
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot synthesis of core-crosslinked star polymers from poly(methyl methacrylate) with unsaturated chain ends and encapsulation of UV absorbing molecules
Yichao
Zheng
a,
Cheng
Wang
 a,
Hong Tho
Le
a,
Michelle Jia Qi
Lua
a,
Hiroshi
Niino
b,
Shunsuke
Chatani
b and
Atsushi
Goto
*a
a,
Hong Tho
Le
a,
Michelle Jia Qi
Lua
a,
Hiroshi
Niino
b,
Shunsuke
Chatani
b and
Atsushi
Goto
*a
aSchool of Chemistry, Chemical Engineering and Biotechnology, Nanyang Technological University, 62 Nanyang Drive, 637459, Singapore. E-mail: agoto@ntu.edu.sg
bOtake R&D Center, Mitsubishi Chemical Corporation, 20-1 Miyuki-cho, Otake, Hiroshima 739-0693, Japan
First published on 17th February 2026
Abstract
Core-crosslinked star polymers were synthesized via a “grafting-through” approach. PMMA–PA block copolymer arms were first prepared via addition–fragmentation chain transfer polymerizations of styrene or acrylate monomers using PMMA–Y, where PA is polystyrene or polyacrylate and PMMA–Y is poly(methyl methacrylate) with an unsaturated chain end. The PMMA–PA block copolymers were used as macroinitiators in the polymerizations of a crosslinkable monomer, generating core-crosslinked star polymers. Using PMMA–Y with different molecular weights and functional acrylates, various star polymers were obtained. A “one-pot” synthesis of star polymers was also achieved. The PMMA–PA macroinitiators were not purified but directly used in the star polymer synthesis, enabling facile synthesis of star polymers, which is industrially preferred. Star polymers with different core sizes and crosslinking densities were also obtained. As a demonstration, a PMMA–PTHFA core-crosslinked star polymer was synthesized and used for encapsulating a UV absorber (UVA) molecule, i.e., (2-(2-hydroxy-5-methylphenyl)benzotriazole) that is commonly used in coating and cosmetic formulations, where PTHFA is poly(tetrahydrofurfuryl acrylate). The UVA-containing star polymer was embedded in a PMMA matrix, and the obtained film showed UV-cut properties. Notably, the obtained film significantly suppressed leaching of the UVA due to the encapsulation of the UVA in the star polymer even at high temperature without impeding the visual appearance (transparency) of the film, which is attractive for possible industrial applications.
Introduction
Star-shaped polymers consist of a central core and multiple arm chains, finding various applications such as bioimaging, catalysis, energy storage, and agricultural applications.1–12 A variety of synthetic approaches has been developed to access star polymers, including arm-first, core-first, and grafting-through approaches. These approaches often utilize controlled polymerization techniques such as living radical polymerization (also known as reversible deactivation radical polymerization (RDRP))1,8,13–21 and ring-opening polymerization (ROP).22–24 Among RDRP techniques, reversible addition–fragmentation chain transfer (RAFT) polymerization and atom transfer radical polymerization (ATRP) are widely employed to prepare multi-arm star polymers with controlled molecular weights and arm numbers. Using controlled polymerizations, block copolymers are also fabricated in arm chains.1,7,12 Each segment in the block copolymer can bear functionality, encapsulating external molecules in the inner segment and dispersing the star polymer by the outer segment, for example. For the block copolymer star synthesis, multiple steps are usually required, and simplified processes are desired. For encapsulation, optical clarity of the resultant materials and suppression of additive leaching are also key requirements for practical applications.We previously synthesized core-crosslinked star polymers via a “grafting-through” approach using reversible complexation mediated polymerization (RCMP),25 which is an organocatalyzed living radical polymerization. A polymer-iodide (polymer–I) is used as a dormant species, and organic molecules such as tetrabutylammonium iodide (Bu4N+I−) are used as catalysts.26,27 RCMP is attractive for no use of special capping agents, odorous compounds, or heavy metal catalysts and its amenability to a wide range of monomers. We prepared homopolymer or block copolymer macroinitiators via RCMP and purified (step 1) and then carried out the chain extension of the macroinitiators using crosslinkable divinyl monomers, generating core-crosslinked star polymers (step 2).25
For the synthesis of block copolymer macroinitiators, we also utilized poly(methyl methacrylate) (PMMA) bearing an unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C group at the chain end (PMMA–Y with Y = CH2CH(CH2)COOCH3) (Fig. 1) as a precursor. We conducted polymerizations of monomer A (such as styrene and acrylates) in the presence of PMMA–Y, where addition fragmentation chain transfer (AFCT)28–30 operated between PMMA–Y and a polymer A radical (PA˙) to generate a PMMA radical (PMMA˙) and polymer A with an unsaturated chain end (PA–Y) (Scheme 1a). The generated PMMA˙ can propagate with monomer A to generate a PMMA–PA–I block copolymer macroinitiator. Additionally, the generated PA–Y may act as a macromonomer to give a branch chain in the macroinitiator (Scheme 1b). While PMMA–I generally lacks long-term stability upon storage, PMMA–Y does not bear iodide at the chain end and is stable upon storage, which is an advantage of the use of PMMA–Y as a starting compound. We previously prepared PMMA–PBA–I block copolymer macroinitiators from PMMA–Y31 and used the obtained macroinitiators and a crosslinkable monomer to synthesize core-crosslinked PMMA–PBA star polymers,25 where PBA is poly(butyl acrylate).
C group at the chain end (PMMA–Y with Y = CH2CH(CH2)COOCH3) (Fig. 1) as a precursor. We conducted polymerizations of monomer A (such as styrene and acrylates) in the presence of PMMA–Y, where addition fragmentation chain transfer (AFCT)28–30 operated between PMMA–Y and a polymer A radical (PA˙) to generate a PMMA radical (PMMA˙) and polymer A with an unsaturated chain end (PA–Y) (Scheme 1a). The generated PMMA˙ can propagate with monomer A to generate a PMMA–PA–I block copolymer macroinitiator. Additionally, the generated PA–Y may act as a macromonomer to give a branch chain in the macroinitiator (Scheme 1b). While PMMA–I generally lacks long-term stability upon storage, PMMA–Y does not bear iodide at the chain end and is stable upon storage, which is an advantage of the use of PMMA–Y as a starting compound. We previously prepared PMMA–PBA–I block copolymer macroinitiators from PMMA–Y31 and used the obtained macroinitiators and a crosslinkable monomer to synthesize core-crosslinked PMMA–PBA star polymers,25 where PBA is poly(butyl acrylate).
 | ||
| Fig. 1 Structures of the monomers, crosslinkable monomer, PMMA–Y, and UV absorber studied in this work. | ||
 | ||
| Scheme 1 (a) Addition–fragmentation chain transfer of PA˙ and PMMA–Y and (b) propagation of PMMA–PA˙ with the PA–Y macromonomer. | ||
In the present work, we further explored the synthesis of core-crosslinked PMMA–PBA star polymers from PMMA–Y (Scheme 2). We explored the use of PMMA–Y with different molecular weights (Mn = 3900 and 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) to obtain stars with different molecular weights in the PMMA segment, where Mn is the number-average molecular weight. We also newly explored a “one-pot” synthesis of stars (Scheme 2b) instead of the previously studied “two-pot’ synthesis, where the synthesis and purification of the macroinitiator (step 1) and the synthesis of the star (step 2) were separated. In the present work, PMMA–PBA–I macroinitiators were synthesized, and without purification, crosslinkable monomers were directly added to the same mixture, generating core-crosslinked star polymers in the same pot. This one-pot synthesis can avoid tedious purification of the macroinitiators, which is industrially preferred. Besides butyl acrylate (BA), we used other functional acrylates and styrene (Fig. 1) and prepared the relevant block copolymer macroinitiators from PMMA–Y. As an application, we further studied encapsulation of a guest molecule, i.e., 2-(2-hydroxy-5-methylphenyl)benzotriazole (Fig. 1), in a core-crosslinked PMMA–PTHFA star polymer, where PTHFA is poly(tetrahydrofurfuryl acrylate). 2-(2-Hydroxy-5-methylphenyl)benzotriazole is a commercially used UV absorber (UVA) that is added to coating formulations to enhance the durability of the coatings against UV exposure. We blended the UVA-encapsulated core-crosslinked PMMA–PTHFA star polymer with neat PMMA, formed a transparent PMMA thin film, and demonstrated an efficient UV absorption capability of the film and significantly suppressed leaching of the UVA from the film due to the encapsulation of the UVA in the star polymer. The use of stable PMMA–Y species, together with a one-pot star polymer synthesis, simplifies operation, which is industrially attractive. Effective encapsulation and suppression of additive leaching without compromising optical transparency would be useful for applications.
000) to obtain stars with different molecular weights in the PMMA segment, where Mn is the number-average molecular weight. We also newly explored a “one-pot” synthesis of stars (Scheme 2b) instead of the previously studied “two-pot’ synthesis, where the synthesis and purification of the macroinitiator (step 1) and the synthesis of the star (step 2) were separated. In the present work, PMMA–PBA–I macroinitiators were synthesized, and without purification, crosslinkable monomers were directly added to the same mixture, generating core-crosslinked star polymers in the same pot. This one-pot synthesis can avoid tedious purification of the macroinitiators, which is industrially preferred. Besides butyl acrylate (BA), we used other functional acrylates and styrene (Fig. 1) and prepared the relevant block copolymer macroinitiators from PMMA–Y. As an application, we further studied encapsulation of a guest molecule, i.e., 2-(2-hydroxy-5-methylphenyl)benzotriazole (Fig. 1), in a core-crosslinked PMMA–PTHFA star polymer, where PTHFA is poly(tetrahydrofurfuryl acrylate). 2-(2-Hydroxy-5-methylphenyl)benzotriazole is a commercially used UV absorber (UVA) that is added to coating formulations to enhance the durability of the coatings against UV exposure. We blended the UVA-encapsulated core-crosslinked PMMA–PTHFA star polymer with neat PMMA, formed a transparent PMMA thin film, and demonstrated an efficient UV absorption capability of the film and significantly suppressed leaching of the UVA from the film due to the encapsulation of the UVA in the star polymer. The use of stable PMMA–Y species, together with a one-pot star polymer synthesis, simplifies operation, which is industrially attractive. Effective encapsulation and suppression of additive leaching without compromising optical transparency would be useful for applications.
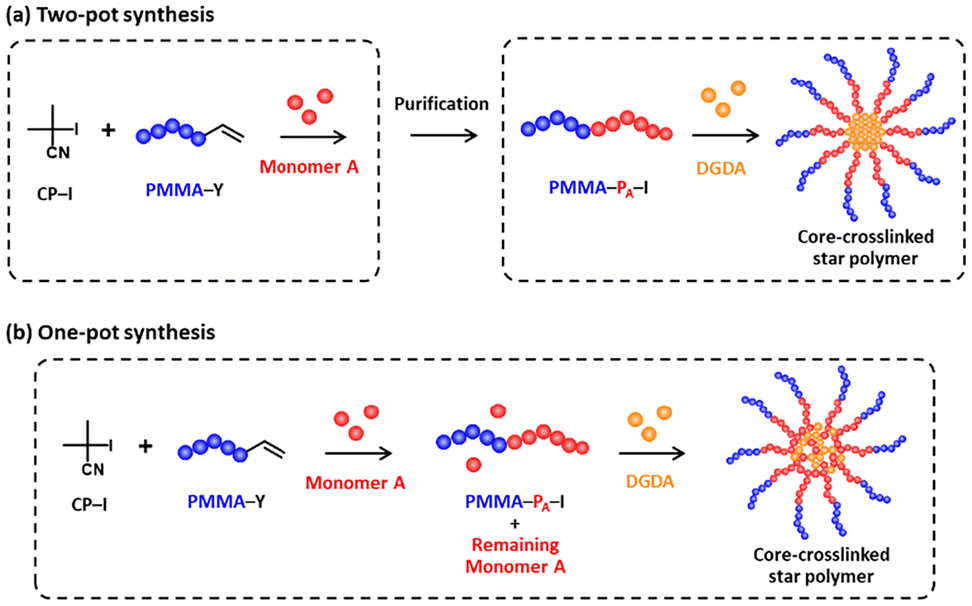 | ||
| Scheme 2 (a) Two-pot synthesis and (b) one-pot synthesis of core-crosslinked PMMA–PA star polymers from PMMA–Y. | ||
Results and discussion
Two-pot synthesis of core-crosslinked PMMA–PBA star polymers from PMMA–Y with two different molecular weights
Core-crosslinked PMMA–PBA star polymers were synthesized via a two-pot process (Scheme 2a). We first prepared a PMMA–PBA–I block copolymer from PMMA–Y with Mn = 3900. We heated a mixture of BA (200 equiv.), 2-iodo-2-methylpropionitrile (CP–I) (1 equiv.) as an initiating dormant species, PMMA–Y (Mn = 3900 and Đ = 1.67) (1 equiv.) as a macroinitiator precursor, and BNI (8 equiv.) as a catalyst at 110 °C (Table 1 (entry 1)), where Đ (= Mw/Mn) is dispersity and Mw is the weight-average molecular weight. After 24 h of polymerization, the monomer conversion reached 83% (as determined using 1H NMR), yielding a PMMA–PBA–I block copolymer with Mn = 22000 and Đ = 2.28 after purification (reprecipitation in a methanol/water mixed solvent (8/2 (v/v)) (a non-solvent)). The Mn and Mw values are PMMA-calibrated gel permeation chromatography (GPC) values (not absolute values). 1H NMR analysis (Fig. S1 in the SI) showed that, after the polymerization, the unsaturated chain end of PMMA–Y nearly completely disappeared, suggesting that the PMMA chain nearly fully extended to a PMMA–PBA block copolymer. The 1H NMR analysis also showed that the generated PBA–Y with an unsaturated chain end (PA–Y shown in Scheme 1) was also largely consumed as a macromonomer, suggesting that the PBA–Y was largely incorporated in the PMMA–PBA block copolymer.
