
Discovery and optimization of pyrrolopyrimidines as highly potent, selective and brain-penetrant LRRK2 inhibitors
Jeffrey M.
Axten
 *a,
Xiao
Ding
b,
Luigi Piero
Stasi
b,
Baowei
Zhao
b,
Yingxia
Sang
b,
Ming-Hsun
Ho
a,
Lizhen
Wang
b,
Minhua
Zhang
b,
Xianjun
Guo
b,
Chengfang
Tan
b,
Xu
Feng
b,
Colin
Edge
c,
Klara
Valko
c,
Yi
Li
b,
Kelly
Dong
b,
Xiaoming
Guan
b,
Nico
Zinn
d,
F.
David Tattersall
a,
Feng
Ren
b,
Dai-Shi
Su
a and
Alastair D.
Reith
c
*a,
Xiao
Ding
b,
Luigi Piero
Stasi
b,
Baowei
Zhao
b,
Yingxia
Sang
b,
Ming-Hsun
Ho
a,
Lizhen
Wang
b,
Minhua
Zhang
b,
Xianjun
Guo
b,
Chengfang
Tan
b,
Xu
Feng
b,
Colin
Edge
c,
Klara
Valko
c,
Yi
Li
b,
Kelly
Dong
b,
Xiaoming
Guan
b,
Nico
Zinn
d,
F.
David Tattersall
a,
Feng
Ren
b,
Dai-Shi
Su
a and
Alastair D.
Reith
c
aGSK Research and Development, 1250 S. Collegeville Road, Collegeville, Pennsylvania 19426, USA. E-mail: jeffrey.m.axten@gsk.com
bGSK Research and Development, 898 Halei Road, Zhangjiang Hi-Tech Park, Pudong, Shanghai 201203, PR China
cGSK Research and Development Medicines Research Centre, Gunnels Wood Road, Stevenage, SG1 2NY, UK
dOmics Technologies, Cellzome a GSK Company, Meyerhofstrasse 1, D-69117 Heidelberg, Germany
First published on 17th November 2025
Abstract
Leucine-rich repeat kinase 2 (LRRK2) is a promising therapeutic target for Parkinson's disease. We report herein the discovery of pyrrolopyrimidine analogs as potent and selective LRRK2 kinase inhibitors. Elucidation of the structure–activity relationship (SAR) of the kinase-inhibitor-focused screening lead compound 1 led to the development of compound 39 (GSK3357679) that shows excellent cellular potency, oral bioavailability, brain-penetration, and excellent PK/PD correlation in animal studies. The SAR optimization of the biological and pharmacokinetic profiles of the compounds are described. The pharmacodynamic characteristics for extended oral dosing studies in rodents are also presented.
Introduction
Parkinson's disease is a progressive neurodegenerative disorder characterized by the loss of dopaminergic neurons in the substantia nigra region of the midbrain, resulting in cardinal symptoms of bradykinesia, resting tremor and rigidity. An array of non-motor symptoms, including fatigue, pain, psychosis and dementia, are also characteristic features in the course of disease progression.1 Current primary treatment strategies for Parkinson's disease are focused on compensating for lowered dopamine concentrations in the brain by replacement, blocking reuptake or degradation, or direct agonism of dopamine receptors. Unfortunately, the efficacy of such symptomatic drugs decreases over time and are associated with significant side effects, such as dyskinesia. Moreover, their use does not alter the underlying neurodegenerative disease process.2A variety of approaches are being pursued in the search for novel treatments that may slow or halt disease progression in Parkinson's patients.3 Prominent amongst these is the modulation of LRRK2 kinase activity based on clinical genetic studies and strong human target validation of LRRK2 in Parkinson's disease.4–6 Mendelian genetic studies have shown that coding mutations in LRRK2 that elevate the intrinsic protein kinase activity of LRRK2 are considered pathogenic for Parkinson's disease.7 The most common pathogenic LRRK2 G2019S mutation is frequently found in some ethnic groups (e.g. Ashkenazi Jews and Berber Arabs) and in 1–3% of sporadic Parkinson's disease patients in Caucasian populations. Genome-wide association studies (GWAS) have also identified LRRK2 SNPs associated with sporadic Parkinson's disease, which has further stimulated interest in the development of small molecule inhibitors of LRRK2 kinase activity as potential disease modifying drugs for Parkinson's disease.8 Representative structurally diverse inhibitors are shown in Chart 1. Clinical evaluation of LRRK2 modulators as potential disease modifying drugs for Parkinson's disease is underway with both small molecule kinase inhibitors and LRRK2 ASOs in clinical trials with some results from phase I clinical trials reported.9,10
 | ||
| Chart 1 Representative structurally diverse brain-penetrant selective LRRK2 inhibitors. | ||
Results and discussion
Initial hits were identified in a screen of GSK's kinase inhibitor-focused set of compounds for lead discovery using a homogenous time-resolved fluorescence (HTRF) assay that measured inhibition of phosphorylation of the peptide substrate LRRKtide by baculoviral recombinant 6His-Tev-LRRK2 (1326–2527).11,12 Compound 1, a pyrrolopyrimidine analog, is presented as a good starting point for our chemistry effort based on its reasonable potency in SHSY5Y cells measuring inhibition of pS935 (IC50 = 159 nM), kinase selectivity, and physicochemical properties (Table 1). Compound 1 also possesses good in vitro clearance in liver microsomes. To facilitate chemistry efforts, we utilized a LRRK2 homology model and predicted the docking pose of compound 1 in the ATP binding pocket (Fig. 1).13 The model suggested that the pyrrolopyrimidine scaffold formed multiple hydrogen bonding interactions with the backbone of Glu1948 and Ala1950 in the hinge domain. The morpholino amide side chain is exposed to the solvent front. The flexible sidechain on Arg1957 was close to the phenyl ring, suggesting that we could improve the potency by introducing a polar group or hydrogen bond acceptor on the ligand. Replacement of the phenyl ring with other heterocycles led to the discovery of pyrazole regioisomers 2 and 3 (Table 1). This modification improved not only the physicochemical properties (chromlog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D and PFI), but also the cell potency. The analysis of the compound 2 docking pose suggested that the tetrahydropyran ring was twisted in order to fit into the pocket and we hypothesized that the potency was hampered by higher strain energy. Compound 3 was designed to favorably orient the substituent and lower the strain energy. Compound 3 showed a 20-fold improvement in potency compared with compound 1 with an IC50 value of 8 nM, possibly due to additional hydrogen bond interactions. Although 3 had lower solubility, the intrinsic clearance in human liver microsomes was decreased. Our effort was therefore shifted to focus on the optimization of compound 3, which will be described in more detail below.
D and PFI), but also the cell potency. The analysis of the compound 2 docking pose suggested that the tetrahydropyran ring was twisted in order to fit into the pocket and we hypothesized that the potency was hampered by higher strain energy. Compound 3 was designed to favorably orient the substituent and lower the strain energy. Compound 3 showed a 20-fold improvement in potency compared with compound 1 with an IC50 value of 8 nM, possibly due to additional hydrogen bond interactions. Although 3 had lower solubility, the intrinsic clearance in human liver microsomes was decreased. Our effort was therefore shifted to focus on the optimization of compound 3, which will be described in more detail below.
| Cmpd | Structure | SHSY5Y pS935 pIC50/IC50a (nM) | Kinase selectivityb | ChromlogD7.4c |
PFId | Pgp/BCRPe ERf | h/r LM Clig (mL min−1 g−1) | FaSSIF solubilityh (μg mL−1) |
|---|---|---|---|---|---|---|---|---|
|
a All the pIC50 values represent the average of at least two determinations.
b Standard radioactivity-based enzymatic assays against a panel of 140 kinases at 1 μM, quoted as numbers of kinase displaying >50% inhibition.
c Measured chromlogD at pH = 7.4.
d Property forecast index, summation of chromlogD7.4 and the number of aromatic rings.
e MDCKII-MDR1 transduced with the BacMam2-BCRP cell line.
f Efflux ratio, A → B (apical to basolateral) with GF120918/A → B without GF120918.
g Human and rat liver microsomal clearance.
h FaSSIF Solubility was measured after 4 hours of incubation.
i Not determined.
|
||||||||
| 1 |

|
6.8/158.5 | 12/140 | 4.5 | 7.5 | 3.1 | 1.1/1.3 | 127.3 |
| 2 |
|
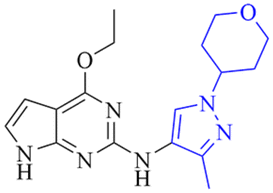
6.7/199.5 | NDi | 3.7 | 6.7 | 2.5 | 1.4/10.8 | 18.6 |
| 3 |

|
8.1/7.9 | 13/140 | 3.5 | 6.5 | 3.4 | <0.78/5.5 | 7.9 |
 | ||
| Fig. 1 Predicted docking pose of 1 (cyan), 2 (magenta) and 3 (orange) in the LRRK2 homology model (gray). Intermolecular hydrogen-bond interactions are shown as yellow dashed lines. | ||
The initial SAR was focused on the tetrahydropyran ring. Substitutions of the tetrahydropyran (4 and 5) and pyran isomers (6 and 7) were well tolerated in terms of potency with modest improvement of solubility (Table 2). Ring contraction to tetrahydrofurans (8 and 9) and oxetane 10 afforded similar potency and good solubility. A heteroatom switch to nitrogen provided compounds 11–13 with significant improvement of solubility with a loss of potency. Exploring ring extension or replacement with alkyl chains was not fruitful (compounds 14–20).
|
|
||||||
|---|---|---|---|---|---|---|
| Cmpd | R1 | SHSY5Y pS935 pIC50/IC50b | clogD7.4c (PFI)d | LM Cli h/re (mL min−1 g−1) | Pgp/Bcrpf PRg PPh (nm s−1) h | FaSSIF solubilityi (μg mL−1) |
|
a All the compounds were prepared and tested as single unknown enantiomers unless otherwise noted.
b All the pIC50 values represent the average of at least two determinations.
c Measured chromlogD at pH = 7.4.
d PFI = chromlogDpH7.4 + # Ar.
e Human and rat liver microsomal clearance.
f MDCKII-MDR1 transduced with the BacMam2-BCRP cell line.
g Permeability ratio, A → B (apical to basolateral) with GF120918/A → B without GF120918.
h Passive permeability, A → B with GF120918.
i FaSSIF solubility was measured after 4 h of incubation.
j Not determined.
|
||||||
| 3 |

|
8.1/7.9 | 3.5 (6.5) | <0.78/5.5 | 3.4/214 | 3.9 |
| 4 |

|
8.1/7.9 | 4.2 (7.2) | 0.9/10.8 | 2.1/300 | 19.7 |
| 5 |
|
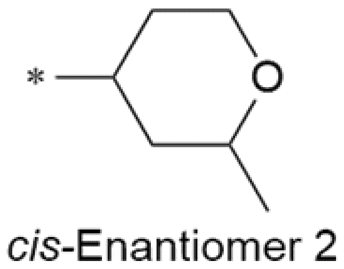
8.0/10 | 4.2 (7.2) | 1.0/9.7 | 1.6/349 | 14 |
| 6 |

|
8.1/7.9 | 3.9 (6.9) | 1.6/33.6 | 2.3/409 | 112 |
| 7 |

|
7.9/12.6 | 4.0 (7.0) | <0.78/8.2 | 2.4/386 | 114.2 |
| 8 |

|
7.6/25.1 | 3.6 (6.6) | <0.78/9.5 | 3.5/204 | 5.1 |
| 9 |

|
8.0/10 | 3.6 (6.6) | <0.78/9.8 | 2.8/288 | 6.2 |
| 10 |

|
7.7/25.1 | 3.0 (6.0) | 1.2/15.1 | 4.0/202 | 158.9 |
| 11 |

|
6.8/158.5 | 2.2 (5.2) | <0.78/3.9 | 6.5/127 | 244.4 |
| 12 |

|
7.2/63.1 | 2.2 (5.2) | <0.78/3.8 | 1.7/183 | 227.5 |
| 13 |

|
7.2/63.1 | NDj | <0.78/2.7 | NDj | 298.1 |
| 14 |

|
8.0/10.0 | 3.5 (6.5) | 1.0/12.2 | 2.6/233 | 70.4 |
| 15 |

|
7.7/20.0 | 3.5 (6.5) | 2.2/30.8 | 3.3/476 | 153.1 |
| 16 |

|
7.3/50.1 | 3.3 (6.3) | <0.78/4.0 | 1.6/410 | 92 |
| 17 |

|
7.8/15.8 | 3.6 (6.6) | 1.2/10.0 | 2.95/365 | 130.4 |
| 18 |

|
7.7/20.0 | 3.0 (5.0) | 1.1/2.7 | 5.84/260 | 155.7 |
| 19 |

|
7.4/39.8 | 3.8 (6.8) | 17.5/28.5 | 4.42/363 | 69.7 |
| 20 |

|
6.8/158.5 | 5.0 (8.0) | <0.78/5.5 | 1.67/322 | 10.8 |

To assess the brain penetration of our analogs, we orally dosed the compounds to rats and measured the brain, plasma, and kidney concentrations of the compounds. In addition, we determined the free fraction of the compounds in tissue vs. plasma (Kp,uu; Table 3). In general, compounds in this series are not Pgp substrates as determined in the measurement of the apical to basolateral ratio and passive permeability in MDCKII-MDR1 cells transduced with BacMam2-BCRP. Compounds 12, 16, and 18 showed low exposure in the brain and blood, with sub-optimal Kp,uu values of <0.2.
| Cmpd | DNAUC0–7hb (ng h mL−1)/(mg kg−1) | K p (Br/Bl)c | Kd/Bld | Fubr/Fuble (%) | Brain Kp,u,uf | ||
|---|---|---|---|---|---|---|---|
| Blood | Brain | Kidneys | |||||
| a 2.0 mg kg−1, single oral gavage administration to male Han-Wistar rats. b DNAUC = dose normalized area under the curve. c K p (Br/Bl) = brain DNAUC0–7h/blood DNAUC0–7h. d Kd/Bl = kidney DNAUC0–7h/blood DNAUC0–7h. e Free unbound fractions measured in rat brain and blood. f K p,uu = Kp × Fubr/Fubl. g Not determined. h 30 mg kg−1, P.O.(0−24 h). | |||||||
| 12 | 57.0 | 33.7 | 1770 | 0.59 | 31.1 | 3.8/21.4 | 0.11 |
| 16 | 86.2 | 21.0 | NDg | 0.24 | 1.0h | 12.8/9.0 | 0.17 |
| 18 | 88.2 | 5.55 | 133 | 0.06 | 1.51 | 7.8/14.5 | 0.03 |
Because of the balanced potency and solubility of compound 13, we further investigated the piperidine ring (Table 4). Introduction of a fluorine atom at the 3-position of the piperidine ring was tolerated (21 and 22) with loss of FaSSIF solubility. One diastereomer (22 (3S, 4S)) showed more potency than the other (21 (3R, 4R)) by ∼0.5log units. The solubility was regained with the substitution of a methoxyethyl side chain (23 and 24), but both compounds showed no improvement in terms of Kp,uu (Table 5). Replacement of the side chain with cyclopropyl alkyls (25–28) caused a loss of potency and solubility. Introduction of oxygen atoms into the alkyl ring delivered a series of oxetane analogs (29–32). All showed good potency, solubility, and excellent in vitro clearance; however, they are Pgp substrates and showed higher efflux ratios. Among them, compound 32 showed the most promising profile, in consideration of target potency and solubility.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Cmpd | R1 | SHSY5Y pS935 pIC50/IC50b | clogD7.4c (PFI)d |
pKae | LM Cli h/rf (mL min−1 g−1) | Pgp/Bcrpg PRh PPi (nm s−1) h | FaSSIF solubilityj (μg mL−1) |
|
a All the compounds were prepared and tested as single unknown enantiomers unless chirality is designated.
b All the pIC50 values represent the average of at least two determinations.
c Measured chromlogD7.4.
d Property forecast index (PFI) = chromlogD7.4 + # Ar rings.
e Using predicted pKa values from ACD software, version 11.0.
f Human and rat liver microsomal clearance.
g MDCKII-MDR1 transduced with the BacMam2-BCRP cell line.
h Permeability ratio, A → B (apical to basolateral) with GF120918/A → B without GF120918.
i Passive Permeability, A → B with GF120918.
j FaSSIF Solubility was measured after 4 h of incubation.
k Not determined.
l MDCKII-MDR1 cell line.
|
|||||||
| 13 |

|
7.2/63.1 | NDk | 8.8 | <0.78/2.7 | NDk | 298.1 |
| 21 |

|
7.5/31.6 | 3.8(6.8) | 7.3 | 1.1/11.8 | 2.1/233 | 18.6 |
| 22 |

|
8.0/10.0 | 3.9(6.9) | 7.3 | 1.0/8.3 | 1.7/210 | 15.7 |
| 23 |

|
7.1/79.4 | 4.0(7.0) | 7.2 | 0.68/2.1 | 1.4/494l | 230.4 |
| 24 |

|
7.8/15.8 | 4.0(7.0) | 7.2 | <0.59/2.6 | 1.7/572l | 334.7 |
| 25 |

|
6.7/199.5 | 5.0(8.0) | 8.0 | 4.1/11.5 | 1.5/402 | 69.2 |
| 26 |

|
7.1/79.4 | 5.0(8.0) | 8.0 | 2.9/8.1 | 1.4/383 | 100.0 |
| 27 |

|
7.6/25.1 | 5.3(8.3) | 7.3 | 2.9/7.1 | 1.5/342 | 16.9 |
| 28 |

|
7.2/63.1 | 5.3(8.3) | 7.3 | 2.2/6.6 | 1.4/345 | 27.8 |
| 29 |

|
7.6/25.1 | 3.4(6.4) | 5.6 | <0.78/<0.78 | 7.5/274 | 477.7 |
| 30 |

|
7.2/63.1 | 3.4(6.4) | 5.6 | <0.78/<0.78 | 5.0/147 | 340.6 |
| 31 |

|
7.2/63.1 | 2.6(5.6) | 5.6 | <0.59/<0.68 | 8.2/99 | 285.3 |
| 32 |

|
8.1/7.9 | 3.0(6.0) | 5.6 | <0.59/<0.68 | 5.7/90 | 574.0 |

| Cmpd | DNAUC0–7hb (ng h mL−1)/(mg kg−1) | K p (Br/Bl)c | Kd/Bld | Fubr/Fuble (%) | Brain Kp,uuf | ||
|---|---|---|---|---|---|---|---|
| Blood | Brain | Kidneys | |||||
| a 2.0 mg kg−1, single oral gavage administration to male Han-Wistar rats. b DNAUC = dose normalized area under the curve. c K p (Br/Bl) = brain DNAUC0–7h/blood DNAUC0–7h. d Kd/Bl = kidney DNAUC0–7h/blood DNAUC0–7h. e Free unbound fractions measured in rat brain and blood. f K p,uu = Kp × Fubr/Fubl. | |||||||
| 23 | 197 | 62.0 | 1032 | 0.32 | 5.2 | 3.4/5.7 | 0.19 |
| 24 | 360 | 58.3 | 1546 | 0.16 | 4.3 | 3.4/3.0 | 0.18 |
Leveraging the favorable profile of oxetane analogs 29–32, we shifted our SAR study to the pyrazole ring (Table 6). Replacement of the methyl group in the pyrazole ring with ethyl and difluoro methyl provided analogs 33–36 with decreased potency and solubility presumably due to increased lipophilicity. Introducing the cyano group in 37–38 maintained the potency compared with the parent compound 32. Chlorine-substituted analogs 39–42 provided a more balanced and the most promising overall profile. Among them, compounds 39 and 40 showed excellent potency (pIC50: 7.8 and 7.7, respectively), FaSSIF solubility (118.5 and 266.5 μg mL−1, respectively), low intrinsic clearance, and were not Pgp substrates.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Cmpd | R1 | R2 | SHSY5Y pS935 pIC50/IC50b | clogD7.4c (PFI)d | pKae | LM Cli h/rf (mL min−1 g−1) | Pgpg PRh PPi (nm s−1) | FaSSIF solubilityj (μg mL−1) |
|
a All the compounds were prepared and tested as single unknown enantiomers unless otherwise noted.
b All the pIC50 values represent the average of at least two determinations.
c Measured chromlogD7.4.
d PFI = chromlogDpH7.4 + # Ar.
e Predicted pKa values from ACD software, version 11.0.
f Human and rat liver microsomal clearance.
g MDCKII-MDR1 cell line.
h Permeability ratio, A → B (apical to basolateral) with GF120918/A → B without GF120918.
i Passive permeability, A → B with GF120918.
j FaSSIF solubility was measured after 4 h of incubation.
k Not determined.
|
||||||||
| 33 |

|
CH2CH3 | 6.8/158.5 | NDk | 5.6 | <0.59/1.3 | 2.3/354 | 97.7 |
| 34 |

|
CH2CH3 | 7.4/39.8 | NDk | 5.6 | <0.59/1.1 | 2.8/432 | 52.6 |
| 35 |

|
CHF2 | 7.5/31.6 | 4.7(7.7) | 5.5 | <0.59/2.8 | 1.2/582 | 5.4 |
| 36 |

|
CHF2 | 6.8/158.5 | 4.7(7.7) | 5.5 | <0.59/2.2 | 1.3/529 | 11.3 |
| 37 |

|
CN | 8.1/7.9 | 4.3(7.3) | 5.3 | 0.66/4.0 | 1.5/457 | 72.5 |
| 38 |

|
CN | 7.8/15.8 | 4.4(7.4) | 5.3 | <0.59/3.2 | 1.5/390 | 24.0 |
| 39 (GSK3357679) |

|
Cl | 7.8/20.0 | 4.4(7.4) | 4.9 | <0.59/3.7 | 1.2/415 | 118.5 |
| 40 |

|
Cl | 7.7/20.0 | 4.3(7.3) | 4.9 | <0.59/2.3 | 1.1/371 | 266.5 |
| 41 |

|
Cl | 7.2/63.1 | 3.6(6.6) | 4.9 | <0.13/4.8 | 1.9/286 | 49.3 |
| 42 |

|
Cl | 8.0/10.0 | 3.6(6.6) | 4.9 | <0.59/2.4 | 1.7/333 | 34.7 |

Based on the overall activity and property profiles, we further evaluated compounds 39 and 40. Both compounds demonstrated favorable kinase selectivity and excellent in vitro and in vivo PK profiles in rats and dogs (Table 7). In the in vitro studies, compound 39 exhibited lower hepatocyte clearance and lower human protein binding. In the in vivo PK studies, compound 39 showed lower clearance, longer T1/2, higher exposure, and better bioavailability than 40. Both compounds 39 and 40 showed excellent Kp,uu, but analog 39 has a much higher exposure in the plasma (about 7-fold) and brain (1.5-fold) than compound 40 in rats (Table 8). Therefore, compound 39 was selected for further evaluation in vivo in rodents.
| Cmpd | In vitro | In vivo | ||||||
|---|---|---|---|---|---|---|---|---|
| Kinase selectivitya | Hep Cli(h/r/d)b (mL min−1 g−1) | Dog BPBc (%) | Rate/dogf DNAUC0–24h (ng h mL−1)/(mg kg−1) | Rat/dog CL (L h−1 kg−1) | Rat/dog Vss (L kg−1) | Rat/dog T1/2 (h) | Rat/dog F% | |
| a Kinobead profiling – number/total of kinases within 100-fold potency compared with LRRK2. b Clearance in human, rat and dog hepatocytes. c Dog blood protein binding. d DNAUC = dose normalized area under the curve after I.V., T1/2 = half-life, CL = blood clearance, Vss = volume of distribution, and F = oral bioavailability. e 1.0 mg kg−1, I.V. to male Han-Wistar rats. f 1.0 mg kg−1 I.V., 2.0 mg kg−1 P.O. to male Beagle dogs, I.V. followed by P.O. with a 7-day washout in between. g Rat P.O. 2 mg kg−1, blood DNAUC0–7h. | ||||||||
| 39 | 3/365 | <0.4/3.0/2.7 | 2.9 | 3001/602 | 5.6/29.0 | 0.89/17.8 | 2.5/14.0 | 100g/85 |
| 40 | 9/368 | 0.9/3.1/2.4 | 2.7 | 905/3557 | 17.9/4.9 | 1.46/3.3 | 1.1/10.0 | 47g/76 |
| Cmpd | DNAUC0–7hb (ng h mL−1)/(mg kg−1) | K p (Br/Bl)c | Kd/Bld | Fubr/Fuble (%) | Brain Kp,uuf | ||
|---|---|---|---|---|---|---|---|
| Blood | Brain | Kidneys | |||||
| a 2.0 mg kg−1, single oral gavage administration to male Han-Wistar rats. b DNAUC = dose normalized area under the curve. c K p (Br/Bl) = brain DNAUC0–7h/blood DNAUC0–7h. d Kd/Bl = kidney DNAUC0–7h/blood DNAUC0–7h. e Free unbound fractions measured in rat brain and blood. f K p,uu = Kp × Fubr/Fubl. | |||||||
| 39 | 3024 | 483 | 4995 | 0.16 | 1.7 | 1.8/0.7 | 0.41 |
| 40 | 427 | 321 | 1902 | 0.75 | 4.5 | 1.6/2.4 | 0.50 |
The level of LRRK2 pS935 phosphorylation was used to measure in vivo pharmacodynamics. This approach was previously established as a surrogate measure of LRRK2 kinase activity.14,15 Compound 39 in rats (5 to 30 mg kg−1 p.o.; 4 hour pretreatment) and mice (3 to 30 mg kg−1 p.o.; 1 hour pretreatment) dose-dependently inhibited the levels of pS935 in brain tissue with near complete inhibition at 10 and 30 mg kg−1 in both species. In kidney and lung tissues, the levels of pS935 LRRK2 were near-maximally inhibited at all doses following administration of 39 in both rats and mice (Fig. 2A and 3A). The total LRRK2 was also dose-dependently reduced in both species (Fig. S1). In rat and mouse time-course studies, compound 39 (3 mg kg−1 p.o.) time-dependently inhibited pS935 LRRK2 with maximum effects observed at 1 hour after administration (Fig. 2B and 3B). The inhibition observed in lung and kidney tissues tended to be greater than in brain. In all studies, the levels of inhibition of pS935 LRRK2 closely tracked concentrations of the compounds in the tissues. Modest effects on the total LRRK2 levels were observed in mice and rats in the time-course studies. The maximum decreases observed were: brain 5%, kidneys 29% and lungs 17% in rats and brain 17%, kidneys 24% and lungs 12% in mice (Fig. S2).
 | ||
| Fig. 2 Dose–response (A) and time course (B) of the pharmacodynamic effects of compound 39 in rats. LRRK2 pS935 levels (red solid symbols and lines) were measured by an ELISA assay to assess LRRK2 kinase inhibition in rat brain, kidneys and lungs after oral administration of a single dose of 5, 10, and 30 mg kg−1 of compound 39 (A, 4 h pretreatment) or 3 mg kg−1 p.o. of compound 39 (B). Compound levels in the tissues at each time point (blue dashed lines and open symbols) correlated with the pharmacodynamic effect. N = 3 rats per time point. | ||
 | ||
| Fig. 3 Dose–response (A) and time course (B) of the pharmacodynamic effects of compound 39 in mice. LRRK2 pS935 levels (red solid lines) were measured by an ELISA assay as a measure of LRRK2 kinase inhibition in rat brain, kidneys and lungs after oral administration of a single dose of 3, 10, and 30 mg kg−1 of compound 39 (A, 1 h pretreatment) or 3 mg kg−1 p.o. (B). Compound levels in the tissues at each time point (blue dashed lines) correlated with the pharmacodynamic effect. N = 3 mice per data point. | ||
Following dosing compound 39 in mice at 5 and 10 mg kg−1 p.o. b.i.d. for 4 days, pS935 LRRK2 levels were inhibited by greater than 90% in the brain, kidneys and lungs at 1 h after the last dose. At 16 hours after the last dose, pS935 LRRK2 levels were inhibited by ≤ 20% in all tissues (data not shown).
In addition, we have explored the characteristics of compound 39 following dosing in mice at 10 and 15 mg kg−1 p.o. b.i.d for two weeks (Table 9). Twelve hours after the last dose, inhibition of pS935 LRRK2 and exposure of the compound were similar to 12 hours after a single dose. The total LRRK2 levels were only modestly (<20%) decreased after either a single dose or 14 days' dosing in the brain and lungs with greater decreases (up to 34% after 14 days' dosing) observed in the kidneys. This suggests that compound 39 has favorable characteristics for extended dosing to obtain sustained LRRK2 kinase inhibition in mouse tissues, including the brain.
| Dose (mg kg−1) | Tissue | pS935 LRRK2 inhibition (%) | Total LRRK2 inhibition (%) | [Compound 39] (ng g−1) | |||
|---|---|---|---|---|---|---|---|
| Single dose | 2 weeks dosing | Single dose | 2 weeks dosing | Single dose | 2 weeks dosing | ||
| a Tissues were sampled 12 hours after the single or last dose. | |||||||
| 10 | Brain | 45 | 42 | 19 | 12 | 42 | 40 |
| 15 | 78 | 75 | 10 | 19 | 164 | 175 | |
| 10 | Lungs | 35 | 51 | −2 | 6 | 101 | 125 |
| 15 | 76 | 80 | 8 | 9 | 595 | 489 | |
| 10 | Kidneys | 66 | 63 | 24 | 18 | 316 | 440 |
| 15 | 88 | 94 | 19 | 34 | 1521 | 1483 | |
Chemistry
Compounds 39 and 40 were prepared in a straightforward manner according to the route depicted in Scheme 1. All other analogs can be prepared similarly (see the SI). Reduction of the commercially available Boc-protected 3-fluoro-4-oxopiperidine with sodium borohydride afforded the cis alcohol 43. Chiral HPLC separation delivered the enantiomers 44a and 44b. The Mitsunobu reaction of 44a or 44b with 4-nitropyrazole afforded the desired key intermediates 45a and 45b. Chlorination followed by deprotection of the Boc group provided piperidine analogs 47a and 47b. Reductive amination followed by the reduction of the nitro group afforded 49a and 49b. Palladium-catalyzed coupling of 49a and 49b with pyrrolopyrimidine 50 (easily accessed via the displacement of chlorine with sodium ethoxide) delivered the final compounds 39 and 40, respectively. | ||
| Scheme 1 Synthesis of pyrrolopyrimidine LRRK2 inhibitors 39 and 40. Reagents and conditions: (i) NaBH4, MeOH, 0 °C, 2 h; (ii) chiral separation, Chiralpak IC; (iii) (3R,4S)-tert-butyl 3-fluoro-4-hydroxypiperidine-1-carboxylate (44a) or (3S,4R)-tert-butyl 3-fluoro-4-hydroxypiperidine-1-carboxylate (44b), PPh3, DIAD, THF, rt, overnight; (iv) LiHMDS, C2Cl6, THF, −78 °C, 2 h; (v) 8 M HCl in dioxane, MeOH, rt, 2 h; (vi) oxetan-3-one, NaBH(OAc)3, DCE, rt, overnight; (vii) iron powder and NH4Cl, EtOH/H2O, rt for 49a and 50 °C for 49b, overnight; (viii) 2-chloro-4-ethoxy-7H-pyrrolo[2,3-d]pyrimidine, X-phos, Pd2(dba)3, K2CO3, dioxane, 110 °C, overnight. | ||
Conclusions
In summary, we report the discovery of pyrrolopyrimidine analogs as potent and selective LRRK2 inhibitors that were derived from the kinase-inhibitor-focused screening hit 1. Optimization of compound 1 led to 39 in desired property space that shows excellent cellular potency, kinase selectivity, oral bioavailability, brain penetration, and excellent PK/PD correlation in animal studies. Compound 39 (GSK3357679) can serve as a useful new in vitro and in vivo tool molecule to investigate the role of LRRK2 in Parkinson's disease in cell and rodent models.16,17Ethical statement
All studies were conducted in accordance with the GSK Policy on the Care, Welfare and Treatment of Laboratory Animals and were reviewed by the Institutional Animal Care and Use Committee at GSK (Shanghai) or Wuxi Apptec (Shanghai and Suzhou sites).Author contributions
J. M. A.: writing – review and editing and visualization. X. D., B. Z., Y. S., L. W., M. Z., X. G., C. T., X. F., and Y. L.: methodology and investigation. L. P. S.: supervision, conceptualization, investigation, and formal analysis. M. H., C. E., and K. V.: investigation, formal analysis, and visualization. K. D.: investigation and formal analysis. X. G.: supervision. N. Z.: visualization, data curation, and formal analysis. F. D. T.: methodology, investigation, visualization, and supervision. F. R.: methodology, investigation, and supervision. D. S.: writing – original draft, review and editing, and visualization. A. D. R.: conceptualization, supervision, and writing – review and editing.Conflicts of interest
All authors are either current or former employees of GSK and may hold stock in GSK. All authors declare that they have no other known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Data availability
Supplemental figures, methods for assays and animal studies, experimental procedures for compounds 3–42, and kinase selectivity data for compounds 39 and 40 can be found in the supplementary information (SI).Supplementary information is available. See DOI: https://doi.org/10.1039/d5md00856e.
Acknowledgements
We thank Zehong Wan, Changhui Sun, Qian Liu, and Hailong Wang for assistance with the supervision, methodology, and experimental assays. We are also grateful to Mark Schultz, Ross Biddulph, and Shenaz Bunally for assistance with the data integrity review.References
- L. V. Kalia and A. E. Lang, Parkinson's disease, Lancet, 2015, 386, 896–912, DOI:10.1016/S0140-6736(14)61393-3.
- W. Poewe, K. Seppi, C. Tanner, G. M. Halliday, P. Brundin, J. Volkmann, A. E. Schrag and A. E. Lang, Parkinson disease, Nat. Rev. Dis. Primers, 2017, 3, 17013, DOI:10.1038/nrdp.2017.13.
- A. Elkouzi, V. Vedam-Mai, R. S. Eisinger and M. S. Okun, Nat. Rev. Neurol., 2019, 15, 204–223, DOI:10.1038/s41582-019-0155-7.
- A. B. West, D. J. Moore, S. Biskup and T. M. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16842–16847, DOI:10.1073/pnas.0507360102.
- M. Taylor and D. R. Alessi, Curr. Opin. Cell Biol., 2020, 63, 102–113, DOI:10.1016/j.ceb.2020.01.001.
- G. R. Jeong and B. D. Lee, Cells, 2020, 9, 2565, DOI:10.3390/cells9122565.
- W. P. G. Gilks, P. M. Abou-Sleiman, S. Gandhi, S. Jain, A. Singleton, A. J. Lees, K. Shaw, K. P. Bhatia, V. Bonifati, N. P. Quinn, J. Lynch, D. G. Healy, J. L. Holton, T. Revesz and N. W. Wood, Lancet, 2005, 365, 415–416, DOI:10.1016/S0140-6736(05)17830-1.
- E. M. Rocha, M. T. Keeney, R. Di Maio, B. R. De Miranda and J. T. Greenamyre, Trends Neurosci., 2022, 45, 224–236, DOI:10.1016/j.tins.2021.12.002.
- J. M. Taymans, M. Fell, T. Greenamyre, W. D. Hirst, A. Mamais, S. Padmanabhan, I. Peter, H. Rideout and A. Thaler, NPJ Parkinsons Dis., 2023, 9, 104, DOI:10.1038/s41531-023-00544-7.
- D. Jennings, S. Huntwork-Rodriguez, M. F. J. M. Vissers, V. M. Daryani, D. Diaz, M. S. Goo, J. J. Chen, R. Maciuca, K. Fraser, O. S. Mabrouk, J. van de Wetering de Rooij, J. A. A. C. Heuberger, G. J. Groeneveld, M. T. Borin, A. Cruz-Herranz, D. Graham, K. Scearce-Levie, J. De Vicente, A. G. Henry, P. Chin, C. Ho and M. D. Troyer, Mov. Disord., 2023, 38, 386–398, DOI:10.1002/mds.29297.
- X. Ding, X. Dai, K. Long, C. Peng, D. Andreotti, P. Bamborough, A. J. Eatherton, C. Edge, K. S. Jandu, P. L. Nichols, O. J. Philps, L. P. Stasi, Z. Wan, J. N. Xiang, K. Dong, P. Dossang, M. H. Ho, Y. Li, L. Mensah, X. Guan and F. Ren, Bioorg. Med. Chem. Lett., 2017, 27, 4034–4038, DOI:10.1016/j.bmcl.2017.07.052.
- A. D. Reith, P. Bamborough, K. Jandu, D. Andreotti, L. Mensah, P. Dossang, H. G. Choi, X. Deng, J. Zhang, D. R. Alessi and N. S. Gray, Bioorg. Med. Chem. Lett., 2012, 22, 5625–5629, DOI:10.1016/j.bmcl.2012.06.104.
- X. Ding, L. P. Stasi, M. H. Ho, B. Zhao, H. Wang, K. Long, Q. Xu, Y. Sang, C. Sun, H. Hu, H. Yu, Z. Wan, L. Wang, C. Edge, Q. Liu, Y. Li, K. Dong, X. Guan, F. D. Tattersall, A. D. Reith and F. Ren, Bioorg. Med. Chem. Lett., 2018, 28, 1615–1620, DOI:10.1016/j.bmcl.2018.03.045.
- N. Dzamko, M. Deak, F. Hentati, A. D. Reith, A. R. Prescott, D. R. Alessi and R. J. Nichols, Biochem. J., 2010, 430, 405–413, DOI:10.1042/BJ20100784.
- L. Delbroek, K. Van Kolen, L. Steegmans, R. de Cunha, W. Mandemakers, G. Daneels, P. J. De Bock, J. Zhang, K. Gevaert, B. De Strooper, D. R. Alessi, P. Verstreken and D. W. Moechars, J. Pharm. Biomed. Anal., 2013, 76, 49–58, DOI:10.1016/j.jpba.2012.12.002.
- A. Tasegian, F. Singh, I. G. Ganley, A. D. Reith and D. R. Alessi, Biochem. J., 2021, 478, 3555–3573, DOI:10.1042/BCJ20210375.
- F. Singh, A. R. Prescott, P. Rosewell, G. Ball, A. D. Reith and I. G. Ganley, eLife, 2021, 10, e67604, DOI:10.7554/eLife.67604.
| This journal is © The Royal Society of Chemistry 2026 |