
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of p-aminobenzylamine derivatives as dual non-covalent inhibitors of the transmembrane host proteases TMPRSS2 and HAT proteases with anti-viral potential
Hélène
Carvaillo†
a,
Ashok
Dussol†
b,
Nancy
Chaaya
b,
Sara
Kadri
b,
Feryel
Soualmia
 b,
Nicolas
Masurier
*a and
Chahrazade
El Amri
*b
b,
Nicolas
Masurier
*a and
Chahrazade
El Amri
*b
aInstitut des Biomolécules Max Mousseron, Université de Montpellier, CNRS, ENSCM, F-34093 Montpellier, France. E-mail: nicolas.masurier@umontpellier.fr
bSorbonne Université, Faculty of Sciences and Engineering, IBPS, UMR 8263 CNRS-SU, INSERM U1345, Development Adaptation and Ageing, F-75252 Paris, France. E-mail: chahrazade.el_amri@sorbonne-universite.fr
First published on 28th October 2025
Abstract
TMPRSS2 and HAT (or TMPRSS11D) are host serine proteases critically involved in the entry of several respiratory viruses, including SARS-CoV-2. To our knowledge, no dual inhibitors targeting both enzymes have been reported to date. Here, we describe a series of para-aminobenzylamine derivatives acting as potent dual TMPRSS2/HAT non-covalent inhibitors. In SARS-CoV-2 infection assays in lung epithelial cells, four compounds demonstrated significant antiviral activity without cytotoxicity at tested doses. Drug-likeness profiling confirmed compliance with Lipinski's and Veber's rules, as well as favourable solubility and microsomal stability. These findings highlight a novel chemical series with potential as broad-spectrum antivirals targeting host proteases.
1. Introduction
Since its discovery and spread around the world, the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has placed a significant burden on global public health. To combat the resulting COVID-19 pandemic, vaccines, antibodies, and antiviral medications have been developed and several of them have been authorized for clinical use.1 However, SARS-CoV-2 variants, particularly Omicron subvariants, have shown an ability to evade immune responses from both antibody-mediated neutralization and vaccine protection.2–4 In addition, antiviral agents targeting either the RNA-dependent RNA polymerase (RdRp), such as remdesivir,5 molnupiravir,6 azvudine,7 or the main viral protease (Mpro), such as nirmatrelvir8 and ensitrelvir,9 have been also authorized for COVID-19 treatment. Nonetheless, studies have demonstrated that mutations in SARS-CoV-2 RdRp can lead to resistance against remdesivir,10 and Mpro mutations can cause resistance to nirmatrelvir,11 indicating that drug resistance is an imminent challenge.Unlike viral proteins, which mutate frequently during SARS-CoV-2 evolution, host proteins are more conserved and could serve as therapeutic targets for broad-spectrum antiviral compounds.12 Consequently, there is growing interest in host-targeted therapies that inhibit viral replication by disrupting host cell processes essential for viral entry and propagation. These proteases play a crucial role in the activation of viral glycoproteins, facilitating viral entry into host cells.13 By cleaving viral envelope proteins, these enzymes enable the fusion of viral and cellular membranes, a critical step in the viral life cycle. Thus, regarding respiratory viruses, one promising approach to fight them involves targeting host cell proteases expressed in the human airway.14 Among these proteases, transmembrane protease serine 2 (TMPRSS2) is particularly important for respiratory viruses, including influenza and coronaviruses like SARS-CoV-2.15 TMPRSS2 belongs to the type II membrane-anchored serine proteases (TTSPs) and was first identified in prostate cancer, where its expression is increased in the prostate epithelium.16 Its involvement in influenza A and B virus infection was subsequently demonstrated.17 It is implicated in the cleavage of haemagglutinin (HA), enabling membrane fusion of the influenza virus envelope and the endosome and finally the entry of the virus in the host cell. It has been demonstrated that inhibition of TMPRSS2 suppresses influenza virus replication in cell cultures, confirming its key role in influenza virus infection.14,18 In addition, the Spike protein of SARS-CoV-2 and of Middle East respiratory syndrome coronavirus (MERS-CoV) is also mainly cleaved by TMPRSS215 and TMPRSS2-KO mice infected with these two viruses presented a reduced viral replication.19
It has also been demonstrated that broad spectrum serine protease inhibitors, like camostat or nafamostat, were able to reduce viral replication of SARS-CoV-2 or MERS-CoV in vitro and in vivo.20–22 Further works showed that these inhibitors act as covalent inhibitors, after the nucleophilic attack of their ester function by the catalytic serine of several host proteases, including TMPRSS2.23–25 The structures of these compounds are shown in Fig. 1. However, these compounds did not hold promise in COVID-19 clinical trials, probably due to their short plasma half-lives.26,27
 | ||
| Fig. 1 Structure of some previously reported inhibitors of host cell proteases expressed in the human airway. | ||
Using a repositioning strategy, several other covalent inhibitors have also been evaluated against this protease.28 Substrate-based ketobenzothiazole inhibitors of TMPRSS2 (such as compounds MM3122 or N0385, Fig. 1) have been also developed and showed strong inhibition of SARS-CoV-2 entry and replication in vivo.29 However, these pseudopeptide compounds are not orally available and are moderately selective. Indeed, these compounds are able to inhibit, in the nanomolar or micromolar range, several other proteases, including Hepatocyte Growth Factor Activator (HGFA), hepsin and factor Xa, which could lead to side effects. In addition to TMPRSS2, many other proteases such as furin, cathepsins (especially cathepsin L), TMPRSS11D (also named HAT for Human Airway Trypsin-like protease), TMPRSS4, TMPRSS11A, TMPRSS11E, matriptase, and neutrophil elastase have been also implicated in respiratory virus infections.13,30 Among these proteases, HAT cleaves the Spike SARS-CoV-2 protein at a different site to TMPRSS2 and favour the fusion of the virus envelope with the host cell membrane.31 The HAT protein also cleaves the HA of influenza viruses and promotes their entry.17 A series of substrate analogues of this enzyme, such as compound 1 (Fig. 1), have been developed and shown nanomolar inhibition potency against HAT, but with low selectivity, notably against the coagulation proteases thrombin and factor Xa or the fibrinolytic enzyme plasmin.32
On the other hand, dual-target drugs offer a promising approach to combating viral infections by simultaneously inhibiting multiple viral targets and counteracting associated inflammation, resulting in synergistic antiviral effects.33 This strategy presents advantages over traditional drug combination therapy, as it avoids the complexities of drug–drug interactions, potentially leading to improved safety profiles. By targeting two mechanisms with a single molecule, dual-target drugs can achieve enhanced efficacy, while minimizing the risks associated with administering multiple separate compounds. This strategy has been already applied with success against the SARS-CoV-2.34–36 Notably, compound 212-148 (Fig. 1), an inhibitor targeting both TMPRSS2 and cathepsin L, two host cell proteases, have demonstrated potent antiviral activity against various SARS-CoV-2 variants.37 Some other compounds have also shown potency against two or more cell host proteases, such as the natural pseudo-tetrapeptide omicsynin B4 (Fig. 1) which displayed IC50 in the sub-micromolar range against TMPRSS2 and in the sub-nanomolar range against cathepsin L,38 or diminazen, a bis-benzamidine derivative (Fig. 1), able to inhibit both TMPRSS2 and furin with IC50s of 1.35 and 13.2 μM, respectively.39
Due to the critical role played by TMPRSS2 and HAT proteases in the entry process of several viruses, including SARS-CoV2, targeting these two proteases could be therefore of interest as an anti-corona and influenza antiviral strategy, but also in the fight against respiratory viruses more generally. To our knowledge, such compounds have not yet been reported. In order to identify molecules with dual TMPRSS2 and HAT inhibition activity, a series of para-aminobenzylamine-based compounds, initially designed as Kallikrein-6 inhibitors, were evaluated against these two host serine proteases. The most active compounds were then tested on human lung carcinoma cells infected with SARS-CoV2 to assess their antiviral activity and potential toxicity. Finally, key physicochemical and in vitro pharmacokinetic properties were evaluated, for the most promising compounds.
2. Results and discussion
2.1. Chemistry
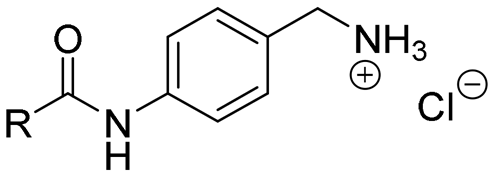
The para-aminobenzylamine derivatives were synthesized following our previously reported methodology.40 Briefly, compound 3 was obtained in two steps from 4-nitrobenzylamine hydrochloride 2, involving Boc protection of the amine and reduction of the nitro group (Scheme 1). It was then coupled with various carboxylic acid derivatives, including salicylic, naphthoic, indole or 4-oxodihydroquinoline acids, using EDCI (N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride) to afford compounds 4–22 as hydrochloride salts after Boc deprotection with HCl in dioxane. Compound 23 was synthesized from 1-naphthoic acid, 1-Boc-4-aminopiperidine and EDCI, followed by Boc removal.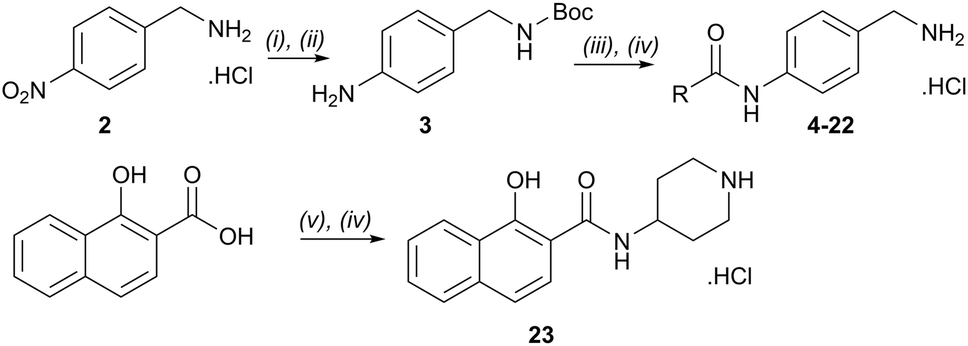 | ||
| Scheme 1 Reagents and conditions: (i) Boc2O, TEA, DCM, RT, 2 h, 80%; (ii) H2, 10% Pd/C, AcOEt, RT, 4 h, qt; (iii) ArCO2H, EDCI, THF, reflux, 12 h, 12–82%; (iv) 6 N HCl gas in 1,4-dioxane, RT, 1.5 h, 44–99%; (v) 4-Boc-1-aminopiperidine, EDCI, THF, reflux 12 h, 23%. R groups are described in Table 1. | ||
2.2. In vitro TMPRSS2 and HAT inhibition activity
TMPRSS2 and HAT are both trypsin-like serine proteases, meaning they preferentially cleave peptide bonds after basic residues, such as arginine or lysine. This substrate specificity arises from the presence of a negatively charged aspartate residue in the enzyme's S1 pocket, which stabilizes interactions with positively charged substrates. Given this mechanism, inhibitors containing a basic functional group can effectively mimic substrate binding, thereby enhancing their affinity for the enzyme's active site.41 We recently evaluated a series of benzylamine derivatives against human kallikrein 6, another trypsin-like serine protease that shares this conserved S1 pocket architecture.40 Owing to these structural and mechanistic similarities, we hypothesized that the same scaffold might also inhibit TMPRSS2 and HAT. To investigate this possibility and explore their inhibitory potential against TMPRSS2 and HAT, these compounds were first screened at a single dose of 10 μM against both proteases and those exhibiting more than 20% inhibition were retained for IC50 determination to assess their potency. The results are shown in Table 1.|
|
|||
|---|---|---|---|
| Cpd | R | IC50 (μM) | |
| TMPRSS2 | HAT | ||
| a NI: no inhibition (% inhibition at 10 μM < 20%). | |||
| 4 |
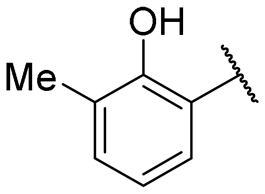
|
NIa | 42.1 ± 2.9 |
| 5 |
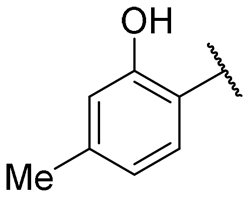
|
4.9 ± 0.2 | 18.9 ± 1.0 |
| 6 |
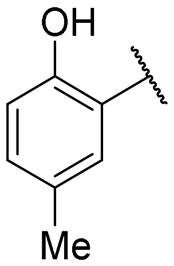
|
40.5 ± 4.0 | 2.4 ± 0.3 |
| 7 |
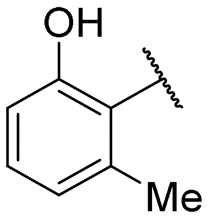
|
NI | NI |
| 8 |
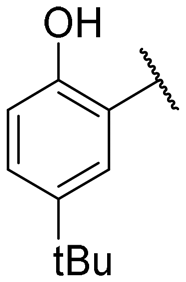
|
13.1 ± 0.4 | NI |
| 9 |
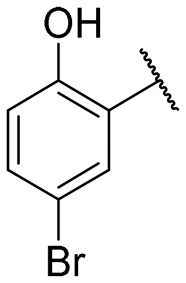
|
NI | NI |
| 10 |
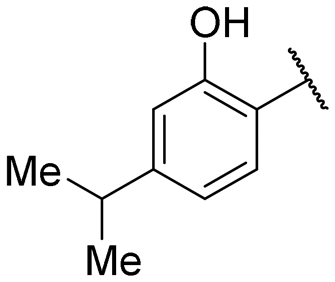
|
1.5 ± 0.1 | 11.6 ± 0.6 |
| 11 |
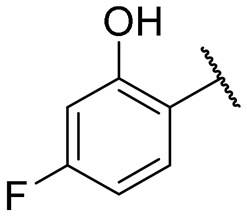
|
NI | NI |
| 12 |
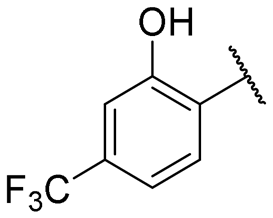
|
30.2 ± 2.0 | NI |
| 13 |
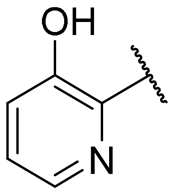
|
NI | NI |
| 14 |
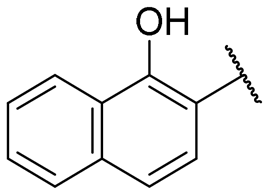
|
12.5 ± 2.7 | 30.3 ± 2.6 |
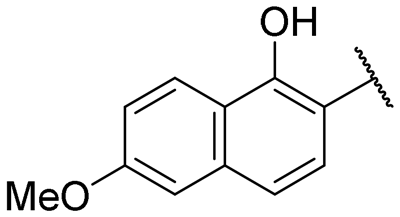
| 15 |
|
20.6 ± 1.5 | NI |
| 16 |
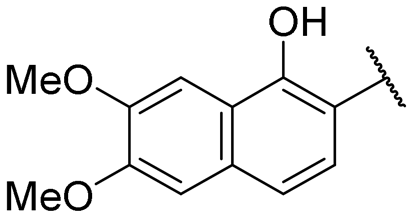
|
NI | NI |
| 17 |
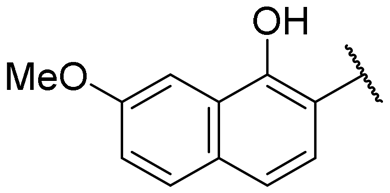
|
6.0 ± 0.5 | NI |
| 18 |
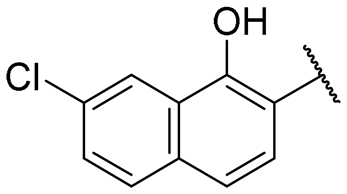
|
NI | NI |
| 19 |
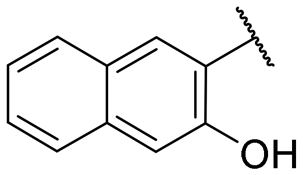
|
2.1 ± 0.1 | NI |
| 20 |
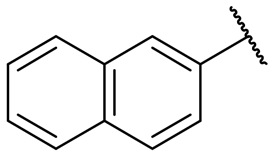
|
NI | NI |
| 21 |
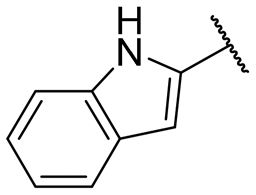
|
NI | NI |
| 22 |
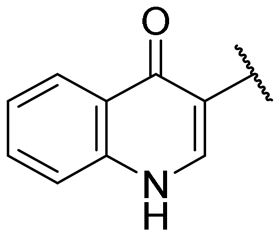
|
NI | NI |
| 23 |
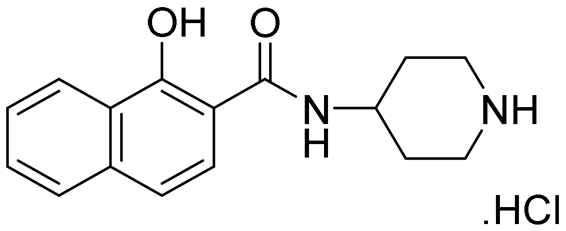
|
NI | NI |
| Camostat | — | 0.0042 (ref. 42) | 0.151 (ref. 41) |
Regarding TMPRSS2 inhibition, several compounds exhibited promising potency. Firstly, the introduction of a methyl group on the salicylamide moiety at position 5 or 4 led to compounds with moderate to good inhibitory potency (IC50 of 40.5 and 4.9 μM for compounds 6 and 5, respectively), whereas substitution at position 3 or 6 was not tolerated (compounds 4 and 7). Introducing a bulkier group at position 4 or 5, such as a tert-butyl (compound 8) or an isopropyl group (compound 10), enhanced potency (IC50 of 13.1 μM and 1.5 μM, respectively), whereas the presence of a halogen at position 5 or 4 abolished activity (compounds 9 and 11). A trifluoromethyl group at position 4 (compound 12) decreased activity compared to the methyl analogue 5, and a hydroxypyridine moiety was unfavourable (compound 13).
A series of naphthol derivatives also displayed some potency against this protease. While the 1-naphthol derivative 14 exhibited good potency (IC50 of 12.5 μM), the introduction of a methoxy group or a chlorine at position 7 decreased or abolished activity (compounds 15 and 18, respectively). The 6-methoxy isomer 17 showed better activity (IC50 of 6.0 μM), whereas the 5,6-dimethoxy derivative 16 was inactive. Interestingly, shifting the position of substitution of the hydroxy and amide groups from positions 1,2 to positions 2,3 increased potency (IC50 of 2.1 μM for compound 19, compared to 12.5 μM for compound 14). The removal of the hydroxy group (compound 20) or replacing the naphthol moiety with an indole or a quinolone group was unfavourable (compounds 21 to 22). Finally, replacement of the benzylamine part of the molecule with a piperidine group was not tolerated (compound 23). Thus, among these series of derivatives, compound 10 showed the highest activity, followed by compounds 19, 5 and 17, with IC50 values ranging between 1.5 and 6.0 μM.
Regarding the second protease target HAT, this series of compounds is generally less potent. Inhibitory activity was observed with 3-methyl, 4-methyl, 5-methyl, 4-isopropyl salicylamide derivatives, as well as with the 1-naphthol compound (compounds 4, 5, 6, 10 and 14, respectively). All other modifications resulted in a loss of inhibition. The 5-methyl derivative 6 exhibited the highest potency of all evaluated compounds (IC50 of 2.4 μM), followed by the 4-isopropyl and the 4-methyl derivatives (IC50 of 11.6 μM and 18.9 μM for compounds 10 and 5, respectively). Thus, from this study, four compounds displayed dual activity against TMPRSS2 and HAT, namely compounds 5, 6, 10 and 14, with compound 10 as the most potent one.
2.3. Mechanism of inhibition and selectivity
Mechanistic studies were performed only on hit compounds, i.e. compounds 5, 6, 10 and 14. For all compounds, the reversibility of inhibition was assessed using the jump dilution method, which distinguishes between covalent and non-covalent inhibitors.43 Whatever the compound tested, a 1/100 dilution of the enzyme-inhibitor complex allowed restoring more than 80% of the enzyme activity. Hence, all hit compounds are reversible inhibitors.Dixon plots were then used to determine the type of inhibition and the inhibition constant (Ki). For compounds 5, 6 and 14, the plots are consistent with competitive inhibition (confirmed by Lineweaver-Burk representations, data not shown). As an example, Dixon plots obtained with compound 6 against HAT protease and 10 against TMPRSS2 are shown in Fig. 2. The plots showed straight lines that intersect at the same point above the abscissa axis, confirming a competitive inhibition mode (Fig. 2A). This suggests that compounds 6 and 10 bind respectively to HAT and TMPRSS2 active sites with Ki values of 1.4 μM for 6 against HAT and 0.7 μM for 10 against TMPRSS2.
 | ||
| Fig. 2 Mechanism of inhibition towards HAT and TMPRSS2 serine proteases. A. Dixon plot for inhibitor 6 tested at different concentrations using 22.5 pM of HAT with substrate Boc-VPR-AMC at different concentrations (25 μM, red; 50 μM, blue; 100 μM, green; 200 μM, grey). B. Dixon plot for inhibitor 10 tested at different concentrations using 5 nM of TMPRSS2 with substrate Boc-QAR-AMC at different concentrations (15.62 μM, black; 62.5 μM, blue; 250 μM, green; 1000 μM, red). | ||
The selected compounds were then tested against a set of representative serine proteases, including matriptase (another transmembrane protease), thrombin, plasmin and cathepsin L, which represent highly abundant serine proteases, at a single dose of 10 μM to evaluate their selectivity profile. For comparison, their inhibitory potency against TMPRSS2 and HAT, the two targeted proteases, was also determined at this concentration, as well as their activity against KLK6, since these compounds were initially designed to target this enzyme. The results are presented in Table 2.
As anticipated, most of these compounds exhibited strong KLK6 inhibitory activity (more than 50% inhibition), except for compound 6, which displayed only 25% inhibition. Interestingly, no or low inhibition was observed against matriptase, cathepsin L and thrombin for all compounds. Compounds 5, 10 and 14 showed also good inhibitory potency against plasmin (inhibition greater than 70%), except for compound 6 which showed no inhibition at the tested concentration. Thus, even though compound 6 is not the most active compound against TMPRSS2 and HAT, it displayed good selectivity against the tested panel.
2.4. Molecular modelling
To gain insight into the structural basis of inhibition properties and potential binding interactions of the most active compound, we conducted a molecular docking study using Molegro Virtual Docker 6.0.A docking simulation of compound 10 with the target protease TMPRSS2 (PDB ID: 7MEQ) was performed. As illustrated in Fig. 3A and B, compound 10 fits well within the active site of the protease. Notably, its benzylamine moiety is oriented toward the S1 pocket, consistent with the trypsin-like nature of TMPRSS2, which preferentially accommodates basic functional groups in this pocket. The primary amine establishes close polar contacts with the side chains of Asp435 and Ser436, as well as with the backbone of Gly464. The phenol group interacts with the catalytic Ser441 side chain and with the amidic NH of Cys437. Additionally, the carbonyl group of the inhibitor may interact with two water molecules present in the active site of the co-crystallized TMPRSS2 with nafamostat. Finally, the isopropyl chain of 10 is oriented toward a hydrophobic region of the protease, mainly composed of Val275, Val280 and Leu302, which may explain why the protease also accommodates large hydrophobic groups such as naphthyl groups found in compounds 14, 15, 17 and 19.
 | ||
| Fig. 3 Pose prediction by molecular modeling of compound 10 in TMPRSS2 (A and B) and of compound 6 in HAT (C and D). More relevant interactions were depicted by yellow dashed lines. The code color for the atoms is as follows: carbon of the inhibitor in green, nitrogen in blue, oxygen in red, sulfur in yellow, and hydrogen in white. The side chains that make no contact with compound 10 or 6, as well as hydrogen of the protein are not shown. | ||
Then, molecular docking was performed with compound 6 and HAT protease (PDB ID: 8VIS). The best pose revealed that the inhibitor could fit into the active site of the protease, with the benzylamine moiety occupying the S1 pocket (Fig. 3C and D). The primary amine interacts with the side chains of Asp362 and the carbonyl groups of Lys398 and of the main chain of Gln392. The NH group of the amide bond in compound 6 forms a hydrogen bond with the carbonyl group of Cys364, while the phenol group of the inhibitor interacts with several amino acids of the active site, including Asp367, Gly366, and the catalytic Ser368. Additionally, the entrance to the S1 pocket appears narrower than that of TMPRSS2, which may explain the protease's intolerance toward inhibitors bearing bulkier substituents, such as a naphthol group.
2.5. Evaluation of anti-SARS CoV-2 activity and cytotoxicity
All compounds (4–23) were tested for their antiviral activity against live SARS-CoV-2 virus in the human lung cell line A549. These compounds were evaluated at a single dose of 10 μM for their antiviral potency against the SARS-CoV-2 strain D614G.This strain emerged in Europe in February 2020 and this mutation conferred significantly increased infectivity compared to the first early clinical isolate.44 Infection of the cells with this virus induces cell lysis, a phenomenon known as the Cytopathic Effect (CPE). By measuring cell viability 48 hours after infection, the protective effect of the compounds was determined and expressed as the percentage of infection inhibition compared to untreated cells. As internal controls, remdesivir and camostat were included as reference compounds. In parallel, the cytotoxicity of the samples was assessed in non-infected A549 cells. The results are shown in Fig. 4.
 | ||
| Fig. 4 Anti SARS-CoV-2 activity (Viral ToxGlo® Assay) and cytotoxicity (Cell Titer 96® AQueous One Solution Assay) of compounds 4–23 in A549 cell-based assays at a single dose of 10 μM. Results for infected cells are shown in orange and non-infected cells in blue. | ||
Among the tested compounds, compounds 8, 10 and 17 exhibited noticeable antiviral activity in infected cells at 10 μM, with 73 to 83% inhibition, while displaying low or even no toxicity in non-infected cells. These compounds, although slightly less effective than remdesivir, show similar activity to camostat (85% inhibition in investigated conditions). Additionally, although less potent, molecules 19 and 15 demonstrated good infection inhibition efficacy (56% and 49% inhibition, respectively) with low or no cytotoxicity. Interestingly, the best inhibitors of TMPRSS2 (IC50 below 10 μM), generally displayed important antiviral activity, as illustrated by compounds 10, 17 and 19, which showed infection inhibition percentages greater than 56%.
Compounds with moderate inhibition properties against this protease (IC50 between 10 and 30 μM) also demonstrated moderate to good antiviral potency, as seen with compounds 8, 14 and 15, which exhibited infection inhibition percentages ranging from 44% to 73%. Finally, all compounds that exhibited low or no activity against TMPRSS2 and/or HAT demonstrated only weak antiviral effect in the cell-based assay, with inhibition percentages generally below 40%, except for compound 18. However, the apparent activity of this compound may be misleading, as it also showed high cytotoxicity in A549 cells, causing 33% cell death at 10 μM.
Based on this assay, compounds 8, 10, 15, 17 and 19 displayed the most potent antiviral activity. These compounds were subsequently selected for EC50 determination, using the same experimental protocol and a range of compound concentrations between 0.6 and 40 μM. In parallel, their potential cell cytotoxicity (CC50) was evaluated using a cell proliferation assay.
In the case of compound 19, the EC50 could not be determined due to its cytotoxicity at the highest concentrations tested. For all other compounds, EC50 values were successfully determined, and the results are presented in Table 3. Among the para-aminobenzylamine compounds tested, compound 8 exhibited the strongest antiviral activity (EC50 of 3.32 μM), followed by compound 17 (EC50 = 5.3 μM). Notably, although compound 8 showed the highest activity in this assay, its inhibition of TMPRSS2 was only moderate (IC50 = 13 μM), and no detectable inhibition was observed against HAT. These findings suggest that the antiviral effect of compound 8 may involve an alternate mechanism of action beyond direct inhibition of these two proteases.
No cytotoxicity was observed for these compounds, resulting in a favourable selectivity index. Compounds 15 and 10 showed slightly lower antiviral activity, with EC50 between 8 and 10 μM, and no noticeable cytotoxicity was detected, even at the highest concentration tested.
2.6. Physicochemical properties and drug-likeness of selected compounds
The evaluation of physicochemical and pharmacokinetic properties of synthesized compounds is crucial in the early stages of drug discovery. Chemoinformatics analysis of the most potent compounds, namely 8, 10, 15 and 17, revealed that all derivatives complied with Lipinski's rule of five and Veber's rules (Table 4), suggesting their suitability for oral administration.| Cpd | MW (Da) | RB | HBD | HBA | clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P P |
PSA (Å2) | Solubilitya (μM) | RLMbt1/2 (min) |
|---|---|---|---|---|---|---|---|---|
| Abbreviations: HBA, hydrogen bond acceptors; HBD, hydrogen bond donors; MW, molecular weight; PSA, polar surface area.a Thermodynamic aqueous solubility in PBS at pH 7.4.b Rat liver microsomal stability. | ||||||||
| 8 | 298.4 | 5 | 3 | 3 | 2.88 | 75.3 | 265 ± 51 | 63 |
| 10 | 284.3 | 5 | 3 | 3 | 2.73 | 75.3 | 47 ± 11 | 22 |
| 15 | 322.3 | 5 | 3 | 4 | 2.74 | 84.6 | 746 ± 37 | 63 |
| 17 | 322.3 | 5 | 3 | 4 | 2.71 | 84.6 | 254 ± 9 | 53 |
The aqueous solubility at pH 7.4 was assessed, indicating that the isopropyl derivative 10 was moderately soluble, while the tert-butylphenol 8 and the two methoxynaphthol derivatives 17 and 15 exhibited higher solubility, with compound 15 being the most soluble. Finally, to further investigate the drug-likeness of these compounds, rat liver microsomal (RLM) stability was investigated.
All compounds showed moderate to good overall microsomal stability; the isopropyl derivative 10 exhibited the shortest half-life (22 min), whereas compounds 8 and 15 showed the highest microsomal stability, with half-lives of 63 min.
Conclusions
As new coronavirus variants continue to emerge with mutations that may impact transmissibility and immune escape, there is a pressing need for effective broad-spectrum antivirals to complement vaccination strategies and strengthen pandemic preparedness. In this context, we report for the first time a series of para-aminobenzylamine derivatives acting as dual, non-covalent inhibitors of TMPRSS2 and HAT, two host serine proteases involved in the entry mechanisms of SARS-CoV-2 and other respiratory viruses, as well as in virus-associated inflammatory responses. Dual targeting offers a promising approach to combating viral infections by simultaneously inhibiting multiple targets and counteracting associated inflammation, resulting in synergistic antiviral effects.A SAR study revealed that introducing bulky substituents on the salicylamide moiety or its replacement with a naphthol group enhanced TMPRSS2 inhibition, whereas smaller substituents on the phenol ring favoured HAT inhibition. Molecular docking supported these findings, highlighting key interactions in the active sites of both proteases and providing a structural rationale for their differing tolerance to steric hindrance. Among the tested derivatives, compounds 5, 6, 10, and 14 displayed dual activity against TMPRSS2 and HAT, with compound 10 emerging as the most potent TMPRSS2 inhibitor and compound 6 as the most selective.
In antiviral assays using the SARS-CoV-2 D614G strain in A549 lung carcinoma cells, five compounds (8, 10, 15, 17, and 19) significantly inhibited viral infection. Notably, compound 8 demonstrated the highest antiviral activity (EC50 = 3.32 μM) without detectable cytotoxicity in human cells. Finally, all lead compounds exhibited drug-like physicochemical properties and complied with key oral bioavailability rules. Most compounds showed moderate to good microsomal stability, with compound 15 standing out, due to its good aqueous solubility and favourable metabolic stability.
Altogether, this study presents the first report of para-aminobenzylamine derivatives as dual TMPRSS2/HAT inhibitors with promising antiviral activity against SARS-CoV-2. These findings lay the groundwork for further optimization and development of broad-spectrum antiviral agents targeting host proteases involved in respiratory virus entry.
Materials and methods
Compounds 4 to 23 were synthesized according to our previous reported procedures and their physical characteristics were in agreement with the published data.40 The A549 cells were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA).Enzyme activity assays
Recombinant human proteases HAT and TMPRSS2, are supplied by Bio-Techne® and CliniSciences®, respectively. All proteases tested to evaluate the selectivity profile, as well as camostat, a reference inhibitor of TMPRSS2, were provided by Bio-Techne®. Fluorogenic peptide substrates Boc-VPR-AMC (100 μM) and Boc-QAR-AMC (100 μM) carrying the fluorophore 7-amino-4-methylcoumarin (AMC) were used for enzyme kinetics assays of HAT/TMPRSS11D (22.5 pM), and TMPRSS2 (5 nM) in Tris 50 mM, NaCl 150 mM, Brij-35 0.05%, pH 8.Inhibition assays were performed in duplicate across a range of inhibitor concentrations. Enzymes and inhibitors were preincubated for 15 minutes prior to substrate addition. Initial reaction rates in control samples (V0) were defined as 100% enzymatic activity. Inhibitory effects were determined by comparing initial rates in the presence of compounds (Vi) to V0, with inhibition calculated as: % Inhibition = 100 × (1 − Vi/V0). IC50 values (compound concentration yielding 50% inhibition) were determined by fitting data to the following equation: % Inhibition = 100 × [I]0/(IC50 + [I]0). Reversibility was evaluated by the jump dilution method.43 The reaction mixtures were diluted 100-fold after 15 min preincubation of the enzyme with inhibitor. Aliquots of reaction mixtures (1 μL) were added to 99 μL of the buffer containing the fluorogenic substrate (experimental conditions identical to the routine protocol) before determination of remaining activity. For non-covalent inhibitors, the mechanisms of inhibition were determined by varying substrate and inhibitor concentrations and using classical Lineweaver−Burk representation and Dixon plot to extract inhibition constants Ki.
Molecular docking
Initial 3D structures of compounds 10 and 6 were built with ChemDraw and refined by molecular mechanics with the MM2 force field using HyperChem pro 6.0. Docking was performed using Molegro Virtual Docker 6.0 software, using the structure of TMPRSS2 (PDB ID: 7MEQ) and HAT (PDB ID: 8VIS). The ligand molecules were docked into the active site of TMPRSS2 for compound 10 and of HAT for compound 6. The docking parameters were as follows: Grid resolution: 0.30 Å, Max iterations: 1500, Population size: 50, Energy threshold: 100.00, Simplex evolution: 300 (Max steps) and 1.00 (Neighbor distance factor). The post-docking optimizations of ligand conformation and H-bonds were applied. The docking poses were analysed using built-in facilities of MVD software, and the docking results were visualized using the PyMol software.In vitro evaluation of the antiviral effect of compounds on cell lines infected by SARS-CoV-2
In 96-well plates, the compounds are diluted to 10 μM for the single dose assay in RPMI and incubated with A549 cells for 2 hours at 37 °C. For EC50 determination of the selected compounds, serial dilutions were prepared starting from 40 μM down to 0.6 μM. The cells were then infected with SARS-CoV-2 (D614G) at a MOI of 10−2. All assays were performed in triplicate. The plates included control wells containing infected cells, non-infected cells, and pre-treated cells with the reference compounds remdesivir and camostat. The plates were incubated at 37 °C with 5% CO2 for 48 hours. At 48 hours post-infection, the cytopathic effect (CPE) of the virus was observed using an Evos® microscope, and cell viability was quantified using the Viral ToxGlo™ Assay kit (Promega) according to the manufacturer's instructions. Signal detection was carried out with an EnVision® microplate reader (PerkinElmer). The percentage of infection inhibition for each tested compound was calculated using the following formula: % inhibition = [(Infected treated cell signal − Infected untreated cell signal)/(Uninfected untreated cell signal − Infected untreated cell signal)] × 100.Cytotoxicity
The compounds, at the same concentrations than those used for the antiviral assay (at 10 μM or between 0.6 and 40 μM) are incubated with A549 cells in 96-well plates at 37 °C with 5% CO2 for 48 hours. The plates include control wells containing untreated cells as well as cells treated with the reference compounds remdesivir and camostat at the same concentrations. Cell viability is quantified using the Promega “CellTiter 96® AQueous One Solution Assay” kit, according to the manufacturer's instructions. The percentage of toxicity for each tested compound was calculated using the following formula: % Toxicity = [1 − (treated cell signal/untreated cell signal)] × 100.ADME predictions
The compounds were drawn using ChemDraw, copied in a SMILES format and uploaded to Swiss ADME (https://www.swissadme.ch/). The following parameters were then calculated: physicochemical Properties (MW, Num. rotatable bonds, Num. H bond acceptors, Num. H-bond donors and TPSA); lipophilicity (Consensus Log Po/w) and druglikeness (Lipinski, Veber).Determination of solubility in PBS
Aqueous solubility of selected compounds was determined using the shake-flask method as previously reported.45Metabolic stability
The tested compounds (final concentration: 1 μM) were incubated in triplicate with a solution of rat liver microsomes RLM (0.5 mg mL−1 final concentration) in presence of NADPH (1 mM final concentration) at 37 °C, 400 rpm. Aliquots were collected at 5, 10, 20, 30, and 40 min and proteins were precipitated by the addition of cold ACN. T0 and negative control samples were collected in the same way, from a microsomal solution deactivated with ACN. The samples were centrifuged at 15 °C and 3000 rpm for 15 min. The supernatants were collected and analysed by UPLC-MS. The degradation percentage was calculated based on the T0 samples. Remaining compound percentages were plotted against time and the slope of the resulting curve was determined. The data were calculated from the following equation: t1/2 = (ln2)/k, where k is the first-order degradation constant (the slope of the logarithm of the compound concentration versus incubation time).
Author contributions
HC: synthesis and structural characterization of compounds, solubility and microsomal stability determinations. AD, NC, SK, FS: in vitro inhibition activity evaluation, mechanism of inhibition and selectivity determination; CEA: conceptualization, supervision, review and editing. NM: supervision, writing, review and editing. All authors reviewed the results and approved the final version of the manuscript.Conflicts of interest
The authors declare that there are no conflicts of interests.Data availability
All relevant data are within the manuscript.Acknowledgements
We would like to thank the UAR 3725 Cemipai for the antiviral assays and the SynBio3 platform (IBMM, UMR 5247 Montpellier) which was supported by GIS IBISA and whose equipment was publicly funded through the I-SITE Excellence Program of the University of Montpellier, under the Investissements d'Avenir France 2030. We are also grateful to Sorbonne Université, Université de Montpellier, the Centre National de la Recherche Scientifique (CNRS) and the Institut National pour la Recherche Médicale (INSERM) for their financial support. We thank the French Ministry of Research and Education for the PhD fellowship granted to A. Dussol, and the French National Research Agency (ANR Restore 21-CE18-0032) for the PhD fellowship of H. Carvaillo and the research engineer contract of N. Chaaya. This work was also supported by the COST Action ProteoCure, CA20113, supported by COST (European Cooperation in Science and Technology).References
- F. Nazir, A. John Kombe Kombe, Z. Khalid, S. Bibi, H. Zhang, S. Wu and T. Jin, Mol. Cell. Probes, 2024, 77, 101973 CrossRef CAS PubMed.
- X.-J. Zhao, B. Ji, C. Shang, D.-Y. Li, S. Zhang, H.-J. Gu, H.-H. Peng, C. Qian, C.-L. Zhang, C. Shi, Y. Shen, J.-J. Chen, Q. Xu, C.-L. Lv, B.-G. Jiang, H. Wang, X. Li, G.-L. Wang and L.-Q. Fang, iScience, 2024, 27, 110283 CrossRef CAS.
- Q. Wang, S. Iketani, Z. Li, L. Liu, Y. Guo, Y. Huang, A. D. Bowen, M. Liu, M. Wang, J. Yu, R. Valdez, A. S. Lauring, Z. Sheng, H. H. Wang, A. Gordon, L. Liu and D. D. Ho, Cell, 2023, 186, 279–286.e8 CrossRef CAS.
- Y. Cao, A. Yisimayi, F. Jian, W. Song, T. Xiao, L. Wang, S. Du, J. Wang, Q. Li, X. Chen, Y. Yu, P. Wang, Z. Zhang, P. Liu, R. An, X. Hao, Y. Wang, J. Wang, R. Feng, H. Sun, L. Zhao, W. Zhang, D. Zhao, J. Zheng, L. Yu, C. Li, N. Zhang, R. Wang, X. Niu, S. Yang, X. Song, Y. Chai, Y. Hu, Y. Shi, L. Zheng, Z. Li, Q. Gu, F. Shao, W. Huang, R. Jin, Z. Shen, Y. Wang, X. Wang, J. Xiao and X. S. Xie, Nature, 2022, 608, 593–602 CrossRef CAS PubMed.
- J. H. Beigel, K. M. Tomashek, L. E. Dodd, A. K. Mehta, B. S. Zingman, A. C. Kalil, E. Hohmann, H. Y. Chu, A. Luetkemeyer, S. Kline, D. L. de Castilla, R. W. Finberg, K. Dierberg, V. Tapson, L. Hsieh, T. F. Patterson, R. Paredes, D. A. Sweeney, W. R. Short, G. Touloumi, D. C. Lye, N. Ohmagari, M. Oh, G. M. Ruiz-Palacios, T. Benfield, G. Fätkenheuer, M. G. Kortepeter, R. L. Atmar, C. B. Creech, J. Lundgren, A. G. Babiker, S. Pett, J. D. Neaton, T. H. Burgess, T. Bonnett, M. Green, M. Makowski, A. Osinusi, S. Nayak and H. C. Lane, N. Engl. J. Med., 2020, 383, 1813–1826 CrossRef CAS PubMed.
- A. J. Bernal, M. M. G. da Silva, D. B. Musungaie, E. Kovalchuk, A. Gonzalez, V. D. Reyes, A. Martín-Quirós, Y. Caraco, A. Williams-Diaz, M. L. Brown, J. Du, A. Pedley, C. Assaid, J. Strizki, J. A. Grobler, H. H. Shamsuddin, R. Tipping, H. Wan, A. Paschke, J. R. Butterton, M. G. Johnson and C. D. Anda, N. Engl. J. Med., 2022, 386, 509–520 CrossRef CAS.
- B. Yu and J. Chang, Signal Transduction Targeted Ther., 2020, 5, 236 CrossRef CAS PubMed.
- D. R. Owen, C. M. N. Allerton, A. S. Anderson, L. Aschenbrenner, M. Avery, S. Berritt, B. Boras, R. D. Cardin, A. Carlo, K. J. Coffman, A. Dantonio, L. Di, H. Eng, R. Ferre, K. S. Gajiwala, S. A. Gibson, S. E. Greasley, B. L. Hurst, E. P. Kadar, A. S. Kalgutkar, J. C. Lee, J. Lee, W. Liu, S. W. Mason, S. Noell, J. J. Novak, R. S. Obach, K. Ogilvie, N. C. Patel, M. Pettersson, D. K. Rai, M. R. Reese, M. F. Sammons, J. G. Sathish, R. S. P. Singh, C. M. Steppan, A. E. Stewart, J. B. Tuttle, L. Updyke, P. R. Verhoest, L. Wei, Q. Yang and Y. Zhu, Science, 2021, 374, 1586–1593 CrossRef CAS.
- Y. Unoh, S. Uehara, K. Nakahara, H. Nobori, Y. Yamatsu, S. Yamamoto, Y. Maruyama, Y. Taoda, K. Kasamatsu, T. Suto, K. Kouki, A. Nakahashi, S. Kawashima, T. Sanaki, S. Toba, K. Uemura, T. Mizutare, S. Ando, M. Sasaki, Y. Orba, H. Sawa, A. Sato, T. Sato, T. Kato and Y. Tachibana, J. Med. Chem., 2022, 65, 6499–6512 CrossRef CAS PubMed.
- S. Gandhi, J. Klein, A. J. Robertson, M. A. Peña-Hernández, M. J. Lin, P. Roychoudhury, P. Lu, J. Fournier, D. Ferguson, S. A. K. Mohamed Bakhash, M. Catherine Muenker, A. Srivathsan, E. A. Wunder, N. Kerantzas, W. Wang, B. Lindenbach, A. Pyle, C. B. Wilen, O. Ogbuagu, A. L. Greninger, A. Iwasaki, W. L. Schulz and A. I. Ko, Nat. Commun., 2022, 13, 1547 CrossRef CAS PubMed.
- S. Iketani, H. Mohri, B. Culbertson, S. J. Hong, Y. Duan, M. I. Luck, M. K. Annavajhala, Y. Guo, Z. Sheng, A.-C. Uhlemann, S. P. Goff, Y. Sabo, H. Yang, A. Chavez and D. D. Ho, Nature, 2023, 613, 558–564 CrossRef CAS PubMed.
- C. Gil, T. Ginex, I. Maestro, V. Nozal, L. Barrado-Gil, M. Á. Cuesta-Geijo, J. Urquiza, D. Ramírez, C. Alonso, N. E. Campillo and A. Martinez, J. Med. Chem., 2020, 63, 12359–12386 CrossRef CAS PubMed.
- C. B. Jackson, M. Farzan, B. Chen and H. Choe, Nat. Rev. Mol. Cell Biol., 2022, 23, 3–20 CrossRef CAS PubMed.
- M. Laporte and L. Naesens, Curr. Opin. Virol., 2017, 24, 16–24 CrossRef CAS.
- D. Bestle, M. R. Heindl, H. Limburg, T. Van Lam van, O. Pilgram, H. Moulton, D. A. Stein, K. Hardes, M. Eickmann, O. Dolnik, C. Rohde, H.-D. Klenk, W. Garten, T. Steinmetzer and E. Böttcher-Friebertshäuser, Life Sci. Alliance, 2020, 3, e202000786 CrossRef.
- B. Lin, C. Ferguson, J. T. White, S. Wang, R. Vessella, L. D. True, L. Hood and P. S. Nelson, Cancer Res., 1999, 59, 4180–4184 CAS.
- E. Böttcher-Friebertshäuser, C. Freuer, F. Sielaff, S. Schmidt, M. Eickmann, J. Uhlendorff, T. Steinmetzer, H.-D. Klenk and W. Garten, J. Virol., 2010, 84, 5605–5614 CrossRef.
- K. Baby, M. P. Vithalkar, S. G. Dastidar, C. Mukhopadhyay, R. Hamdy, S. S. M. Soliman and Y. Nayak, Scientifica, 2025, 2025, 3687892 CrossRef PubMed.
- N. Iwata-Yoshikawa, T. Okamura, Y. Shimizu, H. Hasegawa, M. Takeda and N. Nagata, J. Virol., 2019, 93, e01815–e01818 CAS.
- P. Breining, A. L. Frølund, J. F. Højen, J. D. Gunst, N. B. Staerke, E. Saedder, M. Cases-Thomas, P. Little, L. P. Nielsen, O. S. Søgaard and M. Kjolby, Basic Clin. Pharmacol. Toxicol., 2021, 128, 204–212 CrossRef CAS.
- K. Li, D. K. Meyerholz, J. A. Bartlett and P. B. McCray, mBio, 2021, 12(4) DOI:10.1128/mbio.00970-21.
- M. Yamamoto, S. Matsuyama, X. Li, M. Takeda, Y. Kawaguchi, J.-I. Inoue and Z. Matsuda, Antimicrob. Agents Chemother., 2016, 60, 6532–6539 CrossRef CAS PubMed.
- G. Sun, Y. Sui, Y. Zhou, J. Ya, C. Yuan, L. Jiang and M. Huang, J. Virol., 2021, 95(19) DOI:10.1128/jvi.00861-21.
- Y. Zhou, J. Wu, G. Xue, J. Li, L. Jiang and M. Huang, Biophys. J., 2022, 121, 3940–3949 CrossRef CAS.
- T. Yang, W. Yu, D. Guo, J. Li, X. Wang, Y. Song, Y. Cheng, Y. Luo, J. Yang, W. Ouyang and J. Hu, Phys. Chem. Chem. Phys., 2025, 27, 18539–18554 RSC.
- S. V. Zhuravel, O. K. Khmelnitskiy, O. O. Burlaka, A. I. Gritsan, B. M. Goloshchekin, S. Kim and K. Y. Hong, EClinicalMedicine, 2021, 41, 101169 CrossRef PubMed.
- J. D. Gunst, N. B. Staerke, M. H. Pahus, L. H. Kristensen, J. Bodilsen, N. Lohse, L. S. Dalgaard, D. Brønnum, O. Fröbert, B. Hønge, I. S. Johansen, I. Monrad, C. Erikstrup, R. Rosendal, E. Vilstrup, T. Mariager, D. G. Bove, R. Offersen, S. Shakar, S. Cajander, N. P. Jørgensen, S. S. Sritharan, P. Breining, S. Jespersen, K. L. Mortensen, M. L. Jensen, L. Kolte, G. S. Frattari, C. S. Larsen, M. Storgaard, L. P. Nielsen, M. Tolstrup, E. A. Sædder, L. J. Østergaard, H. T. T. Ngo, M. H. Jensen, J. F. Højen, M. Kjolby and O. S. Søgaard, EClinicalMedicine, 2021, 35, 100849 CrossRef PubMed.
- A. F. A. Moumbock, H. T. T. Tran, E. Lamy and S. Günther, Arch. Pharm., 2023, 356, 2200371 CrossRef CAS PubMed.
- M. Mahoney, V. C. Damalanka, M. A. Tartell, D. hee Chung, A. L. Lourenço, D. Pwee, A. E. Mayer Bridwell, M. Hoffmann, J. Voss, P. Karmakar, N. P. Azouz, A. M. Klingler, P. W. Rothlauf, C. E. Thompson, M. Lee, L. Klampfer, C. L. Stallings, M. E. Rothenberg, S. Pöhlmann, S. P. J. Whelan, A. J. O'Donoghue, C. S. Craik and J. W. Janetka, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2108728118 CrossRef CAS.
- M. Kishimoto, K. Uemura, T. Sanaki, A. Sato, W. W. Hall, H. Kariwa, Y. Orba, H. Sawa and M. Sasaki, Viruses, 2021, 13, 384 CrossRef CAS.
- A. M. Tharappel, S. K. Samrat, Z. Li and H. Li, ACS Infect. Dis., 2020, 6, 2844–2865 CrossRef CAS PubMed.
- F. Sielaff, E. Böttcher-Friebertshäuser, D. Meyer, S. M. Saupe, I. M. Volk, W. Garten and T. Steinmetzer, Bioorg. Med. Chem. Lett., 2011, 21, 4860–4864 CrossRef CAS PubMed.
- Z. A. Shyr, Y.-S. Cheng, D. C. Lo and W. Zheng, Drug Discovery Today, 2021, 26, 2367–2376 CrossRef CAS PubMed.
- W. Zhang, L. Xiao, D. Li, Y. Hu and W. Yu, J. Med. Chem., 2024, 67, 11522–11542 CrossRef CAS.
- S. Toba, K. Uemura, T. Sanaki, S. Kusakabe, K. Konishi, S. Miki, Y. Maruyama, A. Iimuro, T. Shishido, M. Sasaki, Y. Orba, W. W. Hall, H. Sawa and A. Sato, Virology, 2025, 612, 110661 CrossRef CAS PubMed.
- H. Shuai, J. Qiao, C. Yoon, G. Zhang, Y. Hou, X. Xia, L. Wang, X. Deng, Y. Wang, Q. Li, L. Du, Y. Liu, M. Zhou, H. T. Wong, J. Hu, H. Liu, B. Hu, D. Wang, J. Su, Y. Pan, Y. Ye, Y. Chen, Z. Fang, Z. Xia, Y. Chai, J. Shi, Y. Wang, T. Zhu, H. Zhang, S. Yuan, J. Zhou, J. F.-W. Chan, K.-Y. Yuen, C. Xu, J. Lei, S. Yang and H. Chu, Nat. Commun., 2025, 16, 6541 CrossRef CAS PubMed.
- H. Wang, Q. Yang, X. Liu, Z. Xu, M. Shao, D. Li, Y. Duan, J. Tang, X. Yu, Y. Zhang, A. Hao, Y. Wang, J. Chen, C. Zhu, L. Guddat, H. Chen, L. Zhang, X. Chen, B. Jiang, L. Sun, Z. Rao and H. Yang, Nat. Commun., 2023, 14, 7574 CrossRef CAS.
- Y. Li, K. Wang, H. Sun, S. Wu, H. Wang, Y. Shi, X. Li, H. Yan, G. Yang, M. Wu, Y. Li, X. Ding, S. Si, J. Jiang, Y. Du, Y. Li and B. Hong, Antiviral Res., 2023, 214, 105606 CrossRef CAS PubMed.
- Y.-M. Xu, M. C. Inacio, M. X. Liu and A. A. L. Gunatilaka, Curr. Res. Chem. Biol., 2022, 2, 100023 CrossRef CAS PubMed.
- S. Aït Amiri, C. Deboux, F. Soualmia, N. Chaaya, M. Louet, E. Duplus, S. Betuing, B. Nait Oumesmar, N. Masurier and C. El Amri, J. Med. Chem., 2021, 64, 5667–5688 CrossRef PubMed.
- B. J. Fraser, R. P. Wilson, S. Ferková, O. Ilyassov, J. Lac, A. Dong, Y.-Y. Li, A. Seitova, Y. Li, Z. Hejazi, T. M. G. Kenney, L. Z. Penn, A. Edwards, R. Leduc, P.-L. Boudreault, G. B. Morin, F. Bénard and C. H. Arrowsmith, Nat. Commun., 2025, 16, 4351 CrossRef CAS PubMed.
- M. Hoffmann, H. Hofmann-Winkler, J. C. Smith, N. Krüger, P. Arora, L. K. Sørensen, O. S. Søgaard, J. B. Hasselstrøm, M. Winkler, T. Hempel, L. Raich, S. Olsson, O. Danov, D. Jonigk, T. Yamazoe, K. Yamatsuta, H. Mizuno, S. Ludwig, F. Noé, M. Kjolby, A. Braun, J. M. Sheltzer and S. Pöhlmann, EBioMedicine, 2021, 65, 103255 CrossRef CAS PubMed.
- R. A. Copeland, A. Basavapathruni, M. Moyer and M. P. Scott, Anal. Biochem., 2011, 416, 206–210 CrossRef CAS PubMed.
- B. Korber, W. M. Fischer, S. Gnanakaran, H. Yoon, J. Theiler, W. Abfalterer, N. Hengartner, E. E. Giorgi, T. Bhattacharya, B. Foley, K. M. Hastie, M. D. Parker, D. G. Partridge, C. M. Evans, T. M. Freeman, T. I. de Silva, A. Angyal, R. L. Brown, L. Carrilero, L. R. Green, D. C. Groves, K. J. Johnson, A. J. Keeley, B. B. Lindsey, P. J. Parsons, M. Raza, S. Rowland-Jones, N. Smith, R. M. Tucker, D. Wang, M. D. Wyles, C. McDanal, L. G. Perez, H. Tang, A. Moon-Walker, S. P. Whelan, C. C. LaBranche, E. O. Saphire and D. C. Montefiori, Cell, 2020, 182, 812–827.e19 CrossRef CAS PubMed.
- P. Lagardère, R. Mustière, N. Amanzougaghene, S. Hutter, M. Casanova, J.-F. Franetich, S. Tajeri, A. Malzert-Fréon, S. Corvaisier, N. Azas, P. Vanelle, P. Verhaeghe, N. Primas, D. Mazier, N. Masurier and V. Lisowski, Eur. J. Med. Chem., 2023, 249, 115115 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work and should be considered co-first authors. |
| This journal is © The Royal Society of Chemistry 2026 |