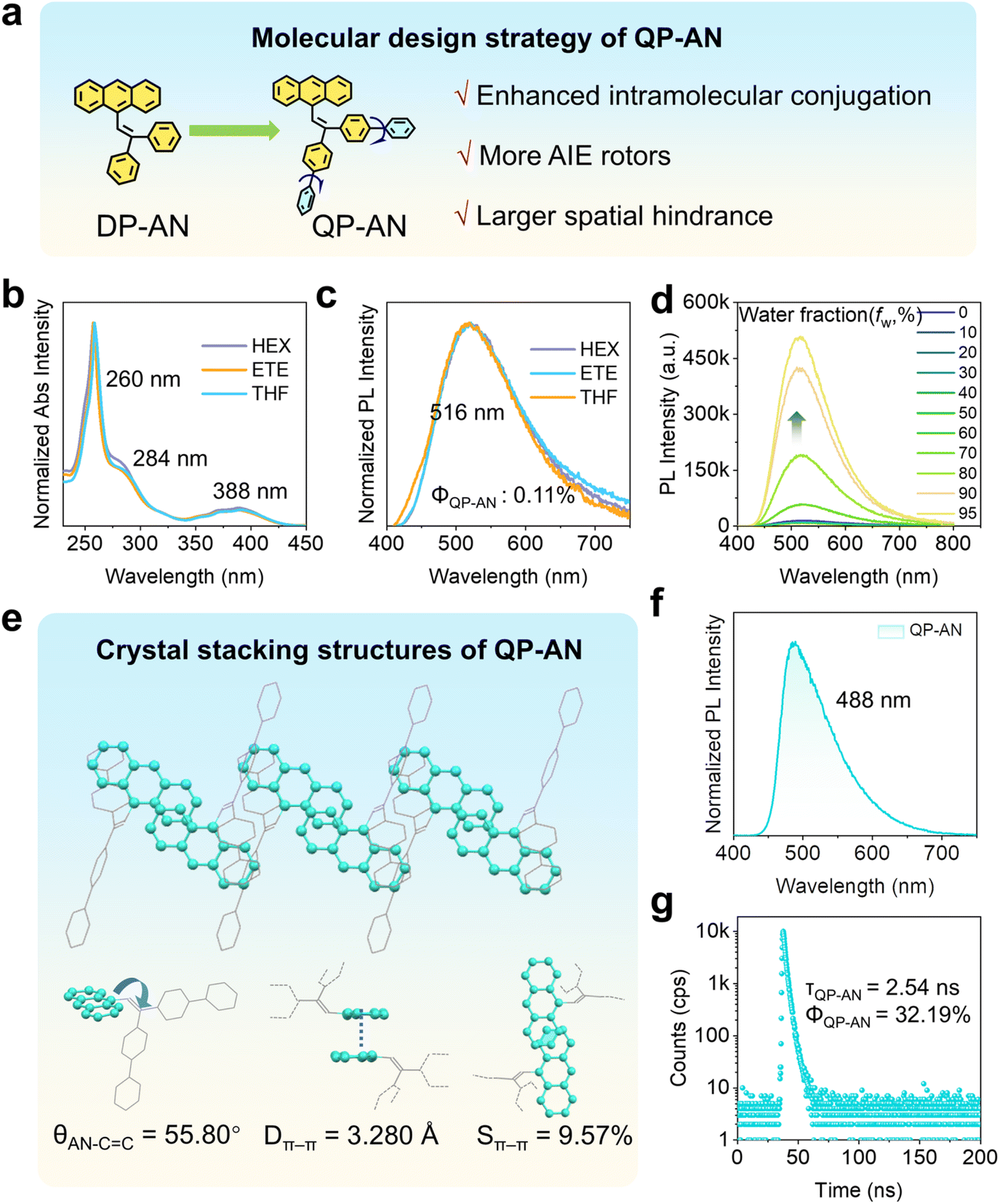 | ||
| Fig. 1 (a) Molecular structure and design strategy of QP-AN. (b) UV-vis absorption spectra and (c) PL spectra of QP-AN in different monodispersed dilute solutions. HEX = hexane, ETE = ethyl ether, and THF = tetrahydrofuran. (d) PL spectra of QP-AN in THF/H2O mixtures with varying water fractions (fw, %). (e) Molecular stacking motifs of QP-AN crystals. (f) PL spectra and (g) time-resolved PL spectra of QP-AN crystals. | ||
We first systematically investigated the photophysical properties of QP-AN in its monodispersed solution state. As shown in Fig. 1b and c, the absorption and PL spectra in different solvents exhibit a characteristic anthracene absorption at 388 nm and an emission peaking at 516 nm with a localized-excited (LE) character, as revealed by the unchanged PL spectra in solvents with varying polarity. Both are red-shifted compared to DP-AN (Fig. S4), consistent with the extended π-conjugation resulting from the biphenyl substituents. The vibronic fine structure of anthracene in QP-AN is significantly diminished due to electronic delocalization through the conjugated vinyl bridge. Time-resolved PL spectra reveal an ultrashort lifetime of 0.06 ns monitored at 516 nm (Fig. S5), arising from strong quenching induced by molecular motions in solution. Photoluminescence quantum yield (Φ) is markedly reduced to only 0.11% in tetrahydrofuran (THF) solution (Table S1), approximately tenfold lower than that of DP-AN, likely due to enhanced nonradiative decay pathways facilitated by the outer phenyl rings. Notably, in a THF/H2O mixture with 95% water content, the emission intensity of QP-AN increases by more than 35-fold compared to the pure THF solution, demonstrating the effectiveness of biphenyl substitution in activating AIE behaviours (Fig. 1d).
To evaluate how the molecular structure of QP-AN affects its packing arrangement, we performed single-crystal structure analysis. The results reveal that QP-AN, like DP-AN, exhibits discrete π–π dimer stacking in the crystal. However, compared to DP-AN, its Dπ–π is reduced to 3.280 Å, while Sπ–π decreases to 9.57% (Fig. 1e). Beyond these packing metrics, the dihedral angle between the anthracene core and the adjacent double bond in QP-AN is notably reduced to 55.80°, in contrast to the 70.33° observed for DP-AN, significantly enhancing its intramolecular planarity and conjugation. The PL spectra of the crystals exhibit a distinct blue shift relative to solution-state emission, with the peak maximum shifting to 488 nm (Fig. 1f). This shift is likely due to the conformational rigidity imposed by the crystal lattice, which restricts molecular structural relaxation and reduces effective conjugation. Structural and photophysical analysis together suggest that the introduction of biphenyls preserves the discrete dimer packing mode while weakening π–π interactions within dimers. As a result, QP-AN does not display excimer emission in the crystal. Temperature-dependent PL measurements on QP-AN crystals (Fig. S6), showing an emission peak that remains nearly unchanged with temperature, strongly corroborate this conclusion. This behaviour contrasts with that of typical excimers, which usually exhibit a pronounced blue shift and decreased intensity as the temperature increases.56,57 This conclusion is further supported by its fluorescence lifetime of 2.54 ns (Fig. 1g), which is significantly shorter than that of typical anthracene excimer crystals.58,59 Concurrently, the Φ increases markedly to 32.19% in the crystalline state, confirming that QP-AN exhibits AIE behaviour. Therefore, QP-AN serves as an excellent model for investigating AIE-dominated emission properties in the absence of excimer interference under high-pressure.
To gain further insights into the pressure dependence of the fluorescence properties, we conducted in situ high-pressure absorption, PL, and time-resolved PL measurements on QP-AN crystals using a diamond anvil cell (DAC) (Fig. 2a, b and S2). The absorption spectra exhibit a continuous red-shift with increasing pressure. From ambient pressure to 0.62 GPa, the PL spectra of QP-AN show a slight red-shift and weak quenching. Notably, beyond 0.62 GPa, an anomalous PIEE appears, reaching its maximum at 2.49 GPa, which is visually apparent in the corresponding fluorescence images (Fig. 2c). The PL intensity of the QP-AN crystal at 2.49 GPa increases by 54.94% relative to that at 0.62 GPa. Beyond this point, the emission exhibits a further red-shift accompanied by pronounced quenching. The complete pressure-responsive emission evolution is illustrated by the plots of PL wavelength and PL intensity against pressure (Fig. 2d). Remarkably, upon releasing the pressure back to ambient conditions, the PL peak position largely recovers to its initial value, indicating good reversibility (Fig. S7).
 | ||
| Fig. 2 (a) In situ high-pressure PL spectra and (b) time-resolved PL spectra of QP-AN crystals at various pressures from 1 atm to 3.50 GPa. (c) Corresponding fluorescence photographs of QP-AN crystals under pressures ranging from 1 atm to 3.50 GPa. (d) Pressure-dependent plots of maximum emission wavelength and PL intensity. (e) Pressure-dependent kr and knr of QP-AN crystals. | ||
In situ time-resolved PL measurements revealed that the fluorescence lifetime of QP-AN crystals steadily increases from 1 atm to 3.00 GPa. In contrast, DP-AN shows typical pressure-induced emission quenching in this pressure range (Fig. S3), with its lifetime increasing slightly up to 2.00 GPa and then decreasing at higher pressures (Fig. S8a). Based on these results, we calculated the radiative (kr) and nonradiative (knr) rate constants of both QP-AN and DP-AN crystals under varying pressures (Fig. 2e, S8b, and Table S2). For both systems, kr gradually decreases with pressure. However, their nonradiative behaviour diverges significantly: knr for QP-AN initially rises from 1 atm to 0.31 GPa, and then declines sharply from 0.31 to 3.50 GPa. In contrast, knr for DP-AN consistently increases across the entire pressure range (Table S3). These findings demonstrate that within 0.62–2.02 GPa, nonradiative loss in QP-AN crystals is efficiently suppressed, likely due to sterically hindered packing that prevents excimer formation under pressure.
To uncover the structural origin of the pressure-dependent PL changes in QP-AN, we conducted in situ synchrotron angular-dispersive X-ray diffraction (ADXRD) measurements. With increasing pressure, all Bragg diffraction peaks gradually broaden and shift to higher angles due to lattice contraction (Fig. S9). Notably, no new peaks appear or disappear, ruling out the possibility of phase transitions. Analysis of the unit cell parameters reveals continuous volume contraction and progressive reductions in all three crystallographic axes (Fig. 3a and b). Upon decompression, the diffraction peaks return to their original positions, demonstrating structural reversibility and stability of the QP-AN crystals. To probe the structural basis of the observed emission enhancement, we focused on the packing structure at 2.00 GPa (near the maximum PL intensity) and compared it with that at ambient pressure (Fig. 3c). The results show that, compared to ambient pressure, Dπ–π decreases from 3.280 Å to 3.065 Å under pressure (a 6.55% reduction). However, unlike typical systems where compression enhances π–π overlap,52,54,60 QP-AN exhibits a decrease in Sπ–π from 9.57% to 6.83% (a 28.63% reduction). This dissociation primarily arises from lateral slippage along the short axis of anthracene, which effectively disrupts π-stacking and disfavours excimer formation.
 | ||
| Fig. 3 (a) Compression ratios of the crystallographic axes and (b) evolution of unit cell volume of QP-AN crystals under pressure (V0 and B0 denote the volume and the bulk modulus of elasticity under ambient pressure). (c) Dimeric geometry and (d) calculated NTOs and TDM maps of QP-AN crystals at 1 atm and 2.00 GPa. Carbon atoms are numbered sequentially by fragment for TDM analysis, and hydrogen atoms are omitted due to their negligible contribution to the transitions of interest. | ||
To substantiate this inference and further elucidate the excited-state characteristics of QP-AN crystals under pressure, we extracted the dimer geometry from the crystal structure and performed energy level and natural transition orbital (NTO) calculations (Fig. 3d). At 2.00 GPa, the energy level reveals a pronounced decrease, consistent with the experimental PL behaviour. The calculated energy levels of monomers and dimers are relatively close (Fig. S10) in contrast to the distinct energy separation observed for DP-AN (Fig. S11). Moreover, the excited-state electron density of QP-AN becomes more delocalized over the side groups upon compression. These results confirm that the excited-state transition in QP-AN is governed by localized intramolecular characteristics rather than excimer-like interactions.
To further analyse the contributions of different molecular fragments to the excitation process, we performed transition density matrix (TDM) calculations (Fig. 3e, S12, Tables S4 and S5). All carbon atoms were sequentially numbered by fragment. At 1 atm, QP-AN exhibits primarily localized excitation on the anthracene (AN) core (73.36%), with a notably low intermolecular π–π charge-transfer (CT) component (4.61%). This stands in stark contrast to the significantly higher intermolecular CT (19.92%) and lower LE (62.53%) observed in the excimer-forming DP-AN, thus underscoring the monomer-like characteristics of QP-AN. Upon compression to 2.00 GPa, the LE component decreases to 69.08%, while contributions from intramolecular charge transfer (ICT) between the AN core and the biphenyl side groups (QP) increase from 20.70% to 24.45%. In contrast, the intermolecular π–π CT component slightly decreases from 4.61% to 4.41%. These results confirm that, under pressure, the QP-AN dimer undergoes further π–π dissociation, resulting in monomer-like emission. The absence of excimer formation indicates that the dominant nonradiative decay pathways associated with excimers are effectively suppressed under high pressure. It is also noteworthy that the pressure-enhanced ICT character may intrinsically lower radiative transition probability, consistent with the experimentally observed decrease in kr.
We further evaluated the free volume fractions (FVFs) of QP-AN crystals at ambient pressure and 2.00 GPa. Compression markedly reduces the FVF from 33.16% to 26.18% (Fig. S13), indicating significant unit cell shrinkage and tighter intermolecular packing. Analysis of the crystal structures reveals that the C–H⋯π interactions between the outer phenyl ring of QP-AN and neighbouring molecules are compressed from 2.877 Å/2.910 Å to 2.555 Å/2.635 Å under pressure (Fig. S14). Meanwhile, both the number and strength of intermolecular contacts involving the biphenyl unit increase substantially, including additional C–H⋯π interactions (∼3.5 Å) and C⋯C contacts (2.3–3.0 Å). These strengthened interactions further restrict the rotational and vibrational motions of the phenyl rings, thereby efficiently suppressing nonradiative decay pathways. Hirshfeld surface analysis supports this conclusion, showing a notable expansion of red regions, corresponding to C–H⋯π interactions, on the biphenyl side groups at 2.00 GPa (Fig. S14). Taken together, these findings demonstrate that the enhanced intermolecular interactions under pressure restrict biphenyl motions, effectively activating the AIE effect.
To probe the effect of pressure on intramolecular rigidity and the suppression of molecular motion at the molecular level, we conducted in situ high-pressure infrared (IR) spectroscopy on QP-AN crystals from ambient pressure to 4.78 GPa (Fig. S16 and S17). The absorption bands at ∼3060 cm−1 and 750 cm−1 were assigned to aromatic C–H stretching (ν(C–H)) and out-of-plane bending (γ(C–H)) vibrations, respectively. Below 2.02 GPa, both modes remain sharp and well-resolved, indicating minimal structural constraint. However, the observed decrease in emission intensity below 0.56 GPa suggests that pressure-induced shortening of intermolecular distances may subtly enhance certain molecular motions. This is likely due to the relatively loose molecular packing at lower pressures, which facilitates certain non-radiative vibrational modes through local structural rearrangements or pressure-activated quenching pathways.45,61 Between 0.56 and 2.02 GPa, all vibrational modes exhibit a progressive blue shift, indicating C–H bond shortening and increasing conformational rigidity. This restriction of intramolecular motion contributes to AIE activation and a reduction in nonradiative losses, consistent with the observed emission enhancement. Beyond 2.02 GPa, the C–H vibrational bands broaden and shift rapidly to higher wavenumbers, indicating distortion and deformation of the C–H⋯π network.62 As structural rigidity approaches its limit, nonradiative decay pathways are no longer effectively suppressed. Meanwhile, kr begins to decrease, likely due to enhanced ICT character and pressure-induced molecular distortion, reducing the radiative transition ability of QP-AN and ultimately causing second-stage emission quenching. Additionally, at higher pressures, molecules are forced into closer proximity and distortion, which may promote new excimer-like interactions between adjacent biphenyl side groups. These low-emissive excited-state traps, characterized by intrinsically low radiative ability, could compete with LE emission and further diminish overall emission output.
Building on the above findings, we here propose the PIEE mechanism for QP-AN: the synergistic effect between suppression of dark-state excimer formation and activation of AIE (Fig. 4). The extended side groups in QP-AN prevent the formation of tightly bound dimers with strong π–π interactions, resulting in a “geometrically frustrated” dimer characterized by an unusually small Dπ–π (3.280 Å) but minimal Sπ–π (9.57%). While a short Dπ–π would typically favour strong π–π interactions, the limited Sπ–π indicates insufficient orbital overlap—two seemingly conflicting structural features that jointly suppress excimer formation. Under compression, the anthracene planes are forced closer together, but steric repulsion prevents an increase in π–π overlap, instead leading to partial dissociation of π–π stacking and blocking dark-state anthracene excimer formation, which is associated with dominated nonradiative decay pathways. Meanwhile, enhanced intermolecular interactions under pressure rigidify the biphenyl rotors, thereby activating the AIE effect and contributing to a continuous decrease in the knr. While kr also decreases slightly, likely due to enhanced ICT, the drop in knr outweighs the decline in kr, resulting in a net increase in emission intensity. This behaviour contrasts with DP-AN, where strong π–π interactions dominate and knr increases sharply under pressure, leading to emission quenching. These comparative results confirm that the emission of AIE systems with excimers tends to quench under high pressure, whereas excimer-free AIE systems can exhibit a pronounced emission enhancement.
 | ||
| Fig. 4 Synergistic mechanism for pressure-induced emission enhancement in QP-AN crystals: suppression of dark-state excimer formation and activation of the AIE effect. | ||
To further confirm the critical role of the “geometrically frustrated” dimer in pressure-induced emission enhancement, we investigated a second polymorph of QP-AN, B-QP-AN, which adopts a distinct packing motif. SCXRD reveals that B-QP-AN also forms discrete dimers, but with a lower Sπ–π of 3.63%, a larger Dπ–π of 3.606 Å, and a significantly larger torsion angle of 75.30° between the anthracene core and the vinyl bridge (Fig. S18a). At ambient pressure, B-QP-AN crystals exhibit slightly blue-shifted emission and a longer lifetime of 2.89 ns compared with QP-AN crystals (Fig. S18b and c). However, under a pressure ranging from 1 atm to 3.98 GPa, the absorption and PL spectra of B-QP-AN show a continuous red-shift and notable emission quenching, respectively (Fig. S19a–d). Meanwhile, the lifetime of B-QP-AN decreases continuously with increasing pressure (Fig. S19e). An analysis of rate constants reveals that the kr of B-QP-AN crystals decreases drastically, while the knr concomitantly increases with pressure (Table S6 and Fig. S19f). This trend is opposite to QP-AN, where both kr and knr decrease under compression, highlighting that B-QP-AN undergoes pressure-induced activation of nonradiative decay pathways, resulting in fluorescence quenching. Under compression, the relatively relaxed Dπ–π permits a continuous increase in overlap area, promoting dark-state excimer formation rather than suppressing it. Upon pressure release, the absorption and emission spectra of B-QP-AN revert to their initial states, demonstrating reversibility (Fig. S20a and b). These results confirm that the “geometrically frustrated” dimer configuration, defined by a tightly compressed Dπ–π and minimal Sπ–π, is a crucial structural feature for achieving pressure-induced emission enhancement in AIE systems.
Conclusions
In summary, we designed QP-AN, an anthracene-based AIE-active molecule, to achieve PIEE in π-stacked crystals. Structural analysis reveals that the short interplanar distance (Dπ–π = 3.280 Å) in QP-AN induces strong interlayer repulsion, leading to a dissociation in π–π overlap (Sπ–π decreasing from 9.57% to 6.83%) upon compression. This unconventional packing mode effectively suppresses dark-state anthracene excimer formation under high pressure. Meanwhile, the introduction of biphenyl side groups activates the AIE effect by restricting molecular motion, thereby further reducing nonradiative decay pathways. The synergy between excimer suppression and AIE activation enables QP-AN to exhibit pronounced emission enhancement and a prolonged excited-state lifetime. These findings unveil a novel structural strategy for tuning π–π interactions under high pressure and provide guidance for developing high-performance pressure-responsive luminescent materials.Author contributions
Conceptualization: Y. D., H. L. and B. Y. Data curation: X. Z., D. Y., R. G., L. Z. and H.-L. Y. Chemical synthesis and product analysis: X. Y. DFT calculations: X. Y., Y. D. and H. L. Supervision: H. L., K. W., and B. Y. Funding acquisition: Y. D., H. L. and B. Y. Writing – original draft: X. Y. Writing – review & editing: Y. D., K. W, H. L. and B. Y.Conflicts of interest
The authors state that there are no conflicts to declare.Data availability
CCDC 2486652 (B-QP-AN) and 2486653 (QP-AN) contain the supplementary crystallographic data for this paper.63a,bAll other data, including details on compounds' synthesis methods, photophysical property characterization, and theoretical calculations, supporting the findings of this study are available within the article and its supplementary information (SI), as well as available from the corresponding authors upon reasonable request. Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5sc07579c.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (52373183, 52573196, 12574242, and 52103201), the National Key Research and Development Program of China (No. 2020YFA0714603), and Special Construction Project Fund for Shandong Province Taishan Scholars. The authors thank the staff from Shanghai Synchrotron Radiation Facility (SSRF) at BL15U1.Notes and references
- Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS PubMed.
- M. A. Stuart, W. T. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- M. Grzelczak, L. M. Liz-Marzan and R. Klajn, Chem. Soc. Rev., 2019, 48, 1342–1361 RSC.
- Y. Wang, X. Lu, W. Yang, T. Wen, L. Yang, X. Ren, L. Wang, Z. Lin and Y. Zhao, J. Am. Chem. Soc., 2015, 137, 11144–11149 CrossRef CAS PubMed.
- H. Li, Z. Wang, L. Chen, J. Wu, H. Huang, K. Yang, Y. Wang, L. Su and G. Yang, J. Phys. Chem. B, 2015, 119, 14245–14251 CrossRef CAS PubMed.
- F. Bai, K. Bian, X. Huang, Z. Wang and H. Fan, Chem. Rev., 2019, 119, 7673–7717 CrossRef CAS PubMed.
- D. Laniel, B. Winkler, T. Fedotenko, A. Pakhomova, S. Chariton, V. Milman, V. Prakapenka, L. Dubrovinsky and N. Dubrovinskaia, Phys. Rev. Lett., 2020, 124, 216001 CrossRef CAS PubMed.
- X. Wang, X. Yang, Y. Wang, X. Tang, H. Zheng, P. Zhang, D. Gao, G. Che, Z. Wang, A. Guan, J. F. Xiang, M. Tang, X. Dong, K. Li and H. K. Mao, J. Am. Chem. Soc., 2022, 144, 21837–21842 CrossRef CAS PubMed.
- P. Zhang, D. Gao, X. Tang, X. Yang, H. Zheng, Y. Wang, X. Wang, J. Xu, Z. Wang, J. Liu, X. Wang, J. Ju, M. Tang, X. Dong, K. Li and H. K. Mao, J. Am. Chem. Soc., 2023, 145, 6845–6852 CrossRef CAS PubMed.
- Z. Ma, Z. Wang, M. Teng, Z. Xu and X. Jia, ChemPhysChem, 2015, 16, 1811–1828 CrossRef CAS.
- Y. Zhang, M. Qile, J. Sun, M. Xu, K. Wang, F. Cao, W. Li, Q. Song, B. Zou and C. Zhang, J. Mater. Chem. C, 2016, 4, 9954–9960 RSC.
- C. Feng, K. Wang, Y. Xu, L. Liu, B. Zou and P. Lu, Chem. Commun., 2016, 52, 3836–3839 RSC.
- F. Gao, H. Song, Z. Li, Y. Qin, X. Li, Z. Yao, J. Fan, X. Wu, W. Li and X. Bu, Angew. Chem., Int. Ed., 2023, 62, e202218675 CrossRef CAS PubMed.
- K. Nagura, S. Saito, H. Yusa, H. Yamawaki, H. Fujihisa, H. Sato, Y. Shimoikeda and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 10322–10325 CrossRef CAS PubMed.
- J. Zou, Y. Fang, Y. Shen, Y. Xia, K. Wang, C. Zhang and Y. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202207426 CrossRef CAS.
- W. J. Oldham, Y.-J. Miao, R. J. Lachicotte and G. C. Bazan, J. Am. Chem. Soc., 1998, 120, 419–420 CrossRef CAS.
- J. Cornil, D. A. dos Santos, X. Crispin, R. Silbey and J. L. Brédas, J. Am. Chem. Soc., 1998, 120, 1289–1299 CrossRef CAS.
- T. Hayashi, N. Mataga, Y. Sakata, S. Misumi, M. Morita and J. Tanaka, J. Am. Chem. Soc., 2002, 98, 5910–5913 CrossRef.
- M. Shimizu and T. Hiyama, Chem.–Asian J., 2010, 5, 1516–1531 CrossRef CAS PubMed.
- S. P. Jagtap, S. Mukhopadhyay, V. Coropceanu, G. L. Brizius, J. L. Bredas and D. M. Collard, J. Am. Chem. Soc., 2012, 134, 7176–7185 CrossRef CAS PubMed.
- Q. Zhang, J. Su, D. Feng, Z. Wei, X. Zou and H. C. Zhou, J. Am. Chem. Soc., 2015, 137, 10064–10067 CrossRef CAS PubMed.
- Z. Man, Z. Lv, Z. Xu, Q. Liao, J. Liu, Y. Liu, L. Fu, M. Liu, S. Bai and H. Fu, Adv. Funct. Mater., 2020, 30, 2000105 CrossRef CAS.
- H. Hu, X. Cheng, Z. Ma, R. P. Sijbesma and Z. Ma, J. Am. Chem. Soc., 2022, 144, 9971–9979 CrossRef CAS PubMed.
- S. Wakako, Y. Hori, T. Kinoshita, T. Saiki, X. Qi, K. Hasegawa, Y. Imai, T. Mori, K. Nakagawa and G. Fukuhara, ACS Macro Lett., 2023, 12, 1389–1395 CrossRef CAS.
- D. J. Kiebala, R. Style, D. Vanhecke, C. Calvino, C. Weder and S. Schrettl, Adv. Funct. Mater., 2023, 33, 202304938 Search PubMed.
- H. Shi, Y. Wang, Y. Zhang, J. Yan and T. Meng, Opt. Laser Technol., 2025, 184, 112504 CrossRef.
- Z. Ma, Z. Wang, X. Meng, Z. Ma, Z. Xu, Y. Ma and X. Jia, Angew. Chem., Int. Ed., 2016, 55, 519–522 CrossRef CAS PubMed.
- Z. Luo, Y. Liu, Y. Liu, C. Li, Y. Li, Q. Li, Y. Wei, L. Zhang, B. Xu, X. Chang and Z. Quan, Adv. Mater., 2022, 34, e2200607 CrossRef PubMed.
- D. Zhao, S. Li, Y. Su, J. Qin, G. Xiao, Y. Shang, X. Yin, P. Lv, F. Wang, J. Yang, Z. Liu, F. Lan, Q. Zeng, L. Zhang, F. Gao and B. Zou, Nat. Commun., 2025, 16, 6203 CrossRef CAS.
- L. Bai, P. Bose, Q. Gao, Y. Li, R. Ganguly and Y. Zhao, J. Am. Chem. Soc., 2017, 139, 436–441 CrossRef CAS PubMed.
- Y. Hu, D. Yang, D. Ma, C. Qi and S. Huang, Adv. Funct. Mater., 2023, 34, 202310861 Search PubMed.
- Y. Hong, J. W. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- W. Wu, S. Ye, L. Huang, L. Xiao, Y. Fu, Q. Huang, G. Yu, Y. Liu, J. Qin, Q. Li and Z. Li, J. Mater. Chem., 2012, 22, 6374–6382 RSC.
- J. Xiong, H. Feng, J. Sun, W. Xie, D. Yang, M. Liu and Y. Zheng, J. Am. Chem. Soc., 2016, 138, 11469–11472 CrossRef CAS PubMed.
- H. Feng, Y. Yuan, J. Xiong, Y. Zheng and B. Z. Tang, Chem. Soc. Rev., 2018, 47, 7452–7476 RSC.
- J. Yang, M. Fang and Z. Li, Aggregate, 2020, 1, 6–18 CrossRef.
- Y. Gu, N. Li, G. Shao, K. Wang and B. Zou, J. Phys. Chem. Lett., 2020, 11, 678–682 CrossRef CAS PubMed.
- V. Kachwal and I. R. Laskar, Top. Curr. Chem., 2021, 379, 28 CrossRef CAS PubMed.
- S. Tong, J. Dai, J. Sun, Y. Liu, X. Ma, Z. Liu, T. Ma, J. Tan, Z. Yao, S. Wang, H. Zheng, K. Wang, F. Hong, X. Yu, C. Gao and X. Gu, Nat. Commun., 2022, 13, 5234 CrossRef CAS.
- Z. Li, F. Jiang, M. Yu, S. Li, L. Chen and M. Hong, Nat. Commun., 2022, 13, 2142 CrossRef CAS.
- D. Liu, D. Dong, T. Liu, S. Liu, Z. Yao, Q. Li, B. Liu, R. Liu, L. Yue, X. Yin, Z. Li, J. Niu, N. Yu, Z. Zhang and B. Liu, ACS Mater. Lett., 2025, 7, 1746–1753 CrossRef CAS.
- Z. Fu, K. Wang and B. Zou, Chin. Chem. Lett., 2019, 30, 1883–1894 CrossRef CAS.
- A. Li, J. Wang and Z. Li, Cell Rep. Phys. Sci., 2025, 6, 102733 CrossRef CAS.
- B. Zou, Y. Han, Z. Yang, Q. Wang, G. Wang, G. Zhang, Y. Li and C. Liu, APL Mater., 2024, 12, 030601 CrossRef CAS.
- H. Yuan, K. Wang, K. Yang, B. Liu and B. Zou, J. Phys. Chem. Lett., 2014, 5, 2968–2973 CrossRef CAS PubMed.
- H. Liu, D. Cong, B. Li, L. Ye, Y. Ge, X. Tang, Y. Shen, Y. Wen, J. Wang, C. Zhou and B. Yang, Cryst. Growth Des., 2017, 17, 2945–2949 CrossRef CAS.
- Y. Ge, Y. Wen, H. Liu, T. Lu, Y. Yu, X. Zhang, B. Li, S.-T. Zhang, W. Li and B. Yang, J. Mater. Chem. C, 2020, 8, 11830–11838 RSC.
- J. Ochi, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2023, 62, e202214397 CrossRef CAS PubMed.
- Y. Zheng, P. Chen, Z. Niu and E. Wang, Spectrochim. Acta, Part A, 2024, 312, 124035 CrossRef CAS.
- X. Yang, Y. Dai, H. Liu, K. Wang, H. L. Yan, X. Yu, Z. A. Xia, M. Wu, S. Zhang, G. Xiao, B. Zou and B. Yang, J. Am. Chem. Soc., 2025, 147, 5300–5309 CrossRef CAS PubMed.
- Y. Dai, H. Liu, T. Geng, F. Ke, S. Niu, K. Wang, Y. Qi, B. Zou, B. Yang, W. L. Mao and Y. Lin, J. Mater. Chem. C, 2021, 9, 934–938 RSC.
- Y. Dai, H. Liu, T. Geng, R. Duan, X. Li, Y. Liu, W. Liu, B.-G. He, L. Sui, K. Wang, B. Zou, B. Yang and Y. Qi, J. Mater. Chem. C, 2023, 11, 4892–4898 RSC.
- C. Wang, X. Yang, R. Duan, X. Li, Y. Liu, W. Liu, B.-g. He, S. Zhang, B. Yang, K. Wang, B. Zou, Y. Qi, Q. Li, H. Liu and Y. Dai, Dyes Pigm., 2024, 223, 111953 CrossRef CAS.
- Q. Liao, A. Li, A. Huang, J. Wang, K. Chang, H. Li, P. Yao, C. Zhong, P. Xie, J. Wang, Z. Li and Q. Li, Chem. Sci., 2024, 15, 4364–4373 RSC.
- Y. Shen, H. Liu, J. G. Cao, S. T. Zhang, W. J. Li and B. Yang, Phys. Chem. Chem. Phys., 2019, 21, 14511–14515 RSC.
- S. Wang, H. Liu, S. Zhao, Q. Wu, Z. Yang, D. Yang, Y. Lv, Q. Su, S. Zhang and B. Yang, Chem. Sci., 2025, 16, 3275–3284 RSC.
- L. S. Kaanumalle, C. L. Gibb, B. C. Gibb and V. Ramamurthy, J. Am. Chem. Soc., 2005, 127, 3674–3675 CrossRef CAS PubMed.
- H. Liu, L. Yao, B. Li, X. Chen, Y. Gao, S. Zhang, W. Li, P. Lu, B. Yang and Y. Ma, Chem. Commun., 2016, 52, 7356–7359 RSC.
- S. Hu, Z. Yao, X. Ma, L. Yue, L. Chen, R. Liu, P. Wang, H. Li, S. T. Zhang, D. Yao, T. Cui, B. Zou and G. Zou, J. Phys. Chem. Lett., 2022, 13, 1290–1299 CrossRef CAS PubMed.
- Y. Gu, H. Liu, R. Qiu, Z. Liu, C. Wang, T. Katsura, H. Zhang, M. Wu, M. Yao, H. Zheng, K. Li, Y. Wang, K. Wang, B. Yang, Y. Ma and B. Zou, J. Phys. Chem. Lett., 2019, 10, 5557–5562 CrossRef CAS PubMed.
- Y. Gu, K. Wang, Y. Dai, G. Xiao, Y. Ma, Y. Qiao and B. Zou, J. Phys. Chem. Lett., 2017, 8, 4191–4196 CrossRef CAS.
- (a) CCDC 2486652: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2pgklb; (b) CCDC 2486653: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2pgkmc.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |