
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Adamantane-based inhibitors of the influenza A M2 proton channel: structure-based design, biological evaluation, and synthetic approaches
Marianna
Stampolaki
 ab,
Maria-Eleni
Kouridaki
a,
Kyriakos
Georgiou
a and
Antonios
Kolocouris
*a
ab,
Maria-Eleni
Kouridaki
a,
Kyriakos
Georgiou
a and
Antonios
Kolocouris
*a
aLaboratory of Medicinal Chemistry, Section of Pharmaceutical Chemistry, Department of Pharmacy, National and Kapodistrian University of Athens, Panepistimiopolis-Zografou, Athens, 15771, Greece. E-mail: ankol@pharm.uoa.gr
bDepartment of NMR Based Structural Biology, Max Planck Institute for Multidisciplinary Sciences, Am Fassberg 11, Göttingen, Germany
First published on 23rd February 2026
Abstract
The influenza A matrix 2 (AM2) protein, a prototype of viroporins, conducts protons along a chain of water molecules and ionizable side chains, including histidine-37. Solid-state NMR (ssNMR) and high-resolution X-ray crystal structures have been obtained for AM2 wild-type (WT) constructs in complex with adamantanamines, as well as for mutant AM2 channels that confer resistance to amantadine and adamantanamines across different pH levels. For the structure of AM2 S31N channels in complex with second-generation adamantane derivatives, consisting of an amantadine analog linked to an aryl group via a methylene bridge, X-ray crystal structures are still unavailable. These complexes have been studied in some detail to date, primarily using solution NMR spectroscopy in micelles or ssNMR in lipid bilayers, providing insights into the inhibition mechanisms of these drugs. These findings, when combined with advances in computational methods, can inform the design and synthesis of adamantane-based blockers targeting WT and mutant AM2 channels. The most popular testing assays were presented. Selected synthetic chemistry routes leading to complex adamantanamines, other saturated polycyclic amines, and second-generation adamantane-based inhibitors were provided. Extensive and long-term research on the druggable M2 channel has provided the scientific society with fundamental tools of structure-based drug design, a synthetic chemistry toolbox, and a library of adamantane-based compounds that can be useful antivirals due to the frequent viral AM2 mutations.
Introduction
Purpose of the review article
Previous review articles have been published, including: (a) The structure and function of the influenza A M2 (AM2) proton channel1–7 and its role in virus assembly and budding.8 (b) The molecular basis of inhibition of AM2 viroporin function by adamantanamines.4,6,7,9,10 (c) The structure of adamantanamines and other saturated polycyclic amines that block AM2 wild-type (WT) (with serine at position 31), as well as adamantane-based drugs, that block mutant AM2-mediated proton currents studied through electrophysiology (EP).11–17 (d) The synthetic routes for selected adamantanamines.18,19 (e) The structures of other compounds that inhibit influenza A virus replication.20–22 We reviewed the development of adamantane- and other polycyclic amine-based blockers targeting either AM2 WT or mutant AM2 proteins and their associated viruses based on key experimental structures of AM2 constructs. Our discussion included the assays used to measure potency and the structure-based drug design methods. Finally, we reported selected synthetic routes for adamantanamines, saturated polycyclic amines, and second-generation adamantane-based derivatives, that consist of an amantadine analog linked to an aryl group via a methylene bridge.Functional role of influenza AM2 protein
The AM2 proton channel plays a crucial role in the life cycle of the influenza A virus. During the early phase of infection, the virus enters the host cell through endocytosis. The AM2 channel is activated in response to the lowered pH within the endosome, which leads to the transport of protons into the viral interior. This acidification of the interior of the virus triggers the dissociation of the viral RNA from the M1 protein,23,24 resulting in virus uncoating. This processs, faciltated by a conformational change in hemagglutinin, leads to the fusion of the membranes of the virus and the host cell endosome.25,26 During the later stages of virus replication, during virus cellular exit, the AM2 protein maintains the integrity of the hemagglutinin during virus assembly in the trans-Golgi network. This function is inhibited by amantadine or rimantadine (see Scheme 1).25 The AM2 protein helps to maintain the high pH of the trans-Golgi network, thereby preventing the premature conformational change of hemagglutinin that occurs at low pH levels and triggers hemagglutinin-induced fusion.27 The AM2 facilitates viral exit by curving the host cell membrane, enabling membrane scission and virus budding.28,29 | ||
| Scheme 1 Chemical structures of amantadine and racemic rimantadine. | ||
The AM2 is a 97-residue membrane protein that assembles and functions as a homo-tetramer, allowing it to conduct protons when activated by low pH, a process essential for the viral life cycle.30–35 It features a short unstructured N-terminal extracellular ectodomain, made up of residues 1–21,36,37 followed by a single transmembrane (TM) domain, made up of residues residues 22–46, known as AM2TM. Finally, it contains a long C-terminal intracellular tail, made us of residues 47–97, which is in contact with the M1 protein.38
The proton-selective ion channel of AM2 is located within the AM2TM domain.39 This domain is the minimal structure required for tetramerization, selective proton transport,30,32,33 and amantadine binding.40–44 The conductance rate of the AM2TM domain and the blocking of the proton current by amantadine, as measured with EP,45,46 correspond to those of the full-length AM2 protein (AM2FL).39,47 Near the C-terminal of the TM bundle, there is a region that features an interruption by the H37xxxW41 sequence in the C-terminal half of the TM helix. In this region, polar and single charged residues face the channel, while and hydrophobic residues are oriented towards the lipid bilayer.48 In the AM2TM domain, residue H37 plays a key role in activation as the proton-conducting residue of the channel.49,50 At low pH in the viral endosome, the imidazole rings in the four H37 residues causes the channel to open due to electrostatic repulsion and destabilization of helix–helix packing. W41 is the residues that controls, the gate blocking, and proton flow when the channel is in the alkaline pH state.51,52 Besides the well known gating role of W41, V27 has been suggested to create a secondary gate.51,52 The cavity between V27 and H37 contains a binding site for amantadine.53 Water molecules within the channel pore create a hydrogen-bonded water network along the 17 Å stretch between the V27 valve and H37 tetrad.54,55 Functional56 and structural studies54,55 have demonstrated that the pore is formed by the TM-helix bundle and is lined by V27, S31, G34, H37, W41, D44, and R45, i.e., includes all the polar residues of the TM sequence.
The amphipathic helix (AH), consisting of residues 47–62, is positioned between the TM helix and the M1-binding segment of AM2. The AHs are located at the inner membrane interface and are essential for viral budding.8 It is believed that AHs create the membrane curvature necessary for viral budding.57,58 The TM and AH bundle comprises the AM2TM + AH, collectively known as the conductance domain (CD) AM2CD. In synthetic lipid bilayers, AM2CD appears to contain all the necessary residues and environmental interactions to achieve the detailed conductance properties of the native protein found in oocytes.39
Experimental structures of AM2 bound with adamantane-based derivatives
Wild-type AM2 protein
The structure of the apo-AM2TM48 or apo-AM2CD59 was studied using oriented sample (OS) ssNMR in phospholipid bilayers at pH 7, showing a fourfold symmetric homotetramer; the structure with PDB ID 2L0J59 was deposited for AM2CD. The charge of H37 tetrad was +2 charged between pH 6–7.5, and the charges may be extensively delocalized over a dimer-of-dimers structure. The kink in the TM helix occurs around G34, like the kinked TM-domain structure in the AM2TM protein, with the presence of amantadine.60 At pH 8, the imidazole rings of the four H37 residues are unprotonated. The first protonation of H37 forms an imidazole–imidazolium dimer with a strong hydrogen bond between bridged imidazole rings. The second protonation forms two imidazole–imidazolium dimers.48,59 At pH 7.5, most of the H37 side chains existed as imidazole–imidazolium dimer. At pH 5.0, all the H37 are protonated.The first X-ray crystal structures of samples of apo-AM2TM WT or AMTM WT with amantadine,61 showed a tetrameric left-handed bundle, which was consistent with a previous solid-state NMR (ssNMR) structure of the apo-AM2TM (PDB ID 1NYJ62) in phospholipid bilayers. While the apo-AM2TM WT crystal structure had a 2.05 Å resolution and an I33M mutation (PDB ID 3BKD61), the latter crystal structure (PDB ID 3C9J61), which was suggested to correspond to a complex with amantadine inside the TM pore, had a 3.5 Å resolution and the stabilizing mutation G34A.61,63 However, the 3.5 Å resolution of this structure did not ensure the presence of amantadine (having a diameter of ∼3.4 Å). Additionally, the 3.5 Å resolution of PDB ID 3C9J61 did not allow for examining the critical role of water in drug binding. One of the proposed proton conduction mechanisms is mediated by ordered water molecules arranged in wires inside the channel, which play a role in the conduction of protons through the AM2 pore.64,65 In another work, a potential allosteric binding outside the pore was suggested in the complex of rimantadine with AM2CD in micelles (PDB ID 2RLF66) using solution NMR in micelles. In this site, the ammonium group of rimantadine in the secondary binding site is in contact with the polar sidechain of D44, whereas the adamantyl group of the drug forms hydrophobic interactions with I42 from one of the TM helices and with L40 and L43 from the adjacent helix. However, this structure was solved with a 200-fold excess of rimantadine, while the micelles did not allow proper folding, narrowing the tetrameric bundle in the N-end and disturbing the rimantadine entrance to the channel. Nevertheless, in the later ssNMR structure of a sample of AM2TM with a perdeuterated amantadine in phospholipid bilayers (PDB ID 2KQT67) obtained using experimental restraints from a previous structure (PDB ID 2H9560), amantadine was found inside the pore between V27 and H37, as previously suggested.42,52,60 Perdeuterated amantadine68 was used in complex with AM2AH in dimyristoyl-sn-glycero-3-phosphocholine (DMPC) bilayers, and it was shown with OS ssNMR that amantadine binds close to S31 inside the pore, as was shown in the PDB ID 2KQT structure.67 Additionally, it was also shown using ssNMR and CD3-labeled rimantadine that rimantadine has its amino group towards the C-end.69 Thus, the pharmacologically relevant drug-binding site is in the TM pore of the fully functional AM2CD. Other studies with AM2CD and rimantadine using ssNMR in phospholipid bilayers70 or solution NMR in micelles (PDB ID 2LJC71) also showed the presence of rimantadine inside the AM2 pore.
Eventually, the most direct evidence of the pore binding site and water wire role for the stabilization of channel blockers by EP inside the pore came from higher resolution crystallographic studies of the AM2TM in the presence of adamantanamine. Indeed, water-mediated interactions in receptor binding sites can play key roles in drug binding and inhibitor design. Experimental structures with high resolution made the detection of crystallographically important waters possible. The detection of crystallographically important waters demonstrated the channel inhibition by inhibitors targeting these key waters. This was shown in structures of AM2TM WT bound to amantadine (PDB ID 6BKK;72 2.00 Å), rimantadine (PDB ID 6BKL;72 2.00 Å), or a spiro-adamantanamine inhibitor (PDB ID 6BMZ;72 2.63 Å), see Fig. 1. These X-ray structures of AM2TM ion channel with bound inhibitors reveal that the ammonium group of the drugs is directed toward H37 and binds to water-lined sites that are hypothesized to stabilize transient hydronium ions formed in the proton-conduction mechanism. As supported by molecular dynamics (MD) simulations, the ammonium group is stabilized through three hydrogen bonds with proximal waters, creating a layer at the level of the A30 layer, followed by the G34-water layer and the H37 tetrad. The placement of the adamantanamine molecules within the pore requires a displacement of waters near V27 by the hydrophobic adamantane ring of the above adamantanamine molecules, thus disturbing the AM2 water network.72 The MD simulations72 of the AM2TM WT, with the three adamantanamine drugs predicted with accuracy the position of the ligands and waters inside the pore in the X-ray crystal structure of the AM2TM WT complexes. In another work, the X-ray structures of AM2TM WT pore bound to (R)-rimantadine (PDB ID 6US973) and (S)-rimantadine (PDB ID 6US873) were solved with a 2.00 Å resolution, showing only slight differences in the hydration of each enantiomer.
 | ||
| Fig. 1 X-ray crystal structures of the AM2TM proton channel bound to drugs and inhibitors.72 Top, left to right: (A) AM2TM bound to rimantadine (PDB ID 6BKL72). (B) AM2TM bound to amantadine (PDB ID 6BKK72). (C) AM2TM bound to a spiramic adamantanamine (PDB ID 6BMZ72). This figure has been adapted from ref. 6 with permission from ELSEVIER, copyright 2025. | ||
Mutant AM2 proteins
Amantadine resistance mutations are found close to the drug-binding site located at the pore-facing positions (V27A, A30T, S31N, and G34E), at the interhelical interfaces at the N-terminal half of the channel (L26F), and mutations outside the drug-binding site lying at the interhelical interfaces (L38F, D44). It was suggested74 that a mutated, drug-resistant protein has a larger binding pocket for the drug, as shown with MD simulations.75 Hence, despite binding to the channel, the drug remains sufficiently mobile so as not to exert a proton-blocking positive electrostatic hindrance as a simple mechanism for proton flow blocking in AM2 WT by amantadine.75A solution NMR structure of AM2 (19–49) S31N in complex with M2J33276 in micelles (PDB ID 2LY076 was reported. M2J33276 is a representative second-generation adamantane-based inhibitor that blocks M2 S31N-mediated current and replication of M2 S31N virus (Fig. 2); it can be considered a second-generation adamantane-based compound. Such compounds have been developed to inhibit influenza A M2 S31N which became the major epidemic virus after 2008. Indeed, in 2009, the emergence of the novel pandemic H1N1 influenza virus (A/H1N1/pdm09) led to a dramatic spike in hospitalizations and deaths in the United States, particularly among young adults.77 These drugs consist of amantadine connected through a methylene linker with aryl groups, e.g., the amantadine-5-(thiophenyl)oxazolyl (M2J33276), amantadine-5-phenyloxazolyl (M2J35276), or amantadine-5-cyclopropyloxazolyl78 conjugates. With the structure PDB ID 2LY0,76 it was shown that these amantadine–aryl conjugates blocked AM2AH S31N with an orientation of the amantadine amino and aryl groups towards the N-end, as also demonstrated in the ssNMR study of M2J352,79 compared to adamantanamines, which point their amino groups towards the C-end of the AM2 WT channel according to the observations from ssNMR80 and X-ray structures.72,73 M2J332,76 and M2J35276 were able to block AM2 S31N by EP, and virus replication but did not block AM2 WT by EP and virus replication. Interestingly, the amantadine–3-bromo-thiophenyl conjugate (cmp11 in ref. 81) was a double AM2 S31N and WT blocker by EP. The solution NMR structure of the amantadine–3-bromo-thiophenyl conjugate with AM2 (19–49) WT (PDB ID MUW81) or AM2 (19–49) S31N (PDB ID 2MUV81) in micelles showed an orientation of the amantadine amino and aryl groups towards the N-end in AM2 (19–49) S31N and towards the C-end in AM2 WT.81
 | ||
| Fig. 2 AM2 (19–49) S31N drug inhibition mechanism.76 Left part: binding site of M2WJ332 from the solution NMR structure (PDB ID 2LY076): the side chains V27, A30, H37, W41, and the molecule are shown as colored sticks; other side chains are shown as transparent space filling; the backbone of the protein (one monomer was omitted for clarity) is shown as opaque ribbons. Right part: zoom in showing the drug–protein interactions: one of the N31 side chains forms bidentate interactions with the drug (the carbonyls from another two N31 residues can form a water-mediated hydrogen bond with the ammonium from the drug, which is not shown). This figure has been adapted from ref. 6 with permission from ELSEVIER, copyright 2025. | ||
X-ray crystallography was used to solve the structures of a spiranic adamantanamine inhibitor bound to AM2TM V27A (PDB ID 6NV182), shown in Fig. 3. This spiranic adamantanamine inhibitor was a triple blocker by EP of AM2 WT, V27A, and L26F proteins.83 The X-ray structure of the spiranic adamantanamine with AM2TM WT (PDB ID 6BMZ72) is shown in Fig. 1. The X-ray structures with PDB IDs 6BMZ72 and 6NV182 revealed the mechanism of amantadine resistance. As shown in the crystal structure, compared to the AM2TM WT (Fig. 3B), the AM2TM V27A channel pore was wider at its N-terminus because of the V27A mutation, which removed V27 side chain hydrophobic interactions that are important for binding of amantadine and rimantadine (Fig. 3A). The spiranic adamantanamine inhibitor, which is longer than amantadine, shifted its binding site in the pore depending on which residue is present at position 27, thus blocking effective proton conductance in the AM2 WT and V27A mutant channels. The MD simulations of the AM2TM V27A–spiranic adamantanamine complex agreed with the experimental structures.82 The same argument can be applied to explain the EP-based blocking of the wide-diameter AM2TM L26F mutant by this spiranic adamantanamine.
 | ||
| Fig. 3 The mechanism of amantadine drug resistance in the AM2 V27A mutant channel and the mechanism of dual inhibition by the shown spiranic adamantanamine can be rationalized through the crystal structures of this adamantanamine with (A) AM2TM WT (PDB ID 6BMZ72) and (B) AM2TM V27A (PDB ID 6NV182). This figure has been adapted from ref. 6 with permission from ELSEVIER, copyright 2025. | ||
Resistance of influenza A virus to adamantane-based drugs
The most common AM2 inhibitors are amantadine derivatives. In 1966, amantadine was first approved as a prophylactic drug against Asian influenza.84,85 After a few years, it was announced86 that dyskinesia symptoms of a Parkinson's disease patient were remarkably improved while amantadine was administered for the prevention of flu. The mechanism of action of amantadine against these targets results from the blockage of ion channels either by direct interaction with the channel's pore (influenza AM2) or by stabilization of closed states of the channel (NMDA receptor).87 Since 1985, AM2 has been identified as the primary target of amantadine.40 Resistance to AM2 WT proton channel drugs, amantadine and rimantadine, is associated with mutations in the TM domain of the AM2 WT protein.103 The homo-tetrameric structure of the AM2 WT channel places constraints on the types of drug-resistant mutations that can be accommodated.74,88 A few amantadine- or rimantadine-resistant mutations have been observed in transmissible viruses,89–91 which are L26F, V27A, A30T, G34E, and S31N, located in the TM pore of AM2 WT,89–94 although other mutations can easily be observed in vitro. The mutations that cause the greatest decrease in inhibition are S31N and V27A, which increase the polarity of the pore. The vast majority, 95% of resistant viruses bear the S31N substitution in AM2, 1% have V27A, and L26F, A30T, and G34E are rare.95,96 The AM2 S31N mutant is one of the most conserved in viral proteins among currently circulating influenza A viruses, which happens to maintain nearly identical channel function as the AM2 WT but is resistant to amantadine, rimantadine, and adamantanamines. The presence of L26F, V27A, and particularly S31N in influenza A viruses circulating worldwide pushes the search for novel ion channel blockers with stronger, preferably resistance-overcoming activity.Recently, the World Health Organization reported that influenza activity has globally increased since October 2025, with influenza A virus being predominant among the viruses (WHO, 10 December 2025, Disease Outbreak News; Seasonal influenza-Global situation). Currently circulating in humans are subtype A(H1N1) and A(H3N2) influenza viruses. Both subtypes include the S31N mutation in the M2 sequence. A recent work,97 showed that prolonged treatment with the neuraminidase (NA) inhibitors like the commonly used for influenza treatment, oseltamivir, can cause resistant mutations at hemagglutinin (HA) alone or in combination with resistant mutants at NA as a compensatory mechanism. In that way, the virus reduces receptor binding and restores functional balance between HA and NA. It was shown97 that these mutations can also confer cross-resistance to other NA inhibitors, including zanamivir and peramivir, in the A(H1N1)pdm09 virus. Considering (a) the prevalence of M2 S31N strains and (b) the ability of the virus to develop mutations in both HA and NA and become resistant to the currently used NA inhibitors, the design of new second-generation M2 inhibitors is becoming increasingly relevant. In another work,98 the second-generation drug resistance mechanism of the virus towards second-generation M2 channel blockers was profiled in vitro. Serial viral passage experiments were performed98 in the A/California/07/2009 (H1N1) viral strain under persistent drug selection pressure to identify resistant mutants against such blockers. The results suggested98 that these S31N inhibitors have a higher genetic barrier to drug resistance than amantadine in cell culture. It was shown that the evolved mutations are all located at the N-terminal drug binding site of AM2 S31N, and they have either a direct or indirect effect on the channel pore, which leads to either reduced or complete loss of drug sensitivity. More specifically, those mutations introduced either a structural hindrance for the new agents as their aryl groups lie in this region or change the polarity of that site of the tetramer, affecting their entrance to the pore.99,100 This information is quite important for optimizing the structure of new selective S31N inhibitors.
Methods to detect inhibition of wild-type and mutant AM2 viruses by adamantane-based drugs
Cell antiviral assays
Compounds have been tested as inhibitors of viral replication using cell assays. These are most often the miniplaque assay,101–104 and the cytopathic effect (CPE) inhibition assay,105 performed in Madin-Darby canine kidney (MDCK) cells against influenza virus strains. Mutant AM2 viruses can be produced using reverse genetics.106 For example, using a miniplaque assay, the (R)- and (S)-enantiomers of rimantadine showed indistinguishable potency against the amantadine-sensitive A/Solomon Island/3/2006 (H1N1) strain. Both (R)- and (S)-rimantadine inhibit viral replication with similar EC50 values of 19.62 and 24.44 nM, respectively (Fig. 4).73 | ||
| Fig. 4 Cellular antiviral assay results of (R)- and (S)-rimantadine against the amantadine-sensitive A/Solomon Island/3/2006 (H1N1) strain. The antiviral potency was determined in a plaque assay. The EC50 values are the mean ± standard deviation of two independent repeats. This figure has been reproduced from ref. 73 with permission from the American Chemical Society, copyright 2021. | ||
An Escherichia coli-based assay was used to measure the H+ conductivity of AM2 WT107 or mutants M2108,109 in the presence of a potential inhibitor, since bacteria that constitutively express a pH-sensitive green fluorescent protein—pHluorin, can be used to analyze the membrane permeation to H+. Specifically, the emission at 520 nm of pHluorin has two excitation maxima: 390 nm and 466 nm, whose ratio changes as a function of pH. The fluorescence obtained when exciting at 390 nm and 466 nm upon injection of a concentrated acid solution into the buffer was monitored. Any bacteria that express an H+-conducting channel will exhibit a dramatic change in the fluorescence ratio, in contrast to control bacteria that contain an empty vector or when an inhibitor is present.
Assays that measure direct binding
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25. The AM2 construct system is titrated using a drug dissolved in the same lipid environment. The developed methods mostly explore the thermodynamics of direct ligand binding to AM2 binding. For example, they have applied circular dichroism (CD) spectroscopy, which monitors the ellipticity ratio at 223 vs. 209 nm (θ223/θ209),110 or analytic ultracentrifugation (AUC),110 or isothermal titration calorimetry (ITC) to AM2TM peptides in DPC micelles (Fig. 5).39,111
:25. The AM2 construct system is titrated using a drug dissolved in the same lipid environment. The developed methods mostly explore the thermodynamics of direct ligand binding to AM2 binding. For example, they have applied circular dichroism (CD) spectroscopy, which monitors the ellipticity ratio at 223 vs. 209 nm (θ223/θ209),110 or analytic ultracentrifugation (AUC),110 or isothermal titration calorimetry (ITC) to AM2TM peptides in DPC micelles (Fig. 5).39,111
 | ||
| Fig. 5 Representative ITC plots for enantiomers 2-R, 2-S. | ||
An assay112 was developed that uses the quenching of W41 fluorescence by H37 protonation below pH 6 in the AM2FL channel reconstituted in LDAO (N,N-dimethyldodecylamine-N-oxide) detergent. The quenching of W41 fluorescence was reversed by amantadine and analogs, and was used in testing adamantanamines against AM2 WT.113 Another assay measured the thiol-disulfide equilibria of AM2TM or AM2FL in phospholipid bilayers by CD114 or surface plasmon resonance (SPR).115 A series of SPR experiments was used to accurately measure the affinity of amantadine and rimantadine to AM2TM WT embedded in DMPC liposomes. It was found that this class of drugs can bind to AM2 with two different binding dissociation constants (Kd) in the order of 10−4 (Kd = 0.91 μM) and 10−7 M (Kd = 0.40 mM).115 It was shown that a high-affinity binding site corresponds to the AM2 ion channel pore-binding site, which is responsible for the pharmacological activity elicited by amantadine drug and its analogs, in agreement with the ssNMR study of amantadine bound to AM2TM WT (PDB ID 2KQT67). It was also found that the low-affinity site corresponds to the secondary binding site, which can be attributed to the lipid face of the pore, in agreement with the solution NMR structures of AM2CD WT in micelles (PDB IDs 2RLF66 and 2LJC71).
In another assay, the proton flux mediated by AM2TM or AM2CD was measured in small liposomes composed of unilamellar 2:1 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC)/1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycine (POPG) vesicles.39 An in vitro functional assay, which is an indirect liposome dye release assay for viroporin activity, was also reported.103 However, the most straightforward way to determine the blocking effect of M2-mediated proton current is electrophysiology with AM2FL expressed in oocytes, published early for amantadine116 and further performed for amantadine analogs.117 The compounds are tested with a two-electrode voltage-clamp (TEVC) assay using X. laevis frog oocytes microinjected with RNA expressing the AM2 protein.118–120 Numerous studies have been performed.62,80,100,101,121–138 It was shown that drugs can block AM2-mediated proton current when the koff/kon are, correspondingly, slow off/fast on or slow off/slow on, thus leading to favorable Kd = koff/kon values.101,105,106,139 Only when Kd (TEVC) = koff/kon is small enough, an in vitro antiviral activity is observed.101,106 If the koff is too high, then while the compound can block the M2 channel, it does not inhibit the virus in vitro, and this was the case for compound 8 (see Table 2) against AM2 V27A.101 The potency of the inhibitors was usually expressed as the inhibition percentage of the AM2 current observed after 2 min. However, studies performed independently with second-generation amantadine-based drugs against AM2 S31N,106,139 rimantadine analogs against AM2 WT,105 or spiranic adamantanamines against V27A101 showed that the 2-minute measurements of the %-AM2 blocking effect (usually applied for testing a large number of compounds76,81,139–141) and filtering the most important for subsequent antiviral assays were not efficient. This is due to the presence of compounds with, e.g., very slow on- and slow off-rates for entry (measured as association and dissociation rate constants, kon and koff, respectively), for which characterization of the blocking effect needs more than 2 min, e.g., 5 min. For example, in ref. 105 for a rimantadine analog, only 27% blocking was observed at 2 min against Udorn AM2 WT, but low micromolar EC50 against M2 WT viruses. Despite the very slow binding kon rate, the ligand exhibited high antiviral potency, which requires, as a biological effect, much longer exposure times than EP experiments. For such compounds, after 2 min or 5 min, the percentage of proton current inhibition was progressively increased. Thus, in ref. 105 it was striking to observe that the ligand against Udorn AM2 WT exhibited 27% blocking at 2 min, 38% at 5 min, and 61% at 10 min. It must be clarified that the measurements at 2, 5, or 10 min correspond to measurements before the establishment of equilibrium due to very slow on- and off-rates for entry, which characterizes bulky ligands, and the period can't be extended considerably due to the difficulty of maintaining oocytes at low pH for extended periods. Hence, the %-blocking/IC50 values determined by the TEVC procedure can be significantly lower than those measured with in vitro antiviral assays, where the system can reach the equilibrium block state, or compared with Kd values at equilibrium measured with ITC105 or SPR.115 ssNMR studies have also been performed to investigate the binding interactions of rimantadine and of amantadine and other adamantanamines with AM2 (22–62) WT.83,142–146
An interesting work refers to cell-based antiviral assays and EP, which were applied to evaluate the biological potency of rimantadine enantiomers.105,147 It was observed that both enantiomers have similar %-channel blockage, binding kon and koff, Kd, and antiviral potency (EC50) against AM2 WT.105 These results showed no significant difference between the two rimantadine enantiomers compared to what has been previously suggested with ssNMR at −10 °C, i.e., that the two enantiomers have different affinity against AM2CD WT.148 In a subsequent work,73 much more accurate binding kinetics were performed compared to previous ones105 measurements (Table 1, Fig. 6).
| rac-Rimantadine | (R)-Rimantadine | (S)-Rimantadine | Amantadine | |
|---|---|---|---|---|
| Concentration tested | 50 μM | 50 μM | 50 μM | 100 μM |
| k on (min−1 M−1) | 19600 ± 300 |
20800 ± 700 |
22500 ± 300 |
20500 ± 300 |
| k off (min−1) | (9.1 ± 0.8) × 10−4 | (9 ± 2) × 10−4 | (8.8 ± 0.8) × 10−4 | (119 ± 2) × 10−4 |
| K d = koff/kon (nM) | 46 ± 4 | 41 ± 9 | 39 ± 4 | 580 ± 20 |
 | ||
| Fig. 6 Rimantadine enantiomers and amantadine binding kinetics against Udorn M2 WT were determined using a combined application of inward current and washout procedure TEVC assay.73 (A) Racemic rimantadine, (B) (R)-rimantadine, (C) (S)-rimantadine, or (D) amantadine was applied to oocytes for 5 to 7 min, and after the inward current reached its maximum, a washout protocol was applied to the oocytes. During the washout, pH 8.5, pulses were applied to make sure the current went to baseline to ensure the oocyte quality. The blue bar above the recording trace indicates the period in which the pH 5.5 Barth solution was applied; the red bar indicates the period in which compounds in the pH 5.5 Barth solution were applied. Representative recording traces are shown on the left side of each panel. The best-fit values are shown in Table 1. This figure has been reproduced from ref. 73 with permission from the American Chemical Society, copyright 2021. | ||
It was concluded that the slight differences in hydration for the (R)- and (S)-rimantadine enantiomers observed in structures of M2TM WT with PDB ID 6US973 and PDB ID 6US873 are not relevant to drug binding or channel inhibition. Additionally, it did not result in a difference in potency or binding kinetics as was shown by similar values of kon/koff, and Kd for (R)- and (S)-rimantadine in the TEVC assay (Table 1, Fig. 6).139,149
:tetramer ratio ∼ 3:1 mol), in DPhPC lipid bilayers at pH 7.8. In this alkaline pH, the His37 tetrad has a neutral charge. Proton resonances at 14.3, 11.7 ppm for the His37 Nδ1, Nε2 of apo-AM2CD WT were recorded in the (H)NH spectra due to His-His+ dimer-of-dimers formation (Nδ1–H⋯Nε2 and Nδ1⋯H–Nε2 forms) and showed an intermolecular coupling (JNN = 8.9 ± 0.3 Hz) due to hydrogen bonding between Nε and Nδ of adjacent His37.144,150 When rimantadine was added to AM2CD WT in DPhPC bilayers or in VM bilayers or in DPhPC-30% cholesterol bilayers at 4 °C, the spectra did not show pore binding at all, as observed previously with AM2CD WT (residues 21–61) reconstituted in VM at 30 °C using a drug:tetramer ratio 5:1.83 In the presence of rimantadine with AM2CD WT pore at 40 °C, compared to the apo-AM2CD WT signals, was observed intensity reduction of the side chain His37 proton peaks at 14.3 and 11.7 ppm in the (H)NH spectra and the His37-Nε2 shifted upfield, while disruption of the JNN hydrogen bond between His37 Nε and Nδ was observed, in accordance with its pore-blocking mode of AM2CD WT inhibition. While binding took place over three days at 40 °C, complete binding was seen in just a few hours at 55 °C, offering a novel NMR assay for the detection of inhibition by adamantanamines. The slow kinetics reported for rimantadine by NMR are opposed to the fast blocking of AM2CD WT (residues 18–60)-induced proton currents by either rimantadine or amantadine observed in liposomal proton flux assays,116,151 respectively. At pH 6, however, i.e., around the pH found in the Golgi lumen (pH 6.0–6.7),152 where the AM2 channel adopts its activated +3 charge state, the rimantadine pore-binding kinetics were faster.
3D (H)CαNH NMR was applied to study the potencies against influenza A with the M2 WT channel of several adamantanamines142 and some cage amine analogs, and the AM2 L26F, V27A, A30T, G34E mutants. For example, the compound shown in Fig. 7 exhibited a blockage of AM2 WT, L26F, and V27A channels by EP assays, while some adamantanamine analogs blocked AM2 WT and AM2 L26F channels by EP (see structures in Table 2).153
 | ||
| Fig. 7 Slices from 3D (H)CαNH spectra (15N–13Cα projection) of AM2CD V27A and mutant proteins in complex with the cage amine triple blocker (protein was embedded in DPhPC bilayers with a lipid to protein tetramer molar ratio of 24:1 at pH 7.8) and results from 500 ns MD simulations for AM2TM-cage amine complexes embedded in POPC bilayers utilizing the CHARMM36m force field.154 Panel on the left side: the NMR signals of the apo-protein are shown in red, and the signals of the complexes of AM2CD with the respective compound are shown in blue (at the top left side, in the inset, is shown clearly the pair of signals of G34, which suggests the presence of two conformational states: either due to a slow equilibrium between a kink and non-kink state at G34 or a dimer-of-dimers structure of M2CD). Panel on the right side: average structure of AM2TM V27A from MD simulations; grey cartoon is used to display the AM2TM tetramer, stick representations were used for ligands, waters, and amino acid side chains indicated and cartoon representations was used for the protein backbone (carbons are depicted with grey color for amino acids and ruby color for the ligand, red for oxygen, blue for nitrogen) while the yellow dotted line represents hydrogen bonding between the drug and water molecules. This figure has been adapted from ref. 6 with permission from ELSEVIER, copyright 2025. | ||


|
|
|
|
|
||
|---|---|---|---|---|---|
| a Plaque reduction assay against influenza A Udorn virus. b Cytophathic effect assay against WSN/33 S31 virus. c Plaque reduction assay against A/Hong Kong/8/68 (H3N2). d Plaque reduction assay against influenza M2 WT A/HK/7/87 virus. e Isochronic (2 min) from TEVC studies in oocytes for IC50 values. f Isochronic (2 min) from TEVC studies against M2 from Udorn virus in HEK 293 T cells for IC50 values. g 10.2 μM (3 min) from TEVC studies against M2 from A/California/09/2009 (H1N1) M2: S31 virus; n.a., not active. | |||||
| A/M2 S31 (M2 WT) | EC50 (or IC50 in TEVC) | 18.7 μMa | 0.03 μM,b,105 0.01 μM,a,105 0.012 μM,d,105 9.3 μMe,105 | 0.34 μM,b 10.2g,167 | 0.07 μM,b 30.8%-blockinge |
| A/M2 V27A | 0.3 μMa | n.a. | n.a. | n.a. | |
| A/M2 S31N | n.a. | n.a. | n.a. | n.a. | |
| A/M2 L26F | 5.6 μMa | 0.79 μMb 0.55 μM | 8.75 μMb | 0.74 μMb | |


The cage amine triple blocker in Fig. 7,153 while having length equal to rimantadine, binds and blocks the AM2 V27A channel due to its larger volume, as well as AM2 L26F and AM2 WT, as was shown by 500 ns-MD simulations with the CHARMM36m force field154 of AM2CD V27A, AM2CD L26F, and AM2CD WT embedded in a 150 POPC lipid bilayer. This compound's ammonium group was oriented steadily towards the C-terminus in AM2CD WT, V27A, and L26F channels during the MD simulations, and the ligand was stabilized within the AM2 pore by forming an average of three hydrogen bonds with the water that was present between the ligand's ammonium group and His37. Furthermore, by displacing loosely bound waters close to the top of the pore (N-terminal side), the large cage alkyl group stabilized the complex through interactions between the tetramethyl-cyclobutane ring and residue 27 side chains,44,155 and successfully blocked the passage of water molecules, preventing protons from entering the pore and generating conduction. The MAS ssNMR provided information on the contacts of the triple blocker in Fig. 7 with AM2CD V27A (residues 18–60), AM2CD L26F, and AM2CD WT. When the drug was added to the mutant channels, there was a noticeable increase in peak resolution as opposed to the relatively wide peaks in the apo-AM2 V27A and apo-AM2 L26F. This suggests that the protein's conformational flexibility was decreased during the formation of the drug–protein complex. Fig. 7 shows some representative results for the AM2CD V27A complex.
Selected reports of adamantanamines and assays used to measure AM2 inhibition
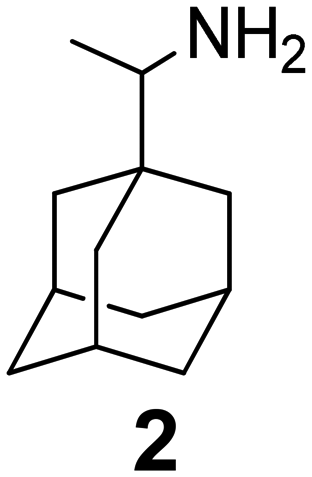
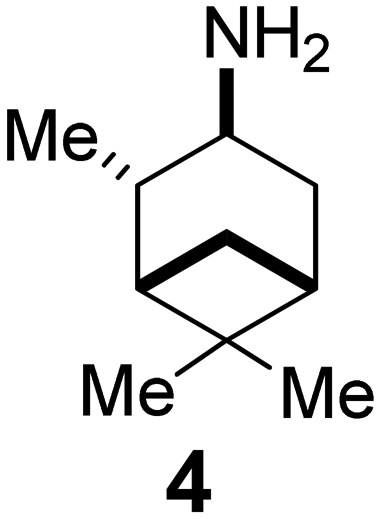
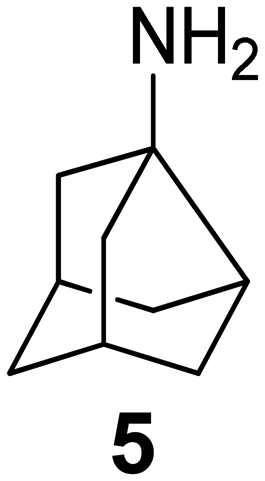
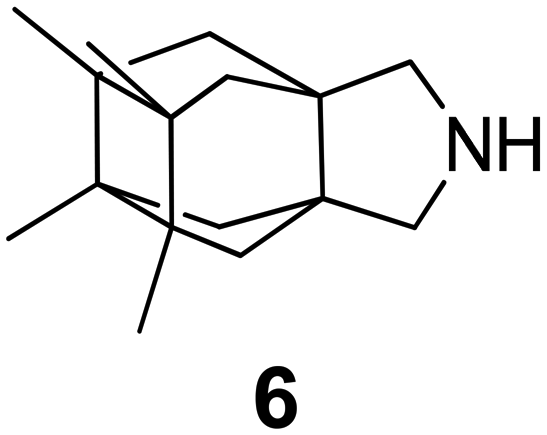
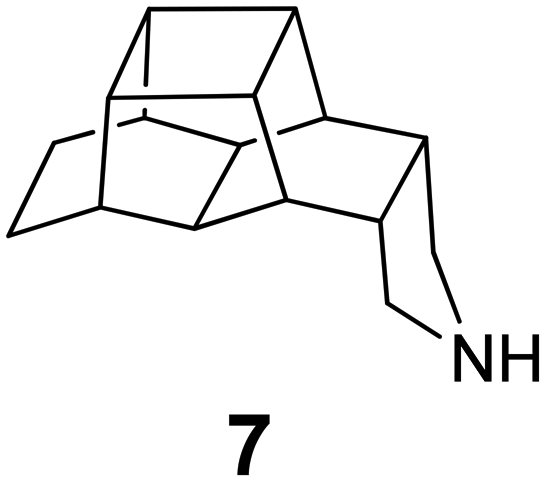
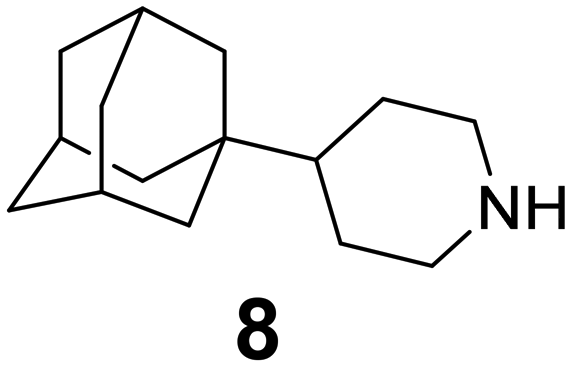
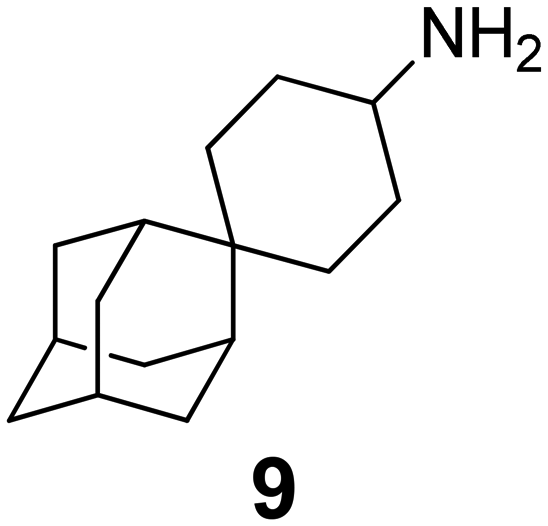
In Table 2, selected compounds and activities against AM2 WT and its AM2 mutant channels were measured with EP, and the in vitro cell-based activities against the corresponding viruses; the activities of amantadine (1) and rimantadine (2), BL-1743 (3),111,117,156 pinanamine (4),157 and noramantadine 5, which all block the AM2 WT channel by EP and inhibit in vitro the replication of the influenza AM2 WT virus.They also showed the activities of the tetramethyl-3-azapentacyclo[7.2.1.15,8.01,5.07,10] tridecane (6),158 which blocks both the AM2 WT- and AM2 V27A-proton mediated current and inhibits in vitro AM2 WT, V27A, and L26F viruses. The piperidine derivative 8 blocks the AM2 WT and V27A by EP and inhibits in vitro the corresponding viruses.101 The azatetracyclo[5.2.1.15,8.01,5]undecane (7)125 blocks AM2 V27A by EP, while the spiranic amine 9159 and tetramethyl-3-azapentacyclo[7.2.1.15,8.01,5.07,10]tridecane (6)158 are blockers of the AM2 WT, V27A, L26F channels and inhibited in vitro the corresponding viruses.
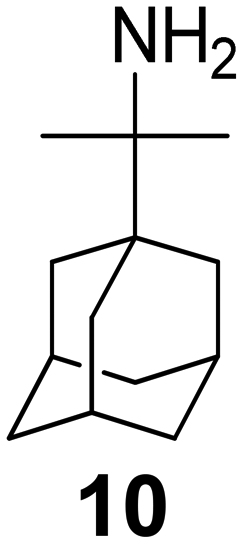
The rimantadine analog 10, the spiropyrrolidine[2,2′]adamantane (11), and the diamantanamine 12 block the AM2 WT and AM2 L26F channels by EP.142 The adamantanamines and saturated polycyclic amines 1–12 did not block the M2 S31N channel.
These adamantanamines are important agents since they can block AM2 WT, V27A, and L26F viruses. Other lipophilic and saturated polycyclic amines that block these AM2 channels have been reported.138,160–164 These compounds can be repurposed if the predominant influenza A M2 S31N, causing the current epidemics, can be changed to one of these channels that are sensitive to adamantanamines.
Structure-based drug design methods targeting wild-type and mutant AM2 channels
Protein channels and water-filled pores present particularly challenging targets for drug design.168 MD simulations for adamantanamine binding to the AM2 protein have been performed, and some of them were applied for structure-based drug design purposes. For example, MD simulations of ligand–AM2TM complexes were used to design and synthesize spiro-piperidine inhibitors (e.g., compound 9 in Table 2) that block the M2 WT, M2 V27A, and M2 L26F channels.159 The design of AM2 blockers would benefit from the application of methods and the generation of models that can accurately calculate the relative binding energies of ligands targeting the AM2 channel.Generally, the molecular mechanics-Poisson–Boltzmann surface area (MM-PBSA) method cannot calculate accurate binding free energies and can only provide a good correlation between calculated and experimental binding free energies for a set of ligands with a Kd value range of 1000.169,170 In contrast, binding free energy methods based on statistical mechanics, e.g., free energy perturbation or thermodynamic integration coupled with MD simulations (FEP/MD or TI/MD, respectively),171,172 have proven capable of predicting relative binding free energy with an accuracy of ∼1 kcal mol−1 in optimal cases. ITC was used to measure Kd values of several adamantanamines against the closed state of AM2TM Weybridge strain (with V28I, L38F mutations compared to Udorn) at pH 8; ∼1:57 peptide/DPC lipid was used to maintain tetramers.170 It was found Kd = 0.74 μM for amantadine.170 It was shown that the application of MM-PBSA provided a fair assessment of the relative importance of the binding free energy components of adamantanamines in their complexes with AM2TM WT after calculating binding free energy decomposition.173 In this work, it was revealed that the calculated enthalpy components can be used to prioritize adamantanamines in agreement with their experimental binding affinities measured with ITC.
For a set of rimantadine analogs,134 binding affinities were measured using ITC in DPC micelles against the closed state of AM2TM WT and S31N protein channel at pH 8, blocking potency by TEVC against the AM2FL WT and S31N protein channel, and antiviral potency using cell-based assays against virus replication. The rimantadine analogs bear progressively larger alkyl groups (with dimethyl, diethyl, and dipropyl in the carbon bridge, see structures of compounds 10, 13, 14, in Table 4). The results showed that compared to amantadine (1) with Kd = 2.7 μM by ITC and rimantadine with Kd ∼ 0.33 μM by ITC, the dimethyl analog 10 had enhanced affinity against AM2TM WT (Udorn sequence) by ITC (Kd = 0.13 μM), and against AM2FL according to the kinetic binding measurements by EP. This was also confirmed by alchemical relative binding free energy calculations with the FEP/MD method, with OPLS2005 provided with the Desmond software. It was found that the diethyl and dipropyl rimantadine analogs showed reduced affinity and potency (Table 3).134
| Complex | ITC | TEVC | CPE |
|---|---|---|---|
| K d (μM) | k on, koff (M−1 s−1), Kd (μM) | EC50 (μM) | |
| 2-R + M2 WT | 0.32 | 412, 0.0013, 3.2 | 0.05 |
| 2-S + M2 WT | 0.34 | 407, 0.0016, 3.9 | 0.06 |
| 10 + M2 WT | 0.13 | 230, 0.003, 13 | 0.01 |
| 13 + M2 WT | 4.59 | — | 0.46 |
| 14 + M2 WT | 3.43 | 34, 0.003, 13 μM | 1.07 |
|
|
||||
|---|---|---|---|---|
| Ligand | K d | ΔG | ΔH | TΔS |
| a Racemic mixture. b Racemic mixtures resulted from the protonation of N-methyl spiro[pyrrolidine-2,2′-adamantane]. | ||||
| 1 | 2.17 ± 0.52 | −7.77 ± 0.14 | −6.66 ± 0.50 | 1.11 ± 0.52 |
| 2 | 0.51 ± 0.26 | −8.64 ± 0.30 | −7.60 ± 0.28 | 1.04 ± 0.41 |
| 2-R | 0.34 ± 0.12 | −8.88 ± 0.21 | −7.73 ± 0.28 | 1.15 ± 0.35 |
| 2-S | 0.32 ± 0.16 | −8.97 ± 0.26 | −7.54 ± 0.34 | 1.42 ± 0.43 |
| 15 | 6.33 ± 1.53 | −7.14 ± 0.14 | −3.60 ± 0.31 | 3.54 ± 0.34 |
| 16 | 1.60 ± 0.34 | −7.96 ± 0.13 | −7.03 ± 0.42 | 0.93 ± 0.44 |
| 10 | 0.89 ± 0.19 | −8.31 ± 0.13 | −6.79 ± 0.26 | 1.51 ± 0.29 |
| 13 | 0.62 ± 0.14 | −8.52 ± 0.13 | −7.14 ± 0.21 | 1.38 ± 0.25 |
| 14 | 0.63 ± 0.17 | −8.53 ± 0.16 | −7.62 ± 0.30 | 0.91 ± 0.34 |
| 11 | 0.36 ± 0.22 | −8.90 ± 0.43 | −5.02 ± 0.41 | 3.88 ± 0.60 |
| 17 | 0.93 ± 0.36 | −8.28 ± 0.23 | −3.82 ± 0.28 | 4.46 ± 0.36 |
| 18 | 1.30 ± 0.43 | −8.08 ± 0.20 | −5.08 ± 0.31 | 3.00 ± 0.37 |

Similarly, the antiviral potency (EC50) evaluation showed the same ranking. In ref. 137 was also shown that the calculated relative binding free energies with FEP/MD calculations was ΔΔGFEP/MD (3 → 2-R) = 0.62 ± 0.14 kcal mol−1 with ΔΔGITC (3 → 2-R) = 0.33 ± 0.50 kcal mol−1, while ΔΔGFEP/MD (3 → 2-S) = 0.68 ± 0.15 kcal mol−1 with ΔΔGITC (3 → 2-R) = 0.42 ± 0.48 kcal mol−1. These FEP/MD calculation results supported that rimantadine enantiomers, although chiral molecules, have equal binding affinities against a chiral receptor, i.e., the AM2TM WT protein. The MD simulations in ref. 134 showed that in diethyl and propyl analogs (13 and 14, respectively, see Table 3), the alkyl groups appear to better occupy the space between the ligand and the pore walls. For these compounds' binding, the ITC data indicated that the constrained motion and the resulting entropy cost of binding are important quantities and reduced the binding affinities of 13 and 14 compared to 10 (see Table 1 in ref. 134). The proton blocking activity of the adamantanamines is consistent with the fact that no water molecules were detected in the area above the adamantane core (i.e., toward the N-terminus) in the MD simulations.
A few adamantanamines (1, 2-R, 2-S, 3–10; Table 4) in complex with AM2TM WT were studied at acidic pH174 and alkaline pH175 with alchemical calculations using the FEP/MD method with OPLS2005 force field provided with the Desmond software, and relative free energies were calculated using the Bennett acceptance ratio (BAR).171,172 The Kd values were used as experimental probes for the FEP/MD calculations of alchemical relative binding free energies.
While the set of 12 compounds in Table 4 had a very narrow range of binding free energies of 8 kJ mol−1 (1.9 kcal mol−1), i.e., Kd values for the close state of AM2TM WT (Udorn strain) at pH 8 that differed by a factor of 20 (Table 4), the FEP/MD method performed with high correlation found (r = 0.94, p < 0.001, PI = 0.74) between calculated and experimental binding free energies.175 The correlation was also good in FEP/MD simulations of AM2TM at acidic pH using experimental Kd values measured at acidic pH against the open conformation of AM2FL.119 As mentioned previously, the experimental assay was based on inhibition by adamantanamines of the quenching of W41 fluorescence by H37 protonation below pH 6 in AM2FL in a detergent environment.118
Subsequently, the experimental binding free energies computed based on the Kd values measured with ITC were measured against the closed form of AM2TM tetramer in DPC micelles for a larger set of 27 adamantanamines, and the antiviral potencies (IC50) with whole cell assays were also measured.169 The range of the experimental Kd values was ∼44, and the range of the antiviral potencies (IC50) values was ∼750. A good correlation (r = 0.76) was found between their corresponding binding free energy, computed using Kd or IC50 values. MD simulations with Amber19sb force field (ff19sb)176 or CHARMM36m force field154 and different experimental starting structures of AM2TM were used to investigate the binding mode of adamantanamines that bind strongly, moderately, or tightly to AM2TM embedded in DMPC, DPPC (dipalmitoyl-sn-glycero-3-phosphocholine), POPC, or a virus (VM) membrane (Fig. 8).
 | ||
| Fig. 8 Representative structures from MD simulations with ff19sb176 of compounds (R)-rimantadine, its dimethyl and dipropyl adduct (see compounds 10, 14 in Table 4) in complex with AM2TM WT in DMPC bilayers. In the figure, the protein is shown with a white cartoon and the ligands with sticks and red color. The water molecules are shown with sticks. White is used for hydrogen, red for oxygen, and blue for nitrogen; yellow dotted lines show the hydrogen bonds. | ||
FEP/MD NPT simulations with the OPLS2005 force field provided with the Desmond software and the BAR estimator,171,172 TI/MD NVT simulations with ff19sb176 and the multistate BAR (MBAR) estimator177 were applied for the AM2TM WT–adamantanamine complexes embedded in DMPC, DPPC, and POPC, to precisely predict experimental relative binding free energies measured with ITC. Dual topology and single topology alchemical transformations were used with TI/MD and FEP/MD, respectively. The pair of ligands considered bears subtle changes in the ligands' structures. With all lipids, it was found that both approaches exhibited a very good correlation between the calculated and experimental relative binding free energies (r = 0.77–0.87, MUE = 0.36–0.92 kcal mol−1), with FEP/BAR performing the worst in DMPC bilayers and TI/MBAR performing the best. The experimental binding free energies were also calculated using antiviral potencies, compared to Kd values, and both FEP/BAR (r = 0.83, MUE = 0.75 kcal mol−1) and TI/MBAR (r = 0.69, MUE = 0.77 kcal mol−1) performed well also in this case.169
It should be noted that FEP/MD accuracy depends heavily on force fields and sampling, and that these methods are not a substitute for experimental validation, especially given the complexities of protonation states and water networks in AM2.
Synthetic routes to selected adamantane-based and saturated polycyclic amines targeting AM2 channels
General
Adamantane derivatives have been extensively used in medicinal chemistry and can be found in several drugs.19,178–181 After the original publication of amantadine's antiviral activity by du Pont de Nemours in 1964,182 early work in the synthesis of amantadine analogs was reported by the same company in 1970 and 1971183,184 and Philips-Duphar Research Laboratories in 1971.185,186 One of the most significant contributions was the synthesis of over 300 compounds from a lab in Greece, synthesized between 1994 and 2003, see, for example, ref. 113, 137, 142, 145 and 187–199. Most were tested initially for their in vitro potency against influenza A. We mentioned here a few works, including selected synthesis of adamantanamines, saturated polycyclic amines, including pinanamine or camphor analogs.111,134,146,153,159,160,162,163,200–208 We started with the presentation of the simpler adamantanamines synthesized with a more scalable route, followed by bulkier cage alkyl amines and saturated polycyclic amines to explore modification of the common adamantane ring. We presented in a distinct section the second-generation AM2 S31N inhibitors that can become available through simple synthetic procedures. Elegant syntheses, including radical functionalization methods, have been developed to access various substituted adamantanes, 1,2-didubstituted adamantanes, and diamondoids.209,210 Selected syntheses of various adamantanamines and other adamantane derivatives have been reviewed.18,19Rimantadine analogs
Extensive structure–activity relationships (SARs) about the in vitro potency have been performed for over 40 years, as mentioned previously in numerous publications, see for example, ref. 208 and 211. These references include selected examples from the literature regarding the synthesis of amantadine and rimantadine analogs, which have been tested and found potent against influenza A. Adamantane derivatives have been extensively used in medicinal chemistry and can be found in several drugs.19,178–181 Elegant syntheses, including radical functionalization methods, have been developed to access various substituted adamantanes, 1,2-didubstituted adamantanes, and diamondoids.209,210 Nevertheless, selected syntheses of various adamantane derivatives have been recently reviewed in ref. 19.For the preparation of 2-rimantadine analogs 24, 25, 32, key intermediates were the 2-adamantyl methyl ketone 19 or the 2-cyanoadamantane 29, as is described in Scheme 2.212
 | ||
| Scheme 2 Synthesis of acyclic 2-rimantadine analogs 24, 25, 32. | ||
The cyclic rimantadine analogs 37, 38, and 43, 44 (Scheme 3) were also synthesized with key intermediates the cyclic alcohols 34 and 40, obtained from the reaction of the acid chloride 33 with the dimagnesium reagent BrMg(CH2)4MgBr167,213,214 and with 1-adamantyl-lithium (generated from 1-bromoadamantane 39 with lithium) with cyclohexanone, respectively.18,215 Primary tert-alkyl amines 38, 44 were obtained from azides 36, 41, respectively, and a work was published that provided routes to produce primary tert-alkyl amines from corresponding alcohols, improving steps d for the preparation of 36 from 41.214
 | ||
| Scheme 3 Synthesis of cyclic rimantadine analogs 37, 38, 43, 44. | ||
Important intermediates for the preparation of 50 were the aminoalcohols 45 and 46 (Scheme 4). Compound 45 was obtained from the reaction of 2-pyridinyl lithium with 2-adamantanone 13, and its protonated form was subjected to catalytic hydrogenation over PtO2 catalyst to provide compound 46.216 Compounds 8 and 54 were prepared from adamantane carboxylic acid 51 by treatment with [bis(trifluoroacetoxy)iodo]benzene, followed by catalytic hydrogenation over H2, PtO2 (Scheme 4).
 | ||
| Scheme 4 Synthesis of cyclic rimantadine analogs 8 and analogs 48, 50, 54. | ||
Spiranic adamantanamines
Synthesis of BL-1743 (3) was accomplished with the reduction of commercially available 3,3-pentamethylene glutarimide (55) with LiAlH4 in refluxing THF to give 3-azaspiro[5,5]undecane (51), which was subjected to a nucleophilic substitution with 2-methylthio-2-imidazoline to furnish BL-1743 (3).The rigidity and crowding around the 2-position of the adamantane nucleus often make chemical transformations difficult and the synthesis of rigid adamantane derivatives challenging. A few synthetic pathways are described below.
2-Nitroadamantane 57, which was prepared originally as described in ref. 131, was used for the synthesis of spiropyrrolidine 11; the latter was prepared through a Michael condensation of 57 with ethyl acrylate to afford nitroester 58, which was hydrogenated over Ni to afford spiro[pyrrolidine-2,2′-adamantan]-5-one (59) that was reduced with LiALH4 to compound 11217,218 (Scheme 5).
 | ||
| Scheme 5 Synthesis of 3-azaspiro[5,5]undecane (5) and its 4,5-dihydro-1H-imidazol-2-yl derivative (BL-1743; 3), spiropyrrolidine 11, spiro[cyclopropane-1,2′-adamantan]-2-amines 63, 64 and -methanamines 66, 67, spiro[cyclobutane-2,2′-adamantan]-2-amine 73, 69 and spiro[cyclopentane-3,2′-adamantan]-2-amine 79, and its and spiro[adamantane-2,1′-cyclohexan]-4′-amine (82). | ||
The key intermediate for the synthesis of the spiro[cyclopropane-1,2′-adamantan]-2-amines 63, 64 and methanamines 66, 67 (Scheme 5) was ethyl spiro[cyclopropane-1,2′-adamantane]-2-carboxylate 61, obtained from the [2 + 1]cycloaddition reaction of ethyl diazoacetate with 2-methyleneadamantane 60 in the presence of copper-bronze and purified through saponification and conversion to acid 62. Routine transformations were performed to prepare 63, 64, 66, 67.
The synthesis of spiro[cyclobutane-2,2′-adamantan]-2-amines 69 or 72 and spiro[cyclopentane-3,2′-adamantan]-2-amines 79 was also accomplished. For the synthesis of these amines, the spirocyclobutanone 68 or spirocyclopentanone 77 were used, respectively, the latter obtained from the dinitrile 75, which was transformed to the cyanoenamine 76 through the Thorpe–Ziegler reaction (Scheme 5).212 Key reaction for the synthesis of cyclohexanamine 82 was a Diels–Alder reaction between 2-adamantanecarboxaldehyde and methyl vinyl ketone to afford cyclohexenone 80.
1,2-Annulated heterocycles
So far, we have presented adamantane derivatives with substitutions at the 1- or 2- position of the adamantane ring. Here are examples of 1,2-annulated adamantano-pyrrolidines and adamantano-piperidines that have been synthesized but not yet tested against influenza A strains. These synthetic ways show how to access disubstituted adamantane derivatives, which might be important for drug design, as different substitutions of the adamantane cage can affect the potency of M2 channel blockers. Synthetic approaches towards the 1,2-substitution pattern on the adamantane framework have been presented.210,219 Selected works containing a directed C–H functionalization step have been reviewed in ref. 210. Additionally, the synthesis of various 1,2-annulated derivatives with antiviral potency has been reviewed.220 As illustrated in Scheme 6, the 2-oxo-1-adamantane acetic acid 83 or 2-oxo-1-adamantane carboxylic acid 89 or 2-oxo-1-adamantanepropanoic acid 94 were the key starting materials to afford the 1,2-annulated adamantano-pyrrolidines 88 or 93 or the adamantano-piperidine 97 or 101 (Scheme 6). For the synthesis of 107, the oxetane derivative 102 first reacted with the triphenyldibromophosphorane, which was prepared in situ by the addition of Br2 to a solution of triphenylphosphine. The resulting dibromide derivative 103 was treated with sodium cyanide in DMSO to obtain the dinitrile 104, which was then converted to lactam 106 and amine 107 (Scheme 6). | ||
| Scheme 6 Synthetic procedures for the preparation of the 1,2-annulated adamantanepyrroldines and piperidines 88 or 93 and 97, 101, 107, and 113. | ||
The synthesis of cyclopentanamine 113 in Scheme 6 starts from the tetrahydrofuran 108, which was converted to dinitrile 110. By employment of the Thorpe–Ziegler reaction, dinitrile 110 underwent an intramolecular condensation catalyzed by LDA to form the enamine 111. Subsequent acid-promoted hydrolysis of the latter gave rise to the racemic cyclic ketone 112, which was converted to the corresponding oxime, which was hydrogenated over RANEY® Ni to provide the cyclopentanamine 113.
Saturated polycyclic amines
The saturated polycyclic amines, e.g., 7,8,9,10-tetramethyl-3-azapentacyclo[7.2.1.15,8.01,5.07,10]tridecane (6)158 and heptacyclo[8.6.1.02,5.03,11.04,9.06,17.012,16]heptadecane (7)166 (Table 2) were potent triple AM2 WT, L26F, V27A blockers and inhibitors in vitro of the corresponding viruses. For their synthesis, fascinating reaction sequences were applied (Scheme 7). | ||
| Scheme 7 Synthetic procedure for the preparation of the polycyclic amines 6, 7. | ||
For the synthesis of the aesthetically appealing amines 6 that include one four-membered ring, three five-membered rings, and two six-membered rings, the known diacid 114 was used, easily available from the cycloaddition reaction between acetylene dicarboxylic acid and 2,3-dimethylbutadiene. The reaction of 114 with urea at 180 °C for 30 min yielded the corresponding imide, which was subjected to photolysis at rt in acetone with a 125 W Hg lamp for 2 days to furnish tetracyclic imide 116, which was reduced with sodium bis(2-methoxyethoxy)aluminum hydride to amine 6.158
The starting material for the synthesis of 7 was the imide 118, prepared from maleimide with cycloheptatriene. The Diels–Alder reaction between 118 and 5,5-dimethoxy-1,2,3,4-tetrachlorocyclopentadiene furnished 120. Then, a one-pot dechlorination/acetal deprotection reaction with metallic sodium in liquid ammonia/acidic medium, and a one-pot decarbonylation produced 121 from which 7 was furnished.166
Further synthetic studies aimed to develop polycyclic pyrrolidine scaffolds with potential inhibitory activity against the AM2 channel. The known diacids 124a,b were heated with urea at 180 °C for 30 min to give the corresponding imides 125a,b, which were subsequently reduced to the secondary amines 126a,b in good overall yields.166 Subsequent reductive alkylation of 126a,b with formaldehyde and NaCNBH3 produced the tertiary amines 127a,b. Because guanidines derived from similar amine precursors had shown AM2 inhibitory properties, compounds 128a,b were synthesized from 126a,b using 1H-pyrazole-1-carboxamidine hydrochloride (Scheme 8). Molecular and structural investigations have demonstrated that substituting V27 with a smaller residue, such as alanine (AM2 V27A mutant), disrupts the hydrophobic constriction created by this position and expands the pore near the N-terminal end by roughly 2 Å.166 Compounds 125a and 127a exhibited stronger inhibition of the M2-V27A mutant channel compared with their smaller analogues 125b and 127b. Molecular dynamics simulations predicted that bulkier, more hydrophobic ligands could occupy this enlarged pore more efficiently. Thus, two extended analogues of 127a, namely 127c and 127d, were designed and synthesized following the same route.166
 | ||
| Scheme 8 Synthetic procedure for the preparation of the polycyclic pyrrolidine and guanidine derivatives 124a–d–128a–d.166 | ||
To obtain the extended analogues 127c,d, novel diacids 124c,d were required. These were synthesized from readily available diketones 129c,d through the multi-step sequence shown in Scheme 9.166 The approach, previously optimized for bisnoradamantane scaffolds, involved conversion of 129c,d to the corresponding bis-hydrazones 130c,d under reflux in ethanol with triethylamine and hydrazine hydrate, followed by iodination with I2/TMG (tetramethylguanidine) to yield the bis-vinyl iodides 131c,d. Subsequent Pd(0)-catalyzed carbonylation (Pd(OAc)2/PPh3, CO, Et3N, MeOH, 70 °C) furnished the diesters 132c,d as mixtures of syn/anti isomers. Catalytic hydrogenation (Pd/C, H2 30 atm) produced diols 133c,d, which, upon double deprotonation with HMDS/n-BuLi and treatment with I2 at −68 °C in THF, afforded 134c,d. Finally, hydrolysis of 134c,d provided the target diacids 124c,d in 63% and 80% yield, respectively.166
 | ||
| Scheme 9 Synthetic procedure for the preparation of diacids 124c,d.208 | ||
Design and synthesis of adamantane-based drugs that lead to selective AM2 S31N inhibitors
After the observation that pinanamine (4)165 blocks M2 WT-mediated proton current (Table 2), it was found128,130 that linking pinanamine with a heterocycle, e.g., imidazole, through a CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) or CH2 linker provided pinanamine–aryl conjugates, e.g., compound 135, which are very potent against influenza A M2 WT and M2 S31N viruses in vitro by blocking the corresponding M2 channels (Scheme 10). The %-blocking efficiency at 2 min of 5-methyl-imidazole was less than 30%, suggesting a slow entry blocking compound.101,105,139
or CH2 linker provided pinanamine–aryl conjugates, e.g., compound 135, which are very potent against influenza A M2 WT and M2 S31N viruses in vitro by blocking the corresponding M2 channels (Scheme 10). The %-blocking efficiency at 2 min of 5-methyl-imidazole was less than 30%, suggesting a slow entry blocking compound.101,105,139
 | ||
| Scheme 10 Chemical structure of pinanamine–aryl conjugate 135 (ref. 228) and amantadine–aryl conjugates 136–145,76,81,140,229 which block both M2 WT and M2 S31N channels according to EP and inhibit in vitro the M2 WT and M2 S31N viruses (in 137 A is 2-thiophenyl, in 138 A is 2-chloro-3-thiophenyl, in 139 A is 3-methoxy-2-thiophenyl, in 140 A is phenyl and in 141 R is cyclopropyl). | ||
It was then found that linking amantadine (1) with a heterocycle can produce potent compounds acting against AM2 S31N in both EP and antiviral assays. Using MD simulations and solution NMR in DPC micelles, it was shown that compound 137 (M2J332) in Scheme 10, with a 2-(2-thiophenyl)-isoxazol-3-yl group, blocks the AM2 S31N channel.76 The adamantyl group of the drug is bound in the pore between AM2 N31 and G34. In contrast, the aryl group projects towards the N-terminus through the V27 side chains (Fig. 2). This orientation on the AM2 S31N channel is opposite to the orientation of amantadine on the M2 WT channel, i.e., with the amino group facing H37 towards the C-end (Fig. 1).72,80,221 Using ssNMR in DMPC lipid bilayers, this position and orientation were also suggested in AM2TM S31N for compound 140 (M2J352).79 It was also found that compounds 144, 145140 bearing one of the simpler aryl groups used, i.e., correspondingly the 3-bromothiophenyl, 3-iodothiophenyl, block both AM2 WT and AM2 S31N channels. Using NMR in micelles and MD simulations, it was found that the dual inhibitors 144 and 145140 are oriented with the aryl head group toward the N-end in the AM2AH S31N pore and toward C-end in the AM2AH WT pore.81 For these amantadine–aryl conjugates, between 2013 and 2018, extensive SAR investigations on in vitro activity and EP-based blocking potency were conducted,78,133,135,136,222–226 and/or binding kinetics78,136 were performed by modifying the adamantyl group and the aryl head group.76,81,139–141 The kinetics of a ligand binding to its protein target are seen as increasingly important for in vivo efficacy in drug discovery227 and were critical for amantadine–aryl conjugates.139,141 Targeted optimization of binding kinetics was difficult to achieve and required systematic studies, as did those for AM2 channels, to increase understanding of the molecular interactions involved.139 It was shown78,136 that, similarly to amantadine analogs against AM2 WT73,147 or against V27A,101 the amantadine–aryl conjugates should have fast kon, slow koff (e.g., compound 141) or slow kon, slow koff (e.g., compound 142) that have low Kd (= koff/kon) values to achieve well in vitro antiviral potency.
Then, in 2020,106 the effect of subtle modifications on the structure of an amantadine–aryl conjugate inhibitor on binding kinetics for AM2 WT and AM2 S31N channels was studied using synthetic amantadine variant-CH2-aryl derivatives 136, 143, 146–149 (Scheme 11) as sensitive chemical probes for blocking AM2 S31N and AM2 WT channels as well as virus replication in cell culture. Notably, only the binding kinetics for the AM2 S31N channel were very dependent on the length between the adamantane moiety and the first ring of the aryl head group, as observed in 146 and 147, and the girth and length of the adamantane adduct, as observed in 148 and 149. The study of 136, 137, 146–149 with MD simulations in AM2TM S31N and binding free energy calculations (MM-PBSA) showed that all compounds bind in to the AM2 S31N channel with the adamantyl group positioned between V27 and G34 the aryl group projecting out of the channel (toward the N-end) with phenyl (or isoxazole in 137) embedded in the V27 cluster. In this outward binding configuration, an elongation of the ligand by only one methylene in rimantadine 146 or using diamantane as well as triamantane instead of adamantane in 148 and 149, respectively, caused an incomplete entry and facilitated exit, abolishing effective block compared to the amantadine derivatives 136 and 137. In the active M2 S31N blockers 136 and 137, the phenyl and isoxazolyl head groups achieve a deeper binding position, corresponding to measured high kon/low koff and high kon/high koff rate constants, compared to inactive 146–149, which have much lower kon and higher koff. The MD simulations also showed that compounds 136, 146–149 each can block the M2TM WT channel by binding in the longer area from V27–H37, in the inward orientation, with high kon and low koff rate constants leading to insufficient block. Infection of cell cultures by influenza virus containing AM2 WT or AM2 S31N is inhibited by 136, 146–149, and 137, respectively. While 136 and 137 block infection through the AM2 block mechanism in the AM2 S31N variant, 146–149 may block AM2 S31N virus replication in cell culture through the lysosomotropic effect.106
 | ||
| Scheme 11 Structures of compounds 136, 146–149, and 137 were used as chemical probes to investigate binding to M2 WT and M2 S31N channels. | ||
For the most potent conjugates, it was shown that 3-hydroxylation of the 1-adamantyl group, e.g., in compound 142 compared to 141, improved the drug metabolism and pharmacokinetics (DMPK) profile, e.g., the microsomal stability in rat and mouse microsomes against cytochromes, the membrane permeability in Caco-2 cells, and increased recovery. Two potent and promising lead candidates for further development as antiviral drugs are 144 and 145141 which have high mouse and human liver microsomal stability (T1/2 > 145 min) and membrane permeability (>200 nm s−1) also inhibit both currently circulating oseltamivir-sensitive and -resistant human influenza A viruses (H1N1 and H3N2) with EC50 values ranging from 0.4 to 2.8 μM and a selectivity index of >100.141
In a recent study,230 based on the structure of the amantadine–aryl conjugates 136, 137 that inhibit AM2 S31N, amantadine was replaced by 16 other adamantanamines. Thus, 36 new compounds were synthesized and tested against AM2 WT and the five amantadine-resistant viruses bearing the AM2 with the mutation L26F, or V27A, or A30T, S31N, or G34E, aiming at identifying inhibitors against multiple M2 mutant-amantadine-resistant viruses. From this study, 16 compounds were identified that inhibited in vitro influenza A viruses with AM2 WT or L26F channels. However, compounds 146–149, which are conjugates of a rimantadine analog or the diamantylamine or the 4-(1-adamantyl)benzenamine with the 2-hydroxy-4-methoxyphenyl group, were in vitro inhibitors against the three influenza A viruses M2 WT or L26F or S31N, while compound 147 also inhibited in vitro the M2 G34E virus, and compound 148 also inhibited in vitro the AM2 A30T virus. Using EP, it was shown that compound 147 was an efficient blocker of the AM2 WT and AM2 L26F channels, compound 148 blocked the AM2 WT channel, and compound 149 blocked the AM2 WT, L26F, and V27A channels. For these compounds, a preliminary drug metabolism and pharmacokinetics study was conducted.230
In another study231 amantadine–aryl conjugates were tested against four amantadine resistant M2 mutants among avian and human influenza A H5N1 strains circulating between 2002 and 2019: the single AM2 S31N and V27A mutants, and the S31N/L26I and S31N/V27A double mutants S31N/L26I and S31N/V27A double mutants. Utilizing TEVC assays, structurally diverse M2 inhibitors were screened against these single and double mutant channels. Compound 139231 (Scheme 10) was found to significantly block all three M2 mutant channels and in vitro replication: AM2 S31N, AM2 S31N/L26I, and AM2 S31N/V27A.
EP studies combined with MD simulations were applied to rationalize the resistance149,232 of AM2 S31N viruses to amantadine–aryl conjugates (e.g., of compound 142) and intriguingly149,232 to mutation L46P outside the M2 S31N channel. A few examples of procedures applied for the synthesis of amantadine–aryl conjugates76,81,139,141,233,234 are shown in Scheme 12.
 | ||
| Scheme 12 Synthetic procedures leading to amantadine–aryl conjugates, e.g., 137 (M2J332),76140 (M2J352),76138,81141,235 as potent blockers of the M2 S31N channel and virus replication. | ||
Interestingly, AM2 S31N blockers, such as pinanyl-thiophenyl oximes 150, have been synthesized as potent blockers of AM2 S31N, having a novel structure132 using (−)-isopinocampheol as raw material (Scheme 13).
 | ||
| Scheme 13 Synthetic procedures lead to pinanyl-thiophenyl oximes, which are potent blockers of AM2 S31N channel and virus replication.132 Reagents and conditions: (a) PDC, CH2Cl2, rt, overnight; (b) TosMIC, t-BuOK, DMSO, 60 °C, 48 h; (c) H2SO4, CH3COOH, reflux, overnight; (a) → (c) 36.4%; (d) N,O-dimethylhydroxylamine hydrochloride, TBTU, triethylamine, CH3CN, rt, 1 h; (e) ArLi, ether, 0 °C, 30 min; −78 °C, 3 h; (f) SOCl2, cat. DMF, reflux, 2 h; (g) substituted thiophene, SnCl4, dry CH2Cl2, 0 °C, 30 min; (h) pyridine, NH2OH·HCl, 80 °C, 24 h. | ||
Propellanamine analogs of adamantanamines
The tricyclic [4.3.3]propellan-8-amines were synthesized208 as conformationally constrained analogues of amantadine. These rigid polycyclic systems were conceived to explore how subtle variations in cage topology affect channel blocking and NMDA receptor binding. The synthesis began with a modified Weiss–Cook reaction between cyclohexane-1,2-dione (151) and dimethyl 3-oxoglutarate (152) to afford the diketone 153, which was subsequently converted into the key propellanedione intermediate (Scheme 14).208 Diastereoselective reduction with L-selectride followed by Wolff–Kishner reduction and acetal manipulation furnished the diastereomerically pure alcohol anti-161 (Scheme 15).208 Further Mitsunobu inversion and SN2 azidation yielded the diastereomeric primary amines syn-158 and anti-158, which were evaluated for both NMDA receptor affinity and anti-influenza A activity. Interestingly, despite structural resemblance to amantadine and comparable binding to the 1-(1-phenylcyclohexyl)piperidine (PCP) site (Ki ≈ 11 μM), neither diastereomer inhibited influenza A replication, demonstrating that modifying the cage geometry separates antiviral from NMDA antagonistic activity. | ||
| Scheme 14 Synthetic route toward diastereomeric [4.3.3]propellan-8-amines.166 | ||
 | ||
| Scheme 15 Diastereoselective synthesis of alcohol anti-161 from monoketal 154.208 | ||
Conclusions
Combining structural studies with advancements in computational methods, can help design and synthesize adamantane-based blockers that target wild-type or/and mutant AM2 channels. The key structural design principles for saturated polycyclic amine-based blockers against AM2 WT, V27A, and L26F are grounded inadvancements the fundamental mechanism of AM2 proton blocking. These principles involve incorporating features that enable a part of the molecule specifically, the carbon framework connected to an amine group-to function as a proton mimic. This allows these inhibitors to occupy the primary proton-binding site within the channel. Second-generation adamantanamines are known to occlude the AM2 S31 pore and block proton transport. Interestingly many saturated adamantanamines inhibit S31N strains, although the mechanism behind this inhibition has not yet been identified.199 Examples of synthetic chemistry that have led to the development of complex adamantanamines, saturated polycyclic amines, and second-generation adamantane-based inhibitors are presented. An extensive and long-term study of the druggable M2 channel has provided the scientific community with essential tools for structure-based drug design, a synthetic chemistry toolkit, and a library of adamantane-based compounds that show promise as effective antivirals, especially given the frequent mutations of the AM2 protein in viruses (Table 5).


|
Conflicts of interest
The authors declare no conflict of interest.Data availability
Data are available upon request.Acknowledgements
AK thanks Chiesi Hellas for supporting this research (SERG No 10354).References
- M. Hong and W. F. DeGrado, Structural Basis for Proton Conduction and Inhibition by the Influenza M2 Protein, Protein Sci., 2012, 21(11), 1620–1633, DOI:10.1002/pro.2158.
- H. X. Zhou and T. A. Cross, Modeling the Membrane Environment Has Implications for Membrane Protein Structure and Function: Influenza A M2 Protein, Protein Sci., 2013, 22(4), 381–394, DOI:10.1002/pro.2232.
- R. M. Pielak and J. J. Chou, Influenza M2 Proton Channels, Biochim. Biophys. Acta, Biomembr., 2011, 522–529, DOI:10.1016/j.bbamem.2010.04.015.
- J. Wang, J. X. Qiu, C. Soto and W. F. Degrado, Structural and Dynamic Mechanisms for the Function and Inhibition of the M2 Proton Channel from Influenza A Virus, Curr. Opin. Struct. Biol., 2011, 21(1), 68–80, DOI:10.1016/j.sbi.2010.12.002.
- K. Georgiou and A. Kolocouris, Conformational Heterogeneity and Structural Features for Function of the Prototype Viroporin Influenza AM2, Biochim. Biophys. Acta, Biomembr., 2025, 1867(1), 184387, DOI:10.1016/j.bbamem.2024.184387.
- K. Georgiou, D. Kolokouris and A. Kolocouris, Molecular Biophysics and Inhibition Mechanism of Influenza Virus A M2 Viroporin by Adamantane-Based Drugs – Challenges in Designing Antiviral Agents, J. Struct. Biol.: X, 2025, 11, 100122, DOI:10.1016/j.yjsbx.2025.100122.
- E. Aledavood, B. Selmi, C. Estarellas and M. Masetti, From Acid Activation Mechanisms of Proton Conduction to Design of Inhibitors of the M2 Proton Channel of Influenza A Virus, Front. Mol. Biosci., 2022, 8, 796229, DOI:10.3389/fmolb.2021.796229.
- J. S. Rossman and R. A. Lamb, Influenza Virus Assembly and Budding, Virology, 2011, 411(2), 229–236, DOI:10.1016/j.virol.2010.12.003.
- S. E. Mtambo, D. G. Amoako, A. M. Somboro, C. Agoni, M. M. Lawal, N. S. Gumede, R. B. Khan and H. M. Kumalo, Influenza Viruses: Harnessing the Crucial Role of the M2 Ion-Channel and Neuraminidase toward Inhibitor Design, Molecules, 2021, 26(4), 880–905, DOI:10.3390/molecules26040880.
- J. Wang, M2 As a Target to Combat Influenza Drug Resistance: What Does the Evidence Say?, Future Virol., 2016, 11(1), 1–4, DOI:10.2217/fvl.15.95.
- P. H. Jalily, M. C. Duncan, D. Fedida, J. Wang and I. Tietjen, Put a Cork in It: Plugging the M2 Viral Ion Channel to Sink Influenza, Antiviral Res., 2020, 178, 104780, DOI:10.1016/j.antiviral.2020.104780.
- M. D. Duque, E. Torres, E. Valverde, M. Barniol, S. Guardiola, M. Rey and S. Vázquez, Inhibitors of the M2 channel of influenza A virus, Recent advances in pharmaceutical sciences, ed. D. Muñoz-Torrero, Transworld research network, 2011, vol. 661 Search PubMed.
- J. Wang, F. Li and C. Ma, Recent Progress in Designing Inhibitors That Target the Drug-Resistant M2 Proton Channels from the Influenza A Viruses, Biopolymers, 2015, 104(4), 291–309, DOI:10.1002/bip.22623.
- G. Kumar and K. A. Sakharam, Tackling Influenza A Virus by M2 Ion Channel Blockers: Latest Progress and Limitations, Eur. J. Med. Chem., 2024, 267, 116172, DOI:10.1016/j.ejmech.2024.116172.
- P. H. Jalily, M. C. Duncan, D. Fedida, J. Wang and I. Tietjen, Put a Cork in It: Plugging the M2 Viral Ion Channel to Sink Influenza, Antiviral Res., 2020, 178, 104780, DOI:10.1016/j.antiviral.2020.104780.
- V. A. Shiryaev and Yu. N. Klimochkin, Viroporins: Important Targets for the Development of New Inhibitors of Viral Replication, Russ. Chem. Bull., 2025, 74(5), 1203–1220, DOI:10.1007/s11172-025-4616-4.
- V. A. Shiryaev and Y. N. Klimochkin, Heterocyclic Inhibitors of Viroporins in the Design of Antiviral Compounds, Chem. Heterocycl. Compd., 2020, 56(6), 626–635, DOI:10.1007/s10593-020-02712-6.
- V. Pardali, E. Giannakopoulou, A. Konstantinidi, A. Kolocouris and G. Zoidis, 1,2-Annulated Adamantane Heterocyclic Derivatives as Anti-Influenza α Virus Agents, Croat. Chem. Acta, 2019, 92(2), 211–218, DOI:10.5562/cca3540.
- C. Dane, A. P. Montgomery and M. Kassiou, The Adamantane Scaffold: Beyond a Lipophilic Moiety, Eur. J. Med. Chem., 2025, 291, 117592, DOI:10.1016/j.ejmech.2025.117592.
- S. Peng, Y. Wang, Z. Gao and B. Xia, Viroporins beyond Pathogenesis: Ion Channel Properties as the Key to Unlocking a Neglected Antiviral Target, Pharmacol. Res., 2025, 219, 107863, DOI:10.1016/j.phrs.2025.107863.
- Y. Ma, E. Frutos-Beltrán, D. Kang, C. Pannecouque, E. De Clercq, L. Menéndez-Arias, X. Liu and P. Zhan, Medicinal Chemistry Strategies for Discovering Antivirals Effective against Drug-Resistant Viruses, Chem. Soc. Rev., 2021, 50(7), 4514–4540, 10.1039/D0CS01084G.
- W.-J. Shin and B. L. Seong, Novel Antiviral Drug Discovery Strategies to Tackle Drug-Resistant Mutants of Influenza Virus Strains, Expert Opin. Drug Discovery, 2019, 14(2), 153–168, DOI:10.1080/17460441.2019.1560261.
- K. Martin and A. Helenius, Nuclear Transport of Influenza Virus Ribonucleoproteins: The Viral Matrix Protein (M1) Promotes Export and Inhibits Import, Cell, 1991, 67(1), 117–130, DOI:10.1016/0092-8674(91)90576-K.
- L. H. Pinto, L. J. Holsinger and R. A. Lamb, Influenza Virus M2 Protein Has Ion Channel Activity, Cell, 1992, 69(3), 517–528, DOI:10.1016/0092-8674(92)90452-I.
- F. Ciampor, P. M. Bayley, M. V. Nermut, E. M. A. Hirst, R. J. Sugrue and A. J. Hay, Evidence That the Amantadine-Induced, M2-Mediated Conversion of Influenza A Virus Hemagglutinin to the Low PH Conformation Occurs in an Acidic Trans Golgi Compartment, Virology, 1992, 188, 14–24, DOI:10.1016/0042-6822(92)90730-D.
- S. Grambas and A. J. Hay, Maturation of Influenza a Virus Hemagglutinin-Estimates of the PH Encountered during Transport and Its Regulation by the M2 Protein, Virology, 1992, 190(1), 11–18, DOI:10.1016/0042-6822(92)91187-Y.
- R. J. Sugrue, G. Bahadur, M. C. Zambon, M. Hall-Smith, A. R. Douglas and A. J. Hay, Specific Structural Alteration of the Influenza Haemagglutinin by Amantadine, EMBO J., 1990, 9(11), 3469–3476, DOI:10.1002/j.1460-2075.1990.tb07555.x.
- D. P. Nayak, R. A. Balogun, H. Yamada, Z. H. Zhou and S. Barman, Influenza Virus Morphogenesis and Budding, Virus Res., 2009, 143(2), 147–161, DOI:10.1016/j.virusres.2009.05.010.
- J. S. Rossman and R. A. Lamb, Influenza Virus Assembly and Budding, Virology, 2011, 411(2), 229–236, DOI:10.1016/j.virol.2010.12.003.
- R. J. Sugrue and A. J. Hay, Structural Characteristics of the M2 Protein of Influenza a Viruses: Evidence That It Forms a Tetrameric Channel, Virology, 1991, 180(2), 617–624, DOI:10.1016/0042-6822(91)90075-M.
- L. H. Pinto and R. A. Lamb, Controlling Influenza Virus Replication by Inhibiting Its Proton Channel, Mol. BioSyst., 2007, 3(1), 18–23, 10.1039/b611613m.
- I. V. Chizhmakov, F. M. Geraghty, D. C. Ogden, A. Hayhurst, M. Antoniou and A. J. Hay, Selective Proton Permeability and PH Regulation of the Influenza Virus M2 Channel Expressed in Mouse Erythroleukaemia Cells, J. Physiol., 1996, 494(Pt 2), 329–336, DOI:10.1113/jphysiol.1996.sp021495.
- C. Wang, R. A. Lamb and L. H. Pinto, Direct Measurement of the Influenza A Virus M2 Protein Ion Channel Activity in Mammalian Cells, Virology, 1994, 205(1), 133–140, DOI:10.1006/viro.1994.1628.
- R. A. Lamb, S. L. Zebedee and C. D. Richardson, Influenza Virus M2 Protein Is an Integral Membrane Protein Expressed on the Infected-Cell Surface, Cell, 1985, 40(3), 627–633, DOI:10.1016/0092-8674(85)90211-9.
- J. S. Rossman, X. Jing, G. P. Leser, V. Balannik, L. H. Pinto and R. A. Lamb, Influenza Virus M2 Ion Channel Protein Is Necessary for Filamentous Virion Formation, J. Virol., 2010, 84(10), 5078–5088, DOI:10.1128/JVI.00119-10.
- E. K. Park, M. R. Castrucci, A. Portner and Y. Kawaoka, The M2 Ectodomain Is Important for Its Incorporation into Influenza A Virions, J. Virol., 1998, 72(3), 2449–2455, DOI:10.1128/jvi.72.3.2449-2455.1998.
- L. H. Pinto and R. A. Lamb, Controlling Influenza Virus Replication by Inhibiting Its Proton Channel, Mol. BioSyst., 2007, 3(1), 18–23, 10.1039/b611613m.
- B. J. Chen, G. P. Leser, D. Jackson and R. A. Lamb, The Influenza Virus M2 Protein Cytoplasmic Tail Interacts with the M1 Protein and Influences Virus Assembly at the Site of Virus Budding, J. Virol., 2008, 82(20), 10059–10070, DOI:10.1128/jvi.01184-08.
- C. Ma, A. L. Polishchuk, Y. Ohigashi, A. L. Stouffer, A. Schon, E. Magavern, X. Jing, J. D. Lear, E. Freire, R. A. Lamb, W. F. DeGrado and L. H. Pinto, Identification of the Functional Core of the Influenza A Virus A/M2 Proton-Selective Ion Channel, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(30), 12283–12288, DOI:10.1073/pnas.0905726106.
- A. J. Hay, A. J. Wolstenholme, J. J. Skehel and M. H. Smith, The Molecular Basis of the Specific Anti-Influenza Action of Amantadine, EMBO J., 1985, 4(11), 3021–3024, DOI:10.1002/j.1460-2075.1985.tb04038.x.
- S. D. Cady, K. Schmidt-Rohr, J. Wang, C. S. Soto, W. F. DeGrado and M. Hong, Structure of the Amantadine Binding Site of Influenza M2 Proton Channels in Lipid Bilayers, Nature, 2010, 463(7281), 689–692, DOI:10.1038/nature08722.
- K. C. Duff, P. J. Gilchrist, A. M. Saxena and J. P. Bradshaw, Neutron Diffraction Reveals the Site of Amantadine Blockade in the Influenza A M2 Ion Channel, Virology, 1994, 202(1), 287–293, DOI:10.1006/viro.1994.1345.
- D. Salom, B. R. Hill, J. D. Lear and W. F. DeGrado, PH-Dependent Tetramerization and Amantadine Binding of the Transmembrane Helix of M2 from the Influenza A Virus, Biochemistry, 2000, 39(46), 14160–14170, DOI:10.1021/bi001799u.
- A. L. Stouffer, R. Acharya, D. Salom, A. S. Levine, L. Di Costanzo, C. S. Soto, V. Tereshko, V. Nanda, S. Stayrook and W. F. DeGrado, Structural Basis for the Function and Inhibition of an Influenza Virus Proton Channel, Nature, 2008, 451(7178), 596–599, DOI:10.1038/nature06528.
- K. C. Duff and R. H. Ashley, The Transmembrane Domain of Influenza A M2 Protein Forms Amantadine-Sensitive Proton Channels in Planar Lipid Bilayers, Virology, 1992, 190(1), 485–489, DOI:10.1016/0042-6822(92)91239-Q.
- C. Wang, K. Takeuchi, L. H. Pinto and R. A. Lamb, Ion Channel Activity of Influenza A Virus M2 Protein: Characterization of the Amantadine Block, J. Virol., 1993, 67(9), 5585–5594, DOI:10.1128/jvi.67.9.5585-5594.1993.
- R. Liang, J. M. J. Swanson, J. J. Madsen, M. Hong, W. F. De Grado and G. A. Voth, Acid Activation Mechanism of the Influenza A M2 Proton Channel, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(45), E6955–E6964, DOI:10.1073/pnas.1615471113.
- J. Hu, R. Fu, K. Nishimura, L. Zhang, H. X. Zhou, D. D. Busath, V. Vijayvergiya and T. A. Cross, Histidines, Heart of the Hydrogen Ion Channel from Influenza A Virus: Toward an Understanding of Conductance and Proton Selectivity, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(18), 6865–6870, DOI:10.1073/pnas.0601944103.
- C. Wang, R. A. Lamb and L. H. Pinto, Activation of the M2 Ion Channel of Influenza Virus: A Role for the Transmembrane Domain Histidine Residue, Biophys. J., 1995, 69(4), 1363–1371, DOI:10.1016/S0006-3495(95)80003-2.
- J. Hu, R. Fu, K. Nishimura, L. Zhang, H. X. Zhou, D. D. Busath, V. Vijayvergiya and T. A. Cross, Histidines, Heart of the Hydrogen Ion Channel from Influenza A Virus: Toward an Understanding of Conductance and Proton Selectivity, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(18), 6865–6870, DOI:10.1073/pnas.0601944103.
- Y. Tang, F. Zaitseva, R. A. Lamb and L. H. Pinto, The Gate of the Influenza Virus M2 Proton Channel Is Formed by a Single Tryptophan Residue, J. Biol. Chem., 2002, 277(42), 39880–39886, DOI:10.1074/jbc.M206582200.
- M. Yi, T. A. Cross and H. X. Zhou, A Secondary Gate as a Mechanism for Inhibition of the M2 Proton Channel by Amantadine, J. Phys. Chem. B, 2008, 112(27), 7977–7979, DOI:10.1021/jp800171m.
- K. Nishimura, S. Kim, L. Zhang and T. A. Cross, The Closed State of a H+ Channel Helical Bundle Combining Precise Orientational and Distance Restraints from Solid State NMR, Biochemistry, 2002, 41(44), 13170–13177, DOI:10.1021/bi0262799.
- J. L. Thomaston, M. Alfonso-Prieto, R. A. Woldeyes, J. S. Fraser, M. L. Klein, G. Fiorin and W. F. DeGrado, High-Resolution Structures of the M2 Channel from Influenza A Virus Reveal Dynamic Pathways for Proton Stabilization and Transduction, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(46), 14260–14265, DOI:10.1073/pnas.1518493112.
- J. L. Thomaston, R. A. Woldeyes, T. Nakane, A. Yamashita, T. Tanaka, K. Koiwai, A. S. Brewster, B. A. Barad, Y. Chen, T. Lemmin, M. Uervirojnangkoorn, T. Arima, J. Kobayashi, T. Masuda, M. Suzuki, M. Sugahara, N. K. Sauter, R. Tanaka, O. Nureki, K. Tono, Y. Joti, E. Nango, S. Iwata, F. Yumoto, J. S. Fraser and W. F. DeGrado, XFEL Structures of the Influenza M2 Proton Channel: Room Temperature Water Networks and Insights into Proton Conduction, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13357–13362, DOI:10.1073/pnas.1705624114.
- L. J. Holsinger, D. Nichani, L. H. Pinto and R. A. Lamb, Influenza A Virus M2 Ion Channel Protein: A Structure-Function Analysis, J. Virol., 1994, 68(3), 1551–1563, DOI:10.1128/jvi.68.3.1551-1563.1994.
- M. F. McCown and A. Pekosz, Distinct Domains of the Influenza a Virus M2 Protein Cytoplasmic Tail Mediate Binding to the M1 Protein and Facilitate Infectious Virus Production, J. Virol., 2006, 80(16), 8178–8189, DOI:10.1128/JVI.00627-06.
- J. S. Rossman, X. Jing, G. P. Leser and R. A. Lamb, Influenza Virus M2 Protein Mediates ESCRT-Independent Membrane Scission, Cell, 2010, 142(6), 902–913, DOI:10.1016/j.cell.2010.08.029.
- M. Sharma, M. Yi, H. Dong, H. Qin, E. Peterson, D. D. Busath, H.-X. Zhou and T. A. Cross, Insight into the Mechanism of the Influenza A Proton Channel from a Structure in a Lipid Bilayer, Science, 2010, 330(6003), 509–512, DOI:10.1126/science.1191750.
- J. Hu, T. Asbury, S. Achuthan, C. Li, R. Bertram, J. R. Quine, R. Fu and T. A. Cross, Backbone Structure of the Amantadine-Blocked Trans-Membrane Domain M2 Proton Channel from Influenza A Virus, Biophys. J., 2007, 92(12), 4335–4343, DOI:10.1529/biophysj.106.090183.
- A. L. Stouffer, R. Acharya, D. Salom, A. S. Levine, L. Di Costanzo, C. S. Soto, V. Tereshko, V. Nanda, S. Stayrook and W. F. DeGrado, Structural Basis for the Function and Inhibition of an Influenza Virus Proton Channel, Nature, 2008, 451(7178), 596–599, DOI:10.1038/nature06528.
- K. Nishimura, S. Kim, L. Zhang and T. A. Cross, The Closed State of a H+ Channel Helical Bundle Combining Precise Orientational and Distance Restraints from Solid State NMR, Biochemistry, 2002, 41(44), 13170–13177, DOI:10.1021/bi0262799.
- R. Acharya, V. Carnevale, G. Fiorin, B. G. Levine, A. L. Polishchuk, V. Balannik, I. Samish, R. A. Lamb, L. H. Pinto, W. F. DeGrado and M. L. Klein, Structure and Mechanism of Proton Transport through the Transmembrane Tetrameric M2 Protein Bundle of the Influenza A Virus, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(34), 15075–15080, DOI:10.1073/pnas.1007071107.
- R. Acharya, V. Carnevale, G. Fiorin, B. G. Levine, A. L. Polishchuk, V. Balannik, I. Samish, R. A. Lamb, L. H. Pinto, W. F. DeGrado and M. L. Klein, Structure and Mechanism of Proton Transport through the Transmembrane Tetrameric M2 Protein Bundle of the Influenza A Virus, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(34), 15075–15080, DOI:10.1073/pnas.1007071107.
- J. L. Thomaston, M. Alfonso-Prieto, R. A. Woldeyes, J. S. Fraser, M. L. Klein, G. Fiorin and W. F. DeGrado, High-Resolution Structures of the M2 Channel from Influenza A Virus Reveal Dynamic Pathways for Proton Stabilization and Transduction, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(46), 14260–14265, DOI:10.1073/pnas.1518493112.
- J. R. Schnell and J. J. Chou, Structure and Mechanism of the M2 Proton Channel of Influenza A Virus, Nature, 2008, 451(7178), 591–595, DOI:10.1038/nature06531.
- S. D. Cady, K. Schmidt-Rohr, J. Wang, C. S. Soto, W. F. DeGrado and M. Hong, Structure of the Amantadine Binding Site of Influenza M2 Proton Channels in Lipid Bilayers, Nature, 2010, 463(7281), 689–692, DOI:10.1038/nature08722.
- S. Cady, T. Wang and M. Hong, Membrane-Dependent Effects of a Cytoplasmic Helix on the Structure and Drug Binding of the Influenza Virus M2 Protein, J. Am. Chem. Soc., 2011, 133(30), 11572–11579, DOI:10.1021/ja202051n.
- R. M. Pielak, J. R. Schnell and J. J. Chou, Mechanism of Drug Inhibition and Drug Resistance of Influenza A M2 Channel, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(18), 7379–7384, DOI:10.1073/pnas.0902548106.
- L. B. Andreas, M. T. Eddy, R. M. Pielak, J. Chou and R. G. Griffin, Magic Angle Spinning NMR Investigation of Influenza A M218−60: Support for an Allosteric Mechanism of Inhibition, J. Am. Chem. Soc., 2010, 132(32), 10958–10960, DOI:10.1021/ja101537p.
- R. M. Pielak, K. Oxenoid and J. J. Chou, Structural Investigation of Rimantadine Inhibition of the AM2-BM2 Chimera Channel of Influenza Viruses, Structure, 2011, 19(11), 1655–1663, DOI:10.1016/j.str.2011.09.003.
- J. L. Thomaston, N. F. Polizzi, A. Konstantinidi, J. Wang, A. Kolocouris and W. F. Degrado, Inhibitors of the M2 Proton Channel Engage and Disrupt Transmembrane Networks of Hydrogen-Bonded Waters, J. Am. Chem. Soc., 2018, 140(45), 15219–15226, DOI:10.1021/jacs.8b06741.
- J. L. Thomaston, M. L. Samways, A. Konstantinidi, C. Ma, Y. Hu, H. E. Bruce Macdonald, J. Wang, J. W. Essex, W. F. Degrado and A. Kolocouris, Rimantadine Binds to and Inhibits the Influenza A M2 Proton Channel without Enantiomeric Specificity, Biochemistry, 2021, 60(32), 2471–2482, DOI:10.1021/acs.biochem.1c00437.
- P. Astrahan and I. T. Arkin, Resistance Characteristics of Influenza to Amino-Adamantyls, Biochim. Biophys. Acta, Biomembr., 2011, 1808(2), 547–553, DOI:10.1016/j.bbamem.2010.06.018.
- H. Leonov, P. Astrahan, M. Krugliak and I. T. Arkin, How Do Aminoadamantanes Block the Influenza M2 Channel, and How Does Resistance Develop?, J. Am. Chem. Soc., 2011, 133(25), 9903–9911, DOI:10.1021/ja202288m.
- J. J. Wang, Y. Wu, C. Ma, G. Fiorin, L. H. Pinto, R. A. Lamb, M. L. Klein and W. F. Degrado, Structure and Inhibition of the Drug-Resistant S31N Mutant of the M2 Ion Channel of Influenza A Virus, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(4), 1315–1320, DOI:10.1073/pnas.1216526110.
- S. S. Shrestha, D. L. Swerdlow, R. H. Borse, V. S. Prabhu, L. Finelli, C. Y. Atkins, K. Owusu-Edusei, B. Bell, P. S. Mead, M. Biggerstaff, L. Brammer, H. Davidson, D. Jernigan, M. A. Jhung, L. A. Kamimoto, T. L. Merlin, M. Nowell, S. C. Redd, C. Reed, A. Schuchat and M. I. Meltzer, Estimating the Burden of 2009 Pandemic Influenza A (H1N1) in the United States (April 2009-April 2010), Clin. Infect. Dis., 2011, 52(Supplement 1), S75–S82, DOI:10.1093/cid/ciq012.
- Y. Wang, Y. Hu, S. Xu, Y. Zhang, R. Musharrafieh, R. K. Hau, C. Ma and J. Wang, In Vitro Pharmacokinetic Optimizations of AM2-S31N Channel Blockers Led to the Discovery of Slow-Binding Inhibitors with Potent Antiviral Activity against Drug-Resistant Influenza A Viruses, J. Med. Chem., 2018, 61(3), 1074–1085, DOI:10.1021/acs.jmedchem.7b01536.
- J. K. Williams, D. Tietze, J. Wang, Y. Wu, W. F. Degrado and M. Hong, Drug-Induced Conformational and Dynamical Changes of the S31N Mutant of the Influenza M2 Proton Channel Investigated by Solid-State NMR, J. Am. Chem. Soc., 2013, 135(26), 9885–9897, DOI:10.1021/ja4041412.
- S. D. Cady, J. Wang, Y. Wu, W. F. Degrado and M. Hong, Specific Binding of Adamantane Drugs and Direction of Their Polar Amines in the Pore of the Influenza M2 Transmembrane Domain in Lipid Bilayers and Dodecylphosphocholine Micelles Determined by NMR Spectroscopy, J. Am. Chem. Soc., 2011, 133(12), 4274–4284, DOI:10.1021/ja102581n.
- Y. Wu, B. Canturk, H. Jo, C. Ma, E. Gianti, M. L. Klein, L. H. Pinto, R. A. Lamb, G. Fiorin, J. Wang and W. F. Degrado, Flipping in the Pore: Discovery of Dual Inhibitors That Bind in Different Orientations to the Wild-Type versus the Amantadine-Resistant S31n Mutant of the Influenza a Virus M2 Proton Channel, J. Am. Chem. Soc., 2014, 136(52), 17987–17995, DOI:10.1021/ja508461m.
- J. L. Thomaston, A. Konstantinidi, L. Liu, G. Lambrinidis, J. Tan, M. Caffrey, J. Wang, W. F. Degrado and A. Kolocouris, X-Ray Crystal Structures of the Influenza M2 Proton Channel Drug-Resistant V27A Mutant Bound to a Spiro-Adamantyl Amine Inhibitor Reveal the Mechanism of Adamantane Resistance, Biochemistry, 2020, 59(4), 627–634, DOI:10.1021/acs.biochem.9b00971.
- S. Cady, T. Wang and M. Hong, Membrane-Dependent Effects of a Cytoplasmic Helix on the Structure and Drug Binding of the Influenza Virus M2 Protein, J. Am. Chem. Soc., 2011, 133(30), 11572–11579, DOI:10.1021/ja202051n.
- W. L. Davies, R. R. Grunert, R. F. Haff, J. W. Mcgahen, E. M. Neumayer, M. Paulshock, J. C. Watts, T. R. Wood, E. C. Hermann and C. E. Hoffmann, Antiviral Activity of 1-Adamantanamine (Amantadine), Science, 1964, 144(3620), 862–863, DOI:10.1126/science.144.3620.862.
- H. A. Wendel, M. T. Snyder and S. Pell, Trial of Amantadine in Epidemic Influenza, Clin. Pharmacol. Ther., 1966, 7(1), 38–43, DOI:10.1002/cpt19667138.
- R. S. Schwab, Amantadine in the Treatment of Parkinson's Disease, JAMA, J. Am. Med. Assoc., 1969, 208(7), 1168, DOI:10.1001/jama.1969.03160070046011.
- T. A. Blanpied, R. J. Clarke and J. W. Johnson, Amantadine Inhibits NMDA Receptors by Accelerating Channel Closure during Channel Block, J. Neurosci., 2005, 25(13), 3312–3322, DOI:10.1523/JNEUROSCI.4262-04.2005.
- A. L. Stouffer, C. Ma, L. Cristian, Y. Ohigashi, R. A. Lamb, J. D. Lear, L. H. Pinto and W. F. DeGrado, The Interplay of Functional Tuning, Drug Resistance, and Thermodynamic Stability in the Evolution of the M2 Proton Channel from the Influenza A Virus, Structure, 2008, 16(7), 1067–1076, DOI:10.1016/j.str.2008.04.011.
- Y. Furuse, A. Suzuki, T. Kamigaki and H. Oshitani, Evolution of the M Gene of the Influenza A Virus in Different Host Species: Large-Scale Sequence Analysis, Virol. J., 2009, 6(1), 67, DOI:10.1186/1743-422X-6-67.
- R. A. Bright, M. J. Medina, X. Xu, G. Perez-Oronoz, T. R. Wallis, X. M. Davis, L. Povinelli, N. J. Cox and A. I. Klimov, Incidence of Adamantane Resistance among Influenza A (H3N2) Viruses Isolated Worldwide from 1994 to 2005: A Cause for Concern, Lancet, 2005, 366(9492), 1175–1181, DOI:10.1016/S0140-6736(05)67338-2.
- Y. Lan, Y. Zhang, L. Dong, D. Wang, W. Huang, L. Xin, L. Yang, X. Zhao, Z. Li, W. Wang, X. Li, C. Xu, L. Yang, J. Guo, M. Wang, Y. Peng, Y. Gao, Y. Guo, L. Wen, T. Jiang and Y. Shu, A Comprehensive Surveillance of Adamantane Resistance among Human Influenza A Virus Isolated from Mainland China between 1956 and 2009, Antiviral Ther., 2010, 15(6), 853–859, DOI:10.3851/IMP1656.
- Y. Abed, N. Goyette and G. Boivin, Generation and Characterization of Recombinant Influenza A (H1N1) Viruses Harboring Amantadine Resistance Mutations, Antimicrob. Agents Chemother., 2005, 49(2), 556–559, DOI:10.1128/AAC.49.2.556-559.2005.
- A. N. Brown, J. J. McSharry, Q. Weng, E. M. Driebe, D. M. Engelthaler, K. Sheff, P. S. Keim, J. Nguyen and G. L. Drusano, In Vitro System for Modeling Influenza A Virus Resistance under Drug Pressure, Antimicrob. Agents Chemother., 2010, 54(8), 3442–3450, DOI:10.1128/AAC.01385-09.
- D. Li, R. Saito, Y. Suzuki, I. Sato, H. Zaraket, C. Dapat, I. M. Caperig-Dapat and H. Suzuki, In Vivo and in Vitro Alterations in Influenza A/H3N2 Virus M2 and Hemagglutinin Genes: Effect of Passage in MDCK-SIAT1 Cells and Conventional MDCK Cells, J. Clin. Microbiol., 2009, 47(2), 466–468, DOI:10.1128/JCM.00892-08.
- G. Dong, C. Peng, J. Luo, C. Wang, L. Han, B. Wu, G. Ji and H. He, Adamantane-Resistant Influenza a Viruses in the World (1902-2013): Frequency and Distribution of M2 Gene Mutations, PLoS One, 2015, 10(3), 1–20, DOI:10.1371/journal.pone.0119115.
- V. Garcia and S. Aris-Brosou, Comparative Dynamics and Distribution of Influenza Drug Resistance Acquisition to Protein M2 and Neuraminidase Inhibitors, Mol. Biol. Evol., 2014, 31(2), 355–363, DOI:10.1093/molbev/mst204.
- L. Zhang, Y. Shao, X. Zou, Y. Cui, S. Ding, Y. Chen, Q. Wu, S. Mu, X. Li, Q. Yang, B. Cao and T. Deng, A Primary Oseltamivir-Resistant Mutation in Influenza Hemagglutinin and Its Implications for Antiviral Resistance Surveillance, Nat. Commun., 2025, 16(1), 11442, DOI:10.1038/s41467-025-66307-5.
- R. Musharrafieh, C. Ma and J. Wang, Profiling the in Vitro Drug-Resistance Mechanism of Influenza A Viruses towards the AM2-S31N Proton Channel Blockers, Antiviral Res., 2018, 153, 10–22, DOI:10.1016/j.antiviral.2018.03.002.
- R. Musharrafieh, P. I. Lagarias, C. Ma, G. S. Tan, A. Kolocouris and J. Wang, The L46P Mutant Confers a Novel Allosteric Mechanism of Resistance Toward the Influenza A Virus M2 S31N Proton Channel Blockers, Mol. Pharmacol., 2019, 96(2), 148–157, DOI:10.1124/mol.119.116640.
- R. Musharrafieh, P. Lagarias, C. Ma, R. Hau, A. Romano, G. Lambrinidis, A. Kolocouris and J. Wang, Investigation of the Drug Resistance Mechanism of M2-S31N Channel Blockers through Biomolecular Simulations and Viral Passage Experiments, ACS Pharmacol. Transl. Sci., 2020, 3(4), 666–675, DOI:10.1021/acsptsci.0c00018.
- M. Barniol-Xicota, S. Gazzarrini, E. Torres, Y. Hu, J. Wang, L. Naesens, A. Moroni and S. Vázquez, Slow but Steady Wins the Race: Dissimilarities among New Dual Inhibitors of the Wild-Type and the V27A Mutant M2 Channels of Influenza A Virus, J. Med. Chem., 2017, 60(9), 3727–3738, DOI:10.1021/acs.jmedchem.6b01758.
- N. Kolocouris, A. Kolocouris, G. B. Foscolos, G. Fytas, J. Neyts, E. Padalko, J. Balzarini, R. Snoeck, G. Andrei and E. De Clercq, Synthesis and Antiviral Activity Evaluation of Some New Aminoadamantane Derivatives. 2, J. Med. Chem., 1996, 39(17), 3307–3318, DOI:10.1021/jm950891z.
- C. Scott, J. Kankanala, T. L. Foster, D. H. Goldhill, P. Bao, K. Simmons, M. Pingen, M. Bentham, E. Atkins, E. Loundras, R. Elderfield, J. K. Claridge, J. Thompson, P. R. Stilwell, R. Tathineni, C. S. McKimmie, P. Targett-Adams, J. R. Schnell, G. P. Cook, S. Evans, W. S. Barclay, R. Foster and S. Griffin, Site-Directed M2 Proton Channel Inhibitors Enable Synergistic Combination Therapy for Rimantadine-Resistant Pandemic Influenza, PLoS Pathog., 2020, 16(8), e1008716, DOI:10.1371/JOURNAL.PPAT.1008716.
- P. E. Aldrich, E. C. Hermann, W. E. Meier, M. Paulshock, W. W. Prichard, J. A. Synder and J. C. Watts, Antiviral Agents. 2. Structure-Activity Relations of Compounds Related to 1-Adamantanamine, J. Med. Chem., 1971, 14(6), 535–543, DOI:10.1021/jm00288a019.
- A. Drakopoulos, C. Tzitzoglaki, K. McGuire, A. Hoffmann, C. Ma, K. Freudenberger, A. Konstantinidi, D. Kolocouris, J. Hutterer, G. Gauglitz, J. Wang, M. Schmidtke, D. D. Busath, A. Kolocouris, D. Kolokouris, C. Ma, K. Freudenberger, J. Hutterer, G. Gauglitz, J. Wang, M. Schmidtke, D. D. Busath and A. Kolocouris, Unraveling the Binding, Proton Blockage, and Inhibition of Influenza M2 WT and S31N by Rimantadine Variants, ACS Med. Chem. Lett., 2018, 9(3), 198–203, DOI:10.1021/acsmedchemlett.7b00458.
- C. Tzitzoglaki, K. McGuire, P. Lagarias, A. Konstantinidi, A. Hoffmann, N. A. Fokina, C. C. Ma, I. P. Papanastasiou, P. R. Schreiner, S. Vázquez, M. Schmidtke, J. Wang, D. D. Busath, A. Kolocouris, S. Vazquez, M. Schmidtke, J. Wang, D. D. Busath and A. Kolocouris, Chemical Probes for Blocking of Influenza A M2 Wild-Type and S31N Channels, ACS Chem. Biol., 2020, 15(9), 2331–2337, DOI:10.1021/acschembio.0c00553.
- P. Astrahan, R. Flitman-Tene, E. R. Bennett, M. Krugliak, C. Gilon and I. T. Arkin, Quantitative Analysis of Influenza M2 Channel Blockers, Biochim. Biophys. Acta, Biomembr., 2011, 1808(1), 394–398, DOI:10.1016/j.bbamem.2010.08.021.
- P. Santner, J. M. D. S. Martins, J. S. Laursen, L. Behrendt, L. Riber, C. A. Olsen, I. T. Arkin, J. R. Winther, M. Willemoës and K. Lindorff-Larsen, A Robust Proton Flux (PHlux) Assay for Studying the Function and Inhibition of the Influenza A M2 Proton Channel, Biochemistry, 2018, 57(41), 5949–5956, DOI:10.1021/acs.biochem.8b00721.
- P. Santner, J. M. D. S. Martins, C. Kampmeyer, R. Hartmann-Petersen, J. S. Laursen, A. Stein, C. A. Olsen, I. T. Arkin, J. R. Winther, M. Willemoës and K. Lindorff-Larsen, Random Mutagenesis Analysis of the Influenza A M2 Proton Channel Reveals Novel Resistance Mutants, Biochemistry, 2018, 57(41), 5957–5968, DOI:10.1021/acs.biochem.8b00722.
- D. Salom, B. R. Hill, J. D. Lear and W. F. DeGrado, PH-Dependent Tetramerization and Amantadine Binding of the Transmembrane Helix of M2 from the Influenza A Virus, Biochemistry, 2000, 39(46), 14160–14170, DOI:10.1021/bi001799u.
- V. Balannik, J. Wang, Y. Ohigashi, X. Jing, E. Magavern, R. A. Lamb, W. F. DeGrado and L. H. Pinto, Design and Pharmacological Characterization of Inhibitors of Amantadine-Resistant Mutants of the M2 Ion Channel of Influenza A Virus, Biochemistry, 2009, 48(50), 11872–11882, DOI:10.1021/bi9014488.
- P. E. Czabotar, S. R. Martin and A. J. Hay, Studies of Structural Changes in the M2 Proton Channel of Influenza a Virus by Tryptophan Fluorescence, Virus Res., 2004, 99(1), 57–61, DOI:10.1016/j.virusres.2003.10.004.
- I. Stylianakis, A. Kolocouris, N. Kolocouris, G. Fytas, G. B. Foscolos, E. Padalko, J. Neyts and E. De Clercq, Spiro[Pyrrolidine-2,2′-Adamantanes]: Synthesis, Anti-Influenza Virus Activity and Conformational Properties, Bioorg. Med. Chem. Lett., 2003, 13(10), 1699–1703, DOI:10.1016/S0960-894X(03)00231-2.
- L. Cristian, J. D. Lear and W. F. DeGrado, Use of Thiol-Disulfide Equilibria to Measure the Energetics of Assembly of Transmembrane Helices in Phospholipid Bilayers, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(25), 14772–14777, DOI:10.1073/pnas.2536751100.
- M. R. Rosenberg and M. G. Casarotto, Coexistence of Two Adamantane Binding Sites in the Influenza A M2 Ion Channel, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(31), 13866–13871, DOI:10.1073/pnas.1002051107.
- R. M. Pielak, J. R. Schnell and J. J. Chou, Mechanism of Drug Inhibition and Drug Resistance of Influenza A M2 Channel, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(18), 7379–7384, DOI:10.1073/pnas.0902548106.
- Q. Tu, L. H. Pinto, G. Luo, M. A. Shaughnessy, D. Mullaney, S. Kurtz, M. Krystal and R. A. Lamb, Characterization of Inhibition of M2 Ion Channel Activity by BL-1743, an Inhibitor of Influenza A Virus, J. Virol., 1996, 70(7), 4246–4252, DOI:10.1128/jvi.70.7.4246-4252.1996.
- L. H. Pinto, G. R. Dieckmann, C. S. Gandhi, C. G. Papworth, J. Braman, M. A. Shaughnessy, J. D. Lear, R. A. Lamb and W. F. DeGrado, A Functionally Defined Model for the M2 Proton Channel of Influenza A Virus Suggests a Mechanism for Its Ion Selectivity, Proc. Natl. Acad. Sci. U. S. A., 1997, 94(21), 11301–11306, DOI:10.1073/pnas.94.21.11301.
- C. Wang, R. A. Lamb and L. H. Pinto, Direct Measurement of the Influenza A Virus M2 Protein Ion Channel Activity in Mammalian Cells, Virology, 1994, 205(1), 133–140, DOI:10.1006/viro.1994.1628.
- K. Shimbo, D. L. Brassard, R. A. Lamb and L. H. Pinto, Ion Selectivity and Activation of the M2 Ion Channel of Influenza Virus, Biophys. J., 1996, 70(3), 1335–1346, DOI:10.1016/S0006-3495(96)79690-X.
- M. D. Duque, C. Ma, E. Torres, J. Wang, L. Naesens, J. Juárez-Jiménez, P. Camps, F. J. Luque, W. F. DeGrado, R. A. Lamb, L. H. Pinto and S. Vázquez, Exploring the Size Limit of Templates for Inhibitors of the M2 Ion Channel of Influenza A Virus, J. Med. Chem., 2011, 54(8), 2646–2657, DOI:10.1021/jm101334y.
- E. Torres, R. Fernández, S. Miquet, M. Font-Bardia, E. Vanderlinden, L. Naesens and S. Vázquez, Synthesis and Anti-Influenza a Virus Activity of 2,2-Dialkylamantadines and Related Compounds, ACS Med. Chem. Lett., 2012, 3(12), 1065–1069, DOI:10.1021/ml300279b.
- E. Torres, R. Leiva, S. Gazzarrini, M. Rey-Carrizo, M. Frigolé-Vivas, A. Moroni, L. Naesens and S. Vázquez, Azapropellanes with Anti-Influenza a Virus Activity, ACS Med. Chem. Lett., 2014, 5(7), 831–836, DOI:10.1021/ml500108s.
- M. Rey-Carrizo, S. Gazzarrini, S. Llabrés, M. Frigolé-Vivas, J. Juárez-Jiménez, M. Font-Bardia, L. Naesens, A. Moroni, F. J. Luque and S. Vázquez, New Polycyclic Dual Inhibitors of the Wild Type and the V27A Mutant M2 Channel of the Influenza A Virus with Unexpected Binding Mode, Eur. J. Med. Chem., 2015, 96, 318–329, DOI:10.1016/j.ejmech.2015.04.030.
- M. Rey-carrizo, E. Torres, C. Ma, M. Barniol-xicota, J. Wang, Y. Wu, L. Naesens, W. F. Degrado, R. A. Lamb, L. H. Pinto, S. Vázquez and S. Va, 3-Azatetracyclo[5.2.1.15,8.01,5]Undecane Derivatives: From Wild-Type Inhibitors of the M2 Ion Channel of Influenza A Virus to Derivatives with Potent Activity against the V27A Mutant, J. Med. Chem., 2013, 56, 9265–9274, DOI:10.1021/jm401340p.
- K. L. McGuire, S. Wallentine, N. A. Gordon, G. Mohl, M. D. Jensen, R. Harrison and D. D. Busath, Divalent Copper Complexes as Influenza a M2 S31N Blockers, Biophys. J., 2016, 110(3), 445, DOI:10.1016/j.bpj.2015.11.2395.
- N. A. Gordon, K. L. McGuire, S. K. Wallentine, G. A. Mohl, J. D. Lynch, R. G. Harrison and D. D. Busath, Divalent Copper Complexes as Influenza A M2 Inhibitors, Antiviral Res., 2017, 147, 100–106, DOI:10.1016/j.antiviral.2017.10.009.
- X. Zhao, Y. Jie, M. R. Rosenberg, J. Wan, S. Zeng, W. Cui, Y. Xiao, Z. Li, Z. Tu, M. G. Casarotto and W. Hu, Design and Synthesis of Pinanamine Derivatives as Anti-Influenza A M2 Ion Channel Inhibitors, Antiviral Res., 2012, 96(2), 91–99, DOI:10.1016/j.antiviral.2012.09.001.
- X. Zhao, Z. W. Zhang, W. Cui, S. Chen, Y. Zhou, J. Dong, Y. Jie, J. Wan, Y. Xu and W. Hu, Identification of Camphor Derivatives as Novel M2 Ion Channel Inhibitors of Influenza A Virus, MedChemComm, 2015, 6(4), 727–731, 10.1039/c4md00515e.
- J. Dong, S. Chen, R. Li, W. Cui, H. Jiang, Y. Ling, Z. Yang and W. Hu, Imidazole-Based Pinanamine Derivatives: Discovery of Dual Inhibitors of the Wild-Type and Drug-Resistant Mutant of the Influenza A Virus, Eur. J. Med. Chem., 2016, 108, 605–615, DOI:10.1016/j.ejmech.2015.12.013.
- X. Zhao, R. Li, Y. Zhou, M. Xiao, C. Ma, Z. Yang, S. Zeng, Q. Du, C. Yang, H. Jiang, Y. Hu, K. Wang, C. K. P. Mok, P. Sun, J. Dong, W. Cui, J. Wang, Y. Tu, Z. Yang and W. Hu, Discovery of Highly Potent Pinanamine-Based Inhibitors against Amantadine- and Oseltamivir-Resistant Influenza A Viruses, J. Med. Chem., 2018, 61(12), 5187–5198, DOI:10.1021/acs.jmedchem.8b00042.
- J. Dong, M. Xiao, Q. Ma, G. Zhang, W. Zhao, M. Kong, Y. Zhang, L. Qiu and W. Hu, Design and Synthesis of Pinane Oxime Derivatives as Novel Anti-Influenza Agents, Bioorg. Chem., 2020, 102, 104–106, DOI:10.1016/j.bioorg.2020.104106.
- C. Tzitzoglaki, K. McGuire, P. Lagarias, A. Konstantinidi, A. Hoffmann, N. A. Fokina, C. C. Ma, I. P. Papanastasiou, P. R. Schreiner, S. Vázquez, M. Schmidtke, J. Wang, D. D. Busath, A. Kolocouris, S. Vazquez, M. Schmidtke, J. Wang, D. D. Busath and A. Kolocouris, Chemical Probes for Blocking of Influenza A M2 Wild-Type and S31N Channels, ACS Chem. Biol., 2020, 15(9), 2331–2337, DOI:10.1021/acschembio.0c00553.
- A. Drakopoulos, C. Tzitzoglaki, K. McGuire, A. Hoffmann, C. Ma, K. Freudenberger, A. Konstantinidi, D. Kolocouris, J. Hutterer, G. Gauglitz, J. Wang, M. Schmidtke, D. D. Busath, A. Kolocouris, D. Kolokouris, C. Ma, K. Freudenberger, J. Hutterer, G. Gauglitz, J. Wang, M. Schmidtke, D. D. Busath and A. Kolocouris, Unraveling the Binding, Proton Blockage, and Inhibition of Influenza M2 WT and S31N by Rimantadine Variants, ACS Med. Chem. Lett., 2018, 9(3), 198–203, DOI:10.1021/acsmedchemlett.7b00458.
- J. Wang, C. Ma, J. Wang, H. Jo, B. Canturk, G. Fiorin, L. H. Pinto, R. A. Lamb, M. L. Klein and W. F. DeGrado, Discovery of Novel Dual Inhibitors of the Wild-Type and the Most Prevalent Drug-Resistant Mutant, S31N, of the M2 Proton Channel from Influenza A Virus, J. Med. Chem., 2013, 56(7), 2804–2812, DOI:10.1021/jm301538e.
- Y. Hu, R. K. Hau, Y. Wang, P. Tuohy, Y. Zhang, S. Xu, C. Ma and J. Wang, Structure-Property Relationship Studies of Influenza A Virus AM2-S31N Proton Channel Blockers, ACS Med. Chem. Lett., 2018, 9(11), 1111–1116, DOI:10.1021/acsmedchemlett.8b00336.
- A. Drakopoulos, C. Tzitzoglaki, C. C. Ma, K. Freudenberger, A. Hoffmann, Y. Hu, G. G. Gauglitz, M. Schmidtke, J. Wang and A. Kolocouris, Affinity of Rimantadine Enantiomers against Influenza A/M2 Protein Revisited, ACS Med. Chem. Lett., 2017, 8(2), 145–150, DOI:10.1021/acsmedchemlett.6b00311.
- W. Hu, S. Zeng, C. Li, Y. Jie, Z. Li and L. Chen, Identification of Hits as Matrix-2 Protein Inhibitors through the Focused Screening of a Small Primary Amine Library, J. Med. Chem., 2010, 53(9), 3831–3834, DOI:10.1021/jm901664a.
- Y. Wang, Y. Hu, S. Xu, Y. Zhang, R. Musharrafieh, R. K. Hau, C. Ma and J. Wang, In Vitro Pharmacokinetic Optimizations of AM2-S31N Channel Blockers Led to the Discovery of Slow-Binding Inhibitors with Potent Antiviral Activity against Drug-Resistant Influenza A Viruses, J. Med. Chem., 2018, 61(3), 1074–1085, DOI:10.1021/acs.jmedchem.7b01536.
- J. Wang, C. Ma, J. Wang, H. Jo, B. Canturk, G. Fiorin, L. H. Pinto, R. A. Lamb, M. L. Klein and W. F. DeGrado, Discovery of Novel Dual Inhibitors of the Wild-Type and the Most Prevalent Drug-Resistant Mutant, S31N, of the M2 Proton Channel from Influenza A Virus, J. Med. Chem., 2013, 56(7), 2804–2812, DOI:10.1021/jm301538e.
- Y. Hu, R. K. Hau, Y. Wang, P. Tuohy, Y. Zhang, S. Xu, C. Ma and J. Wang, Structure-Property Relationship Studies of Influenza A Virus AM2-S31N Proton Channel Blockers, ACS Med. Chem. Lett., 2018, 9(11), 1111–1116, DOI:10.1021/acsmedchemlett.8b00336.
- M. Stampolaki, A. Hoffmann, K. Tekwani, K. Georgiou, C. Tzitzoglaki, C. Ma, S. Becker, P. Schmerer, K. Döring, I. Stylianakis, A. L. Turcu, J. Wang, S. Vázquez, L. B. Andreas, M. Schmidtke and A. Kolocouris, A Study of the Activity of Adamantyl Amines against Mutant Influenza A M2 Channels Identified a Polycyclic Cage Amine Triple Blocker, Explored by Molecular Dynamics Simulations and Solid-State NMR**, ChemMedChem, 2023, 18(16), e202300182, DOI:10.1002/cmdc.202300182.
- J. Wang, S. D. Cady, V. Balannik, L. H. Pinto, W. F. DeGrado and M. Hong, Discovery of Spiro-Piperidine Inhibitors and Their Modulation of the Dynamics of the M2 Proton Channel from Influenza A Virus, J. Am. Chem. Soc., 2009, 131(23), 8066–8076, DOI:10.1021/ja900063s.
- K. T. Movellan, M. Wegstroth, K. Overkamp, A. Leonov, S. Becker and L. B. Andreas, Imidazole-Imidazole Hydrogen Bonding in the PH-Sensing Histidine Side Chains of Influenza A M2, J. Am. Chem. Soc., 2020, 142(6), 2704–2708, DOI:10.1021/jacs.9b10984.
- A. Kolocouris, C. Tzitzoglaki, F. B. Johnson, R. Zell, A. K. Wright, T. A. Cross, I. Tietjen, D. Fedida and D. D. Busath, Aminoadamantanes with Persistent in Vitro Efficacy against H1N1 (2009) Influenza A, J. Med. Chem., 2014, 57(11), 4629–4639, DOI:10.1021/jm500598u.
- C. Tzitzoglaki, A. Wright, K. Freudenberger, A. Hoffmann, I. Tietjen, I. Stylianakis, F. Kolarov, D. Fedida, M. Schmidtke, G. Gauglitz, T. A. Cross and A. Kolocouris, Binding and Proton Blockage by Amantadine Variants of the Influenza M2WT and M2S31N Explained, J. Med. Chem., 2017, 60(5), 1716–1733, DOI:10.1021/acs.jmedchem.6b01115.
- A. Drakopoulos, C. Tzitzoglaki, C. C. Ma, K. Freudenberger, A. Hoffmann, Y. Hu, G. G. Gauglitz, M. Schmidtke, J. Wang and A. Kolocouris, Affinity of Rimantadine Enantiomers against Influenza A/M2 Protein Revisited, ACS Med. Chem. Lett., 2017, 8(2), 145–150, DOI:10.1021/acsmedchemlett.6b00311.
- A. K. Wright, P. Batsomboon, J. Dai, I. Hung, H. X. Zhou, G. B. Dudley and T. A. Cross, Differential Binding of Rimantadine Enantiomers to Influenza A M2 Proton Channel, J. Am. Chem. Soc., 2016, 138(5), 1506–1509, DOI:10.1021/jacs.5b13129.
- R. Musharrafieh, P. Lagarias, C. Ma, R. Hau, A. Romano, G. Lambrinidis, A. Kolocouris and J. Wang, Investigation of the Drug Resistance Mechanism of M2-S31N Channel Blockers through Biomolecular Simulations and Viral Passage Experiments, ACS Pharmacol. Transl. Sci., 2020, 3(4), 666–675, DOI:10.1021/acsptsci.0c00018.
- K. Tekwani Movellan, M. Wegstroth, K. Overkamp, A. Leonov, S. Becker and L. B. Andreas, Real-Time Tracking of Drug Binding to Influenza A M2 Reveals a High Energy Barrier, J. Struct. Biol.: X, 2023, 8, 100090, DOI:10.1016/j.yjsbx.2023.100090.
- C. Ma, A. L. Polishchuk, Y. Ohigashi, A. L. Stouffer, A. Schön, E. Magavern, X. Jing, J. D. Lear, E. Freire, R. A. Lamb, W. F. DeGrado and L. H. Pinto, Identification of the Functional Core of the Influenza A Virus A/M2 Proton-Selective Ion Channel, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(30), 12283–12288, DOI:10.1073/pnas.0905726106.
- S. Kellokumpu, Golgi PH, Ion and Redox Homeostasis: How Much Do They Really Matter?, Front. Cell Dev. Biol., 2019, 7, 93, DOI:10.3389/fcell.2019.00093.
- M. Rey-Carrizo, M. Barniol-Xicota, C. Ma, M. Frigolé-Vivas, E. Torres, L. Naesens, S. Llabrés, J. Juárez-Jiménez, F. J. Luque, W. F. Degrado, R. A. Lamb, L. H. Pinto and S. Vázquez, Easily Accessible Polycyclic Amines That Inhibit the Wild-Type and Amantadine-Resistant Mutants of the M2 Channel of Influenza A Virus, J. Med. Chem., 2014, 57(13), 5738–5747, DOI:10.1021/jm5005804.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. De Groot, H. Grubmüller and A. D. MacKerell, CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins, Nat. Methods, 2016, 14(1), 71–73, DOI:10.1038/nmeth.4067.
- J. L. Thomaston, N. F. Polizzi, A. Konstantinidi, J. Wang, A. Kolocouris and W. F. Degrado, Inhibitors of the M2 Proton Channel Engage and Disrupt Transmembrane Networks of Hydrogen-Bonded Waters, J. Am. Chem. Soc., 2018,(45), 15219–15226, DOI:10.1021/jacs.8b06741.
- S. Kurtz, G. Luo, K. M. Hahnenberger, C. Brooks, O. Gecha, K. Ingalls, K. I. Numata and M. Krystal, Growth Impairment Resulting from Expression of Influenza Virus M2 Protein in Saccharomyces Cerevisiae: Identification of a Novel Inhibitor of Influenza Virus, Antimicrob. Agents Chemother., 1995, 39(10), 2204–2209, DOI:10.1128/AAC.39.10.2204.
- G. Zoidis, C. Fytas, I. Papanastasiou, G. B. Foscolos, G. Fytas, E. Padalko, E. De Clercq, L. Naesens, J. Neyts and N. Kolocouris, Heterocyclic Rimantadine Analogues with Antiviral Activity, Bioorg. Med. Chem., 2006, 14(10), 3341–3348, DOI:10.1016/j.bmc.2005.12.056.
- M. Rey-Carrizo, M. Barniol-Xicota, C. Ma, M. Frigolé-Vivas, E. Torres, L. Naesens, S. Llabrés, J. Juárez-Jiménez, F. J. Luque, W. F. Degrado, R. A. Lamb, L. H. Pinto and S. Vázquez, Easily Accessible Polycyclic Amines That Inhibit the Wild-Type and Amantadine-Resistant Mutants of the M2 Channel of Influenza A Virus, J. Med. Chem., 2014, 57(13), 5738–5747, DOI:10.1021/jm5005804.
- J. Wang, C. Ma, G. Fiorin, V. Carnevale, T. Wang, F. Hu, R. A. Lamb, L. H. Pinto, M. Hong, M. L. Klein and W. F. Degrado, Molecular Dynamics Simulation Directed Rational Design of Inhibitors Targeting Drug-Resistant Mutants of Influenza A Virus M2, J. Am. Chem. Soc., 2011, 133(32), 12834–12841, DOI:10.1021/ja204969m.
- P. Camps, M. D. Duque, S. Vázquez, L. Naesens, E. De Clercq, F. X. Sureda, M. López-Querol, A. Camins, M. Pallàs, S. R. Prathalingam, J. M. Kelly, V. Romero, D. Ivorra and D. Cortés, Synthesis and Pharmacological Evaluation of Several Ring-Contracted Amantadine Analogs, Bioorg. Med. Chem., 2008, 16(23), 9925–9936, DOI:10.1016/j.bmc.2008.10.028.
- M. D. Duque, P. Camps, L. Profire, S. Montaner, S. Vázquez, F. X. Sureda, J. Mallol, M. López-Querol, L. Naesens, E. De Clercq, S. Radhika Prathalingam and J. M. Kelly, Synthesis and Pharmacological Evaluation of (2-Oxaadamant-1-Yl)Amines, Bioorg. Med. Chem., 2009, 17(8), 3198–3206, DOI:10.1016/j.bmc.2009.02.007.
- M. Barniol-Xicota, S. Gazzarrini, E. Torres, Y. Hu, J. Wang, L. Naesens, A. Moroni and S. Vázquez, Slow but Steady Wins the Race: Dissimilarities among New Dual Inhibitors of the Wild-Type and the V27A Mutant M2 Channels of Influenza A Virus, J. Med. Chem., 2017, 60(9), 3727–3738, DOI:10.1021/acs.jmedchem.6b01758.
- E. Torres, R. Fernández, S. Miquet, M. Font-Bardia, E. Vanderlinden, L. Naesens and S. Vázquez, Synthesis and Anti-Influenza a Virus Activity of 2,2-Dialkylamantadines and Related Compounds, ACS Med. Chem. Lett., 2012, 3(12), 1065–1069, DOI:10.1021/ml300279b.
- E. Valverde, F. X. Sureda and S. Vázquez, Novel Benzopolycyclic Amines with NMDA Receptor Antagonist Activity, Bioorg. Med. Chem., 2014, 22(9), 2678–2683, DOI:10.1016/j.bmc.2014.03.025.
- W. Hu, S. Zeng, C. Li, Y. Jie, Z. Li and L. Chen, Identification of Hits as Matrix-2 Protein Inhibitors through the Focused Screening of a Small Primary Amine Library, J. Med. Chem., 2010, 53(9), 3831–3834, DOI:10.1021/jm901664a.
- M. Rey-Carrizo, S. Gazzarrini, S. Llabrés, M. Frigolé-Vivas, J. Juárez-Jiménez, M. Font-Bardia, L. Naesens, A. Moroni, F. J. Luque and S. Vázquez, New Polycyclic Dual Inhibitors of the Wild Type and the V27A Mutant M2 Channel of the Influenza A Virus with Unexpected Binding Mode, Eur. J. Med. Chem., 2015, 96, 318–329, DOI:10.1016/j.ejmech.2015.04.030.
- C. Tzitzoglaki, A. Wright, K. Freudenberger, A. Hoffmann, I. Tietjen, I. Stylianakis, F. Kolarov, D. Fedida, M. Schmidtke, G. Gauglitz, T. A. Cross and A. Kolocouris, Binding and Proton Blockage by Amantadine Variants of the Influenza M2WT and M2S31N Explained, J. Med. Chem., 2017, 60(5), 1716–1733, DOI:10.1021/acs.jmedchem.6b01115.
- F. Spyrakis, M. H. Ahmed, A. S. Bayden, P. Cozzini, A. Mozzarelli and G. E. Kellogg, The Roles of Water in the Protein Matrix: A Largely Untapped Resource for Drug Discovery, J. Med. Chem., 2017, 60(16), 6781–6827, DOI:10.1021/acs.jmedchem.7b00057.
- P. A. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan, W. Wang, O. Donini, P. Cieplak, J. Srinivasan, D. A. Case and T. E. Cheatham, Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models, Acc. Chem. Res., 2000, 33(12), 889–897, DOI:10.1021/ar000033j.
- E. Wang, H. Sun, J. Wang, Z. Wang, H. Liu, J. Z. H. Zhang and T. Hou, End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design, Chem. Rev., 2019, 9478–9508, DOI:10.1021/acs.chemrev.9b00055.
- C. H. Bennett, Efficient Estimation of Free Energy Differences from Monte Carlo Data, J. Comput. Phys., 1976, 22(2), 245–268, DOI:10.1016/0021-9991(76)90078-4.
- M. R. Shirts and J. D. Chodera, Statistically Optimal Analysis of Samples from Multiple Equilibrium States, J. Chem. Phys., 2008, 129(12), 124105, DOI:10.1063/1.2978177.
- N. Homeyer, H. Ioannidis, F. Kolarov, G. G. Gauglitz, C. Zikos, A. Kolocouris and H. Gohlke, Interpreting Thermodynamic Profiles of Aminoadamantane Compounds Inhibiting the M2 Proton Channel of Influenza A by Free Energy Calculations, J. Chem. Inf. Model., 2016, 56(1), 110–126, DOI:10.1021/acs.jcim.5b00467.
- P. Gkeka, A.-S. Eleftheratos, A. Kolocouris, Z. Cournia, S. Eleftheratos, A. Kolocouris and Z. Cournia, Free Energy Calculations Reveal the Origin of Binding Preference for Aminoadamantane Blockers of Influenza A/M2TM Pore, J. Chem. Theory Comput., 2013, 9(2), 1272–1281, DOI:10.1021/ct300899n.
- H. Ioannidis, A. Drakopoulos, C. Tzitzoglaki, N. Homeyer, F. Kolarov, P. Gkeka, K. Freudenberger, C. Liolios, G. G. Gauglitz, Z. Cournia, H. Gohlke and A. Kolocouris, Alchemical Free Energy Calculations and Isothermal Titration Calorimetry Measurements of Aminoadamantanes Bound to the Closed State of Influenza A/M2TM, J. Chem. Inf. Model., 2016, 56(5), 862–876, DOI:10.1021/acs.jcim.6b00079.
- C. Tian, K. Kasavajhala, K. A. A. Belfon, L. Raguette, H. Huang, A. N. Migues, J. Bickel, Y. Wang, J. Pincay, Q. Wu and C. Simmerling, Ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution, J. Chem. Theory Comput., 2020, 16(1), 528–552, DOI:10.1021/acs.jctc.9b00591.
- M. R. Shirts and J. D. Chodera, Statistically Optimal Analysis of Samples from Multiple Equilibrium States, J. Chem. Phys., 2008, 129(12), 124105, DOI:10.1063/1.2978177.
- L. Wanka, K. Iqbal and P. R. Schreiner, The Lipophilic Bullet Hits the Targets: Medicinal Chemistry of Adamantane Derivatives, Chem. Rev., 2013, 113(5), 3516–3604, DOI:10.1021/cr100264t.
- G. Lamoureux and G. Artavia, Use of the Adamantane Structure in Medicinal Chemistry, Curr. Med. Chem., 2010, 17(26), 2967–2978, DOI:10.2174/092986710792065027.
- A. Štimac, M. Šekutor, K. Mlinarić-Majerski, L. Frkanec and R. Frkanec, Adamantane in Drug Delivery Systems and Surface Recognition, Molecules, 2017, 22(2), 297, DOI:10.3390/molecules22020297.
- J. Liu, D. Obando, V. Liao, T. Lifa and R. Codd, The Many Faces of the Adamantyl Group in Drug Design, Eur. J. Med. Chem., 2011, 46(6), 1949–1963, DOI:10.1016/j.ejmech.2011.01.047.
- W. L. Davies, R. R. Grunert, R. F. Haff, J. W. Mcgahen, E. M. Neumayer, M. Paulshock, J. C. Watts, T. R. Wood, E. C. Hermann and C. E. Hoffmann, Antiviral Activity of 1-Adamantanamine (Amantadine), Science, 1964, 144(3620), 862–863, DOI:10.1126/science.144.3620.862.
- J. G. Whitney, W. A. Gregory, J. C. Kauer, J. R. Roland, J. A. Snyder, R. E. Benson and E. C. Hermann, Antiviral Agents. I. Bicyclo[2.2.2]Octan- and -Oct-2-Enamines, J. Med. Chem., 1970, 13(2), 254–260, DOI:10.1021/jm00296a021.
- P. E. Aldrich, E. C. Hermann, W. E. Meier, M. Paulshock, W. W. Prichard, J. A. Snyder and J. C. Watts, Antiviral Agents. 2. Structure-Activity Relationships of Compounds Related to 1-Adamantanamine, J. Med. Chem., 1971, 14(6), 535–543, DOI:10.1021/jm00288a019.
- K. Lundahl, J. Schut, J. L. M. A. Schlatmann, G. B. Paerels, A. Peters and N. V. Philips-Duphar, Synthesis and Antiviral Activities of Adamantane Spiro Compounds. 1. Adamantane and Analogous Spiro-3′-Pyrrolidines, J. Med. Chem., 1972, 15(2), 129–132, DOI:10.1021/jm00272a003.
- R. Van Hes, A. Smit, T. Kralt and A. Peters, Synthesis and Antiviral Activities of Adamantane Spiro Compounds. 21, J. Med. Chem., 1972, 15(2), 132–136, DOI:10.1021/jm00272a004.
- A. Kolocouris, K. Dimas, C. Pannecouque, M. Witvrouw, G. B. Foscolos, G. Stamatiou, G. Fytas, G. Zoidis, N. Kolocouris, G. Andrei, R. Snoeck and E. De Clercq, New 2-(1-Adamantylcarbonyl)Pyridine and 1-Acetyladamantane Thiosemicarbazones-Thiocarbonohydrazones: Cell Growth Inhibitory, Antiviral and Antimicrobial Activity Evaluation, Bioorg. Med. Chem. Lett., 2002, 12(5), 723–727, DOI:10.1016/S0960-894X(01)00838-1.
- N. Kolocouris, A. Kolocouris, G. B. Foscolos, G. Fytas, J. Neyts, E. Padalko, J. Balzarini, R. Snoeck, G. Andrei and E. De Clercq, Synthesis and Antiviral Activity Evaluation of Some New Aminoadamantane Derivatives. 2, J. Med. Chem., 1996, 39(17), 3307–3318, DOI:10.1021/jm950891z.
- G. Zoidis, C. Fytas, I. Papanastasiou, G. B. Foscolos, G. Fytas, E. Padalko, E. De Clercq, L. Naesens, J. Neyts and N. Kolocouris, Heterocyclic Rimantadine Analogues with Antiviral Activity, Bioorg. Med. Chem., 2006, 14(10), 3341–3348, DOI:10.1016/j.bmc.2005.12.056.
- D. Tataridis, G. Fytas, A. Kolocouris, C. Fytas, N. Kolocouris, G. B. Foscolos, E. Padalko, J. Neyts and E. De Clercq, Influence of an Additional 2-Amino Substituent of the 1-Aminoethyl Pharmacophore Group on the Potency of Rimantadine against Influenza Virus A, Bioorg. Med. Chem. Lett., 2007, 17(3), 692–696, DOI:10.1016/j.bmcl.2006.10.092.
- D. Setaki, D. Tataridis, G. Stamatiou, A. Kolocouris, G. B. Foscolos, G. Fytas, N. Kolocouris, E. Padalko, J. Neyts and E. D. Clercq, Synthesis, Conformational Characteristics and Anti-Influenza Virus A Activity of Some 2-Adamantylsubstituted Azacycles, Bioorg. Chem., 2006, 34(5), 248–273, DOI:10.1016/j.bioorg.2006.05.004.
- G. Stamatiou, G. B. Foscolos, G. Fytas, A. Kolocouris, N. Kolocouris, C. Pannecouque, M. Witvrouw, E. Padalko, J. Neyts and E. D. Clercq, Heterocyclic Rimantadine Analogues with Antiviral Activity, Bioorg. Med. Chem., 2003, 11(24), 5485–5492, DOI:10.1016/j.bmc.2003.09.024.
- G. Zoidis, N. Kolocouris, G. B. Foscolos, A. Kolocouris, G. Fytas, P. Karayannis, E. Padalko, J. Neyts and E. De Clercq, Are the 2-Isomers of the Drug Rimantadine Active Anti-Influenza A Agents?, Antiviral Chem. Chemother., 2003, 14(3), 153–164 CrossRef CAS.
- N. Kolocouris, G. B. Foscolos, A. Kolocouris, P. Marakos, N. Pouli, G. Fytas, S. Ikeda and E. De Clercq, Synthesis and Antiviral Activity Evaluation of Some Aminoadamantane Derivatives, J. Med. Chem., 1994, 37(18), 2896–2902, DOI:10.1021/jm00044a010.
- N. Kolocouris, G. Zoidis, G. B. Foscolos, G. Fytas, S. R. Prathalingham, J. M. Kelly, L. Naesens and E. De Clercq, Design and Synthesis of Bioactive Adamantane Spiro Heterocycles, Bioorg. Med. Chem. Lett., 2007, 17(15), 4358–4362, DOI:10.1016/j.bmcl.2007.04.108.
- A. Kolocouris, D. Tataridis, G. Fytas, T. Mavromoustakos, G. B. Foscolos, N. Kolocouris and E. De Clercq, Synthesis of 2-(2-Adamantyl)Piperidines and Structure Anti-Influenza Virus A Activity Relationship Study Using a Combination of NMR Spectroscopy and Molecular Modeling, Bioorg. Med. Chem. Lett., 1999, 9(24), 3465–3470 CrossRef CAS PubMed.
- G. Stamatiou, A. Kolocouris, N. Kolocouris, G. Fytas, G. B. Foscolos, J. Neyts and E. De Clercq, Novel 3-(2-Adamantyl)Pyrrolidines with Potent Activity against Influenza A Virus - Identification of Aminoadamantane Derivatives Bearing Two Pharmacophoric Amine Groups, Bioorg. Med. Chem. Lett., 2001, 11(16), 2137–2142, DOI:10.1016/S0960-894X(01)00388-2.
- C. Fytas, A. Kolocouris, G. Fytas, G. Zoidis, C. Valmas and C. F. Basler, Influence of an Additional Amino Group on the Potency of Aminoadamantanes against Influenza Virus A. II - Synthesis of Spiropiperazines and in Vitro Activity against Influenza A H3N2 Virus, Bioorg. Chem., 2010, 38(6), 247–251, DOI:10.1016/j.bioorg.2010.09.001.
- I. Tietjen, D. C. Kwan, A. Petrich, R. Zell, I. T. Antoniadou, A. Gavriilidou, C. Tzitzoglaki, M. Rallis, D. Fedida, F. X. Sureda, C. Mestdagh, L. Naesens, S. Chiantia, F. B. Johnson and A. Kolocouris, Antiviral Mechanisms and Preclinical Evaluation of Amantadine Analogs That Continue to Inhibit Influenza A Viruses with M2S31N-Based Drug Resistance, Antiviral Res., 2025, 236, 106104, DOI:10.1016/j.antiviral.2025.106104.
- J. Wang, S. D. Cady, V. Balannik, L. H. Pinto, W. F. DeGrado and M. Hong, Discovery of Spiro-Piperidine Inhibitors and Their Modulation of the Dynamics of the M2 Proton Channel from Influenza A Virus, J. Am. Chem. Soc., 2009, 131(23), 8066–8076, DOI:10.1021/ja900063s.
- M. D. Duque, C. Ma, E. Torres, J. Wang, L. Naesens, J. Juárez-Jiménez, P. Camps, F. J. Luque, W. F. DeGrado, R. A. Lamb, L. H. Pinto and S. Vázquez, Exploring the Size Limit of Templates for Inhibitors of the M2 Ion Channel of Influenza A Virus, J. Med. Chem., 2011, 54(8), 2646–2657, DOI:10.1021/jm101334y.
- E. Torres, R. Leiva, S. Gazzarrini, M. Rey-Carrizo, M. Frigolé-Vivas, A. Moroni, L. Naesens and S. Vázquez, Azapropellanes with Anti-Influenza a Virus Activity, ACS Med. Chem. Lett., 2014, 5(7), 831–836, DOI:10.1021/ml500108s.
- M. Rey-Carrizo, S. Gazzarrini, S. Llabrés, M. Frigolé-Vivas, J. Juárez-Jiménez, M. Font-Bardia, L. Naesens, A. Moroni, F. J. Luque and S. Vázquez, New Polycyclic Dual Inhibitors of the Wild Type and the V27A Mutant M2 Channel of the Influenza A Virus with Unexpected Binding Mode, Eur. J. Med. Chem., 2015, 96, 318–329, DOI:10.1016/j.ejmech.2015.04.030.
- M. Rey-carrizo, E. Torres, C. Ma, M. Barniol-xicota, J. Wang, Y. Wu, L. Naesens, W. F. Degrado, R. A. Lamb, L. H. Pinto, S. Vázquez and S. Va, 3-Azatetracyclo[5.2.1.15,8.01,5]Undecane Derivatives: From Wild-Type Inhibitors of the M2 Ion Channel of Influenza A Virus to Derivatives with Potent Activity against the V27A Mutant, J. Med. Chem., 2013, 56, 9265–9274, DOI:10.1021/jm401340p.
- X. Zhao, Y. Jie, M. R. Rosenberg, J. Wan, S. Zeng, W. Cui, Y. Xiao, Z. Li, Z. Tu, M. G. Casarotto and W. Hu, Design and Synthesis of Pinanamine Derivatives as Anti-Influenza A M2 Ion Channel Inhibitors, Antiviral Res., 2012, 96(2), 91–99, DOI:10.1016/j.antiviral.2012.09.001.
- X. Zhao, Z. W. Zhang, W. Cui, S. Chen, Y. Zhou, J. Dong, Y. Jie, J. Wan, Y. Xu and W. Hu, Identification of Camphor Derivatives as Novel M2 Ion Channel Inhibitors of Influenza A Virus, MedChemComm, 2015, 6(4), 727–731, 10.1039/c4md00515e.
- J. Wang, C. Ma, V. Balannik, L. H. Pinto, R. A. Lamb and W. F. Degrado, Exploring the Requirements for the Hydrophobic Scaffold and Polar Amine in Inhibitors of M2 from Influenza A Virus, ACS Med. Chem. Lett., 2011, 2(4), 307–312, DOI:10.1021/ml100297w.
- H. Torres-Gómez, K. Lehmkuhl, B. Frehland, C. Daniliuc, D. Schepmann, C. Ehrhardt and B. Wünsch, Stereoselective Synthesis and Pharmacological Evaluation of [4.3.3]Propellan-8-Amines as Analogs of Adamantanamines, Bioorg. Med. Chem., 2015, 23(15), 4277–4285, DOI:10.1016/j.bmc.2015.06.030.
- W. K. Weigel, H. T. Dang, A. Feceu and D. B. C. Martin, Direct Radical Functionalization Methods to Access Substituted Adamantanes and Diamondoids, Org. Biomol. Chem., 2022, 20(1), 10–36, 10.1039/D1OB01916C.
- R. Hrdina, Directed C–H Functionalization of the Adamantane Framework, Synthesis, 2019, 51(03), 629–642, DOI:10.1055/s-0037-1610321.
- P. Camps, M. D. Duque, S. Vázquez, L. Naesens, E. De Clercq, F. X. Sureda, M. López-Querol, A. Camins, M. Pallàs, S. R. Prathalingam, J. M. Kelly, V. Romero, D. Ivorra and D. Cortés, Synthesis and Pharmacological Evaluation of Several Ring-Contracted Amantadine Analogs, Bioorg. Med. Chem., 2008, 16(23), 9925–9936, DOI:10.1016/j.bmc.2008.10.028.
- G. Zoidis, N. Kolocouris, G. B. Foscolos, A. Kolocouris, G. Fytas, P. Karayannis, E. Padalko, J. Neyts and E. De Clercq, Are the 2-Isomers of the Drug Rimantadine Active Anti-Influenza A Agents?, Antiviral Chem. Chemother., 2003, 14(3), 153–164 CrossRef CAS.
- G. Fytas, N. Kolocouris, G. Foscolos and A. Vamvakidès, Synthesis and Pharmacological Study of Some Adamantylcyclopentanamines, Eur. J. Med. Chem., 1991, 26(5), 563–566, DOI:10.1016/0223-5234(91)90154-F.
- C. Tzitzoglaki, A. Drakopoulos, A. Konstantinidi, I. Stylianakis, M. Stampolaki and A. Kolocouris, Approaches to Primary Tert-Alkyl Amines as Building Blocks, Tetrahedron, 2019, 75(34), 130408, DOI:10.1016/j.tet.2019.06.016.
- G. Stamatiou, G. B. Foscolos, G. Fytas, A. Kolocouris, N. Kolocouris, C. Pannecouque, M. Witvrouw, E. Padalko, J. Neyts and E. D. Clercq, Heterocyclic Rimantadine Analogues with Antiviral Activity, Bioorg. Med. Chem., 2003, 11(24), 5485–5492, DOI:10.1016/j.bmc.2003.09.024.
- A. Kolocouris, D. Tataridis, G. Fytas, T. Mavromoustakos, G. B. Foscolos, N. Kolocouris and E. De Clercq, Synthesis of 2-(2-Adamantyl)Piperidines and Structure Anti-Influenza Virus A Activity Relationship Study Using a Combination of NMR Spectroscopy and Molecular Modeling, Bioorg. Med. Chem. Lett., 1999, 9(24), 3465–3470 CrossRef CAS PubMed.
- N. Kolocouris, G. B. Foscolos, A. Kolocouris, P. Marakos, N. Pouli, G. Fytas, S. Ikeda and E. De Clercq, Synthesis and Antiviral Activity Evaluation of Some Aminoadamantane Derivatives, J. Med. Chem., 1994, 37(18), 2896–2902, DOI:10.1021/jm00044a010.
- A. Kolocouris, Synthesis of Heterocyclic Aminoadamantanes of Pharmacological Interest, Ph.D. Dissertation, National and Kapodistrian University of Athens, Athens, Greece, 1995 Search PubMed.
- M. Todd and R. Hrdina, Synthesis of 1,2-Disubstituted Adamantane Derivatives by Construction of the Adamantane Framework, Molecules, 2023, 28(22), 7636, DOI:10.3390/molecules28227636.
- V. Pardali, E. Giannakopoulou, A. Konstantinidi, A. Kolocouris and G. Zoidis, 1,2-Annulated Adamantane Heterocyclic Derivatives as Anti-Influenza α Virus Agents, Croat. Chem. Acta, 2019, 92(2), 211–228, DOI:10.5562/cca3540.
- C. Tzitzoglaki, A. Wright, K. Freudenberger, A. Hoffmann, I. Tietjen, I. Stylianakis, F. Kolarov, D. Fedida, M. Schmidtke, G. Gauglitz, T. A. Cross and A. Kolocouris, Binding and Proton Blockage by Amantadine Variants of the Influenza M2 WT and M2 S31N Explained, J. Med. Chem., 2017, 60(5), 1716–1733, DOI:10.1021/acs.jmedchem.6b01115.
- F. Li, C. Ma, W. F. Degrado and J. Wang, Discovery of Highly Potent Inhibitors Targeting the Predominant Drug-Resistant S31N Mutant of the Influenza A Virus M2 Proton Channel, J. Med. Chem., 2016, 59(3), 1207–1216, DOI:10.1021/acs.jmedchem.5b01910.
- F. Li, C. Ma, Y. Hu, Y. Wang and J. Wang, Discovery of Potent Antivirals against Amantadine-Resistant Influenza A Viruses by Targeting the M2-S31N Proton Channel, ACS Infect. Dis., 2016, 2(10), 726–733, DOI:10.1021/acsinfecdis.6b00130.
- J. J. Wang, Y. Wu, C. Ma, G. Fiorin, L. H. Pinto, R. A. Lamb, M. L. Klein and W. F. Degrado, Structure and Inhibition of the Drug-Resistant S31N Mutant of the M2 Ion Channel of Influenza A Virus, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(4), 1315–1320, DOI:10.1073/pnas.1216526110.
- C. Tzitzoglaki, A. Hoffmann, A. L. Turcu, P. Schmerer, C. Ma, G. Laros, C. Liolios, B. José, J. Wang, S. Vázquez, M. Schmidtke and A. Kolocouris, Amantadine Variant – Aryl Conjugates That Inhibit Multiple M2 Mutant – Amantadine Resistant Influenza a Viruses, Eur. J. Med. Chem. Rep., 2022, 6, 100083, DOI:10.1016/j.ejmcr.2022.100083.
- Y. Wu, B. Canturk, H. Jo, C. Ma, E. Gianti, M. L. Klein, L. H. Pinto, R. A. Lamb, G. Fiorin, J. Wang and W. F. Degrado, Flipping in the Pore: Discovery of Dual Inhibitors That Bind in Different Orientations to the Wild-Type versus the Amantadine-Resistant S31n Mutant of the Influenza a Virus M2 Proton Channel, J. Am. Chem. Soc., 2014, 136(52), 17987–17995, DOI:10.1021/ja508461m.
- G. Vauquelin, Effects of Target Binding Kinetics on in Vivo Drug Efficacy: Koff, Kon and Rebinding, Br. J. Pharmacol., 2016, 173, 2319–2934, DOI:10.1111/bph.13504.
- X. Zhao, C. Li, S. Zeng and W. Hu, Discovery of Highly Potent Agents against Influenza A Virus, Eur. J. Med. Chem., 2011, 46(1), 52–57, DOI:10.1016/j.ejmech.2010.10.010.
- F. Li, Y. Hu, Y. Wang, C. Ma and J. Wang, Expeditious Lead Optimization of Isoxazole-Containing Influenza A Virus M2-S31N Inhibitors Using the Suzuki − Miyaura Cross-Coupling Reaction, J. Med. Chem., 2017, 60, 1580–1590, DOI:10.1021/acs.jmedchem.6b01852.
- C. Tzitzoglaki, A. Hoffmann, A. L. Turcu, P. Schmerer, C. Ma, G. Laros, C. Liolios, B. José, J. Wang, S. Vázquez, M. Schmidtke and A. Kolocouris, Amantadine Variant – Aryl Conjugates That Inhibit Multiple M2 Mutant – Amantadine Resistant Influenza a Viruses, Eur. J. Med. Chem. Rep., 2022, 6, 100083, DOI:10.1016/j.ejmcr.2022.100083.
- R. Musharrafieh, C. Ma and J. Wang, Discovery of M2 Channel Blockers Targeting the Drug-Resistant Double Mutants M2-S31N/L26I and M2-S31N/V27A from the Influenza A Viruses, Eur. J. Pharm. Sci., 2020, 141, 105124, DOI:10.1016/j.ejps.2019.105124.
- R. Musharrafieh, P. I. Lagarias, C. Ma, G. S. Tan, A. Kolocouris and J. Wang, The L46P Mutant Confers a Novel Allosteric Mechanism of Resistance Toward the Influenza A Virus M2 S31N Proton Channel Blockers, Mol. Pharmacol., 2019, 96(2), 148–157, DOI:10.1124/mol.119.116640.
- F. Li, C. Ma, W. F. Degrado and J. Wang, Discovery of Highly Potent Inhibitors Targeting the Predominant Drug-Resistant S31N Mutant of the Influenza A Virus M2 Proton Channel, J. Med. Chem., 2016, 59(3), 1207–1216, DOI:10.1021/acs.jmedchem.5b01910.
- F. Li, C. Ma, Y. Hu, Y. Wang and J. Wang, Discovery of Potent Antivirals against Amantadine-Resistant Influenza A Viruses by Targeting the M2-S31N Proton Channel, ACS Infect. Dis., 2016, 2(10), 726–733, DOI:10.1021/acsinfecdis.6b00130.
- F. Li, Y. Hu, Y. Wang, C. Ma and J. Wang, Expeditious Lead Optimization of Isoxazole-Containing Influenza A Virus M2-S31N Inhibitors Using the Suzuki − Miyaura Cross-Coupling Reaction, J. Med. Chem., 2017, 60, 1580–1590, DOI:10.1021/acs.jmedchem.6b01852.
| This journal is © The Royal Society of Chemistry 2026 |