
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemoselective reduction of α-keto aldehydes by rongalite in water: a metal- and hydride-free approach
Madhu
Inapanuri
,
Hari Prasad
Kokatla
 *,
Rambabu
Kandukuri
and
Neetika
Singh
*,
Rambabu
Kandukuri
and
Neetika
Singh
Department of Chemistry, National Institute of Technology Warangal, Warangal, Telangana-506004, India. E-mail: harikokatla@nitw.ac.in
First published on 19th November 2025
Abstract
A chemoselective reduction of α-keto aldehydes using rongalite in water has been developed for the efficient synthesis of α-hydroxymethyl ketones. This transformation exploits the unique reactivity of rongalite to achieve a mild, metal- and hydride-free reduction that proceeds smoothly in the presence of various functional groups. The protocol employs inexpensive rongalite (ca. $0.03 per gram), operates in an aqueous medium under open-air conditions, and requires no inert atmosphere. The reaction is readily scalable to gram quantities and exhibits excellent chemoselectivity and broad substrate scope, tolerating electron-donating, electron-withdrawing, and halogen substituents, as well as hetero aromatics, delivering the desired products in high yields within short reaction times.
Introduction
α-Hydroxymethyl ketones represent an important class of compounds that are widely distributed in natural products and bioactive molecules.1 These acyloins are key structural subunits of farnesyltransferase inhibitors, kurasoins A and B, the enzyme urease,2 antibiotics such as olivomycin A and chromomycin A3,3 inhibitors of amyloid-b (Ab) protein production4 and inducers of apoptosis in oral tumors (Fig. 1).5 | ||
| Fig. 1 Some bioactive molecules that contain α-hydroxymethyl ketones and their derivatives. | ||
Beyond their intrinsic biological significance, these motifs are also recognized as versatile synthetic intermediates.6 They are commonly employed in the preparation of structurally complex molecules and play key roles as chiral auxiliaries, ligands, and catalysts in asymmetric synthesis.7 Given their broad utility, the development of efficient and practical methods for the synthesis of α-hydroxymethyl ketones continues to be a central focus in both pharmaceutical and synthetic chemistry.
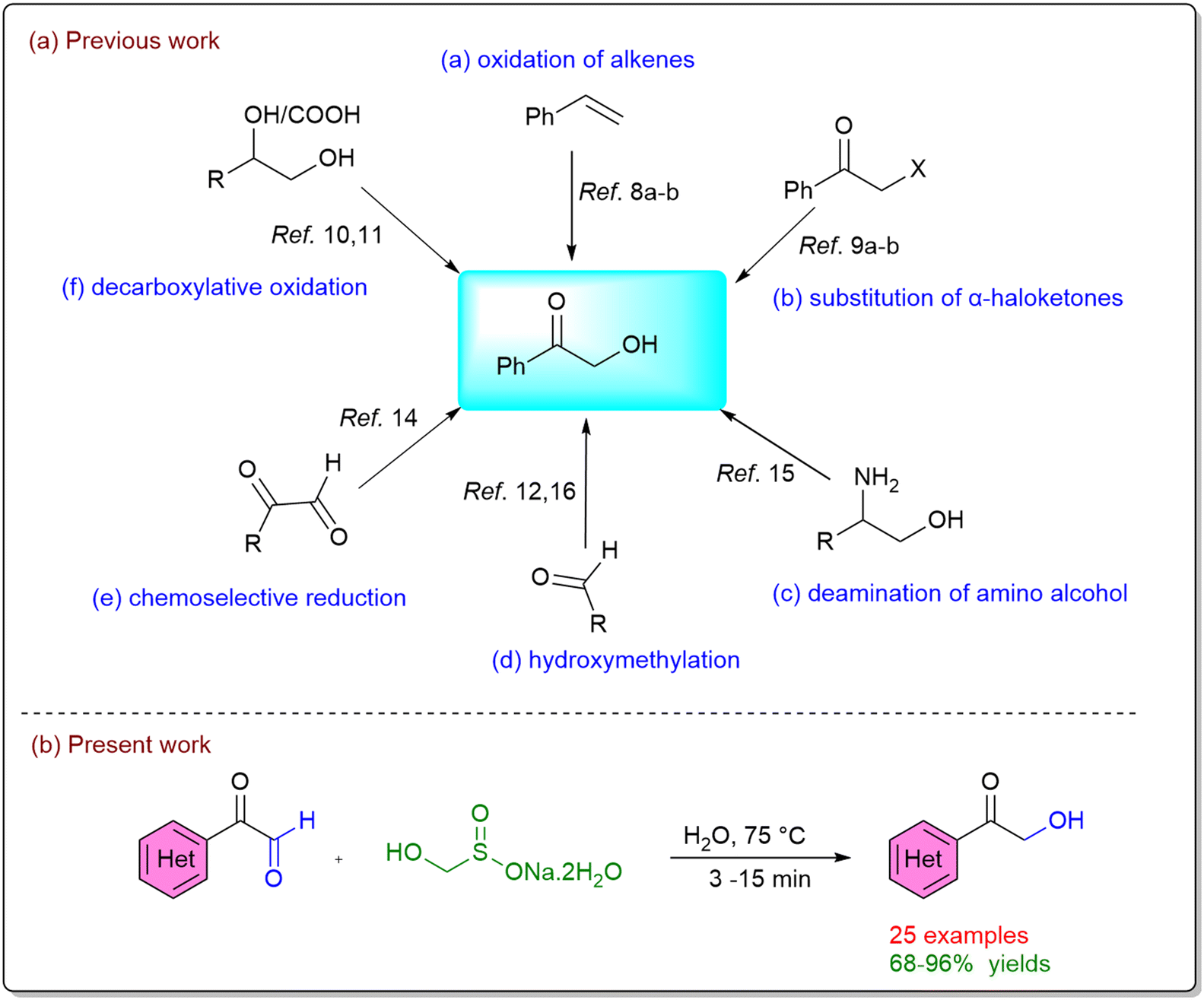
A number of conventional chemical strategies have been explored for their preparation, including iodine-mediated activation and oxidation of alkenes,8 substitution of α-haloketones,9 oxidation of diols,10 decarboxylative oxidation of carboxylic acids,11 and N-heterocyclic carbene (NHC)-catalyzed hydroxymethylation of aldehydes.12 While these approaches are effective, they often suffer from practical drawbacks such as the requirement for toxic or expensive reagents, harsh reaction conditions, and limited product yields. In this context, biocatalytic routes have emerged as attractive alternatives.13 Reported examples include enzymatic reduction of diketones, oxidation of diols,14 transamination of amino alcohols,15 and thiamine diphosphate (ThDP)-dependent hydroxymethylation of aldehydes.16 Although these methods are environmentally benign, their application can be restricted by enzyme availability, cost, pH, and substrate scope. In recent years, the chemoselective reduction of 1,2-dicarbonyl compounds to α-hydroxy carbonyls has gained prominence as a particularly powerful synthetic transformation.17 A central challenge in this area lies in achieving the selective reduction of aldehydes in the presence of ketones, a transformation that is highly relevant to the synthesis of natural products and architecturally complex molecules. The intrinsic higher electrophilicity and lower steric demand of aldehydes compared to ketones usually enable preferential reduction, and this difference has been exploited in the design of selective reducing agents.18 For example, attenuated hydride donors derived from boron or aluminum hydrides have been developed to modulate hydride transfer reactivity, providing improved selectivity.19 Similarly, transition-metal-catalyzed hydrosilylation has been extensively studied.20 Despite these advances, existing methods still suffer from important limitations, such as the requirement for elevated temperatures, extended reaction times, and the formation of undesirable by-products (e.g., silanols in hydrosilylation). These drawbacks significantly restrict their applicability in large-scale or sustainable synthesis. To overcome these challenges, we sought to develop a novel, transition-metal- and hydride-free approach for the chemoselective reduction of 1,2-dicarbonyl compounds (Scheme 1a).
 | ||
| Scheme 1 Chemoselective reduction of α-keto aldehydes. | ||
In this regard, we turned our attention to rongalite (sodium hydroxymethanesulfinate dihydrate, SHM),21 an inexpensive (∼0.03 USD per g) and readily available industrial product that has attracted interest as a green alternative to toxic formaldehyde. Owing to its multifaceted reactivity, rongalite can act as a super electron donor,22 a C1 unit source,23 and a precursor to the sulfoxylate dianion (SO22−).24 In continuation of our efforts to explore the synthetic utility of rongalite as both an electron source and a reagent for hydride-free reductions,25 we herein report a chemoselective, transition-metal- and hydride-free protocol for the preparation of α-hydroxy ketones from the corresponding α-keto aldehydes (Scheme 1b).
Results and discussion
To evaluate our hypothesis for the synthesis of 2-hydroxy-1-phenylethan-1-one 3a, we initially carried out the reaction using phenyl glyoxal 1a (1 mmol) and rongalite 2 in acetonitrile at room temperature for 24 hours. Under these conditions, no transformation was observed, and the starting material remained unchanged (Table 1, entry 1). Interestingly, increasing the temperature to 70 °C initiated the reaction and afforded the desired product 3a, albeit in low yield (15%) after 12 hours (Table 1, entry 2). The structure of compound 3a was confirmed by 1H and 13C NMR spectroscopy, along with HRMS (see the SI).|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Rongalite (equiv.) | Temp. (°C) | Time (h) | Yield (%) |
a All the reactions were conducted on a 1 mmol scale of 1a (1 mmol), 2 (2 mmol) and solvent (3 mL), unless otherwise mentioned.
b Yield where reported is of isolated and purified products.
c Solvent (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 v/v).
d n.r = no reaction. :2 v/v).
d n.r = no reaction.
|
|||||
| 1 | CH3CN | 2.0 | Rt | 24 | n.rd |
| 2 | CH3CN | 2.0 | 70 | 12 | 15 |
| 3 | THF | 2.0 | 70 | 10 | 24 |
| 4 | DMF | 2.0 | 70 | 2 | 31 |
| 5 | DMSO | 2.0 | 70 | 2 | 35 |
| 6 | MeOH | 2.0 | 70 | 15 min | 56 |
| 7 | EtOH | 2.0 | 70 | 10 min | 65 |
| 8 | EtOH + H2Oc | 2.0 | 70 | 3 min | 68 |
| 9 | H2O | 2.0 | 70 | 3 min | 82 |
| 10 | H2O | 2.0 | 75 | 3 min | 85 |
| 11 | H2O | 2.0 | 80 | 3 min | 80 |
| 12 | H2O | 1.5 | 75 | 10 min | 78 |
| 13 | H2O | 2.5 | 75 | 5 min | 70 |

Encouraged by these initial results, we turned our attention to optimizing the reaction conditions to improve yield, and a solvent screening was conducted. Switching the solvent to THF slightly increased the yield to 24% (Table 1, entry 3). Further screening with polar aprotic solvents such as DMF and DMSO resulted in moderately improved yields of 31% and 35%, respectively (Table 1, entries 4 and 5). Notably, the use of polar protic solvents such as methanol and ethanol significantly enhanced the yield to 56% and 65%, respectively, and also reduced the reaction time (Table 1, entries 6 and 7).
These findings can be attributed to the equilibrium nature of glyoxals and the better solubility of rongalite in polar protic media. Based on this insight, we evaluated an EtOH:H2O (8:2) mixture, which led to a further increase in yield to 68% (Table 1, entry 8). Remarkably, performing the reaction in water alone resulted in a rapid transformation within 3 minutes, delivering the product 3a in a high yield (Table 1, entry 9). It is worth mentioning that phenyl glyoxal 1a was sparingly soluble in water at room temperature. However, upon heating to 75 °C, the substrates dissolved progressively, and the reaction proceeded smoothly with rongalite to afford the desired product 3a without any co-solvent. Given the eco-friendly, sustainable, and economical nature of water as a solvent, it was selected as the optimal medium for further optimization studies. Subsequent temperature screening revealed that the reaction proceeds most efficiently at 75 °C (Table 1, entry 10). However, variations in temperature and increasing the equivalents of rongalite did not lead to further improvements in the yield of 3a (Table 1, entries 11–13). With the optimized reaction conditions in hand (Table 1, entry 10), we turned our attention to evaluating the substrate scope of the reaction using a series of structurally and electronically diverse α-keto aldehydes to assess the generality and functional group tolerance of the developed protocol (Table 2). We first examined aromatic α-keto aldehydes bearing electron-donating groups (EDGs) on the phenyl ring. Substrates featuring alkyl (methyl) and alkoxy (methoxy) substituents at the para-, meta-, and ortho- positions smoothly underwent the reaction, affording the corresponding α-hydroxymethyl ketones (3b–3e) in excellent yields ranging from 81 to 90%. Similarly, substrates bearing hydroxy groups or extended electron-donating motifs such as benzo[d][1,3]dioxole (3f–3i) were also well-tolerated, delivering the desired products in 77–96% yield. These results suggest that the presence of EDGs enhances the nucleophilicity of the α-keto carbonyl system, thereby facilitating the transformation. Encouragingly, the reaction also proved efficient for substrates containing halogen substituents. A wide range of halogenated aryl glyoxals, including fluoro, chloro, bromo and iodo (3j–3p) derivatives, were successfully converted into the corresponding products in good yields of 68–85%, regardless of the substitution pattern (ortho, meta, or para).
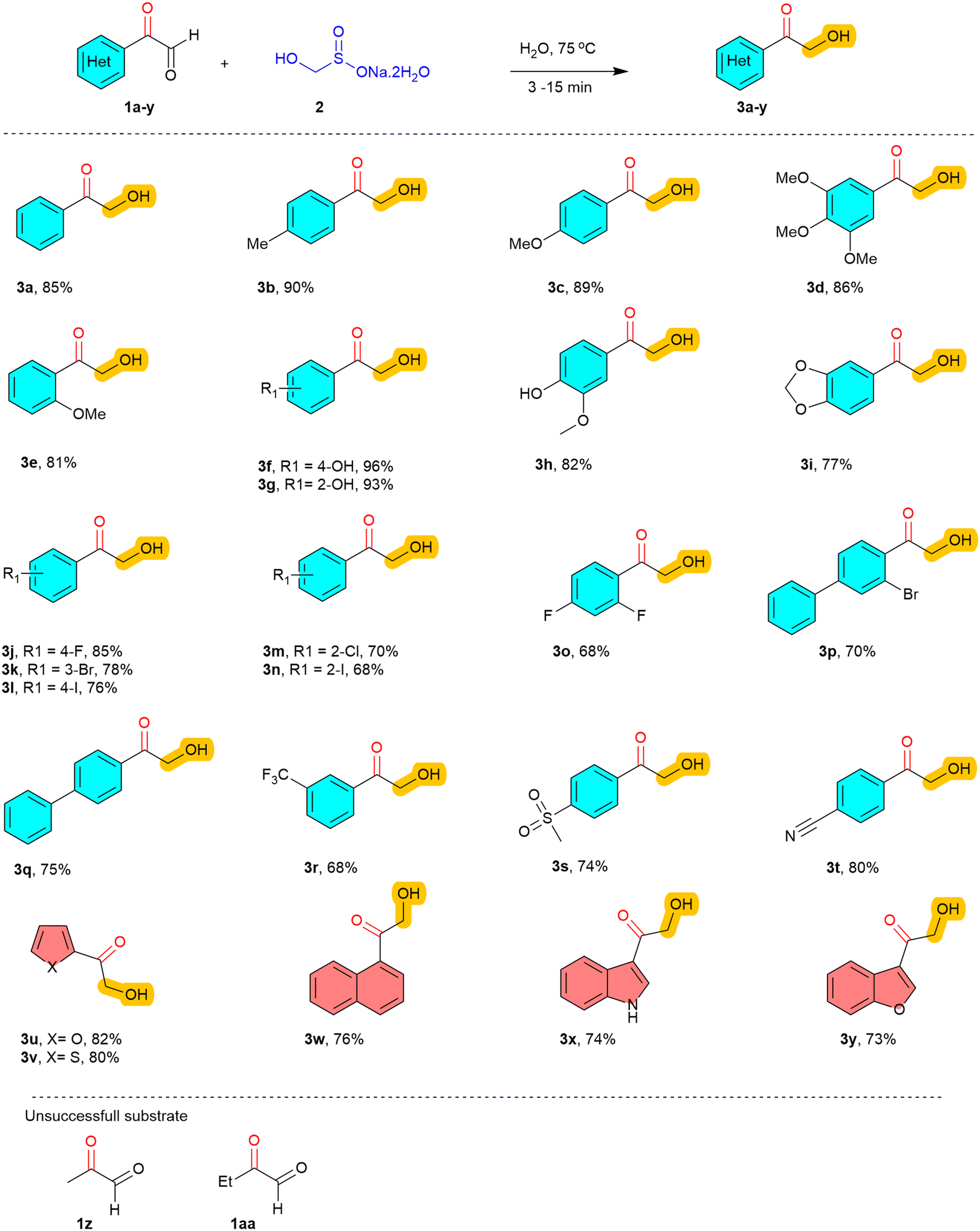
This halogen tolerance not only underscores the robustness of the methodology but also offers synthetic handles for further functionalization via cross-coupling or halogen–metal exchange strategies. We then explored the effect of electron-withdrawing groups (EWGs) on the reactivity of the α-keto aldehyde. Substrates bearing phenyl (3q), trifluoromethyl (3r), sulfonyl (3s), and cyano (3t) substituents all underwent efficient transformation, affording the corresponding products in yields ranging from 68 to 80%. These results indicate that even strongly electron-deficient substrates are amenable to the transformation, albeit with slightly diminished reactivity compared to their electron-rich counterparts. This may be attributed to the decreased electrophilicity at the α-carbon or potential competing interactions with the aqueous solvent system.
Furthermore, the developed protocol was successfully extended to polycyclic and heteroaromatic systems, which are often considered challenging substrates due to potential solubility issues or coordination with functional groups. A naphthyl-derived α-keto aldehyde (3w) gave good yield (76%), demonstrating that sterically hindered and extended π-systems do not significantly impede the reaction. Importantly, heterocyclic substrates containing furan (3u), thiophene (3v), indole (3x), and benzofuran (3y) motifs all reacted smoothly, yielding the desired products in moderate to good yields (73–80%). This tolerance toward heteroatoms highlights the versatility of the method for potential application in medicinal chemistry and complex molecule synthesis. In addition to aromatic and heteroaromatic α-keto-aldehydes, we also examined other aliphatic derivatives such as methyl glyoxal (1z) and ethyl glyoxal (1aa) under the optimized conditions. However, they failed to give the desired α-hydroxyalkyl ketones.
Overall, these results clearly demonstrate the broad substrate scope and functional group compatibility of this transformation, establishing it as a practical and efficient strategy for accessing α-hydroxymethyl ketones from readily available α-keto aldehydes under mild, aqueous, and metal-free conditions. To assess the synthetic practicality and scalability of the developed methodology, we carried out the reaction under gram-scale conditions, which is particularly important for demonstrating potential industrial applicability. A solution of 2-oxo-2-phenylacetaldehyde (1a) (2.00 g, 14.91 mmol) and rongalite (2) (2.36 g, 29.82 mmol) in water (15 mL) was heated at 75 °C under open-air conditions (Scheme 2). As expected, the reaction proceeded efficiently and cleanly, delivering the target product, 2-hydroxy-1-phenylethan-1-one (3a), in 78% isolated yield after a simple workup.
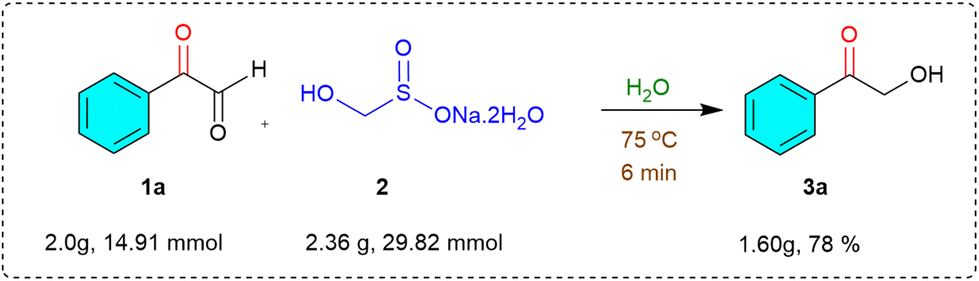 | ||
| Scheme 2 Gram scale synthesis. | ||
To gain insight into the mechanism underlying this chemoselective reduction, we performed a series of control experiments aimed at probing the role of rongalite and the possible involvement of radical intermediates (Scheme 3).
 | ||
| Scheme 3 Control experiments. | ||
Initially, a blank experiment was conducted using phenyl glyoxal 1a (1.0 mmol) in water (3 mL) at 75 °C, in the absence of rongalite 2. Under these conditions, no formation of the desired product 2-hydroxy-1-phenylethan-1-one 3a was observed. This clearly indicates that rongalite is essential for promoting the reduction process. To investigate whether the reaction proceeds via a radical pathway, we conducted further control experiments in the presence of radical scavengers, namely TEMPO and hydroquinone. In these experiments, 2-oxo-2-phenylacetaldehyde 1a (1.0 mmol) was treated with rongalite 2 (2.0 mmol) and either TEMPO or hydroquinone (3 mmol) in water (3 mL) at 75 °C. In both cases, the formation of product 3a was significantly suppressed, with only 10–15% yield obtained. This reduction in yield in the presence of radical inhibitors strongly suggests that the reaction proceeds via a radical mechanism. Moreover, HRMS analysis (see the SI, S61) of the reaction mixture revealed the formation of a TEMPO substrate adduct (4) m/z = 330.1664 (Scheme 3c); collectively, these observations point toward a rongalite-mediated radical reduction mechanism, wherein the α-keto aldehyde is likely reduced through single-electron transfer (SET) processes initiated by the sulfoxylate species generated from rongalite under aqueous thermal conditions. Based on the results from control experiments and insights from previous literature,22 a plausible radical-based mechanism is proposed as shown in Scheme 4. Under the reaction conditions, rongalite undergoes thermal decomposition to generate formaldehyde and the bisulfite anion (HSO2−).
 | ||
| Scheme 4 Plausible mechanism. | ||
The HSO2− species then donates a single electron to the α-keto aldehyde 1a, producing the ketyl radical anion I. Protonation of intermediate I yields the ketyl radical II, which undergoes a second single-electron reduction by another equivalent of HSO2− to form the carbanion intermediate III. Finally, protonation of III furnishes the desired product, 2-hydroxy-1-phenylethan-1-one 3a.
Conclusions
In conclusion, a transition-metal- and hydride-free protocol has been developed for the chemoselective reduction of α-keto aldehydes, affording diversely substituted α-hydroxymethyl ketones in 68–96% yields using rongalite as the reducing agent. Rongalite, an inexpensive and readily available industrial reagent (ca. $0.03 per g), acts as a radical-based, hydride-free reducing source. This strategy effectively overcomes the drawbacks of previously reported methods, such as the formation of hazardous byproducts, prolonged reaction times, high temperature requirements, and poor chemoselectivity. Moreover, the reaction proceeds efficiently in an aqueous medium, highlighting its green and sustainable character. The synthetic utility of the protocol was further demonstrated by a gram-scale transformation, which delivered the desired product in 78% yield.Conflicts of interest
The authors declare no competing financial interest.Data availability
The data underlying this study are available in the published article and its supplementary information (SI). Supplementary information: experimental procedure details, characterization data and copies of 1H NMR, 13C NMR, 19F and HRMS spectra. See DOI: https://doi.org/10.1039/d5ob01653c.Acknowledgements
The author H. P. K. is grateful to the Anusandhan National Research Foundation (EEQ/2022/000511 & SCP/2022/000273) and for the DST-FIST grant (SR/FST/CSII/2018/65) awarded to the Department of Chemistry, NIT Warangal, for financial support of this work. The authors also thank the CRIF Centre, NIT Warangal, for NMR and HRMS analysis and NIT Warangal for providing infrastructure support.References
- (a) M. Martínez, Las Plantas Medicinales de México, Ediciones Botas, México D. F., 1989, p. 43 Search PubMed; (b) S. Escandón-Rivera, M. González-Andrade, R. Bye, E. Linares, A. Navarrete and R. Mata, J. Nat. Prod., 2012, 75, 968–974 CrossRef PubMed; (c) J.-H. Su, S.-W. Tseng, M.-C. Lu, L.-L. Liu, Y. Chou and P.-J. Sung, J. Nat. Prod., 2011, 74, 2005–2009 CrossRef CAS PubMed.
- (a) R. Uchida, K. Shiomi, T. Sunazuka, J. Inokoshi, A. Nishizawa, T. Hirose, H. Tanaka, Y. Iwai and S. Omura, J. Antibiot., 1996, 49, 886 CrossRef CAS PubMed; (b) T. Tanaka, M. Kawase and S. Tani, Bioorg. Med. Chem., 2004, 12, 501–505 CrossRef CAS PubMed.
- W. R. Roush, K. Briner, B. S. Kesler, M. Murphy and D. J. Gustin, J. Org. Chem., 1996, 61, 6098–6105 CrossRef CAS.
- O. B. Wallace, D. W. Smith, M. S. Deshpande, C. Polson and K. M. Felsenstein, Bioorg. Med. Chem. Lett., 2003, 13, 1203–1207 CrossRef CAS PubMed.
- M. Kawase, H. Sakagami, K. Kusama, N. Motohashi and S. Saito, Bioorg. Med. Chem. Lett., 1999, 9, 3113–3117 CrossRef CAS PubMed.
- (a) T. Li, Z. Tang, H. Wei, Z. Tan, P. Liu, J. Li, Y. Zheng, J. Lin, W. Liu, H. Jiang, H. Liu, L. Zhu and Y. Ma, Green Chem., 2020, 22, 6809–6814 RSC; (b) T. Li, Z. Tan, Z. Tang, P. Liu, H. Liu, L. Zhu and Y. Ma, Green Chem., 2022, 24, 5064–5069 RSC; (c) H. J. Jo, J. H. Kim, Y. N. Kim, P. W. Seo, C. Y. Kim, J. W. Kim, H. N. Yu, H. Cheon, E. Y. Lee, J. S. Kim and J. B. Park, Green Chem., 2022, 24, 218–226 RSC; (d) X. Y. Lu, Y. W. Liu, Y. Q. Yang, S. S. Wang, Q. Wang, X. Y. Wang, Z. H. Yan, J. Cheng, C. Liu, X. Yang, H. Luo, S. Yang, J. R. Gou, L. Z. Ye, L. N. Lu, Z. D. Zhang, Y. Guo, Y. Nie, J. P. Lin, S. Li, C. G. Tian, T. Cai, B. Z. Zhuo, H. W. Ma, W. Wang, Y. H. Ma, Y. J. Liu, Y. Li and H. F. Jiang, Nat. Commun., 2019, 10, 1378 CrossRef; (e) J. Zhou, X. Tian, Q. Yang, Z. Zhang, C. Chen, Z. Cui, Y. Ji, U. Schwaneberg, B. Chen and T. Tan, Chem. Catal., 2022, 2, 2675–2690 CAS; (f) B. R. Lukito, B. S. Sekar, S. Wu and Z. Li, Adv. Synth. Catal., 2019, 361, 3560–3568 CrossRef CAS.
- (a) Z. Zhang and X. Jiang, Org. Lett., 2014, 16, 4400–4403 CrossRef CAS PubMed; (b) B. M. Trost and L. R. Terrell, J. Am. Chem. Soc., 2003, 125, 338–339 CrossRef CAS PubMed; (c) D.-Y. Zheng, T. Zhang, R. Bai, M. Li, R. Wang and Y. Gu, Green Chem., 2023, 25, 915–921 RSC; (d) H. Yan, C. Zhang, J. Han, S. Du, Y. Hua, M. Wang, G. Mei and S. Jia, Org. Lett., 2023, 25, 1918–1923 CrossRef CAS PubMed; (e) S. Sithambaram, Y. Ding, W. Li, X. Shen, F. Gaenzler and S. L. Suib, Green Chem., 2008, 10, 1023–1032 RSC; (f) X. Huo, R. He, X. Zhang and W. Zhang, J. Am. Chem. Soc., 2016, 138, 11093–11096 CrossRef CAS PubMed; (g) X. Meng, X. Bi, G. Chen, B. Chen and P. Zhao, Org. Process Res. Dev., 2018, 22, 1716–1722 CrossRef CAS; (h) L. Ren, J. Grina, D. Moreno, J. F. Blake, J. J. Gaudino, R. Garrey, A. T. Metcalf, M. Burkard, M. Martinson, K. Rasor, H. Chen, B. Dean, S. E. Gould, P. Pacheco, S. Shahidi-Latham, J. Yin, K. West, W. Wang, J. G. Moffat and J. B. Schwarz, J. Med. Chem., 2015, 58, 1976–1991 CrossRef CAS PubMed.
- (a) Y. Zhang, Z. Shen, J. Tang, Y. Zhang, L. Kong and Y. Zhang, Org. Biomol. Chem., 2006, 4, 1478–1482 RSC; (b) X. Wu, Q. Gao, M. Lian, S. Liu and A. Wu, RSC Adv., 2014, 4, 51180–51183 RSC.
- (a) J. Jung, J. Kim, G. Park, Y. You and E. J. Cho, Adv. Synth. Catal., 2016, 358, 74–80 CrossRef CAS; (b) H. Xu, X. Wu, Y. Ding, C. Peng and X. Peng, Asian J. Chem., 2015, 27, 3185–3187 CrossRef CAS.
- A. Palav, B. Misal, P. Ganwir, P. Badani and G. Chaturbhuj, Tetrahedron Lett., 2021, 73, 153094 CrossRef CAS.
- Z. Sun, M. K. Zaman, S. N. Khan and Y. Cai, Synlett, 2023, 34, 2029–2033 CrossRef.
- (a) A. Nikolaou, G. Kokotos and V. Magrioti, Tetrahedron, 2016, 72, 7628–7632 CrossRef CAS; (b) N. Kuhl and F. Glorius, Chem. Commun., 2011, 47, 573–575 RSC.
- P. Hoyos, J. V. Sinisterra, F. Molinari, A. R. Alcántara and P. Domínguez de María, Acc. Chem. Res., 2010, 43, 288–299 CrossRef CAS.
- P. D. Gennaro, S. Bernasconi, F. Orsini, E. Corretto and G. Sello, Tetrahedron: Asymmetry, 2010, 21, 1885–1889 CrossRef.
- J. D. Zhang, J. W. Zhao, L. L. Gao, J. Zhao, H. H. Chang and W. L. Wei, ChemCatChem, 2019, 11, 5032–5037 CrossRef CAS.
- (a) X. Zhang, H. Wei, X. Wei, T. Qi, X. Zong, Z. Liu, J. Qin, X. Gao, G. Zheng and Q. Ma, Green Chem., 2023, 25, 4713–4721 RSC; (b) Y. Zhang, Y. Li, Y. Chen, W. Liu, Q. Zhao, J. Feng, P. Yao, Q. Wu and D. Zhu, ACS Catal., 2024, 14, 9687–9700 CrossRef CAS.
- (a) M. B. Smith and J. March, March's Advanced Organic Chemistry, Wiley Interscience, New Jersey, 6th edn, 2007 Search PubMed; (b) J. Seyden-Penne, Reduction by the Alumino and Borohydrides in Organic Synthesis, Wiley-VCH, 2nd edn, 1997 Search PubMed.
- H. C. Brown and S. Krishnamurthy, Tetrahedron, 1979, 35, 567–574 CrossRef CAS.
- (a) D. E. Ward and C. K. Rhee, Can. J. Chem., 1989, 67, 1206–1210 CrossRef CAS; (b) B. C. Ranu, Synlett, 1993, 12, 885–892 CrossRef; (c) B. Zeynizadeh, Bull. Chem. Soc. Jpn., 2003, 76, 317–322 CrossRef CAS; (d) S. Krishnamurthy, J. Org. Chem., 1981, 46, 4628–4632 CrossRef CAS; (e) H. C. Brown and S. U. Kulkarni, J. Org. Chem., 1977, 42, 4169–4173 CrossRef CAS.
- (a) B. Marciniec, Hydrosilylation, Springer, 2009 CrossRef; (b) S. Yumino, T. Hashimoto, A. Tahara and H. Nagashima, Chem. Lett., 2014, 43, 1829–1831 CrossRef.
- (a) S. Kotha and P. Khedkar, Chem. Rev., 2012, 112, 1650–1680 CrossRef CAS PubMed; (b) S. Kotha and M. Meshram, Chem. Rec., 2019, 19, 2480–2504 CrossRef CAS PubMed; (c) S. Kotha, P. Khedkar and Y. Dommaraju, Tetrahedron Lett., 2019, 60, 631–6480 CrossRef CAS; (d) R. Ali, ChemistrySelect, 2020, 5, 10795–10815 CrossRef CAS; (e) H.-Y. Wang, X.-L. Chen, Y.-D. Wu and A.-X. Wu, Chin. J. Chem., 2023, 41, 3388–3400 CrossRef CAS.
- (a) S. Golla and H. P. Kokatla, J. Org. Chem., 2022, 87, 9915–9925 CrossRef CAS; (b) S. Golla, S. Poshala, R. Pawar and H. P. Kokatla, Tetrahedron Lett., 2020, 61, 151539 CrossRef CAS; (c) S. Poshala, S. Thunga, S. Manchala and H. P. Kokatla, ChemistrySelect, 2018, 3, 13759–13764 CrossRef CAS; (d) M. Wang, B.-C. Tang, J.-C. Xiang, X.-L. Chen, J.-T. Ma, Y.-D. Wu and A.-X. Wu, Org. Lett., 2019, 21, 8934–8937 CrossRef CAS PubMed; (e) F. Yu, R. Mao, M. Yu, X. Gu and Y. Wang, J. Org. Chem., 2019, 84, 9946–9956 CrossRef CAS PubMed; (f) J. K. Laha and P. Gupta, J. Org. Chem., 2022, 87, 4204–4214 CrossRef CAS; (g) M. Wang, H.-Y. Ren, S. Jiang and B.-T. Zhao, Org. Lett., 2025, 27, 12024–12029 CrossRef CAS PubMed; (h) M. Wang, H.-Y. Ren, S. Jiang, Y. Pan, X.-Y. Pu, X.-L. Zhang and B.-T. Zhao, Adv. Synth. Catal., 2025, 367, e9604 CrossRef CAS.
- (a) S. Thunga, M. Inapanuri, N. Singh and H. P. Kokatla, J. Org. Chem., 2024, 89, 18313–18321 CrossRef CAS PubMed; (b) S. Thunga, N. Singh, N. Anugu and H. P. Kokatla, J. Org. Chem., 2024, 89, 18649–18653 CrossRef CAS PubMed; (c) S. Thunga, N. Singh, M. Inapanuri and H. P. Kokatla, Org. Biomol. Chem., 2024, 22, 8787–8792 RSC; (d) X.-L. Chen, C.-Y. Wu, J.-T. Ma, S.-Y. Zhuang, Z.-C. Yu, Y.-D. Wu and A.-X. Wu, Org. Lett., 2022, 24, 223–227 CrossRef CAS PubMed; (e) M. Wang, J.-C. Xiang, Y. Cheng, Y.-D. Wu and A.-X. Wu, Org. Lett., 2016, 18, 524–527 CrossRef CAS PubMed; (f) Y. Jain, M. Kumari, H. Laddha and R. Gupta, ChemistrySelect, 2019, 4, 7015–7026 CrossRef CAS.
- (a) A. Shavnya, S. B. Coffey, K. D. Hesp, S. C. Ross and A. S. Tsai, Org. Lett., 2016, 18, 5848–5851 CrossRef CAS; (b) W. Zhang and M. Luo, Chem. Commun., 2016, 52, 2980–2983 RSC; (c) M. Wang, B.-C. Tang, J.-G. Wang, J.-C. Xiang, A.-Y. Guan, P.-P. Huang, W.-Y. Guo, Y.-D. Wu and A.-X. Wu, Chem. Commun., 2018, 54, 7641–7644 RSC; (d) X.-L. Chen, B.-C. Tang, C. He, J.-T. Ma, S.-Y. Zhuang, Y.-D. Wu and A.-X. Wu, Chem. Commun., 2020, 56, 13653–13656 RSC; (e) J. Wang, K. Li, X. Luo, Z. Wu, T. Gu, S. Liao, X. Lin, B. Yang, Y. Liu, W. Fang and X. Zhou, Org. Biomol. Chem., 2019, 17, 8721–8725 RSC; (f) M. Wang and X. Jiang, Chem. Rec., 2021, 21, 3338–3355 CrossRef CAS.
- (a) S. Golla, S. Jalagam, S. Poshala and H. P. Kokatla, Org. Biomol. Chem., 2022, 20, 4926–4932 RSC; (b) S. Golla, N. Anugu, S. Jalagam and H. P. Kokatla, Org. Biomol. Chem., 2022, 20, 808–816 RSC.
| This journal is © The Royal Society of Chemistry 2026 |