
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral Lewis base/Lewis acid/Brønsted acid cooperative catalysis enabled regio- and enantioselective electrophilic sulfenylation/semipinacol rearrangement of allenols
Zheng-Wei
Wei†
a,
Qing-Yun
Cai†
a,
Li-Miao
Yang
a,
Yu-Xuan
Huo
a,
Ze-Long
Li
a,
Xue-Qing
Gong
 *a and
Zhi-Min
Chen
*ab
*a and
Zhi-Min
Chen
*ab
aState Key Laboratory of Synergistic Chem-Bio Synthesis, School of Chemistry and Chemical Engineering, Shanghai Key Laboratory for Molecular Engineering of Chiral Drugs, Shanghai Jiao Tong University, Shanghai 200240, P. R. China. E-mail: chenzhimin221@sjtu.edu.cn; xqgong@sjtu.edu.cn
bKey Laboratory of Drug-Targeting and Drug Delivery System of the Education Ministry, Sichuan University, Chengdu 610041, P. R. China
First published on 10th December 2025
Abstract
A chiral sulfide/B(C6F5)3/phosphoric acid co-catalyzed highly regio- and enantioselective tandem electrophilic sulfenylation/semipinacol rearrangement of allenols is developed for the first time. A variety of chiral organosulfur compounds bearing all-carbon quaternary stereocenters are efficiently synthesized via this cooperative catalysis. The products enable the facile synthesis of trisubstituted alkenes and thioester compounds through a one-step derivatization process. Mechanistic experiments demonstrate that B(C6F5)3 and phosphoric acid cooperate to form a Lewis acid-assisted Brønsted acid (LBA) system, which significantly enhances catalytic reactivity. Density functional theory (DFT) calculations indicate that π⋯π and C–H⋯π interactions play a crucial role in determining the observed enantioselectivity.
Introduction
Catalytic asymmetric electrophilic addition/functionalization reactions of unsaturated hydrocarbons represent important synthetic strategies for constructing complex chiral compounds.1–5 These transformations enable the efficient formation of chiral stereocenters while simultaneously introducing two distinct functional groups. A prominent example is the catalytic asymmetric electrophilic sulfenofunctionalization of unsaturated hydrocarbons, which serves as a direct and effective approach for generating chiral organosulfur compounds.6,7 In particular, the catalytic asymmetric sulfenofunctionalization of alkenes has been extensively studied and well developed by Denmark,8–12 Zhao,13–15 Chen16–20 and other groups21–23 (Scheme 1a). In contrast, the catalytic asymmetric sulfenofunctionalization of allenes, which enables access to synthetically valuable organosulfur compounds containing both alkenes and chiral stereocenters simultaneously, remains undeveloped. Although Zhao's group attempted to explore this transformation in 2021, they were unable to achieve enantioselective control (Scheme 1b).24 We believe that this reaction presents the following three challenges. First, the sulfenium ion may form a thiiranium ion intermediate with the allenes in a regioselective manner.25,26 Second, the resulting thiiranium ion intermediate differs from that formed with alkenes; it tends to be less stable and more susceptible to racemization, thereby hindering enantioselectivity.24 Third, different allene substrates may undergo varying reaction mechanisms. For example, an allene bearing four distinct substituents might proceed through a kinetic resolution pathway, which could significantly influence both reaction efficiency and selectivity.27 Therefore, achieving highly enantioselective sulfenofunctionalization of allenes remains a significant challenge in asymmetric catalysis (Scheme 1b).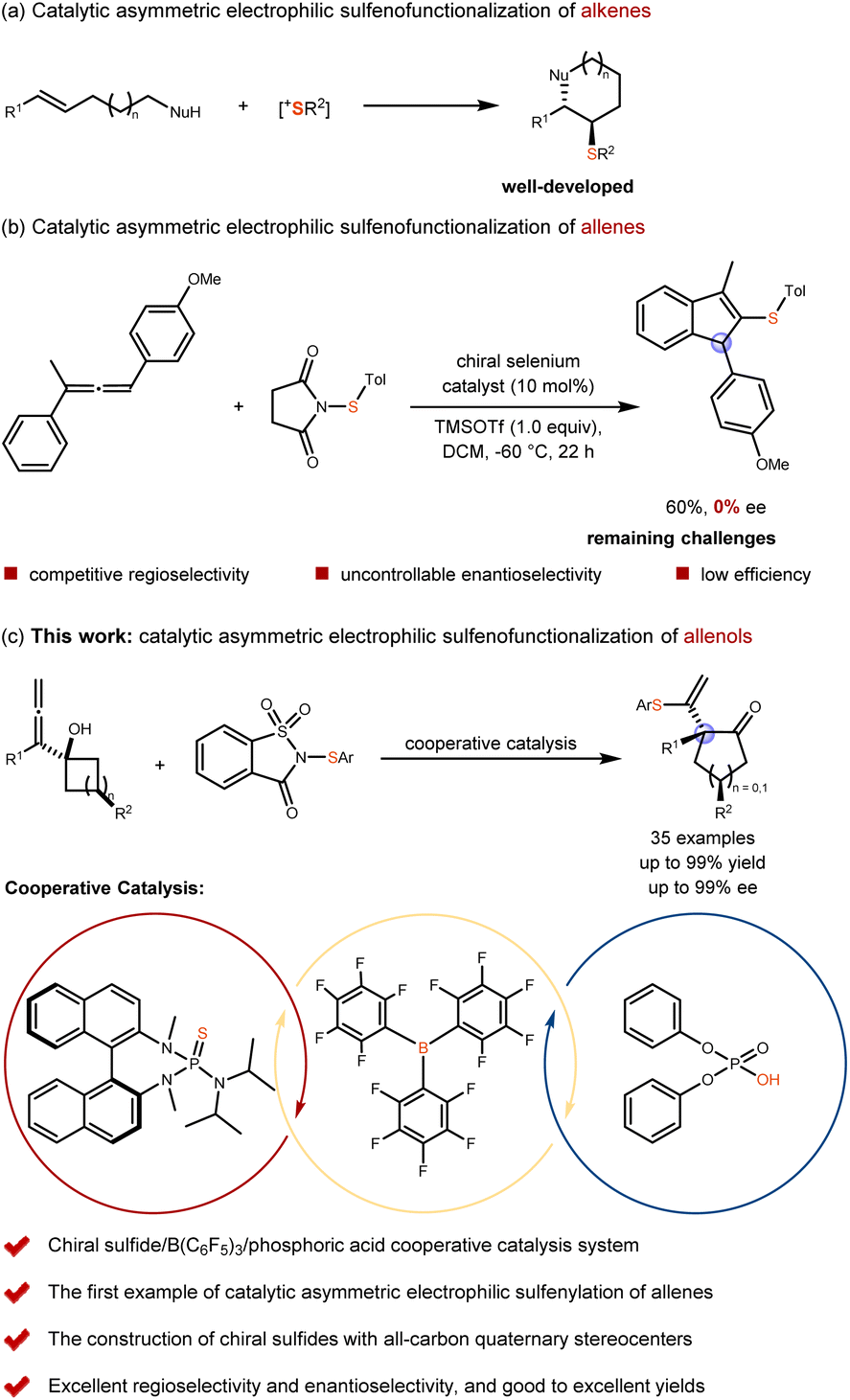 | ||
| Scheme 1 Design of organocatalytic regio- and enantioselective sulfenylation/semipinacol rearrangement of allenols. | ||
Our research group has been engaged in the investigation of catalytic asymmetric electrophilic sulfenylation addition16–20 and electrophilic sulfenylation substitution reactions28–32 involving unsaturated hydrocarbons, as well as selenylation addition and substitution reactions.33–36 For example, we have recently achieved the organocatalytic asymmetric selenylation/semipinacol rearrangement of allenols.35 Building upon these research interests, we aim to explore the catalytic asymmetric electrophilic sulfenofunctionalization of allenes. We believe that the key challenges associated with this reaction can be effectively addressed through the following three strategies. First, selecting a suitable allene substrate, such as a terminal allene can help simultaneously avoid issues related to regioselectivity and unclear reaction mechanisms, particularly those involving kinetic resolution processes. Second, developing a mild and synergistic catalytic system with high efficiency is crucial. Such a system would not only prevent substrate decomposition and undesirable side reactions but also facilitate the formation of enantioselective and stable thiiranium ion intermediates. Third, accelerating the nucleophilic capture process while minimizing racemization is essential for achieving high enantioselectivity in the desired products. For example, intramolecular small-ring expansion processes are known to proceed rapidly under suitable conditions.37–48
Herein, we report a tandem electrophilic sulfenylation/semipinacol rearrangement of allenols, catalyzed synergistically by chiral sulfide, B(C6F5)3, and phosphoric acid. This novel and mild synergistic catalytic system enables both high enantioselectivity and reaction efficiency (Scheme 1c).
Results and discussion
Reaction condition optimization
Under standard reaction conditions, allenyl cyclobutanol 2a was selected as the model substrate, and saccharin-derived 2-(trifluoromethyl)phenylthio compound 3a served as the sulfenylating reagent. The chiral BINAM-derived sulfide (S)-1a was employed as the Lewis base catalyst, while tris(pentafluorophenyl)borane B(C6F5)3 and diphenyl phosphate (DPP) were used as acid catalysts. Additionally, 5 Å molecular sieves (MS) were incorporated as an additive, and chlorobenzene (PhCl) was utilized as the solvent. The reaction was conducted at 0 °C for 12 hours under an argon atmosphere, affording the desired product 4a in 99% yield with 96% enantiomeric excess (ee) (Table 1, entry 1).|
|
|||
|---|---|---|---|
| Entry | Variation of standard conditions | Yield (%) | ee (%) |
| a Standard conditions: the reaction was performed with 2a (0.05 mmol), 3a (0.06 mmol), (S)-1a (0.005 mmol), B(C6F5)3 (0.005 mmol), DPP (0.005 mmol), and 5 Å MS (15 mg) in PhCl (1 mL) at 0 °C for 12 h under Ar. Isolated yields are shown. The ee values were determined by Supercritical Fluid Chromatography (SFC). | |||
| 1 | Standard conditions | 99 | 96 |
| 2 | No B(C6F5)3 | N.R. | — |
| 3 | BF3·Et2O instead of B(C6F5)3 | 37 | 95 |
| 4 | Sc(OTf)2 instead of B(C6F5)3 | 24 | 93 |
| 5 | No DPP | 18 | 89 |
| 6 | (R)-CPA instead of DPP | 97 | 97 |
| 7 | (S)-CPA instead of DPP | 84 | 97 |
| 8 | pTSA instead of DPP | 45 | 95 |
| 9 | No (S)-1a | N.R. | — |
| 10 | (S)-1b instead of (S)-1a | N.R. | — |
| 11 | (S)-1c instead of (S)-1a | 34 | 20 |
| 12 | (S)-1d instead of (S)-1a | N.R. | — |
| 13 | DCM instead of PhCl | 17 | 88 |
| 14 | CH3CN instead of PhCl | N.R. | — |
| 15 | No 5 Å MS | 49 | 95 |
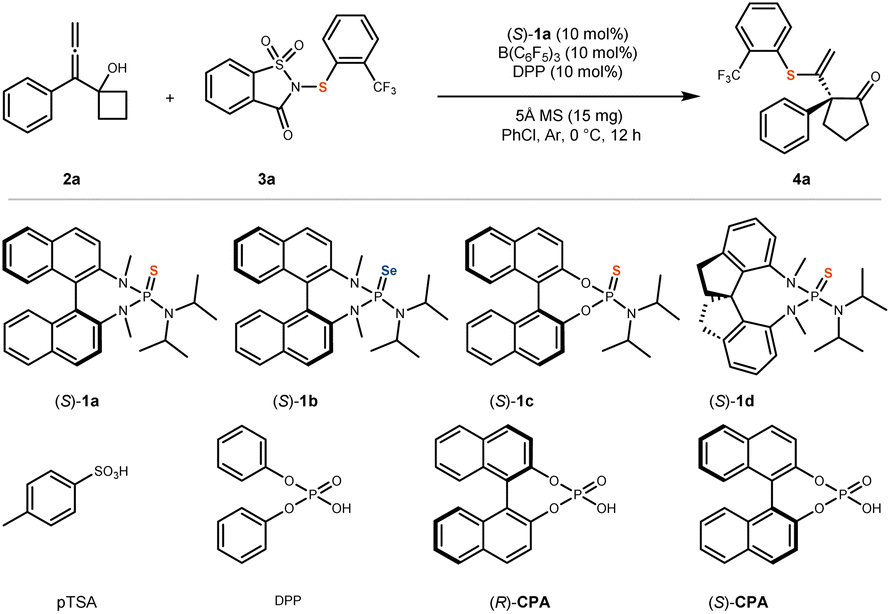
A control experiment revealed that the reaction did not proceed in the absence of B(C6F5)3, confirming its essential role in promoting the transformation (entry 2). When alternative Lewis acids were employed in place of B(C6F5)3, a substantial decrease in the yield of 4a was observed (entries 3 and 4). Similarly, the omission of DPP also led to a significant reduction in product yield (entry 5). Subsequently, chiral phosphoric acids (R)-CPA and (S)-CPA were tested as substitutes for DPP, resulting in a slight improvement in the enantioselectivity of 4a (entries 6 and 7). However, the use of p-toluenesulfonic acid (pTSA) instead of DPP caused a marked decrease in the yield of 4a (entry 8). Optimization of the Lewis acid and Brønsted acid components demonstrated that the combination of B(C6F5)3 and DPP significantly enhances reactivity, albeit with a minor impact on enantioselectivity. Based on previous studies,49–54 we propose that this reaction proceeds via a Lewis acid-assisted Brønsted acid (LBA) catalytic mechanism, wherein the acidity of the Brønsted acid is enhanced, thereby promoting reactivity. Furthermore, the absence of the chiral Lewis base catalyst (S)-1a completely suppressed the reaction, confirming its indispensable role and the absence of background reactivity (entry 9). When alternative Lewis base catalysts were employed in place of (S)-1a, both the yield and enantioselectivity of 4a were significantly diminished, or the reaction failed to proceed altogether (entries 10, 11, and 12), underscoring the critical importance of (S)-1a for both reactivity and enantiocontrol. Substitution of PhCl with other solvents similarly resulted in a notable reduction in yield and enantioselectivity or complete reaction failure (entries 13 and 14), highlighting the necessity of chlorobenzene as the optimal solvent. Moreover, the exclusion of 5 Å molecular sieves led to a considerable decrease in yield, indicating that the additive plays a beneficial role in enhancing reactivity (entry 15). Based on these findings, the optimized reaction conditions were established for further substrate scope investigations (For additional condition screening, please refer to SI.
Substrate scope
With the optimized reaction conditions established, we proceeded to investigate the scope of allenols and sulfenylating reagents. Most of the corresponding products were obtained in excellent yields and enantioselectivities (Scheme 2). Our observations indicated that electron-withdrawing groups and weakly electron-donating groups at the para-position of the aromatic moiety exerted only minor effects on enantioselectivity (4a–4i). However, strongly electron-donating groups, such as the methoxy group, significantly diminished the enantioselectivity (4j). It was observed that electron-rich substrate 2j undergoes racemic background reaction, which may constitute the primary factor contributing to the decrease in enantioselectivity (For the details, please refer to SI). Notably, the absolute configuration of product 4d was determined to be (R) by X-ray crystallography.55 Subsequently, we found that substituents such as –F, –OCF3, and –OMe located at the meta-position of the aromatic ring afforded products with good yields and enantioselectivities (4k–4m). Additionally, multi-substituted aromatic groups, including 2-naphthyl allenols, also provided favorable results (4n–4p). Due to the electron-donating nature of the 1,3-benzodioxole group, the corresponding product 4o was only obtained with 89% ee. Notably, the reaction conditions were compatible with heterocyclic systems such as 2-thiophene, affording product 4q in 99% yield and 84% ee. The observed sluggish reactivity in substrates 2g, 2k, and 2n can be attributed to the presence of electron-withdrawing groups, which diminish the electron density on the allene moiety. This reduction hinders the formation of the thiiranium ion intermediate, consequently leading to a decrease in the overall reaction rate. Furthermore, substrates bearing substituents on the cyclobutanol moieties were also evaluated. Since these substrates exist as mixtures of diastereoisomers, the corresponding products also exhibited diastereomeric ratios. Nevertheless, the major diastereomers were obtained in moderate to good yields and with high enantioselectivity (4r–4y). The absolute configuration of product 4t was determined to be (R, S) by X-ray crystallography.55 However, the cyclopropanol substrate was found to be less suitable, resulting in the formation of product 4z in 78% yield and 47% ee. Notably, sulfenylating reagents bearing meta- and para-trifluoromethyl substituents showed a marked decrease in enantioselectivity, highlighting the importance of steric effects in influencing the stereochemical outcome (4aa–4ab). Therefore, we examined a sulfenylating reagent containing 2,6-biisopropyl groups and obtained product 4ac in 85% yield and 98% ee. In contrast, using the most common sulfenylating reagent without any substituent on the phenyl ring resulted in product 4ad with only 43% yield and 67% ee, further supporting the role of steric hindrance in enhancing enantioselectivity. The relatively low yield can be attributed to the formation of a disulfenylated by-product 4ad′ within the system (For the details, please refer to SI). However, when alkyl substrate 2aa was employed with an ortho-trifluoromethyl-substituted sulfenylating reagent, the reaction failed to proceed, likely due to steric hindrance from the trifluoromethyl group. Through systematic optimization studies on alkyl substrates (For the details, please refer to SI, Table S2), we identified the 3,5-difluorosubstituted sulfenylating reagent as an effective sulfur source, affording a range of the corresponding alkyl-substituted products with moderate to good yields and high enantioselectivity (4ae–4ai).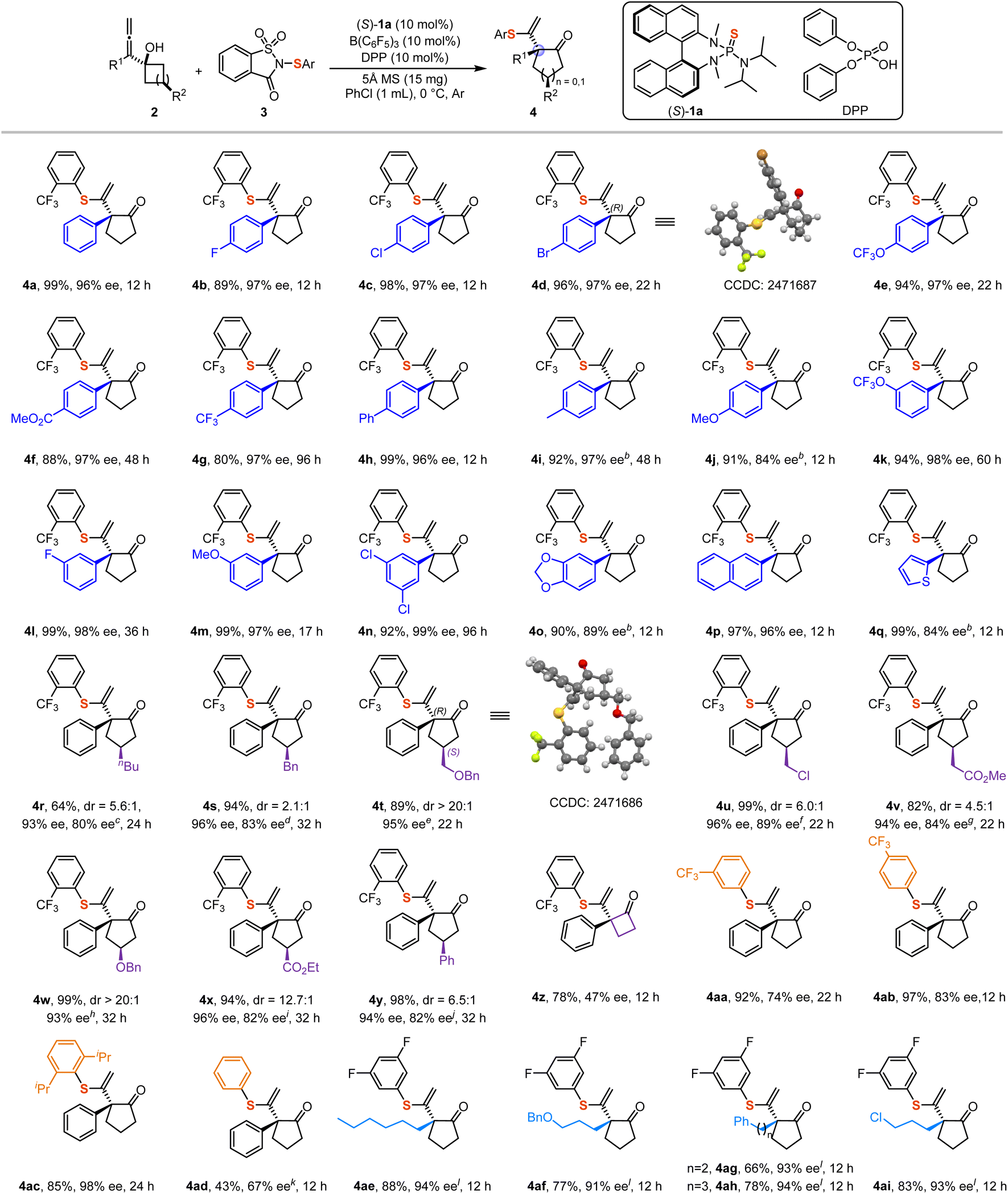 | ||
Scheme 2 Scope of the reaction.a a Reaction conditions: the reaction was performed with 2 (0.1 mmol), 3 (0.12 mmol), (S)-1a (0.01 mmol), B(C6F5)3 (0.01 mmol), DPP (0.01 mmol), and 5 Å MS (15 mg) in PhCl (1 mL) at 0 °C for 12–96 h under Ar. Isolated yields are shown. The ee values were determined by SFC or High-Performance Liquid Chromatography (HPLC). The cis![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :trans ratios of substrates 2r–2y were determined by 1H NMR analysis of the isolated compounds. The diastereomeric ratios of products 4r–4y were determined based on the isolated yields of isomers. bThe reaction was performed at −30 °C. c2r, cis:trans = 2.9:1. d2s, cis:trans = 1.7:1. e2t, cis, single isomer. f2u, cis:trans = 4.4:1. g2v, cis:trans = 2.9:1. h2w, cis:trans = 12.5:1. i2x, cis:trans = 12.5:1. j2y, cis:trans = 5.0:1. KA disulfenylated by-product 4ad’ was obtained in this reaction. Further details are provided in SI. l(R)-CPA (0.01 mmol) was used to instead of DPP. :trans ratios of substrates 2r–2y were determined by 1H NMR analysis of the isolated compounds. The diastereomeric ratios of products 4r–4y were determined based on the isolated yields of isomers. bThe reaction was performed at −30 °C. c2r, cis:trans = 2.9:1. d2s, cis:trans = 1.7:1. e2t, cis, single isomer. f2u, cis:trans = 4.4:1. g2v, cis:trans = 2.9:1. h2w, cis:trans = 12.5:1. i2x, cis:trans = 12.5:1. j2y, cis:trans = 5.0:1. KA disulfenylated by-product 4ad’ was obtained in this reaction. Further details are provided in SI. l(R)-CPA (0.01 mmol) was used to instead of DPP. | ||
Gram-scale reaction and synthetic applications
To demonstrate the synthetic applicability of this catalytic system, a gram-scale reaction employing substrate 2a was conducted under standard conditions, affording product 4a in 93% yield and 96% ee (Scheme 3a). Subsequently, structural modifications were carried out on the terminal alkenes moiety of 4a (Scheme 3b). Electrophilic sulfenylation, selenylation, and bromination reactions of the alkenes were successfully performed, yielding the corresponding trisubstituted alkene derivatives in moderate yields (compounds 5a–7a). However, a slight decrease in enantioselectivity was observed for compound 5a. We hypothesize that this reduction may be attributed to the preferential conversion of the (S)-configured enantiomer of 4a into a disulfenylated side product. The configuration of the trisubstituted alkene in the products was determined to be of the (E)-configuration, based on single-crystal X-ray structural analysis of the racemic compound 7a (For the details, please refer to SI).55 Furthermore, ozonolysis of the olefin efficiently afforded the 1,3-dicarbonyl thioester compound 8a in 95% yield with 96% ee. | ||
| Scheme 3 Gram-scale reaction and synthetic applications. Unless otherwise noted, isolated yields are shown, and the ee values were determined by SFC. For detailed conditions, please refer to SI. aReaction conditions: the reaction was performed with 2a (5.4 mmol), 3a (6.5 mmol), (S)-1a (0.54 mmol), B(C6F5)3 (0.54 mmol), DPP (0.54 mmol), and 5 Å MS (810 mg) in PhCl (54 mL) at 0 °C for 23 h under Ar. bFurther transformations of 4a. Reaction conditions: (i) 4a (0.1 mmol), 3e (0.12 mmol), B(C6F5)3 (0.1 mmol) and DPP (0.1 mmol) in DCM at rt for 1 h; (ii) 4a (0.1 mmol), 3i (0.12 mmol) and B(C6F5)3 (0.1 mmol) in DCM at rt for 1 h; (iii) 4a (0.1 mmol), NBS (0.105 mmol), B(C6F5)3 (0.1 mmol) and 5 Å MS (810 mg) in toluene at rt for 0.5 h; (iv) 4a (0.1 mmol) in DCM was bubbled with ozone at −78 °C for 1 minute. | ||
Mechanistic studies
Based on previous studies of the Lewis acid-assisted Brønsted acid (LBA) system54 and electrophilic sulfenylation reactions,6 a plausible intermediate cp-1 is proposed in Scheme 4. To verify the formation of cp-1, 31P NMR and 19F NMR analyses (Scheme 4a) as well as 1H NMR titration experiments (Scheme 4b) were conducted. When DPP and B(C6F5)3 were mixed in a 1:1 molar ratio in deuterated chloroform (CDCl3), new resonances appeared in both the 31P and 19F NMR spectra, indicating interactions between B(C6F5)3 and DPP and suggesting the possible formation of an LBA system. Upon addition of equimolar amounts of DPP, B(C6F5)3, and 3a in CDCl3, only minor chemical shifts were observed for the phosphorus atom of DPP and for the fluorine atoms at the ortho and para positions of B(C6F5)3, implying interactions among the three components. Building upon our previous findings,28,32 we hypothesize the presence of hydrogen bonding between 3a and the LBA system. To investigate this interaction, 1H NMR titration experiments were performed. With the concentration of B(C6F5)3 and DPP held constant at a 1:1 ratio, incremental addition of 3a resulted in a consistent downfield shift of the resonance signal corresponding to the proton of DPP (indicated by the red arrow), which indicates the presence of hydrogen bonding. Collectively, these results support the likely presence of cp-1 in the catalytic system.
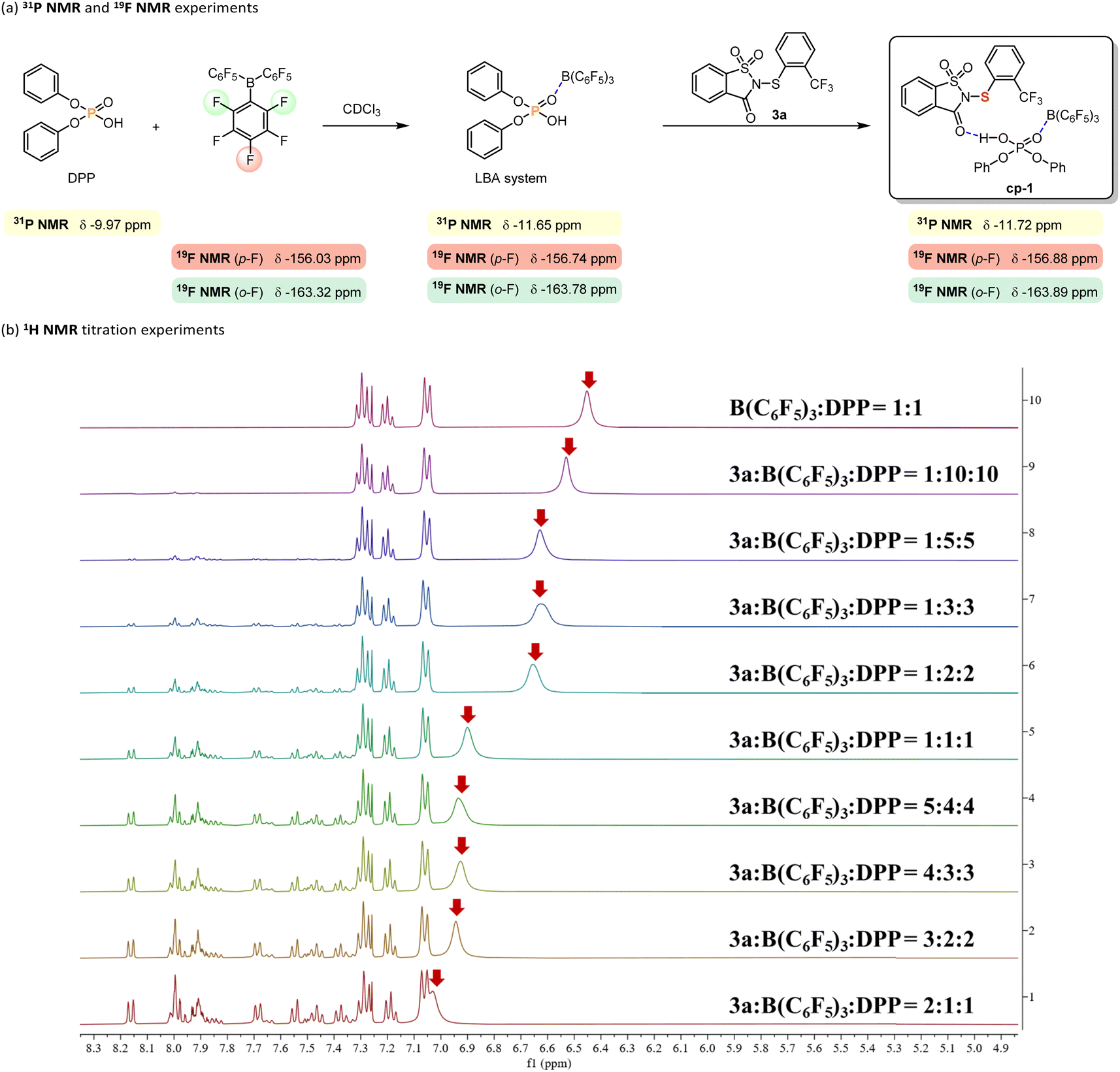 | ||
| Scheme 4 Mechanistic experiments. | ||
To elucidate the origin of the high enantioselectivity observed in the reaction, DFT calculations were performed on the reaction pathways at the ωB97X-D/def2-TZVPP-SMD//ωB97X-D/6-31G(d)-IEFPCM level of theory,56–59 using 3a as the model sulfenylating reagent (the computational details are provided in SI). As shown in Scheme 5, the chiral catalyst (S)-1a initially engages cp-1 through a chalcogen bond, forming the activated intermediate Int-1. Electrophilic sulfenylation subsequently occurs at the Re-face of substrate 2a, proceeding via transition state TS-1-major to yield the thiiranium ion intermediate Int-2, which exhibits high regioselectivity and enantioselectivity. It is followed by a rapid intramolecular ring-expansion rearrangement that affords the desired product 4a and concurrently regenerates the LBA system. The computed transition state structure TS-1-major, which leads to the major enantiomer of product 4a, is more stable than TS-1-minor by 5.5 kcal mol−1; this is consistent with the experimentally observed enantioselectivity. Independent Gradient Model based on Hirshfeld partition (IGMH) weak interaction analysis60–63 revealed the presence of a π⋯π interaction64 between the aryl ring of the sulfenylating reagent and that of substrate 2a in TS-1-major, along with C–H⋯π interactions between the aryl ring of LBA and the naphthalene ring of (S)-1a. In contrast, in TS-1-minor, this π⋯π interaction is disrupted due to steric repulsion between the ortho-trifluoromethyl substituent and (S)-1a. Additionally, the C–H⋯π interactions are absent owing to the unfavorable orientation of the hydroxy group in 2a relative to the naphthalene ring of (S)-1a.
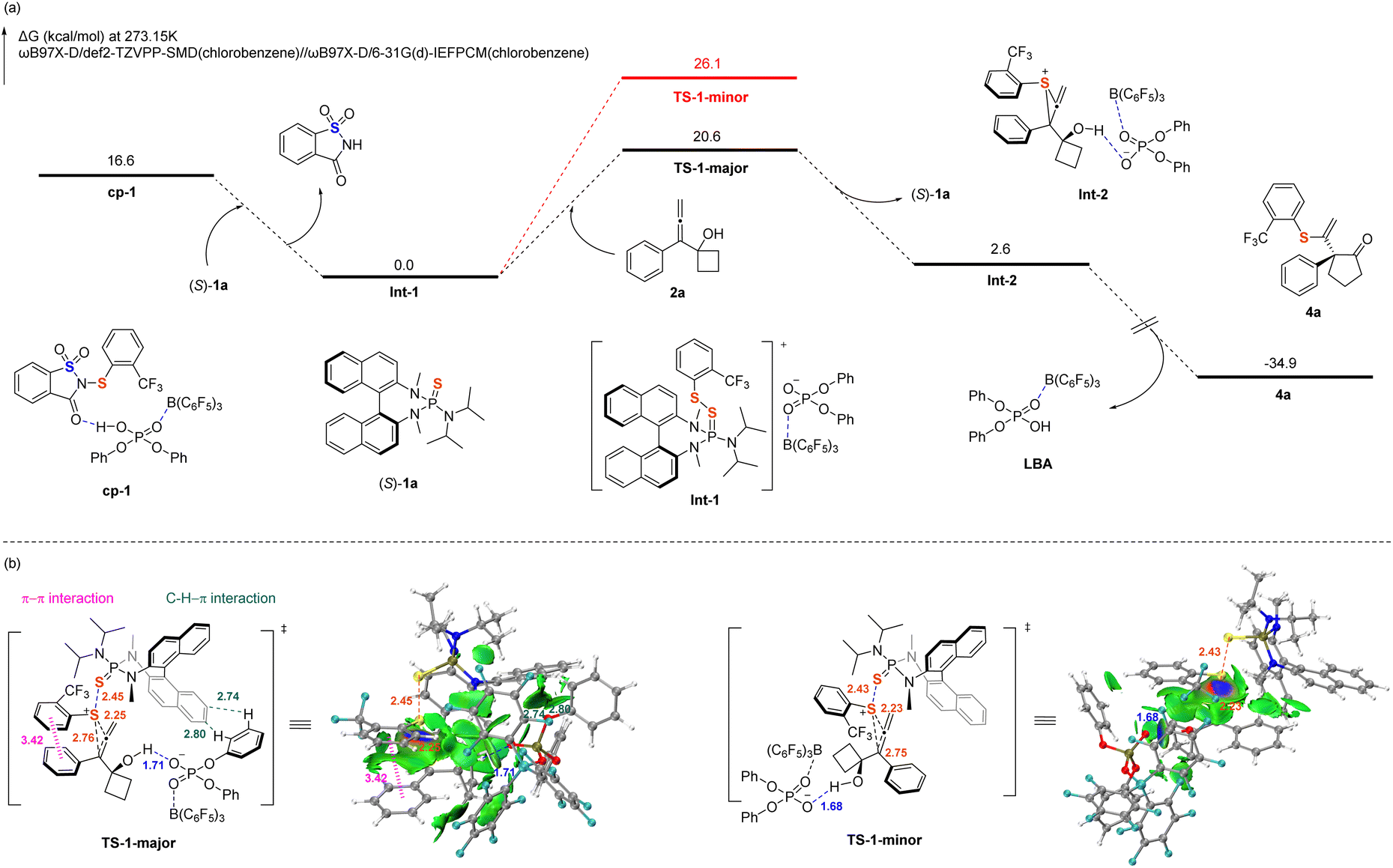 | ||
| Scheme 5 Computational studies on the reaction pathway and the origin of enantioselectivity. (a) Gibbs free energies profiles. (b) Independent Gradient Model based on Hirshfeld partition (IGMH) weak interaction analysis of two transition states. Bond lengths are given in Å. Pink dashed line: π–π interaction. Blue dashed line: hydrogen-bonding interaction. Green dashed line: C–H⋯π interaction. | ||
Conclusion
In conclusion, we have successfully developed a highly regio- and enantioselective electrophilic sulfenylation/semipinacol rearrangement of allenols, which is enabled for the first time through cooperative catalysis involving a chiral Lewis base, an achiral Lewis acid, and an achiral Brønsted acid. This method allows for the synthesis of a wide range of chiral organosulfur compounds containing all-carbon quaternary stereocenters, with high yields and excellent enantioselectivity. The sulfur-substituted terminal alkene moiety present in the products can be readily converted into trisubstituted alkenes and thioester derivatives. DFT studies indicate that steric repulsion between the ortho-trifluoromethyl substituent of the sulfenylating reagent and the catalyst plays a crucial role in achieving high enantioselectivity. Additionally, studies on other sulfenylation and selenylation reactions based on this cooperative catalytic system are currently underway in our laboratory.Author contributions
Z.-M. Chen directed the project. Z.-W. Wei completed the reaction optimization, substrate scope exploration, mechanistic studies and synthetic applications. L.-M. Yang, Y.-X. Huo, and Z.-L. Li helped to synthesize various substrates and analyze the data. Q.-Y. Cai and X.-Q. Gong achieved DFT calculations. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
CCDC 2471687 (4d), 2471686 (4t), 2473436 ((rac)-7a), 2473437 (8a) contain the supplementary crystallographic data for this paper.55a–dSupplementary information: Full experimental details, 1H, 13C, 19F NMR, SFC spectra, HPLC spectra, X-ray data and DFT calculations. See DOI: https://doi.org/10.1039/d5sc06981e.
Acknowledgements
Z.-M. Chen thank the National Natural Science Foundation of China (No. 22471158 and 22071149), the Natural Science Foundation of Shanghai (23ZR1428200), Autonomous Project of State Key Laboratory of Synergistic Chem-Bio Synthesis (sklscbs202562), and the Fundamental Research Funds for the Central Universities (YG2024QNB27) for financial support. We gratefully thank Prof. Yong-Qiang Tu (Shanghai Jiao Tong University) for helpful suggestions and comments on this manuscript.Notes and references
- K. D. Ashtekar, A. Jaganathan, B. Borhan and D. C. Whitehead, Enantioselective halofunctionalization of alkenes, Org. React., 2004, 105, 1 Search PubMed.
- S. E. Denmark, W. E. Kuester and M. T. Burk, Catalytic, Asymmetric Halofunctionalization of Alkenes—A Critical Perspective, Angew. Chem., Int. Ed., 2012, 51, 10938–10953 CrossRef CAS PubMed.
- H. Zhang, S. Lin and E. N. Jacobsen, Enantioselective Selenocyclization via Dynamic Kinetic Resolution of Seleniranium Ions by Hydrogen-Bond Donor Catalysts, J. Am. Chem. Soc., 2014, 136, 16485–16488 CrossRef CAS.
- E. M. Mumford, B. N. Hemric and S. E. Denmark, Catalytic, Enantioselective Syn-Oxyamination of Alkenes, J. Am. Chem. Soc., 2021, 143, 13408–13417 CrossRef CAS PubMed.
- N. Huang, J. Luo, L. Liao and X. Zhao, Catalytic Enantioselective Aminative Difunctionalization of Alkenes, J. Am. Chem. Soc., 2024, 146, 7029–7038 CrossRef CAS PubMed.
- A. Matviitsuk, J. L. Panger and S. E. Denmark, Catalytic, Enantioselective Sulfenofunctionalization of Alkenes: Development and Recent Advances, Angew. Chem., Int. Ed., 2020, 59, 19796–19819 CrossRef CAS PubMed.
- L. Liao and X. Zhao, Indane-Based Chiral Aryl Chalcogenide Catalysts: Development and Applications in Asymmetric Electrophilic Reactions, Acc. Chem. Res., 2022, 55, 2439–2453 CrossRef CAS.
- S. E. Denmark, D. J. P. Kornfilt and T. Vogler, Catalytic Asymmetric Thiofunctionalization of Unactivated Alkenes, J. Am. Chem. Soc., 2011, 133, 15308–15311 CrossRef CAS PubMed.
- S. E. Denmark, E. Hartmann, D. J. P. Kornfilt and H. Wang, Mechanistic, crystallographic, and computational studies on the catalytic, enantioselective sulfenofunctionalization of alkenes, Nat. Chem., 2014, 6, 1056–1064 CrossRef CAS.
- A. Roth and S. E. Denmark, Enantioselective, Lewis Base-Catalyzed, Intermolecular Sulfenoamination of Alkenes, J. Am. Chem. Soc., 2019, 141, 13767–13771 CrossRef CAS PubMed.
- Z. Tao, K. A. Robb, K. Zhao and S. E. Denmark, Enantioselective, Lewis Base-Catalyzed Sulfenocyclization of Polyenes, J. Am. Chem. Soc., 2018, 140, 3569–3573 CrossRef CAS PubMed.
- A. Matviitsuk, J. L. Panger and S. E. Denmark, Enantioselective Inter- and Intramolecular Sulfenofunctionalization of Unactivated Cyclic and (Z)-Alkenes, ACS Catal., 2022, 12, 7377–7385 CrossRef CAS PubMed.
- X. Liu, R. An, X. Zhang, J. Luo and X. Zhao, Enantioselective Trifluoromethylthiolating Lactonization Catalyzed by an Indane-Based Chiral Sulfide, Angew. Chem., Int. Ed., 2016, 55, 5846–5850 CrossRef CAS PubMed.
- J. Luo, Q. Cao, X. Cao and X. Zhao, Selenide-catalyzed enantioselective synthesis of trifluoromethylthiolated tetrahydronaphthalenes by merging desymmetrization and trifluoromethylthiolation, Nat. Commun., 2018, 9, 527 CrossRef.
- Y. Liang and X. Zhao, Enantioselective Construction of Chiral Sulfides via Catalytic Electrophilic Azidothiolation and Oxythiolation of N-Allyl Sulfonamides, ACS Catal., 2019, 9, 6896–6902 CrossRef CAS.
- Y.-Y. Xie, Z.-M. Chen, H.-Y. Luo, H. Shao, Y.-Q. Tu, X. Bao, R.-F. Cao, S.-Y. Zhang and J.-M. Tian, Lewis Base/Brønsted Acid Co-catalyzed Enantioselective Sulfenylation/Semipinacol Rearrangement of Di- and Trisubstituted Allylic Alcohols, Angew. Chem., Int. Ed., 2019, 58, 12491–12496 CrossRef CAS PubMed.
- X.-D. Liu, Y. Luo, X. Huo, H.-Y. Luo, R.-F. Cao and Z.-M. Chen, Chiral Sulfide/Phosphoric Acid Cocatalyzed Enantioselective Intermolecular Oxysulfenylation of Alkenes with Phenol and Alcohol O-Nucleophiles, CCS Chem., 2022, 4, 3342–3354 CrossRef CAS.
- X.-D. Liu, A.-H. Ye and Z.-M. Chen, Catalytic Enantioselective Intermolecular Three-Component Sulfenylative Difunctionalizations of 1,3-Dienes, ACS Catal., 2023, 13, 2715–2722 CrossRef CAS.
- H.-Y. Luo, Y.-Y. Xie, X.-F. Song, J.-W. Dong, D. Zhu and Z.-M. Chen, Lewis base-catalyzed asymmetric sulfenylation of alkenes: construction of sulfenylated lactones and application to the formal syntheses of (−)-nicotlactone B and (−)-galbacin, Chem. Commun., 2019, 55, 9367–9370 RSC.
- H.-Y. Luo, J.-W. Dong, Y.-Y. Xie, X.-F. Song, D. Zhu, T. Ding, Y. Liu and Z.-M. Chen, Lewis Base/Brønsted Acid Co-Catalyzed Asymmetric Thiolation of Alkenes with Acid-Controlled Divergent Regioselectivity, Chem. Eur J., 2019, 25, 15411–15418 CrossRef CAS PubMed.
- H. Guan, H. Wang, D. Huang and Y. Shi, Enantioselective oxysulfenylation and oxyselenenylation of olefins catalyzed by chiral Brønsted acids, Tetrahedron Lett., 2012, 68, 2728–2735 CrossRef CAS.
- L. Li, Z. Li, D. Huang, H. Wang and Y. Shi, Chiral phosphoric acid catalyzed enantioselective sulfamination of amino-alkenes, RSC Adv., 2013, 3, 4523–4525 RSC.
- J.-J. Wang, H. Yang, B.-B. Gou, L. Zhou and J. Chen, Enantioselective Organocatalytic Sulfenylation of β-Naphthols, J. Org. Chem., 2018, 83, 4730–4738 CrossRef CAS.
- Q. Jiang, H. Li and X. Zhao, Catalytic Electrophilic Thiocarbocyclization of Allenes, Org. Lett., 2021, 23, 8777–8782 CrossRef CAS PubMed.
- S. Ma, Electrophilic Addition and Cyclization Reactions of Allenes, Acc. Chem. Res., 2009, 42, 1679–1688 CrossRef CAS PubMed.
- J. Ye and S. Ma, Palladium-Catalyzed Cyclization Reactions of Allenes in the Presence of Unsaturated Carbon–Carbon Bonds, Acc. Chem. Res., 2014, 47, 989–1000 CrossRef CAS PubMed.
- G. Liu, X. Yang, P. Gu, M. Wang, X. Zhang and X.-Q. Dong, Challenging Task of Ni-Catalyzed Highly Regio-/Enantioselective Semihydrogenation of Racemic Tetrasubstituted Allenes via a Kinetic Resolution Process, J. Am. Chem. Soc., 2024, 146, 7419–7430 CrossRef CAS.
- H.-Y. Luo, Z.-H. Li, D. Zhu, Q. Yang, R.-F. Cao, T.-M. Ding and Z.-M. Chen, Chiral Selenide/Achiral Sulfonic Acid Cocatalyzed Atroposelective Sulfenylation of Biaryl Phenols via a Desymmetrization/Kinetic Resolution Sequence, J. Am. Chem. Soc., 2022, 144, 2943–2952 CrossRef CAS PubMed.
- R.-F. Cao and Z.-M. Chen, Catalytic asymmetric synthesis of sulfur-containing atropisomers by C-S bond formations, Sci. Chi. Chem., 2023, 66, 3331–3346 CrossRef CAS.
- D. Zhu, L. Yu, H.-Y. Luo, X.-S. Xue and Z.-M. Chen, Atroposelective Electrophilic Sulfenylation of N-Aryl Aminoquinone Derivatives Catalyzed by Chiral SPINOL-Derived Sulfide, Angew. Chem., Int. Ed., 2022, 61, e202211782 CrossRef CAS PubMed.
- D. Zhu, T. Mu, Z.-L. Li, H.-Y. Luo, R.-F. Cao, X.-S. Xue and Z.-M. Chen, Enantioselective Synthesis of Planar-Chiral Sulfur-Containing Cyclophanes by Chiral Sulfide Catalyzed Electrophilic Sulfenylation of Arenes, Angew. Chem., Int. Ed., 2024, 63, e202318625 CrossRef CAS PubMed.
- X.-Y. Zhang, D. Zhu, R.-F. Cao, Y.-X. Huo, T.-M. Ding and Z.-M. Chen, Enantioselective synthesis of inherently chiral sulfur-containing calix[4]arenes via chiral sulfide catalyzed desymmetrizing aromatic sulfenylation, Nat. Commun., 2024, 15, 9929 CrossRef CAS PubMed.
- R.-F. Cao, L. Yu, Y.-X. Huo, Y. Li, X.-S. Xue and Z.-M. Chen, Chiral Lewis Base Catalyzed Enantioselective Selenocyclization of 1,1-Disubstituted Alkenes: Asymmetric Synthesis of Selenium-Containing 4H-3,1-Benzoxazines, Org. Lett., 2022, 24, 4093–4098 CrossRef CAS PubMed.
- L.-L. Chen, R.-F. Cao, H. Ke and Z.-M. Chen, Lewis Base Catalyzed Selenofunctionalization of Alkynes with Acid-Controlled Divergent Chemoselectivity, Chin. J. Chem., 2024, 42, 1623–1629 CrossRef CAS.
- R.-F. Cao, R. Su, Z.-W. Wei, Z.-L. Li, D. Zhu, Y.-X. Huo, X.-S. Xue and Z.-M. Chen, Chiral sulfide and achiral sulfonic acid cocatalyzed enantioselective electrophilic tandem selenylation semipinacol rearrangement of allenols, Nat. Commun., 2025, 16, 2147 CrossRef CAS PubMed.
- R.-F. Cao, Z.-W. Wei, S.-K. Li, T.-M. Ding, H. Ke and Z.-M. Chen, Organocatalytic asymmetric electrophilic tandem selenylation semipinacol rearrangement of 1-(1-arylvinyl)cyclobutanols, Chem. Commun., 2025, 61, 8532–8535 RSC.
- X.-M. Zhang, B.-S. Li, S.-H. Wang, K. Zhang, F.-M. Zhang and Y.-Q. Tu, Recent development and applications of semipinacol rearrangement reactions, Chem. Sci., 2021, 12, 9262–9274 RSC.
- B. Wang and Y. Q. Tu, Stereoselective Construction of Quaternary Carbon Stereocenters via a Semipinacol Rearrangement Strategy, Acc. Chem. Res., 2011, 44, 1207–1222 CrossRef CAS PubMed.
- Q.-W. Zhang, C.-A. Fan, H.-J. Zhang, Y.-Q. Tu, Y.-M. Zhao, P. Gu and Z.-M. Chen, Brønsted Acid Catalyzed Enantioselective Semipinacol Rearrangement for the Synthesis of Chiral Spiroethers, Angew. Chem., Int. Ed., 2009, 48, 8572–8574 CrossRef CAS.
- J.-W. Dong, T. Ding, S.-Y. Zhang, Z.-M. Chen and Y.-Q. Tu, A Facile Approach to Oximes and Ethers by a Tandem NO+-Initiated Semipinacol Rearrangement and H-Elimination, Angew. Chem., Int. Ed., 2018, 57, 13192–13196 CrossRef CAS PubMed.
- Y.-P. Wang, K. Fang, Y.-Q. Tu, J.-J. Yin, Q. Zhao and T. Ke, An efficient approach to angular tricyclic molecular architecture via Nazarov-like cyclization and double ring-expansion cascade, Nat. Commun., 2022, 13, 2335 CrossRef CAS.
- T. Zheng, R. Chen, J. Huang, T. P. Gonçalves, K.-W. Huang and Y.-Y. Yeung, Cross-assembly confined bifunctional catalysis via non-covalent interactions for asymmetric halogenation, Chem, 2023, 9, 1255–1269 CAS.
- F. Romanov-Michailidis, L. Guénée and A. Alexakis, Enantioselective Organocatalytic Fluorination-Induced Wagner–Meerwein Rearrangement, Angew. Chem., Int. Ed., 2013, 52, 9266–9270 CrossRef CAS PubMed.
- M. A. S. Blackburn, C. C. Wagen, M. R. Bodrogean, P. M. Tadross, A. J. Bendelsmith, D. A. Kutateladze and E. N. Jacobsen, Dual-Hydrogen-Bond Donor and Brønsted Acid Cocatalysis Enables Highly Enantioselective Protio-Semipinacol Rearrangement Reactions, J. Am. Chem. Soc., 2023, 145, 15036–15042 CrossRef CAS PubMed.
- J. Lai and J. P. Reid, A Bulky Imidodiphosphorimidate Brønsted Acid Enables Highly Enantioselective Prins-semipinacol Rearrangements, ACS Catal., 2024, 14, 8518–8527 CrossRef CAS.
- Q. Yin and S.-L. You, Asymmetric Chlorination/Ring Expansion for the Synthesis of α-Quaternary Cycloalkanones, Org. Lett., 2014, 16, 1810–1813 CrossRef CAS PubMed.
- Q. Feng, Q. Wang and J. Zhu, Oxidative rearrangement of 1,1-disubstituted alkenes to ketones, Science, 2023, 379, 1363–1368 CrossRef CAS.
- Q. Feng, C.-X. Liu, Q. Wang and J. Zhu, Palladium-Based Dyotropic Rearrangement Enables A Triple Functionalization of Gem-Disubstituted Alkenes: An Unusual Fluorolactonization Reaction, Angew. Chem., Int. Ed., 2024, 63, e202316393 CrossRef CAS PubMed.
- H. Yamamoto and K. Futatsugi, “Designer Acids”: Combined Acid Catalysis for Asymmetric Synthesis, Angew. Chem., Int. Ed., 2005, 44, 1924–1942 CrossRef CAS PubMed.
- H. Ishibashi, K. Ishihara and H. Yamamoto, Chiral Proton Donor Reagents: Tin Tetrachloride—Coordinated Optically Active Binaphthol Derivatives, Chem. Rec., 2002, 2, 177–188 CrossRef CAS PubMed.
- O. Kanno, W. Kuriyama, Z. J. Wang and F. D. Toste, Regio- and Enantioselective Hydroamination of Dienes by Gold(I)/Menthol Cooperative Catalysis, Angew. Chem., Int. Ed., 2011, 50, 9919–9922 CrossRef CAS PubMed.
- C. H. Cheon, O. Kanno and F. D. Toste, Chiral Brønsted Acid from a Cationic Gold(I) Complex: Catalytic Enantioselective Protonation of Silyl Enol Ethers of Ketones, J. Am. Chem. Soc., 2011, 133, 13248–13251 CrossRef CAS PubMed.
- J. Y. See, H. Yang, Y. Zhao, M. W. Wong, Z. Ke and Y.-Y. Yeung, Desymmetrizing Enantio- and Diastereoselective Selenoetherification through Supramolecular Catalysis, ACS Catal., 2018, 8, 850–858 CrossRef CAS.
- M. Hatano, Y. Goto, A. Izumiseki, M. Akakura and K. Ishihara, Boron Tribromide-Assisted Chiral Phosphoric Acid Catalyst for a Highly Enantioselective Diels–Alder Reaction of 1,2-Dihydropyridines, J. Am. Chem. Soc., 2015, 137, 13472–13475 CrossRef CAS PubMed.
- (a) CCDC 2471687: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2nyzvg; (b) CCDC 2471686: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2nyztf; (c) CCDC 2473436: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2p0t8t; (d) CCDC 2473437: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2p0t9v.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comb. Chem., 2001, 22, 976–984 CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- T. Lu and F.-W. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- T. Lu, A comprehensive electron wavefunction analysis toolbox for chemists, Multiwfn, J. Chem. Phys., 2024, 161, 082503 CrossRef CAS.
- T. Lu and Q.-X. Chen, Visualization Analysis of Weak Interactions in Chemical Systems. In Comprehensive Computational Chemistry, Elsevier, Oxford, 2024, vol. 2, pp. 240–264 Search PubMed.
- T. Lu and Q.-X. Chen, Independent gradient model based on Hirshfeld partition: A new method for visual study of interactions in chemical systems, J. Comput. Chem., 2022, 43, 539 CrossRef CAS PubMed.
- S. E. Wheeler, Understanding substituent effects in noncovalent interactions involving aromatic rings, Acc. Chem. Res., 2013, 46, 1029–1038 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally: Z.-W. Wei and Q.-Y. Cai. |
| This journal is © The Royal Society of Chemistry 2026 |