| Entry | Monomer (M) | R–Ia | Azo or peroxide | M n of PMMA–Y | [M]0/[R–I]0/[PMMA–Y]0/[BNI]0/[azo or peroxide]0 (equiv.) | Solvent | T (°C) | t (h) | Monomer conv.b (%) |
M
nc |
Đc |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Alkyl iodide. b Determined with 1H NMR. c PMMA-calibrated GPC values of PMMA–PA–I block copolymers after purification (reprecipitation). The GPC eluent was THF for entries 1–4 and DMF for entries 5 and 6. d BuAc = butyl acetate and diglyme = diethylene glycol dimethyl ether. e PBZ = tert-butyl peroxybenzoate and V40 = 1,1′-azobis(cyclohexane-1-carbonitrile). | |||||||||||
| 1 | BA | CP–I | None | 3900 | 200/1/1/8/0 | Bulk | 110 | 24 | 83 | 22000 |
2.28 |
| 2 | BA | CP–I | None | 12000 |
200/1/1/8/0 | Bulk | 110 | 24 | 74 | 23000 |
2.19 |
| 3 | MEA | CP–I | None | 3900 | 200/1/1/4/0 | 20% BuAcd | 110 | 20 | 68 | 15000 |
1.61 |
| 4 | St | EPh–I | PBZe | 3900 | 200/1/1/0/0.75 | Bulk | 120 | 1 | 59 | 14000 |
1.42 |
| 5 | THFA | CP–I | V40e | 3900 | 200/1/1/8/0.6 | 10% BuAcd | 100 | 1 | 62 | 14000 |
2.04 |
| 6 | PEGA | CP–I | V40e | 3900 | 50/1/1/2/0.75 | 20% Diglymed | 100 | 4 | 71 | 13000 |
1.65 |
We used the purified PMMA–PBA–I as a macroinitiator. We conducted polymerization of a crosslinkable monomer, i.e., diethylene glycol diacrylate (DGDA) (Fig. 1) (40 equiv.), with the purified PMMA–PBA–I (1 equiv.) and BNI (8 equiv.) in butyl acetate (BuAc) (70 wt%) (solvent) at 110 °C (Table 2 (entry 1)). Fig. 2a shows the GPC chromatograms. The peak of the macroinitiator gradually decayed over time, while higher molecular weight species were generated and their molecular weight and peak intensity increased over time, suggesting that multiple macroinitiators were crosslinked through the polymerization of DGDA to form a core-crosslinked star polymer. After 24 h, the monomer (DGDA) conversion reached 47%, affording a star polymer with Mp = 510000, where Mp is the peak-top molecular weight. The conversion of the macroinitiator (R–I) to a star polymer was determined by the GPC peak resolution method (Fig. S2 in the SI). After 24 h, 43% of the macroinitiator was successfully converted to the star polymer. The remaining macroinitiator fraction (57%) would be dead polymer chains (without the iodide chain end) formed during both macroinitiator and star syntheses. The dead polymer formation during the macroinitiator synthesis is indicated by the high dispersity (Đ = 2.28) of the macroinitiator. The star polymer was easily and nearly completely separated from the remaining macroinitiator by reprecipitation in methanol, which is a good solvent for the macroinitiator and a non-solvent for the star polymer, as confirmed using GPC (Fig. S3 in the SI). The purified star polymer was analyzed using dynamic light scattering (DLS) in tetrahydrofuran (THF) (Fig. 2c, red solid line). A single peak was observed in the DLS intensity distribution curve, suggesting insignificant aggregation of the star (insignificant inter-core coupling). The DLS intensity-average diameter was 71 nm, which was larger than that (6 nm) of the PMMA–PBA–I macroinitiator (Fig. 2c, red dashed line), confirming the formation of a multi-arm star.
 | ||
| Fig. 2 GPC chromatograms (THF eluent) for star polymer synthesis using (a) purified PMMA–PBA–I (synthesized from PMMA–Y with Mn = 3900) (Table 2 (entry 1)) at 0 h (red line), 12 h (blue line), and 24 h (green line) and (b) purified PMMA–PBA–I (synthesized from PMMA–Y with Mn = 12000) (Table 2 (entry 2)) at 0 h (red line), 24 h (blue line), and 48 h (green line). (c) DLS size distribution curves (by intensity) of purified PMMA–PBA–I (synthesized from PMMA–Y with Mn = 3900) (red dashed line) (Table 1 (entry 1)) and purified PMMA–PBA star polymers synthesized from PMMA–Y with Mn = 3900 (red solid line) (24 h in Table 2 (entry 2)) and 12000 (blue solid line) (48 h in Table 2 (entry 2)). The DLS solvent was THF. | ||
| Entry | R–Ia (PMMA–PA−I) | M n of PMMA–Y | M n of R–I | [DGDA]0/[R–I]0/[BNI]0 (equiv.) | Solvent (wt%) | T (°C) | t (h) | DGDA conv.b (%) | R–I conv.c (%) | Star polymer before purification | Star polymer after purification | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
M
pd (star) |
M
pd (star) |
Intensity-average diameter in DLSe (nm) | Size distribution index in DLSf | ||||||||||
| a Macroinitiator (Table 2 (entries 1–6)) synthesized in Table 1 (entries 1–6), respectively. b Determined with 1H NMR. c Conversion of the macroinitiator (R–I) determined from the peak resolution of the GPC chromatogram. d PMMA-calibrated GPC peak-top molecular weight of the star polymer. The GPC eluent was THF for entries 1–4 and DMF for entries 5 and 6. e Determined from the DLS size distribution (by intensity) of the purified star. The DLS solvent was THF for entries 1–4 and DMF for entries 5 and 6. f The size distribution index in DLS is defined as [(standard deviation)/(mean particle size)]2. g BuAc = butyl acetate and diglyme = diethylene glycol dimethyl ether. | |||||||||||||
| 1 | PMMA–PBA–I | 3900 | 22000 |
40/1/8 | 70% BuAcg | 110 | 24 | 47 | 43 | 510000 |
530000 |
71 | 0.140 |
| 2 | PMMA–PBA–I | 12000 |
23000 |
40/1/8 | 70% BuAcg | 110 | 24 | 25 | 18 | 520000 |
NA | NA | NA |
| 48 | 35 | 25 | 650000 |
650000 |
88 | 0.215 | |||||||
| 3 | PMMA–PMEA–I | 3900 | 15000 |
40/1/4 | 65% BuAcg | 110 | 32 | 34 | 43 | 370000 |
370000 |
63 | 0.163 |
| 4 | PMMA–PSt–I | 3900 | 14000 |
80/1/8 | 60% Toluene | 110 | 20 | 34 | 35 | 450000 |
470000 |
57 | 0.181 |
| 5 | PMMA–PTHFA–I | 3900 | 14000 |
40/1/4 | 70% BuAcg | 110 | 28 | 62 | 36 | 280000 |
290000 |
82 | 0.170 |
| 6 | PMMA–PPEGA–I | 3900 | 13000 |
20/1/4 | 65% Diglymeg | 110 | 16 | 77 | 40 | 1100000 |
1200000 |
140 | 0.107 |
We also analyzed the purified star polymer using static light scattering (SLS) to determine its absolute weight-average molecular weight (Mw,ab (star)) (for this and other selected samples (Table 3)). We conducted the SLS analysis of this sample in THF and determined the Mw,ab (star) value to be 1300000 (Table 3 (entry 1)). The number of arms per star polymer (narm) was determined according to narm = (weight fractions of the arms in the star polymer) × Mw,ab (star)/Mw (arm), where Mw (arm) is the Mw value (= 50000) of the PMMA–PBA–I macroinitiator (arm polymer) determined using PMMA-calibrated GPC. We used the PMMA-calibrated GPC value (Mw) as a rough estimate of the molecular weight of arm polymers, because the Mw value determined using SLS may not be very accurate for the relatively low-molecular weight linear polymers. The narm value was estimated to be 18. We also analyzed the purified star polymer using a transmission electron microscope (TEM) (Fig. S4 in the SI). We observed individual stars with core–shell structures (appearing as individual particles with darker cores surrounded with lighter shells in the TEM images), suggesting insignificant inter-core coupling.
| Entry | Star polymer |
M
wa (arm) (GPC) |
M
w,abb (star) (SLS) |
dn/dcc (mL g−1) |
n
armd |
|---|---|---|---|---|---|
| a PMMA-calibrated GPC Mw (not Mn) value of the purified arm (macroinitiator) for entries 1 and 2 and the unpurified arm (macroinitiator) for entries 3–5. The GPC eluent was THF for entries 1–4 and DMF for entry 5. b Molecular weight of the purified star determined using static light scattering (SLS). The SLS solvent was THF for entries 1–4 and DMF for entry 5. c The determination of dn/dc is described in the SI. d Number of arms per star polymer = (weight fraction of arm polymer) × Mw,ab (star)/Mw (arm). | |||||
| 1 | PMMA–PBA star (Table 2, entry 1) | 50000 |
1300000 |
0.0577 | 18 |
| 2 | PMMA–PBA star (Table 2, entry 2 (48 h)) | 50000 |
2400000 |
0.0403 | 31 |
| 3 | PMMA–PBA star (Table 4, entry 1-2) (one-pot synthesis) | 45000 |
1100000 |
0.0514 | 14 |
| 4 | PMMA–PBA star (Table 4, entry 2-2) (one-pot synthesis) | 38000 |
2300000 |
0.0558 | 26 |
| 5 | PMMA–PTHFA star (Table 5, entry 1-2) (one-pot synthesis) | 28000 |
7100000 |
0.0589 | 80 |
Using PMMA–Y with a higher molecular weight (Mn = 12000 and Đ = 1.67), we also prepared a PMMA–PBA–I block copolymer (Mn = 23000 and Đ = 2.19 after purification) (Table 1 (entry 2)). We then carried out a polymerization of DGDA (40 equiv.) with the purified PMMA–PBA–I (Table 2 (entry 2)). After 24 h, the monomer (DGDA) conversion reached 25%, and 18% of the macroinitiator was converted to a star polymer (Fig. 2b and Fig. S5 in the SI). The macroinitiator conversions (18% for 24 h) were lower than that (43% for 24 h) using the PMMA–PBA–I macroinitiator (Mn = 22000) prepared from PMMA–Y with a lower molecular weight (Mn = 3900) (Table 2 (entry 1)). Since the Mn values of the two PMMA–PBA–I macroinitiators (23000 and 22000) were similar, the steric hindrance in the star formation would be similar, and the exact reason for the lower (or slower) macroinitiator conversion is unclear at this moment. After 48 h, the monomer and macroinitiator conversion increased to 35% and 25%, respectively, generating a star polymer with Mp = 650000 (Fig. 2b and Fig. S6 in the SI). After reprecipitation, the DLS intensity-average diameter of the purified star polymer (Fig. S7 in the SI) was 88 nm (Fig. 2c, blue line). The Mw,ab (star) and narm values were 2400000 and 31, respectively (Table 3 (entry 2)). Thus, we obtained core-crosslinked PMMA–PBA star polymers from PMMA–Y precursors with two different molecular weights.
Two-pot synthesis of star polymers using functional acrylates and styrene from PMMA–Y (Mn = 3900)
To expand the monomer scope, we used four other monomers to synthesize PMMA–PA–I macroinitiators from PMMA–Y (Mn = 3900). We studied three hydrophobic monomers, i.e., 2-methoxyethyl acrylate (MEA), styrene (St), and tetrahydrofurfuryl acrylate (THFA) (Table 1 (entries 3–5)), and an amphiphilic monomer, i.e., poly(ethylene glycol) methyl ether acrylate (PEGA) (average Mn = 300) (Table 1 (entry 6)). A radical initiator (tert-butyl peroxybenzoate (PBZ) for St and 1,1′-azobis(cyclohexane-1-carbonitrile) (V40) for THFA and PEGA) was added to increase the polymerization rate. Peroxide and azo initiators are often used to increase the polymerization rate, as previously reported for RCMP26 and other living radical polymerizations.32 The four PMMA–PA–I block copolymer macroinitiators (Mn = 13000–15000 after purification) were generated at moderate to high monomer conversions (59–71%) (Table 1 (entries 3–6)). The macroinitiators were subsequently used in polymerizations of DGDA, yielding core-crosslinked star polymers with Mp = 280000–1100000 (Table 2 (entries 3–6), Fig. 3, and Fig. S8–S15 in the SI). The macroinitiator conversions (35–43%) (Table 2 (entries 3–6)) were similar to that (43%) observed in the PMMA–PBA star synthesis with PMMA–Y (Mn = 3900) (Table 2 (entry 1)). The purified stars showed single DLS peaks (Fig. S16 in the SI). Thus, we successfully obtained core-crosslinked PMMA–PA star polymers in all cases, suggesting a wide monomer scope in the present star polymer synthesis.
 | ||
| Fig. 3 GPC chromatograms for star polymer synthesis from (a) purified PMMA–PMEA–I (Table 2 (entry 3)), (b) purified PMMA–PSt–I (Table 2 (entry 4)), (c) purified PMMA–PTHFA–I (Table 2 (entry 5)), and (d) purified PMMA–PPEGA–I (Table 2 (entry 6)) at t = 0 (red line) and 16–32 h (blue line). The GPC eluent was THF for (a) and (b) and DMF for (c) and (d). | ||
One-pot synthesis of PMMA–PBA star polymers from PMMA–Y (Mn = 3900)
In the PMMA–PBA star polymer synthesis described above, we synthesized the PMMA–PBA–I macroinitiator in one pot, purified the macroinitiator, and synthesized the star polymer in another pot using the purified macroinitiator, which is a “two-pot” synthesis. We also conducted a “one-pot” synthesis (Scheme 2b). Namely, we first synthesized a PMMA–PBA–I macroinitiator, and in the same reaction pot, we added the crosslinkable DGDA monomer, generating a star polymer in the same pot. A certain amount of BA remained after the macroinitiator synthesis, which was copolymerized with DGDA, generating a diluted crosslinked core. The amount of the remaining BA depends on the polymerization time (monomer conversion) in the macroinitiator synthesis and determines how much the crosslinked core is diluted.We heated a mixture of BA (200 equiv.), CP–I (1 equiv.), PMMA–Y (Mn = 3900) (1 equiv.), and BNI (8 equiv.) at 110 °C for 24 h (Table 4 (entry 1-1)). The monomer conversion was 83%, and a PMMA–PBA–I block copolymer with Mn = 21000 and Đ = 2.15 was generated. To the same reaction mixture, we added DGDA (40 equiv.), BNI (4 equiv.), and BuAc (70 wt% of BuAc and 30 wt% of the initial BA and added DGDA in total), and the polymerization continued at 110 °C (Table 4 (entry 1-2) and Fig. 4a, red line). Because of the presence of the unreacted BA (17%) upon the addition of DGDA, the core of the generated star contained both BA (non-crosslinkable monomer) and DGDA (crosslinkable monomer) units and had somewhat diluted crosslinking. After another 24 h (48 h in total) of polymerization, the peak in the GPC chromatogram only slightly shifted to a higher molecular weight region (Fig. 4a, blue line), and the Mn value increased from 21000 (at the addition of DGDA) to 27000 (after another 24 h). As compared to the two-pot system (Table 2 (entry 1)), where 43% of the macroinitiator was converted to star polymers after 24 h of polymerization, the crosslinking in the one-pot system was delayed due to the presence of non-crosslinkable BA monomer units. After another 48 h (72 h in total) of polymerization, the DGDA conversion reached 50%, and the BA conversion reached 91% (increased from 83% upon the addition of DGDA), meaning that 8% of the initial BA was incorporated in the star core. 35% of the macroinitiator (Fig. S17 in the SI) was converted to a star polymer with Mp = 470000. The macroinitiator conversion (35%) in the one-pot system was slightly lower than but still similar to that (43%) in the two-pot system (Table 2 (entry 1)). The DLS intensity-average diameter (Fig. 4c, red line) of the purified star (Fig. S18 in the SI) was 58 nm. The Mw,ab (star) and narm values were 1100000 and 14, respectively (Table 3 (entry 3)), showing the generation of a similar size star in the one-pot system to that in the two-pot system (Mw,ab (star) = 1300000 and narm = 18) (Table 3 (entry 1)).
 | ||
| Fig. 4 GPC chromatograms (THF eluent) for one-pot synthesis of PMMA–PBA star polymers via (a) polymerization of BA for 24 h (red line) and subsequent polymerization of DGDA for +24 h (blue line) and +48 h (green line) (Table 4 (entry 1-2)) and (b) polymerization of BA for 16 h (red line) and subsequent polymerization of DGDA for +24 h (blue line) and +48 h (green line) (Table 4 (entry 2-2)). (c) DLS size distribution curves (by intensity) of purified PMMA–PBA star polymers synthesized in the 24 + 48 h (red line) and 16 + 48 h (blue line) systems. The DLS solvent was THF. | ||
| Entry | Ma | R–Ib | [M]0/[R–I]0/[PMMA–Y]0/[BNI]0 (equiv.) | Solvent | T (°C) | t (h) | BA conv.c (%) | DGDA conv.c (%) | R–I conv.d (%) |
M
ne |
Đ | Star polymer before purification | Star polymer after purification | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
M
pf (star) |
M
pf (star) |
Intensity-average diameter in DLSg (nm) | Size distribution index in DLSh | ||||||||||||
| a M = monomer. b CP–I for entries 1-1 and 2-1 and the macroinitiator (obtained in entries 1-1 and 2-1) for entries 1-2 and 2-2. c Determined with 1H NMR. d Conversion of the macroinitiator (R–I) determined from the peak resolution of the GPC chromatogram. e PMMA-calibrated GPC (THF eluent) value before purification. f PMMA-calibrated GPC (THF eluent) peak-top molecular weight of the star polymer. g Determined from the DLS size distribution (by intensity) of the purified star. The DLS solvent was THF. h The size distribution index in DLS is defined as [(standard deviation)/(mean particle size)]2. i BuAc = butyl acetate (70 wt% of BuAc and 30 wt% of the initial BA and added DGDA in total). | |||||||||||||||
| 1-1 | BA | CP–I | 200/1/1/8 | Bulk | 110 | 24 | 83 | NA | NA | 21000 |
2.15 | NA | NA | NA | NA |
| 1-2 | DGDA | (PMMA–PBA–I in situ generated) | +40/0/0/4 | BuAci | 110 | +24 | 89 | 31 | NA | 27000 |
2.49 | NA | NA | NA | NA |
| +48 | 91 | 50 | 35 | NA | NA | 470000 |
480000 |
58 | 0.171 | ||||||
| 2-1 | BA | CP–I | 200/1/1/8 | Bulk | 110 | 16 | 68 | NA | NA | 18000 |
2.11 | NA | NA | NA | NA |
| 2-2 | DGDA | (PMMA–PBA–I in situ generated) | +40/0/0/4 | BuAci | 110 | +24 | 83 | 42 | NA | 25000 |
2.61 | NA | NA | NA | NA |
| +48 | 91 | 62 | 40 | NA | NA | 580000 |
580000 |
91 | 0.266 | ||||||
Using the same molar ratio of BA (200 equiv.), CP–I (1 equiv.), PMMA–Y (1 equiv.), and BNI (8 equiv.) at the same temperature of 110 °C, we conducted the polymerization for a shorter time of 16 h instead of 24 h (Table 4 (entry 2-1)), reaching a lower monomer (BA) conversion (68%) and yielding a shorter PMMA–PBA–I block copolymer with Mn = 18000 and Đ = 2.11. To the same reaction mixture, we added DGDA (40 equiv.) and BuAc (70 wt% of BuAc and 30 wt% of the initial BA and added DGDA in total) and continued the polymerization at 110 °C (Table 4 (entry 2-2) and Fig. 4b). After another 48 h (64 h in total) of polymerization, the DGDA conversion reached 62%, and the BA conversion reached 91% (increased from 68% upon the addition of DGDA), meaning that 23% of the initial BA was incorporated in the star core. 40% of the macroinitiator (Fig. S19 in the SI) was converted to a star polymer with Mp = 580000. The DLS intensity-average diameter (Fig. 4c, blue line) of the purified star (Fig. S20 in the SI) was 91 nm. Both the Mp (580000) and DLS intensity-average diameter (91 nm) of the star in the 16 + 48 h system were larger than those (470000 and 58 nm) in the 24 + 48 h system. Because of the presence of more unreacted BA (32%) upon the addition of DGDA in the 16 + 48 h system, the star formation would be further delayed, which would afford a star polymer with a larger core with more diluted crosslinking. The Mw,ab (star) and narm values were 2300000 and 26, respectively (Table 3 (entry 4)), which are larger than those in the 24 + 48 h system (Mw,ab (star) = 1100000 and narm = 14) (Table 3 (entry 3)). Due to the shorter PMMA–PBA macroinitiator generated at 16 h of the BA polymerization, there was less steric hindrance among the arm chains during the star formation, which would allow a larger number of arms to be involved in the star. The Mw,ab (star) (2300000) and narm (26) values in this one-pot system (Table 3 (entry 4)) are also larger than those (130000 and 18, respectively) in the corresponding two-pot system (Table 3 (entry 1)) because of the larger core with diluted crosslinking and the lower molecular weight (less steric hindrance) of the macroinitiator in this one-pot system.
In the one-pot system, to avoid macro-gelation, the molecular weight of the block copolymer macroinitiator needs to be high enough to fully shield the crosslinked core and avoid star–star coupling. Thus, the monomer (BA) conversion in the macroinitiator synthesis needs to be high enough to yield a sufficiently high molecular-weight macroinitiator, as attained in the present studies (Table 3 (entries 3 and 4)) where macro-gelation did not occur.
Encapsulation of a UV absorber (UVA) in a PMMA–PTHFA star polymer and a compatibility test with the PMMA matrix
For most of the commercially available UV absorbing films, organic small UVA molecules are embedded in polymer matrices. However, UVA molecules can diffuse and leach out from the polymer matrices, lowering the durability.33 The diffusion of UVA molecules may be suppressed by encapsulating UVA molecules in the present core-crosslinked star polymers. Based on this motivation, we prepared a PMMA–PTHFA star polymer from PMMA–Y via the one-pot synthesis, encapsulated UVA molecules, and embedded the obtained UVA-encapsulated star polymer in a PMMA matrix. The PMMA block of the star arm is responsible for the compatibility with the PMMA matrix, while the PTHFA block is responsible for associating with UVA molecules. In addition, the refractive index of THFA (1.460) is close to that of MMA (1.414). Therefore, blending PTHFA with PMMA will not significantly lower the transparency of the film.We heated a mixture of THFA (200 equiv.), CP–I (1 equiv.), PMMA–Y (Mn = 3900) (1 equiv.), BNI (8 equiv.), V40 (0.6 equiv.), and BuAc (10 wt% of BuAc and 90 wt% of THFA) at 100 °C for 1 h (Table 5 (entry 1-1)). The monomer conversion reached 65%, and a PMMA–PTHFA–I block copolymer with Mn = 14000 and Đ = 1.99 was generated. To the same reaction mixture, we added DGDA (40 equiv.) and BuAc (70 wt% of BuAc and 30 wt% of the initial THFA and added DGDA in total) and continued the polymerization at 100 °C (Table 5 (entry 1-2) and Fig. 5a). After another 2 h (3 h in total), the DGDA conversion reached 83%, and the THFA conversion reached 92% (increased from 65% upon the addition of DGDA). Both monomers reached high conversion, which is desirable for industrial applications. 30% of the macroinitiator (Fig. S21 in the SI) was converted to a star polymer with Mp = 1200000. We purified the star polymer by reprecipitation in methanol. The purified star polymer (Fig. S22 in the SI) had a DLS intensity-average diameter of 145 nm (Fig. 5b). The Mw,ab (star) and narm values were 7100000 and 80, respectively (Table 3 (entry 5)). The narm value (80) of the PMMA–PFMA star was larger than those (14–26) of the PMMA–PBA stars synthesized in the one-pot system (Table 3 (entries 3 and 4)), which would be partly because of the larger amount of the remaining monomer after the macroinitiator synthesis (hence larger core size) and the lower molecular weight of the macroinitiator (hence less steric hindrance) in the PMMA–PFMA system (65% FMA conversion vs. 68–83% BA conversions, and Mw = 28000 (PMMA–PFMA macroinitiator) vs. 38000–45000 (PMMA–PBA macroinitiators)), although the exact reason for the observed relatively large difference in the narm values between the PMMA–PFMA star and PMMA–PBA star systems is unclear at this moment.
 | ||
| Fig. 5 (a) GPC chromatograms (DMF eluent) for one-pot synthesis of PMMA–PTHFA star polymers via polymerization of THFA for 1 h (red line) and subsequent polymerization of DGDA for +2 h (blue line) (Table 5 (entries 1-1 and 1-2)). (b) DLS size distribution curves (by intensity) of the isolated PMMA–PTHFA star polymers. The DLS solvent was DMF. (c) UV-vis transmittance values of the films (Table 6 (entries 1–3)). (d) Photos and schematic illustrations of the films (Table 6 (entries 1–3)). | ||
| Entry | Ma | R–Ib | [M]0/[R–I]0/[PMMA–Y]0/[BNI]0/[V40]0 (equiv.) | Solvent | T (°C) | t (h) | THFA conv.c (%) | DGDA conv.c (%) | R–I conv.d (%) |
M
ne |
Đ | Star polymer after purification | Star polymer after purification | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
M
pf (star) |
M
pf (star) |
Intensity-average diameter in DLSg (nm) | Size distribution index in DLSh | ||||||||||||
| a M = monomer. b CP–I for entry 1-1 and macroinitiator (obtained in entry 1-1) for entry 1-2. c Determined with 1H NMR. d Conversion of the macroinitiator (R–I) determined from the peak resolution of the GPC chromatogram. e PMMA-calibrated GPC (DMF eluent) value before purification. f PMMA-calibrated (DMF eluent) peak-top molecular weight of the star. g Determined from the DLS size distribution (by intensity) of the purified star. The DLS solvent was DMF. h The size distribution index in DLS is defined as [(standard deviation)/(mean particle size)]2. i BuAc = butyl acetate (10 wt% of BuAc and 90 wt% of THFA for entry 1-1 and 70 wt% of BuAc and 30 wt% of the initial THFA and added DGDA in total for entry 1-2). | |||||||||||||||
| 1-1 | THFA | CP–I | 200/1/1/8/0.6 | BuAci | 100 | 1 | 65 | NA | NA | 14000 |
1.99 | NA | NA | NA | NA |
| 1-2 | DGDA | (PMMA–PTHFA–I in situ generated) | +40/0/0/0/0 | BuAci | 100 | +2 | 92 | 83 | 30 | NA | NA | 1200000 |
1200000 |
145 | 0.120 |
We encapsulated 2-(2-hydroxy-5-methylphenyl)benzotriazole as a UVA in the purified PMMA–PTHFA star polymer. The UVA was encapsulated possibly via polar–polar interaction between the polar UVA and the polar THF moiety in the star polymer (FMA unit) and hydrogen bonding between the OH group in the UVA and the THF (ether) moiety in the star polymer. We first dissolved the star polymer in THF at 50 °C and added the UVA until the UVA was saturated in the solution to maximize the encapsulation. The solution was stirred for 2 h to allow the diffusion of the UVA into the star polymer. To this solution, hexane, which is a non-solvent for the star polymer, was added dropwise to slowly precipitate the UVA-encapsulated star polymer. We compared the weight of the original star polymer with that of the UVA-encapsulated star polymer and calculated the encapsulation efficiency to be 40 wt% (0.40 g of the UVA in 1.00 g of the UVA-encapsulated star polymer) (Table S1 (entry 1) in the SI). (The encapsulation efficiency was determined gravimetrically: encapsulation efficiency (%) = [(mass of star polymer after encapsulation) − (mass of star polymer before encapsulation)]/(mass of star polymer after encapsulation).) We also used the purified PMMA–PTHFA star polymer with Mp = 280000 prepared via the two-pot synthesis (Table 2 (entry 5)) to encapsulate the UVA (Table S1 (entry 2) in the SI). The encapsulation efficiency (13 wt%) was much lower than that (40 wt%) of the star polymer prepared via the one-pot synthesis, probably because the one-pot synthesis afforded a larger star polymer with a larger core size due to the diluted crosslinking, which would increase the encapsulation capacity.
We prepared three stock solutions of commercially available PMMA (Mn = 51000 and Đ = 1.88), the non-encapsulated UVA, and the UVA-encapsulated star polymer (prepared via the one-pot synthesis) with 40 wt% encapsulation efficiency, which were dissolved in BuAc. We combined them, cast the obtained solutions in moulds, dried the cast solutions, and prepared approximately 100 µm thick films. We obtained a film of pure PMMA (100 wt%) (Table 6 (entry 1)), a film of the non-encapsulated UVA (7 wt%) embedded in PMMA (93 wt%) (Table 6 (entry 2)), and a film of the UVA-encapsulated star (17 wt%) embedded in PMMA (83 wt%), which corresponds to 7 wt% of the UVA and 93 wt% of the polymer in total (Table 6 (entry 3)). The pure PMMA film (Table 6 (entry 1) and Fig. 5c, red line) had more than 90% transmittance above 300 nm. By dosing the non-encapsulated UVA (Table 6 (entry 2) and Fig. 5c, blue line), the transmittance reduced to 0% below 380 nm (UV region), while it was maintained at a high level (>71%) above 420 nm (visible light region). The film with the encapsulated UVA (Table 6 (entry 3)) also showed 0% transmittance below 380 nm and >79% transmittance above 420 nm. Thus, we obtained efficient UV-absorbing films containing the non-encapsulated (Table 6 (entry 2)) and encapsulated (Table 6 (entry 3)) UVA with no transmittance below 380 nm and high transmittance above 420 nm. Visually, all three thin films had similar transparency (Fig. 5d). To study the leaching of the UVA, we heated the two films containing the non-encapsulated UVA (Table 6 (entry 2)) and the encapsulated UVA (Table 6 (entry 3)) at 150 °C, which is above the glass transition temperature (110 °C) of the PMMA matrix, for 3 h. After cooling to room temperature, the films were subsequently immersed in methanol for 30 min to dissolve the leached UVA in methanol. From the absorption of the UVA in the methanol solution, we determined the amount of the leached UVA (Fig. S23 and Table S2 in the SI) to be 2.5% for the non-encapsulated UVA (2.5% of the originally embedded amount of the UVA) (Table 6 (entry 2)) and only 0.04% for the encapsulated UVA (only 0.04% of the originally embedded amount of the UVA) (Table 6 (entry 3)). We repeated the heating–cooling–immersion process three times. The total amount of the leached UVA in the three cycles was 8.4% for the non-encapsulated UVA and only 0.19% for the encapsulated UVA, showing consistently low UVA leaching under heating (high thermal stability of the encapsulation). Therefore, by taking advantage of the encapsulated UVA in the star polymer, we successfully prepared the UV-absorbing film with suppressed leaching but without impeding the visual appearance (transparency) of the film.
| Entry | Non-encapsulated UVA (g) | UVA-encapsulated star (40 wt% UVA) (g) | PMMA matrixa (g) | UVA/polymerb (wt%) |
|---|---|---|---|---|
|
a PMMA with Mn = 51000 and Đ = 1.88.
b Weight ratio of the UVA (the sum of the encapsulated UVA and the non-encapsulated UVA) and the polymer (the sum of the star polymer and the PMMA matrix).
|
||||
| 1 | 0 | 0 | 0.30 | 0/100 |
| 2 | 0.022 | 0 | 0.30 | 7/93 |
| 3 | 0 | 0.060 | 0.30 | 7/93 |
Conclusions
Core-crosslinked star polymers were successfully synthesized using PMMA–PA block copolymer macroinitiators prepared from PMMA–Y with different molecular weights (Mn = 3900 and 12000). The second block (PA) monomers encompassed styrene, BA, and functional acrylates. The use of stable PMMA–Y (instead of PMMA–I) eases operation and is industrially preferred. A “one-pot” synthesis of a PMMA–PA star polymer was also achieved, which can avoid tedious purification of the macroinitiators and is further industrially preferred. Star polymers with different core sizes and crosslinking densities were obtained by varying the amount of the remaining second block (PA) monomer in the star formation. As an application, a PMMA–PTHFA core-crosslinked star polymer was synthesized in “one-pot” and used for encapsulating a UVA. The UVA-containing star polymer was embedded in a PMMA matrix, and the obtained film showed good UV-cut properties with high transmittance in the visible region. The obtained film significantly suppressed leaching of the UVA due to the encapsulation of the UVA in the star polymer even at high temperature without impeding the visual appearance (transparency) of the film.
Conflicts of interest
There are no conflicts to declare.Data availability
Supplementary information (SI): experimental section, 1H NMR data, GPC chromatograms, preparation of UVA encapsulated star polymers, and determination of the amount of UVA from films. See DOI: https://doi.org/10.1039/d5py01227a.Acknowledgements
This work was partly supported by a Nanyang Technological University start-up grant (SUG) for A. Goto.References
- S. Harrisson, R. Whitfield, A. Anastasaki and K. Matyjaszewski, Nat. Rev. Methods Primers, 2025, 5, 2 CrossRef CAS.
- M. Kalina, B. Nouri and K. Almdal, RSC Adv., 2025, 15, 27700–27722 RSC.
- T. Haino and N. Nitta, ChemPlusChem, 2024, 89, e202400014 CrossRef CAS PubMed.
- G. Shao, A. Li, Y. Liu, B. Yuan and W. Zhang, Macromolecules, 2024, 57, 830–846 CrossRef CAS.
- S. Nakagawa and N. Yoshie, Polym. Chem., 2022, 13, 2074–2107 RSC.
- M. Ohira, T. Katashima, M. Naito, D. Aoki, Y. Yoshikawa, H. Iwase, S. Takata, K. Miyata, U. Chung, T. Sakai, M. Shibayama and X. Li, Adv. Mater., 2022, 34, 2108818 CrossRef CAS PubMed.
- Y. Zhang, L. Fu, S. J. Jeon, J. Yan, J. P. Giraldo, K. Matyjaszewski, R. D. Tilton and G. V. Lowry, ACS Nano, 2022, 16, 4467–4478 CrossRef CAS PubMed.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- A. B. Cook and S. Perrier, Adv. Funct. Mater., 2020, 30, 1901001 CrossRef CAS.
- N. J. Oldenhuis, K. P. Qin, S. Wang, H. Z. Ye, E. A. Alt, A. P. Willard, T. Van Voorhis, S. L. Craig and J. A. Johnson, Angew. Chem., Int. Ed., 2020, 59, 2784–2792 CrossRef CAS PubMed.
- H. Sun, C. P. Kabb, M. B. Sims and B. S. Sumerlin, Prog. Polym. Sci., 2019, 89, 61–75 CrossRef CAS.
- J. M. Ren, T. G. McKenzie, Q. Fu, E. H. Wong, J. Xu, Z. An, S. Shanmugam, T. P. Davis, C. Boyer and G. G. Qiao, Chem. Rev., 2016, 116, 6743–6836 CrossRef CAS PubMed.
- A. Hochół, I. Zaborniak, M. Bednarenko, A. Pellis, K. Matyjaszewski and P. Chmielarz, ACS Polym. Au, 2025, 5, 827–852 CrossRef PubMed.
- S. Zhu, W. Kong, S. Lian, A. Shen, S. P. Armes and Z. An, Nat. Synth., 2025, 4, 15–30 CrossRef CAS.
- R. W. Hughes, M. E. Lott, R. A. Olson S and B. S. Sumerlin, Prog. Polym. Sci., 2024, 156, 101871 CrossRef CAS.
- C. Boyer, M. Kamigaito, K. Satoh and G. Moad, Prog. Polym. Sci., 2023, 138, 101648 CrossRef CAS.
- Y. Lee, C. Boyer and M. S. Kwon, Chem. Soc. Rev., 2023, 52, 3035–3097 RSC.
- G. K. K. Clothier, T. R. Guimarães, S. W. Thompson, J. Y. Rho, S. Perrier, G. Moad and P. B. Zetterlund, Chem. Soc. Rev., 2023, 52, 3438–3469 RSC.
- Y. Zhou, J. Li, T. Wang, Y. Wu and Z. Luo, Prog. Polym. Sci., 2022, 130, 101555 CrossRef CAS.
- H. Dau, G. R. Jones, E. Tsogtgerel, D. Nguyen, A. Keyes, Y. Liu, H. Rauf, E. Ordonez, V. Puchelle, H. B. Alhan, C. Zhao and E. Harth, Chem. Rev., 2022, 122, 14471–14553 CrossRef CAS PubMed.
- J. Wan, B. Fan and S. H. Thang, Chem. Sci., 2022, 13, 4192–4224 Search PubMed.
- M. A. Ntrivala, A. C. Pitsavas, K. Lazaridou, Z. Baziakou, D. Karavasili, M. Paradimitriou, C. Ntagkopoulou, E. Balla and D. N. Bikiaris, Eur. Polym. J., 2025, 234, 114033 CrossRef CAS.
- C. Shi, E. C. Quinn, W. T. Diment and E. Y.-X. Chen, Chem. Rev., 2024, 124, 4393–4478 CrossRef CAS PubMed.
- Z. Li, Y. Shen and Z. Li, Macromolecules, 2024, 57, 1919–1940 CrossRef CAS.
- Y. Zheng, J. Sarkar, H. Niino, S. Chatani, S. Y. Hsu and A. Goto, Polym. Chem., 2021, 12, 4043–4051 RSC.
- A. Goto, A. Ohtsuki, H. Ohfuji, M. Tanishima and H. Kaji, J. Am. Chem. Soc., 2013, 135, 11131–11139 Search PubMed.
- C.-G. Wang, A. M. L. Chong, H. M. Pan, J. Sarkar, X. T. Tay and A. Goto, Polym. Chem., 2020, 11, 5559–5571 Search PubMed.
- J. P. Heuts and N. M. Smeets, Polym. Chem., 2011, 2, 2407–2423 RSC.
- B. Yamada, P. B. Zetterlund and E. Sato, Prog. Polym. Sci., 2006, 31, 835–877 Search PubMed.
- J. Krstina, G. Moad, E. Rizzardo, C. L. Winzor, C. T. Berge and M. Fryd, Macromolecules, 1995, 28, 5381–5385 CrossRef CAS.
- J. J. Chang, L. Xiao, C. G. Wang, H. Niino, S. Chatani and A. Goto, Polym. Chem., 2018, 9, 4848–4855 RSC.
- A. Goto and T. Fukuda, Prog. Polym. Sci., 2004, 29, 329–385 CrossRef CAS.
- C. Queant, V. Landry, P. Blanchet and D. Schorr, J. Coat. Technol. Res., 2017, 14, 1411–1422 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |