
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtonation-driven structural deformation in a conformationally twisted pyridyl-linked AIEgen: a platform to detect polyamines and nicotine
Madhuparna
Chakraborty
,
Avanigadda Madhu
Niharika
,
Zubair Khalid
Baig
 and
Manab
Chakravarty
*
and
Manab
Chakravarty
*
Department of Chemistry, Birla Institute of Technology and Science-Pilani, Hyderabad Campus, Hyderabad, 500078, India. E-mail: manab@hyderabad.bits-pilani.ac.in
First published on 3rd December 2025
Abstract
Stimuli-responsive fluorescent probes have emerged as key tools in chemical sensing and environmental monitoring due to their high sensitivity and real-time detection capabilities. However, conventional probes often rely on limited mechanisms, restricting their adaptability and scope. Addressing this challenge, this study introduces a bis-pyridyl anthracene-based fluorophore, TB4Py, as an AIEgen that exhibits reversible acidofluorochromic behavior in both solution and solid states through a rare structural deformation event. Upon protonation, TB4Py undergoes a significant and reversible distortion from planarity within its anthracene core, distinguishing it from conventional fluorescent probes. Such a structural deformation with an anthracene ring bend angle of ∼40.5° drives a redshift in emission and pronounced fluorescence (FL) quenching. This weakly emissive platform offers a visually distinct and reliable turn-on response for detecting aliphatic polyamines/biogenic amines (PAs/BAs) and nicotine (NIC) through a pronounced blueshift in color change (∼73 nm), enabling detection in both solution and solid states. The solution state detection was relatively more selective towards putrescine. While multiple mono/polyamine vapors responded to the solid state, the emission was relatively more intense for PAs. Incorporating distant, dual pyridine units significantly amplifies the sensitivity and selectivity of TB4Py. This advancement is exemplified through practical applications, such as detecting fish spoilage, highlighting its utility in food safety and quality monitoring. This study emphasizes the largely untapped potential of structural deformation in stimuli-responsive fluorescent probes, offering novel approaches to enhance chemical sensing technologies.
Introduction
Stimuli-responsive fluorescent organic molecules represent an intriguing research field that primarily focuses on the development of optoelectronic materials.1–5 The notable merit of such research is that it circumvents the need for further chemical modifications to create molecules displaying various emission behaviors.6,7 The enormous endeavour was dedicated to designing molecules that hold various functional groups/cores to enable the creation of various tunable supramolecular interactions (C–H⋯π, C–H⋯N, C–H⋯O, etc.), leading to luminescent materials that respond to temperature (thermofluorochromic),8–11 mechanical force/pressure (mechanofluorochromic),12–14 chemical vapor (vapochromic)15,16 and so on. Among such materials, acidofluorochromism is promoted to display reversible emission switching by various molecular materials, which respond differently to acid/base stimuli.17,18 The protonation–deprotonation process affects the molecular conformation and frontier molecular orbitals (FMOs), which are primarily responsible for shifting the emission.19 The pyridyl groups with an sp2 nitrogen atom with a free lone pair of electrons are capable of producing multiple noncovalent interactions as H-bond acceptors and are often linked to various emitters to access stimuli-responsive materials, more importantly, acidofluorochromic ones.19,20 The effect is very much operative in the probe's solution/solid state with acid/alkali vapor and can be easily recognized visually with very good reversibility. Such materials are appropriate for pH detection in various microenvironments.21–23 Apart from typical amine vapor sensing, the application has been successfully extended to testing crucial biogenic amines (BAs), such as putrescine (PUT) and cadaverine (CAD), which are key indicators of the freshness of protein-rich foods.24–26The non-emissive protonated forms of 1,2-dihydroxyquinoxaline turned into respective emissive deprotonated forms of A and B (Fig. 1) and were utilized to detect ammonia and many volatile BA vapors. Both A and B have been effectively used to monitor the spoilage of protein-rich food items visually.26,27
 | ||
| Fig. 1 Earlier reported 1,2-dihydroxyquinoxaline (A) and (B), imidazole-based donor–π–acceptor (D–π–A) (C), and anthracene-linked unsymmetrically (D) and symmetrically (E) and (F) substituted π-conjugated pH-sensitive fluorophores. | ||
Building on these precedents, imidazole-based donor–π–acceptor (D–π–A) fluorophores (Fig. 1C)28a have emerged as versatile optoelectronic materials, valued for their bipolar character and tunable photophysical properties. The multiple nitrogen sites within the imidazole core allow for selective protonation, enabling efficient acidochromic sensing with reversible emission switching. Incorporation of imidazo[1,5-a]pyridine donors and rigid π-spacers, such as diethylfluorene, enhances dual-state emission and aggregation-induced emission (AIE) properties.28b These design strategies yield fluorophores with high quantum efficiency, pronounced pH responsiveness, and excellent stability, making them strong candidates for sensing, environmental monitoring, and next-generation optoelectronic applications. In this context, previously investigated pyridyl-π-conjugates of type D (Fig. 1) exhibited efficient reversible acidochromic behavior.20
Moreover, symmetrical divinylanthracene (DVA)-based pyridyl-π-conjugates are well-established AIEgens and multifunctional dyes.28c,29 Specifically, compounds E and F exhibit notable capabilities as AIEgens and solid-state emitters,28c,29 demonstrating tunable emission spectra in response to external stimuli such as mechanical stress, temperature, and acid vapors. This versatility highlights their attractiveness for sensing applications and the development of smart materials. While both compounds have been reported to detect volatile amine vapors, their capability to sense critical biogenic amines (BAs) remains unexplored.
Previous studies on compound E revealed no significant structural changes in the anthracene core upon protonation,28c and similarly, compound F, which initially adopts a Z conformation, did not undergo conformational alterations after protonation.29 Despite several reports on protonation–deprotonation using pyridyl chromophores, we are unaware of reports where structural deformation occurs upon protonation. Furthermore, these observations suggest that protonation-induced structural changes are predominantly localized to the pyridyl moiety rather than the anthracene core, and there are no significant reports on anthracene core deformation (twisted) upon pyridyl ring protonation. This insight opens avenues for fine-tuning sensor design, leveraging these molecular systems to enhance selectivity and sensitivity for BA detection, which is crucial for applications in food safety and medical diagnostics.
Thus, with our current focus on developing an efficient solid-state emitting acidochromic platform,20,30,31 we herein present a new class of conformationally twisted π-system, TB4Py, centrally supported by a small thiophene ring, which is symmetrically substituted with anthracene-π-pyridyl units, hanging on both sides. This TB4Py is recognized as a decent AIEgen with yellowish-green emission, but it becomes weakly emissive (red) upon protonation. The protonation of this dye resulted in notable structural disruption in the anthracene core due to enhanced electronic conjugations from the middle ring of anthracene to the pyridinium core, as corroborated by the DFT (density functional theory)-optimized structure. Moreover, the system demonstrated a selective and rapid response to nicotine (NIC) and biogenic amines (BAs) in both solution and solid-state forms, governed by protonation–deprotonation processes. The reversibility between distorted and planar anthracenyl cores, resulting in on–off signals, is utilized in detecting selective BAs and NIC in different phases. Analytical techniques, such as FT-IR, 1H-NMR, and PXRD, were employed to substantiate these findings, highlighting the role of protonation dynamics in sensor selectivity. This newly designed molecule is conveniently used to monitor fish spoilage and has potential real-life applications.
Design strategy of pyridyl-based molecular probes
This study investigates the effect of incorporating dual pyridine units (TB4Py) with two active sites, in contrast to the mono-pyridyl motif (T4Py, D in Fig. 1), on the sensor's responsiveness to biogenic amines (BAs) and nicotine (NIC) in different phases.Therefore, with this thought, a symmetrical π-conjugated bis-pyridyl anthracene-based molecule (Fig. 2) was rationally designed considering the (a) introduction of the dual pyridyl unit for better-extended π-conjugations through the distinguished anthracene fluorophore, (b) incorporation of the tiny stable thiophene donor as a central core to facilitate the conformational twist and enhance solid-state emission before protonation, and (c) utilization of the basicity of a couple of pyridyl rings through the lone pair of the N-atom, which can be easily protonated and result in weak emission. The stabilization of the excited state via extended conjugations, followed by molecular coplanarity, creates a smaller energy gap, leading to non-radiative relaxations.32
 | ||
| Fig. 2 Strategic design of symmetrically substituted anthracene-linked pyridyl π-conjugates. | ||
Results and discussion
Synthesis and characterization
The dual-emissive TB4Py compound was synthesized by combining a thiophene unit and anthracenyl hydroxyl-methylphosphonate. The thiophene moiety, known for its reactivity towards electrophilic aromatic substitution reactions, enabled a direct one-step Friedel–Crafts-type arylation with anthracenyl hydroxyl-methylphosphonate (AnP).33This metal-free synthetic approach, conducted at 83 °C, resulted in the formation of TBP, which was subsequently utilized for synthesizing TB4Py through simple Horner–Wadsworth–Emmons (HWE) reactions (Scheme 1) with a satisfactory yield. All the synthesized compounds are well characterized by FT-IR, multinuclear-1H, 13C, and 31P NMR, and high-resolution mass spectrometry (HR-MS). Thermal gravimetric analysis (TGA, Fig. S1) of these compounds shows stability up to 160 °C.
 | ||
| Scheme 1 Synthesis of thiophene anthranyl bis-phosphonate (TBP) and bis pyridyl anthracene π-conjugate (TB4Py). | ||
Photophysical studies
To explore the solvatochromic behavior of the TB4Py fluorophore, its absorption and emission properties were investigated in a range of solvents with different polarities. The absorption spectra displayed a band near λmax ∼ 405–409 nm, indicating a π–π* transition (Fig. S2a), and an emission band near λmax ∼ 485–495 nm (Fig. S2b) with a stronger emission in non-polar solvents. The emission with structured bands was noticed in a few solvents, as presented in Fig. S2b. However, no impressive solvent dependent absorption and emission shift was observed for TB4Py (mentioned in Table S1), indicating the inefficacy of the pyridyl ring in generating a significant donor–acceptor property in the molecule.Aggregation-induced emission (AIE)-property of TB4Py
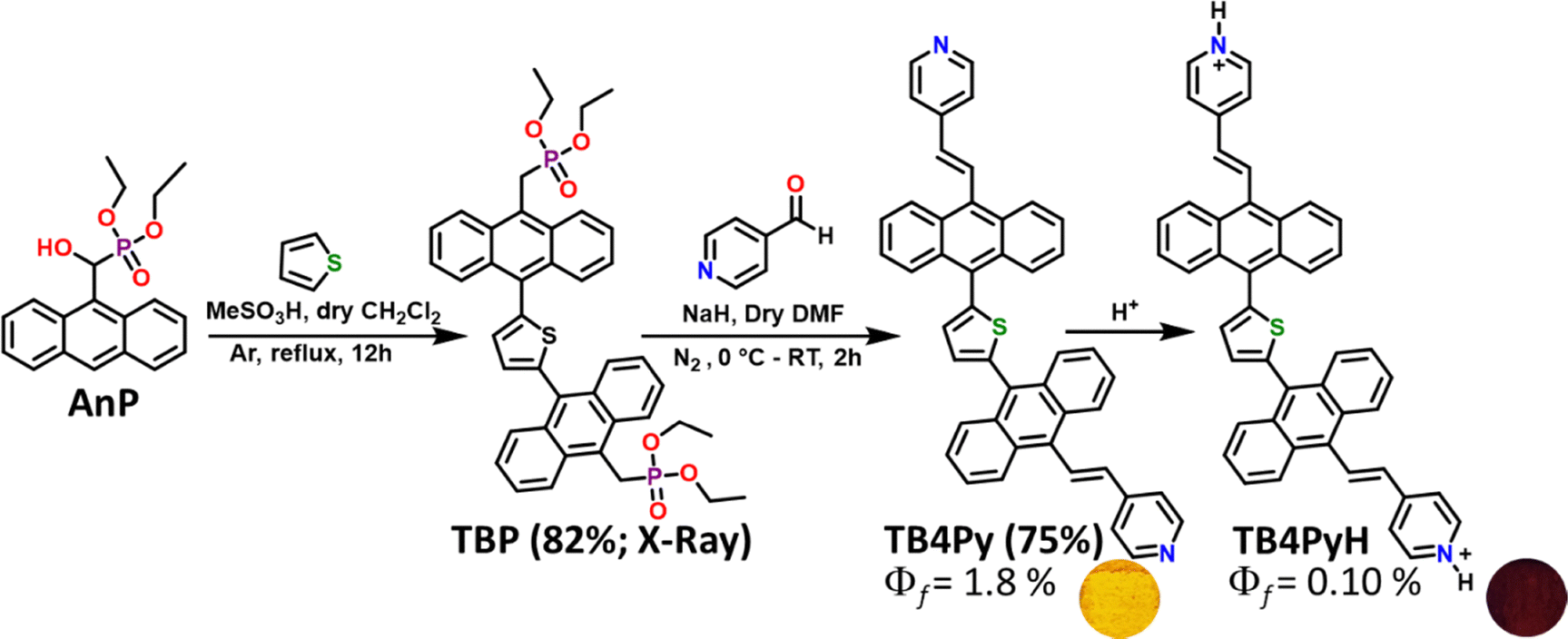
As we observed a poor emission of TB4Py in a highly polar and water-miscible solvent, acetonitrile (MeCN), the AIE property of TB4Py was further investigated by measuring absorption and emission using the MeCN/H2O solvent medium with gradual increase of water [fraction of water fw (v/v)] in a 10 µM acetonitrile solution of the probe (Fig. 3a and b).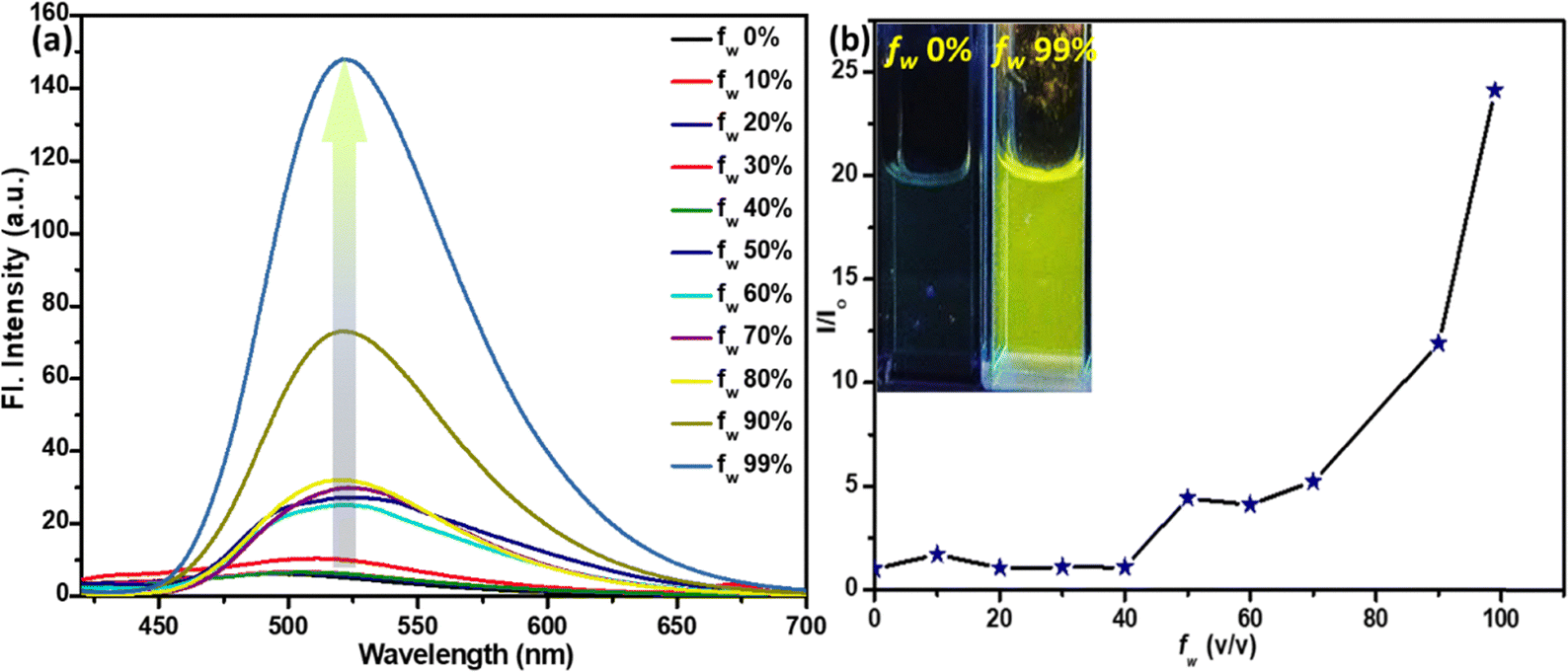 | ||
| Fig. 3 (a) The emission spectra of TB4Py, λex = 406 nm; (b) the I/I0 plot [I0: Fl. intensity before the addition of water, I: Fl. intensity after the addition of water]. The concentration of the probe is 10 µM. Inset: the image was taken at fw = 0% and fw = 99% for TB4Py under a 365 nm UV lamp. | ||
Notably, an apparent increase in the FL intensity was observed after fw ∼ 80%, reaching a maximum enhancement at fw ∼ 99% (24–25 times) with a redshifted emission (λem = 522 nm) (Fig. S2c shows the corresponding absorption spectra). The twisted molecular conformation does not allow a close-packed aggregate; instead, it forms an aggregate, which enables restricted intramolecular motion to display enhanced emission. To further probe the aggregate characteristics, dynamic light scattering (DLS) measurements were performed, revealing an average particle size of ∼557 nm (Fig. S3), confirming the formation of aggregates in the aqueous medium. To further substantiate the AIE phenomenon, viscochromism studies were performed, which revealed a sudden enhancement in fluorescence intensity at fg = 90% (Fig. S4), providing additional evidence for the restricted intramolecular motion mechanism that underlies the enhanced emission characteristics. However, the observed fluorescence intensity reduction at fg = 99% is likely attributed to the formation of precipitates, which can disrupt the optimal aggregate structure and reduce the emission efficiency.
Furthermore, molecules with a couple of conjugated anthracene rings with a central thiophene core will induce a strong hydrophobic microenvironment, possibly favoring the radiative emission. The redshifts in both absorption and emission indicate a possible J-type aggregate formation. Time-resolved fluorescence lifetime measurements further corroborate the AIE behavior, revealing a marked increase in the average lifetime in the aggregated state (fw = 99%; τ = 0.66 ns) compared to that in pure solution (fw = 0% in MeCN; τ = 0.03 ns). This enhanced excited-state lifetime (Fig. S5 and Table S2) arises from the effective restriction of intramolecular motions (RIM) and the concomitant suppression of non-radiative decay channels upon aggregation. The extended lifetime thus signifies that molecular motions responsible for energy dissipation are efficiently constrained, thereby promoting radiative decay and strengthening the emission. These findings provide direct evidence supporting the proposed AIE mechanism.
Protonation-induced photophysical changes in the solution/solid phases
To investigate the protonation capability of TB4Py initially, the HCl concentration was gradually increased. A noticeable change in emission from green to a faded orange was observed under a 365 nm UV lamp. Interestingly, the original color was restored upon adding diamines or polyamines (PAs), suggesting that protonation and deprotonation occur through the lone pairs on the pyridyl nitrogen atoms. To further explore this observation and bis-pyridyl TB4Py, UV-vis (Fig. S6a) and fluorimetric titration (Fig. 4a) were carried out upon incremental addition of HCl solution (10−2 M in water) from 0 µM to 175 µM to the probe TB4Py (10 µM in 1,4-dioxane). A reduction in the emission intensity at λmax = 496 nm was noticed, and gradually, this signal was broadened and shifted to λmax ∼ 570 nm, which became prominent after adding 100 µM HCl solution. Such a change remained intact even after adding 175 µM of HCl. This broad and shifted signal signifies a stable protonated product (TB4PyH) formation in the solution state. Such a protonated species can be easily deprotonated, maintaining an equilibrium between them (Fig. 4a). | ||
| Fig. 4 The emission spectra of TB4Py in (a) solution (10 µM in 1,4-dioxane) upon the gradual addition of 10−2 M HCl [λex = 407 nm] and (b) the solid state before and after exposure to 50 µL of HCl vapor for 30 minutes in a closed vial. | ||
Next, the protonation impact on the optical properties of the solid-state fluorophore was observed. The efficient yellow-emitting TB4Py (λem = 598 nm; φf = 1.8%) platform was exposed to HCl vapor for 30 minutes, resulting in a redshift (13 nm) and quenched emission (negligible emission with estimated Φf = 0.10%; Fig. 4b).
For comparison, the monopyridyl analog D was realized to display a 108 nm redshift upon protonation in the solid state.20 Such an enormous difference in the protonation between mono- and bis-pyridyl analogs indicates a substantial difference in their molecular structure after protonation. The absorption spectrum of the TB4Py solid was also significantly redshifted by 70 nm after protonation (Fig. S6b). The emergence of distinct, single emission peaks near λem ∼ 611 nm in the solid state (Fig. 4b) suggests that all pyridyl nitrogen atoms in TB4Py are completely protonated. The protonation of the pyridyl units in TB4Py is further supported by 1H-NMR spectroscopy, which reveals the formation of a stable protonated TB4PyH salt. The spectrum exhibits a distinct broad signal near δ9.03, assignable to the two newly added protons (Fig. S8). Additionally, the observed downfield shifts (∼δ0.25) of other aromatic proton signals indicate an electron-withdrawing effect within the molecule. Moreover, the FL quenching of TB4Py upon protonation can be attributed to the deformed Z-shaped conformation that is adapted to maintain better electronic conjugations (discussed later) after forming pyridinium cations. This modulation of photophysical properties, based on a simple protonation–deprotonation process, renders TB4PyH as an ideal candidate for sensing toxic amine vapors, with potential applications in environmental monitoring and industrial safety.
Experimental support for the deformed molecular conformation after protonation
The theoretical outcome of the deformed molecular structure upon protonation was validated by 2D-NMR, specifically the NOESY (nuclear Overhauser effect spectroscopy) experiment (Fig. S8a), which demonstrates correlations between protons with close spatial proximity. The bent anthracene is anticipated to exhibit better through-space interactions in TB4PYH, whereas they would be far less pronounced in the planar state in TB4PY. In the deprotonated state (TB4Py), the NOESY spectrum exhibited limited through-space correlations between the pyridinyl proton and other aromatic protons, indicating a relatively planar geometry of the anthracene core. However, upon protonation (TB4PyH), a distinct increase in cross-peak intensities was observed, signifying enhanced spatial proximity between the protonated pyridinyl proton and multiple protons on the anthracene framework. This emergence of new or intensified NOE interactions suggests a notable structural deformation, wherein protonation likely induces a bending of the anthracene ring to minimize electrostatic repulsion and stabilize the pyridinium cores. The experimental evidence from NOESY thus aligns well with the DFT-optimized geometry, which reveals a clear deviation from planarity of the anthracene core after protonation. Furthermore, a closer look at the 1D-1H-NMR spectra of both protonated and deprotonated compounds reveals that the coupling constant between two anthracenyl protons is reduced after protonation (Fig. S8b), indicating a change in the dihedral angles between the two hydrogen atoms, which originates from the deformed anthracenyl structure.The response towards BAs/nicotine in the solution phase
The mono-pyridyl probe D (T4Py, Fig. 1) was established as a potential platform for acidofluorochromism, but the response against other vital amines was not explored.20 In this study, we extend this concept to a bispyridyl system (TB4Py) to investigate its acidofluorochromic response. Comparative studies were conducted on TB4PyH (protonated TB4Py) in solution with various amines, including polyamines (PAs, 1 mM in DMAc), nicotine (1 mM in water), and myosmine (1 mM in 1,4-dioxane; Fig. S7). Notably, myosmine is another naturally occurring tobacco alkaloid, structurally similar to nicotine. The emission shift upon deprotonation was significant (maximum shift of 73 nm) in many cases, demonstrating the influence of TB4Py's structural features, particularly its two pyridinium units, which provide multiple reactive sites and enhance its acid-responsive behaviour (Fig. 5a and Table 1). Among various amines screened (listed in Fig. 5), significant, blue-shifted emission was identified for almost all the PAs; in particular, a notable change from non-emissive brown to emissive green (original deprotonated TB4Py) was distinguished only for putrescine (PUT) and nicotine (NIC) with a maximum blueshift of 73 nm and 72 nm, respectively (Table 1 and Fig. 5a). The myosmine solution was unable to deprotonate the probe. It could not restore the emission of the probe, specifying that the higher basicity (pKa ∼ 8) of nicotine compared to myosmine (pKa = 6.66) is most important for the proton exchange. Thus, this probe is specific to detect nicotine. | ||
| Fig. 5 The emission spectra of (a) TB4PyH (10 µM in 1,4-dioxane) after the addition of various biogenic amines (1 mM in DMAc)/nicotine (1 mM in water) in the solution state; (1) ammonia; (2) butylamine (BuA); (3) aniline; (4) triphenylamine; (5) diphenylamine; (6) ethylenediamine (EDA); (7) triethylamine (TEA); (8) 2-phenylethylamine (PEA); (9) tryptamine; (10) N-ethyldiisopropylamine; (11) 1,3-diaminopropane; (12) putrescine; (13) cadaverine; (14) 1,6-diaminohexane; (15) spermidine; (16) spermine; and (17) nicotine; [λex = 407–420 nm]; TB4PyH [10 µM in 1,4-dioxane] upon gradual addition of (b) PUT (1 mM) [λex = 406 nm] and (c) NIC (1 mM), with different volumes (in µL); λex = 411–418 nm. | ||
| Amine | TB4PyH [λex (nm)] | TB4PyH [λem (nm)] | TB4PyH [Δλ (nm): 570 − λem] |
|---|---|---|---|
| TB4PyH | 425 | 570 | — |
| BuA | 421 | 571 | 1 |
| EDA | 414 | 512 | 58 |
| Aniline | 412 | 533 | 37 |
| TPA | 423 | 541 | 29 |
| DPA | 424 | 560 | 10 |
| TEA | 424 | 560 | 10 |
| PEA | 423 | 528 | 42 |
| Tryptamine | 413 | 548 | 25 |
| DIPEA | 413 | 546 | 24 |
| 1,3-DAP | 423 | 569 | 1 |
| Putrescine | 411 | 497 | 73 |
| Cadaverine | 412 | 516 | 54 |
| 1,6-DAH | 419 | 555 | 15 |
| Spermidine | 413 | 552 | 18 |
| Spermine | 415 | 514 | 56 |
| Nicotine | 407 | 498 | 72 |
Notably, the green emission intensity of TB4Py was considerably diminished upon protonation. A slight greenish emission was also observed for a few other PAs, such as EDA, CAD, and spermine. These outcomes reveal that NIC and PUT were most suitable for abstracting the protons from TB4PyH and achieving distinct color changes. Such a difference in the degree of deprotonation responses could arise due to several reasons, including the basicity of the amines, chain length, and stability of the protonated diamines through the proton transfer process. Upon protonation, the nitrogen atoms in the pyridine rings become positively charged and act as hydrogen bond donors. These protonated nitrogen atoms interact with the lone pairs on the nitrogen atoms in diamines/BAs (like PUT) or NIC, forming hydrogen bonds, resulting in specific and robust binding, as substantiated by detailed 1H NMR spectral analysis (Fig. 7a, vide infra).
Also, an equilibrium between protonated and deprotonated states mainly governs the regaining of the color change. However, the FL intensity does not fully recover to its original state, likely due to the formation of salts between the amine and acid. Moreover, dual pyridine units in structurally deformed TB4PyH significantly boost its binding capabilities.
The selective emission response observed in the PUT and NIC experiments motivated us to perform fluorescence titration experiments with PUT and NIC (Fig. 5b and c), which revealed a gradual color shift from a faint orange to green upon the addition of these analytes. TB4PyH achieved a remarkable detection limit of 2–3 µM for both PUT and NIC (Fig. 5b and c), showcasing its decent sensitivity. The experimental detection limits appeared to be 0.11 and 0.25 µM for PUT and NIC, respectively (using 3.3σ/k; considering lower concentrations; Fig. S9).
The deprotonation of TB4PyH in solution, following the gradual addition of PUT and NIC, is further reflected in the absorption spectrum, which exhibits a blue shift of 19 nm (Fig. S10). The increased basicity of the amine and higher acidic character of the probe in the excited state facilitate this protonation–deprotonation process, leading to a superior emission shift. Putrescine differs slightly from cadaverine and spermidine in terms of its basicity and nucleophilicity. Furthermore, the relatively smaller chain length in putrescine would favour the geometry (cyclic transition state) formed during the proton transfer.
The response towards PA vapor/nicotine in the solid phase
Based on the encouraging results from the solution phase, we further investigated the response of the fluorophores to various amine vapors in the solid state. This approach prioritizes operational simplicity and holds promise for practical applications. Protonated fluorophore-coated thin film glass coverslips were employed for vapor sensing experiments. Notably, exposure to 30 µL of various amine vapors within a sealed 200 mL jar for 30 minutes at 298 K led to a distinct color change in the fluorophore.Initially, the fluorophore TB4Py in the solid state exhibited significant differences in the ground state, as reflected in the absorption spectra (Fig. S11). The UV absorption of solid-state TB4Py corresponds to a yellow dye with an absorption maximum (λmax) at 454 nm. Upon exposure to HCl vapor, TB4Py underwent protonation, resulting in a red solid [protonated form (TB4PyH)] and displaying a λmax shift to 524 nm, indicating a substantial 70 nm red shift. Interestingly, when exposed to putrescine or nicotine vapor, the emission was shifted and displayed a λmax at 461 nm, reverting to a yellow dye and demonstrating a blueshift in the solid-state absorption spectra. Furthermore, this change was accompanied by a significant blueshift in emission, resembling the emission of the AIE state, and an increase in absolute φf (%) for most biogenic amines (BAs) (Fig. 6 and Table S3).
 | ||
| Fig. 6 (a) Normalized FL spectra of TB4PyH thin film before and after exposure to different amine vapors (λex: 432 nm). (b) Emission change in the thin film state (1) TB4PyH; after exposing to (2) ammonia; (3) butylamine; (4) ethylenediamine; (5) 1,3-DAP; (6) PUT; (7) CAD; (8) 1,6-diaminohexane; (9) 2-phenylethylamine; (10) N-ethyl diisopropylamine; (11) SPM; and (12) spermine; All the images were taken under a 365 nm UV lamp. | ||
The different optical responses observed for ammonia and BAs highlight variations in binding affinities. For ammonia, a blueshift from red to yellow (74 nm) emphasizes a moderate interaction with the protonated pyridyl group of TB4PyH. In contrast, the BAs exhibit a pronounced blueshift from red to green within the 96–99 nm range. The enhanced response observed with BAs can be attributed to the stronger interactions between the pyridyl proton of TB4Py and the N-atoms of the two amine groups, facilitating more effective neutralization of the protonated pyridyl part. Additionally, two H-bonding interactions between the probe and the –NH2 functionality likely play a crucial role. In contrast, the weaker interactions of monoamines, due to the presence of only a single amine group, limit their ability to fully restore the probe's original electronic environment. These optical changes, noticeable under ambient light, provide a visually distinctive and easily detectable signal, highlighting the practical applications of TB4Py for vapor-based amine sensing.
Also, incorporating dual pyridine units in the newly designed molecular sensor significantly impacts the responses against amine vapor compared to the previously reported mono-pyridyl anthracene-based fluorophore (T4Py). The mono-pyridyl T4PyH was relatively more emissive (φf = 3%) with a 108 nm redshift after protonation.20 Both side protonations in TB4PyH change the electronic distribution by pulling the thiophene electron cloud toward two pyridinium rings, resulting in almost fully quenched redshifted emission. The structural deformation in the protonated form of TB4Py (noticed from the DFT-optimized structure of TB4PyH) also offers new insight into this study.
Mechanistic insight into the solid-state emission change
The superior responses by nicotine and PUT motivated us to explore the mechanistic routes using only these two amines, which responded similarly. To validate the proton transfer mechanism from TB4PyH to nicotine (NIC), an FT-IR spectroscopic study was performed. The protonation of TB4Py resulted in the emergence of a new broad absorption band in the region of 3380–2400 cm−1, corresponding to the two N–H stretching, which was absent in TB4Py (Fig. S12). Furthermore, pyridyl ring protonation caused a significant downshift in the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching signal from 1583 cm−1 to 1497 cm−1 and the –CN stretching signal from 1681 cm−1 to 1616 cm−1. This shift indicates electronic rearrangement within the π-conjugated system, where protonation alters the electron density distribution, enhances the delocalization of π-electrons, and weakens the bond force constant, reducing the stretching frequencies. Hence, structural deformation is very likely to occur. The subsequent addition of NIC to TB4PyH resulted in a new broad absorption band at 3439 cm−1, which may indicate protonation in the nitrogen of nicotine. Such stretching may arise from a combined protonation of the pyridine and pyrrolidine rings in NIC, leading to overlapping N+–H stretching frequencies contributing to a broad absorption peak. Simultaneously, this observation is consistent with the deprotonation of TB4PyH, accompanied by a reverse upshift in the –CC stretching peak from 1497 cm−1 to 1583 cm−1 and the –CN stretching peak from 1616 cm−1 to 1681 cm−1, suggesting the restoration of the electronic environment in TB4Py upon proton transfer to NIC. These spectral changes strongly support the dynamic proton transfer mechanism between TB4PyH and NIC, emphasizing the molecular interactions and electronic state modulations involved in this process. The combined shifts in both stretching frequencies and peak intensities highlight the sensitivity of the system to proton transfer. Furthermore, a careful comparison between the FT-IR spectra of TB4PY and TB4PYH indicates that the aromatic CC stretching frequencies (cm−1) shifted from 1584 to 1496, and 1498 to 1430. The in plane CH bending frequency (cm−1) also showed a shift from 1116 to 1042 and 1207 to 1192; a few more CH out-of-plane frequency shifts were observed from 967 to 976 and 749 to 757. All these IR stretching shifts clearly indicate the change in the aromatic rings in terms of their connectivity.
C stretching signal from 1583 cm−1 to 1497 cm−1 and the –CN stretching signal from 1681 cm−1 to 1616 cm−1. This shift indicates electronic rearrangement within the π-conjugated system, where protonation alters the electron density distribution, enhances the delocalization of π-electrons, and weakens the bond force constant, reducing the stretching frequencies. Hence, structural deformation is very likely to occur. The subsequent addition of NIC to TB4PyH resulted in a new broad absorption band at 3439 cm−1, which may indicate protonation in the nitrogen of nicotine. Such stretching may arise from a combined protonation of the pyridine and pyrrolidine rings in NIC, leading to overlapping N+–H stretching frequencies contributing to a broad absorption peak. Simultaneously, this observation is consistent with the deprotonation of TB4PyH, accompanied by a reverse upshift in the –CC stretching peak from 1497 cm−1 to 1583 cm−1 and the –CN stretching peak from 1616 cm−1 to 1681 cm−1, suggesting the restoration of the electronic environment in TB4Py upon proton transfer to NIC. These spectral changes strongly support the dynamic proton transfer mechanism between TB4PyH and NIC, emphasizing the molecular interactions and electronic state modulations involved in this process. The combined shifts in both stretching frequencies and peak intensities highlight the sensitivity of the system to proton transfer. Furthermore, a careful comparison between the FT-IR spectra of TB4PY and TB4PYH indicates that the aromatic CC stretching frequencies (cm−1) shifted from 1584 to 1496, and 1498 to 1430. The in plane CH bending frequency (cm−1) also showed a shift from 1116 to 1042 and 1207 to 1192; a few more CH out-of-plane frequency shifts were observed from 967 to 976 and 749 to 757. All these IR stretching shifts clearly indicate the change in the aromatic rings in terms of their connectivity.
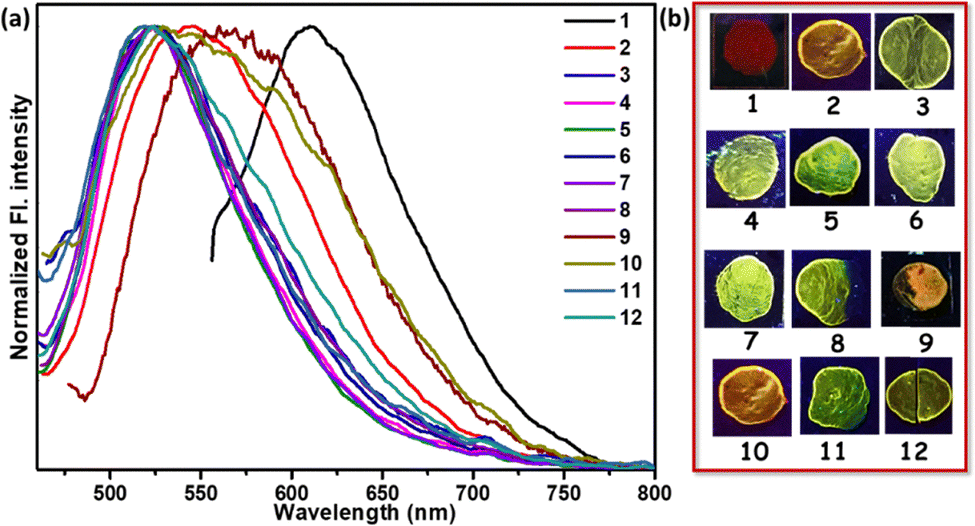
Furthermore, the proton exchange mechanism is supported by a 1H NMR study. TB4PyH+ displayed the downfield shifting (∼0.25 ppm; Fig. 7a) of the relevant signals, including the olefinic (trans) H2CCH (downfield shift from δ7.05 to δ7.30), proving the presence of pyridinium ions. Upon gradual NIC addition (0.2–1 eq. in DMSO-d6, Fig. 7a), the downfield-shifted signals of the probe began to return gradually to their original position, matching TB4PY. The clear and gradual upfield shifts in the olefinic (trans) H2CCH signal upon slow addition of NIC solution were observed from δ7.30 → δ7.16 (0.2 equiv. NIC) → δ7.05 (0.4 equiv. NIC). However, the original signals from nicotine were shifted (∼0.235 ppm) downfield, signifying the protonation of the nitrogen atom in nicotine, as indicated in Fig. 7a. This sequential shift pattern strongly validates the fact that the proton is transferred from TB4PyH to nicotine (a stronger base than myosmine), highlighting the dynamic nature of their binding and interaction processes. Of note, one of the identified coupling constants (3JHH) due to trans-CHCH differs between TB4Py (16.8 Hz) and TB4PyH+ (16.4 Hz), specifying a change in the dihedral angle between interconvertible protonated and deprotonated forms.
 | ||
| Fig. 7 (a) The partial 1H-NMR spectra (only aromatic part) of TB4Py, TB4PyH, and after the addition of 0.2 equiv., 0.4 equiv., and 1 equiv. of NIC, dissolved in DMSO-d6; (b) changes in the PXRD pattern of TB4Py before and after HCl and PUT vapor exposure for 15 minutes. | ||
We also monitored the proton transfer in the case of PUT using 1H NMR, revealing similar signal shifts described in Fig. S13.
To acquire an in-depth understanding of the phase transition and the emission changes upon protonation–deprotonation of TB4Py, powder X-ray diffraction (PXRD) analysis was performed. Initially, both fluorophores exhibited a well-ordered crystalline phase in the pristine state; however, upon fumigation with HCl vapor, the disappearance of almost all sharp signals indicates the formation of a new amorphous phase (Fig. 7b).
This transformation is likely attributed to the protonation of the molecule, leading to the formation of an effective intermolecular interaction between the counter chloride ion and the protonated species. Additionally, the morphological change was confirmed by scanning electron microscopy (SEM) (Fig. S14), indicating that the crystalline properties of the original sample were altered to an amorphous phase upon exposure to acid vapor. This amorphous phase eventually reverted to its initial crystalline phase upon exposure to amine vapor, with a reversibility of more than five cycles (Fig. S15). However, the PXRD patterns of the deprotonated form (TB4PyD) with a few more strong/weak diffraction signals indicate the presence of amine salts.
Theoretical investigations and experimental support
A comparative study of the molecular geometry of TBP (experimentally determined from single-crystal X-ray data), TB4Py, and TB4PyH was conducted to gain deeper insights into the observed phenomena at the molecular level. The DFT (density functional theory) optimized molecular structures (gas phase) of TB4Py and TB4PyH were generated in the ground state. The experimentally determined crystal structure of phosphonate TBP revealed an initial twisted geometry with an interplanar angle of ∼80° (Fig. 8c) between the thiophene and anthracene rings. We were delighted to note the anthracene–thiophene torsion angle of 88.68° in TB4Py, like the TBP system. It supports the DFT-optimized molecular structure, which further exhibits a puckered conformation with significant torsion angles, particularly between the anthracene and pyridine rings (71.30°). The anthracene ring in experimentally determined TBP and optimized TB4Py was almost planar, without any distortion/bending, as evidenced by maintaining C–C bond distances in the range of 1.354–1.445 Å (Fig. 8a and c), which is still suitable for a twisted molecular conformation and enhanced emission. Interestingly, the theoretically determined structure of TB4PyH adopted a deformed Z-shaped conformation upon protonation with a notable ∼40.6° bend angle in each terminal phenyl ring of the anthracene ring (Fig. 8b). Of note, the centered thiophene ring intends to attain planarity with both the anthracene rings (interplanar angle of 3.97°), achieving tub-shaped anthracenyl rings facing different sites and, overall, attained Z-shaped conformation (Fig. 8b). The interplanar angle between the anthracene and pyridine rings was reduced to 31.18° in TB4PyH (after protonation) from 71.30°, detected in TB4Py. | ||
| Fig. 8 (A) The DFT optimized [CAM-B3LYP/6-31G(d) level of theory] ground state (S0) molecular structure for (a) TB4Py and (b) TB4PyH; (c) experimentally (single crystal X-ray) determined molecular structure of the bisphosphonate TAP; (B) Frontier molecular orbital (FMO) contributions of TB4Py and TB4PyH using the CAM-B3LYP/6-31G(d) level of theory. | ||
The C–C bond lengths (1.387–1.478 Å) were significantly increased in TB4PyH, specifying strong electronic conjugations and stabilizing the system considerably. The substantially improved coplanarity of the molecule upon protonation is likely responsible for the observed redshifted emission and lower quantum yield. We further noticed a significantly shorter C–C bond (1.358 and 1.360 Å) outside the anthracene ring in TB4PYH, compared to TB4PY, which showed the same bonds with a length of 1.483 Å, indicating the presence of a quinoid form. Such quinoid forms in the bending acene were proposed before as a cause of fluorescence quenching.34 Such bending in the anthracene ring was also earlier investigated theoretically, which showed the enhancement in chemical reactivity35 and electronic properties were significantly altered.36 Notably, the bending angle of the anthracene ring is 40.6°; such a high bending angle is beyond expectation. Earlier theoretical studies on the bending of the anthracene ring35,36 support our findings regarding optical properties.
The frontier molecular orbital (FMO) distributions and energy levels of TB4Py in its protonated and deprotonated forms are determined based on optimized geometries. Before protonation, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are predominantly localized on the divinyl anthracene ring, exhibiting a substantial band gap of 5.473 eV (Fig. 8b). The electronic distributions remain similar even after protonation, but the energies of both the HOMO and LUMO were increased due to the distorted structure. However, a reduction from 5.473 eV to 4.423 eV in the energy band gap is observed after protonation. This ∼1.05 eV band gap reduction is responsible for a red shift in the absorption and emission spectra of TB4Py. Earlier calculations also showed a smaller energy gap between the singlet ground state and the triplet state of molecular anthracene in its bent form.36 The emission could also be attributed to such a lower energy gap in TB4PYH.
On-site detection of nicotine using paper strips and detection of the freshness of fish
A straightforward and cost-effective approach utilizing fluorophore-dispersed paper strips was implemented for the facile detection of nicotine (Fig. 9a). Subsequently, 5 µL of varying nicotine concentrations in aqueous solutions was dropped onto the emissive platform. A control experiment used only the solvent to monitor potential solvent-induced shifts in wavelength. This control experiment demonstrated unaltered emission, thereby eliminating the solvent as a cause of the observed color change. A visually detectable color switching was observed up to a concentration of 10 µM. All images were taken under a 365 nm ultraviolet lamp. | ||
| Fig. 9 (a) Images taken after treating TB4PyH dye-dispersed paper with various nicotine concentrations under a 365 nm UV lamp, (b) application of the probe in detecting the freshness of fish and chicken. | ||
Considering the urge to provide fresh food to consumers, fluorophores can be utilized to determine the freshness of fish samples through visually detectable color changes caused by the protonation–deprotonation effect. To provide a handy solution, drop-cast Whatman filter paper was used inside a sealed glass jar containing fresh fish samples. Furthermore, a control jar was used to eliminate the effect of moisture on color variation. Variations in wavelength shifts were observed at different time intervals under a 365 nm UV lamp (Fig. 9b). The initial shifts from red to yellow were first observed after 19 hours, and then the shifts became more pronounced after 23 hours. Therefore, such a prominent color change makes the probe ideal for use as a handy food freshness indicator.
Conclusions
In summary, we report here a new structurally twisted thiophene-centered bianthracenyl bis-phosphonate that produces a bis-pyridyl anthracenyl π-conjugate as a new AIEgen. This emissive bis-pyridyl fluorophore was quickly and reversibly responsive to acid in both solution and the solid state, displaying almost no emission. Upon treatment with various amines, this non-emissive salt regains its emission, and the response was visually detectable only for putrescine and nicotine with a 2–3 µM sensitivity because they could exhibit a significant original emission. We substantiated the fact with the support of 1H NMR studies. The regaining of green emission was also observed upon exposing amine vapor to the non-emissive solid protonated probe, where most of the diamines could retrieve the original emission. Unlike previous reports, the protonated bipyridyl compound showed a deformation in the molecular structure (DFT-optimized) where both the anthracene rings are bent, ∼40.6° (the highest bent beyond thought). Such a distortion occurs because the core thiophene ring is in conjugation with the central ring of anthracene. A notable Z-type molecular structure was observed, where two anthracene rings were bent and oriented in different directions, and the associated emission quenching was described. The experimentally determined X-ray structure of the precursor phosphonate somewhat supports the theoretically determined structures. Furthermore, real-world application was demonstrated to detect the freshness of fish by monitoring the color change from red to yellow. Such a finding of the anthracene ring's bending effect in stimuli-responsive systems would serve as a design principle for acidofluorochromic molecules and offer various on-site applications.Experimental section
Materials and general conditions
All the chemicals were purchased from Merck. The solvents for column chromatography were purchased from Finar. Spectroscopic grade solvents were purchased from Finar and used for UV-vis and fluorescence spectroscopy. All the photophysical studies were performed at room temperature, 298 ± 2 K. The 1H, 13C, and 31P NMR spectra were recorded on Bruker 400 MHz spectrometers with operating frequencies of 101 MHz for 13C. Chemical shifts (δ) are reported in ppm relative to the residual solvent signal (δ2.50, δ7.26 for 1H NMR and δ39.5, δ77.0 for 13C NMR).Diethyl ((10-(thiophen-2-yl)anthracen-9-yl)methyl)phosphonate32,33. This compound was synthesized following earlier reported methods.
1H NMR (400 MHz, CDCl3) δ8.41 (d, J = 8.9 Hz, 2H), 7.89 (d, J = 8.8 Hz, 2H), 7.63–7.54 (m, 3H), 7.42 (dd, J = 8.7, 6.5 Hz, 2H), 7.31 (dd, J = 5.2, 3.4 Hz, 1H), 7.19 (m, 1H), 4.29 (d, J = 22.5 Hz, 2H), 4.08–3.83 (m, 4H), 1.17 (t, J = 7.1 Hz, 6H).
Synthesis of tetraethyl ((thiophene-2,5-diylbis(anthracene-10,9-diyl))bis(methylene))bis(phosphonate) (TBP)
In a 100 mL round-bottom flask, diethyl(anthracene-9-yl(hydroxy)methyl)-phosphonate (1 g, 2.904 mmol) was dissolved in 20 mL of dry 1,2-dichloroethane at ice temperature under a nitrogen atmosphere followed by the dropwise addition of methanesulfonic acid (0.566 mL, 8.71 mmol) and thiophene (0.115 mL, 1.45 mmol). The reaction mixture was heated to reflux for 12 hours at 90 °C. The reaction was monitored by TLC. Upon completion, the reaction mixture was allowed to cool to room temperature, quenched with water, and extracted with dichloromethane (DCM, 30 mL × 3). The organic layer was dried over anhydrous sodium sulfate and concentrated using a rotary evaporator. The compound TBP was purified using column chromatography (100–200 mesh-sized silica gel) using 60% ethylacetate-hexane to obtain a pale yellow solid. The single crystals are obtained from a mixture of 60% of ethyl acetate–hexane.TBP: yield 82% (1.47 g), m.p. 265–270 °C, IR (ν cm−1, in KBr): 3346, 1638, 1566, 1473, 1392, 1317, 1235, 1121, 1079, 980. 1H NMR (400 MHz, CDCl3) δ8.36–8.34 (m, 4H), 8.13–8.11 (m, 4H), 7.56–7.47 (m, 8H), 7.27 (s, 2H), 4.24 (d, J = 22.8 Hz, 4H), 3.96–3.78 (m, 8H), 1.07 (t, J = 7.2 Hz, 12H). 13C NMR (101 MHz, CDCl3) δ140.7, 131.7, 130.2, 130.1, 129.0, 128.9, 127.4, 125.8, 125.7, 125.2, 62.3 (d, J = 6.8 Hz), 27.4 (d, J = 140.9 Hz), 16.3 3 (d, J = 5.9 Hz); HR-MS for C42H42O6P2S calc 737.2256, found 737.2262. X-ray structure was determined for this compound. CCDC No. 2412726.37
Synthesis of 2,5-bis(10-((E)-2-(pyridin-4-yl)vinyl)anthracen-9-yl)thiophene (TB4Py)
In a 50 mL round-bottomed flask tetraethyl ((thiophene-2,5-diylbis(anthracene-10,9-diyl))bis(methylene))bis(phosphonate) (0.20 g, 0.27 mmol) was dissolved in 20 mL of dry DMF under a nitrogen atmosphere at ice temperature, followed by the addition of NaH dispersed in 60% paraffin oil (0.052 g, 2.17 mmol) and stirred for 15 minutes. 4-Pyridinecarboxaldehyde (0.059 mL, 0.62 mmol) was added slowly to the reaction mixture and stirred for 2 h. The completion of the reaction was monitored by TLC. The resulting reaction mixture was quenched using water, extracted with ethyl acetate (20 mL × 3), dried over anhydrous sodium sulfate, and concentrated using a rotary evaporator. The compound TB4Py was purified using column chromatography (100–200 mesh-sized silica gel) using 50% ethyl acetate–hexane to obtain a yellow solid.TB4Py: yield 75% (0.130 g), m.p. 265–270 °C, IR (ν cm−1, in KBr): 3065, 2921, 2851, 1710, 1590, 1414, 1375, 1261, 1215, 1028, 966. 1H NMR (400 MHz, CDCl3) δ8.64–8.63 (m, 4H), 8.30–8.28 (m, 4H), 8.17–8.13 (m, 6H), 7.55–7.46 (m, 13H), 7.33 (s, 1H), 6.89 (d, J = 16.4 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ150.3, 144.4, 140.5, 135.4, 133.1, 131.6, 130.06, 129.8, 129.3, 127.2, 126.06, 125.8, 125.7, 121.08, 128.02. HR-MS for C46H30N2S, calc 643.2208, found 643.2193.
TB4PyH: yield: quantitative yield of 100% (0.130 g), m.p. 286–288 °C, IR (ν cm−1, in KBr): 3371, 3065, 2920, 2850, 1709, 1617, 1497, 1435, 1367, 1195, 966. 1H NMR (400 MHz, DMSO-d6) δ9.03 (bs, 2H), 8.93–8.89 (m, 3H), 8.47–8.42 (m, 8H), 8.18–8.16 (m, 5H), 7.73–7.67 (m, 10H), 7.57 (s, 2H), 7.30 (d, J = 16.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ153.6, 141.9, 140.01, 137.8, 133.7, 132.8, 131.4, 131.0, 129.3, 129.1, 127.3, 127.02 (one signal merged), 126.3, 124.62. HR-MS for C46H30N2S, calc 644.2275, found 644.2181.
Single crystal measurement
All measurements were carried out using a Rigaku XtaLAB P200 diffractometer using multi-layer mirror monochromated Cu-Kα radiation (λ = 1.54184 Å). The measurements were taken at a temperature of −173 ± 11 °C up to a maximum 2θ value of 149.81°. CrysAlisPro was used to collect and process the data (Rigaku Oxford Diffraction). Cu-Kα radiation has a linear absorption coefficient, µ, of 18.391 cm−1. The transmission factors ranged from 0.227 to 0.593 when an empirical absorption correction was implemented. The data were corrected for the Lorentz and polarization effects. The structure was solved using direct methods (SIR2011)31 and expanded using Fourier techniques. The non-hydrogen atoms were refined anisotropically. Hydrogen atoms were purified using the riding model. All calculations were performed using the Olex2 crystallographic software package except for the refinement, which was performed using SHELXL Version 2014/7.Methods and measurements
All the fluorophores were synthesized using the earlier reported method. The electronic absorption spectra were recorded with a UV 3600 Plus (Shimadzu). The FL spectra were recorded using a Hitachi spectrofluorometer (F7000) using a 1 cm path-length quartz cuvette. Origin Pro 8.5 software was used to plot the obtained data.Preparation of solutions
All the stock solutions of the fluorophores (10−3 M) were prepared in 1,4-dioxane. Stock solutions of the investigated amines (10−3 M) were prepared in N,N-dimethylacetamide (DMAc). A separate 10−3 M stock solution of nicotine was prepared in Milli-Q water.Absorption and FL studies
Absorption studies were carried out with the sample (2 mL, 10 µM) in a quartz cuvette (1 cm × 1 cm). The wavelength range was kept within 550 to 200 nm. The emission spectra of the same sample were instantaneously recorded in the range of 415 to 800 nm, with a PMT voltage of 400 eV and excitation slit/emission slit 5.0.Absolute quantum yield measurement
The solid-state absolute quantum yield measurement used a calibrated integrating sphere method with an absolute error of ±0.25%.Time-resolved decay measurement
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 counts. Ludox solution (an aqueous dispersion of colloidal silica) was used to measure the IRF for all solution samples. The solution state decay measurements were performed using a 10 µM solution of TB4Py before and after the incremental addition of different water fractions. All measurements were achieved at room temperature (298 K). The decay fitting was completed, keeping the χ2 value close to unity.
000 counts. Ludox solution (an aqueous dispersion of colloidal silica) was used to measure the IRF for all solution samples. The solution state decay measurements were performed using a 10 µM solution of TB4Py before and after the incremental addition of different water fractions. All measurements were achieved at room temperature (298 K). The decay fitting was completed, keeping the χ2 value close to unity.
where σ represents the standard deviation of the TB4PyH solution (10 µM), and K is the slope of the plot obtained from the Fl. titration experiment.
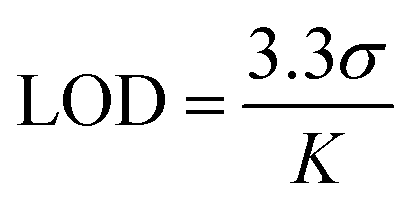
Theoretical studies
See the details in the SI.Paper strip sensing study
For the solid-state sensing study, a solid TB4PyH sample was dispersed on strips of Whatman filter paper (15 cm × 4.5 cm) and dropped into different concentrations of nicotine (2 µL) solution in water. The image was taken using a OnePlus Nord 2T 5G (f/0.8, ISO200) after drying at room temperature. However, the blue-shifted emission was instantaneous. Other information is articulated in the SI in detail.Conflicts of interest
There are no conflicts to declare.Data availability
This study was carried out using publicly available data that are mentioned in the manuscript.The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: data analysis scripts. See DOI: https://doi.org/10.1039/d5ma00546a.
CCDC 2412726 contains the supplementary crystallographic data for this paper.37
Acknowledgements
We thank SERB [CRG/2022/001499] and LSRB [389/FSH&ABB/2021] for financial support. BITS-Pilani Hyderabad Campus X-ray diffraction and NMR facilities are acknowledged. Madhuparna thanks BITS-Pilani Hyderabad Campus for her fellowship. The support from the DST-FIST HR-MS facility SR/FST/CS-I/2020/158 and DSR-PURSE is also acknowledged.References
- D. Yan, Z. Wang and Z. Zhang, Acc. Chem. Res., 2022, 55, 1047–1058 CrossRef CAS PubMed.
- C. Zeng, J. Dai, T. Yang, Z. Wang, Y. Gao, J. Xia, Y. Chen and M. Sun, Dyes Pigm., 2024, 222, 111906 CrossRef CAS.
- Z. M. Png, C.-G. Wang, J. C. C. Yeo, J. J. C. Lee, N. E. Surat’man, Y. L. Tan, H. Liu, P. Wang, B. H. Tan, J. W. Xu, X. J. Loh and Q. Zhu, Mol. Syst. Des. Eng., 2023, 8, 1097–1129 RSC.
- Y. Huang, L. Ning, X. Zhang, Q. Zhou, Q. Gong and Q. Zhang, Chem. Soc. Rev., 2024, 53, 1090–1166 RSC.
- S. Rana, S. Vaidyanathan and S. Patel, J. Mater. Chem. C, 2024, 12, 14148–14164 RSC.
- X. He and P. Wei, Chem. Soc. Rev., 2024, 53, 6636–6653 RSC.
- X. Huang, L. Qian, Y. Zhou, M. Liu, Y. Cheng and H. Wu, J. Mater. Chem. C, 2018, 6, 5075–5096 RSC.
- B. Prusti, P. K. Samanta, N. J. English and M. Chakravarty, Chem. Commun., 2021, 57, 12321–12324 RSC.
- N. Phonchai, C. Khanantong, F. Kielar, R. Traiphol and N. Traiphol, ACS Appl. Nano Mater., 2019, 2, 4489–4498 CrossRef CAS.
- Z. Liu, Z. Jiang, C. Xu, B. Chen and G. Zhu, Dyes Pigm., 2021, 186, 108990 CrossRef CAS.
- X. Chen, X. Zhang and G. Zhang, Chem. Commun., 2015, 51, 161–163 RSC.
- M. Charaborty and M. Chakravarty, Mater. Today Chem., 2024, 35, 101836 CrossRef.
- B. Prusti, P. Sarkar, S. Pati and M. Chakravarty, J. Mater. Chem. C, 2021, 9, 9555–9570 RSC.
- F. Khan, M. Mahmoudi, P. K. Gupta, D. Volyniuk, J. V. Grazulevicius and R. Misra, J. Phys. Chem. C, 2023, 127, 1643–1654 CrossRef CAS.
- E. Li, K. Jie, M. Liu, X. Sheng, W. Zhu and F. Huang, Chem. Soc. Rev., 2020, 49, 1517–1544 RSC.
- B. Prusti and M. Chakravarty, Dyes Pigm., 2020, 181, 108543 CrossRef CAS.
- B. Razavi, H. Roghani-Mamaqani and M. Salami-Kalajahi, Sci. Rep., 2022, 12, 9412 CrossRef CAS PubMed.
- Y. Xie, Y. Pan, F. Xiao, Y. Lei, Y. Zhou, M. Liu, W. Gao, X. Huang and H. Wu, Dyes Pigm., 2021, 188, 109217 CrossRef CAS.
- J. Xiong, K. Wang, Z. Yao, B. Zou, J. Xu and X.-H. Bu, ACS Appl. Mater. Interfaces, 2018, 10, 5819–5827 CrossRef CAS PubMed.
- S. Bhuin, P. K. Samanta and M. Chakravarty, Dyes Pigm., 2022, 197, 109944 CrossRef CAS.
- Z. Wang, X. Cheng, A. Qin, H. Zhang, J. Z. Sun and B. Z. Tang, J. Phys. Chem. B, 2018, 122, 2165–2176 CrossRef CAS PubMed.
- R. Kumari and M. D. Milton, Dyes Pigm., 2022, 205, 110474 CrossRef CAS.
- (a) H. Nawaz, S. Chen, X. Zhang, X. Li, T. You, J. Zhang and F. Xu, ACS Nano, 2023, 17, 3996–4008 CrossRef CAS PubMed; (b) Z. Yang, W. Qin, J. W. Y. Lam, S. Chen, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Chem. Sci., 2013, 4, 3725–3730 RSC.
- S. Rochat and T. M. Swager, Angew. Chem., Int. Ed., 2014, 53, 9792–9796 CrossRef CAS PubMed.
- G. Das, B. Garai, T. Prakasam, F. Benyettou, S. Varghese, S. K. Sharma, F. Gándara, R. Pasricha, M. Baias, R. Jagannathan, N. Saleh, M. Elhabiri, M. A. Olson and A. Trabolsi, Nat. Commun., 2022, 13, 3904 CrossRef CAS PubMed.
- P. Alam, N. L. C. Leung, H. Su, Z. Qiu, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. – Eur. J., 2017, 23, 14911–14917 CrossRef CAS PubMed.
- X. Chen, Y. Tu, S. Cheng, X. Guo, T. Lu, Y. Guo, X. Huang, Y. Xiong and B. Z. Tang, Chem. Eng. J., 2022, 450, 137928 CrossRef CAS.
- (a) B. P. Debata, S. Patel and S. Vaidyanathan, J. Mater. Chem. C, 2025, 13, 10413–10433 RSC; (b) S. Rana, S. R. Nayak, S. Patel and S. Vaidyanathan, J. Mater. Chem. C, 2024, 12, 765 RSC; (c) Y. Dong, J. Zhang, X. Tan, L. Wang, J. Chen, B. Li, L. Ye, B. Xu, B. Zou and W. Tian, J. Mater. Chem. C, 2013, 1, 7554 RSC.
- Q. Sun, H. Wang, X. Xu, Y. Lu, S. Xue, H. Zhang and W. Yang, Dyes Pigm., 2018, 149, 407–414 CrossRef CAS.
- B. Prusti and M. Chakravarty, ACS Omega, 2019, 4, 16963–16971 CrossRef CAS PubMed.
- M. Z. K. Baig, B. Prusti and M. Chakravarty, J. Mater. Chem. C, 2019, 7, 3735–3739 RSC.
- V. D. Singh, A. K. Kushwaha and R. S. Singh, Dyes Pigm., 2021, 187, 109117 CrossRef CAS.
- M. Chakraborty and M. Chakravarty, Mater. Adv., 2021, 2, 6418–6427 RSC.
- (a) G. Povie, Y. Segawa, T. Nishihara, Y. Miyauchi and K. Itami, Science, 2017, 356, 172 CrossRef CAS PubMed; (b) G. J. Bodwell, Nat. Chem., 2014, 6, 383–385 CrossRef CAS PubMed; (c) M. R. Golder and R. Jasti, Acc. Chem. Res., 2015, 48, 557–566 CrossRef CAS PubMed.
- S. Biswas, C. S. Qiu, L. N. Dawe, Y. Zhao and G. J. Bodwell, Angew. Chem., Int. Ed., 2019, 58, 9166–9170 CrossRef CAS PubMed.
- A. M. Armon, A. Bedi, V. Borin, I. Schapiro and O. Gidron, Eur. J. Org. Chem., 2021, 5424–5429 CrossRef CAS PubMed.
- CCDC 2412726: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2lzmw3.
| This journal is © The Royal Society of Chemistry 2026 |