
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePoly(ethyl glyoxylate)-derived self-immolative elastomers
Anna L.
Watson†
 a,
Chuanfeng
Li†
b,
Adnan
Sharif
a,
Elizabeth R.
Gillies
*bc and
Helen
Tran
*ad
a,
Chuanfeng
Li†
b,
Adnan
Sharif
a,
Elizabeth R.
Gillies
*bc and
Helen
Tran
*ad
aDepartment of Chemistry, University of Toronto, Toronto, Ontario M5S 3H6, Canada. E-mail: tran@utoronto.ca
bDepartment of Chemistry, The University of Western Ontario, 1151 Richmond Street, London, Ontario N6A 5B7, Canada. E-mail: egillie@uwo.ca
cDepartment of Chemical and Biochemical Engineering, The University of Western Ontario, 1151 Richmond Street, London, Ontario N6A 5B9, Canada
dDepartment of Chemical Engineering & Applied Chemistry, University of Toronto, Toronto, Ontario, Canada
First published on 19th November 2025
Abstract
The development of degradable elastic materials has become important to reduce waste and develop transient devices. Most degradable elastomers have issues with uncontrolled and random degradation and poor storage stability. Self-immolative polymers (SIPs) can offer stabilization and triggered depolymerization through stimuli-responsive end-caps. In this paper, we describe the crosslinking of poly(ethyl glyoxylate) (PEtG), a SIP with UV and acid labile end-caps, to create an elastic polymer network. The material withstood strains up to 100 percent before failure in our pull to break tests and was able to withstand up to 10 repeated strains of 20 percent with little change to the stress strain curve. The material was then exposed to degradation conditions where UV light triggered partial degradation and 1 molar hydrochloric acid degraded it fully. The controlled degradability and mechanical properties of this material represent a step towards sustainable transient devices.
Introduction
Self-immolative polymers (SIPs), notable for their ability to depolymerize from head to tail after end-cap cleavage, are well suited to applications such as tissue engineering, drug delivery, and transient electronics, where it is important for the material to degrade in a controlled manner.1–3 While there are various degradable polymers, the ability to control and trigger their degradation is limited, with most relying on gradual and random breakdown in the presence of water or enzymes,4–7 creating a variety of end products and oligomers. In contrast, the incorporation of stimuli-responsive end-caps onto low ceiling temperature SIPs enables them to depolymerize back to monomers on command.8 Depending on the end-caps used, they can respond to a wide array of stimuli including changes in pH,9,10 light,11–13 force,14 enzymes15–17 and temperature.18–21While SIPs have been used in a wide variety of applications such as drug delivery3,15,22 and signal amplification in sensors,23–25 there are few examples of their use in bulk materials. Of these, the materials are either rigid and crystalline,26 or stretchable but not elastic.27 For example, the Phillips group was able to selectively depolymerize a solid state poly(benzyl ether) in the presence of polystyrene and polyethylene,26 and the White, Sottos and Moore groups were able to maintain mechanical properties of a cyclic PPA and carbon fiber composite after rounds of depolymerization and reprocessing.28 In biomedical applications such as tissue engineering or wearable electronics, it is important for the materials used to conform to their surroundings and match the mechanical properties of the tissues they are in contact with.29–32
In addition to elastic and stretchable mechanical properties, it is important that the materials have benign degradation products. Many SIPs have degradation products that are likely toxic, including quinone methides33 and o-phthalaldehyde.34 Polyglyoxylates comprise a promising class of SIPs popularized by the Gillies lab.35 In particular, poly(ethyl glyoxylate) (PEtG) and its derivatives such as polyglyoxylamides have been shown to exhibit low toxicity to cells and environmental organisms.36–38 In addition, PEtG has pendent ester moieties allowing for easy crosslinking along the backbone.
In this work, we crosslinked PEtG with a diamine to create an elastic polymer network. By using o-nitrobenzyl and triphenylmethyl (trityl) end-caps on the PEtG, the resulting elastomers can be degraded on demand with either acidic pH or ultraviolet (UV) light. For demonstration purposes, trityl was selected as a moderate acidic pH sensitive end-cap. We demonstrate the network's stretchable properties with pull to break stress strain curves and its elastic properties with cycling tests. Stress relaxation tests show modest deformation before plateauing. Finally, we exposed the materials to UV light and acidic pH to demonstrate its degradability.
Results and discussion
Materials synthesis and characterization
PEtG (Fig. 1A) was synthesized by polymerization of ethyl glyoxylate at −20 °C using NEt3 as a proton transfer catalyst39 (Fig. S1). 4,5-Dimethoxy-2-nitrobenzyl alcohol was used as the initiator to enable depolymerization to be triggered by UV light.40 The polymers were end-capped with pH-labile trityl ethers by reaction with trityl trifluoroacetate. Two different chain lengths were targeted to examine the resulting effects on the material properties. Size exclusion chromatography (SEC) analysis in THF relative to poly(methyl methacrylate) (PMMA) standards indicated that one polymer had a number average molar mass (Mn) of 4.7 kg mol−1 and a dispersity (Đ) of 1.63, while the other polymer had an Mn of 7.9 kg mol−1 and Đ of 1.49. 1H NMR spectroscopic analyses showed a degree of polymerization (DPn) of 54 for the 4.7 kg mol−1 PEtG and 89 for the 7.9 kg mol−1 PEtG, in reasonable agreement with the SEC analysis. These polymers are referred to as the 5k and 8k polymers in the subsequent discussion.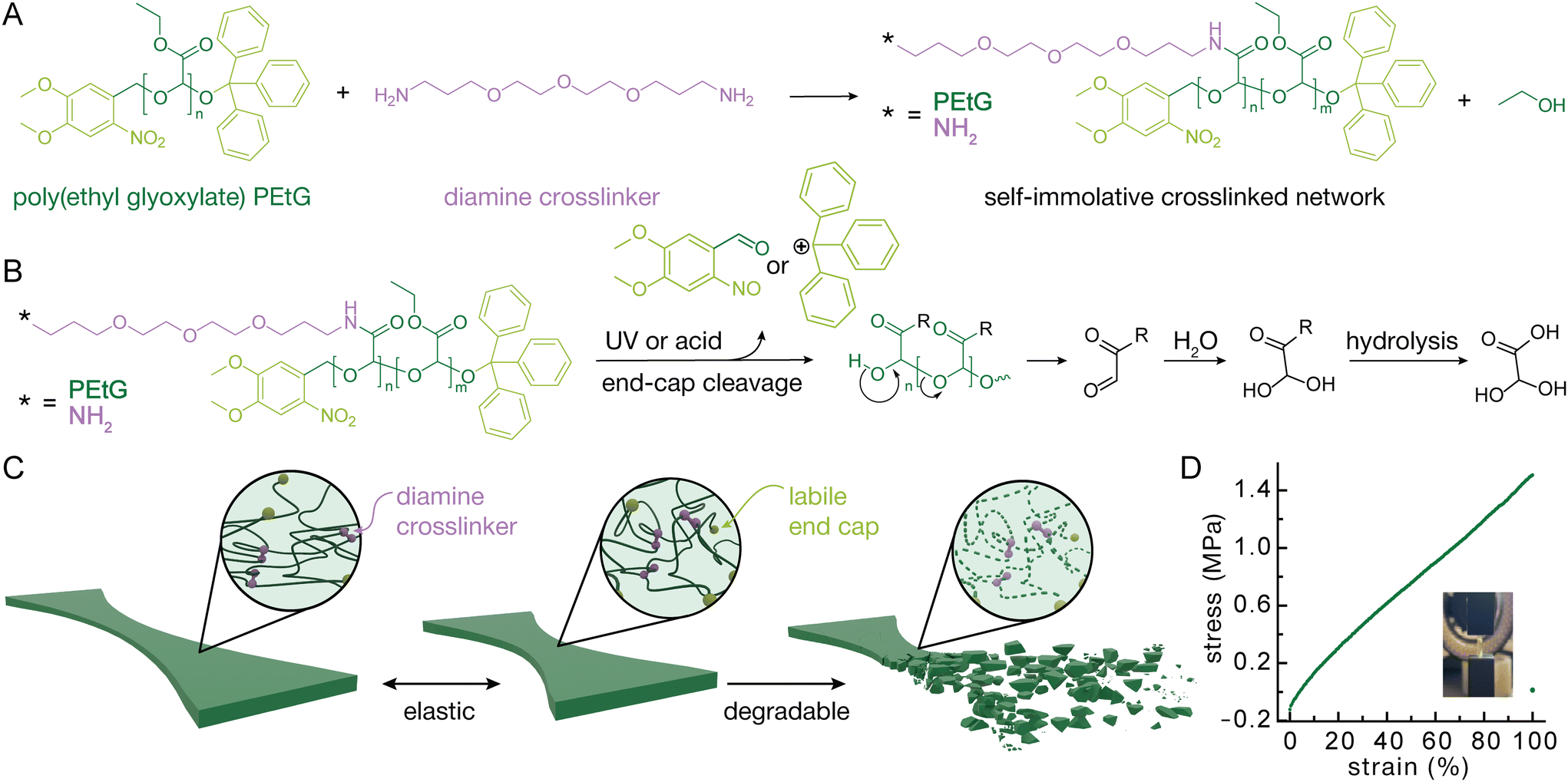 | ||
| Fig. 1 (A) Crosslinking of PEtG with a diamine to create crosslinked polymer network. (B) Depolymerization triggered by UV light or acid and subsequent hydrolysis of the product. (C) Scheme to show the stretching and degradation of the bulk material. (D) Representative example stress strain curve for the crosslinked material. | ||
After synthesis the PEtG was crosslinked with 4,7,10-trioxa-1,13-tridecanediamine through amidation reactions on the pendent esters.41 This crosslinking reaction was confirmed through Fourier transform infrared (FTIR) spectroscopy based on the appearance of an amide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch at 1650 cm−1 and N–H stretch at 3400 cm−1 (Fig. 2A). The crosslinking was performed in a 37 mm by 14 mm by 3 mm Teflon mold as seen in Fig. S2E. The resulting strip was then cut into smaller strips using a 3D printed jig to provide samples for tensile testing (Fig. S2).
O stretch at 1650 cm−1 and N–H stretch at 3400 cm−1 (Fig. 2A). The crosslinking was performed in a 37 mm by 14 mm by 3 mm Teflon mold as seen in Fig. S2E. The resulting strip was then cut into smaller strips using a 3D printed jig to provide samples for tensile testing (Fig. S2).
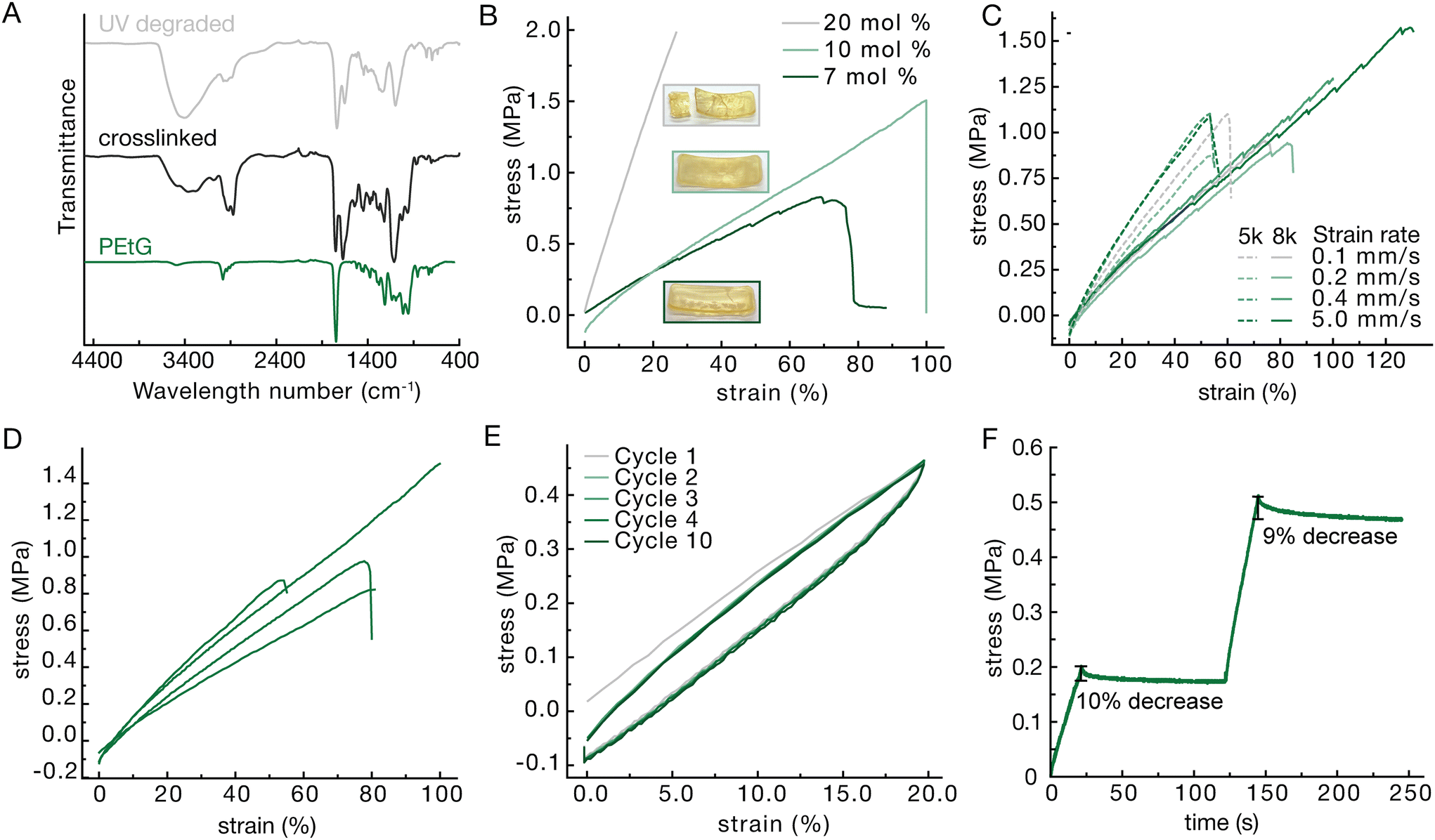 | ||
| Fig. 2 (A) FTIR spectroscopy of PEtG alone (green), crosslinked (black) and UV degraded after crosslinking (grey). (B) Stress strain curve of 5k PEtG crosslinked with 7, 10 and 20 percent diamine to ethyl ester moiety. (C) Stress strain curves for 5k and 8k PEtG crosslinked with 10 mol percent diamine at strain rates from 0.1 to 5.0 mm s−1. (D) Four different batches of 5k PETG crosslinked with 10 mol percent diamine for comparison. (E) 10 cycles straining the 5k PEtG crosslinked with 10 mol percent diamine to 20 percent. (F) Stress relaxation test for 5k PEtG crosslinked with 10 mol percent diamine strained to 10 percent and held 100 s then to 20 percent and held for 100 s. | ||
Mechanical properties
Limited starting material created a need for customized sample preparation. Most American Society for Testing and Materials (ASTM) guides call for dog bones that are multiple centimeters long and therefore commercial dies to cut samples are all quite large. Having materials that are already quite soft, the samples needed to be thick enough for the instrument to read a force output in the working range of the load cells. This would require large amounts of polymer material for each test. To create consistent samples, a 3D printed scaffold was used to hold two razorblades in shape. Another piece was printed to slot into the top to hold the sample and create a guide for the cutting lines (Fig. S2). The resulting sample was a strip narrower in the middle and thicker on the edges to encourage fracture in the center similar to a dog bone. Additionally, some material needed to be cut from either end to remove thicker ends created by the meniscus induced in the Teflon mold.Variations in polymer length, crosslinking density, and strain rate were explored to give an understanding of the variety of material properties that could be achieved. Crosslinking density was explored first as it generally correlates with the moduli of polymer networks.42 Materials were tested at 7, 10 and 20 mole percent crosslinker with respect to pendent ethyl ester (Fig. 2B). Lower crosslinking percentages did not provide enough structure to create solid materials with 7 percent being the lowest that could be tested as a solid, giving a Young's modulus of 1.1 MPa. At higher percentages of crosslinker, the materials became brittle. For example, 20 percent crosslinker resulted in materials with a Young's modulus of 7.6 MPa while only reaching 27 percent elongation at break. Additionally, the material fractured during the crosslinking making it difficult to find enough unbroken area to create a testable sample. Thus, 10 percent crosslinker was found to provide a good balance of the desired elastic properties while not being too brittle and giving a Young's modulus of 1.4 MPa. Strain rate was not found to have much of an effect on the stress strain curve in the range of 0.1 mm s−1 to 5 mm s−1 and no consistent trends were observed indicating a lower tendency to creep (Fig. 2C). While there was some batch-to-batch variation (Fig. 2D), the 5k polymer tended to provide more consistent materials so it was chosen to be used in further testing. In comparing networks made using 8k PEtG with the 5k there did seem to be a small decrease in the Young's modulus, but this is within the error associated with batch-to-batch variation – four batches made separately from 5k PEtG showed Young's modulus of 1.38 ± 0.250 MPa and coefficient of variation of 18.1%. Since the same mole percent of crosslinker relative to the pendent ethyl esters of PEtG was used in both materials, it was expected that the materials would behave similarly.
To better understand the mechanical properties of the 10% crosslinked 5k material, it was tested for stress strain cycling as well as stress relaxation. The materials were able to maintain their integrity for up to ten cycles at 20 percent elongation. While there was a reduction in modulus from the 1st to the 2nd cycles, this can be attributed to the Mullin's effect wherein the polymer strands rearrange, absorbing some of the force in the first loading cycle.43 There is some hysteresis seen in the loop, meaning the material is not perfectly elastic over the time scale used, however a rest of 20 s between cycles allows for almost a full recovery and a very similar hysteresis loop over 10 cycles (Fig. 2E). The materials also exhibit some stress relaxation over the course of 100 seconds (Fig. 2F). At both 10 percent and 20 percent strain there is first a sharp dip and then a plateau where the material requires 10 percent or 9 percent less force, respectively, to maintain the strain.
UV and acid degradation
Exposure to UV light degraded both the mechanical properties and the surface of the polymer network. A single 4 h exposure caused a sharp and then more gradual decrease in Young's modulus over the course of days (Fig. 3A). As expected, the 370 nm UV light cleaved the o-nitrobenzyl ether group, initiating the depolymerization of the polymer backbone (Fig. 1B) and weakening the bulk material. Notably the Young's modulus decreased from 1.4 MPa to 0.5 MPa over the course of 12 days (Fig. 3A and B). Scanning electron microscopy (SEM) indicated some surface structure arising from the material preparation before exposure to UV light, which seemed to be lost after, indicating surface degradation (Fig. 3D). Due to the photo darkening and the thickness of tested materials, the UV light was likely unable to fully penetrate the center, which drastically slowed the degradation of the material. On the other hand, the acid labile trityl group may also contribute to absorb the UV light and further reduce the penetration efficiency. This might be remedied with thinner films or a pH sensitive end-cap without absorptions in similar wavelength regions. | ||
| Fig. 3 (A) Stress strain curve showing pull to break tests before UV exposure, 6 days after UV exposure and 12 days after UV exposure (B) and the respective Young's moduli. (C) DART-MS of the 5k 10 mol percent crosslinked PEtG after acid exposure. (D) SEM of the 5k 10 mole percent crosslinked PEtG before and after UV exposure. (E) Photographs at timepoints during exposure to varied concentrations of hydrochloric acid. | ||
Exposure to acid had a more immediate and substantial effect on the network structure with complete degradation observed by 16 hours at all acid concentrations (Fig. 3E). This complete degradation was likely facilitated by the swelling of the network in acetone, such that the acid was able to penetrate through the material, cleaving the trityl end-caps and initiating depolymerization. Even concentrations as low as 1 M hydrochloric acid (HCl) were capable of inducing fast degradation and it is expected that even lower concentrations would degrade the material, albeit at a slower rate. The materials were also exposed to trifluoracetic acid (TFA) with similar concentrations degrading the material more quickly (Fig. S5). Direct analysis in real time mass spectrometry (DART-MS) indicated the degradation into multiple expected products including fragments of the ethyl glyoxylate monomer derivatives, their hydrates, and fragments related to the end-caps, such as nitrotoluene and the triphenylmethyl cation (Fig. 3C).
Monoamine degradation
To better understand and monitor the degradation, PEtG was exposed to a monoamine (2-methoxyethylamine) to create a control with solution processability. As expected, the reaction resulted in a viscous liquid, similar to the starting material as no crosslinking occurred. The resulting material dissolved readily in CD2Cl2, which we used to monitor the degradation via nuclear magnetic resonance (NMR) spectroscopy. An aldehyde peak, which is indicative of the expected depolymerization products, emerged in the first day after exposure to UV light (Fig. 4B and C). Over the course of 14 days the ratio of the integral of the aldehyde peak to that of the backbone peaks increased as expected, though the actual integration of the aldehyde peak plateaued (Fig. S4B). This plateau effect is likely due to similar penetration issues as were observed in the crosslinked material. Additionally, ethyl glyoxylate, the main depolymerization product, is volatile and may evaporate unless exposed to water which traps the molecule in the non-volatile hydrate form. | ||
| Fig. 4 (A) Reaction scheme of PEtG with monoamine to create a soluble sample for NMR analysis. (B) Depolymerization of PEtG with appended monoamine on exposure to UV light. (C) Zooms of NMR spectra showing the appearance of an aldehyde peak. (D) Ratio of integration of the aldehyde peak to the backbone peak after UV exposure on day 1, 7 and 14. | ||
Conclusions
In conclusion, we developed a triggerable SIP network with stretchable and elastic properties. The linear polymer was crosslinked to form a network by a simple treatment with a diamine. This allowed for control over the crosslink density and consequent material properties. The material was demonstrated to change very little over the course of 10 cycles of stress to 20 percent strain. In addition, stress relaxation studies showed a quick drop in strain before plateauing for a total of a 10 percent decrease in stress. These tests demonstrated the first instance of a stretchable and elastic SIP material. In addition to studying the mechanical properties, the material's ability to degrade was also explored. It was degraded through the cleavage of o-nitrobenzyl or trityl end-caps with UV light or acid respectively, which triggered the depolymerization of poly(ethyl glyoxylate). While depolymerization didn't proceed to completion using UV light, submersion in an HCl solution allowed for rapid and complete degradation. We envision that the polymer degradation properties can be further improved by replacing the trityl end-cap with non-aromatic pH sensitive end-caps (e.g. ethyl vinyl ether) to enhance UV penetration of the materials. In addition, different depolymerization stimuli can be incorporated by varying the end-cap. We anticipate that mechanical properties can be further tuned using different diamine crosslinkers. This is a step towards transient devices that can be degraded and potentially recycled.Author contributions
A. W. performed conceptualization (co-lead), investigation and methodology for characterization of mechanical properties, data visualization (co-lead), writing of the original draft (co-lead), review & editing of the original draft (support). C. L. performed conceptualization (co-lead), investigation and methodology for synthesis of materials, writing of the original draft (co-lead), review & editing or the original draft (support). A. S. performed methodology for material sample preparation. E. R. G. performed conceptualization (co-lead), funding acquisition (co-lead), project administration (co-lead), provided resources (co-lead), supervision (co-lead), wrote – review & edited the original draft (co-lead). H. T. performed conceptualization (co-lead), funding acquisition (co-lead), project administration (co-lead), provided resources (co-lead), supervision (co-lead), wrote – review & edited the original draft (co-lead).Conflicts of interest
There are no conflicts of interest to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5lp00310e.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council (NSERC) of Canada (H. T.: RGPIN2021-03554; E. R. G.: RGPIN2021-03950) the Canadian Foundation for Innovation (H. T.: JELF-41743, CFI, grant #43160), and the Canada Research Chairs program (E. R. G. CRC-2020-00101). We thank the Centre for Research and Applications in Fluidic Technologies (CRAFT), and the Province of Ontario for funding. The authors gratefully acknowledge the Department of Chemistry and the Department of Chemical Engineering & Applied Chemistry at the University of Toronto for their support. In particular, the authors acknowledge Darcy Burns in the CSICOMP NMR facility at the University of Toronto and Ilya Gourevich in the Centre for Nanostructure Imaging at the University of Toronto for their help.References
- R. W. Lenz and R. H. Marchessault, Biomacromolecules, 2005, 6, 1–8 CrossRef CAS.
- K. K. Fu, Z. Wang, J. Dai, M. Carter and L. Hu, Chem. Mater., 2016, 28, 3527–3539 CrossRef CAS.
- K. Wang, X. Xiao, Y. Liu, Q. Zong, Y. Tu and Y. Yuan, Biomaterials, 2022, 289, 121803 CrossRef CAS.
- M. Shahriari, M. Zahiri, K. Abnous, S. M. Taghdisi, M. Ramezani and M. Alibolandi, J. Controlled Release, 2019, 308, 172–189 CrossRef CAS.
- B. Kong, Y. Chen, R. Liu, X. Liu, C. Liu, Z. Shao, L. Xiong, X. Liu, W. Sun and S. Mi, Nat. Commun., 2020, 11, 1435 CrossRef CAS PubMed.
- Y. S. Choi, Y.-Y. Hsueh, J. Koo, Q. Yang, R. Avila, B. Hu, Z. Xie, G. Lee, Z. Ning, C. Liu, Y. Xu, Y. J. Lee, W. Zhao, J. Fang, Y. Deng, S. M. Lee, A. Vázquez-Guardado, I. Stepien, Y. Yan, J. W. Song, C. Haney, Y. S. Oh, W. Liu, H.-J. Yoon, A. Banks, M. R. MacEwan, G. A. Ameer, W. Z. Ray, Y. Huang, T. Xie, C. K. Franz, S. Li and J. A. Rogers, Nat. Commun., 2020, 11, 5990 Search PubMed.
- C. J. Stubbs, J. C. Worch, H. Prydderch, Z. Wang, R. T. Mathers, A. V. Dobrynin, M. L. Becker and A. P. Dove, J. Am. Chem. Soc., 2022, 144, 1243–1250 CrossRef CAS PubMed.
- J. Gong, B. Tavsanli and E. R. Gillies, Annu. Rev. Mater. Res., 2024, 54, 47–73 CrossRef CAS.
- R. J. Amir, N. Pessah, M. Shamis and D. Shabat, Angew. Chem., Int. Ed., 2003, 42, 4494–4499 CrossRef CAS PubMed.
- M. F. Nichol, K. D. Clark, N. D. Dolinski and J. R. de Alaniz, Polym. Chem., 2019, 10, 4914–4919 RSC.
- J. Steinkoenig, M. M. Zieger, H. Mutlu and C. Barner-Kowollik, Macromolecules, 2017, 50, 5385–5391 CrossRef CAS.
- J. Olejniczak, V. A. N. Huu, J. Lux, M. Grossman, S. He and A. Almutairi, Chem. Commun., 2015, 51, 16980–16983 RSC.
- V. Kumar, J. T. Harris, A. Ribbe, M. Franc, Y. Bae, A. J. McNeil and S. Thayumanavan, ACS Macro Lett., 2020, 9, 377–381 CrossRef CAS PubMed.
- X. Hu, T. Zeng, C. C. Husic and M. J. Robb, J. Am. Chem. Soc., 2019, 141, 15018–15023 CrossRef CAS PubMed.
- F. M. H. de Groot, W. J. Loos, R. Koekkoek, L. W. A. van Berkom, G. F. Busscher, A. E. Seelen, C. Albrecht, P. de Bruijn and H. W. Scheeren, J. Org. Chem., 2001, 66, 8815–8830 CrossRef CAS.
- R. Weinstain, P. S. Baran and D. Shabat, Bioconjugate Chem., 2009, 20, 1783–1791 CrossRef CAS PubMed.
- G. G. Lewis, J. S. Robbins and S. T. Phillips, Anal. Chem., 2013, 85, 10432–10439 CrossRef CAS.
- M. F. Nichol, K. D. Clark, N. D. Dolinski and J. R. de Alaniz, Polym. Chem., 2019, 10, 4914–4919 RSC.
- B. Fan, J. F. Trant, G. Hemery, O. Sandre and E. R. Gillies, Chem. Commun., 2017, 53, 12068–12071 RSC.
- R. E. Yardley and E. R. Gillies, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1868–1877 CrossRef CAS.
- G. I. Peterson, D. C. Church, N. A. Yakelis and A. J. Boydston, Polymer, 2014, 55, 5980–5985 CrossRef CAS.
- A. Dey, J. Jeon, B. Yoon, Y. Li and J. H. Park, J. Controlled Release, 2023, 358, 555–565 CrossRef CAS PubMed.
- M. E. Roth, O. Green, S. Gnaim and D. Shabat, Chem. Rev., 2016, 116, 1309–1352 CrossRef CAS.
- Q. Zong, J. Li, Q. Xu, Y. Liu, K. Wang and Y. Yuan, Nat. Commun., 2024, 15, 7558 CrossRef CAS.
- A. Sagi, R. Weinstain, N. Karton and D. Shabat, J. Am. Chem. Soc., 2008, 130, 5434–5435 CrossRef CAS PubMed.
- M. S. Baker, H. Kim, M. G. Olah, G. G. Lewis and S. T. Phillips, Green Chem., 2015, 17, 4541–4545 RSC.
- S. M. Heuchan, J. P. MacDonald, L. A. Bauman, B. Fan, H. A. L. Henry and E. R. Gillies, ACS Omega, 2018, 3, 18603–18612 CrossRef CAS PubMed.
- E. M. Lloyd, H. Lopez Hernandez, E. C. Feinberg, M. Yourdkhani, E. K. Zen, E. B. Mejia, N. R. Sottos, J. S. Moore and S. R. White, Chem. Mater., 2019, 31, 398–406 CrossRef CAS.
- A. Kumar, K. C. Nune, L. E. Murr and R. D. K. Misra, Int. Mater. Rev., 2016, 61, 20–45 CrossRef CAS.
- K. Lim, H. Seo, W. G. Chung, H. Song, M. Oh, S. Y. Ryu, Y. Kim and J.-U. Park, Commun. Mater., 2024, 5, 49 CrossRef CAS.
- K. K. Fu, Z. Wang, J. Dai, M. Carter and L. Hu, Chem. Mater., 2016, 28, 3527–3539 CrossRef CAS.
- C. L. C. Chan, J. M. Taylor and E. C. Davidson, Nat. Synth., 2022, 1, 592–600 CrossRef CAS.
- T. J. Monks and D. C. Jones, Curr. Drug Metab., 2002, 3, 425–438 CrossRef CAS.
- S. E. Anderson, C. Umbright, R. Sellamuthu, K. Fluharty, M. Kashon, J. Franko, L. G. Jackson, V. J. Johnson and P. Joseph, Toxicol. Sci., 2010, 115, 435–443 CrossRef CAS PubMed.
- B. Fan, J. F. Trant, A. D. Wong and E. R. Gillies, J. Am. Chem. Soc., 2014, 136, 10116–10123 CrossRef CAS PubMed.
- B. Belloncle, C. Bunel, L. Menu-Bouaouiche, O. Lesouhaitier and F. Burel, J. Polym. Environ., 2012, 20, 726–731 CrossRef CAS.
- J. Gong, A. Borecki and E. R. Gillies, Biomacromolecules, 2023, 24, 3629–3637 Search PubMed.
- J. D. Pardy, B. Tavsanli, Q. E. A. Sirianni and E. R. Gillies, Chem. – Eur. J., 2024, 30, e202401324 CrossRef CAS.
- X. Mei and E. R. Gillies, Macromol. Rapid Commun., 2025, 46, e00419 CrossRef CAS.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed.
- S. A. Smith, B. Rossi Herling, C. Zhang, M. A. Beach, S. L. Y. Teo, E. R. Gillies, A. P. R. Johnston and G. K. Such, Biomacromolecules, 2023, 24, 4958–4969 CrossRef CAS.
- K. Kato, Y. Ikeda and K. Ito, ACS Macro Lett., 2019, 8, 700–704 CrossRef CAS.
- J. Diani, B. Fayolle and P. Gilormini, Eur. Polym. J., 2009, 45, 601–612 CrossRef CAS.
Footnote |
| † Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |