
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA hydrogen-bonded organic framework possessing one-dimensional wide channels surrounded by the naphthalenediimide plane
Yuzuki
Murata
a,
Taito
Hashimoto
a,
Ryusei
Oketani
 a,
Miki
Naruoka
b,
Rajendra Prasad
Paitandi
c,
Norimitsu
Tohnai
b,
Shu
Seki
c and
Ichiro
Hisaki
*a
a,
Miki
Naruoka
b,
Rajendra Prasad
Paitandi
c,
Norimitsu
Tohnai
b,
Shu
Seki
c and
Ichiro
Hisaki
*a
aDivision of Chemistry, Graduate School of Engineering Science, The University of Osaka, 1-3 Machikaneyama, Toyonaka, Osaka 560-8531, Japan. E-mail: i.hisaki.es@osaka-u.ac.jp
bDivision of Applied Chemistry, Graduate School of Engineering, The University of Osaka, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan
cDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto 615-8510, Japan
First published on 2nd December 2025
Abstract
π-Conjugated molecule-based porous organic frameworks that possess both one-dimensionally π-stacked columnar domains and pore channels surrounded by the π-conjugated surface are a sophisticated platform for optoelectronic materials responsive to chemical stimuli. In this paper, we report a hydrogen-bonded organic framework (HOF) with a wide inclusion channel whose surfaces are composed of the π-conjugated plane of naphthalenediimide (NDI). The Brunauer–Emmett–Teller surface area was determined to be 1410 m2 g−1. Slip-stacking of a hydrogen-bonded two-dimensional network composed of tetracarboxylic acid NDITA with an NDI core provides both electron-conductive π-stacked NDI domains and pore channels surrounded by the NDI surface. Since aromatic solvents can come into contact with the NDI moieties in the pores, the HOF exhibits solvent-dependent photophysical behaviors. In particular, inclusion of dimethoxybenzene (DMB) into the pores enhances charge-transfer interactions, resulting in significant changes in absorption and emission spectra as well as electron conductivity.
Introduction
Two-dimensional network porous frameworks constructed from π-conjugated molecules, such as metal–organic frameworks (MOFs)1–4 and covalent organic frameworks (COFs),5–8 are attracting interest as promising candidates for semiconductors, photocatalysts, and electrocatalysts. Well overlapped two-dimensional (2D) network sheets can provide both columnar domains formed by π⋯π stacking of the π-conjugated cores and one-dimensional (1D) pores along the stacking directions. The former can serve as a pathway for charge carriers and the latter allows guest molecule access from outside of the framework through the channels. However, such π-stacked structures prevent aromatic guest molecules from interacting with the π-orbitals of the building block molecules, resulting in poor guest-responsivity of electronic properties (Fig. 1a). To achieve efficient host–guest interactions, the channel walls of the framework should be composed of π-conjugated surfaces.9,10 Such frameworks can be achieved using non-coplanar and robust molecules, including triptycene derivatives.11–14 In the system, on the other hand, no pathway for charge transport is formed. Therefore, the framework is required to exhibit both the channel surrounded by π-conjugated planes and the π-stacked columnar structure. In this context, the porous framework formed by partially stacked π-conjugated molecules meets the above requirements (Fig. 1b).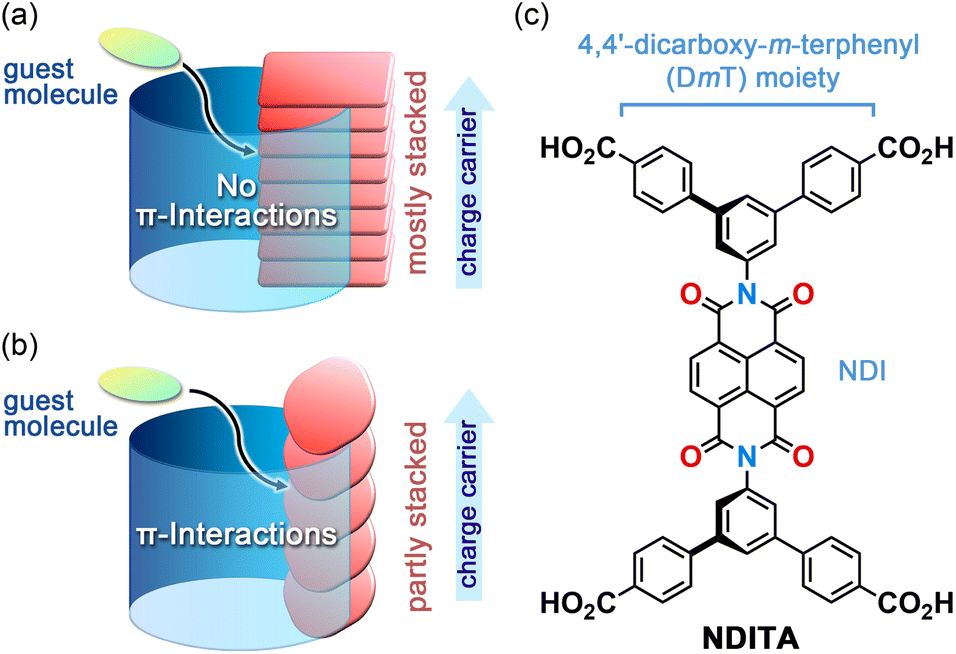 | ||
| Fig. 1 Porous frameworks with (a) mostly stacked and (b) partly stacked π-conjugated building block molecules. (c) Bis(4,4′-dicarboxy-m-terphenyl)naphthalenediimide (NDITA). | ||
For precise structural characterization of the porous frameworks, those formed through reversible bonds, such as dynamic covalent bonds15 and hydrogen bonds, are preferable because of their single crystallinity suitable for single-crystal X-ray diffraction (SCXRD) analysis. In particular, hydrogen-bonded organic frameworks (HOFs) have been intensively investigated from this point of view,16–25 although excellent single-crystalline COFs were also reported recently.26–28
Naphthalenediimide (NDI) is one of the most typical n-type semiconductor molecules, and NDI-based MOFs29 and COFs30,31 have been actively researched from the perspectives of redox and photoelectronic properties.32,33 On the other hand, only a handful of NDI-based HOFs were reported.34–42 Luo and coworkers reported a photochromic and electrochromic HOF based on NDI.34 Ono, Mathevet, Adachi and coworkers reported an electron-conductive HOF using a cyclopentanol-modified NDI derivative.37 Champness and coworkers reported the photoinduced radical formation of NDI-based HOFs.41 Nevertheless, electron-conductive, stimuli-responsive NDI-HOFs with large void spaces are unexplored.
In this study, we report on the structure and properties of an NDI-based HOF that possesses large voids. The HOF NDITA-1 is composed of bis(4,4′-dicarboxy-m-terphenyl)naphthalenediimide (NDITA), a derivative of NDI modified with 4,4′-dicarboxy-m-terphenyl (DmT) moieties (Fig. 1c). Since NDITA has an orthogonal conformation between the DmT and NDI moieties, slip-stacking of the molecule resulted in the formation of a layered HOF that possesses π-stacked columnar domains and pore channels surrounded by π-conjugated planes. These structural factors allowed the HOF to exhibit electron conductivity through the stacked NDI parts and aromatic guest responsivity due to interactions between the exposed NDI plane and the guests. These results suggest that the HOF with π-conjugated moieties partially exposed to the pore surface and partially stacked with neighboring π-conjugated moieties is a promising platform to develop materials that enable the dynamic modulation of electronic properties by guest molecules.
Results and discussion
Crystallography of HOF NDITA-1
NDITA was synthesized according to the literature (Scheme S1 and Fig. S1).43,44 The synthesized compound was recrystallized by slow evaporation of a solution of NDITA dissolved in N,N′-dimethylformamide (DMF) and 1,2,4-trichlorobenzene (TCB) at 140 °C, yielding the solvated HOF NDITA-1(TCB) as needle crystals with an average dimension of 10 µm × 10 µm × 150 µm (Fig. S2). A first attempt of SCXRD analysis was conducted using an in-house diffractometer equipped with a Cu-tube. However, the analysis failed because of only a small number of weak diffraction peaks. Consequently, the crystal structure of the HOF was successfully revealed using focused and high-flux synchrotron X-ray radiation (Fig. 2 and Table S1).† An NDITA molecule with the inversion center crystallized into the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The DmT moieties have a nearly planar conformation due to packing forces in the crystal: dihedral angles between the central benzene ring (A) and the peripheral rings (B and C) are 15.1° and 2.0°, respectively (Fig. 2a), which significantly differ from the theoretically optimized structure (Fig. S4 and Table S2). The NDI core and DmT moieties have almost orthogonal arrangements: the dihedral angle between ring A and NDI is 86.2°. The orthogonal conformation is frequently observed in other crystalline systems of diaryl NDI derivatives.29–33NDITA forms a honeycomb-like sql-topological network sheet through intermolecular H-bonded dimerization of the peripheral carboxy groups: the O⋯O distances are 2.64 and 2.60 Å (Fig. 2b). The resultant sheet structure has a hexagonal aperture with dimensions of 26.0 Å × 31.5 Å. The sheets are slip-stacked with an interlayer distance of 3.20 Å, resulting in a layered framework with 1D channel spaces with an aperture size of 22.6 Å × 12.8 Å (Fig. 2c and d). It is noteworthy that two of the six inside walls of the channel are composed of the NDI molecular plane. By stacking 2D sheets, a half part of the π-conjugated plane of NDI overlaps with the adjacent π-plane of NDI with an intermolecular distance of 3.43 Å and the other half part is exposed within the channel. Therefore, it is expected that guest molecules can approach the π-conjugated plane of NDI directly and add perturbation to the electronic state of NDITA in the HOF. In the channel, disordered solvent molecules (TCB) are accommodated with a host
. The DmT moieties have a nearly planar conformation due to packing forces in the crystal: dihedral angles between the central benzene ring (A) and the peripheral rings (B and C) are 15.1° and 2.0°, respectively (Fig. 2a), which significantly differ from the theoretically optimized structure (Fig. S4 and Table S2). The NDI core and DmT moieties have almost orthogonal arrangements: the dihedral angle between ring A and NDI is 86.2°. The orthogonal conformation is frequently observed in other crystalline systems of diaryl NDI derivatives.29–33NDITA forms a honeycomb-like sql-topological network sheet through intermolecular H-bonded dimerization of the peripheral carboxy groups: the O⋯O distances are 2.64 and 2.60 Å (Fig. 2b). The resultant sheet structure has a hexagonal aperture with dimensions of 26.0 Å × 31.5 Å. The sheets are slip-stacked with an interlayer distance of 3.20 Å, resulting in a layered framework with 1D channel spaces with an aperture size of 22.6 Å × 12.8 Å (Fig. 2c and d). It is noteworthy that two of the six inside walls of the channel are composed of the NDI molecular plane. By stacking 2D sheets, a half part of the π-conjugated plane of NDI overlaps with the adjacent π-plane of NDI with an intermolecular distance of 3.43 Å and the other half part is exposed within the channel. Therefore, it is expected that guest molecules can approach the π-conjugated plane of NDI directly and add perturbation to the electronic state of NDITA in the HOF. In the channel, disordered solvent molecules (TCB) are accommodated with a host![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :guest ratio of 1:6/7 (Fig. S5). The void ratio of the solvent accessible volume was calculated to be 57.4% using PLATON with a probe radius of 1.2 Å.45
:guest ratio of 1:6/7 (Fig. S5). The void ratio of the solvent accessible volume was calculated to be 57.4% using PLATON with a probe radius of 1.2 Å.45
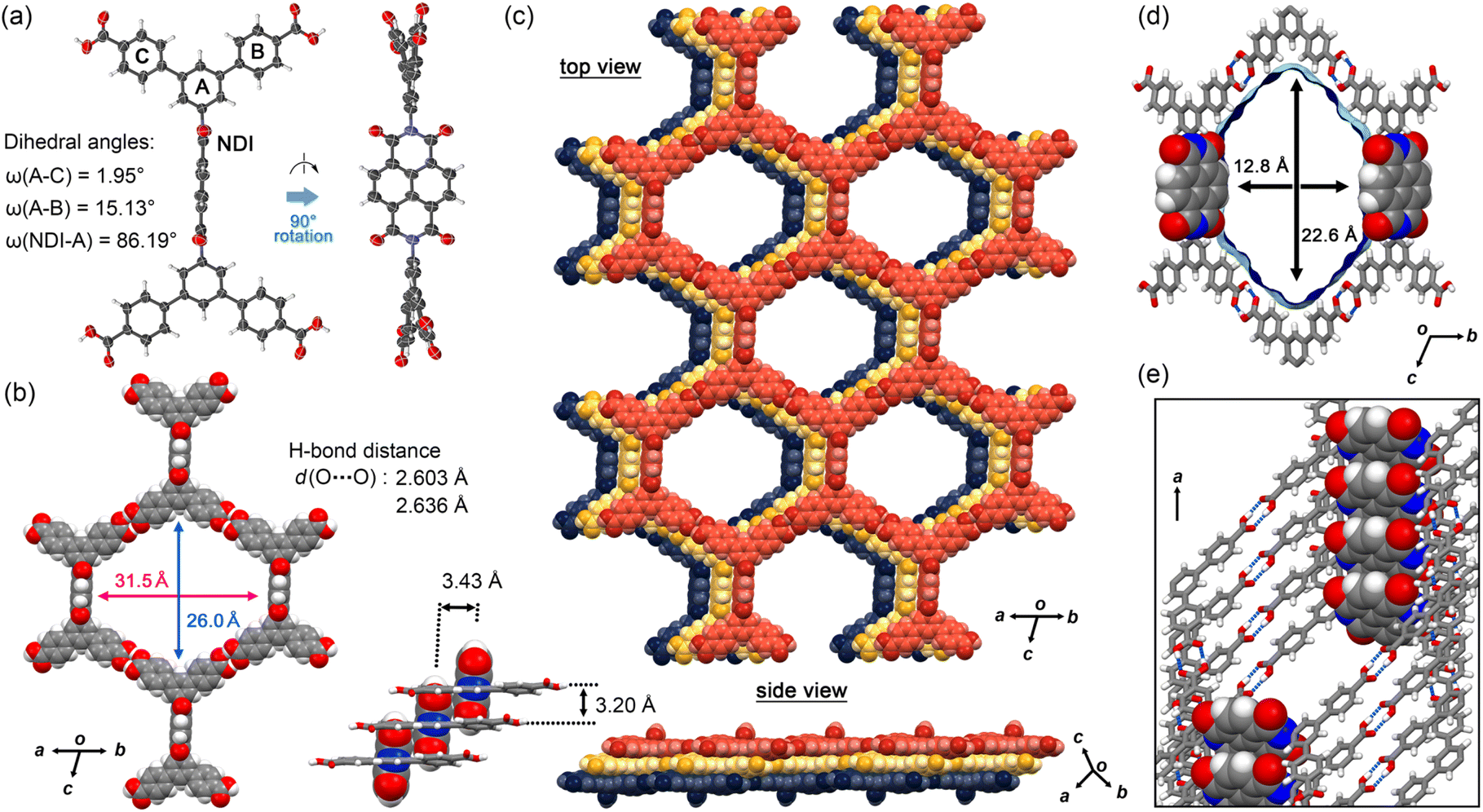 | ||
| Fig. 2 Crystal structure of HOF NDITA-1(TCB). (a) Molecular conformation in a nearly orthogonal fashion. (b) Motif of the sql-network. (c) Selected layered structure composed of three sheets. (Inset) Interlayer distances between the sql-sheets and those between NDI cores. (d) Visualized surfaces of the solvent accessible void. (e) The channel wall composed of the π-conjugated plane of NDI. | ||
The present stacking manner is governed by molecular geometry with the orthogonal conformation of NDI and DmT moieties as well as effective CH/O interactions between the aromatic hydrogen atom of the DmT group and the oxygen atom of the NDI core (Fig. S3). This slip-stacking manner of NDITA in the HOF enables the LUMO of the NDI core to have a preferable overlap with the same phase. However, no mixing of the MOs was observed (Fig. S11 and S12).
Thermal stability
Thermal gravimetry (TG) analysis on bulk crystals of NDITA-1(TCB) showed a weight loss of 60% by heating up to 200 °C due to the release of the guest molecules (Fig. 3a). Another weight loss observed at approximately 300 °C was probably due to thermal decomposition of the molecules. The structural changes of HOF NDITA-1(TCB) were monitored by variable-temperature powder X-ray diffraction (VT-PXRD) analysis on the bulk crystals (Fig. 3b). An ambiguous profile was observed below 100 °C due to the significantly low electron diffraction contrast provided by the low density frameworks composed of light-weight elements and scattering of diffracted X-rays by disordered TCB molecules inside the channel, which is often observed for the solvated HOF crystals that include chlorinated aromatic solvent molecules.46,47 Diffraction peaks started to appear at 100 °C and became apparent at higher temperatures. The diffraction pattern is similar to that simulated from the SCXRD data, although they are not exactly matched. For example, characteristic peaks at 4.10°, 5.03° and 5.11° corresponding to the (0 0 1), (0 1 0) and (0 1 −1) planes, respectively, in the crystal structure of NDITA-1(TCB) were replaced by one prominent peak at 4.61° and a weak peak at 4.08°. These changes in the pattern were probably caused by the slight deformation of the framework by slipping of the layers during and/or after release of the guest solvent molecules. Single crystals of NDITA-1(TCB) heated at 130 °C under vacuum conditions retained needle-like crystalline morphology, while transparency was declining (Fig. S6), indicating subtle changes of the framework. We attempted SCXRD analysis and Rietveld analysis using these activated crystals, both of which failed to give a precise crystal structure.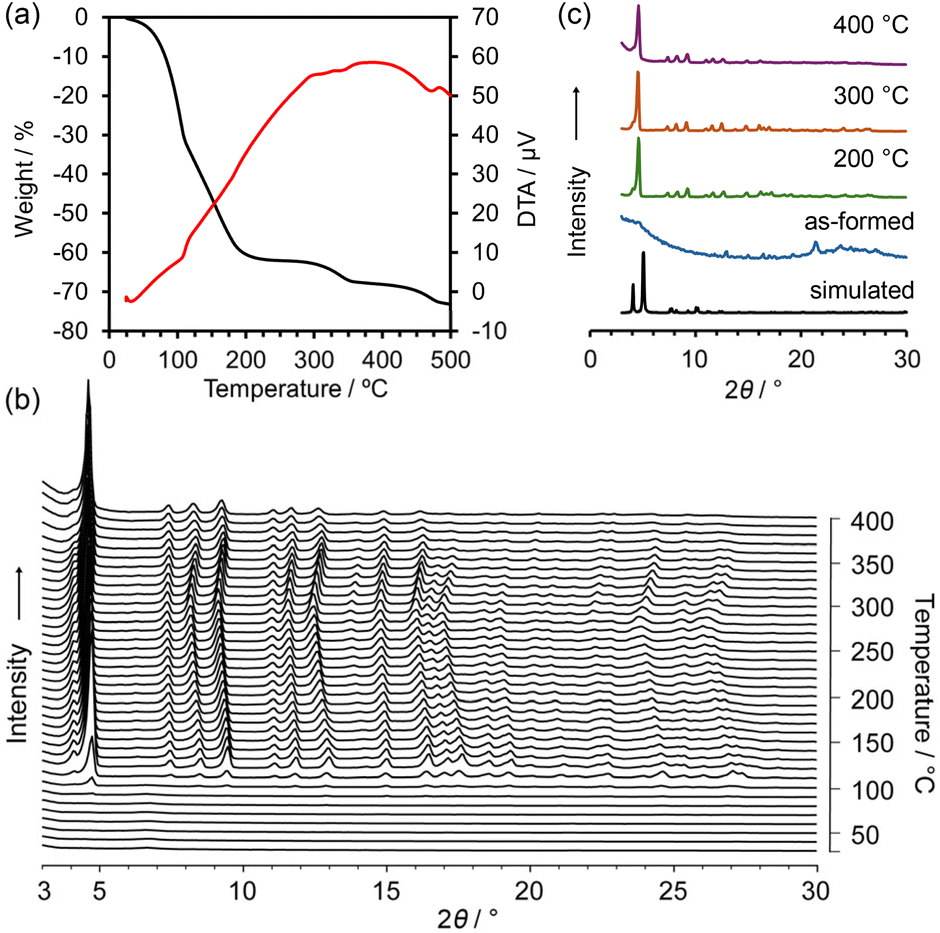 | ||
| Fig. 3 Thermal analysis of NDITA-1(TCB). (a) TG-DA curves. (b) VT-PXRD patterns. (c) Representative PXRD patterns of the VT experiment. | ||
Upon further heating, the diffraction peaks were slightly shifted to small-angle regions and then were shifted back to the wide-angle regions at temperatures higher than 300 °C. In more detail, some diffraction peaks, such as at 7.50°, 11.1° and 15.0°, showed less shift than others, such as at 9.47°, 13.0° and 16.5°, indicating anisotropic structural deformation of the framework. The diffraction pattern remained even when heated up to 400 °C.
Porosity
According to the thermal analysis described above, activation of HOF NDITA-1(TCB) was first attempted by heating under reduced pressure conditions. However, the resultant samples showed relatively weak diffraction peaks and only a subtle amount of gas uptake. Therefore, activation was conducted using a supercritical fluid of carbon dioxide (for detailed conditions, see the SI). The activated HOF NDITA-1a showed a diffraction pattern closer to that of NDITA-1(TCB) compared to that observed during the VT-PXRD measurements, indicating that the nearly original framework structure was retained (Fig. S7). The complete removal of guest solvent molecules was confirmed by the 1H NMR spectrum of the sample dissolved in DMSO-d6 solution.N2 and CO2 sorption experiments were conducted at 77 K and 195 K, respectively, using freshly activated samples (Fig. 4a). The N2 adsorption isotherm exhibited a sharp rise at a relative pressure (P/P0) below 0.01, followed by a small step, and reached a plateau at P/P0 of 0.1. The uptake amount was 350 cm3 (STP) per g at a P/P0 of 0.9. The Brunauer–Emmett–Teller (BET) surface area of NDITA-1a was calculated to be 1410 m2 g−1 (Fig. S8). CO2 adsorption isotherms at 195 K exhibit a sigmoidal shape with a hysteresis at P/P0 ∼ 0.15, indicating that interactions between the pore surface and CO2 are less attractive or a certain structural change was derived by the adsorption. The total amount of uptake was 192 cm3 (STP) per g. The pore diameter was estimated using the non-local density functional theory (NLDFT) method for the N2 adsorption isotherm with a peak at 1.88 nm (Fig. 4b). Unfortunately, the sample after being subjected to gas sorption experiments showed a decrease in the diffraction intensity in the PXRD pattern and the uptake amount of gas, indicating a fragile nature of the activated framework.
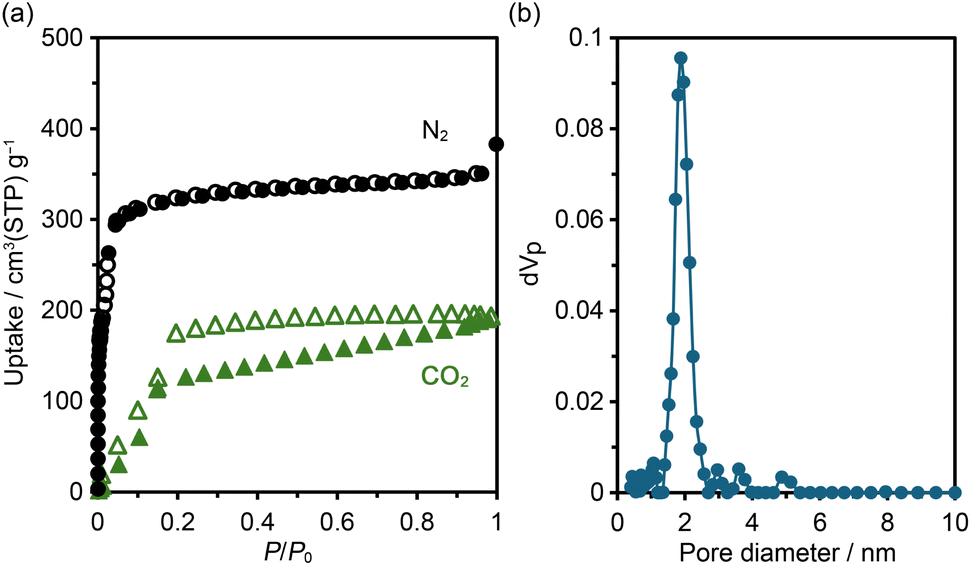 | ||
| Fig. 4 (a) Gas sorption isotherms for N2 at 77 K (black) and CO2 at 195 K (green). (b) Pore width distribution using the NLDFT model for the N2 adsorption isotherm. | ||
Guest solvent-dependent photophysical properties
NDITA showed absorption bands at 270, 360, and 380 nm in DMF solution (Fig. 5). The observed lowest-energy band was attributed to the transition from HOMO−2 to LUMO at 383.9 nm (f = 0.629), corresponding to the π–π* transition of the NDI moiety, according to the TDDFT calculation of NDITA (Fig. S9 and S10). On the other hand, the calculated HOMO–LUMO transition at 450.9 nm, which corresponds to an intramolecular CT transition from DmT to NDI moieties, is symmetrically forbidden (f = 0.018) and was not observed experimentally. A fluorescence spectrum showed a band at 436 nm. | ||
| Fig. 5 Absorption (black solid line), fluorescence (cyan solid line), and excitation (cyan dotted line) spectra of NDITA in DMF solution. Calculated one-electron transition oscillations are shown with orange bars in arbitrary units. The fluorescence spectrum was measured at the excitation wavelength of 381 nm. The excitation spectrum was recorded at 430 nm. | ||
Then, spectroscopic analyses on the aromatic guest molecule-included HOFs were conducted in a solid state. An amorphous solid of NDITA shows an absorption band at 380 nm with a broad shoulder from 450 to 650 nm (Fig. 6). The HOF activated using a supercritical fluid of carbon dioxide also showed quite a similar absorption band to the amorphous solid (Fig. S13), indicating that the amorphous solid and activated HOF have similar molecular stacking orientations in short distance ranges. The as-formed crystalline powder of NDITA-1(TCB) showed a band at ca. 450–500 nm, originating from intermolecular interactions between the framework and TCB molecules. The guest-included HOFs were prepared by immersing bulk crystals of NDITA-1(TCB) in the corresponding solvents, affording exchange of the included guest molecules. When the HOF was immersed in o-dimethoxybenzene (DMB), an obvious color change from pale yellow to brown was observed. A diffuse reflectance spectrum of the DMB-immersed HOF showed a band at around 550 nm, which appeared in a lower energy region than that of NDITA-1(TCB) and the amorphous solid. The band was caused by charge transfer (CT) interactions between electron-deficient NDI and electron-rich DMB. Other solvents such as 1,2,4-trimethylbenzene (TMB) and benzonitrile (PhCN) showed no significant color changes.
 | ||
| Fig. 6 (a) Photographs of solvent-immersed bulk crystals of the HOF NDITA-1; (i) amorphous solid, (ii) as-formed NDITA-1(TCB), (iii) TMB-immersed HOF, (iv) DMB-immersed HOF and (v) PhCN-immersed HOF. (b) Normalized diffuse reflectance spectra of the amorphous solid (black), NDITA-1(TCB) (green) and DMB-immersed HOF (grey). The spectra were adjusted to zero at 650 nm. | ||
The solvent-immersed HOFs also exhibited solvent-dependent fluorescence changes (Fig. 7); although the intensity was very weak, the fluorescence quantum yield was less than 0.5%. The amorphous solid exhibited an emission band at 583 nm accompanied by a shoulder at around 655 nm. The band was red-shifted by 147 nm compared to that in DMF solution (Fig. 5). The as-formed HOF NDITA-1(TCB) showed two emission bands at 589 and 627 nm. TMB-immersed HOF showed the emission maximum at 631 nm with shoulders at 580 and 680 nm. The PhCN-immersed HOF exhibited a band at 650 nm with a shoulder at 600 nm. The observed red-shifted band for the PhCN-immersed HOF indicates the existence of attractive interactions presumably between the electron-rich DmT moieties of NDITA and the electron-deficient PhCN molecule. Although the DMB-immersed HOF showed almost no emission (Fig. S14), the magnified emission spectrum showed a band at 680 nm, which is consistent with the formation of a CT complex.48 It is reported that volatile organic compound (VOC)-induced fluorescence color changes of NDI-based porous coordination polymers (PCPs) are correlated with the ionic potential of VOC absorbed in the PCP.49,50 In the present system, on the other hand, the relationship between the color changes and the ionization potential of the aromatic solvent is ambiguous, while Gutmann donor number may show a better correlation with the emission maximum wavelength (Table S3). Bulk crystals of NDITA-1(TCB) immersed in solvents were also subjected to PXRD measurements to confirm whether structural changes occurred or not upon immersion (Fig. S15). The HOFs immersed in TMB, DMB, and PhCN showed obvious diffraction profiles similar to the activated sample and NDITA-1(TCB), although the diffraction of NDITA-1(TCB) is very weak due to TCB molecules being disordered in the pore, as we have explained earlier. It should be noted that the DMB-immersed HOF showed a new peak at 2θ ∼ 6° (with a d space of 14.9), which was not observed in the pattern of NDITA-1(TCB). This indicates that the CT interaction between DMB and NDI slightly perturbed the structure or periodicity of the framework.
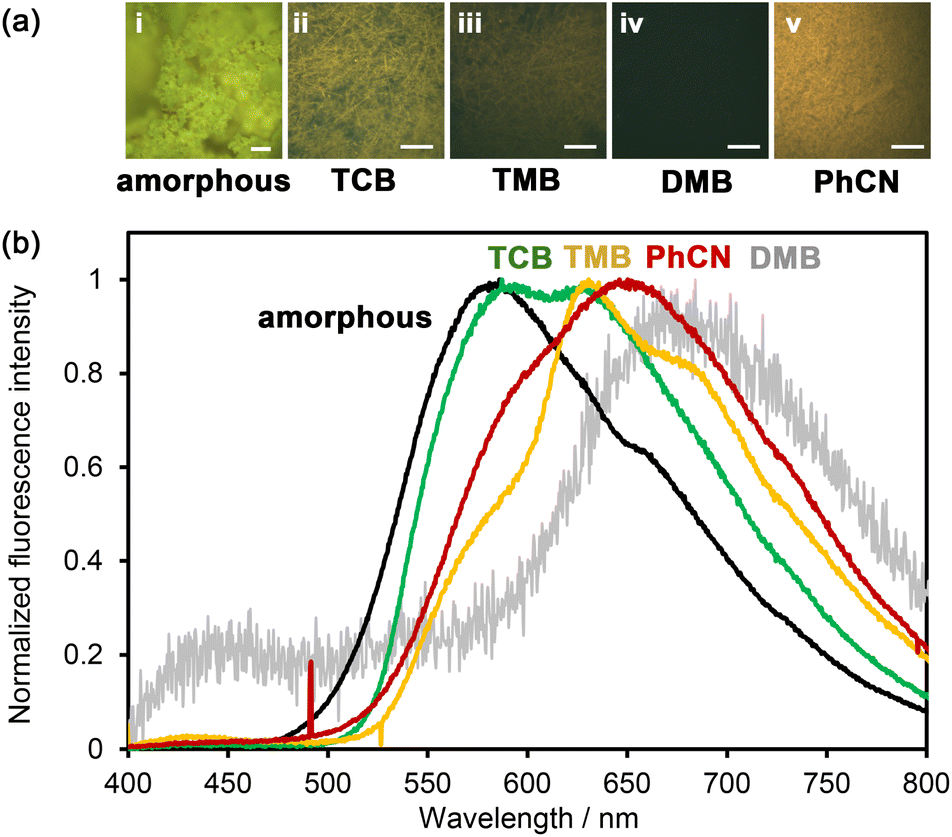 | ||
| Fig. 7 (a) Photographs of NDITA solids under UV light (365 nm); (i) amorphous solid, (ii) as-formed NDITA-1(TCB), (iii) TMB-immersed HOF, (iv) DMB-immersed HOF and (v) PhCN-immersed HOF. Scale bar: 50 µm. (b) Normalized solid-state fluorescence spectra of the amorphous solid (black), as-formed NDITA-1(TCB) (green), TMB-immersed HOF (yellow), DMB-immersed HOF (grey) and PhCN-immersed HOF (red). | ||
To investigate the conductivity of the HOF, bulk NDITA-1 crystals that include solvent molecules (TCB, TMB, DMB, and PhCN) in the pores were subjected to flash-photolysis time-resolved microwave conductivity (FP-TRMC) measurements51,52 with excitation at 355 nm (Fig. 8). NDITA-1(DMB) showed the largest conductivity among them due to effective charge separation: the ΦΣµ value (9.8 × 10−9 m2 V−1 s−1) is four times larger than that of the amorphous solid of NDITA (Fig. S16), two times larger than those of other HOFs (Table 1). The ΦΣµ values of the related NDI-based materials are summarized in Table S4. The measurement on the SF6-treated sample showed a smaller value, indicating that the charge carrier is an electron (Fig. S17). It is also remarkable that the conductivity transient of the PhCN-immersed HOF showed no decay even after 90 µs (Fig. S18), indicating that charge carriers may be accumulated in PhCN.
 | ||
| Fig. 8 Comparison of conductivity transients observed for the bulk crystals of NDITA-1 immersed in TCB (green), TMB (yellow), DMB (grey) and PhCN (red) upon excitation of 355 nm laser pulses at 9.1 × 1015 photons per cm2. | ||
| NDITA-1(DMB) | NDITA-1(TCB) | NDITA-1(TMB) | NDITA-1(PhCN) | |
|---|---|---|---|---|
| Photoconductivity/10−9 m2 V−1 s−1 | 9.8 | 5.0 | 3.6 | 4.6 |
Conclusions
In summary, we constructed a naphthalenediimide (NDI)-based HOF NDITA-1 using tetratopic carboxylic acid (NDITA) that has a NDI core and two dicarboxy-m-terphenyl (DmT) moieties in a nearly orthogonal conformation. The HOF has a slip-stacking structure of H-bonded 2D porous sheets. In the HOF, half of the NDI surface participates in the formation of 1D stacked columnar domains that act as the electron conducting pathway, while the other half is exposed inside the void channel. Since aromatic solvents can come into contact with the NDI moieties, the HOF exhibits solvent-responsive photophysical and electronic behaviors. In particular, the HOF encapsulating electron-rich dimethoxybenzene (DMB) in the void enhances charge-transfer interactions, resulting in significant changes in color from pale yellow to brown and quenching of the emission. Furthermore, FP-TRMC measurements on the solvent-immersed HOFs revealed that the DMB-immersed HOF showed the highest electron conductivity among solvent-immersed HOFs in the present systems. These results indicate that the HOF with π-conjugated moieties partly exposed to the channel surface and partly stacked with neighboring π-conjugated moieties can contribute to the development of materials that enable the dynamic modulation of electronic properties by guest molecules.Author contributions
Y. M. synthesized and characterized all materials, evaluated the properties of the materials and co-wrote the paper. R. O. performed the theoretical calculations. R. P. P. and S. S. performed conductivity measurements. M. N. and N. T. performed activation of HOFs under critical CO2 and gas sorption experiments. T. H. and I. H. planned the research and analysed the data. I. H. supervised and co-wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: details of synthesis, characterization, spectroscopic data, theoretical calculation and crystallographic data. See DOI: https://doi.org/10.1039/d5sc07488f.CCDC 2478400 contains the supplementary crystallographic data for this paper.53
Acknowledgements
This work was supported by KAKENHI (JP23H04029, JP24K01468, JP25H01672 and JP25H02042) from JSPS and MEXT Japan. Synchrotron X-ray diffraction data were collected at BL40XU and BL41XU at SPring-8 with approval of the Japan Synchrotron Radiation Research Institute (JASRI, proposal no. 2024A1208, 2024B1717 and 2025A1137). The authors thank Dr K. Ichiyanagi, Dr T. Sasaki, and Dr S. Baba at JASRI for synchrotron radiation experiments and Prof. Dr T. Kusamoto and Dr R. Matsuoka at the University of Osaka for fluorescence measurements. The authors sincerely thank the reviewers for their constructive and important comments.Notes and references
- L. S. Xie, G. Skorupskii and M. Dincă, Chem. Rev., 2020, 120, 8536–8580 CrossRef CAS PubMed.
- M. Wang, R. Dong and X. Feng, Chem. Soc. Rev., 2020, 50, 2764–2793 RSC.
- J. Liu, G. Xing and L. Chen, Acc. Chem. Res., 2024, 57, 1032–1045 CrossRef CAS PubMed.
- Y. Lu, P. Samorì and X. Feng, Acc. Chem. Res., 2024, 57, 1985–1996 CrossRef CAS PubMed.
- R.-R. Liang, S.-Y. Jiang, A. Ru-Hun and X. Zhao, Chem. Soc. Rev., 2020, 49, 3920–3951 RSC.
- X. Li, P. Yadav and K. P. Loh, Chem. Soc. Rev., 2020, 49, 4835–4866 RSC.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8841–8933 CrossRef PubMed.
- A. M. Evans, M. J. Strauss, A. R. Corcos, Z. Hirani, W. Ji, L. S. Hamachi, X. Aguilar-Enriquez, A. D. Chavez, B. J. Smith and W. R. Dichtel, Chem. Rev., 2022, 122, 442–564 CrossRef CAS PubMed.
- P. Sozzani, S. Bracco, A. Comotti, L. Ferretti and R. Simonutti, Angew. Chem., Int. Ed., 2005, 44, 1816–1820 CrossRef CAS PubMed.
- X.-N. Han, Y. Han and C.-F. Chen, Nat. Commun., 2021, 12, 6378 CrossRef CAS.
- W. Yan, X. Yu, T. Yan, D. Wu, E. Ning, Y. Qi, Y.-F. Han and Q. Li, Chem. Commun., 2017, 53, 3677–3680 RSC.
- R. Zhang, H. Daglar, C. Tang, P. Li, L. Feng, H. Han, G. Wu, B. N. Limketkai, Y. Wu, S. Yang, A. X.-Y. Chen, C. L. Stern, C. D. Malliakas, R. Q. Snurr and J. F. Stoddart, Nat. Chem., 2024, 16, 1982–1988 CrossRef CAS.
- M. Mastalerz and I. M. Oppel, Angew. Chem., Int. Ed., 2012, 51, 5252–5255 CrossRef CAS PubMed.
- A. Pulido, L. Chen, T. Kaczorowski, D. Holden, M. A. Little, S. Y. Chong, B. J. Slater, D. P. McMahon, B. Bonillo, C. J. Stackhouse, A. Stephenson, C. M. Kane, R. Clowes, T. Hasell, A. I. Cooper and G. M. Day, Nature, 2017, 543, 657–664 CrossRef CAS PubMed.
- T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L.-H. Li, Y. Wang, J. Su, J. Li, X. Wang, W. D. Wang, W. Wang, J. Sun and O. M. Yaghi, Science, 2018, 361, 48–52 CrossRef CAS PubMed.
- R.-B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Chem. Soc. Rev., 2019, 48, 1362–1389 RSC.
- I. Hisaki, C. Xin, K. Takahashi and T. Nakamura, Angew. Chem., Int. Ed., 2019, 58, 11160–11170 CrossRef CAS.
- X. Song, Y. Wang, C. Wang, D. Wang, G. Zhuang, K. O. Kirlikovali, P. Li and O. K. Farha, J. Am. Chem. Soc., 2022, 144, 10663–10687 CrossRef CAS.
- M. R. di Nunzio, I. Hisaki and A. Douhal, J. Photochem. Photobiol., C, 2021, 47, 100418 CrossRef CAS.
- P. Li, M. R. Ryder and J. F. Stoddart, Acc. Mater. Res., 2020, 1, 77–87 CrossRef CAS.
- Z. Xiong, S. Xiang, Y. Lv, B. Chen and Z. Zhang, Adv. Funct. Mater., 2024, 34, 2403635 CrossRef CAS.
- X. Liu, G. Liu, T. Fu, K. Ding, J. Guo, Z. Wang, W. Xia and H. Shangguan, Adv. Sci., 2024, 11, 2400101 CrossRef CAS PubMed.
- L. Chen, B. Zhang, L. Chen, H. Liu, Y. Hu and S. Qiao, Mater. Adv., 2022, 3, 3680–3708 RSC.
- B. Wang, R.-B. Lin, Z. Zhang, S. Xiang and B. Chen, J. Am. Chem. Soc., 2020, 142, 14399–14416 CrossRef CAS PubMed.
- Y. Suzuki and I. Hisaki, Polym. J., 2024, 56, 1–16 CrossRef CAS.
- J. Han, J. Feng, J. Kang, J.-M. Chen, X.-Y. Du, S.-Y. Ding, L. Liang and W. Wang, Science, 2024, 383, 1014–1019 CrossRef CAS PubMed.
- B. Yu, R.-B. Lin, G. Xu, Z.-H. Fu, H. Wu, W. Zhou, S. Lu, Q.-W. Li, Y. Jin, J.-H. Li, Z. Zhang, H. Wang, Z. Yan, X. Liu, K. Wang, B. Chen and J. Jiang, Nat. Chem., 2024, 16, 114–121 CrossRef CAS PubMed.
- L. Yi, Y. Gao, S. Luo, T. Wang and H. Deng, J. Am. Chem. Soc., 2024, 146, 19643–19648 CrossRef CAS.
- Y. Zhou and L. Han, Coord. Chem. Rev., 2021, 430, 213665 CrossRef CAS.
- D.-H. Yang, Z.-Q. Yao, D. Wu, Y.-H. Zhang, Z. Zhou and X.-H. Bu, J. Mater. Chem. A, 2016, 4, 18621–18627 RSC.
- S. Royuela, E. Martínez-Periñán, M. P. Arrieta, M. M. Ramos, F. Zamora, E. Lorenzo and J. L. Segura, Chem. Commun., 2020, 56, 1267–1270 RSC.
- C. He, W. Dai, Y. Zhao and J.-J. Liu, Dalton Trans., 2025, 54, 15–37 RSC.
- L. Li, J.-G. Zeng, N.-N. Zhang, Y.-T. Yu, S.-H. Li and Y. Hua, Inorg. Chem. Front., 2025, 12, 11–38 RSC.
- L. Wang, L. Yang, L. Gong, R. Krishna, Z. Gao, Y. Tao, W. Yin, Z. Xu and F. Luo, Chem. Eng. J., 2020, 383, 123117 CrossRef CAS.
- Y. Wu, X. Mao, M. Zhang, X. Zhao, R. Xue, S. Di, W. Huang, L. Wang, Y. Li and Y. Li, Adv. Mater., 2021, 33, 2106079 CrossRef CAS PubMed.
- H. Zhao, Z. Zhou, X. Feng, C. Liu, H. Wu, W. Zhou and H. Wang, Nano Res., 2023, 16, 8809–8816 CrossRef CAS.
- K. Imaoka, H. S. Kim, Y. Yamamoto, S. Fukutomi, L.-M. Chamoreau, L. Qu, H. Iguchi, Y. Tsuchiya, T. Ono, F. Mathevet and C. Adachi, Adv. Funct. Mater., 2024, 34, 2409299 CrossRef CAS.
- K. Wakamatsu, Y. Yamaguchi, S. Furuno, H. Wang and H. Yoshikawa, ChemElectroChem, 2024, 11, e202300607 CrossRef CAS.
- A. R. Y. Almuhana, G. R. F. Orton, C. Rosenberg and N. R. Champness, Chem. Commun., 2024, 60, 452–455 RSC.
- C. Liu, L.-H. Cao, Z.-Y. Zhou, X.-Y. Chen and W. Zhang, ACS Appl. Mater. Interfaces, 2025, 17, 40613–40622 CrossRef CAS PubMed.
- A. R. Y. Almuhana, S. L. Griffin and N. R. Champness, CrystEngComm, 2024, 26, 4643–4648 RSC.
- Q. Li, Y. Zhao, X. Shen, S.-B. Xia and J.-J. Liu, Cryst. Growth Des., 2025, 25, 8002–8009 CrossRef CAS.
- A. Mallick, H. Liang, O. Shekhah, J. Jia, G. Mouchaham, A. Shkurenko, Y. Belmabkhout, H. N. Alshareef and M. Eddaoudi, Chem. Commun., 2020, 56, 1883–1886 RSC.
- X.-Y. Wu, Z.-Y. Li, M.-L. Zhang, J.-F. Lu, Z.-H. Zhu, J. Zhao, S.-J. Liu and H.-R. Wen, Inorg. Chem., 2024, 63, 19897–19905 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- I. Hisaki, S. Nakagawa, N. Ikenaka, Y. Imamura, M. Katouda, M. Tashiro, H. Tsuchida, T. Ogoshi, H. Sato, N. Tohnai and M. Miyata, J. Am. Chem. Soc., 2016, 138, 6617–6628 CrossRef CAS PubMed.
- I. Hisaki, N. Ikenaka, E. Gomez, B. Cohen, N. Tohnai and A. Douhal, Chem.–Eur. J., 2017, 23, 11611–11619 CrossRef CAS.
- F. S. Thuluvanchery, N. Tamaoki and Y. Sagara, Bull. Chem. Soc. Jpn., 2024, 97, uoae135 CrossRef CAS.
- Y. Takashima, V. M. Martínez, S. Furukawa, M. Kondo, S. Shimomura, H. Uehara, M. Nakahama, K. Sugimoto and S. Kitagawa, Nat. Commun., 2011, 2, 168 CrossRef PubMed.
- T. Ono, M. Sugimoto and Y. Hisaeda, J. Am. Chem. Soc., 2015, 137, 9519–9522 CrossRef CAS PubMed.
- S. Seki, A. Saeki, T. Sakurai and D. Sakamaki, Phys. Chem. Chem. Phys., 2014, 16, 11093–11113 RSC.
- A. Saeki, Y. Koizumi, T. Aida and S. Seki, Acc. Chem. Res., 2012, 45, 1193–1202 CrossRef CAS.
- CCDC 2478400: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2p5zd8.
Footnote |
| † Crystal data for NDITA-1(TCB), C54H30N2O12·6(C6H3Cl3); Fw = 1919.4; triclinic, P (#2), Z = 1, a = 4.9531(8) Å, b = 19.0154(15) Å, c = 23.435(2) Å, α = 112.684(8)°, β = 93.637(11)°, γ = 90.887(10)°, V = 2030.5(4) Å3, T = 100 K, D = 1.570 g cm−3, 22250 collected, 9320 unique (Rint = 0.2126) reflections, the final R1 and wR2 values: 0.1618 [I > 2.0σ(I)] and 0.4485 (all data), respectively. CCDC-2478400. |
| This journal is © The Royal Society of Chemistry 2026 |