
Exploring pyrazolidinone and pyrazolidinedione scaffolds for Alzheimer's therapy: multitarget COX-2 inhibitors with anti-amyloid β, anti-tau, antioxidant, and neuroprotective activities
Michael
Emad†
a,
Reham
Waheed†
a,
Zeinab
Mostafa
a,
Sarah S.
Darwish
 ab,
Rosa
Purgatorio
c,
Daniela Valeria
Miniero
d,
Annalisa
De Palma
e,
Tzu-Peng
Cheng
f,
Yu-Cheng
Chen
gh,
Moustafa
Gabr
i,
Ahmed M.
El Kerdawy
jk,
Marco
Catto
c,
Ashraf H.
Abadi
a,
Tsong-Long
Hwang
*fglmn and
Mohammad
Abdel-Halim
*a
ab,
Rosa
Purgatorio
c,
Daniela Valeria
Miniero
d,
Annalisa
De Palma
e,
Tzu-Peng
Cheng
f,
Yu-Cheng
Chen
gh,
Moustafa
Gabr
i,
Ahmed M.
El Kerdawy
jk,
Marco
Catto
c,
Ashraf H.
Abadi
a,
Tsong-Long
Hwang
*fglmn and
Mohammad
Abdel-Halim
*a
aDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy and Biotechnology, German University in Cairo, Cairo 11835, Egypt. E-mail: Mohammad.abdel-halim@guc.edu.eg
bSchool of Life and Medical Sciences, University of Hertfordshire hosted by Global Academic Foundation, New Administrative Capital, 11578 Cairo, Egypt
cDepartment of Pharmacy and Pharmaceutical Sciences, University of Bari Aldo Moro, via E. Orabona 4, 70125 Bari, Italy
dDepartment of Medicine and Surgery, LUM University Giuseppe Degennaro, Torre Rossi, 70010 Casamassima, Italy
eDepartment of Biosciences, Biotechnologies and Environment, University of Bari Aldo Moro, via E. Orabona 4, 70125 Bari, Italy
fGraduate Institute of Natural Products, College of Medicine, Chang Gung University, Taoyuan 333, Taiwan. E-mail: htl@mail.cgust.edu.tw
gCenter for Drug Research and Development, Graduate Institute of Health Industry Technology, College of Human Ecology, Chang Gung University of Science and Technology, Taoyuan 333, Taiwan
hGraduate Institute of Health Industry Technology, College of Human Ecology, Chang Gung University of Science and Technology, Taoyuan, 333324, Taiwan
iDepartment of Radiology, Molecular Imaging Innovations Institute (MI3), Weill Cornell Medicine, New York, NY 10065, USA
jSchool of Health and Care Sciences, College of Health and Science, University of Lincoln, Joseph Banks Laboratories, Green Lane, Lincoln, UK
kDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Cairo University, Kasr El-Aini Street, P.O. Box 11562, Cairo, Egypt
lDepartment of Anesthesiology, Chang Gung Memorial Hospital, Taoyuan 333, Taiwan
mSchool of Chinese Medicine, College of Medicine, National Yang Ming Chiao Tung University, Taipei 112, Taiwan
nDepartment of Chemical Engineering, Ming Chi University of Technology, New Taipei City 243, Taiwan
First published on 10th November 2025
Abstract
COX-2 enzyme is implicated in Alzheimer's disease (AD) through amyloid beta (Aβ) accumulation, tau aggregation, and neuroinflammation. However, clinical outcomes of COX-2 inhibitors in AD have been inconsistent. This study explores a novel series of pyrazolidinones and pyrazolidinediones as selective COX-2 inhibitors. Among these, 4-hydrazonopyrazolidinediones exhibited potent COX-2 inhibition, reducing PGE2 release in a THP-1 cell model. Compounds 15 and 16 demonstrated multitargeting potential by inhibiting Aβ and tau aggregation (PHF6 and R3) and showed significant neuroprotective effects against Aβ and H2O2-induced toxicity in SH-SY5Y cells without cytotoxicity. Additionally, both compounds displayed high permeability in PAMPA and MDCK-MDR1 assays, indicating their potential to cross the blood–brain barrier and reach therapeutic targets. These findings highlight the potential of reviving COX-2 inhibitors as multitargeted therapeutic agents for AD, offering a promising strategy to address multiple pathological aspects of the disease, including neuroinflammation, amyloid aggregation, and tau pathology.
1 Introduction
Cyclooxygenase (COX) enzymes are critical mediators of inflammation and play a central role in the production of prostaglandins and other eicosanoids from arachidonic acid.1 There are two main isoforms of COX: COX-1 and COX-2.2 COX-1 is constitutively expressed in most tissues and is responsible for the production of prostaglandins involved in normal physiological functions, such as gastric cytoprotection and platelet aggregation.3 The second isoform, COX-2, is typically expressed at low levels in most tissues, but its expression can be rapidly upregulated in response to various stimuli, including inflammatory cytokines, growth factors, and oxidative stress.4 Its primary function is to catalyze the conversion of arachidonic acid into cyclic endoperoxide prostaglandin (PGG2), which is further converted to prostaglandin H2 (PGH2) and other prostaglandins PGA, PGE2, PGF2α, and PGD2, as well as thromboxane A2 (TXA2).3 These prostaglandins play a crucial role in the body's response to injury, infection, or other sources of inflammation.5,6 Remarkably, COX-2 plays an important role in immune cell activation and recruitment to the site of inflammation, further amplifying the inflammatory response.3,6 These effects of COX-2 are observed in a wide range of inflammatory conditions, including arthritis,7 cancer,8,9 cardiovascular disease,10 and Alzheimer's disease (AD).11,12COX-2 was reported to play a significant role in the pathogenesis of AD by interacting with amyloid-beta (Aβ) and tau proteins, the two key pathological hallmarks of AD.13 Aβ is derived from amyloid precursor protein (APP) and its aggregation can lead to the formation of toxic oligomers and amyloid plaques, which are associated with neurotoxicity and brain inflammation.14 COX-2 overexpression is linked to increased Aβ accumulation, particularly in the cerebellum, contributing to cognitive deficits.15,16 Moreover, metabolic products of COX-2, such as prostaglandin E2 (PGE2) and TXA2, promote Aβ aggregation and increase the production of Aβ peptides by affecting β- and γ-secretase activities.16,17 Additionally, COX-2 is involved in the hyperphosphorylation of tau protein. Tau is an essential protein for stabilizing and regulating internal microtubules in the brain, which are critically involved in regulating cellular growth and other processes.18 The phosphorylation of tau is controlled by the activities of tau kinases and tau phosphatases.19 However, in AD patients, tau becomes hyperphosphorylated, leading to its detachment from the microtubules and formation of fibers that accumulate as neurofibrillary tangles (NFTs).20 Elevated levels of COX-2 metabolic products, such as prostaglandin F2α (PGF2α) and prostacyclin (PGI2), induce tau hyperphosphorylation, contributing to cognitive deficits and the progression of AD.21 The overexpression of cytokines like tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), IL-18, and IL-6, which is done by the action of COX-2 enzyme, further induces COX-2 expression causing the acceleration of tau phosphorylation.22
COX-2 products and inflammatory mediators not only contribute to the chronic neuroinflammatory state observed in AD,23 but also they can induce oxidative stress, which can severely impact neuronal function and viability.24,25 They also play a role in impairing the synaptic plasticity and disrupting the normal synaptic function, which is essential for cognitive processes and memory formation.26,27
All these findings suggest that COX-2 inhibitors have potential as therapeutic agents for AD. Some notable ones include celecoxib, which has been evaluated in clinical trials for its ability to prevent or slow down the progression of AD. However, results have been controversial, with some studies showing no significant benefit,28 while another study showed that celecoxib can improve cognitive performance and increase regional brain metabolism in people with age-associated memory decline.29 Also, nimesulide has shown promise in reducing neuroinflammation and the overexpression of COX-2, which may help in managing AD symptoms.30 In addition, a study suggested that long-term use of non-selective nonsteroidal anti-inflammatory drugs (NSAIDs), especially ibuprofen, was associated with a lower risk of developing AD.31 Another clinical study showed that patients previously exposed to long-term use of naproxen had a 67% reduced risk of developing AD compared to those given a placebo.32 Indomethacin33 and diclofenac34 were shown to protect mild to moderately impaired patients from further cognitive decline. These results suggest that long-term NSAID use may have a protective effect against the development of AD especially during the very early stages of initial Aβ deposition and microglia activation.35 However, the long-term use of NSAIDs, especially non-selective ones, increases the risk of peptic ulcer disease and acute renal failure.36 Therefore, selective COX-2 inhibitors are considered as better options with higher safety profile compared to non-selective NSAIDs.37 In contrast, other studies showed that long-term clinical trials with both non-selective and COX-2 selective NSAID inhibitors in patients with mild-to-moderate AD did not slow disease progression.38,39 Similarly, a secondary prevention study with the COX-2 selective inhibitor rofecoxib in patients with mild cognitive impairment also yielded negative results.40 Overall, clinical trials have yielded inconclusive results, with some studies suggesting a protective role for NSAIDs in early or preclinical stages of AD, and others showing little to no benefit in patients with established disease. Therefore, researchers are still interested to develop new COX-2 inhibitors that can interact with different mechanisms and targets, thus preventing the progression of AD.12
AD is a multifactorial disease with several pathologic mechanisms.41,42 Notably, many older COX-2 inhibitors were not designed to engage additional pathological processes central to AD, such as Aβ and tau aggregation. Consequently, multitarget-directed ligands (MTDLs), which interact with various biological targets associated with these diseases, may provide a better therapeutic option compared to the traditional “one-target, one-molecule” approach.43 Therefore, our research group surmised that designing compounds that can target multiple pathways, specifically aiming to inhibit COX-2 and independently inhibit Aβ and tau aggregation, might be able to address the complex pathology of AD more effectively than traditional single-target therapies. We hypothesized that expanding the pharmacological profile of COX-2 inhibitors to include additional key pathways involved in AD could enhance their potential as therapeutic tools and improve their success rate in clinical settings.
In this study, we provide a novel approach: the development of COX-2 inhibitors that not only show improved CNS permeability but also function as multitarget-directed ligands (MTDLs), addressing the multifactorial etiology of AD through concurrent anti-inflammatory, anti-amyloid, anti-tau, and antioxidant activities. This strategy aims to overcome the limitations of prior compounds and offers renewed promise for disease modification in AD.
2 Results and discussion
2.1 Compound design
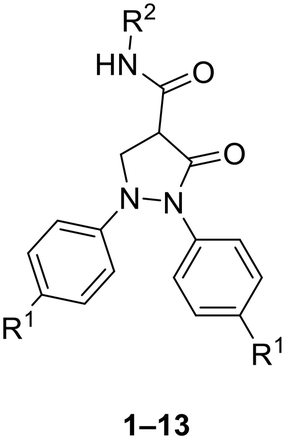
Our designed scaffolds were inspired by the structural features in some selective COX-2 inhibitors and the diaryl pyrazolidinedione COX inhibitor, phenylbutazone,44 where we incorporated key pharmacophoric elements shared among these agents (Fig. 1). | ||
| Fig. 1 Previously reported COX-2 inhibitors and general structures of our novel series. | ||
Since some selective COX-2 inhibitors were reported to have a lactam/lactone ring (carrying one carbonyl) as a core ring such as imrecoxib45 (COX-2 IC50 = 0.018 μM) and compound Y46 (COX-2 IC50 = 0.06 μM) (Fig. 1), we decided to synthesize the first series of compounds as pyrazolidinone analogues of phenylbutazone keeping only one carbonyl on the core ring. In this series, analogues having 1,2-substituted and unsubstituted phenyl and 4-alkyl amides were synthesized (Fig. 1). Given that many potent diaryl heterocycles have been reported to have a third aryl group incorporated into the core, as exemplified by compound Y, we opted to synthesize diaryl pyrazolidinones featuring a third aryl group.46 The third aryl was introduced via amide or arylidene linkage, as shown in Fig. 1.
For the second series of compounds, pyrazolidinedione derivatives were synthesized featuring a 1,2-diphenyl core with a 4-chloro substituent, inspired by the enhanced potency observed in the first series. A third aryl group was incorporated at position 4 through a hydrazone linker introduced via diazo coupling. Structural diversity was achieved by substituting the N-aryl hydrazone with various electron-withdrawing and electron-donating groups at different positions on the aromatic ring.
2.2 Chemistry
The desired 3-pyrazolidinones (1–13) were synthesized through a five-step sequence, as outlined in Scheme 1.47 The synthesis started with the oxidative coupling of the aniline derivatives using manganese(IV) oxide under reflux in toluene for 8 hours, employing a Dean–Stark apparatus to remove the water produced during the reaction. The resulting colored diazenes (A1–A2) were subsequently reduced to the corresponding hydrazines (B1–B2) using Zn/NH4Cl in acetone at room temperature. Cyclization was achieved by reacting the diaryl hydrazine derivatives with 3-chloropropionyl chloride in the presence of potassium carbonate as a base at room temperature. The reaction proceeded over 72 hours, yielding the pyrazolidinone derivatives (C1–C2). | ||
| Scheme 1 Synthetic pathway of target derivatives 1–13. Reagents and conditions: (i) MnO2, toluene, reflux, 8 h; (ii) Zn, NH4Cl, acetone, room temperature, overnight; (iii) 3-chloropropionylchloride, K2CO3, acetone, room temperature, 72 h; (iv) CO2, LDA, THF, −78 °C; (v) HBTU, DCM, TEA, amine derivative, room temperature, 2 h. | ||
Substitution at position 4 of pyrazolidinone derivatives (C1–C2) was achieved by deprotonating the acidic methylene group using lithium diisopropylamide (LDA) at −78 °C for 1 hour. The resulting anion was then treated with carbon dioxide (dry ice), leading to the formation of the 4-carboxy derivatives (D1–D2).47,48 Several amide derivatives (1–13) were synthesized by reacting the carboxylic acid derivatives (D1–D2) with various amines, utilizing hexafluorophosphate benzotriazole tetramethyl uronium (HBTU) as the coupling agent in the presence of triethylamine (TEA), as illustrated in Scheme 1.
The synthesis of compound 14 was achieved through an aldol condensation reaction between the pyrazolidinone derivative (C2) and 3-chlorobenzaldehyde. The reaction was carried out in the presence of potassium hydride as a base under reflux in THF for 3 hours, as depicted in Scheme 2.
 | ||
| Scheme 2 Synthetic pathway of target derivative 14. Reagents and conditions: (i) KH, 3-chlorobenzaldehyde, THF, reflux, 3 h. | ||
The pyrazolidinedione derivative (E2) was synthesized via an addition–elimination reaction, involving the cyclization of the hydrazine derivative (B2) with malonyl chloride as the cyclizing agent and sodium hydride as the base. The reaction was conducted in THF under inert conditions, as illustrated in Scheme 3.
 | ||
| Scheme 3 Synthetic pathway of target derivative 15–39. Reagents and conditions: (i) NaH, THF, ice, 15 min, malonyl dichloride, room temperature, 4 h; (ii) diazonium salt, MeOH, 4 h, room temperature (diazonium salt: aniline derivative, sodium nitrite, acetic acid, propionic acid, hydrochloric acid). | ||
The pyrazolidinedione derivatives (15–39) were synthesized through a two-step process. Initially, various diazonium salts were prepared by treating the corresponding anilines with sodium nitrite in the presence of hydrochloric acid, propionic acid, and acetic acid. These diazonium salts were subsequently reacted with the nucleophilic methylene group at position 4 of the pyrazolidinedione derivative (E2), as outlined in Scheme 3.
The product formed from the coupling of diazonium salts with 1,2-diphenyl-3,5-dioxopyrazolidine may exist in three possible tautomeric forms, as illustrated in Chart 1.49 Based on 1H-NMR measurements, tautomer i can be excluded due to the presence of a highly deshielded singlet observed at 13–14 ppm. Tautomer ii is confirmed by the presence of carbonyl stretching bands at 1650–1750 cm−1 in the IR spectrum, along with the absence of an OH band expected for tautomer iii (Fig. S1).49 Furthermore, all compounds display two distinct signals for the carbonyl carbons of the pyrazolidinedione nucleus in the range of 160–170 ppm in the 13C-NMR spectra.
 | ||
| Chart 1 The three possible tautomers for the product of diazo coupling with 1,2-diphenyl-3,5-dioxopyrazolidine. | ||
2.3 Biological evaluation and molecular modeling
2.3.1.1 Structure–activity relationship for COX-2 inhibition. All the newly synthesized compounds were tested for their in vitro ability to inhibit COX-2 enzyme using a Cayman COX-2 (human) inhibitor screening kit (Tables 1 and 2). Celecoxib was used as a positive control (IC50 = 0.06 μM). Compounds were screened at 30 μM to determine the percent inhibition, and IC50 values were only obtained for compounds with more than 50% inhibition.
|
|
|
|||
|---|---|---|---|---|
| Cpd No. | R1 | R2 | % Inh. at 30 μM | IC50 (μM) |
| a Values are mean ± SEM of three experiments; ND: not determined. | ||||
| 1 | H |

|
21.1 ± 3.2 | ND |
| 2 | H |
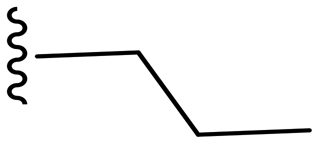
|
20.0 ± 8.3 | ND |
| 3 | H |
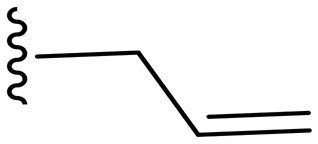
|
20.5 ± 6.5 | ND |
| 4 | H |
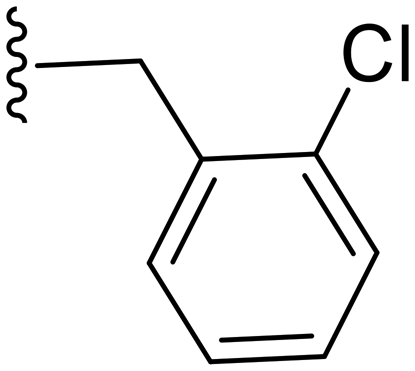
|
21.0 ± 3.2 | ND |
| 5 | H |
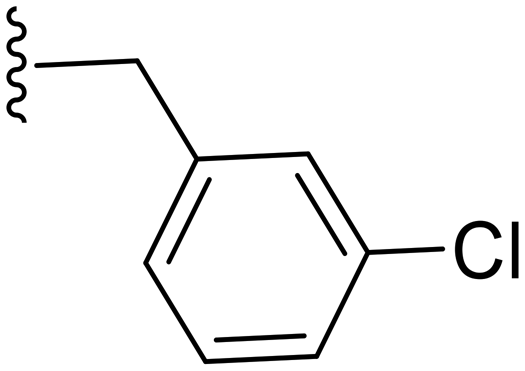
|
21.9 ± 5.6 | ND |
| 6 | H |
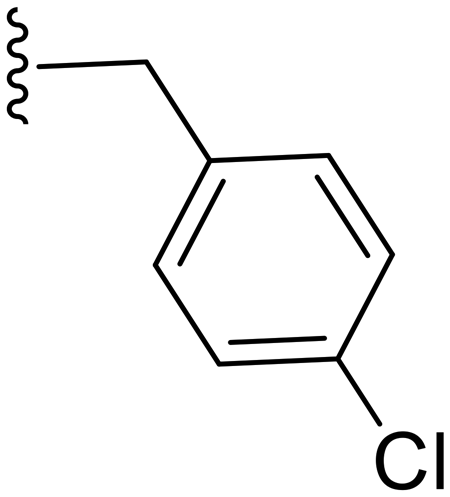
|
27.3 ± 0.4 | ND |
| 7 | Cl |
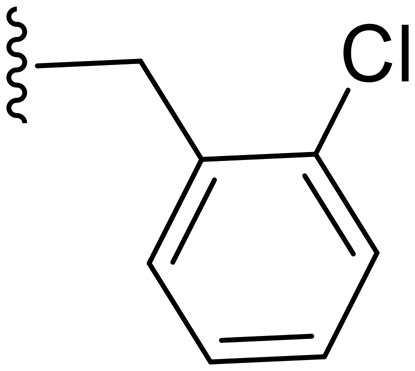
|
9.1 ± 3.2 | ND |
| 8 | Cl |
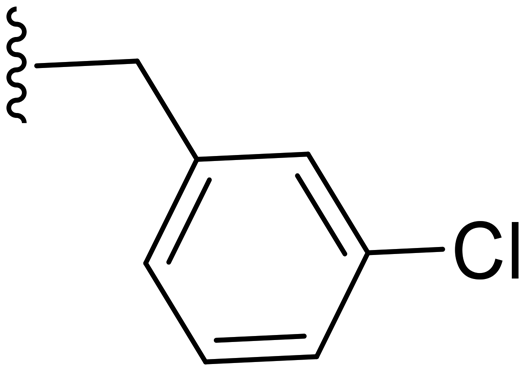
|
74.3 ± 6.8 | 2.82 ± 0.48 |
| 9 | Cl |

|
28.4 ± 2.4 | ND |
| 10 | Cl |

|
75.7 ± 1.9 | 2.29 ± 0.55 |
| 11 | Cl |

|
29.4 ± 4.6 | ND |
| 12 | Cl |

|
26.8 ± 1.3 | ND |
| 13 | Cl |

|
33.9 ± 4.6 | ND |
| 14 | 36.3 ± 5.5 | ND | ||
|
|
|||
|---|---|---|---|
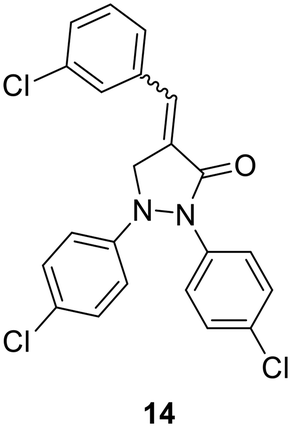
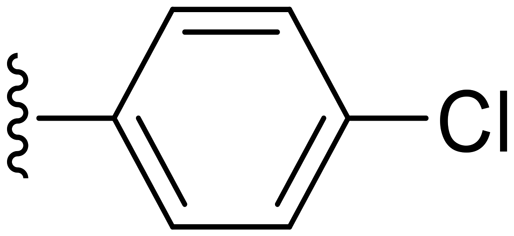
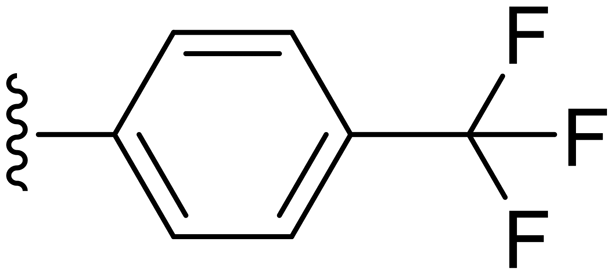
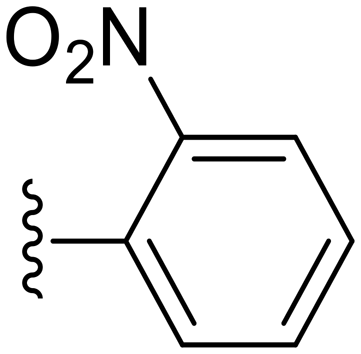
| Cpd No. | R | % Inh. at 30 μM | IC50 (μM) |
| a Values are mean ± SEM of three experiments; ND: not determined. | |||
| 15 |

|
72.4 ± 1.9 | 0.15 ± 0.04 |
| 16 |

|
85.6 ± 3.7 | 0.22 ± 0.02 |
| 17 |

|
76.5 ± 2.0 | 1.31 ±0.78 |
| 18 |

|
33.2 ± 6.0 | ND |
| 19 |

|
57.8 ± 1.8 | 8.18 ± 1.29 |
| 20 |
|
42.4 ± 3.5 | ND |
| 21 |

|
79.7 ± 8.7 | 0.60 ± 0.37 |
| 22 |

|
34.0 ± 2.1 | ND |
| 23 |

|
71.1 ± 7.9 | 3.10 ± 1.34 |
| 24 |

|
81.8 ± 2.4 | 0.70 ± 0.28 |
| 25 |

|
70.0 ± 6.0 | 1.53 ± 0.58 |
| 26 |
|
44.8 ± 3.4 | ND |
| 27 |
|
85.0 ± 2.7 | 0.20 ± 0.07 |
| 28 |

|
92.6 ± 3.8 | 0.22 ± 0.01 |
| 29 |

|
80.3 ± 4.9 | 0.91 ± 0.15 |
| 30 |
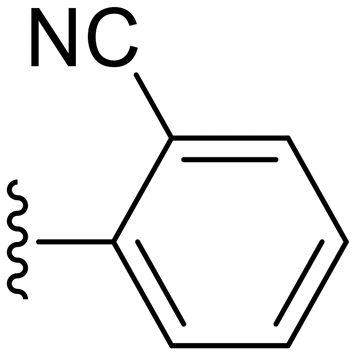
|
74.3 ± 8.5 | 7.34 ± 2.36 |
| 31 |
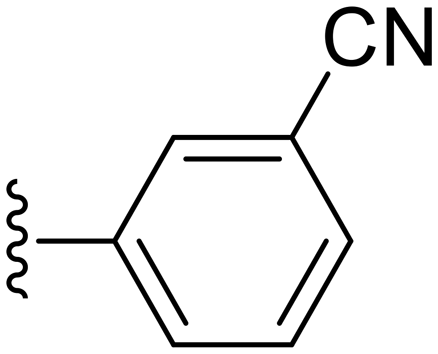
|
90.6 ± 2.0 | 4.57 ± 0.92 |
| 32 |

|
81.6 ± 4.0 | 2.74 ± 0.81 |
| 33 |
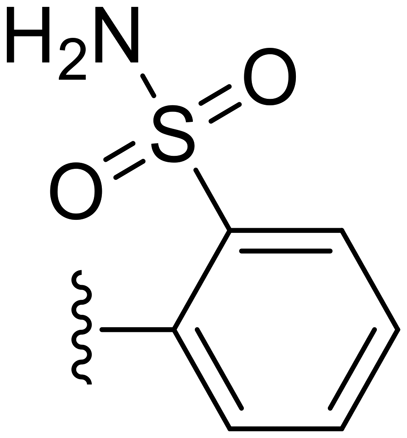
|
73.5 ± 1.0 | 5.93 ± 2.39 |
| 34 |

|
30.0 ± 5.7 | ND |
| 35 |

|
36.5 ± 3.3 | ND |
| 36 |

|
14.5 ± 9.3 | ND |
| 37 |

|
22.2 ± 1.4 | ND |
| 38 |

|
29.2 ± 4.6 | ND |
| 39 |
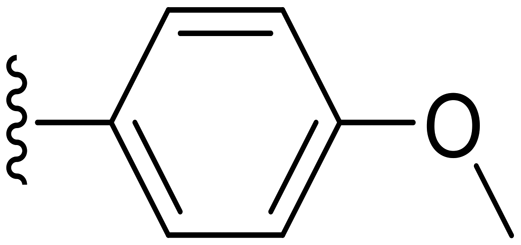
|
29.2 ± 6.8 | ND |
| Celecoxib | 93.7 ± 3.3 | 0.06 ± 0.01 | |

2.3.1.1.1 Structure–activity relationship of COX-2 inhibition by pyrazolidinone derivatives (1–14). Starting with the amide derivatives containing various aliphatic side chains, which share close structural similarity to phenylbutazone, all three compounds (1–3) exhibited diminished activity against COX-2, as shown in Table 1. Similarly, compounds having unsubstituted 1,2-diphenyl with ortho, meta or para chlorobenzyl (4–6) showed no improvement in activity. Incorporating a 4-chloro substituent into the 1,2-diphenyl scaffold significantly improved activity, as evidenced by the comparison between compound 8 and compound 5, with compound 8 showing a COX-2 IC50 value of 2.82 μM (Table 1). However, this modification did not restore the activity of the other chlorobenzyl positional isomers. Consequently, other analogues for 8 having a meta substituted benzyl were synthesized and tested. Compound 10 with a meta-fluorobenzyl was almost equipotent to compound 8 with an IC50 value of 2.29 μM, while compound 11 with a meta-methylbenzyl showed deterioration in activity indicating the importance of the halogen substituent at the meta position. To test the importance of the methylene spacer in compounds 8 and 10, two compounds were synthesized introducing meta substituted anilide instead of benzylamide. Both compounds 12 and 13, with meta-fluoro and meta-chloroanilide respectively, showed a detrimental loss of potency compared to the benzylamide analogues indicating the importance of the methylene spacer (Table 1). Finally, the meta-chloroarylidene analogue (14) was synthesized in order to mimic the meta-chlorobenzylamide in compound 8, yet it showed loss of potency, as displayed in Table 1. In summary, the presented pyrazolidinone analogues demonstrated activity exclusively when the m-chloro and m-fluorobenzyl amide groups were positioned at position 4 while having 1,2-bis(4-chlorophenyl groups).
2.3.1.1.2 Structure–activity relationship of COX-2 inhibition by the 4-hydrazonopyrazolidine-3,5-dione derivatives (15–39). Starting with monosubstitution by halogens, the fluorophenyl derivatives (15–17) showed the highest potency, with IC50 values of 0.15, 0.22 and 1.31 μM, respectively, with the ortho and meta analogues showing superior potency to their para analogue (Table 2). In contrast, when replacing the fluoro substituents with chloro, the potency decreased significantly, where the ortho-chloro (18) and para-chloro (20) derivatives showed abolished activity while the meta-chloro derivative (19) showed an 8-fold decrease in potency with an IC50 value of 8.18 μM compared to its fluoro congener (16), as shown in Table 2. Interestingly, the bromo-substituted derivatives exhibited a distinct pattern. The ortho-bromo derivative (21) retained relatively high potency, with an IC50 of 0.6 μM. However, shifting the bromo substituent to the meta position (22) resulted in a significant loss of activity, while the para-bromo analogue (23) displayed a 5-fold decrease in potency compared to the ortho derivative, with an IC50 of 3.1 μM (Table 2).
The trifluoromethyl substituent showed the same pattern as the fluoro substituent, where compounds 24 and 25 showed high potency with IC50 values of 0.7 and 1.53 μM, respectively, while the para analogue (26) showed abolished activity. Furthermore, the more powerful electron-withdrawing nitro group was utilized which showed a similar pattern as the fluoro and the trifluoromethyl substituents, where the ortho and meta substituted analogues (compound 27, IC50 = 0.2 μM and compound 28, IC50 = 0.22 μM), showed superior potency compared to the para analogue; compound 29 (IC50 = 0.91 μM) (Table 2). Also the cyano group was introduced in all three positions with the para-cyano analogue (32) showing the highest potency among the three compounds with an IC50 value of 2.74 μM (Table 2). Finally, the sulfonyl amino group was introduced in the ortho position (33) and showed moderate potency against COX-2 with an IC50 value of 5.93 μM, as shown in Table 2.
Finally, we tried substituting the third phenyl ring with different alkyl/alkoxy groups (34–39) (Table 2). All the electron-donating groups showed deterioration in activity proving the superiority of using electron-withdrawing groups. Our SAR analysis highlights the importance of electron-withdrawing substituents, particularly fluorine (compounds 15, IC50 = 0.15 μM and 16, IC50 = 0.22 μM) and nitro groups (compounds 27–28), at the ortho and meta positions of the phenyl ring for achieving potent COX-2 inhibition.
Overall, our modifications with pyrazolidine-3,5-dione derivatives proved more successful in producing potent COX-2 inhibitors compared to the pyrazolidinone series, as evidenced by the significantly lower IC50 values and enhanced activity observed across multiple analogues. Fig. S2 presents a summary for the SAR of the pyrazolidinone and pyrazolidine-3,5-dione series (SI).
2.3.1.2 Assessing the selective inhibitory effect of potent COX-2 inhibitors on COX-1. Traditional NSAIDs exert non-selective COX-1/2 inhibition, which leads to side effects (e.g. peptic lesions) mediated by the inhibition of COX-1 in the gastrointestinal mucosa. Thus, we further tested the effect of our most potent COX-2 inhibitors (IC50 < 1 μM) on COX-1 inhibition. The tested compounds generally demonstrated selective inhibition of COX-2. Compounds 15 and 21 displayed outstanding selectivity towards COX-2 with negligible inhibition against COX-1 enzyme (IC50 > 30 μM), as shown in Table 3. Similarly, compounds 16 and 27 showed weak inhibition towards COX-1 with SI values of 88 and 116, respectively (Table 3), with selectivity still shifted towards COX-2 enzyme. Finally, compounds 28–29 exhibited the highest affinity to the COX-1 enzyme (Table 3) with IC50 values of 0.61 μM and 2.29 μM, respectively, showing dual COX-1/COX-2 inhibition (SI = 2–3). These findings support the superiority of electron-withdrawing groups, not only for potent COX-2 inhibition, but also for remarkable selectivity for COX-2 over COX-1.
| Cpd No. | IC50 (μM) (COX-1) | IC50 (μM) (COX-2) | SI |
|---|---|---|---|
| a Values are mean ± SEM of three experiments; ND: not determined. | |||
| 15 | >30 | 0.15 ± 0.04 | >200 |
| 16 | 19.24 ± 2.78 | 0.22 ± 0.02 | 87 |
| 21 | >30 | 0.6 ± 0.37 | >50 |
| 24 | 4.45 ± 1.07 | 0.7 ± 0.28 | 6.35 |
| 27 | 23.26 ± 2.99 | 0.20 ± 0.07 | 116 |
| 28 | 0.61 ± 0.09 | 0.22 ± 0.01 | 2.8 |
| 29 | 2.29 ± 0.69 | 0.91 ± 0.15 | 2.51 |
| Celecoxib | 33 ± 0.5 | 0.06 | 550 |
| Ibuprofen | 25.86 ± 0.59 | >30 | |
2.3.1.3 Molecular modeling. Compounds 15, 16, 21, 24, and 27–29, which showed a promising COX-2 inhibitory activity (Table 2), were selected as representatives to explore the binding pattern of the synthesized compounds in COX-1 and COX-2 active sites utilising molecular docking simulation, and then the most potent and selective COX-2 compounds 15, 16, and 27 were further investigated using molecular dynamics (MD) simulations to confirm their docking-predicted binding pattern and to study their dynamic behaviour.
2.3.1.3.1 Molecular docking simulation. To perform the present molecular docking study, we employed our reported validated molecular docking protocol,50,51 which uses PDB ID 5WBE (COX-1) and 3LN1 (COX-2) crystal structures.52,53 It can proficiently predict the binding mode of different ligands in COX-1 and COX-2 enzymes.50,51
Using the validated molecular docking protocol, the compounds showed promising COX-2 inhibitory activity; 15, 16, 21, 24, and 27–29 were docked in the active sites of COX-1 and COX-2 to study their binding pattern in an attempt to rationalize their COX-1/COX-2 selectivity.
COX-1 and COX-2 active sites share a high degree of sequence identity and topology similarity54–56 and they can be visualized as hydrophobic channels that comprise four hydrophobic regions responsible for binding with arachidonic acid double bonds (Δ5, Δ8, Δ11 and Δ14).57–60 Despite this similarity, the COX-2 active site is approximately 20% larger with an extra hydrophilic side pocket extending from the main pocket.50,51,54,56
In COX-1, all docked compounds showed a common binding pattern involving the accommodation of the pyrazolidinedione ring in the Δ8 double-bond binding region (occupied by the isoxazole ring of co-crystallized mofezolac) surrounded by the hydrophobic side chains of Val349, Ala527, and Leu531, with some compounds interacting with Tyr355 OH through hydrogen bonding by their pyrazolidinedione ring C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group. It directs one of the 4-chlorophenyl moieties towards the Δ11 double-bond binding region interacting through hydrophobic interactions with the surrounding hydrophobic side chains of Leu352, Tyr385, Trp387, and Phe518. Simultaneously, it directs the second 4-chlorophenyl moiety towards the Δ5 double-bond binding region interacting through hydrophobic interactions with the surrounding hydrophobic side chains Leu352, Phe518, and Ile523 and through halogen bonding with Ser516 side chain OH in some compounds. On the other hand, this binding pattern directs the longer arm (substituted phenyl hydrazono moiety) near the entrance of the active site interacting through hydrophobic interactions with the hydrophobic side chains of the amino acids Met113, Val116, Leu117, Leu357, and Leu359 and through non-classical hydrogen bonding with the Val116 side chain in some compounds (Fig. 6A and B) (see SI for 2D interaction diagrams).
O group. It directs one of the 4-chlorophenyl moieties towards the Δ11 double-bond binding region interacting through hydrophobic interactions with the surrounding hydrophobic side chains of Leu352, Tyr385, Trp387, and Phe518. Simultaneously, it directs the second 4-chlorophenyl moiety towards the Δ5 double-bond binding region interacting through hydrophobic interactions with the surrounding hydrophobic side chains Leu352, Phe518, and Ile523 and through halogen bonding with Ser516 side chain OH in some compounds. On the other hand, this binding pattern directs the longer arm (substituted phenyl hydrazono moiety) near the entrance of the active site interacting through hydrophobic interactions with the hydrophobic side chains of the amino acids Met113, Val116, Leu117, Leu357, and Leu359 and through non-classical hydrogen bonding with the Val116 side chain in some compounds (Fig. 6A and B) (see SI for 2D interaction diagrams).
In COX-2, apart from compounds 28 and 29, the docked compounds showed a common reversed binding pattern involving the accommodation of the pyrazolidinedione ring in the Δ8 double-bond binding region (occupied by the pyrazole ring of co-crystallized celecoxib) surrounded by the hydrophobic side chains of Leu338 and Ala513, with some compounds interacting with Tyr341 OH through hydrogen bonding by their pyrazolidinedione ring CO group. It directs one of the 4-chlorophenyl moieties towards the Δ11 double-bond binding region interacting through hydrophobic interactions with the surrounding hydrophobic side chains of Phe367, Leu370, Tyr371, and Trp373. Simultaneously, it directs the second 4-chlorophenyl moiety towards the entrance of the active site (celecoxib CF3 binding region) interacting through hydrophobic interactions with the surrounding hydrophobic side chains of the amino acids Val102, Ile331, Val335, Leu517, and Met521. On the other hand, this binding pattern directs the hydrazono arm through the Δ5 double-bond binding region, interacting through hydrogen bonding with Leu338 in some compounds, fitting the substituted phenyl moiety into the hydrophilic side pocket interacting through hydrophobic interaction with the Val509 side chain and through non-classical hydrogen bonding in some compounds. This binding pattern involving the accommodation of the longer hydrazono arm into the hydrophilic side pocket in the COX-2 active site could attribute the preferential selectivity of compounds 15, 16, 21, 24, and 27 towards COX-2 binding due to the less steric constraints and entropic penalty (in some compounds like compound 15) in comparison to its binding mode in the COX-1 active site (Fig. 6C and D) (see SI for 2D interaction diagrams).
On the other hand, in the COX-2 active site, compounds 28 and 29 showed similar binding patterns to theirs in COX-1 which could rationalize their non-selective binding affinity to both COX isoforms (see SI for 2D interaction diagrams).
In summary, these results indicate that COX1/2 selectivity or non-selectivity in this series is a function of their predicted binding mode in COX-1 and COX-2 active sites. Compounds showing similar binding patterns in both isoforms (28 and 29) show comparable experimental affinity to both enzymes and thus non-selective inhibition (Table 3). On the other hand, compounds (15, 16, 21, 24, and 27) that adopt a different, more sterically favorable binding orientation within the COX-2 active site—which is approximately 20% larger than that of COX-1—exhibited selective inhibition toward the COX-2 isoform (Table 3).
2.3.1.3.2 Molecular dynamics simulations. To further investigate and confirm the binding pattern of the most potent and COX-2 selective compounds 15, 16, and 27 and to study their dynamic behaviour, molecular dynamics (MD) simulations in the active sites of the target isozymes COX-1 and COX-2 were carried out using the GROMACS 2021.3 package.61 The MD simulations were run for 100 ns starting from their predicted molecular docking poses in COX-1 and COX-2 active sites. To analyse the resulting MD trajectories and to assess system stability and simulation quality, the root mean square deviation (RMSD), root mean square fluctuation (RMSF), and radius of gyration (Rg) were calculated and examined.
The RMSD graph (Fig. 2) shows the relative stability of the backbone atoms of 15/COX-2, 16/COX-2, and 27/COX-2 structures during the simulation time with an average RMSD values of 0.218 ± 0.033, 0.215 ± 0.033, and 0.196 ± 0.024 nm, respectively, which are acceptable values for small globular proteins. The abrupt increase in the RMSD values between 90 and 95 ns in 15/COX-2 and 27/COX-2 complexes at the end of the simulations (Fig. 2) could be due to the high fluctuation of the terminal loop region Ala18–Thr70 during this simulation period (Fig. 3). The backbone atoms of 15/COX-1, 16/COX-1, and 27/COX-1 structures showed comparable stability during the simulation time with average RMSD values of 0.223 ± 0.024, 0.244 ± 0.032, and 0.251 ± 0.028 nm, respectively (see SI for the RMSD graph in COX-1).
 | ||
| Fig. 2 RMSD graph for the backbone atoms of 15/COX-2 (orange), 16/COX-2 (blue), and 27/COX-2 (grey) structures from their initial reference frame backbone during 100 ns MD simulations (see SI for the RMSD graph in COX-1). | ||
 | ||
| Fig. 3 RMSF graph for the residues of 15/COX-2 (orange), 16/COX-2 (blue), and 27/COX-2 (grey) structures during 100 ns MD simulation (see SI for the RMSF graph in COX-1). | ||
The RMSF graph (Fig. 3) shows system stability in all COX-2 simulations, as except for the terminal and loop regions, the RMSF values did not exceed 0.1 nm for most residues. Moreover, 15/COX-1, 16/COX-1, and 27/COX-1 structures showed comparable stability throughout the simulation time with RMSF values not exceeding 0.1 nm, apart from the terminal and loop regions (see SI for the RMSF graph in COX-1).
The Rg graph (Fig. 4) shows that COX-2 enzyme was kept well compacted during the simulation time exhibiting a stable Rg with average values of 2.449 ± 0.011 (15/COX-2), 2.440 ± 0.009 (16/COX-2), and 2.428 ± 0.010 (27/COX-2) nm. Moreover, COX-1 enzyme showed comparable compactness during the simulation time exhibiting a stable Rg with average values of 2.425 ± 0.008 (15/COX-1), 2.458 ± 0.009 (16/COX-1), and 2.446 ± 0.018 (27/COX-1) nm (see SI for the Rg graph in COX-1).
 | ||
| Fig. 4 Radius of gyration (Rg) graph for 15/COX-2 (orange), 16/COX-2 (blue), and 27/COX-2 (grey) structures during 100 ns MD simulation (see SI for the Rg graph in COX-1). | ||
Further analysis employing the ligand RMSD graph in Chimera 1.17.1,62 which compares the ligand atoms' positions to the initial pose (0 ns) in the active sites of both isozymes throughout the simulation time, demonstrated the compound pose stability with average RMSD values of 1.664 ± 0.141 (15), 1.498 ± 0.179 (16), and 1.402 ± 0.164 (27) Å in COX-2 enzyme (Fig. 5), as well as 1.598 ± 0.074 (15), 1.524 ± 0.087 (16), and 1.549 ± 0.112 (27) Å in COX-1 enzyme (see SI for the ligands' RMSD graph in COX-1). Noteworthily, after 50 ns simulation, the tested compounds showed a higher stability in the COX-2 active site with average RMSD values of 1.697 ± 0.043 (15), 1.516 ± 0.075 (16), and 1.421 ± 0.063 (27) Å versus 1.613 ± 0.071 (15), 1.543 ± 0.066 (16), and 1.522 ± 0.116 (27) Å in the COX-1 active site. The lower standard deviation indicates the higher ligand pose stability at 50 ns onwards in COX-2 than in COX-1, which agrees with the experimental results that showed the higher potency of the tested compounds on COX-2. It is worth noting as well that the degree of ligand stability in COX-2 enzyme during this time frame represented by the RMSD standard deviation agrees with the order of their COX-2 inhibitory activity (Table 3).
 | ||
| Fig. 5 RMSD graph of compounds 15 (orange), 16 (blue), and 27 (grey) atoms from their initial pose in COX-2 structures during 100 ns MD simulation (see SI for the ligands' RMSD graph in COX-1). | ||
An in-depth study of the binding patterns of the tested compounds in COX-1 and COX-2 active sites throughout the simulations using the cluster analysis tool in Chimera 1.17.1 (ref. 62) showed that, in each isozyme, the tested compounds steadily accommodated a major binding mode during the simulation time (Fig. 6), which is similar to that predicted by the molecular docking study (vide supra) (see SI for the dominant pose for compounds 16 and 27).
 | ||
| Fig. 6 2D diagram (A) and 3D representation (B) showing the dominant binding pattern of compound 15 in the active site of COX-1 (PDB ID: 5WBE). 2D diagram (C) and 3D representation (D) showing the dominant binding pattern of compound 15 in the active sites of COX-2 (PDB ID: 3LN1). | ||
2.3.1.4 Effect on PGE2 release by LPS-stimulated THP-1 cells. In order to confirm the anti-inflammatory ability and cellular activity of our most potent COX-2 inhibitors (compounds 15, IC50 = 0.15 μM and 27, IC50 = 0.2 μM), we investigated their ability to reduce the production of PGE2 in lipopolysaccharide (LPS)-stimulated THP-1 cells. LPS stimulation is known for the activation of COX-2 enzyme, which in turn is responsible for the synthesis of various inflammatory mediators, including PGE2.63
Both compounds were tested for their ability to reduce PGE2 production. As surmised, compound 15 treatment successfully inhibited PGE2 production whereas 60% inhibition was achieved at 3 μM with an IC50 of 1.68 μM, as shown in Fig. 7A. However, compound 27 lacked cellular effect at all tested concentrations. Based on the previous results, we evaluated the effect of compound 15 on the viability of lipopolysaccharide (LPS)-stimulated phorbol 12-myristate 13-acetate (PMA)-differentiated THP-1 cells using the LDH assay to exclude potential cytotoxicity. Compound 15 did not show any significant effect on cell viability of THP-1 cells (Fig. 7B).
 | ||
| Fig. 7 (A) The effect of COX-2 inhibitors 15 and 27 on PGE2 expression in LPS-stimulated THP-1 cells. (B) The effect of 15 on cell viability in THP-1 cells using the LDH assay. | ||
Our results suggest that compound 15 interferes with PGE2 synthesis (IC50 = 1.68 μM) during inflammation due to COX-2 inhibition. These findings also highlight its potential for therapeutic application in treating inflammatory conditions.
2.3.2.1 Aβ aggregation. Inhibiting Aβ aggregation is a well-recognized strategy for treating AD. Accordingly, compounds 15, 16, 21, 24, and 27–29 were assessed for their ability to inhibit Aβ40 self-induced aggregation using a thioflavin T (ThT) fluorescence assay. Quercetin, a well-known in vitro inhibitor of Aβ aggregation, was included as a positive control (Fig. 8). Remarkably, most of the tested compounds outperformed quercetin (except compound 24), achieving inhibition rates between 67% and 96% at 10 μM concentration. Among them, compound 21 (96%) emerged as the most potent Aβ aggregation inhibitor, highlighting the potential of these compounds as therapeutic agents targeting Aβ pathology in AD (Fig. 8).
 | ||
| Fig. 8 Inhibition of amyloid beta-40 (30 μM) aggregation by selected compounds (10 μM). Quercetin was used as a reference compound. Bars represent the mean ± SD of % inhibition. | ||
2.3.2.2 Tau aggregation. Furthermore, we explored the inhibitory activity of the aforementioned compounds in an in vitro ThT-based assay of tau aggregation, using PHF6 as well as R3 tau sequences. PHF6 is a highly repeated hexapeptide fragment (306)VQIVYK(311) that is located at the third repeat (R3) and is present in all tau isoforms.65 Compounds 15, 16, 21, 24, and 27–29 were first evaluated for their ability to inhibit PHF6 aggregation (Fig. 9). Most of the tested compounds demonstrated over 50% inhibition at 10 μM concentration, with several surpassing the activity of quercetin. Notably, compound 24 exhibited the highest inhibition, achieving 85% inhibition, highlighting its superior anti-aggregation potential.
 | ||
| Fig. 9 Inhibition of PHF6 (50 μM) aggregation by selected compounds (10 μM). Quercetin was used as a reference compound. Bars represent the mean ± SD of % inhibition. | ||
The compounds were subsequently tested in the R3 tau aggregation assay at a concentration of 10 μM (Fig. 10). R3 is a 30-amino acid peptide (V306-Q336) that includes the PHF6 sequence and is found in all tau isoforms. For this reason, it is widely used in in vitro assays for mimicking the self-aggregating features of native tau. The commercial R3 peptide was pretreated with 1,1,1-trifluoroethanol (TFE) overnight to dissolve any preformed aggregates and restore its random coil structure. The peptide was then diluted to a final concentration of 25 μM in PBS, and its aggregation was monitored using ThT fluorescence.66 Some of the tested compounds demonstrated over 50% inhibition of R3 tau aggregation, with compounds 24 and 29 achieving more than 80% inhibition. Notably, four of these R3 tau aggregation inhibitors exhibited greater activity than the reference compound, quercetin, as shown in Fig. 10.
 | ||
| Fig. 10 Inhibition of R3 (25 μM) aggregation by selected compounds (10 μM). Quercetin was used as a reference compound. Bars represent the mean ± SD of % inhibition. | ||
Overall, many of the potent COX-2 inhibitors also exhibit strong inhibitory activity against Aß40, PHF6, and R3 tau aggregation, underscoring their potential as multitarget ligands for the treatment of AD. The antiaggregating activity, as measured in the ThT fluorescence-based assay, could be derived from the disruption of the β-sheet arrangement of the peptides, induced by the interaction of molecules with the peptides themselves. Additional biophysical studies would warrant a deeper investigation of the inhibition mechanism.
 | ||
| Fig. 11 (A) Viability of SH-SY5Y cells incubated for 24 h with compounds 15 and 16 tested in the range of concentrations from 1 to 100 μM (MTT assay). Results are reported as % of cell viability referred to untreated cells (CTRL) and are mean ± SD of three independent experiments. One-way analysis of variance (ANOVA) followed by multiple comparison tests (Dunnett's test; GraphPad Prism). (B) Viability of SH-SY5Y cells incubated for 24 h in DMEM serum-free with 400 μM H2O2 in the absence or in the co-presence of increasing concentrations of compounds 15 and 16 (from 5 to 25 μM). Reference drug: quercetin (99 ± 8% of cell viability at 25 μM conc.). Results are reported as % of cell viability referred to untreated cells (black bar) and represent the mean ± SD of three independent experiments. One-way analysis of variance (ANOVA) followed by multiple comparison tests (Dunnett's test). Levels of significance: *p < 0.05, **p < 0.01 referred to H2O2; ####p < 0.0001 referred to untreated cells (GraphPad Prism). (C) Viability of SH-SY5Y cells incubated for 24 h in DMEM serum-free with 20 μM Aβ42 in the absence or in the co-presence of increasing concentrations of compounds 15 and 16 (from 5 to 25 μM). Reference drug: donepezil (95 ± 6% of cell viability at 5 μM conc.). Results are reported as % of cell viability referred to untreated cells (black bar) and represent the mean ± SD of three independent experiments. One-way analysis of variance (ANOVA) followed by multiple comparison tests (Dunnett's test). Levels of significance: **p < 0.01, ***p < 0.001 referred to Aβ; ###p < 0.001 referred to untreated cells (GraphPad Prism). | ||
Next, the cytoprotective effects of compounds 15 and 16 were assessed in two models of neurotoxicity in SH-SY5Y cells. First, their ability to counteract oxidative stress and cytotoxicity induced by H2O2 was investigated. As shown in Fig. 11B, both compounds exhibited significant neuroprotective effects at concentrations as low as 10 μM, effectively mitigating H2O2-induced apoptosis and oxidative damage, indicative of their antioxidant properties. This antioxidant effect is particularly important in AD, as oxidative stress is a key contributor to neuronal damage and the progression of neurodegeneration, highlighting the therapeutic potential of these compounds.67–69 Based on these findings, the compounds were evaluated for their free radical scavenging capacity. Compounds 15 and 16, together with quercetin as a positive control, were tested in the DPPH assay at 50 μM. No radical scavenging activity was detected for either compound, whereas quercetin showed 66% inhibition, thereby excluding this mechanism as a contributor to their antioxidant effect.
In the second model, the capacity of compounds 15 and 16 to enhance cell viability in SH-SY5Y cells treated with Aβ42 was examined. Aβ42 treatment caused a marked reduction in cell viability; however, co-treatment with either compound 15 or 16 significantly improved cell viability at concentrations as low as 5 μM (Fig. 11C). These findings highlight the potential of compounds 15 and 16 to alleviate neurotoxicity through both antioxidant and anti-aggregation mechanisms. When combined with their COX-2 inhibitory activity, which counteracts neuroinflammation in AD, these properties make them of great interest for AD treatment.
Consistent with the findings from the PAMPA assay, both compounds also exhibited high permeability in the MDCK-MDR1 assay (Table 5). The apparent permeability coefficients (Papp) in the absorptive (apical-to-basolateral, A→B) direction were 9.6 × 10−6 cm s−1 for 15 and 10.2 × 10−6 cm s−1 for compound 16 (Table 5). In the secretory (basolateral-to-apical, B→A) direction, the corresponding Papp values were 11.4 × 10−6 cm s−1 and 12.5 × 10−6 cm s−1, yielding efflux ratios of 1.19 and 1.23, respectively (Table 5). These values indicate that neither compound is a significant substrate of P-gp.72
| Parameter | 15 | 16 |
|---|---|---|
| P app (A→B) | 9.6 × 10−6 cm s−1 | 10.2 × 10−6 cm s−1 |
| P app (B→A) | 11.4 × 10−6 cm s−1 | 12.5 × 10−6 cm s−1 |
| Efflux ratio | 1.19 | 1.23 |
3 Conclusion
In this study, we have introduced a series of novel selective COX-2 inhibitors with multitargeted activities, including potent inhibition of Aβ and tau aggregation and antioxidant effects, making them promising candidates for the treatment of AD. These compounds, inspired by the phenylbutazone framework, feature pyrazolidinone or pyrazolidine-3,5-dione scaffolds with an additional third aryl group. The SAR analysis revealed distinct activity profiles. Pyrazolidinone derivatives showed COX-2 inhibition only when m-chloro or m-fluorobenzyl amide groups were positioned at the C4 and paired with 1,2-bis(4-chlorophenyl) groups. Pyrazolidine-3,5-dione derivatives demonstrated a preference for electron-withdrawing substituents, particularly fluorine and nitro groups, at the ortho and meta positions of the third aryl ring, significantly enhancing potency, whereas para substitution and electron-donating groups diminished activity in most cases. Among the series, compound 15 emerged as the most potent and selective COX-2 inhibitor, effectively attenuating PGE2 release in LPS-stimulated THP-1 cells. Additionally, compounds 15, 16, and 29 possessed multi-targeting capabilities by simultaneously inhibiting Aβ and tau aggregation. Importantly, compounds 15 and 16 demonstrated significant neuroprotective effects, counteracting the neurotoxic effects of Aβ and H2O2 in SH-SY5Y cells, highlighting their potential to combat oxidative stress and protein aggregation-related neurodegeneration. Furthermore, both compounds possess a highly possible BBB permeability, as indicated by PAMPA and MDCK-MDR1 assay results, emphasizing their ability to reach the CNS and exert therapeutic effects. The multitarget profile of these compounds addresses key pathological features of AD, including neuroinflammation, protein aggregation, and oxidative stress, which may enhance the therapeutic potential of COX-2 inhibitors. This approach could overcome the mixed results observed in clinical trials of traditional COX-2 inhibitors for AD by providing a broader mechanism of action and improving efficacy.Our compounds offer a distinct mechanistic strategy by simultaneously targeting multiple key drivers of AD progression. By combining selective COX-2 inhibition to reduce neuroinflammation, dual Aβ and tau aggregation inhibition, antioxidant and neuroprotective properties, and potential BBB permeability, our featured series demonstrates unique potential as multitarget-directed anti-AD agents. These attributes strongly support their further evaluation in in vivo models of Alzheimer's disease.
4 Experimental
4.1 Chemistry
Reagents and solvents were sourced from established commercial providers and utilized as received, without further purification. For reactions requiring an inert environment, argon was employed. Analytical-grade solvents were used throughout the procedures. Purification of reaction products was accomplished using column chromatography on silica gel (40–60 μm). Thin-layer chromatography (TLC) with pre-coated silica gel plates (fluorescent) was employed to monitor the reaction progress, with visualization under short-wave ultraviolet light (λ = 254 nm). Nuclear magnetic resonance (NMR) spectroscopy (1H and 13C) was performed on Bruker Fourier 300 and Varian Mercury 400 Plus spectrometers. Chemical shifts (δ) are reported in ppm, with 1H chemical shifts referenced to the residual signal of the solvent (δ 2.50 for DMSO-d6, δ 7.26 for CDCl3) and 13C shifts aligned to the deuterated solvent signals (δ 39.5 for DMSO-d6, δ 77.0 for CDCl3). Coupling constants (J) are expressed in hertz (Hz), and the multiplicity of peaks is described using standard abbreviations: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), dd (doublet of doublet), ddd (doublet of doublet of doublet), and dt (doublet of triplet). Mass spectrometric analysis (UPLC-ESI-MS) was conducted on a Waters ACQUITY Xevo TQD system, comprising an ACQUITY UPLC H-Class platform coupled with a Xevo™ TQD triple-quadrupole mass spectrometer, utilizing an electrospray ionization (ESI) source. Chromatographic separation was achieved using an Acquity BEH C18 column (100 mm × 2.1 mm, 1.7 μm particle size). The mobile phase was a binary system of water with 0.1% trifluoroacetic acid (TFA) (A) and acetonitrile containing 0.1% TFA (B). The gradient method involved an initial 5% B held for 1 minute, increased to 100% over 10 minutes, maintained at 100% for 2 minutes, returned to 5% within 3 minutes, and held for 1 minute. The flow rate was set at 200 μL min−1. The mass spectrometer settings were as follows: capillary voltage of 3.5 kV, cone voltage of 20 V, radio frequency (RF) lens voltage at 2.5 V, source temperature maintained at 150 °C, and desolvation gas temperature at 500 °C. Nitrogen was used as both the desolvation gas and cone gas, at flow rates of 1000 L h−1 and 20 L h−1, respectively. Data acquisition and instrument control were managed using Mass Lynx 4.1 software (Waters). The purity of all synthesized compounds was confirmed by HPLC-MS, with values exceeding 95%. Melting points were determined using a Buchi B-540 capillary melting point apparatus.Diphenyl-diazene (A1). The compound was synthesized according to procedure A using aniline; orange solid; yield: 85%; mp 69 °C; MS (ESI): m/z = 183.9 (M + H)+; CAS Number: 103-33-3.47
Bis-(4-chloro-phenyl)-diazene (A2). The compound was synthesized according to procedure A using 4-chloroaniline; orange solid; yield: 76.27%; mp 64.5–66 °C; MS (ESI): m/z = 251.11 (M + H)+; CAS number: 1602-00-2.73
1,2-Diphenylhydrazine (B1). This compound was synthesized according to procedure B using diazene (A1); yellow solid; yield: 74%; mp 131–132 °C; MS (ESI): m/z = 185.10 (M + H)+; CAS number: 122-66-7.47
N,N′-Bis-(4-chloro-phenyl)-hydrazine (B2). This compound was synthesized according to procedure B using 4-chlorodiazene (A2); yellow solid; yield: 67.75%; mp 97–99 °C; MS (ESI): m/z = 253.13 (M + H)+; CAS number: 953-14-0.73
1,2-Diphenylpyrazolidin-3-one (C1). This compound was synthesized according to procedure C by reacting hydrazine derivative B1; yellowish white solid; yield: 56%; the product was purified using CC (DCM); mp 110.6–113 °C; 1H NMR (400 MHz, CDCl3) δ 7.82–7.77 (m, 2H), 7.34–7.27 (m, 4H), 7.12–7.01 (m, 2H), 7.01–6.96 (m, 2H), 4.02 (t, J = 7.3 Hz, 2H), 2.73 (t, J = 7.3 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 172.29, 150.18, 138.62, 129.70, 129.33, 124.88, 124.14, 118.92, 118.85, 55.41, 30.17; MS (ESI) m/z = 239.01 (M + H)+.
1,2-Bis(4-chlorophenyl)pyrazolidin-3-one (C2). This compound was synthesized according to procedure C by reacting hydrazine derivative B2; yellowish white solid; yield: 60%; the product was purified using CC (DCM/hexane 2
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1); mp 110.6–113 °C; 1H NMR (500 MHz, CDCl3) δ 7.55–7.49 (m, 2H), 7.30–7.26 (m, 2H), 7.26–7.22 (m, 2H), 6.71–6.65 (m, 2H), 3.85 (t, J = 7.3 Hz, 2H), 2.58 (t, J = 7.3 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ 171.24, 148.16, 136.60, 132.05, 131.69, 128.05, 119.80, 119.53, 117.12, 54.54, 31.13; MS (ESI): m/z 307.05 (M + H)+.
:1); mp 110.6–113 °C; 1H NMR (500 MHz, CDCl3) δ 7.55–7.49 (m, 2H), 7.30–7.26 (m, 2H), 7.26–7.22 (m, 2H), 6.71–6.65 (m, 2H), 3.85 (t, J = 7.3 Hz, 2H), 2.58 (t, J = 7.3 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ 171.24, 148.16, 136.60, 132.05, 131.69, 128.05, 119.80, 119.53, 117.12, 54.54, 31.13; MS (ESI): m/z 307.05 (M + H)+.
3-Oxo-1,2-diphenylpyrazolidine-4-carboxylic acid (D1). This compound was synthesized according to procedure D by carboxylation of pyrazolidinone (C1); light brown solid; yield: 80%; the product was purified using CC (DCM/HCOOH 97
:3); mp 141–143.5 °C; 1H NMR (400 MHz, CDCl3) δ 11.82 (s, 1H), 7.79 (d, J = 8.2 Hz, 2H), 7.31–7.28 (m, 4H), 7.12–7.06 (m, 2H), 6.98 (d, J = 8.0 Hz, 2H), 4.35 (t, J = 11.8 Hz, 1H), 4.19 (dd, J = 11.9, 7.8 Hz, 1H), 3.97 (t, J = 7.8 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 168.63, 168.39, 148.63, 137.08, 129.50, 129.24, 129.12, 125.76, 124.57, 118.95, 118.47, 118.44, 57.09, 46.14; MS (ESI) m/z = 283.51 (M + H)+.
1,2-Bis(4-chlorophenyl)-3-oxopyrazolidine-4-carboxylic acid (D2). This compound was synthesized according to procedure D by carboxylation of pyrazolidinone (C2); brown solid; yield: 75%; the product was purified using CC (DCM/HCOOH 97
:3); mp 158.5–160 °C; 1H NMR (400 MHz, CDCl3) δ 11.95 (s, 1H), 7.75–7.65 (m, 2H), 7.34–7.27 (m, 2H), 7.24 (d, J = 2.2 Hz, 2H), 6.92–6.84 (m, 2H), 4.35 (t, J = 11.3 Hz, 1H), 4.14 (dd, J = 11.9, 7.8 Hz, 1H), 3.93 (t, J = 7.8 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 169.02, 167.56, 147.05, 135.43, 130.99, 130.07, 129.62, 129.27, 120.06, 119.74, 57.07, 46.73; MS (ESI): m/z = 350.96 (M + H)+.
1,2-Bis-(4-chloro-phenyl)-pyrazolidine-3,5-dione (E2). This compound was synthesized according to procedure E using 4-chlorohydrazine (B2); brown solid; yield: 66.42%; mp 194–197 °C; MS (ESI): m/z = 321.16 (M + H)+.
:2); 1H NMR (500 MHz, DMSO-d6) δ 8.13 (t, J = 5.6 Hz, 1H), 7.72–7.69 (m, 2H), 7.40–7.33 (m, 2H), 7.31 (tt, J = 7.8, 2.3 Hz, 2H), 7.16–7.09 (m, 1H), 7.05 (td, J = 7.4, 1.2 Hz, 1H), 7.01–6.93 (m, 2H), 4.26 (t, J = 11.5 Hz, 1H), 4.18–4.11 (m, 1H), 3.87 (dd, J = 11.4, 7.5 Hz, 1H), 3.14–3.03 (m, 2H), 1.37–1.27 (m, 4H), 0.86 (d, J = 7.3 Hz, 3H); 13C NMR (126 MHz, DMSO-d6) δ 169.10, 165.78, 149.69, 137.81, 129.24, 129.09, 124.61, 123.55, 118.08, 118.04, 57.35, 48.03, 38.46, 31.01, 19.38, 13.64; MS (ESI) m/z = 338.21 (M + H)+.
:3); mp 118–120 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.10 (t, J = 5.6 Hz, 1H), 7.68 (d, J = 8.2 Hz, 2H), 7.34 (t, J = 7.9 Hz, 2H), 7.29 (t, J = 7.8 Hz, 2H), 7.11 (t, J = 7.4 Hz, 1H), 7.03 (t, J = 7.3 Hz, 1H), 6.96 (d, J = 8.0 Hz, 2H), 4.23 (t, J = 11.5 Hz, 1H), 4.18–4.10 (m, 1H), 3.85 (dd, J = 11.3, 7.5 Hz, 1H), 3.03 (dp, J = 19.3, 6.5 Hz, 2H), 1.37 (dt, J = 16.3, 8.1 Hz, 2H), 0.83 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 169.52, 166.24, 150.13, 138.24, 129.65, 129.50, 125.03, 123.97, 118.51, 118.49, 57.77, 48.47, 41.02, 22.61, 11.70; MS (ESI) m/z = 324.2 (M + H)+.
:3); mp 122–125 °C; 1H NMR (400 MHz, CDCl3) δ 7.80 (d, J = 8.2 Hz, 2H), 7.74–7.60 (m, 1H), 7.33 (t, J = 7.8 Hz, 2H), 7.26 (d, J = 7.8 Hz, 2H), 7.13 (t, J = 7.4 Hz, 1H), 7.05 (t, J = 7.4 Hz, 1H), 6.97 (d, J = 7.9 Hz, 2H), 5.83 (ddt, J = 16.2, 10.7, 5.5 Hz, 1H), 5.27–5.08 (m, 2H), 4.35–4.17 (m, 2H), 3.91 (t, J = 5.7 Hz, 2H), 3.78 (dd, J = 11.4, 8.0 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 169.49, 165.47, 149.17, 137.61, 133.57, 129.33, 129.03, 125.31, 124.15, 118.83, 118.34, 116.53, 76.70, 57.09, 41.74; MS (ESI) m/z = 322.2 (M + H)+.
:1); mp 175–176 °C; 1H NMR (400 MHz, CDCl3) δ 13.52 (s, 1H), 7.97–7.90 (m, 1H), 7.34 (d, J = 7.2 Hz, 4H), 7.32–7.28 (m, 4H), 7.25–7.15 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 160.32, 159.45, 152.08 (d, J(c–f) = 248.2 Hz), 134.96, 133.97, 132.39 (d, J(c–f) = 33.7 Hz), 129.37, 129.24, 128.95 (d, J(c–f) = 8.8 Hz), 127.51 (d, J(c–f) = 7.4 Hz), 125.47 (d, J(c–f) = 3.7 Hz), 123.47, 123.31, 123.18, 118.99, 117.10, 115.96 (d, J(c–f) = 17.6 Hz); MS (ESI): m/z = 443.10 (M + H)+.
:3); mp 204.7–206 °C; 1H NMR (500 MHz, CDCl3) δ 13.41 (s, 1H), 7.47–7.44 (m, 4H), 7.28–7.27 (m, 4H), 7.11–7.08 (m, 4H); 13C NMR (126 MHz, CDCl3) δ 161.47 (d, J = 248.5 Hz), 160.69, 159.77, 136.64 (d, J = 2.7 Hz), 135.61, 126.45, 122.47, 122.16, 118.31 (d, J = 8.3 Hz), 117.85, 116.86 (d, J = 23.5 Hz); MS (ESI): m/z = 443.13 (M + H)+.
:1); mp 195.6–196.2 °C; 1H NMR (400 MHz, CDCl3) δ 13.70 (s, 1H), 7.99 (dd, J = 8.3, 1.5 Hz, 2H), 7.43 (dd, J = 8.1, 1.4 Hz, 2H), 7.39–7.35 (m, 2H), 7.33 (s, 4H), 7.20 (td, J = 7.7, 1.5 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 160.33, 159.28, 137.09, 132.63, 132.28, 129.86, 129.42, 129.23, 128.42, 127.40, 123.79, 117.08, 115.92; MS (ESI): m/z = 459.11 (M + H)+.
:1); mp 190–190.8 °C; 1H NMR (400 MHz, CDCl3) δ 13.31 (s, 1H), 7.58 (t, J = 2.0 Hz, 2H), 7.36–7.32 (m, 8H), 7.26–7.22 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 160.29, 159.41, 141.49, 136.00, 134.91, 132.30, 130.80, 127.11, 123.50, 123.20, 118.17, 116.64, 114.87; MS (ESI): m/z = 459.11 (M + H)+.
:1); mp 226.8–227.1 °C; 1H NMR (500 MHz, CDCl3) δ 13.33 (s, 1H), 7.40–7.32 (m, 4H), 7.28–7.25 (m, 4H), 7.24–7.23 (m, 4H); 13C NMR (126 MHz, CDCl3) δ 160.41, 159.61, 138.94, 134.96, 133.98, 132.70, 129.97, 129.23, 123.43, 117.79, 117.62; MS (ESI): m/z = 459.11 (M + H)+.
:1); mp 210.5–211 °C; 1H NMR (400 MHz, CDCl3) δ 13.56 (s, 1H), 7.90 (dd, J = 8.3, 1.6 Hz, 2H), 7.51 (dd, J = 8.1, 1.3 Hz, 2H), 7.36–7.30 (m, 2H), 7.24 (s, 6H), 7.05 (ddd, J = 8.0, 7.4, 1.6 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 160.35, 159.19, 138.24, 134.94, 133.98, 133.02, 132.62, 132.27, 129.43, 129.27, 129.02, 127.78, 123.52, 123.32, 119.05, 117.43; MS (ESI): m/z = 502.16 (M + H)+.
:1); 1H NMR (400 MHz, CDCl3) δ 13.30 (s, 1H), 7.73 (t, J = 2.0 Hz, 1H), 7.32–7.27 (m, 9H), 6.77 (dd, J = 4.7, 2.9 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 160.49, 159.36, 141.50, 134.72, 130.73, 130.14, 129.43, 129.33, 123.79, 122.76, 119.54, 118.81, 115.36; MS (ESI): m/z = 502.16 (M + H)+.
:1); mp 215–215.7 °C; 1H NMR (400 MHz, CDCl3) δ 13.32 (s, 1H), 7.50 (d, 4H), 7.34 (d, 4H), 7.28 (d, J = 4.5 Hz, 4H); 13C NMR (101 MHz, CDCl3) δ 160.44, 159.61, 139.44, 134.96, 133.98, 132.37, 129.39, 123.48, 120.50, 118.09, 117.76; MS (ESI): m/z = 502.16 (M + H)+.
:5); mp 217.5–218.1 °C; 1H NMR (500 MHz, CDCl3) δ 13.29 (s, 1H), 7.64–7.50 (m, 4H), 7.29–7.26 (m, 2H), 7.25–7.21 (m, 6H); 13C NMR (126 MHz, CDCl3) δ 160.06, 159.14, 142.97, 134.71, 133.70, 132.77, 132.42, 129.37 (d, J(c–f) = 15.9 Hz), 128.80, 128.54, 127.12 (d, J(c–f) = 3.9 Hz), 127.07, 124.77, 123.52, 123.22, 122.61, 119.00, 116.52, 115.46; MS (ESI): m/z = 493.16 (M + H)+.
:1); mp 198.5–199 °C; 1H NMR (400 MHz, CDCl3) δ 13.35 (s, 1H), 8.31 (d, 2H), 7.61 (d, 2H), 7.37–7.33 (m, 3H), 7.33–7.27 (m, 6H); 13C NMR (101 MHz, CDCl3) δ 161.72, 159.69, 158.59, 145.52, 145.16, 134.38, 133.36, 133.09, 132.78, 129.53, 129.41, 126.18, 125.82, 123.69, 123.38, 120.61, 116.48, 115.64; MS (ESI): m/z = 470.13 (M + H)+.
:1); mp 256.1–257.5 °C; 1H NMR (500 MHz, CDCl3) δ 13.68 (s, 1H), 7.95 (dd, J = 8.5, 0.9 Hz, 2H), 7.61–7.55 (m, 4H), 7.24–7.24 (m, 3H), 7.23–7.22 (m, 3H); 13C NMR (126 MHz, CDCl3) δ 159.63, 158.59, 143.01, 134.61, 133.50, 132.63, 132.52, 129.39, 126.24, 123.54, 120.87, 116.15, 114.84, 100.55; MS (ESI): m/z = 450.14 (M + H)+.
:1); mp 222–222.7 °C; 1H NMR (500 MHz, CDCl3) δ 13.24 (s, 1H), 7.58 (dd, 4H), 7.29–7.26 (m, 2H), 7.26–7.20 (m, 6H); 13C NMR (126 MHz, CDCl3) δ 159.69, 158.77, 143.61, 134.51, 133.93, 133.47, 132.92, 129.46, 123.26, 118.15, 116.74, 109.82; MS (ESI): m/z = 450.14 (M + H)+.
:1); mp 267.3–268.9 °C; 1H NMR (400 MHz, CDCl3) δ 13.24 (s, 1H), 7.86 (dd, J = 7.3, 1.7 Hz, 1H), 7.66 (dd, J = 7.8, 1.5 Hz, 5H), 7.43 (dtd, J = 25.6, 7.5, 1.6 Hz, 2H), 7.34–7.29 (m, 4H), 6.18 (s, 2H); MS (ESI): m/z = 504.12 (M + H)+.
:1); 1H NMR (400 MHz, CDCl3) δ 13.43 (s, 1H), 7.40 (d, J = 2.3 Hz, 2H), 7.33 (s, 6H), 7.30–7.26 (m, 4H), 2.40 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 160.87, 159.95, 140.28, 140.21, 135.21, 134.27, 129.34, 129.21, 128.37, 123.42, 123.14, 117.12, 114.16, 21.39; MS (ESI): m/z = 439.16 (M + H)+.
:1); 1H NMR (400 MHz, CDCl3) δ 13.45 (d, J = 34.3 Hz, 1H), 7.50–7.40 (m, 4H), 7.33 (q, J = 2.1 Hz, 4H), 7.31 (s, 4H), 2.38 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 160.96, 160.13, 138.08, 135.28, 134.35, 131.98, 130.41, 129.39, 123.47, 117.82, 116.30, 21.12; MS (ESI): m/z = 439.16 (M + H)+.
:3); 1H NMR (400 MHz, CDCl3) δ 13.33 (s, 1H), 7.28 (s, 1H), 7.15 (d, J = 5.2 Hz, 8H), 7.12–7.06 (m, 3H), 2.50 (q, J = 7.6 Hz, 2H), 1.08 (t, J = 7.5 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 160.98, 160.13, 144.17, 138.23, 135.28, 131.98, 129.32, 129.19, 123.41, 116.85, 115.11, 28.49, 15.38; MS (ESI): m/z = 453.11 (M + H)+.
:1); mp 172.9–173.8 °C; 1H NMR (500 MHz, CDCl3) δ 13.41 (s, 1H), 7.36–7.33 (m, 2H), 7.23 (dd, J = 6.6, 1.0 Hz, 8H), 7.17–7.14 (m, 2H), 2.52 (dd, J = 8.5, 6.8 Hz, 2H), 1.61–1.52 (m, 2H), 0.86 (t, J = 7.3 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 160.93, 160.09, 142.63, 138.22, 135.25, 134.32, 132.30, 131.93, 129.80, 129.30, 129.16, 123.36, 123.08, 116.74, 116.27, 37.56, 24.39; MS (ESI): m/z = 467.19 (M + H)+.
:1); 1H NMR (400 MHz, CDCl3) 13.16 (s, 1H), δ 7.69–7.66 (m, 4H), 7.39 (d, J = 7.6 Hz, 2H), 7.32–7.27 (m, 6H), 1.35 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 153.13, 151.20, 143.43, 135.01, 134.18, 132.12, 129.34, 126.75, 126.39, 123.53, 123.22, 116.60, 114.72, 34.04, 31.51; MS (ESI): m/z = 481.11 (M + H)+.
:1); 1H NMR (400 MHz, CDCl3) δ 13.59 (s, 1H), 7.51–7.45 (m, 2H), 7.32 (d, J = 4.8 Hz, 7H), 7.00–6.93 (m, 2H), 3.84 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 161.14, 160.39, 159.27, 135.39, 134.47, 133.90, 131.90, 129.31, 123.37, 118.34, 115.13, 55.65; MS (ESI): m/z = 455.16 (M + H)+.
4.2 Biology
080, Cayman Chemical, MI, USA). COX/heme/reaction buffer was incubated with DMSO (0.1%, as the control), test compounds, celecoxib, or ibuprofen at 37 °C for 10 min, and then arachidonic acid (substrate) was added and incubated for another 2 min. Next, the saturated stannous chloride solution (6.82 mg ml−1) was added for 15 min at 37 °C (COX-1) or 5 min at 25 °C (COX-2). Afterwards, the sample (1:2000 dilution of the original sample) was transferred to the plate, and then the prostaglandin screening AChE tracer and antiserum was added and incubated for 18 h at 4 °C (COX-1) or 25 °C (COX-2). Then, Ellman's reagent was added and incubated at 25 °C for 1 h. COX activity was assayed at 405 nm on an ELISA reader (Thermo Multiskan Go).
In vitro aggregation assays were performed in triplicate with a spectrofluorimetric assay based on thioflavin T (ThT) fluorescence, using black low-binding, flat-bottomed polystyrene 96-well microtiter plates (Greiner Bio-One GmbH, Frickenhausen, Germany). Incubations and readouts were performed with an Infinite M1000 Pro plate reader (Tecan, Cernusco s.N., Italy) at 440/485 nm of excitation/emission wavelengths. Inhibition data expressed as mean ± SEM were calculated with Prism (version 5.01 for Windows, GraphPad Software, San Diego, CA, USA).
Inhibition of Aβ40 aggregation was run as previously described,74 by incubating at 25 °C peptide (30 μM) and compounds (10 μM) in PBS containing 2% HFIP for 2 h. After the addition of ThT (25 μM) the fluorescence was read and referred to as % of inhibition to a sample of free aggregating peptide.
For tau peptides, PHF6 (50 μM) or R3 (25 μM) peptides were incubated with compounds (10 μM) and ThT (10 μM) in PBS following previously described procedures.66,75 Incubations were conducted at 30 °C for 3 h and 37 °C for 4 h for PHF6 and R3, respectively. ThT fluorescence was read continuously until the plateau of emission was reached.
To assess the antioxidant activity in vitro, DPPH spectrophotometric assay was performed as described in ref. 77. Scavenging of the radical 2,2-diphenyl-1-picrylhydrazyl (DPPH) was determined in clear polystyrene flat-bottomed 96-well microplates with a Tecan Infinite M1000 plate reader (commercial sources: Sigma (Milan, Italy)), by measuring the loss of absorbance at 515 nm of ethanol solutions of DPPH (10 uM) and test compounds (50 uM). The radical scavenging activities were determined for test compounds and reference quercetin as mean of 3 experiments.
The potential neuroprotective effect of the compounds, at the same concentrations reported above, was evaluated with the specific cytotoxic insult of Aβ42 20 μM in DMEM-serum free for 24 h, at 37 °C, 5% CO2. After the treatment, the medium was removed and neuroprotective effect exerted by compounds was measured as cell viability through the MTT assay. Controls were represented by cells treated with Aβ alone and by untreated cells. The results were expressed as the percentage of surviving cells compared to untreated cells as the mean ± SD of three independent experiments. One-way analysis of variance (ANOVA) followed by multiple comparison tests (Dunnett's test) was used.
:3) was added to the acceptor microplates. The filter membrane was filled with 10 μL of porcine brain lipid (PBL) in dodecane (20 mg mL−1). Stock solutions of the tested compounds were diluted with PBS/EtOH (7:3) until 100 μg mL−1 concentration. The donor wells were then filled with 200 μL of tested compound solution and 300 μL of PBS/EtOH (7:3). The donor filter plate was placed on the acceptor plate and incubated for 16 h at 25 °C. After removal of the donor plate, the concentrations of tested compounds were detected in the acceptor, donor and reference wells using a plate reader. Samples were analyzed using three independent runs in three wells.78
000 cells per well on polycarbonate Transwell inserts (Corning, 12-well format) and cultured for 4 days. Monolayer integrity was confirmed by transepithelial electrical resistance (TEER) measurements before proceeding. On the day of the assay, cells were washed and preincubated in HBSS transport buffer (pH 7.4) for 30 minutes at 37 °C. Compounds were added at a final concentration of 10 μM with 0.5% DMSO to either the apical (A) or basolateral (B) chamber to initiate A→B or B→A transport, respectively. Samples (100 μL) were collected from the receiver compartment at 0, 30, 60, and 90 minutes and replaced with fresh buffer. The assay was conducted in triplicate. Compound concentrations in donor and receiver compartments were quantified by LC–MS/MS.
Abbreviations used
| Aβ | Amyloid-beta |
| AD | Alzheimer's disease |
| APP | Amyloid precursor protein |
| COX | Cyclooxygenase; |
| HBTU | Hexafluorophosphate benzotriazole tetramethyl uronium |
| IC50 | Half maximal inhibitory concentration |
| IL | Interleukin |
| LPS | Lipopolysaccharide |
| MTDLs | Multitarget-directed ligands |
| NFTs | Neurofibrillary tangles |
| NSAID | Non-steroidal anti-inflammatory drug |
| PAMPA-BBB | Parallel artificial membrane permeability assay for the blood–brain barrier |
| PMA | Phorbol 12-myristate 13-acetate |
| ROS | Reactive oxygen species |
| PGE2 | Prostaglandin E2 |
| PHF6 | β-sheet rich paired helical filaments |
| PGF2α | Prostaglandin F2α |
| PGI2 | Prostaglandin I2 |
| SI | Selectivity index |
| TEA | Triethylamine |
| TXA2 | Thromboxane A2 |
Author contributions
Michael Emad, investigation, methodology, writing – original draft, writing – review & editing; Reham Waheed, investigation, methodology, writing – original draft, writing – review & editing; Zeinab Mostafa, investigation, methodology, writing – original draft, writing – review & editing; Sarah S. Darwish, investigation, methodology, writing – original draft, writing – review & editing; Rosa Purgatorio, investigation, methodology; Daniela Valeria Miniero, investigation, methodology; Annalisa De Palma, investigation, methodology; Tzu-Peng Cheng, investigation, methodology; Moustafa T. Gabr, investigation, methodology; Ahmed M. El Kerdawy, investigation, methodology; Marco Catto, investigation, methodology, resources, supervision, writing – review & editing, project administration; Ashraf H. Abadi, resources, supervision; Tsong-Long Hwang, conceptualization, resources, supervision, funding acquisition, writing – original draft, writing – review & editing, project administration; Mohammad Abdel-Halim, conceptualization, investigation, methodology, resources, supervision, writing – original draft, writing – review & editing, project administration.Conflicts of interest
The authors declare no competing financial interest.Data availability
The data that support the findings of this study are available from the corresponding authors (MA-H and T-L H), upon reasonable request.Supplementary information: Fig. S1: IR charts of compounds 24 and 31; Fig. S2: (A) first series SAR summary, (B) second series SAR summary, 1H NMR and 13C NMR spectra of representative compounds; Fig. S3–23: molecular docking simulation and molecular dynamics simulation (experimental part); Fig. S24 and 25: cell viability as assessed with PrestoBlue of NHA (A), hBMECs (B), and HepG2 (C) for compounds 15 and 16. See DOI: https://doi.org/10.1039/d5md00802f.
Acknowledgements
This study was supported by the National Science and Technology Council (111-2320-B-255-006-MY3, NSTC 113-2321-B-255-001, and 113-2321-B-182-003), Chang Gung University of Science and Technology (ZRRPF3P0091 and ZRRPF3M0091), and Chang Gung Memorial Hospital (CORPF3Q0011-3, CORPF1P0051-3, CORPF1P0071-3, and BMRP450), Taiwan. The funding sources had no role in this study.References
- J. A. Mitchell and N. S. Kirkby, Br. J. Pharmacol., 2019, 176, 1038–1050 CrossRef CAS PubMed.
- C. A. Rouzer and L. J. Marnett, J. Lipid Res., 2009, 50, S29–34 CrossRef PubMed.
- E. Ricciotti and G. A. FitzGerald, Arterioscler., Thromb., Vasc. Biol., 2011, 31, 986–1000 CrossRef CAS PubMed.
- N. Zidar, K. Odar, D. Glavac, M. Jerse, T. Zupanc and D. Stajer, J. Cell. Mol. Med., 2009, 13, 3753–3763 CrossRef PubMed.
- C. S. Williams, M. Mann and R. N. DuBois, Oncogene, 1999, 18, 7908–7916 CrossRef CAS PubMed.
- I. Morita, Prostaglandins Other Lipid Mediators, 2002, 68-69, 165–175 CrossRef CAS PubMed.
- J. M. Woods, A. Mogollon, M. A. Amin, R. J. Martinez and A. E. Koch, Exp. Mol. Pathol., 2003, 74, 282–290 CrossRef CAS PubMed.
- N. Hashemi Goradel, M. Najafi, E. Salehi, B. Farhood and K. Mortezaee, J. Cell. Physiol., 2019, 234, 5683–5699 CrossRef CAS PubMed.
- B. Liu, L. Qu and S. Yan, Cancer Cell Int., 2015, 15, 106 CrossRef PubMed.
- W. Chen, Y. Zhong, N. Feng, Z. Guo, S. Wang and D. Xing, Mol Med, 2021, 27, 123 CAS.
- L. Chen, H. Deng, H. Cui, J. Fang, Z. Zuo, J. Deng, Y. Li, X. Wang and L. Zhao, Oncotarget, 2018, 9, 7204–7218 CrossRef PubMed.
- N. Moussa and N. Dayoub, Saudi Pharm. J., 2023, 31, 101729 CrossRef CAS PubMed.
- M. P. Murphy and H. LeVine, 3rd, J. Alzheimer's Dis., 2010, 19, 311–323 Search PubMed.
- G. F. Chen, T. H. Xu, Y. Yan, Y. R. Zhou, Y. Jiang, K. Melcher and H. E. Xu, Acta Pharmacol. Sin., 2017, 38, 1205–1235 CrossRef CAS PubMed.
- P. P. Guan and P. Wang, FASEB J., 2019, 33, 13–33 CrossRef CAS PubMed.
- Z. Xiang, L. Ho, S. Yemul, Z. Zhao, W. Qing, P. Pompl, K. Kelley, A. Dang, W. Qing, D. Teplow and G. M. Pasinetti, Gene Expression, 2002, 10, 271–278 CrossRef CAS PubMed.
- P. P. Guan, X. Yu, Y. H. Zou and P. Wang, Cell. Mol. Immunol., 2019, 16, 892–894 CrossRef CAS PubMed.
- N. V. Gorantla, V. G. Landge, P. G. Nagaraju, P. Priyadarshini Cg, E. Balaraman and S. Chinnathambi, ACS Omega, 2019, 4, 16702–16714 CrossRef CAS PubMed.
- W. Noble, D. P. Hanger, C. C. Miller and S. Lovestone, Front. Neurol., 2013, 4, 83 CAS.
- Z. F. Brotzakis, P. R. Lindstedt, R. J. Taylor, D. J. Rinauro, N. C. T. Gallagher, G. J. L. Bernardes and M. Vendruscolo, ACS Cent. Sci., 2021, 7, 1986–1995 CrossRef CAS PubMed.
- Y. Wang, P. P. Guan, X. Yu, Y. S. Guo, Y. J. Zhang, Z. Y. Wang and P. Wang, Oncotarget, 2017, 8, 99296–99311 CrossRef PubMed.
- S. Ghosh, M. D. Wu, S. S. Shaftel, S. Kyrkanides, F. M. LaFerla, J. A. Olschowka and M. K. O'Banion, J. Neurosci., 2013, 33, 5053–5064 CrossRef CAS PubMed.
- D. E. López and S. J. Ballaz, Mol. Neurobiol., 2020, 57, 5167–5176 CrossRef PubMed.
- Z. Liu, T. Li, P. Li, N. Wei, Z. Zhao, H. Liang, X. Ji, W. Chen, M. Xue and J. Wei, Oxid. Med. Cell. Longevity, 2015, 2015, 352723 Search PubMed.
- S. N. Rai, Recent Advances in the Treatment of Neurodegenerative Disorders, Bentham Science Publishers, 2021 Search PubMed.
- H. Y. Jung, D. Y. Yoo, S. M. Nam, J. W. Kim, W. Kim, H. J. Kwon, K. Y. Lee, J. H. Choi, D. W. Kim, Y. S. Yoon, J. K. Seong and I. K. Hwang, Mol. Med. Rep., 2019, 19, 1996–2004 CAS.
- S. N. R. Sangeeta Singh and S. Kumar Singh, Synaptic Plasticity in Neurodegenerative Disorders, CRC Press, Boca Raton, 1st edn, 2024 Search PubMed.
- B. K. Martin, C. Szekely, J. Brandt, S. Piantadosi, J. C. Breitner, S. Craft, D. Evans, R. Green and M. Mullan, Arch. Neurol., 2008, 65, 896–905 CrossRef PubMed.
- G. W. Small, P. Siddarth, D. H. Silverman, L. M. Ercoli, K. J. Miller, H. Lavretsky, S. Y. Bookheimer, S. C. Huang, J. R. Barrio and M. E. Phelps, Am. J. Geriatr. Psychiatry, 2008, 16, 999–1009 CrossRef PubMed.
- P. S. Aisen, J. Schmeidler and G. M. Pasinetti, Neurology, 2002, 58, 1050–1054 CrossRef CAS PubMed.
- S. C. Vlad, D. R. Miller, N. W. Kowall and D. T. Felson, Neurology, 2008, 70, 1672–1677 CrossRef CAS PubMed.
- J. C. Breitner, L. D. Baker, T. J. Montine, C. L. Meinert, C. G. Lyketsos, K. H. Ashe, J. Brandt, S. Craft, D. E. Evans, R. C. Green, M. S. Ismail, B. K. Martin, M. J. Mullan, M. Sabbagh and P. N. Tariot, Alzheimer's Dementia, 2011, 7, 402–411 CrossRef PubMed.
- J. Rogers, L. C. Kirby, S. R. Hempelman, D. L. Berry, P. L. McGeer, A. W. Kaszniak, J. Zalinski, M. Cofield, L. Mansukhani and P. Willson, et al. , Neurology, 1993, 43, 1609–1611 CrossRef CAS PubMed.
- S. Scharf, A. Mander, A. Ugoni, F. Vajda and N. Christophidis, Neurology, 1999, 53, 197–201 CrossRef CAS PubMed.
- B. P. Imbimbo, V. Solfrizzi and F. Panza, Front. Aging Neurosci., 2010, 2, 19 CAS.
- Z. A. Marcum and J. T. Hanlon, Ann. Long-Term Care, 2010, 18, 24–27 Search PubMed.
- E. Rahme and H. Nedjar, Rheumatology, 2007, 46, 435–438 CrossRef CAS PubMed.
- H. Soininen, C. West, J. Robbins and L. Niculescu, Dementia Geriatr. Cognit. Disord., 2007, 23, 8–21 CrossRef CAS PubMed.
- P. S. Aisen, K. L. Davis, J. D. Berg, K. Schafer, K. Campbell, R. G. Thomas, M. F. Weiner, M. R. Farlow, M. Sano, M. Grundman and L. J. Thal, Neurology, 2000, 54, 588–593 CrossRef CAS PubMed.
- P. S. Aisen, K. A. Schafer, M. Grundman, E. Pfeiffer, M. Sano, K. L. Davis, M. R. Farlow, S. Jin, R. G. Thomas and L. J. Thal, JAMA, 2003, 289, 2819–2826 CrossRef CAS PubMed.
- C. R. Jack, Jr., D. A. Bennett, K. Blennow, M. C. Carrillo, B. Dunn, S. B. Haeberlein, D. M. Holtzman, W. Jagust, F. Jessen, J. Karlawish, E. Liu, J. L. Molinuevo, T. Montine, C. Phelps, K. P. Rankin, C. C. Rowe, P. Scheltens, E. Siemers, H. M. Snyder and R. Sperling, Alzheimer's Dementia, 2018, 14, 535–562 CrossRef PubMed.
- S. N. R. Sangeeta Singh and S. Kumar Singh, Neurodegenerative Diseases Translational Models, Mechanisms, and Therapeutics, CRC Press, Boca Raton, 1st edn, 2024 Search PubMed.
- H. Zheng, M. Fridkin and M. Youdim, Pharmaceuticals, 2014, 7, 113–135 CrossRef PubMed.
- M. Worboys and E. Toon, Hist. Philos. Life Sci., 2018, 40, 27 CrossRef PubMed.
- Y. H. Wang, L. L. Zhu, T. L. Li and Q. Zhou, Drug Des., Dev. Ther., 2024, 18, 1711–1725 CrossRef CAS PubMed.
- Y. H. Hyup, J. K. Kwan, S. H. Kang, M. S. Noh, J. Y. Ha, J. K. Kyu, K. M. Min, C. H. Hoon and S. Chung, Bioorg. Med. Chem. Lett., 2003, 13, 413–417 CrossRef PubMed.
- S. A. Mokbel, R. K. Fathalla, L. Y. El-Sharkawy, A. H. Abadi, M. Engel and M. Abdel-Halim, Bioorg. Chem., 2020, 99, 103759 CrossRef CAS PubMed.
- R. A. Wagdy, N. S. Abutaleb, R. K. Fathalla, Y. Elgammal, S. Weck, R. Pal, P. D. Fischer, C. Ducho, A. H. Abadi, M. N. Seleem, M. Engel and M. Abdel-Halim, Eur. J. Med. Chem., 2023, 261, 115789 CrossRef CAS PubMed.
- V. G. Yakutovich, B. L. Moldaver, Y. P. Kitaev and Z. S. Titova, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1968, 17, 838–843 CrossRef.
- E. M. Gedawy, A. E. Kassab and A. M. El Kerdawy, Eur. J. Med. Chem., 2020, 189, 112066 CrossRef CAS PubMed.
- P. A. Halim, H. H. Georgey, M. Y. George, A. M. El Kerdawy and M. F. Said, Bioorg. Chem., 2021, 115, 105253 CrossRef CAS PubMed.
- G. Cingolani, A. Panella, M. G. Perrone, P. Vitale, G. Di Mauro, C. G. Fortuna, R. S. Armen, S. Ferorelli, W. L. Smith and A. Scilimati, Eur. J. Med. Chem., 2017, 138, 661–668 CrossRef CAS PubMed.
- J. L. Wang, D. Limburg, M. J. Graneto, J. Springer, J. R. Hamper, S. Liao, J. L. Pawlitz, R. G. Kurumbail, T. Maziasz, J. J. Talley, J. R. Kiefer and J. Carter, Bioorg. Med. Chem. Lett., 2010, 20, 7159–7163 CrossRef CAS PubMed.
- W. L. Smith, D. L. DeWitt and R. M. Garavito, Annu. Rev. Biochem., 2000, 69, 145–182 CrossRef CAS PubMed.
- L. J. Marnett and A. S. Kalgutkar, Curr. Opin. Chem. Biol., 1998, 2, 482–490 CrossRef CAS PubMed.
- J. R. Kiefer, J. L. Pawlitz, K. T. Moreland, R. A. Stegeman, W. F. Hood, J. K. Gierse, A. M. Stevens, D. C. Goodwin, S. W. Rowlinson, L. J. Marnett, W. C. Stallings and R. G. Kurumbail, Nature, 2000, 405, 97–101 CrossRef CAS PubMed.
- P. Gund and T. Y. Shen, J. Med. Chem., 1977, 20, 1146–1152 CrossRef CAS PubMed.
- C. Luong, A. Miller, J. Barnett, J. Chow, C. Ramesha and M. F. Browner, Nat. Struct. Biol., 1996, 3, 927–933 CrossRef CAS PubMed.
- D. E. Duggan, K. F. Hooke, E. A. Risley, T. Y. Shen and C. G. Arman, J. Pharmacol. Exp. Ther., 1977, 201, 8–13 CrossRef CAS PubMed.
- R. G. Kurumbail, A. M. Stevens, J. K. Gierse, J. J. McDonald, R. A. Stegeman, J. Y. Pak, D. Gildehaus, J. M. Miyashiro, T. D. Penning, K. Seibert, P. C. Isakson and W. C. Stallings, Nature, 1996, 384, 644–648 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1-2, 19–25 CrossRef.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- T. Wang, L. Qin, B. Liu, Y. Liu, B. Wilson, T. E. Eling, R. Langenbach, S. Taniura and J. S. Hong, J. Neurochem., 2004, 88, 939–947 CrossRef CAS PubMed.
- R. M. Nisbet, J. C. Polanco, L. M. Ittner and J. Götz, Acta Neuropathol., 2015, 129, 207–220 CrossRef CAS PubMed.
- I. Aillaud and S. A. Funke, Cell. Mol. Neurobiol., 2023, 43, 951–961 CrossRef CAS PubMed.
- O. M. Al-Saad, M. Gabr, S. S. Darwish, M. Rullo, L. Pisani, D. V. Miniero, G. M. Liuzzi, A. M. Kany, A. K. H. Hirsch, A. H. Abadi, M. Engel, M. Catto and M. Abdel-Halim, Eur. J. Med. Chem., 2024, 269, 116266 CrossRef CAS PubMed.
- M. Hatami, M. Mortazavi, Z. Baseri, B. Khani, M. Rahimi and S. Babaei, J. Evidence-Based Complementary Altern. Med., 2023, 2023, 8056462 CrossRef PubMed.
- U. C. Dash, N. K. Bhol, S. K. Swain, R. R. Samal, P. K. Nayak, V. Raina, S. K. Panda, R. G. Kerry, A. K. Duttaroy and A. B. Jena, Acta Pharm. Sin. B, 2025, 15, 15–34 CrossRef CAS PubMed.
- A. Singh, R. Kukreti, L. Saso and S. Kukreti, Molecules, 2019, 24, 1583 CrossRef CAS PubMed.
- L. Di, E. H. Kerns, K. Fan, O. J. McConnell and G. T. Carter, Eur. J. Med. Chem., 2003, 38, 223–232 CrossRef CAS PubMed.
- M. Kansy, F. Senner and K. Gubernator, J. Med. Chem., 1998, 41, 1007–1010 CrossRef CAS PubMed.
- E. Dolghih and M. P. Jacobson, ACS Chem. Neurosci., 2013, 4, 361–367 CrossRef CAS PubMed.
- K. M. Kutterer, J. M. Davis, G. Singh, Y. Yang, W. Hu, A. Severin, B. A. Rasmussen, G. Krishnamurthy, A. Failli and A. H. Katz, Bioorg. Med. Chem. Lett., 2005, 15, 2527–2531 CrossRef CAS PubMed.
- I. Bolognino, N. Giangregorio, L. Pisani, M. de Candia, R. Purgatorio, A. Tonazzi, C. D. Altomare, S. Cellamare and M. Catto, Molecules, 2019, 24, 4298 CrossRef CAS PubMed.
- M. Campora, C. Canale, E. Gatta, B. Tasso, E. Laurini, A. Relini, S. Pricl, M. Catto and M. Tonelli, ACS Chem. Neurosci., 2021, 12, 447–461 CrossRef CAS PubMed.
- M. de Candia, G. Zaetta, N. Denora, D. Tricarico, M. Majellaro, S. Cellamare and C. D. Altomare, Eur. J. Med. Chem., 2017, 125, 288–298 CrossRef CAS PubMed.
- M. Catto, F. Arnesano, G. Palazzo, A. De Stradis, V. Calo, M. Losacco, R. Purgatorio and F. Campagna, Arch. Biochem. Biophys., 2014, 560, 73–82 CrossRef CAS PubMed.
- Y. T. AlNajjar, M. Gabr, A. K. ElHady, M. Salah, G. Wilms, A. H. Abadi, W. Becker, M. Abdel-Halim and M. Engel, Eur. J. Med. Chem., 2022, 227, 113911 CrossRef CAS PubMed.
Footnote |
| † Authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2026 |