DOI:
10.1039/D5NJ03345D
(Paper)
New J. Chem., 2026,
50, 333-345
The effect of the nature of supports on the selective reduction of CO2 to CO catalysed by a supported single-site heterobimetallic iron–potassium complex
Received
18th August 2025
, Accepted 21st November 2025
First published on 24th November 2025
Abstract
The utilization of CO2 for the production of value-added chemicals has attracted significant interest. Among various strategies, the selective hydrogenation to liquid hydrocarbons via a two-step process, first the reduction of CO2 to CO through the reverse water–gas shift (RWGS) reaction, followed by CO conversion, is particularly promising. In this work, [{(THF)2KFe(OtBu)3}2] is supported on various oxides in order to investigate the influence of the support's properties (neutral, basic, acidic, and redox) on the catalytic performance in the RWGS reaction. A series of catalysts with 1 wt% Fe are synthesized and characterized. Remarkably, catalysts supported on neutral and basic oxides maintain the close Fe–K proximity and exhibit high catalytic activity (conversion around 20%) and full selectivity toward CO (100%). Among these, the FeK/ZrO2-250 catalyst appears to be stable after 40 h on stream, which is supported by XAFS and TEM analyses of the spent catalyst. K+ serves as an electronic promoter, as well as a structural stabilizer of the active Fe site, preventing agglomeration. In contrast, the use of acidic or redox-active supports results in loss of the activity or the selectivity in CO. The formation of methane is attributed to the dissociation of FeK species into Fe nanoparticles and the incorporation of K into the CeO2 lattice or exchange with acid sites of ZSM-5, Nb2O5 or SiO2–Al2O3, thereby disrupting the critical Fe–K synergy. These findings underscore the pivotal role of support selection in preserving the structural and electronic integrity of Fe–K active sites. Supports that inhibit sintering and maintain the FeK single-site configuration are essential for achieving high activity, full CO selectivity, and long-term stability in the RWGS reaction.
1. Introduction
Recently, the catalytic reduction of CO2 to CO via the reverse water gas shift (RWGS) reaction has gained attention from industry and academia due to its increasing importance in the CO2 conversion technology and the interest of using CO as feedstock in the Fischer–Tropsch process, a mature technology for converting this intermediate into valuable chemicals and fuels.1–7 However, due to the high thermodynamic stability of CO2, the RWGS reaction is an endothermic process (ΔH° = 42.1 kJ mol−1) that requires higher temperatures and high H2/CO2 ratios to be optimal.8–11 Although pressure does not influence its equilibrium, the reaction faces competition from undesired methanation and the exothermic Sabatier reaction, which are favored at lower temperatures and pressures (eqn (1)–(3)).12–15 In general, for temperatures below 600 °C, methane formation dominates, whereas higher temperatures (>600 °C) enhance CO selectivity.11,16 In order to reduce energy and capital costs, the reaction temperature shall be kept as low as possible and simultaneously optimize the CO production with minimum CH4 as the undesired byproduct. Therefore, understanding the mechanisms, the structure of the active species and kinetics of the RWGS reaction is crucial for the catalyst design for CO production. These strategies include varying the support, tuning metal–support interactions, adding reducible transition metal oxide promoters, forming bimetallic alloys, adding alkali metals, and enveloping metal particles.17–20 Selecting an appropriate support is important to tune the activity and selectivity of this reaction. Various oxides such as SiO2, ZrO2, TiO2, mullite (aluminum–silicate), Al2O3 and CeO2 have been reported as catalyst supports in the RWGS reaction.11,16,21,22 The authors described that the redox and acid–base properties of the supports can greatly influence CO2 adsorption/activation, as well as the nature of the reaction intermediates and the mechanisms (redox or associative). SiO2 and Al2O3 are neutral and irreducible supports,17 and many studies indicate that these supports indirectly participate in the RWGS reaction by enhancing metal dispersion and CO2 adsorption. For example, when Pt single site catalysts are supported on silica, it is proposed that CO2 adsorbs at the interface between Pt and SiO2via hydrogen bonding with silanol groups.16 This CO2 adsorption strength is enhanced by 0.21 eV compared to unsupported Pt nanoparticles, thus allowing the formation of formate species, an intermediate for CO production.16 Similarly, the use of Al2O3 as an irreducible and amphoteric oxide for the dispersion of Pd single site catalysts provides high selectivity to CO compared to Pd supported on inert multiwall carbon nanotubes (MWCNTs).23 In order to explain this effect of the support, Bobadilla et al.24 proposed that CO2 reacts with the basic hydroxyl groups of Al2O3 to form bicarbonate species which, via hydrogen spillover from Pd, lead to the formation of aluminum formate intermediates that further decompose into CO.| | | CO2 + H2 ⇄ CO + H2O(g) ΔHθ = +41.3 kJ mol−1 | (1) |
| | | CO2 + 4H2 ⇄ CH4 + 2H2O(g) ΔHθ = −164.7 kJ mol−1 | (2) |
| | | CO + 3H2 ⇄ CH4 + H2O(g) ΔHθ = −206.2 kJ mol−1 | (3) |
The effect of an acidic support such as a zeolite (H-ZSM-5) is also reported for the conversion of CO2 to CO.25–27 For example, Wang et al.27 compared CO2 hydrogenation on Rh supported on zeolite such as H-ZSM-5 and pure silica MFI, revealing that neutral silica MFI modified by rhodium exhibits high CO selectivity with high CO2 conversions. However, Rh supported on acidic H-ZSM-5 shows moderate selectivity to CO with the formation of methane under the same conditions. Strong correlations are observed between the nanoporous environment and catalytic selectivity, indicating that the neutral silica MFI minimizes hydrogen spillover and favors the fast desorption of CO to limit total hydrogenation to methane.27 Furthermore, the use of basic support such as zirconia is reported and shows that the selectivity to CO depends on the nature of the metal and metal loading.28–30 Based on literature studies, the nature and properties of the supports thus have a strong effect on the catalytic performance.
Redox supports such as TiO2, CeO2–ZrO2 and CeO2 are known to generate oxygen vacancies, particularly in the presence of an immobilized metal during the activation of H2 and CO2.31 In the case of ceria-based catalysts, CO2 hydrogenation is generally attributed to oxygen storage capacity, redox properties, and the ability to enhance the reaction rates and selectivity.32 CeO2 is reported to exhibit unique synergistic effects when doped with other metals that improve their catalytic activity.33 Using DFT calculations, ceria supported copper and iron catalysts34,35 are both reported and exhibit moderate activity in the RWGS reaction.36 Conversely, ceria supported Ni and Co favor CO2 methanation.37 From these studies, the high activity of metal doped CeO2 is attributed to the increase of the reducibility and consequently the number of oxygen vacancies.38 However, the selectivity in CO is favored when the dopants are well dispersed on the ceria surface. On the other hand, the presence of metal nanoparticles on the ceria surface increases the formation of undesired products such as methane.39,40 Recently, the inherent relationships between the performance of transition metal (Fe, Co, Ni and Cu) doped CeO2 catalysts, the concentration of transition metals doped onto CeO2 surfaces, CO2 binding strength, and the oxygen vacancy formation energy have been explored for the RWGS reaction. At low loading, Fe/CeO2 showed low conversion (10%) and high selectivity in CO (100%) at high temperature (600 °C). However, at temperatures lower than 500 °C, no activity in the RWGS reaction was reported.39 In addition to the effect of support, the presence of alkali metal ions is reported to enhance the metal–support interactions for transition metal-based catalysts, leading to increased stability and thereby increased activity and CO selectivity.36,41,42 The incorporation of potassium into Fe/Al2O3 with a K/Fe ratio of 1 results in a significant increase in CO formation rates compared to unpromoted Fe/Al2O3.43 The increased basicity using K improves the adsorption of CO2 by forming Lewis acid–base pairs that enhance CO2 binding by chemisorption on the surface through electron transfer.36 Thus, the potassium promoter activates a secondary pathway for CO formation, which may be the so-called associative pathway.43 Based on this finding, we recently reported a well-defined supported single-site FeK RWGS catalyst with low Fe loading that essentially provided 100% selectivity to CO at 400 °C. The heterobimetallic FeK complex, [{(THF)2KFe(OtBu)3}]2, containing K as the metal cation, is grafted by surface organometallic chemistry (SOMC)44 onto partially dehydroxylated Al2O3 at 500 °C and characterized by ICP, IR, XPS, EDX, XAFS, and EPR. The material consists of an isolated anionic Fe(II) site coordinated by two tert-butoxide ligands and one anionic surface oxygen, associated with K+. The postulated mechanism (Scheme 1) proceeds through H2 activation to form an iron hydride, which evolves to an iron hydroxycarbonyl intermediate upon insertion of CO2 that by further decomposition leads to CO.45 Interestingly, the Na and Li counterparts exhibit substantially lower activity in the RWGS reaction. Taking into account that the potassium in the proximity of Fe has a significant effect on the activity and selectivity during the adsorption and the activation of CO2, the modification of the nature and the properties (acidity, basicity and redox) of the supports may also affect the activity and the selectivity during CO2 conversion to CO.
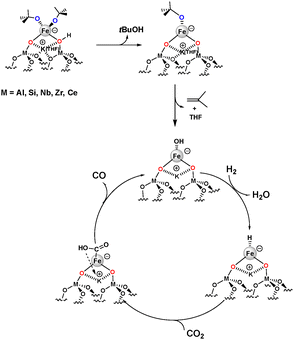 |
| | Scheme 1 Proposed mechanism for selective conversion of CO2 to CO on supported anionic heterobimetallic Fe and K complexes independent of the nature of support (neutral/basic/acidic or redox). | |
Here, this approach has been extended to study the effect of the supports (neutral, basic, acidic and redox) on activity and selectivity in the RWGS reaction. This methodology allows us to study the effect of the combination of the two parameters (K and support properties) on the activity and selectivity to CO, as well as the validation of the proposed mechanism on supported anionic iron single active species. To ensure the aforementioned criteria, neutral (SiO2), basic (MgO–Al2O3 (Pural) and ZrO2), acidic (Nb2O5, SiO2–Al2O3 and μ-H-ZSM-5) and redox (CeO2 and CeZrOx) supports have been selected to immobilize the isolated iron species. The resulting materials have been characterized using ICP, DRIFT, HRTEM, BET, EDX, EPR, XRD and XAFS for the catalyst based on zirconia before and after catalysis. The catalysts have been tested for the reduction of CO2 to CO.
2. Experimental methods
2.1. Chemicals and materials
All experiments have been carried out under a controlled atmosphere using Schlenk and glovebox techniques for organometallic synthesis. For the synthesis and treatment of supported species, reactions have been carried out using high-vacuum lines (ca. 1 mPa) and gloveboxes. Pentane (99%) and THF (99.9%) were purchased from MilliporeSigma, distilled from NaK and degassed using freeze–pump–thaw cycles. FeBr2 (98%) and tBuOK (98%) were purchased from MilliporeSigma and used as received.
2.2. Characterization of materials
All characterization studies have been performed under an inert atmosphere, except for EDX/STEM/HRTEM for which sample preparation is performed in air. Elemental analysis has been carried out at the Mikroanalytisches Labor Pascher, Remagen, Germany, and CREALINS, Villeurbanne, France, using an iCAP 6500 Duo inductively coupled plasma atomic emission spectrometer (Thermo Scientific). All samples sent for elemental analysis have been sealed under high vacuum (10−5 mbar). Infrared spectra have been recorded in diffuse reflectance mode on a Nicolet 6700 FT-IR spectrometer, in an air-tight cell with CaF2 windows under an atmosphere of Ar at room temperature. Each spectrum is composed for 64 scans recorded at a resolution of 4 cm−1. A Micromeritics ASAP 2020 (surface area and porosity analyzer) has been used for the determination of textural properties (e.g., Brunauer–Emmett–Teller, BET surface area). Off-line gas chromatographic analyses have been performed on an HP 5890 series II GC, equipped with an HP5 GC column and an FID detector. High resolution transmission electron microcopy (HRTEM) and energy dispersive X-ray (EDX) spectroscopy have been performed at the Centre Technologique des Microstructures, Université Lyon 1. Prior to EDX/STEM/HRTEM analysis (JEOL 2100F, 200 kV), samples are prepared by placing a suspension spot containing the test material on an ultrathin Ni or Cu grid. Powder X-ray diffraction (XRD) has been recorded at the Institut des Sciences Analytiques Lyon 1. Electron paramagnetic resonance (EPR) spectroscopy has been carried out with a Bruker spectrometer Elexsys E500 using X Band (9.4 GHz) radiation at T = 110–120 K, in the Laboratoire de Chimie, ENS Lyon. Samples for EPR have been prepared in air-tight quartz tubes loaded inside a glove box. For quantitative studies of the paramagnetic phase, double integration of the EPR signal is performed and compared to that of a reference composed of a known amount of vanadyl(IV) sulfate. XAS spectra have been acquired at ESRF, Grenoble, France, using the BM23 beamline at the iron K-edge in the transmission mode between 7.0 and 8.2 keV. Four scans are recorded at room temperature for each sample. Each data set is collected simultaneously with a Fe metal foil and is later aligned according to that reference (first inflection point set at 7112.0 eV). The samples have been packed in an argon-filled glovebox within double air-tight sample holders. The data analyses have been carried out using the program “Athena”46 and the EXAFS fitting program “RoundMidnight”, from the “MAX” package,47 using spherical waves. For each neighboring atom or path i, the refined parameters are Ni, the number of neighbors; Ri (Å), the distance of the Fe center to that neighbor; σi2 (Å2), the Debye–Waller parameter; and ΔE0 (eV) designing the global energy shift. The program FEFF8 has been used to calculate theoretical files for phases and amplitudes based on model clusters of atoms.48
2.3. Catalyst preparation
2.3.1. The molecular iron complexes.
[{(THF)2KFe(OtBu)3}2] has been synthesized as reported in a recent work.45
2.3.2. Preparation of the supports.
All supports (5 g) have been calcined at 500 °C (heating rate: 4 °C min−1) under continuous flow of dry air for 6 hours. Then, 2 g each of SiO2, SiO2–Al2O3 and Al2O3–MgO has been dehydroxylated for 12 h at 700 °C, 500 °C and 500 °C, respectively, which involves a heat treatment under high vacuum. Nb2O5, μ-ZSM-5, CeO2, CeZrOx and ZrO2 have been hydrated (exposed to O2-free water vapor) at 100 °C prior to the dehydroxylation process at 250 °C (12 h) in a glass reactor (500 mL).
2.3.3. Grafting of molecular complexes onto supports.
As described most recently in the case of catalysts, FeK/Al2O3-500,45 a solution of [{(THF)2KFe(OtBu)3}2] (0.25 g) in n-pentane (15 mL) has been added to 3 g of ZrO2-250 to produce a solid containing ca. 1 wt% Fe. The brownish suspension has been stirred for 12 h at room temperature and then filtered through a glass frit. The solid has been dried under high vacuum (10−5 mbar), while the filtrate has been analyzed by GC. The same procedure is used to graft the complex [{(THF)2KFe(OtBu)3}2] onto SiO2-700, Al2O3–MgO-500, Nb2O5-250, μ-ZSM-5250, SiO2–Al2O3-500, CeO2-250 and CeZrOx-250 (all ca. 1 wt% Fe).
2.4. Catalytic testing
Catalysts have been evaluated in a ½″ stainless-steel tubular continuous flow reactor whose outlet is connected to an online GC (Agilent Technologies 7890A). The dead volume is minimized using stainless steel inserts. The GC is equipped with two columns in series: a carbon PLOT column to separate CO2, CO, and CH4 and a PLOT Q column to separate higher hydrocarbons. The gas effluent from the reactor passes through both columns before reaching the detectors. Separated products are detected using a thermal conductivity detector (TCD) and a flame ionization detector (FID), connected in series. The FID is equipped with a methanizer (Jetanizer™). The reactor is charged (250 mg catalyst) in the glovebox and equipped with a 4-way valve, allowing extensive purging of the gas lines with the reactant gas once connected to the reactor host (PID, Micrometrics). The gas flow is controlled using Bronkhorst mass flow controllers. Catalytic tests are performed using a certified gas mixture with a H2/CO2 ratio of 3 (Mélange Crystal, Air Liquide), at a constant flow rate of 3 mL min−1 (measured at room temperature). Prior to starting the reaction, the catalyst has been activated in situ in flowing Ar (Alphagaz 1, Air Liquide) for 2 h at 300 °C. CO2 conversion and product selectivity are calculated using eqn (4)–(6):| | 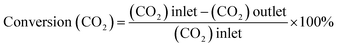 | (4) |
| | 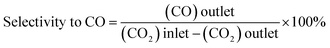 | (5) |
| | 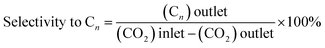 | (6) |
3. Results and discussion
3.1. Supported [{(THF)2KFe(OtBu)3}2] single site on ZrO2-250 through surface organometallic chemistry
3.1.1. Preparation and characterization of ZrO2-250.
Commercial ZrO2 is obtained from MilliporeSigma as white powder with a surface area of 80 m2 g−1 determined by BET using N2 adsorption at 77 K (Table S1). After calcination at 500 °C for 24 hours and hydration at 100 °C with water vapor, ZrO2-250 is obtained by dehydroxylation at 250 °C for 12 hours. The surface area and pore volume of ZrO2-250 support by N2 adsorption at 77 K are found to be 72 m2 g−1 and 0.10 cm3 g−1, respectively, and these values are similar to the one reported in the literature.49–51 Furthermore, the adsorption–desorption isotherm plot (Fig. S1 and Table S2) of the zirconia dehydroxylated at 250 °C follows the type IV adsorption–desorption isotherm which is similar to those already reported in the literature.51–53 X-ray diffraction pattern of ZrO2-250 is presented in Fig. S2 and shows diffraction peaks from 18 to 65° (2θ scale) corresponding to the presence of mainly monoclinic (baddeleyite), mixed with the tetragonal phase based on the match search with the data of the Joint Committee on Power Diffraction Standards (JCPDS). This is similar to the diffraction patterns of ZrO2 reported in the literature.54–56 Peaks at 2θ = 30.32°, 35.20°, 42.84°, 50.56°, 60.04°, 63.00 and 74.52° correspond to the XRD pattern of the tetragonal phase, and peaks at 2θ = 24.5°, 28.20°, 31.5°, 34.56°, 42.10°, 46.5°, 50.50, and 66.04° correspond to the baddeleyite phase.57,58 The surface properties of the commercial ZrO2 remain the same after thermal treatment. The IR spectrum of ZrO2-250 recorded in diffuse reflectance mode (DRIFT) shows hydroxyl groups attributed to a mixture of tetragonal and monoclinic phases. In fact, different zirconia polymorphs possess various surface hydroxyl groups and acid–base properties.59 Bi-bridged hydroxyl only exists on tetragonal zirconia and amorphous zirconia shows obvious H-band hydroxyl, while two types of tri-bridged hydroxyl appear on monoclinic zirconia.59,60 These differences affect surface acidity and basicity: amorphous zirconia has strong Lewis acid sites and monoclinic zirconia has strong Brønsted acid sites, but for tetragonal zirconia, the basicity is predominant.59,61,62 The DRIFT spectrum of ZrO2-250 shows three main bands at 3774, 3738, and 3675 cm−1 assigned to the OH stretches of terminal, 2-fold coordinated (bi-bridged), and 3-fold coordinated (tri-bridged) hydroxyl groups, respectively (Fig. 1a).62–65 This spectrum is similar in situ DRIFT of ZrO2 heated at different temperatures reported in the literature.65,66 The current spectrum is also in good agreement with the theoretical calculations in which terminal hydroxyls are found to vibrate between 3822 and 3743 cm−1, bi-bridged hydroxyls in the range of 3755–3568 cm−1, and tri-bridged hydroxyls between 3647 and 3498 cm−1.65–67 To achieve the grafting and functionalization of surface hydroxides under optimum conditions, their quantification is required. Among the different quantification methods, a chemical titration using Al(iBu)3, a surface reaction with a highly reactive organometallic complex, has been shown to be reliable.68,69 This complex quantitatively reacts in pentane with surface hydroxyl groups of ZrO2-500 by releasing one equivalent of isobutane per OH. The quantification of isobutane by GC shows that ZrO2-250 contains 0.25 mmol OH g−1 that corresponds to 2.1 OH per nm2.
 |
| | Fig. 1 DRIFT spectra of (a) ZrO2-250 and (b) ZrO2-250 after reaction with [{(THF)2KFe(OtBu)3}2], FeK/ZrO2-250. | |
3.1.2. Preparation and characterization of the catalyst, FeK/ZrO2-250.
The supported catalyst is prepared by grafting [{(THF)2KFe(OtBu)3}2] (Fe/OH = 0.7, 1 wt Fe%) from a pentane solution onto zirconia partially dehydroxylated at 250 °C (ZrO2-250). The reaction is proceeded at room temperature for 12 h. The solid is washed with pentane and dried under vacuum, affording a yellowish material. Upon grafting, tBuOH and THF are released and confirmed by GC. The resulting material is characterized by DRIFT, elemental analysis, STEM/EDX, BET, EPR, XRD and XAFS. The DRIFT spectrum reveals that the surface OH groups of ZrO2-250 (ν(O–H): 3795–3660 cm−1) are largely consumed by the grafting reaction (Fig. 1b). Simultaneously, bands corresponding to the organic ligands appear at 3000–2800 cm−1 (ν(C–H)), at 1466 and 1455 cm−1 (δ(CHx)) and at 1380 and 1356 cm−1 (δ(CH3)). The FeK/ZrO2-250 material is further characterized for the determination of textural properties using N2 adsorption at 77 K. The isotherm plot in Fig. S1 and Table S2 show a decrease in both the pore volume and the surface area (0.07 cm3 g−1 and 55.26 m2 g−1, respectively) compared to those of ZrO2-250 (0.11 cm3 g−1 and 71.90 m2 g−1). The decrease in pore volume, pore size and surface area after grafting of the complex onto ZrO2-250 is as expected and previously reported.70–77 The adsorption–desorption plot (type IV) remains the same after grafting of the complex onto ZrO2-250. The X-ray diffraction pattern of FeK/ZrO2-250 (Fig. S2) shows characteristic phases corresponding to monoclinic and tetragonal zirconia. No additional crystalline phases are observed after introducing 1 wt% iron, such as iron oxides or iron particles (iron oxides XRD main peaks, 2θ, Cu λKα: FeO at 36.1, 41.9 and 60.8°; Fe3O4 at 30.1, 35.5, 57.1 and 62.7°; Fe2O3 at 24.1, 33.1, 35.6, 49.5, 54.0, 62.4 and 64.0°). There is no visible difference between the diffraction patterns of the ZrO2-250 support (Fig. S2a) and the grafted material, FeK/ZrO2-250 (Fig. S2b). This result is similar to what is observed in the case of several metal supported catalysts with low metal loading.78–81 The result indicates that the anionic iron complex is highly dispersed on the surface of ZrO2-250 in agreement with HRTEM. This reveals that with very low average density of Fe on the surface of ZrO2-250 (1.6 Fe per nm2), the formation of iron clusters during the grafting at low temperature (25 °C) is limited.
Elemental analysis of the resulting material, FeK/ZrO2-250, reveals Fe, K and C contents of 1.16 wt%, 0.86 wt% and 2.98 wt%, respectively, representing a Fe/K ratio of 1. The amount of grafted Fe, 0.208 mmol g−1 ZrO2-250, represents a grafting stoichiometry (mol Fe per mol OH) of 0.75 in agreement with the DRIFT spectrum of FeK/ZrO2-250 where most OH groups are consumed. Furthermore, the C/Fe ratio of 11.96 is consistent with the loss of one HOtBu ligand per Fe and retention of one THF solvating K+ (expected C/Fe: 12). Based on mass balance analysis, the nature of the support appears to have no effect on the reactivity of the FeK precursor and the resulting structure on the ZrO2-250 surface, FeK/ZrO2-250, since a similar surface complex is also obtained on Al2O3-500, FeK/Al2O3-500.45 The Fe/K ratio of 1 is also confirmed by EDX analysis. Moreover, the EDX mapping and spectrum (Fig. S3) indicate that Fe and K are uniformly dispersed on the surface. No agglomerated phases of Fe or K are observed by HRTEM (Fig. S4). Importantly, the oxidation state in FeK/ZrO2-250 is predominantly Fe(II), based on the EPR analysis (Fig. S5). In fact, EPR spectroscopy of FeK/ZrO2-250 shows evidence of only a small amount of Fe(III) (<0.02%). This small amount is probably due to traces of Fe(III) in the molecular complex [{(THF)2KFe(OtBu)3}2].
The structure of FeK/ZrO2-250 is further studied by X-ray absorption fine structure (XAFS) spectroscopy at the Fe K-edge. The XANES of FeK/ZrO2-250 (Fig. S6) shows a pre-edge at +1.2 (±0.2) eV from the Fe(0) edge with a maximum normalized intensity of 0.10. This suggests that the Fe sites in the supported complex are in a distorted tetrahedral coordination with an iron oxidation state of +2,82,83 as also revealed by EPR spectroscopy (Fig. S5). The EXAFS curve fit for FeK/ZrO2-250 is displayed in Fig. 2, with the resulting parameters in Table 1 (left part, fresh sample). The results are consistent with a Fe center in a distorted tetrahedral geometry, with ca. three oxygen atoms at 2.00(2) Å and one more at 2.35(3) Å. The shorter distance is in the range of Fe–OR bond lengths observed for the [{(THF)2KFe(OtBu)3}2] molecular complex by XRD (average: 1.995 ± 0.09 Å),45 with the EXAFS distance resolution being ca. 0.13 Å (π/2kmax). The longer distance is attributed to a surface oxygen either from a surface hydroxyl group (see Scheme 1), or bridged between two surface Zr atoms. The fit could be further improved by including additional shells of back-scatterers, in particular, two C atoms at 3.04(4) Å, one K atom at 2.87(4) Å and ca. one Zr atom at 3.55(4) Å. However, there is no evidence for a well-defined Fe–Fe path (expected at ca. 3.3 Å in a dimer structure), despite attempts to include it in the model. Based on this fit, a proposed monomeric structure for zirconia-supported [{(THF)2KFe(OtBu)3}2] is displayed in Scheme 2.
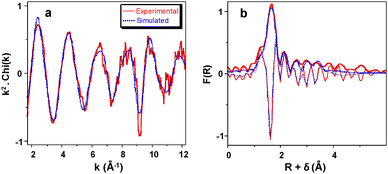 |
| | Fig. 2
k
2-Weighted Fe K-edge EXAFS of FeK/ZrO2-250. (a) k-space; (b) R-space (FT modulus and imaginary part). Solid red lines: experimental data; dashed blue lines: curve fit obtained using spherical wave theory. | |
Table 1 EXAFS curve fit parameters for FeK/ZrO2-250 before and after the catalytic reactiona
| Path |
Fresh sample |
After the catalytic reaction |
|
N
|
R (Å) |
σ
2 (Å2) |
N
|
R (Å) |
σ
2 (Å2) |
|
Errors generated using the EXAFS fitting program “RoundMidnight” are indicated in parentheses. Global fit parameter: S02 = 0.93. Fresh sample: 1.7 ≤ k ≤ 12.2 Å−1; 0.3 ≤ R ≤ 3.6 Å. Used sample: 1.7 ≤ k ≤ 12.1 Å−1; 0.2 ≤ R ≤ 3.4 Å. Fit residual: ρ; quality factor: (Δχ)2/ν.
Short.
Long.
Shell constrained to a parameter above.
|
| Fe–O1b |
2.7(5) |
2.00(2) |
0.0022(9) |
2.9(5) |
2.04(2) |
0.0027(12) |
| Fe–O2c |
1.1(3) |
2.35(3) |
0.0022d |
0.9(4) |
2.34(3) |
0.0027d |
| Fe–K |
1.0 |
2.87(4) |
0.0057(21) |
1.0 |
2.90(4) |
0.0084(20) |
| Fe–C/O |
2.0 |
3.04(4) |
0.0022(12) |
1.9(6) |
3.07(4) |
0.0028(15) |
| Fe–ZrS |
0.9(3) |
3.55(4) |
0.0057d |
1.8(7) |
3.39(4) |
0.0084d |
|
|
|
|
ΔE0 = 6.4 ± 1.0 eV; ρ = 8.2% |
ΔE0 = 3.5 ± 0.9 eV; ρ = 6.4% |
|
|
(Δχ)2/ν = 2.47 (ν = 12/24) |
(Δχ)2/ν = 2.38 (ν = 10/23) |
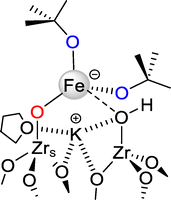 |
| | Scheme 2 Proposed monomeric structure of FeK/ZrO2-250, prepared by grafting dimeric [{(THF)2KFe(OtBu)3}2], 1, onto ZrO2-250. The structure is consistent with the XAFS analysis (the K coordination is proposed analogous to that of molecular complex 1). | |
Overall, the evidence suggests that, in accordance with elemental analysis, the dimeric complex, [{(THF)2KFe(OtBu)3}2], reacts readily with OH groups from zirconia by the protonolysis reaction, affording isolated, bipodal [(THF)K(ZrsO)Fe(OtBu)2] species (Scheme 3). In this species, Fe is close to a K+ counter-cation, and with a labile proton, that may be located on a coordinated oxygen from the zirconia surface, as already reported on alumina.45
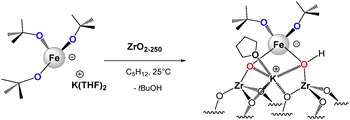 |
| | Scheme 3 Reaction of the heterobimetallic [{(THF)2KFe(OtBu)3}2] complex with ZrO2-250 to produce supported isolated single site [(THF)K(ZrsO)Fe(OtBu)2] species. | |
3.1.3. Catalytic activity in CO2 hydrogenation over FeK/ZrO2-250.
The RWGS activity and CO selectivity of the catalyst, FeK/ZrO2-250, are evaluated in a continuous flow reactor at 30 bar and 400 °C. The catalyst provides high CO2 conversion (22%, well below the equilibrium value of ca. 45% to ensure meaningful kinetic measurements) and selectivity towards CO (100%). This catalyst thus exhibits similar catalytic performance in terms of CO2 conversion (22.5%) and CO selectivity (100%) than its FeK/Al2O3-500 counterpart recently reported (Fig. 3b).45
 |
| | Fig. 3 CO2 conversion (open circles) and CO selectivity (filled circles) in the RWGS reaction catalyzed by (a) FeK/ZrO2-250 and (b) FeK/Al2O3-500 (feed composition: CO2/H2 = 1/3, volumetric flow rate = 3 mL min−1, 400 °C, 30 bar). | |
No significant deactivation is observed over the course of 40 h (Fig. 3a). This high stability is likely due to the presence of well-dispersed isolated single sites, compared with a reported catalyst with a higher Fe loading that evolves to a material with a mixture of single sites and nanoparticles.84 The presence of K+ in proximity to Fe appears to stabilize Fe(II) and provides a favorable site for CO2 adsorption. The EXAFS spectrum at the Fe K-edge of FeK/ZrO2-250 after 40 h on stream is presented in Fig. 4, with the fit resulting parameters shown in Table 1 (right part, after the catalytic reaction). The coordination sphere of iron in sample FeK/ZrO2-250 after the catalytic reaction is very similar to that in the fresh sample with iron single sites firmly grafted to the surface by two Fe–OZrs bonds and in a pseudo-tetrahedral environment (ca. three oxygen atoms at 2.04(2) Å and one at 2.34(3) Å), with a close K atom (Scheme 4). This result suggests that no sintering or oxide phase occurred during the catalytic process.
 |
| | Fig. 4
k
2-Weighted Fe K-edge EXAFS of FeK/ZrO2-250 after the catalytic reaction. (a) k-space; (b) R-space (FT modulus and imaginary part). Solid red lines: experimental data; dashed blue lines: curve fit obtained using spherical wave theory. | |
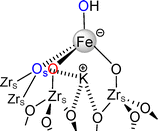 |
| | Scheme 4 Proposed monomeric structure for the iron site after the catalytic reaction, consistent with the XAFS analysis. | |
In addition, HRTEM of FeK/ZrO2-250 performed after catalyst testing (Fig. S7) supports the XAFS result showing that the active single site is robust in a range of reaction conditions. The presence of K+ in proximity to Fe seems to stabilize Fe(II) and to provide a favorable site for CO2 adsorption, leading only to CO formation.
3.2. Preparation and characterization of catalysts, [{(THF)2KFe(OtBu)3}2], supported on different oxide supports
The current approach is extended to other oxide supports in order to study the effect of the acidity and redox properties on the catalytic performance of single site FeK catalysts. All the supports used in this study are pretreated at different temperatures to afford the given surface OH group concentration required for the grafting reaction of [{(THF)2KFe(OtBu)3}2]. Table S1 shows the treatment conditions for each support. The supports are calcined in air at 500 °C followed by a dehydroxylation treatment and are denoted as SiO2-700, Al2O3–MgO-500, Nb2O5-250, μ-ZSM-5250, SiO2–Al2O3-500, CeO2-250 and CeZrOx-250 based on the dehydroxylation temperature used. The supported catalysts are all prepared by grafting [{(THF)2KFe(OtBu)3}2] in pentane solution for 12 h with the same loading (1 wt% Fe).85,86 The resulting materials have been washed with pentane and dried in a high vacuum (10−5 mbar), affording yellowish powder (dark yellow for CeO2-250), as reported in the case of [{(THF)2KFe(OtBu)3}2] supported alumina.45 Upon grafting, tBuOH and THF are released as confirmed by GC analysis. The resulting supported materials, FeK/SiO2-700, FeK/Al2O3–MgO-500, FeK/Nb2O5-250, FeK/μ-ZSM-5-250, FeK/SiO2–Al2O3-500, FeK/CeO2-250 and FeK/CeZrOx-250, have been characterized by BET (Table S2), elemental analysis (Table S3), DRIFT, XRD, STEM/EDX, and EPR (see the SI, Fig. S8–S18). The results confirm the successful grafting of the heterobimetallic [{(THF)2KFe(OtBu)3}2] complex via protonolysis of Fe–OtBu bonds by surface OH groups, as revealed by DRIFT and mass balance analysis. This suggests that well dispersed Fe(II) mononuclear anionic surface species have been obtained on various supports with a similar structure to what was observed in the case of Al2O3-50045 and ZrO2-250 (Scheme 3), with only a difference in the second neighbor of Fe (Si, Al, Zr or Ce).
3.3. Catalytic activities of FeK on various supports in the RWGS reaction
The catalytic activities and CO selectivity of all [{(THF)2KFe(OtBu)3}2] supported materials are evaluated using a continuous-flow reactor at 30 bar and 400 °C. During each catalytic test, 250 mg of catalyst with a same 1 wt% Fe loading is introduced into the flow reactor. Prior to the reaction, the catalysts have been treated under an argon atmosphere at 300 °C for 120 min. The flow rate of the CO2 and H2 mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) is set to 3 mL min−1, and the temperature and pressure are ramped to the desired conditions (400 °C and 30 bar). Table 2 summarizes and compares the catalytic performances after 20 h on stream for all catalysts during CO2 hydrogenation. The catalytic performances as a function of time on stream are presented in Fig. S19 and S20. No significant deactivation is observed for most catalysts (FeK/SiO2-700, FeK/Al2O3-500, FeK/ZrO2-250, FeK/Al2O3–MgO-500 and FeK/Nb2O5-250) over the course of 40 h (Fig. S19 and S20). The high stability of these catalysts is likely due to the presence of well-dispersed isolated single sites and the presence of K+ in proximity to Fe which stabilize Fe(II) and provide a favorable site for CO2 adsorption.43,45,84 However, the catalyst FeK/CeO2-250 showed an evolution of the stability as a function of time on stream.
:3) is set to 3 mL min−1, and the temperature and pressure are ramped to the desired conditions (400 °C and 30 bar). Table 2 summarizes and compares the catalytic performances after 20 h on stream for all catalysts during CO2 hydrogenation. The catalytic performances as a function of time on stream are presented in Fig. S19 and S20. No significant deactivation is observed for most catalysts (FeK/SiO2-700, FeK/Al2O3-500, FeK/ZrO2-250, FeK/Al2O3–MgO-500 and FeK/Nb2O5-250) over the course of 40 h (Fig. S19 and S20). The high stability of these catalysts is likely due to the presence of well-dispersed isolated single sites and the presence of K+ in proximity to Fe which stabilize Fe(II) and provide a favorable site for CO2 adsorption.43,45,84 However, the catalyst FeK/CeO2-250 showed an evolution of the stability as a function of time on stream.
Table 2 Catalytic performance of the different metal oxide supported materials during CO2 hydrogenation under identical conditions
| Catalyst |
Conversion (%) |
Selectivity (%) |
| CO |
CH4 |
C2+ |
| Feed composition: CO2/H2: 1/3, flow = 3 mL min−1, 400 °C, 30 bars, amount = 250 mg. |
| Neutral supports |
FeK/Al2O3-500
45
|
22.5 |
100 |
|
|
|
FeK/SiO2-700
|
18 |
100 |
— |
— |
| Basic supports |
FeK/Al2O3–MgO-500
|
17 |
100 |
— |
— |
|
FeK/ZrO2-250
|
22 |
100 |
— |
— |
| Acidic supports |
FeK/SiO2–Al2O3-50000
|
7.8 |
97 |
3 |
— |
|
FeK/μ-ZSM-5-250
|
10 |
90 |
10 |
— |
|
FeK/Nb2O5-250
|
14 |
100 |
— |
— |
| Redox supports |
FeK/CeO2-250
|
34 |
100–40 |
0–55 |
0–10 |
|
FeK/CeZrOx-250
|
26 |
100 |
|
— |
For acidic supports, the catalysts FeK/SiO2–Al2O3-500, FeK/μ-ZSM-5-250 and FeK/Nb2O5-250 show a lower CO2 conversion compared to other supports (Table 2). The activity of these catalysts decreases when the acidic strength of the supports increases (μ-ZSM-5-250 > SiO2–Al2O3-500 > Nb2O5-250).87–93 For the zeolite support, an ionic exchange process between a Brønsted proton and K+ may have occurred during the catalyst preparation, as already reported for Fe/Y-zeolite and Co/ZSM-5 promoted by K.94–96 This leads to a decrease of the amount of FeK active sites and a loss of the important K/Fe synergy for the RWGS reaction.43,45 However, for neutral or basic supports (Al2O3-500, Al2O3–MgO-500, SiO2-700 or ZrO2-250), no substantial support effect is observed in the RWGS reaction, with similar stable CO2 conversion (with just minor variations) and high CO selectivity (100%). Therefore, the use of basic and neutral supports for single-site catalysts containing tailored potassium promoters remains a promising strategy to tune the activity, selectivity and stability for the RWGS reaction. Interestingly, supports based on distinct elements such as Mg, Al, Si and Zr show virtually the same activity and selectivity in the RWGS reaction. The present data indicate that these elements contribute only to the dispersion and the stability of the active sites by keeping K+ in the proximity of Fe. Such proximity may limit the sintering of Fe single site to Fe nanoparticles, generally known to generate methane.97,98 This finding confirms the critical role of K/Fe proximity in CO2 adsorption and H2 dissociation, as described in the previously proposed mechanism (Scheme 1) and supported by DFT calculations.45 The latter is further supported by the use of ceria as a support. Indeed, when compared to acidic, neutral and basic supports, single-site FeK supported on redox oxide CeO2-250 shows a different behavior and undergoes several evolutions during the time on stream. At initial time (Fig. 5a), the conversion and the selectivity are similar to those obtained on neutral and basic supports (Conv. ca. 20% and Sel. 100%), indicating that Fe and K are in proximity and the selective conversion of CO2 to CO occurs in accordance with the postulated mechanism without any contribution of ceria. After 200 min, a gradual decrease in CO selectivity by formation of methane accompanied by an increase in CO2 conversion is observed (Fig. 5a). This feature can be associated with an evolution of the RWGS active site (isolated anionic FeK) upon the feed exposure, in particular, H2. This is consistent with partial Fe agglomeration under the reaction conditions, resulting in small Fe nanoparticles responsible for the methanation and light hydrocarbon production (C2H6 and C3H8).99 Indeed, it is known that small Fe particles are more selective towards methane during CO2 reduction, while larger Fe particles lead generally to light olefins and alkanes.97,98 The agglomeration of Fe nanoparticles is assumed to pursue until a steady state (after 1300 min), where the conversion (34%) and selectivities remain constant (CO = 38%; CH4 = 54%; C2–C3 = 9%). The evolution of the selectivity from CO towards methane during CO2 conversion has already been reported for Ru/Al2O3 where the authors observe the evolution of the active phase from Ru single site to Ru nanoparticles using HRTEM and DRIFT during the RWGS reaction.99 Another important example is reported in the literature describing the RWGS reaction over Ni supported on ceria with 1% loading, modified or unmodified by K+.100 This study suggests a K-guided selectivity control method based on the regulation of key intermediates HCO*/H3CO* for Ni/CeO2 catalysts. By incorporating K into Ni/CeO2, the CO selectivity of CO2 hydrogenation at 400 °C shifts from around 9% for Ni/CeO2 to approximately 85% for Ni/CeO2 promoted by K. The high stability of the K–Ni/CeO2 catalyst during the selective conversion of CO2 to CO is attributed to the presence of excess of K (K/Ni ratio of 6) which limits the formation of nickel nanoparticles while always keeping a K in the proximity of each Ni. Importantly, Ang et al. reported that at low loadings of Na (0.5 to 2 wt%), Na+ can readily incorporate into the defective CeO2 lattice,101 generating a lattice strain and activating the lattice oxygen, thereby increasing the reducibility of the catalyst. However, beyond the solubility limit of 2 wt%, Na is deposited on the CeO2 surface, thus retarding the reducibility and decreasing the activity in WGS.101 Based on these findings, we propose a similar pathway in the case of FeK/CeO2-250 where the evolution of the stability and selectivity during the RWGS reaction may be explained by the incorporation of K+ into the CeO2-250 lattice, leading to the partial loss of K in the proximity of Fe. The naked Fe single site without K in proximity can agglomerate over time in the presence of H2 at 400 °C to form Fe nanoparticles responsible for the formation of methane. The evolution of the selectivity to CO and methane during the RWGS reaction can thus be correlated with the ratio of FeK single sites and Fe nanoparticles. The local structure of FeK/CeO2-250 before and after catalysis has been investigated using a combination of XRD and HRTEM analyses (Fig. S21 and S22). However, due to the low loading of Fe, no Fe particles on ceria can be detected. Such limitations in XRD and HRTEM analyses have previously been reported for Fe and Cu supported on CeO2, where no sign of Fe and Cu is observed until the loadings exceeded 10 wt%.102–104 In addition, Fe K-edge X-ray spectroscopy of the catalyst FeK/CeO2-250 before and after the catalytic tests cannot lead to any significant changes due to the heavy absorption of Ce.105 Furthermore, when the catalyst, FeK/CeO2-250, is reduced under hydrogen at a higher temperature (500 °C), the amount of the promotor, K, in the lattice of CeO2 may increase and leads to an increase of Fe nanoparticles on the surface and consequently a drop of the selectivity in CO from 45% to 15% due to the methanation reaction (Fig. S23). For comparison, FeK/CeZrOx-250 is tested under the same conditions as FeK/CeO2-250 in the RWGS reaction. The catalyst FeK/CeZrOx-250 exhibits high activity and stability over time on stream with full selectivity in CO (100%) compared to FeK/CeO2-250. Thus, the presence of zirconia on the mixed CeZrOx support seems to inhibit the diffusion phenomenon of K in the case of the FeK/CeZrOx-250 catalyst, resulting in high selectivity and activity in the RWGS reaction due to the synergy between Fe and K. This result reveals that the active site FeK is selectively immobilised on the ZrO2 phase rather than CeO2 in CeZrOx mixed oxide since we demonstrate (vide supra) that FeK/ZrO2-250 is more selective in the RWGS reaction than FeK/CeO2-250. Indeed, previous studies report that under reductive conditions, the structure of ceria–zirconia support is more stable than that of pure CeO2.106–108
 |
| | Fig. 5 CO2 conversion and product selectivity over (a) FeK/CeO2-250 and (b) FeK/CeZrOx-250 (feed composition: CO2/H2: 1/3, flow = 3 mL min−1, 400 °C, 30 bar). | |
Therefore, all the results obtained support the hypothesis that the reduction of CO2 to CO is directed by the presence of K and Fe in proximity and that the supports contribute to the dispersion of the active phase. However, while neutral and basic supports have little influence on performance, ZrO2 provided the best results, maintaining high activity, selectivity, and long-term stability by preserving the Fe–K single site without agglomeration. In contrast, acidic supports degrade the performance due to ion exchange, disrupting Fe–K synergy, and CeO2 causes Fe agglomeration and methane formation, though CeZrO2 mitigates these issues. Mechanistic studies confirm that the cooperative action between Fe(II) and nearby K+ cations is key to the reaction, with the support's role being primarily to stabilize this active site. This work highlights the importance of support-controlled dispersion and stabilization of Fe–K sites for efficient CO2-to-CO conversion.
4. Conclusions
The heterobimetallic complex, [{(THF)2KFe(OtBu)3}2], is successfully grafted with 1 wt% Fe loading onto a variety of solid supports, encompassing a wide spectrum of surface chemistries: neutral (SiO2 and Al2O3), basic (Al2O3–MgO and ZrO2), acidic (SiO2–Al2O3, μ-H-ZSM-5, and Nb2O5), and redox (CeO2 and CeZrOx) oxides. This is achieved via a synthetic strategy based on SOMC for the grafting of reactive species in a stable and well dispersed fashion. Regardless of the support used, the grafted complexes consistently retain a K/Fe ratio of 1, with Fe remaining in its +2 oxidation state. The potassium cations play a stabilizing role, ensuring that anionic Fe–K single sites remains atomically dispersed across the support surfaces. Comprehensive characterization by DRIFT, EPR, ICP, EDX, HRTEM, BET, XRD, and Fe K-edge XAFS (especially for the ZrO2 supported sample) confirm these structural features.
Catalytic tests in the RWGS reaction reveal the critical influence of the support in tuning activity, selectivity, and long-term performances. On neutral and basic oxides, the catalytic behavior is similar, showing minor dependence on the support's properties. However, ZrO2 stands out: FeK/ZrO2-250 demonstrates exceptional RWGS activity and stability. This is attributed to zirconia's unique ability to anchor and disperse Fe–K species without promoting aggregation or sintering. Post-reaction analyses confirm this stability. HRTEM images show no signs of clustering even after 40 hours on stream, while EXAFS spectra reveal no Fe–Fe interactions, indicating that the local environment, defined by Fe–O, Fe–K, and Fe–Zr distances, remains similar throughout the reaction.
In contrast, catalysts supported on acidic oxides (μ-ZSM-5, SiO2–Al2O3, and Nb2O5) are far less effective. These materials likely suffer from ion exchange between K+ and Brønsted acid sites, disrupting the subtle Fe–K synergy essential for catalysis. Redox-active CeO2 initially exhibits promising CO selectivity, but this quickly declines due to structural changes by diffusion of K+ into the ceria lattice and formation of Fe nanoparticles, as previously reported in the literature, shifting the selectivity toward methane. Interestingly, due to the presence of the ZrO2 phase, the mixed oxide, CeZrOx, overcomes this issue by offering a more stable framework, preserving both catalytic performance and selectivity. Mechanistically, the RWGS reaction is found to rely on a cooperative interaction between Fe(II) and K+: Fe(II) facilitates H2 and CO2 activation, forming hydroxycarbonyl intermediates, while nearby K+ promoted CO2 adsorption and transition state stabilization. Across all supports, this Fe–K cooperation, not the inherent properties of the oxide supports, is the principal driver of activity and CO selectivity. Thus, this study highlights the importance of pairing Fe(II) with K+ at the atomic level and choosing the right precursor and support to preserve this synergy. This work offers valuable insight into designing robust, selective RWGS catalysts for CO2 conversion under mild conditions. Importantly, such materials may easily be transformed into bifunctional catalysts by adding a Fischer–Tropsch (FT) active phase (for example, well-defined metallic nanoparticles based on Fe, Co or Ru), yielding hydrocarbons directly from CO2 through consecutive RWGS and FT reactions.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Abdulrahman Adamu Isah and Yahaya Nasiru performed the synthesis and characterization of catalysts and testing of the catalysts. Fadila Hamachi and Jie Pan performed the synthesis and characterization of catalysts and testing of the catalysts based on silica and ZSM-5. Pierre-Yves Dugas conducted the electron microscopy studies. All these tasks were carried out under the supervision of Kai C. Szeto for testing of the catalysts and Aimery De Mallmann for the characterization of materials; Mostafa Taoufik and Cyril Godard conceived the initial idea and designed the project.
Conflicts of interest
The authors declare that they have no known competing financial interest or personal relationships that could have appeared to influence the work reported in this study.
Data availability
The data supporting this article have been included as part of the supplementary information (SI): Supplementary information is available. See DOI: https://doi.org/10.1039/d5nj03345d.
Acknowledgements
A. A. I. and N. Y. thank the Petroleum Technology Development Fund (PTDF). A. D. M. thank Olivier Mathon and Kiril Lomachenko for their help during the recording of the X-ray absorption spectra at ESRF, on beamline BM23 (proposal IN-1134). J. P. acknowledges grant PID2021-128128NB-I00 funded by MICIU/AEI/10.13039/501100011033 and by “FEDER/UE” and the Generalitat de Catalunya (2021SGR00110).
Notes and references
- R. M. Bown, M. Joyce, Q. Zhang, T. R. Reina and M. S. Duyar, Identifying Commercial Opportunities for the Reverse Water Gas Shift Reaction, Energy Technol., 2021, 9, 2100554 CrossRef CAS.
- K. T. Rommens and M. Saeys, Molecular Views on Fischer–Tropsch Synthesis, Chem. Rev., 2023, 123, 5798–5858 CrossRef CAS PubMed.
- T. Lin, Y. An, F. Yu, K. Gong, H. Yu, C. Wang, Y. Sun and L. Zhong, Advances in Selectivity Control for Fischer–Tropsch Synthesis to Fuels and Chemicals with High Carbon Efficiency, ACS Catal., 2022, 12, 12092–12112 CrossRef CAS.
- P. Ebrahimi, A. Kumar and M. Khraisheh, A Review of CeO2 Supported Catalysts for CO2 Reduction to CO through the Reverse Water Gas Shift Reaction, Catalysts, 2022, 12, 1101 CrossRef CAS.
- R. Zhang, Y. Wang, P. Gaspard and N. Kruse, The oscillating Fischer-Tropsch reaction, Science, 2023, 382, 99–103 CrossRef CAS PubMed.
- P. Buchenberg, T. Addanki, D. Franzmann, C. Winkler, F. Lippkau, T. Hamacher, P. Kuhn, H. Heinrichs and M. Blesl, Global Potentials and Costs of Synfuels via Fischer–Tropsch Process, Energies, 2023, 16, 1976 CrossRef CAS.
- M. P. Jones, T. Krexner and A. Bismarck, Repurposing Fischer-Tropsch and natural gas as bridging technologies for the energy revolution, Energy Convers. Manage., 2022, 267, 115882 CrossRef CAS.
- K. Ahmad and S. Upadhyayula, Greenhouse gas CO2 hydrogenation to fuels: a thermodynamic analysis, Environ. Prog. Sustainable Energy, 2019, 38, 98–111 CrossRef CAS.
- A. Wolf, A. Jess and C. Kern, Syngas Production via Reverse Water–Gas Shift Reaction over a Ni–Al2O3 Catalyst: Catalyst Stability, Reaction Kinetics, and Modeling, Chem. Eng. Technol., 2016, 39, 1040–1048 CrossRef CAS.
- E. Portillo, J. Gandara-Loe, T. R. Reina and L. Pastor-Pérez, Is the RWGS a viable route for CO2 conversion to added value products? A techno-economic study to understand the optimal RWGS conditions, Sci. Total Environ., 2023, 857, 159394 CrossRef CAS PubMed.
- X. Su, X. Yang, B. Zhao and Y. Huang, Designing of highly selective and high-temperature endurable RWGS heterogeneous catalysts: recent advances and the future directions, J. Energy Chem., 2017, 26, 854–867 CrossRef.
- I. Hussain, A. A. Jalil, N. S. Hassan and M. Y. S. Hamid, Recent advances in catalytic systems for CO2 conversion to substitute natural gas (SNG): perspective and challenges, J. Energy Chem., 2021, 62, 377–407 CrossRef CAS.
- A. H. Hatta, A. A. Jalil, N. S. Hassan, M. Y. S. Hamid, M. B. Bahari, M. A. Aziz, M. Alhassan, N. Ibrahim, N. W. C. Jusoh and N. H. H. Hairom, A comprehensive review on the advancements in catalyst regeneration strategies for enhanced reactivity in CO methanation, Mater. Today Chem., 2023, 33, 101743 CrossRef CAS.
- W. K. Fan and M. Tahir, Recent trends in developments of active metals and heterogenous materials for catalytic CO2 hydrogenation to renewable methane: a review, J. Environ. Chem. Eng., 2021, 9, 105460 CrossRef CAS.
- D. Schmider, L. Maier and O. Deutschmann, Reaction Kinetics of CO and CO2 Methanation over Nickel, Ind. Eng. Chem. Res., 2021, 60, 5792–5805 CrossRef CAS.
- S. Kattel, B. Yan, J. G. Chen and P. Liu, CO2 hydrogenation on Pt, Pt/SiO2 and Pt/TiO2: importance of synergy between Pt and oxide support, J. Catal., 2016, 343, 115–126 CrossRef CAS.
- M. Zhu, Q. Ge and X. Zhu, Catalytic Reduction of CO2 to CO via Reverse Water Gas Shift Reaction: Recent Advances in the Design of Active and Selective Supported Metal Catalysts, Trans. Tianjin Univ., 2020, 26, 172–187 CrossRef CAS.
- X. Chen, Y. Chen, C. Song, P. Ji, N. Wang, W. Wang and L. Cui, Recent Advances in Supported Metal Catalysts and Oxide Catalysts for the Reverse Water-Gas Shift Reaction, Front. Chem., 2020, 8, 709 CrossRef CAS PubMed.
- M. González-Castaño, B. Dorneanu and H. Arellano-García, The reverse water gas shift reaction: a process systems engineering perspective, React. Chem. Eng., 2021, 6, 954–976 RSC.
- Y. A. Daza and J. N. Kuhn, CO2 conversion by reverse water gas shift catalysis: comparison of catalysts, mechanisms and their consequences for CO2 conversion to liquid fuels, RSC Adv., 2016, 6, 49675–49691 RSC.
- B. Liang, H. Duan, X. Su, X. Chen, Y. Huang, X. Chen, J. J. Delgado and T. Zhang, Promoting role of potassium in the reverse water gas shift reaction on Pt/mullite catalyst, Catal. Today, 2017, 281, 319–326 CrossRef CAS.
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, Optimizing Binding Energies of Key Intermediates for CO2 Hydrogenation to Methanol over Oxide-Supported Copper, J. Am. Chem. Soc., 2016, 138, 12440–12450 CrossRef CAS PubMed.
- J. H. Kwak, L. Kovarik and J. Szanyi, Heterogeneous Catalysis on Atomically Dispersed Supported Metals: CO2 Reduction on Multifunctional Pd Catalysts, ACS Catal., 2013, 3, 2094–2100 CrossRef CAS.
- L. F. Bobadilla, J. L. Santos, S. Ivanova, J. A. Odriozola and A. Urakawa, Unravelling the Role of Oxygen Vacancies in the Mechanism of the Reverse Water–Gas Shift Reaction by Operando DRIFTS and Ultraviolet-Visible Spectroscopy, ACS Catal., 2018, 8, 7455–7467 CrossRef CAS.
- B. Lu, Y. Ju, T. Abe and K. Kawamoto, Dispersion and distribution of bimetallic oxides in SBA-15, and their enhanced activity for reverse water gas shift reaction, Inorg. Chem. Front., 2015, 2, 741–748 RSC.
- B. Lu and K. Kawamoto, Preparation of monodispersed NiO particles in SBA-15, and its enhanced selectivity for reverse water gas shift reaction, J. Environ. Chem. Eng., 2013, 1, 300–309 CrossRef CAS.
- C. Wang, E. Guan, L. Wang, X. Chu, Z. Wu, J. Zhang, Z. Yang, Y. Jiang, L. Zhang, X. Meng, B. C. Gates and F.-S. Xiao, Product Selectivity Controlled by Nanoporous Environments in Zeolite Crystals Enveloping Rhodium Nanoparticle Catalysts for CO2 Hydrogenation, J. Am. Chem. Soc., 2019, 141, 8482–8488 CrossRef CAS PubMed.
- S. Biswas, C. Kundu, A. P. Kulkarni, S. Kattel, S. Giddey and S. Bhattacharya, A Study on CO2 Hydrogenation Using a Ceria–Zirconia Mixed Oxide (CexZr1−xO2)-Supported Fe Catalyst, Ind. Eng. Chem. Res., 2021, 60, 14410–14423 CrossRef CAS.
- B. Yan, B. Zhao, S. Kattel, Q. Wu, S. Yao, D. Su and J. G. Chen, Tuning CO2 hydrogenation selectivity via metal-oxide interfacial sites, J. Catal., 2019, 374, 60–71 CrossRef CAS.
- S. Kattel, W. Yu, X. Yang, B. Yan, Y. Huang, W. Wan, P. Liu and J. G. Chen, CO2 Hydrogenation over Oxide-Supported PtCo Catalysts: The Role of the Oxide Support in Determining the Product Selectivity, Angew. Chem., Int. Ed., 2016, 55, 7968–7973 CrossRef CAS PubMed.
- S. S. Kim, H. H. Lee and S. C. Hong, A study on the effect of support's reducibility on the reverse water-gas shift reaction over Pt catalysts, Appl. Catal., A, 2012, 423–424, 100–107 CAS.
- M. Nolan, S. C. Parker and G. W. Watson, The electronic structure of oxygen vacancy defects at the low index surfaces of ceria, Surf. Sci., 2005, 595, 223–232 CrossRef CAS.
- B. Lu and K. Kawamoto, Preparation of mesoporous CeO2 and monodispersed NiO particles in CeO2, and enhanced selectivity of NiO/CeO2 for reverse water gas shift reaction, Mater. Res. Bull., 2014, 53, 70–78 CrossRef CAS.
- J. Yoshihara and C. T. Campbell, Methanol Synthesis and Reverse Water–Gas Shift Kinetics over Cu(110)
Model Catalysts: Structural Sensitivity, J. Catal., 1996, 161, 776–782 CrossRef CAS.
- H.-J. Li and J.-J. Ho, Density Functional Calculations on the Hydrogenation of Carbon Dioxide on Fe(111) and W(111) Surfaces, J. Phys. Chem. C, 2010, 114, 1194–1200 CrossRef CAS.
- E. Pahija, C. Panaritis, S. Gusarov, J. Shadbahr, F. Bensebaa, G. Patience and D. C. Boffito, Experimental and Computational Synergistic Design of Cu and Fe Catalysts for the Reverse Water–Gas Shift: A Review, ACS Catal., 2022, 12, 6887–6905 CrossRef CAS.
- D. Li, X. Ding, X. Liu, J. Cheng, Z. Jiang and Y. Guo, CO2 hydrogenation to methane over Ni/ZrO2 and Ni/CeO2 catalysts: experimental and DFT studies, J. Mater. Sci., 2023, 58, 12584–12595 CrossRef CAS.
- Z. Wang, Q. Wang, Y. Liao, G. Shen, X. Gong, N. Han, H. Liu and Y. Chen, Comparative Study of CeO2 and Doped CeO2 with Tailored Oxygen Vacancies for CO Oxidation, ChemPhysChem, 2011, 12, 2763–2770 CrossRef CAS PubMed.
- Y. Yu, W. Xia, A. Yu, D. S. A. Simakov and L. Ricardez-Sandoval, Transition-Metal-Doped CeO2 for the Reverse Water–Gas Shift Reaction: An Experimental and Theoretical Study on CO2 Adsorption and Surface Vacancy Effects, ChemSusChem, 2025, 18, e202400681 CrossRef CAS PubMed.
- P. Ebrahimi, A. Kumar and M. Khraisheh, A Review of CeO2 Supported Catalysts for CO2 Reduction to CO through the Reverse Water Gas Shift Reaction, Catalysts, 2022, 12, 1101 CrossRef CAS.
- X. Zhu, M. Shen, L. L. Lobban and R. G. Mallinson, Structural effects of Na promotion for high water gas shift activity on Pt–Na/TiO2, J. Catal., 2011, 278, 123–132 CrossRef CAS.
- M. Yang, S. Li, Y. Wang, J. A. Herron, Y. Xu, L. F. Allard, S. Lee, J. Huang, M. Mavrikakis and M. Flytzani-Stephanopoulos, Catalytically active Au–O(OH)(x)-species stabilized by alkali ions on zeolites and mesoporous oxides, Science, 2014, 346, 1498–1501 CrossRef CAS PubMed.
- J. A. Loiland, M. J. Wulfers, N. S. Marinkovic and R. F. Lobo, Fe/γ-Al2O3 and Fe–K/γ-Al2O3 as reverse water-gas shift catalysts, Catal. Sci. Technol., 2016, 6, 5267–5279 RSC.
- M. K. Samantaray, V. D’Eia, E. Pump, L. Falivene, M. Harb, S. O. Chikh, L. Cavallo and J.-M. Basset, The Comparison between Single Atom Catalysis and Surface Organometallic Catalysis, Chem. Rev., 2020, 120, 734–813 CrossRef CAS PubMed.
- A. A. Isah, O. Ohiro, L. Li, Y. Nasiru, K. C. Szeto, P.-Y. Dugas, A. Benayad, A. De Mallmann, S. L. Scott, B. R. Goldsmith and M. Taoufik, Selective Catalytic Reduction of CO2 to CO by a Single-Site Heterobimetallic Iron–Potassium Complex Supported on Alumina, ACS Catal., 2024, 14, 2418–2428 CrossRef CAS.
- B. Ravel and M. Newville,
ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- A. Michalowicz, J. Moscovici, D. Muller-Bouvet and K. Provost, MAX: Multiplatform Applications for XAFS, J. Phys.: Conf. Ser., 2009, 190, 012034 CrossRef.
- A. L. Ankudinov, B. Ravel, J. J. Rehr and S. D. Conradson, Real-space multiple-scattering calculation and interpretation of X-ray-absorption near-edge structure, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7565–7576 CrossRef CAS.
- H. Zou and Y. S. Lin, Structural and surface chemical properties of sol–gel derived TiO2–ZrO2 oxides, Appl. Catal., A, 2004, 265, 35–42 CrossRef CAS.
- J. M. Dominguez, J. L. Hernandez and G. Sandoval, Surface and catalytic properties of Al2O3–ZrO2 solid solutions prepared by sol–gel methods, Appl. Catal., A, 2000, 197, 119–130 CrossRef CAS.
- A. Chareonlimkun, V. Champreda, A. Shotipruk and N. Laosiripojana, Catalytic conversion of sugarcane bagasse, rice husk and corncob in the presence of TiO2, ZrO2 and mixed-oxide TiO2–ZrO2 under hot compressed water (HCW) condition, Bioresour. Technol., 2010, 101, 4179–4186 CrossRef CAS PubMed.
- L. D’Souza, A. Suchopar, K. Zhu, D. Balyozova, M. Devadas and R. M. Richards, Preparation of thermally stable high surface area mesoporous tetragonal ZrO2 and Pt/ZrO2: an active hydrogenation catalyst, Microporous Mesoporous Mater., 2006, 88, 22–30 CrossRef.
- H. Tel, Y. Altaş, M. Eral, Ş. Sert, B. Çetinkaya and S. İnan, Preparation of ZrO2 and ZrO2–TiO2 microspheres by the sol–gel method and an experimental design approach to their strontium adsorption behaviours, Chem. Eng. J., 2010, 161, 151–160 CrossRef CAS.
- R. D. Purohit, S. Saha and A. K. Tyagi, Combustion synthesis of nanocrystalline ZrO2 powder: XRD, Raman spectroscopy and TEM studies, Mater. Sci. Eng., B, 2006, 130, 57–60 CrossRef CAS.
- R. Srinivasan, B. H. Davis, O. B. Cavin and C. R. Hubbard, Crystallization and Phase Transformation Process in Zirconia: An in situ High-Temperature X-ray Diffraction Study, J. Am. Ceram. Soc., 1992, 75, 1217–1222 CrossRef CAS.
- D. Lopez, K. Suwannakarn, D. Bruce and J. Goodwinjr, Esterification and transesterification on tungstated zirconia: effect of calcination temperature, J. Catal., 2007, 247, 43–50 CrossRef CAS.
- Y. Zhao, W. Li, M. Zhang and K. Tao, A comparison of surface acidic features between tetragonal and monoclinic nanostructured zirconia, Catal. Commun., 2002, 3, 239–245 CrossRef CAS.
- M. T. Nikolajsen, J.-C. Grivel, A. Gaur, L. P. Hansen, L. Baumgarten, N. C. Schjødt, U. V. Mentzel, J.-D. Grunwaldt, J. Sehested, J. M. Christensen and M. Høj, Surface ZnO on zirconia is highly active for high temperature methanol synthesis, J. Catal., 2024, 431, 115389 CrossRef CAS.
- Z.-Y. Ma, C. Yang, W. Wei, W.-H. Li and Y.-H. Sun, Surface properties and CO adsorption on zirconia polymorphs, J. Mol. Catal. A: Chem., 2005, 227, 119–124 CrossRef CAS.
- S. Kouva, K. Honkala, L. Lefferts and J. Kanervo, Review: monoclinic zirconia, its surface sites and their interaction with carbon monoxide, Catal. Sci. Technol., 2015, 5, 3473–3490 RSC.
- Y. Zhao, W. Li, M. Zhang and K. Tao, A comparison of surface acidic features between tetragonal and monoclinic nanostructured zirconia, Catal. Commun., 2002, 3, 239–245 CrossRef CAS.
- K. T. Jung and A. T. Bell, The effects of synthesis and pretreatment conditions on the bulk structure and surface properties of zirconia, J. Mol. Catal. A: Chem., 2000, 163, 27–42 CrossRef CAS.
- S. T. Korhonen, S. M. K. Airaksinen, M. A. Bañares and A. O. I. Krause, Isobutane dehydrogenation on zirconia-, alumina-, and zirconia/alumina-supported chromia catalysts, Appl. Catal., A, 2007, 333, 30–41 CrossRef CAS.
- B. Bachiller-Baeza, I. Rodriguez-Ramos and A. Guerrero-Ruiz, Interaction of Carbon Dioxide with the Surface of Zirconia Polymorphs, Langmuir, 1998, 14, 3556–3564 CrossRef CAS.
- D. M. Kaphan, R. C. Klet, F. A. Perras, M. Pruski, C. Yang, A. J. Kropf and M. Delferro, Surface Organometallic Chemistry of Supported Iridium(III) as a Probe for Organotransition Metal–Support Interactions in C–H Activation, ACS Catal., 2018, 8, 5363–5373 CrossRef CAS.
- S. T. Korhonen, M. Calatayud and A. O. I. Krause, Structure and Stability of Formates and Carbonates on Monoclinic Zirconia: A Combined Study by Density Functional Theory and Infrared Spectroscopy, J. Phys.
Chem. C, 2008, 112, 16096–16102 CrossRef CAS.
- S. T. Korhonen, M. Calatayud and A. O. I. Krause, Stability of Hydroxylated (
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 11) and (01) Surfaces of Monoclinic Zirconia: A Combined Study by DFT and Infrared Spectroscopy, J. Phys. Chem. C, 2008, 112, 6469–6476 CrossRef CAS.
11) and (01) Surfaces of Monoclinic Zirconia: A Combined Study by DFT and Infrared Spectroscopy, J. Phys. Chem. C, 2008, 112, 6469–6476 CrossRef CAS.
- E. Mazoyer, J. Trébosc, A. Baudouin, O. Boyron, J. Pelletier, J. Basset, M. J. Vitorino, C. P. Nicholas, R. M. Gauvin, M. Taoufik and L. Delevoye, Heteronuclear NMR Correlations To Probe the Local Structure of Catalytically Active Surface Aluminum Hydride Species on γ-Alumina, Angew. Chem., Int. Ed., 2010, 49, 9854–9858 CrossRef CAS PubMed.
- C. Larabi, C. Chen, N. Merle, M. Charlin, K. C. Szeto, A. De Mallmann, A. Benayad, K. B. Meziane, A. Kaddouri, H. P. Nguyen and M. Taoufik, Well-defined surface tungstenocarbyne complex through the reaction of [W(CtBu)(CH2tBu)3] with CeO2: a highly stable precatalyst for NOx reduction with NH3, New J. Chem., 2021, 45, 12024–12032 RSC.
- H. Idriss, C. Diagne, J. P. Hindermann, A. Kiennemann and M. A. Barteau, Reactions of Acetaldehyde on CeO2 and CeO2-Supported Catalysts, J. Catal., 1995, 155, 219–237 CrossRef CAS.
- E. S. Bickford, S. Velu and C. Song, Nano-structured CeO2 supported Cu-Pd bimetallic catalysts for the oxygen-assisted water–gas-shift reaction, Catal. Today, 2005, 99, 347–357 CrossRef CAS.
- E. B. Fox, S. Velu, M. H. Engelhard, Y.-H. Chin, J. T. Miller, J. Kropf and C. Song, Characterization of CeO2-supported Cu–Pd bimetallic catalyst for the oxygen-assisted water–gas shift reaction, J. Catal., 2008, 260, 358–370 CrossRef CAS.
- S. Y. Choung, M. Ferrandon and T. Krause, Pt-Re bimetallic supported on CeO2–ZrO2 mixed oxides as water–gas shift catalysts, Catal. Today, 2005, 99, 257–262 CrossRef CAS.
- T. Sakpal and L. Lefferts, Structure-dependent activity of CeO2 supported Ru catalysts for CO2 methanation, J. Catal., 2018, 367, 171–180 CrossRef CAS.
- H. T. T. Nguyen, Y. Kumabe, S. Ueda, K. Kan, M. Ohtani and K. Kobiro, Highly durable Ru catalysts supported on CeO2 nanocomposites for CO2 methanation, Appl. Catal., A, 2019, 577, 35–43 CrossRef CAS.
- S. Zhang and T. Kim, Effects of iron precursor and loading on the catalytic performance of FeOx/CeO2 catalysts for NO reduction by CO, Mol. Catal., 2020, 494, 111123 CAS.
- X. Qi and M. Flytzani-Stephanopoulos, Activity and Stability of Cu−CeO2 Catalysts in High-Temperature Water−Gas Shift for Fuel-Cell Applications, Ind. Eng. Chem. Res., 2004, 43, 3055–3062 CrossRef CAS.
- D. Y. Ebert, N. V. Dorofeeva, A. S. Savel’eva, T. S. Kharlamova, M. A. Salaev, V. A. Svetlichnyi, O. V. Magaev and O. V. Vodyankina, Silica-supported Fe–Mo–O catalysts for selective oxidation of propylene glycol, Catal. Today, 2019, 333, 133–139 CrossRef CAS.
- S. K. Das, S. Majhi, P. Mohanty and K. K. Pant, CO-hydrogenation of syngas to fuel using silica supported Fe–Cu–K catalysts: Effects of active components, Fuel Process. Technol., 2014, 118, 82–89 CrossRef CAS.
- M. P. Maia, M. A. Rodrigues and F. B. Passos, Nitrate catalytic reduction in water using niobia supported palladium–copper catalysts, Catal. Today, 2007, 123, 171–176 CrossRef CAS.
- K. V. R. Chary, K. K. Seela, G. V. Sagar and B. Sreedhar, Characterization and Reactivity of Niobia Supported Copper Oxide Catalysts, J. Phys. Chem. B, 2004, 108, 658–663 CrossRef CAS.
- L. Galoisy, G. Calas and M. A. Arrio, High-resolution XANES spectra of iron in minerals and glasses: structural information from the pre-edge region, Chem. Geol., 2001, 174, 307–319 CrossRef CAS.
- T. E. Westre, P. Kennepohl, J. G. DeWitt, B. Hedman, K. O. Hodgson and E. I. Solomon, A Multiplet Analysis of Fe K-Edge 1s → 3d Pre-Edge Features of Iron Complexes, J. Am. Chem. Soc., 1997, 119, 6297–6314 CrossRef CAS.
- J. A. Loiland, M. J. Wulfers, N. S. Marinkovic and R. F. Lobo, Fe/gamma-Al2O3 and Fe-K/gamma-Al2O3 as reverse water-gas shift catalysts, Catal. Sci. Technol., 2016, 6, 5267–5279 RSC.
- K. C. Szeto, Z. R. Jones, N. Merle, C. Rios, A. Gallo, F. Le Quemener, L. Delevoye, R. M. Gauvin, S. L. Scott and M. Taoufik, A Strong Support Effect in Selective Propane Dehydrogenation Catalyzed by Ga(i-Bu)3 Grafted onto γ-Alumina and Silica, ACS Catal., 2018, 8, 7566–7577 CrossRef CAS.
- in Modern Surface Organometallic Chemistry, ed. J. Basset, R. Psaro, D. Roberto and R. Ugo, Wiley, 1st edn, 2009 Search PubMed.
- M. Paulis, M. Martín, D. B. Soria, A. Díaz, J. A. Odriozola and M. Montes, Preparation and characterization of niobium oxide for the catalytic aldol condensation of acetone, Appl. Catal., A, 1999, 180, 411–420 CrossRef CAS.
- L. Faba, J. Gancedo, J. Quesada, E. Diaz and S. Ordóñez, One-Pot Conversion of Acetone into Mesitylene over Combinations of Acid and Basic Catalysts, ACS Catal., 2021, 11, 11650–11662 CrossRef CAS.
- P. Reif, N. K. Gupta and M. Rose, Highly stable amorphous silica-alumina catalysts for continuous bio-derived mesitylene production under solvent-free conditions, Green Chem., 2023, 25, 1588–1596 RSC.
- I. S. Pieta, M. Ishaq, R. P. K. Wells and J. A. Anderson, Quantitative determination of acid sites on silica–alumina, Appl. Catal., A, 2010, 390, 127–134 CrossRef CAS.
- S. Kotrel, M. P. Rosynek and J. H. Lunsford, Quantification of Acid Sites in H-ZSM-5, H-β, and H-Y Zeolites, J. Catal., 1999, 182, 278–281 CrossRef CAS.
- R. Borade, A. Sayari and A. Adnot, and Serge. Kaliaguine, Characterization of acidity in ZSM-5 zeolites: an X-ray photoelectron and IR spectroscopy study, J. Phys. Chem., 1990, 94, 5989–5994 CrossRef CAS.
- I. E. Wachs, J.-M. Jehng and W. Ueda, Determination of the Chemical Nature of Active Surface Sites Present on Bulk Mixed Metal Oxide Catalysts, J. Phys. Chem. B, 2005, 109, 2275–2284 CrossRef CAS PubMed.
- T. Riedel, M. Claeys, H. Schulz, G. Schaub, S.-S. Nam, K.-W. Jun, M.-J. Choi, G. Kishan and K.-W. Lee, Comparative study of Fischer–Tropsch synthesis with H2/CO and H2/CO2 syngas using Fe- and Co-based catalysts, Appl. Catal., A, 1999, 186, 201–213 CrossRef CAS.
- R. Liu, D. Leshchev, E. Stavitski, M. Juneau, J. N. Agwara and M. D. Porosoff, Selective hydrogenation of CO2 and CO over potassium promoted Co/ZSM-5, Appl. Catal., B, 2021, 284, 119787 CrossRef CAS.
- K. Qin, Y. Men, S. Liu, J. Wang, Z. Li, D. Tian, T. Shi, W. An, X. Pan and L. Li, Direct conversion of carbon dioxide to liquid hydrocarbons over K-modified CoFeOx/zeolite multifunctional catalysts, J. CO2 Util., 2022, 65, 102208 CrossRef CAS.
- T. Franken and A. Heel, Are Fe based catalysts an upcoming alternative to Ni in CO2 methanation at elevated pressure?, J. CO2 Util., 2020, 39, 101175 CrossRef CAS.
- T. Xie, J. Wang, F. Ding, A. Zhang, W. Li, X. Guo and C. Song, CO2 hydrogenation to hydrocarbons over alumina-supported iron catalyst: Effect of support pore size, J. CO2 Util., 2017, 19, 202–208 CrossRef CAS.
- J. H. Kwak, L. Kovarik and J. Szanyi, CO2 Reduction on Supported Ru/Al2O3 Catalysts: Cluster Size Dependence of Product Selectivity, ACS Catal., 2013, 3, 2449–2455 CrossRef CAS.
- Y. Zang, Z. Zhang, J. Qu, F. Gao, J. Gu, T. Wei and X. Lin, K-guided selective regulation mechanism for CO2 hydrogenation over Ni/CeO2 catalyst, J. Colloid Interface Sci., 2024, 658, 167–178 CrossRef CAS PubMed.
- M. L. Ang, U. Oemar, E. T. Saw, L. Mo, Y. Kathiraser, B. H. Chia and S. Kawi, Highly Active Ni/xNa/CeO2 Catalyst for the Water–Gas Shift Reaction: Effect of Sodium on Methane Suppression, ACS Catal., 2014, 4, 3237–3248 CrossRef CAS.
- Z. Gou, C. Huang, G. Zhou, X. Ren, L. Deng, T. Wang and Q. Peng, Coupling and electronic synergistic effects of Fe/CeO2 composite to achieve high efficiency and selectivity for RWGS reaction, J. CO2 Util., 2024, 81, 102728 CrossRef CAS.
- T. C. Peck, G. K. Reddy, M. Jones and C. A. Roberts, Monolayer Detection of Supported Fe and Co Oxides on Ceria To Establish Structure–Activity Relationships for Reduction of NO by CO, J. Phys. Chem. C, 2017, 121, 8435–8443 CrossRef CAS.
- M. Sakai, Y. Nagai, Y. Aoki and N. Takahashi, Investigation into the catalytic reduction of NO at copper–ceria interface active sites, Appl. Catal., A, 2016, 510, 57–63 CrossRef CAS.
- A. Gili, M. F. Bekheet, F. Thimm, B. Bischoff, M. Geske, M. Konrad, S. Praetz, C. Schlesiger, S. Selve, A. Gurlo, F. Rosowski and R. Schomäcker, One-pot synthesis of iron-doped ceria catalysts for tandem carbon dioxide hydrogenation, Catal. Sci. Technol., 2024, 14, 4174–4186 RSC.
- C. Zhang, X.-D. Wen, B.-T. Teng, Y. Zhao and M. Fan, Catalytic effects of Zr doping ion on ceria-based catalyst, Fuel Process. Technol., 2015, 131, 1–6 CrossRef CAS.
- S. Zeng, X. Zhang, X. Fu, L. Zhang, H. Su and H. Pan, Co/CexZr1−xO2 solid-solution catalysts with cubic fluorite structure for carbon dioxide reforming of methane, Appl. Catal., B, 2013, 136–137, 308–316 CrossRef CAS.
- Q. Yu, L. Liu, L. Dong, D. Li, B. Liu, F. Gao, K. Sun, L. Dong and Y. Chen, Effects of Ce/Zr ratio on the reducibility, adsorption and catalytic activity of CuO/CexZr1−xO2/γ-Al2O3 catalysts for NO reduction by CO, Appl. Catal., B, 2010, 96, 350–360 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *b,
Aimery
De Mallmann
a and
Mostafa
Taoufik
*b,
Aimery
De Mallmann
a and
Mostafa
Taoufik











