
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sustainable TPS/PBAT biocomposites tailored with epoxidized soybean oil for improved mechanical properties
Chandramani
Batsh
 ad,
Chandan Kumar
Munagala
ad,
Bitopan
Boro
bd,
Devasish
Chowdhury
bd,
Harsha
Nagar
cd and
Vineet
Aniya
*ad
ad,
Chandan Kumar
Munagala
ad,
Bitopan
Boro
bd,
Devasish
Chowdhury
bd,
Harsha
Nagar
cd and
Vineet
Aniya
*ad
aProcess and Polymer Engineering Lab, Chemical Engineering and Process Technology Department, CSIR-Indian Institute of Chemical Technology, Hyderabad, Telangana 500007, India. E-mail: batshchandramani@gmail.com; chandank635@gmail.com; vineetkumaraniya@gmail.com
bMaterial Nanochemistry Laboratory, Physical Sciences Division, Institute of Advanced Study in Science and Technology, Garchuk, Guwahati 781035, India. E-mail: bitopanboro@rediffmail.com; devasish@iasst.gov.in
cPolymer Science and Engineering Division, CSIR-National Chemical Laboratory, Pune, Maharashtra 411008, India. E-mail: h.nagar@ncl.res.in
dAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India
First published on 19th November 2025
Abstract
The growing demand for eco-friendly substitutes of traditional plastics has further driven the research on compostable and biodegradable films with enhanced functionality. Epoxidized soybean oil (EPSO) was prepared through in situ peracid epoxidation and used as a nontoxic, hydrophobic plasticizer in thermoplastic starch (TPS)/poly(butylene adipate-co-terephthalate) (PBAT) nanocomposites in this study. For better interfacial compatibility and functionality, TPS was filled with nanocrystalline cellulose (NCC) and mixed with EPSO, glycerol, maleic anhydride (MA), rice bran wax (RBW), and stearic acid (SA). The flexible films (50–80 μm) were prepared using blown film extrusion. Structural assessments verified effective soybean oil epoxidation and its homogeneous integration into the polymer matrix. Mechanical analysis indicated that EPSO-modified films, especially 20% glycerol–10% EPSO and 15% glycerol–15% EPSO, exhibited higher tensile strength (13.63 MPa), better sealability, and maintained ductility. Barrier analysis demonstrated these films have lower water vapor permeability and greater hydrophobicity (contact angle up to 108.6°). Thermal analysis (TGA, DSC and FTIR) also confirmed their higher stability and compatibility. Soil burial tests provided proof of excellent biodegradability (>75%), emphasizing the compostability of the films. In general, the synergistic contribution of EPSO and bio-reinforcements resulted in mechanically stable, hydrophobic, and biodegradable films that provide an eco-friendly substitute for single-use packaging and farming purposes.
1. Introduction
Polythene bags, although very popular for their strength and ease of use, have emerged as a major source of environmental pollution because of their non-biodegradable content. They clog up the soil and water for extended periods of time1 and pose a serious danger to wildlife,2 especially marine life,3 which often mistake them for food. Additionally, their manufacturing process depends greatly on fossil fuels, with resultant greenhouse emissions and global warming.4 As plastic consumption continues to rise dramatically, Asia is expected to reach a plastic demand of 321 million tons by 2050 due to rapid surges in population and urbanization.5 Thus, the need for sustainable, biodegradable substitutes has become critical.6 Good candidates for such solutions are biodegradable polymers like poly(butylene adipate-co-terephthalate) (PBAT) and thermoplastic starch (TPS). PBAT is a flexible, mechanically strong biodegradable polyester,7,8 whereas TPS is a renewable starch-based material. However, native starch is brittle and not very flexible, and therefore less attractive industrially.9 Nayak et al.10 proved that adding 30 wt% starch to PBAT gave materials with tensile strength of 4–10 MPa and elongation of 180–390%, whereas simultaneous addition of nanoclays improved stiffness but at the cost of ductility. Olivato et al.11 also used citric acid and maleic anhydride as compatibilizers and attained tensile strength close to 6.5 MPa, although elongation plunged (<20%) due to a lack of suitable interfacial compatibility. Fourati et al.12 subsequently strengthened PBAT/TPS blends with cellulose nanofibrils, with tensile strength gains of up to ∼6 MPa and modulus enhancements of approximately 225%, yet the blends continued to show water absorption of 5–9% and contact angles of less than 90°, verifying their hydrophilic characteristics. Even with several alterations, TPS–PBAT blends retain low tensile strength, reduced elongation, and high moisture sensitivity. Their contact angle tends to be below 90°, reflecting minimal hydrophobicity, which limits their function for long-lasting packaging purposes. This drawback is overcome by adding additives to enhance their processability and flexibility. The one additive most widely used is glycerol, which is also very hydrophilic in nature and hence enhances moisture uptake and decreases the long-term stability of TPS. An alternative additive, chemically synthesized through epoxidation, is epoxidized soybean oil (EPSO),13 which has enhanced hydrophobicity, flexibility, and thermal stability that makes it a good bio-based plasticizer.Herein, we hypothesize that the addition of a second plasticizer would improve the performance of TPS/PBAT composite films having a high TPS content (up to 40%), thereby increasing their carbon content. In particular, the addition of EPSO, a bio-based hydrophobic plasticizer, should enhance interfacial adhesion, mechanical strength, and barrier properties of the films without compromising their biodegradability. In addition, synergistic interaction between NCC, EPSO, RBW, and SA is expected to improve hydrophilicity, water contact angle, and thermal stability. Overall, these changes will facilitate the production of sustainable, compostable films with potential use in packaging and agriculture.
In the previous work reported by our group,14 we blended TPS/PBAT at a ratio of 30/70 and prepared films with tensile strength of 11.01 ± 0.15 and Young's modulus of 1200.94 ± 16 MPa. In our current research work, biodegradable composite films were prepared employing blends of TPS/PBAT in a ratio of 40/60 plasticized with glycerol14 and EPSO.15 Reactive melt extrusion was utilized for plasticization. EPSO was a contributor to the bio-based content along with functioning as a hydrophobic plasticizer, while glycerol acted as a plasticizer and chain extender. The coupling agent of maleic anhydride (MA) was employed to increase interfacial compatibility between the hydrophilic TPS and hydrophobic PBAT. Nanocrystalline cellulose (NCC) was incorporated as a reinforcing filler owing to its high strength and capability of influencing improved mechanical performance through hydrogen bonding and efficient stress transfer.12,17 Rice bran wax (RBW) was added as secondary reinforcement to reduce water permeability caused by cellulose. Stearic acid (SA) was also included to serve as a lubricant, facilitating processing and enhancing the surface texture of the film. The films that were produced showed better mechanical and barrier properties. Addition of NCC greatly improved tensile strength and Young's modulus, with the best performance occurring at certain filler loadings. RBW and compatibilizer addition provided improved hydrophobicity by lowering water absorption and increasing the water contact angle18 – a key requirement for applications needing moisture resistance.19 Blown film extrusion was employed to fabricate the composite into smooth, flexible films with good mechanical strength. Thermal analysis revealed enhanced stability, and biodegradation tests verified the compostability of the films under natural composting conditions. The bio-based content, mechanical strength, hydrophobicity, and compostability rendered these films suitable for use in packaging, agricultural mulch, and wrapping films.
Overall, the biodegradable films prepared in this research using PBAT–TPS blends proved to be a promising and eco-friendly substitute for traditional polythene bags. With the combination of EPSO, NCC, MA, RBW, and SA, composite films have been developed with improved mechanical performance, water resistance, and degradability in an outdoor environment, paving the way toward environmentally friendly packaging solutions.
2. Materials and methods
PBAT was procured from Ecopond® (KB 100 grade, MFI: 3.0–5.0 g per 10 min at 190 °C, 2.16 kg, melting point: 115–125 °C). Corn starch was purchased from Deepak Starch & Chemicals, Hyderabad, India. Glycerol (extra pure, density: 1.257–1.260 g ml−1, refractive index: 1.470–1.475, 98% miscibility in ethanol), and stearic acid (n-octadecanoic acid, melting point: 54 °C, acid value: 194–212) were procured from Finar Chemicals, Gujarat, India. Maleic anhydride (molecular weight: 98.06, melting point: 52–54 °C), hydrogen peroxide, formic acid (molecular weight: 46.03), ethyl acetate and hexane (molecular weight: 86.18, specific gravity at 25 °C: 0.658–0.669) were procured from Avra Chemicals, India. Silica gel (pH∼7.0, 60–120 mesh) and toluene (HPLC grade, refractive index at 20 °C: 1.496–1.497) were procured from SRL Chemicals, India. Nanocrystalline cellulose powder was provided by a nanocellulose pilot plant at the Chemical Engineering and Process Technology Division, CSIR – Indian Institute of Chemical Technology, India. The edible soybean oil used in this study was Fortune soybean oil purchased from Adani Wilmar, India. Rice bran wax was procured from Vijay Impex, India. All the chemicals were used without any further purification.2.1 Methods for the preparation of the composite material
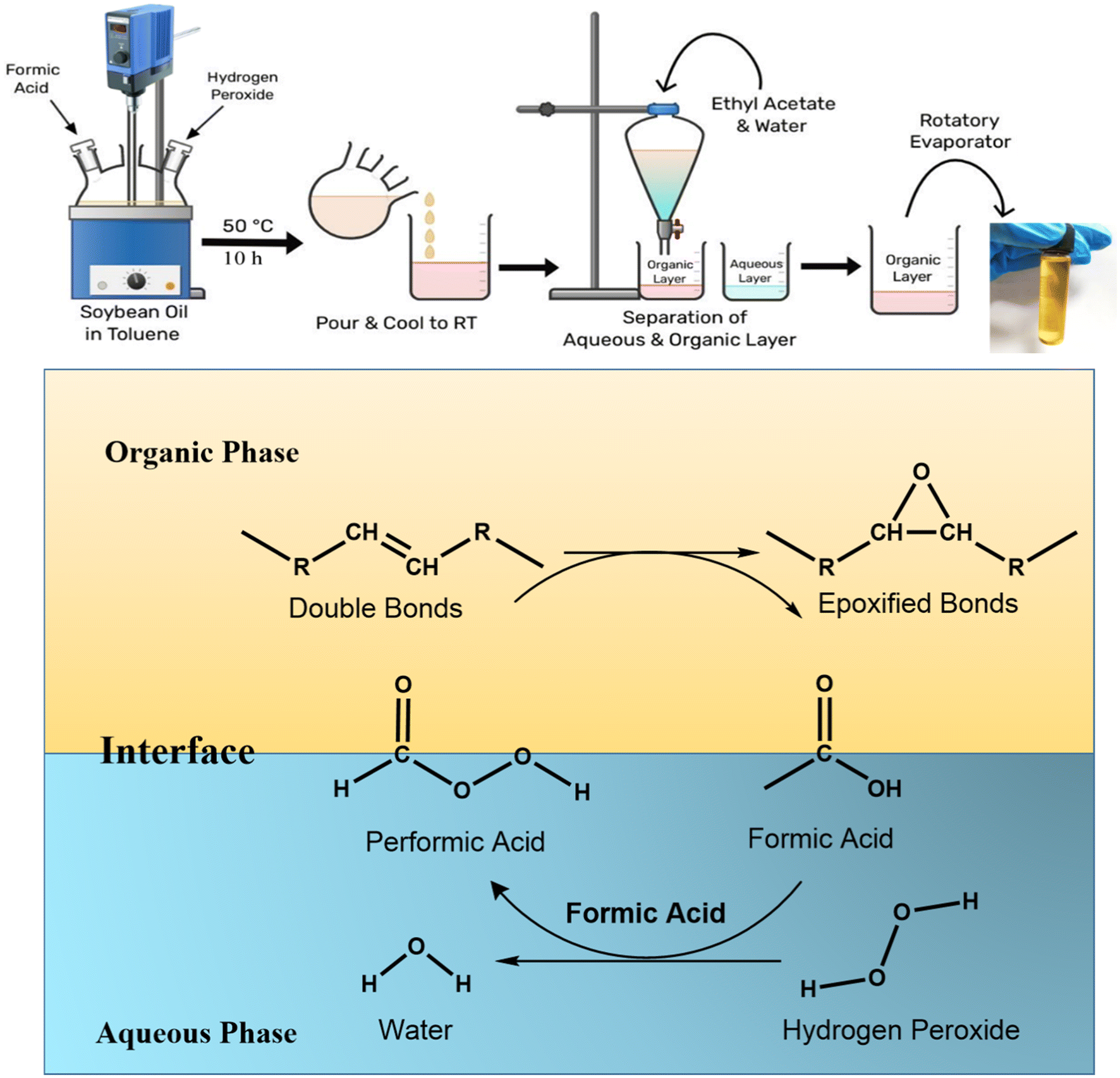 | ||
| Fig. 1 Schematic representation of epoxidation of soybean oil. | ||
| Starch | Glycerol | EPSO | RBW | NCC | MA | SA | |
|---|---|---|---|---|---|---|---|
| TPS | 70% | 30% | — | — | 3% | 1% | 0.5% |
| TPS-G30-E0 | 30% | — | 2% | ||||
| TPS-G25-E5 | 25% | 5% | |||||
| TPS-G20-E10 | 20% | 10% | |||||
| TPS-G15-E15 | 15% | 15% | |||||
| TPS-G10-E20 | 10% | 20% |
The pre-dried constituents were blended with starch using a mechanical kneader fitted with double naben blades to ensure homogeneous mixing and to prevent agglomeration. The uniform pre-mix was subsequently melt-processed in a twin-screw extruder (L/D ratio – 25, eight thermal zones) with the temperature profile set as follows: Z1–Z2 at 135 °C, Z3 at 140 °C, Z4 at 145 °C, Z5–Z7 at 150 °C, and Z8 (die) at 145 °C. The extrusion was conducted at a screw speed of 12 rpm with a feeding rate of 60 g min−1, and vacuum-assisted degassing was applied to remove any residual moisture. The extrudates were pelletized using a titanium–carbide-tipped cutter to obtain pellets of approximately 2 × 3 cm2. The pellets were then oven-dried at 60 °C for 4 h and stored in a vacuum-sealed desiccator at ambient temperature to prevent oxidative degradation prior to subsequent processing.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60 (TPS:PBAT) and thoroughly mixed using a kneader. The mixture was subsequently processed through a twin-screw extruder, following a controlled temperature profile: zones Z1–Z2 at 125 °C, Z3 at 130 °C, Z4 at 135 °C, zones Z5–Z7 at 140 °C, and Z8 (die) maintained at 135 °C. The screw speed was set to 10 rpm—lower than that employed for reinforced TPS preparation—to promote enhanced molecular interactions between the blend components. A vacuum system was engaged during extrusion to remove residual moisture. The extrudates were pelletized using a titanium–carbide-tipped cutter, producing pellets of approximately 2 × 3 cm2. The resulting pellets (TPS-PBAT, TPS-G30-E0-PBAT, TPS-G25-E5-PBAT, TPS-G20-E10-PBAT, TPS-G15-E15-PBAT and TPS-G10-E20-PBAT) were oven-dried at 60 °C for 4 h, and then stored in a vacuum-sealed desiccator at ambient temperature to minimize oxidative degradation prior to further processing.
:60 (TPS:PBAT) and thoroughly mixed using a kneader. The mixture was subsequently processed through a twin-screw extruder, following a controlled temperature profile: zones Z1–Z2 at 125 °C, Z3 at 130 °C, Z4 at 135 °C, zones Z5–Z7 at 140 °C, and Z8 (die) maintained at 135 °C. The screw speed was set to 10 rpm—lower than that employed for reinforced TPS preparation—to promote enhanced molecular interactions between the blend components. A vacuum system was engaged during extrusion to remove residual moisture. The extrudates were pelletized using a titanium–carbide-tipped cutter, producing pellets of approximately 2 × 3 cm2. The resulting pellets (TPS-PBAT, TPS-G30-E0-PBAT, TPS-G25-E5-PBAT, TPS-G20-E10-PBAT, TPS-G15-E15-PBAT and TPS-G10-E20-PBAT) were oven-dried at 60 °C for 4 h, and then stored in a vacuum-sealed desiccator at ambient temperature to minimize oxidative degradation prior to further processing.
 | ||
| Fig. 2 Method showing the preparation of composite thin films. | ||
3. Testing and characterization
The viscosity and shear stress of SO and EPSO were measured by using a First Plus Viscometer manufactured by Lamy Rheology Instruments. The viscosity of oils was determined using LV3 measuring equipment at 30 °C for 60 seconds with a speed setting of 30 rpm. Five specimens were analyzed and the average values calculated were used for viscosity and shear stress.The density and specific gravity values of SO and EPSO were measured by using an Anton Paar density meter (DMA 4500 M) at a temperature of 30 °C. Five specimens were analyzed and the average result was used for the density value, expressed in g cm−3.
To measure the oxirane oxygen content, approximately 1.0 g of each oil sample was accurately weighed and dissolved in 25 mL of glacial acetic acid in a conical flask. After ensuring complete dissolution, 10 mL of 0.1 N hydrochloric acid (HCl) solution was added to the mixture. The solution was stirred thoroughly for 30 minutes to achieve complete homogenization and to allow the reaction to proceed. Following this, a few drops of phenolphthalein indicator were added to the reaction mixture. The resulting solution was then titrated against a standardized 0.1 N sodium hydroxide (NaOH) solution until a persistent pink endpoint was observed. The oxirane oxygen content was calculated as per ASTM D1652–97 by using the following equation:21
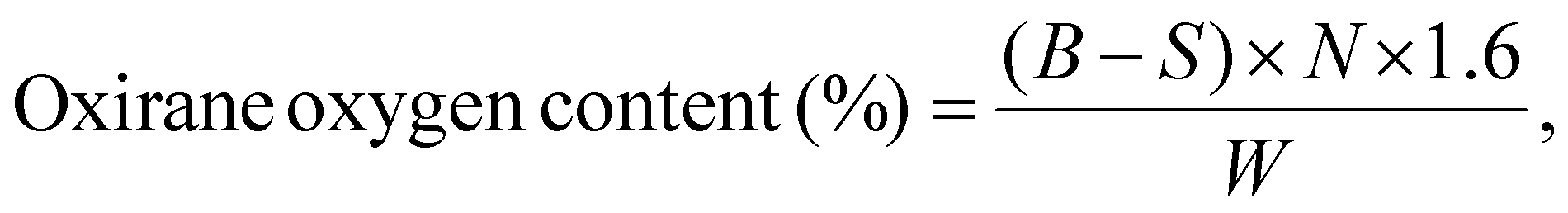 | (1) |
The CHNSO analysis of SO and EPSO was done by using a CHNSO analyzer to check the elemental composition in the oil. The oils were kept in an oven at 50 °C for 4 hours before analysis to remove moisture content that can affect the elemental composition results.
Nuclear magnetic resonance (NMR) spectra were recorded for SO and EPSO to check the formation of epoxy linkages in the unsaturated chain of the oils. The spectra of the oils were recorded at 300 Hz for 1H and 13C NMR with tetramethylsilane used as the internal standard. The oils were kept in an oven at 50 °C for 12 hours before the analysis to remove moisture content. The oils were mixed with chloroform-D (1H NMR peak at 7.26 ppm and 13C NMR peak at 77.16 ppm) for the analysis.
Attenuated Transmission – Fourier Transmission IR (AT-FTIR) spectroscopy was used to analyze the thin films in the wavenumber range of 4000–400 cm−1 to determine the functional groups associated with them. Samples of thin film of 1 × 1 cm2 were cut and then cleaned using cotton to ensure lint- and dust-free conditions and avoid any contamination. The oils were kept in an oven at 50 °C for 4 hours before the analysis to remove moisture content. The thin film samples were conditioned at 27 ± 2 °C under 52% relative humidity for 24 hours prior to testing.
The melt flow index (MFI) values for the composite pellets were determined using an MFI tester procured from Advanced Equipments, India, with measurements undertaken in accordance with the ASTM D-1238 standard. The measurements were conducted at 190 °C with load weights of 2.16 kg. After a pre-heating time of 360 seconds and a rejection time of 60 seconds, three cuts were performed at one-minute intervals. The average value of these three cuts was recorded as the resultant MFI. Three specimens were tested, and the average MFI value was expressed in g per 10 min. All the specimens were stored at a temperature of 27 ± 2 °C under 52% humidity before testing.
Burst tests were performed using a burst tester procured from Advanced Equipments, India, in accordance with ASTM F1140-00. A film of size 15 × 20 cm2 was folded, and the exposed edges were heat-sealed ensuring proper sealing to form a packet. A 1 mm hole was then cut into the middle of the formed packet, which was attached to the air pipe of the burst tester, and subjected to burst testing. Three specimens of each sample were tested, and the average value was recorded as the burst capacity, expressed in psi. All of the film packets were stored at a temperature of 27 ± 2 °C under 52% humidity before testing.
The tensile strength of the films was measured using a Universal Testing Machine (UTM) operated at a maximum load of 2.5 kN and a speed of 12 mm min−1, in accordance with the ASTM D882-02 standard. The thickness of the films was measured using a thickness meter provided by S C Dey and Co., India. The thickness was measured at eight different places of the films and the average value was taken as the final thickness of the films. The stress (kg) versus elongation (%) graph was obtained, and the tensile strength (MPa), elongation at break (%), and Young's modulus (MPa) were calculated. Three specimens of each sample were tested, and the average values were recorded. All the specimens were stored at a temperature of 27 ± 2 °C under 52% humidity for 24 h prior to testing.
The contact angle of the composite films was measured using a Krüss goniometer controlled by Kruss advance software (v1.6.2.0) with gravitational acceleration of 9.0665 m s−2. The drop phase was water and the surrounding phase was air. Three specimens of the same film with sample size of 2 × 5 cm2 were cut and stuck to a glass slide and then tested; the mean contact angle was evaluated. All specimens were stored at a temperature of 27 ± 2 °C under 52% humidity for 24 h prior to testing.
The thermogravimetric analysis of the films was performed using a PerkinElmer thermal analyzer with a heating profile of 35 °C to 750 °C ramped at 10 °C min−1 under a continuous nitrogen flow of 10 mL min−1. The initial mass (nearly 8–10 mg) of the specimen was precisely measured before testing. All the specimens were stored at a temperature of 27 ± 2 °C under 52% humidity for 24 h prior to testing.
The DSC analysis of the films was performed using a PerkinElmer thermal analyzer over a temperature range of 40–400 °C at a heating rate of 20 °C min−1. Film samples of 8–10 mg were prepared and subjected to analysis, and heat-flow curves (Endo Up) were recorded as a function of temperature. From these curves, enthalpy changes were integrated to determine the melting temperature (Tm), crystallization temperature (Tc), crosslinking temperature, decomposition/oxidation temperature and the crystallinity index (χc). The enthalpy change (exothermic and endothermic) and the crystallinity index were calculated using the formulas given below:22
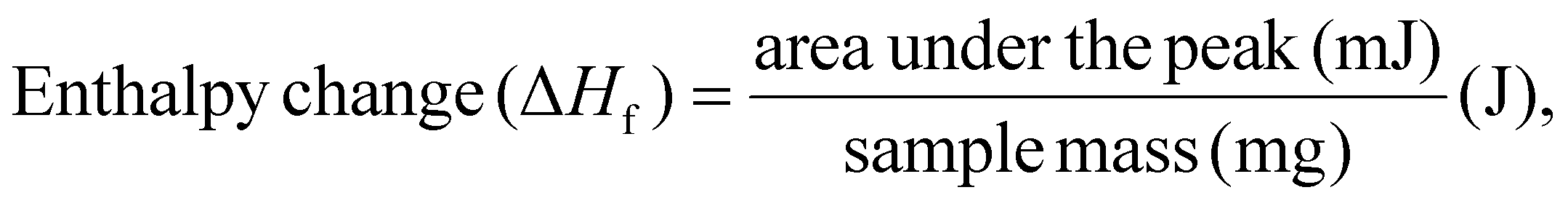 | (2) |
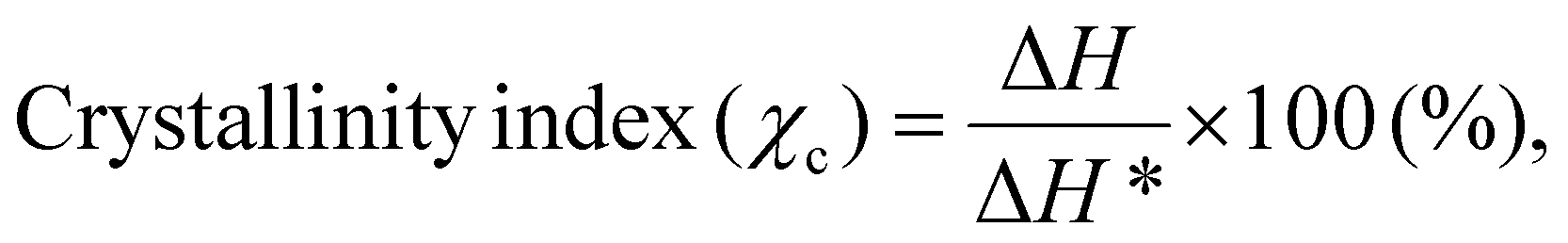 | (3) |
The water vapor permeability test was done following ASTM E96-E96M. 120 g of calcium chloride (CaCl2) was heated at 80 °C for 4 hours in a hot-air oven to remove any excess moisture, setting the internal vapor pressure to zero. Then, the dried CaCl2 was transferred into three 100 mL beakers (40 g per beaker) and covered using thin film samples. The beaker was tightly sealed using Sellotape to prevent the entry of any excess moisture. The thickness of the films was measured and the exposed area of the film was calculated using the diameter of the beaker. Subsequently, the setup was placed in a humidity chamber at 90% RH and 27 °C. The initial weight was measured, and at intervals of 3 h, the weight was measured again for 24 h. The water vapor transmission rate (WVTR) and water vapor permeability (WVP) were calculated using the following equations:23,24
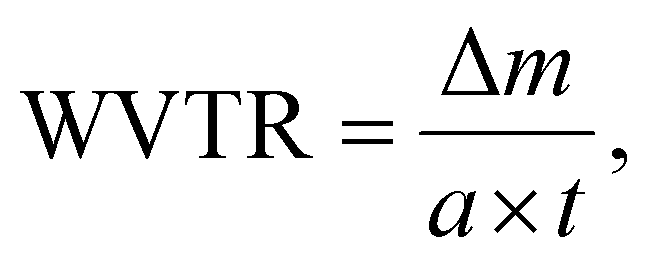 | (4) |
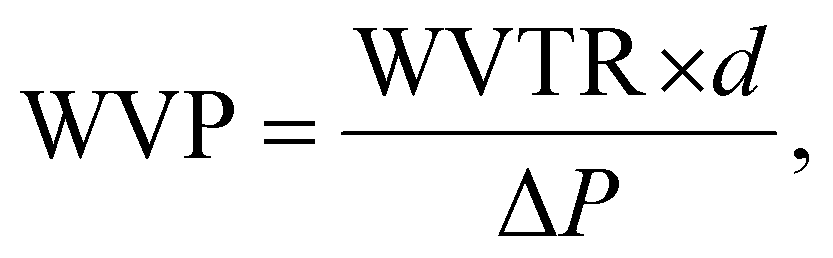 | (5) |
| ΔP = Pwater = Psat × RH, | (6) |
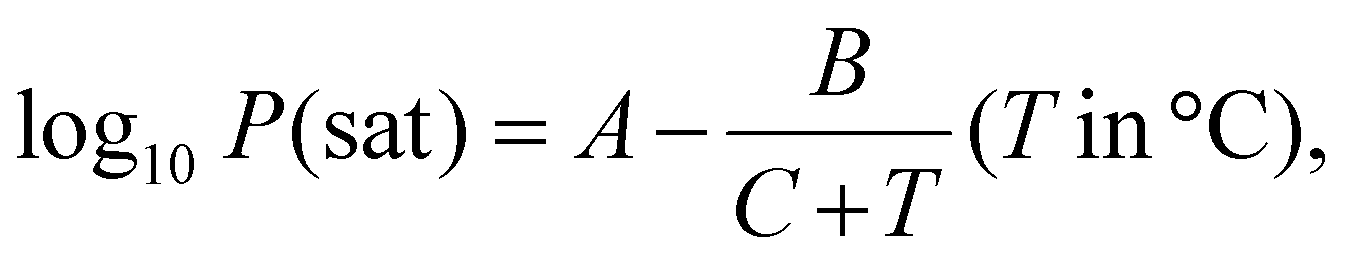 | (7) |
Atomic force microscopy (AFM) images of the films were acquired to check the surface vibration and topology of the films. A thin film of 1 × 1 cm2 was cut, and then it was cleaned using cotton to ensure it was lint-free to avoid any contamination. The sample was conditioned at 27 ± 2 °C under 52% relative humidity for 24 hours prior to testing.
The surface imaging of the films was performed by scanning electron microscopy (SEM) to check for surface cracks and the incorporation of fillers into the compostable polyester. A thin film of 1 × 1 cm2 was cut, and then it was cleaned using cotton to ensure it was lint-free to avoid any contamination during the surface imaging. Additionally, a thin film of 1 × 1 cm2 was cut perpendicular to the surface for cross-sectional imaging. The samples were conditioned at 27 ± 2 °C under 52% relative humidity for 24 hours prior to testing.
For biodegradation studies, each film sample was cut into pieces of nearly 2 × 2 cm2 and accurately weighed using a precision balance. Each sample was then buried at a depth of 10–15 cm in microbe-enriched soil to facilitate biodegradation under natural conditions. The film samples were incubated in the soil for a period of 45 days. After the incubation period, the films were carefully retrieved and gently cleaned to remove any adhering soil and contaminants, and then dried before being reweighed. The extent of biodegradation was determined by calculating the percentage weight loss using the following equation:
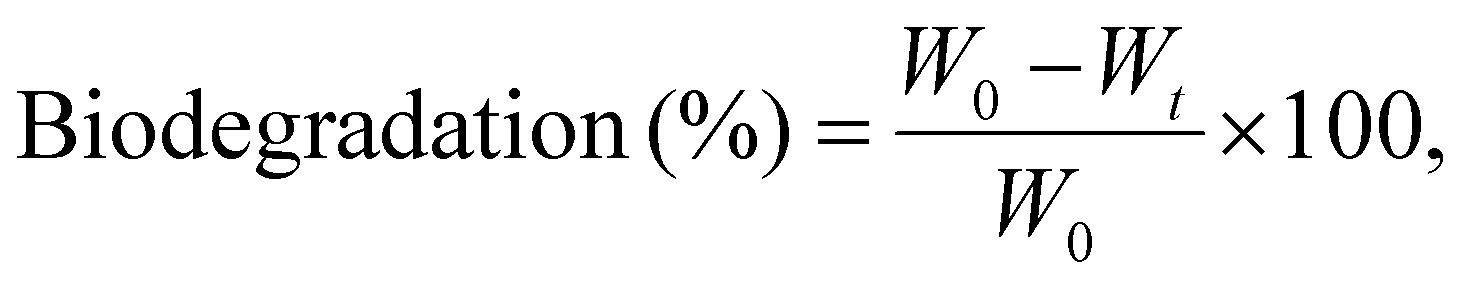 | (8) |
Statistical analysis was performed using one-way analysis of variance (ANOVA), followed by the Student–Newman–Keuls method for all pairwise multiple comparisons. A p-value of less than 0.05 was considered statistically significant. Each experiment was conducted in triplicate in this research work, and the data were expressed as mean ± standard deviation.
4. Results and discussion
SO, containing a high proportion of unsaturated triglycerides, was epoxidized successfully via a peracid-based epoxidation process. The efficiency of the epoxidation reaction was evaluated using different analytical techniques: the oxirane oxygen content test, CHNSO elemental analysis and NMR studies.4.1 Oxirane oxygen test and CHNSO analysis
The oxirane oxygen content, which indicates the presence of epoxy (oxirane) groups formed by the reaction of double bonds with a peracid (formed after formic acid reacts with peroxide), showed a substantial increase from 21.6 to 63.2. This increase confirmed that a significant number of double bonds in the fatty acid chains were successfully converted into epoxy groups, validating the effectiveness of the epoxidation process. Further confirmation was obtained through CHNSO elemental analysis, which tracks the elemental composition (carbon, hydrogen, nitrogen, sulfur, and oxygen) of the sample. A notable increase in the oxygen content from 7.39% (in the edible soybean oil) to 24.32% (in the epoxidized oil) was observed, as shown in Table S1. This considerable increase in oxygen percentage supports the introduction of oxygen-containing epoxy functionalities during epoxidation. Following epoxidation, the viscosity of soybean oil increased dramatically from 572 ± 20 cP to 3760 ± 45 cP, while its shear stress increased from 5207 to 10783 mPa at 30 °C, indicating increased molecular interactions as a result of the epoxy group formation. The addition of these rigid polar oxirane rings decreased the molecular mobility, creating a more cohesive and thicker fluid. Moreover, the density was 0.878 g cm−3 to 0.938 g cm−3, indicating successful integration of oxygen atoms into the molecular structure of the oil.26
The formation of oxirane rings from carbon–carbon double bonds was established using both 1H and 13C nuclear magnetic resonance (NMR) spectroscopy.
4.2 1H-NMR analysis and 13C-NMR analysis
The 1H NMR spectrum (Fig. S1a) of edible soybean oil showed distinctive signals of olefinic protons at δ ∼ 5.30–5.40 ppm corresponding to the protons of the unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. Following epoxidation, these signals were drastically decreased or vanished in the EPSO spectrum, showing the absence of double bonds. At the same time, new signals emerged in the δ ∼ 2.90–3.20 ppm region in the EPSO spectrum. These peaks were due to the protons of the newly formed oxirane rings (epoxy groups), verifying successful epoxidation. Also, minor shifts and changes in the methylene proton region (δ ∼ 1.20–2.80 ppm) also supported the structural alterations in the triglyceride backbone after epoxidation.27
C bonds. Following epoxidation, these signals were drastically decreased or vanished in the EPSO spectrum, showing the absence of double bonds. At the same time, new signals emerged in the δ ∼ 2.90–3.20 ppm region in the EPSO spectrum. These peaks were due to the protons of the newly formed oxirane rings (epoxy groups), verifying successful epoxidation. Also, minor shifts and changes in the methylene proton region (δ ∼ 1.20–2.80 ppm) also supported the structural alterations in the triglyceride backbone after epoxidation.27
The 13C NMR (Fig. S1b) spectrum of SO featured olefinic carbon signals at δ ∼ 127–130 ppm. In the EPSO spectrum, these signals either decreased or disappeared, with new signals appearing at δ ∼ 54–58 ppm corresponding to the carbons of the epoxide rings. This change was a definite sign of the double bond conversion to three-membered oxirane rings. In addition, carbonyl (δ ∼ 173 ppm) and glycerol backbone (δ ∼ 62–72 ppm) signals did not exhibit much change, which suggested that the epoxidation process had selectively taken place at the CC positions without the degradation of the triglyceride backbone.28,29
Together, these results clearly showed the successful epoxidation of soybean oil, providing a modified product enriched with reactive epoxy groups suitable for further applications such as polymer synthesis, plasticization, or resin formulations.
4.3 Melt flow index
The melt flow index (MFI) of each blended composite was measured, and the results are presented in Table S2. An increasing trend in the MFI values was observed with the incremental addition of EPSO. The control sample without EPSO and RBW (secondary reinforcement filler), i.e. TPS–PBAT, exhibited an MFI value of 5.35 g per 10 min at 190 °C under a load of 2.16 kg. Upon incorporating the secondary reinforcement filler, the MFI value slightly increased to 5.52 for TPS-G30-E0-PBAT. Upon incorporation of EPSO at varying concentrations (5% to 20%), the MFI values increased to 6.86, 7.72, and 8.95 g per 10 min for the films TPS-G25-E5-PBAT (5%), TPS-G20-E10-PBAT (10%), and TPS-G15-E15-PBAT (15%), respectively. This increase was attributed to the plasticizing effect of EPSO, which acts as a green plasticizer. EPSO improves the chain mobility and reduces the intermolecular interactions between polymer chains, thereby decreasing melt viscosity and enhancing flow properties. This improved MFI contributes significantly to better processability during blown film extrusion, facilitating smoother flow of the melt through the die and film formation. However, a slight decrease in the MFI to 5.88 g per 10 min was observed for the sample TPS-G10-E20-PBAT at the highest EPSO concentration (20%). This decline was likely due to over-plasticization, which can lead to phase separation or reduced compatibility between the polymer matrix and the plasticizer at excessive concentrations. Such behavior hinders effective stress transfer and reduces the free volume available for chain mobility, resulting in decreased melt flow and alterations of blow properties.4.4 Mechanical properties
Mechanical properties of the blown thin films were assessed using tensile strength, elongation at break, and burst tests. The results are presented in Table S2. TPS–PBAT exhibited well-balanced mechanical characteristics with moderate tensile strength and high elongation, reflecting ductile and flexible film properties. Application of glycerol as a hydrophilic plasticizer enhanced starch flexibility due to increased intermolecular spacing and chain mobility. In TPS-G30-E0-PBAT, the incorporation of RBW, a hydrophobic and semi-crystalline material, resulted in reduced tensile strength (6.21 MPa) but elevated elongation (426%). This is due to the fact that wax interferes with polymer chain packing, decreases stiffness, and improves plastic deformation ability. Reduced interfacial compatibility between hydrophilic and hydrophobic phases might be responsible for strength loss, however. In TPS-G25-E5-PBAT, TPS-G20-E10-PBAT, and TPS-G15-E15-PBAT, EPSO was added in increasing quantities in place of glycerol. EPSO, as a reactive, green plasticizer containing oxirane functional groups, can form non-covalent bonds (e.g., hydrogen bonding) and potential covalent linkages (weak crosslinking) with starch, NCC, and PBAT, hence enhancing film cohesion. In TPS-G25-E5-PBAT, even though EPSO has been added, the tensile strength decreased even more (5.63 MPa), since, at lower levels of EPSO, there would not be complete plasticizer–matrix interactions with reduced glycerol content. In TPS-G20-E10-PBAT, there was remarkable improvement in the tensile strength (13.63 MPa) due to the optimum EPSO content where increased intermolecular interactions, enhanced chain entanglement, and greater compatibility between PBAT and starch resulted in a more robust and consistent network. Nevertheless, elongation decreased to 299%, signifying a compromise in strength and elongation because of an increasingly rigid structure. In TPS-G15-E15-PBAT, minor loss in strength (11.55 MPa) and partial recovery in elongation (325%) were revealed, signifying that redundant EPSO might begin to phase-separate or decrease crosslink density, causing a more plasticized but slightly weaker matrix to form. Failure to acquire a blown film for TPS-G10-E20-PBAT indicated over-plasticization or Phase separation, which degrades melt strength and elasticity—parameters that are the most important for stable bubble development during the process of blown film extrusion. The mechanical properties of the blended composite films are shown in Fig. 3. | ||
| Fig. 3 Mechanical properties of the composite thin films. | ||
The burst pressure of the blown films was determined in order to check their resistance to internal pressure under stress conditions (Table S2). These findings showed that all films had good structural integrity, with TPS-G20-E10-PBAT having the greatest burst strength (16 ± 2 psi), matching well with its increased tensile strength. The addition of EPSO in TPS-G25-E5-PBAT, TPS-G20-E10-PBAT, and TPS-G15-E15-PBAT helped to increase internal cohesion and flexibility of the matrix, improving the resistance of the films to higher pressures prior to rupture. Notably, none of the films ruptured from the heat-sealed areas under testing. This showed very good heat sealability of the films, which was due to the addition of PBAT and plasticizers (glycerol and EPSO) that provided enhanced thermoplastic flow and interfacial adhesion on sealing. The compatibility and partial miscibility of the components of the films ensured strong, homogeneous seals that resist high internal pressure without rupture.
4.5 Barrier properties
The barrier properties of the composite thin films were quantified by analyzing their water vapor permeability (WVP) and water contact angle (WCA). The values of WVP and WCA are shown in Table S3. A distinct decreasing trend of WVP was found going from TPS-PBAT to TPS-G15-E15-PBAT, revealing the enhancement of barrier properties of the films with an increase in EPSO content. The large value of WVP for TPS-PBAT (190.23 g m−2 day−1 cm−1 bar−1) is a result of the hydrophilic characteristics of glycerol and starch, both of which easily associate with water molecules and make it easy for them to diffuse into the matrix of the film. The stepwise decrease in WVP in TPS-G30-E0-PBAT to TPS-G15-E15-PBAT was the result of the addition of RBW (TPS-G30-E0-PBAT) and varying the levels of EPSO in TPS-G25-E5-PBAT to TPS-G15-E15-PBAT. Both RBW and EPSO add hydrophobic long-chain aliphatic moieties and epoxide functional groups, which decreased the overall polarity of the polymer matrix. This decreased the affinity of the film for water molecules and restricted their diffusion within the material. In a similar way, the water contact angle, a measure of surface wettability, increased from 69.7° in TPS-PBAT to 108.6° in TPS-G15-E15-PBAT. The low contact angle of TPS-PBAT suggested a hydrophilic surface, as seen with the presence of glycerol and starch. The minor increase in contact angle of TPS-G30-E0-PBAT (72.4°) is because RBW is hydrophobic. The water contact angle on the film created by a water droplet is shown in Fig. 4. An even greater increase was seen in TPS-G25-E5-PBAT to TPS-G15-E15-PBAT, with readings of over 90°, signifying a shift toward a hydrophobic surface. This increase in hydrophobicity was attributed to the occurrence of epoxide rings and long alkyl chains in EPSO, which lower surface energy and affinity to water. Moreover, the partial incompatibility between hydrophilic starch and hydrophobic EPSO could result in microphase separation, where hydrophobic domains migrate to the surface upon film formation, enhancing water repellency. | ||
| Fig. 4 Contact angle of the composite thin films. | ||
The prepared films exhibited enhanced mechanical and barrier properties compared to films in previously reported works based on TPS-PBAT containing different additives in different ratios (Table 2).
| TPS-PBAT | Tensile strength | Elongation at break | Young's modulus | WVP | Ref. |
|---|---|---|---|---|---|
| 40–60 | 5 | 184 | 57.68 | — | 10 |
| 40–60 | 4.32 ± 0.31 | 264.47 ± 21.33 | — | — | 11 |
| 50–50 | 3.5 ± 2 | 48 ± 5 | 261 ± 15 | 12 | |
| 30–70 | 10.3 ± 0.91 | — | 1200.94 ± 16 | 63.96 ± 1.02 | 14 (previous work) |
| 40–60 | 5.8 ± 0.2 | 520 ± 20 | — | 12.5 ± 0.8 | 30 |
| 40–60 | 6.3 ± 0.3 | 256.8 ± 34.0 | 112.0 ± 34.5 | — | 31 |
| 30–70 | 25.43 | 580.83 | — | — | 32 |
| 50–50 | 8.8 ± 0.3 | 510 ± 27 | — | 415 ± 25 | 33 |
| 40–60 | 11.55 ± 0.35 | 325 ± 26 | — | 117.09 ± 3.88 | Current work |
4.6 Surface morphology topography
The surface morphology and topography of the composite films were analyzed using scanning electron microscopy (SEM) and atomic force microscopy (AFM), as shown in Fig. 5. The micrographs indicated that the different formulations displayed conspicuous differences in surface characteristics, a reflection of the effect of the formulation composition and plasticizer content. The TPS-PBAT film, made up of glycerol-plasticized starch, NCC, PBAT, MA, and SA, had a porous and rugged surface with visible non-uniformity. The occurrence of multiple surface pores and defects was due to the irregular dispersion of NCC in the starch–PBAT matrix. Ineffective interfacial compatibility and low hydrogen bonding between the hydrophilic NCC and hydrophobic PBAT could cause localized aggregation and stress sites in the film formation process, leading to surface vibration and micro-voids. In TPS-G30-E0-PBAT, the addition of RBW resulted in surface uniformity, accompanied by a reduction in surface vibrations. Nevertheless, pores were still visible, suggesting that the presence of wax was insufficient to improve the compatibility or dispersion of NCC into the matrix. SA, which is a long-chain fatty acid, is both a processing aid and dispersion promoter. It assisted in lowering the surface tension and enhancing the dispersion of NCC and RBW in the hydrophobic PBAT matrix, further enabling a smoother surface.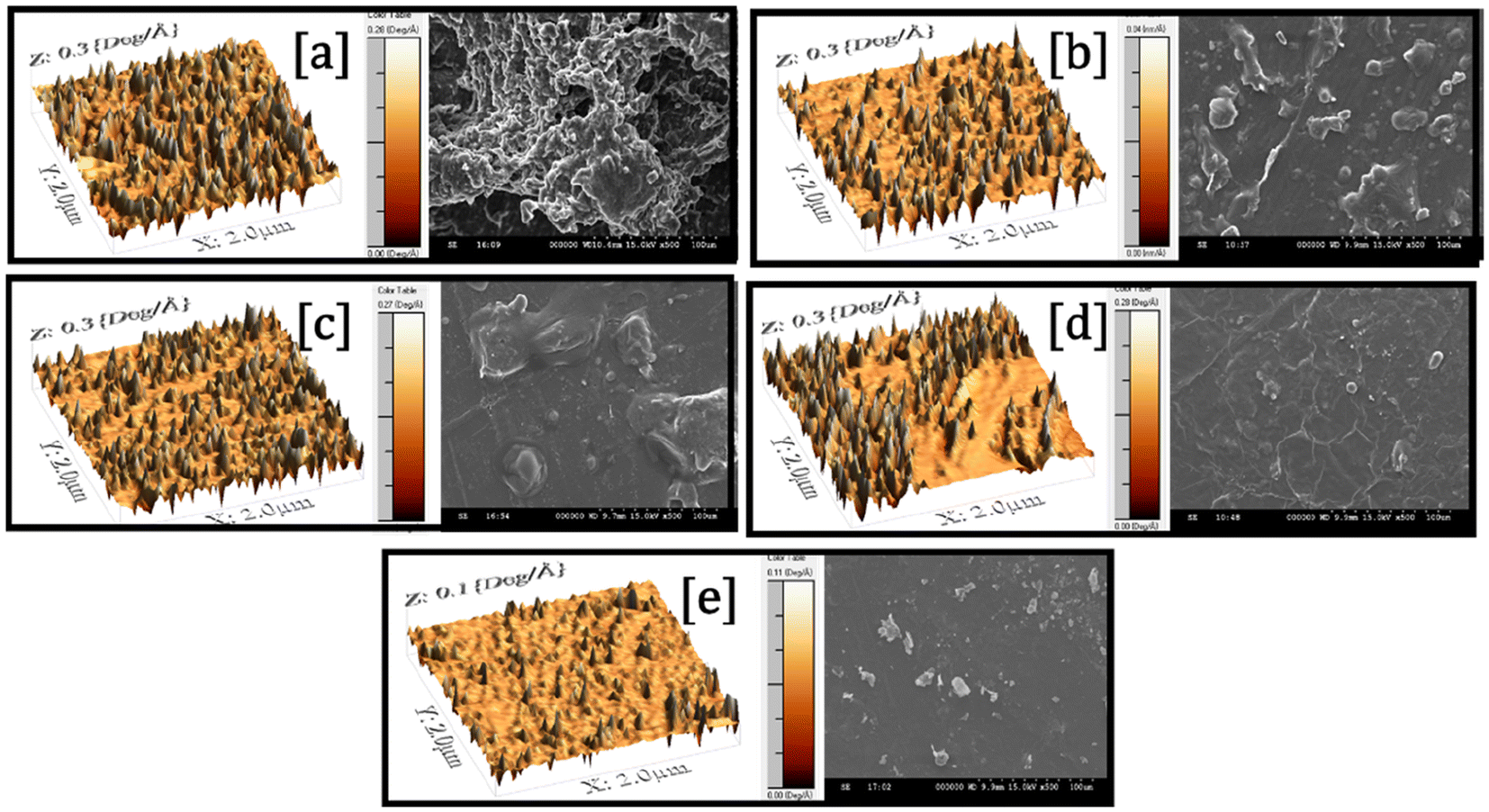 | ||
| Fig. 5 AFM and SEM images of the films (a) TPS-PBAT (b) TPS-G30-E0-PBAT (c) TPS-G25-E5-PBAT (d) TPS-G20-E10-PBAT (e) TPS-G15-E15-PBAT. | ||
With the addition of EPSO to the films TPS-G25-E5-PBAT, TPS-G20-E10-PBAT, and TPS-G15-E15-PBAT, there was a clear decrease in surface porosity and increased smoothness. The oxirane groups in EPSO were capable of interacting with starch and NCC hydroxyl groups through hydrogen bonding or weak covalent interactions, facilitating interfacial compatibility, which resulted in its enhanced dispersion. MA acts as a compatibilizer by grafting onto PBAT chains and creating ester or anhydride linkages with hydroxyl functionality in starch and NCC. This chemical bridging enhances interfacial adhesion and minimizes phase incompatibility, leading to improved morphology and mechanical integrity. Furthermore, the plasticizing effect of EPSO enhances the flexibility and flowability of the polymer matrix during processing, leading to pore closure and surface leveling. Of all the films, TPS-G20-E10-PBAT and TPS-G15-E15-PBAT contained the most even and smooth surfaces with the least porosity. This was because an ideal ratio of EPSO was used, which had a balance of plasticization and compatibility but without causing phase separation.
4.7 Thermal analysis and FTIR
Thermal stability of the composite films was analyzed by thermogravimetric analysis (TGA) and derivative thermogravimetric analysis (dTGA). The TGA curves are plots of the percentage weight loss against temperature, whereas dTGA curves reflect the rate of degradation (shown in Fig. 6), which marks the different thermal events characterizing the decomposition of different components of the blend. All the films had a multi-step thermal degradation profile. The first weight loss, seen at temperatures below 150 °C, was associated with the evaporation of adsorbed water and low molecular weight volatiles. The second degradation process, in the range of 250–350 °C, was primarily the result of thermal decomposition of the hydrophilic moieties like starch, glycerol, and NCC. These degradation processes included dehydration and depolymerization reactions, coupled with fragmentation of the polysaccharide backbone. The third significant degradation region, from 380 °C to 500 °C, was linked to the breakdown of PBAT, EPSO, and lipid additives such as SA and RBW. PBAT was degraded by chain scission via ester bond cleavage, whereas EPSO was degraded via epoxide and fatty acid chain breakdown. From the TGA analysis, it was evident that the TPS-PBAT sample, which contained only glycerol as a plasticizer, displayed the lowest thermal stability, as indicated by the earlier onset of degradation and higher rate of mass loss. This was primarily due to the poor compatibility between starch and PBAT and the high volatility of glycerol. Conversely, the films with RBW and EPSO exhibited improved thermal stability, characterized by a retarded degradation onset and a shift of the dominant decomposition peaks to higher temperatures. Adding RBW to TPS-G30-E0-PBAT made the thermal behavior slightly better by helping to enhance its hydrophobic nature and produce a semi-crystalline thermal transport barrier. A greater improvement was found for TPS-G25-E5-PBAT, TPS-G20-E10-PBAT, and TPS-G15-E15-PBAT with progressively higher content of EPSO. The epoxide groups of EPSO can facilitate secondary interactions or weak covalent bonding with the hydroxyl groups of starch and NCC, hence enhancing compatibility and thermal homogeneity. In addition, MA, when utilized as a compatibilizer, can graft onto PBAT chains and react with starch and NCC hydroxyl groups to produce ester or anhydride linkages, promoting interfacial adhesion and improving the thermal resistance of the matrix. SA further improved thermal stability by enhancing the dispersion of NCC and minimizing the stress concentrations in the matrix, leading to further thermal uniformity. These observations were supported by the dTGA curves. TPS-PBAT showed steep and prominent degradation peaks, which represented fast and inhomogeneous decomposition. Yet, TPS-G20-E10-PBAT and TPS-G15-E15-PBAT showed smoother and wider peaks at elevated temperatures, showing more gradual and homogeneous degradation. This trend verifies the enhanced thermal integration of additives within the matrix as a result of the synergistic effects from the plasticizers.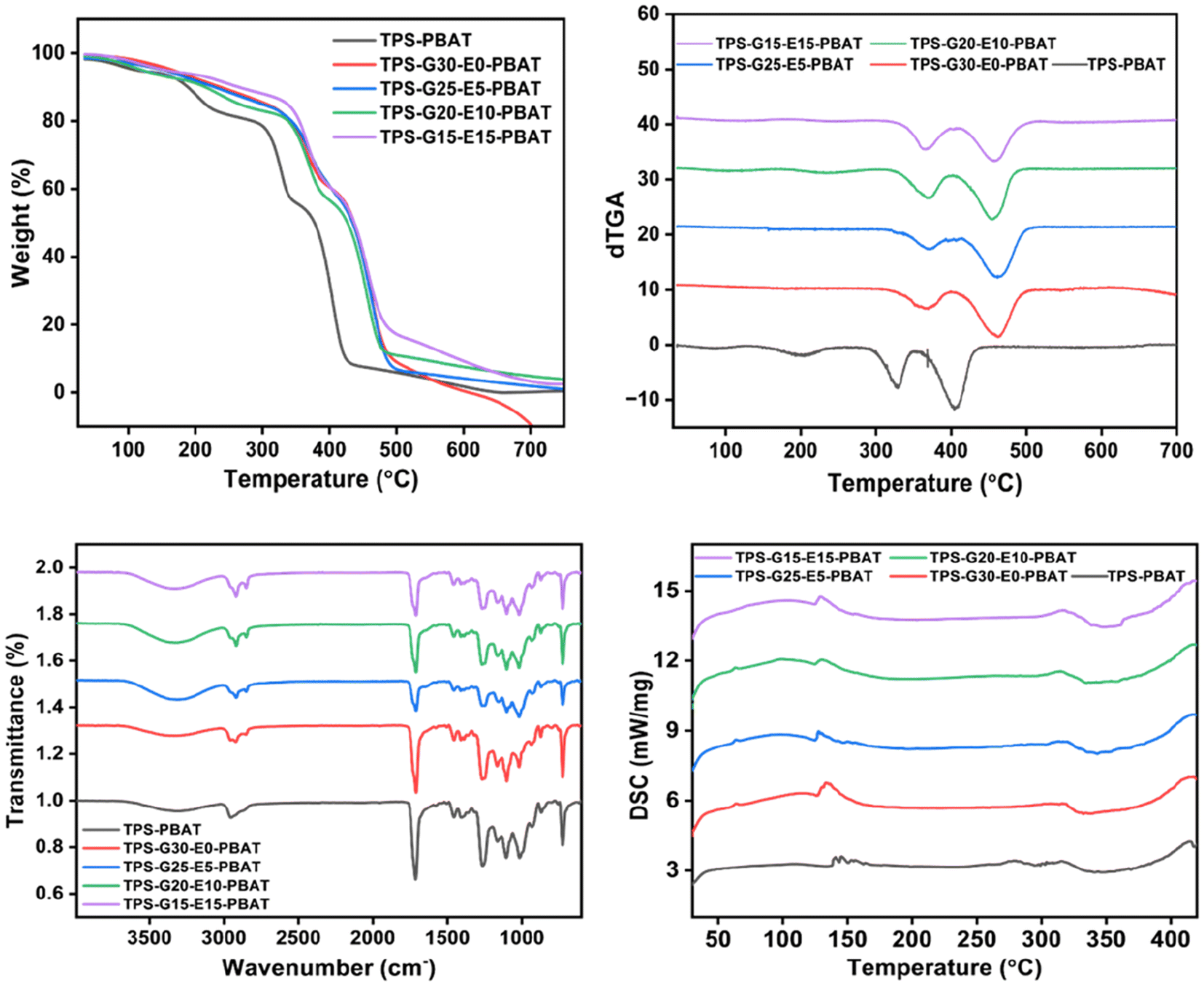 | ||
| Fig. 6 TGA, dTGA, FTIR and DSC graphs of the composite films. | ||
The thermal and structural responses of the composite films were evaluated by Fourier transform infrared spectroscopy (FTIR) and differential scanning calorimetry (DSC). These analytical methods gave information regarding the chemical functional groups, compatibility between the components, and thermal transitions.
The FTIR spectra (Fig. 6) of the films exhibited representative absorption bands that affirmed the existence and interaction among the constituent polymers and additives. All films featured a broad peak in the range 3200–3500 cm−1, attributed to O–H stretching vibrations, essentially from starch, glycerol, and NCC. This wide band decreased in intensity in TPS-G20-E10-PBAT and TPS-G15-E15-PBAT, suggesting that fewer free hydroxyl groups were available, which was probably due to hydrogen bonding and esterification reactions between the epoxy groups of EPSO.34 Peaks at around 2920 and 2850 cm−1 corresponded to C–H stretching of aliphatic chains in PBAT, SA, and EPSO. An intense absorption signal at 1740 cm−1 was attributed to the CO vibration of ester linkages in PBAT and EPSO, which became more intense and well-defined in the films TPS-G25-E5-PBAT, TPS-G20-E10-PBAT and TPS-G15-E15-PBAT, in agreement with the increased interaction between EPSO and the starch matrix. The FTIR spectrum further exhibited intense bands in the range of 1000 to 1200 cm−1, linked with C–O–C and C–O stretching vibrations of starch and PBAT. These bands were more uniform and peak-resolved in the TPS-G20-E10-PBAT and TPS-G15-E15-PBAT films, indicating improved uniformity of distribution and interaction of the components. The presence of MA was thought to enhance covalent bonding between the hydroxyl-abundant starch/NCC phase and the hydrophobic PBAT matrix, thus improving interfacial adhesion. SA also helped in better dispersion of NCC and increased the hydrophobic nature of the matrix, which indirectly contributed towards better molecular alignment and smoothness of the film.35,36
Thermal transitions of the composite films were analyzed by differential scanning calorimetry (DSC) (Fig. 6). A wide endothermic zone at 60–90 °C represented the glass transition temperature (Tg) of the amorphous part of the starch and PBAT matrix. The Tg value of TPS-G20-E10-PBAT and TPS-G15-E15-PBAT was marginally higher than that in TPS-PBAT, reflecting their greater molecular interaction and lower mobility of polymer chains because of the incorporation of EPSO and compatibilizers. This reflects that EPSO is not only a plasticizer but also that it improves chain entanglement through its multifunctional epoxy groups. Sharp and intense peaks at distinct endothermic points between 130 and 200 °C are reflective of the melting points of the crystalline regions in PBAT and lipoid additives like RBW and SA. TPS-G20-E10-PBAT and TPS-G15-E15-PBAT revealed these peaks to be sharper and more intense, pointing towards their enhanced crystallinity and more thermally resistant structures. The enhanced crystallization behavior was likely due to successful plasticization by EPSO, which promoted chain mobility during processing, and subsequent improvement of molecular packing on cooling. The DSC profiles of TPS-PBAT and TPS-G30-E0-PBAT featured wider and less sharp transitions, indicative of poorer miscibility and structural order caused by glycerol-rich plasticization.
4.8 Biodegradation experiments
In the biodegradation experiments (Table 3), a remarkable decrease in the weight of the composite films was noticed, reflecting successful microbial degradation under conditions of soil burial. Of all the films, the TPS-PBAT film had the maximum weight loss of 82.46%, reflecting better biodegradability. This was followed by 79.23% in TPS-G30-E0-PBAT, 78.56% in TPS-G25-E5-PBAT, 76.23% in TPS-G20-E10-PBAT, and 75.11% in TPS-G15-E15-PBAT after 45 days of soil burial. The greater degradation of the TPS-PBAT film was due to the greater content of glycerol, which increased the hydrophilicity of the matrix, thus allowing greater water uptake and penetration by microorganisms. Furthermore, the absence of EPSO in TPS-PBAT minimized the level of crosslinking and hydrophobic interactions, thus making the matrix more prone to enzymatic attack under natural conditions. In contrast, with increasing EPSO content going from TPS-G25-E5-PBAT to TPS-G20-E10-PBAT and TPS-G15-E15-PBAT, the films became hydrophobic and more structurally dense because crosslinked networks were formed between the hydroxyl groups of starch or NCC and the epoxy groups of EPSO. The higher crosslink density and lower water absorption impeded microbial invasion, leading to slightly reduced rates of biodegradation in the EPSO-rich composites. However, all the formulations proved to be highly biodegradable, upholding their place within areas of sustainable use.| TPS-PBAT | TPS-G30-E0-PBAT | TPS-G25-E5-PBAT | TPS-G20-E10-PBAT | TPS-G15-E15-PBAT | |
|---|---|---|---|---|---|
| Before degradation |

|

|

|

|

|
| Degradation after 45 days |

|

|

|

|

|
5. Conclusion
In this reported work, elemental and spectroscopic analyses confirmed the successful synthesis of EPSO via in situ peracid epoxidation of edible soybean oil. Incorporating EPSO as a bio-based plasticizer into TPS-PBAT nanocomposites, reinforced with NCC, RBW and other sustainable additives, material performance was significantly enhanced. Substituting 10–15 wt% of glycerol with EPSO improved processability, interfacial compatibility, and polymer chain mobility, which led to increased mechanical strength (13.63 ± 0.81 MPa), elongation (299 ± 18%), and film integrity.EPSO-plasticized TPS-PBAT films exhibited increased thermal stability, lower water vapor permeability (117.09 ± 3.88), and greater surface hydrophobicity (WCA of 108.6 ± 0.46°), as indicated by higher contact angles, and smoother morphologies as observed using scanning electron microscopy (SEM) and atomic force microscopy (AFM). These enhancements resulted from the reactive, hydrophobic, and lubricating properties of EPSO, which facilitated uniform dispersion and effective molecular interactions within the polymer matrix. The biodegradation rate remained >75%, indicating that EPSO incorporation did not reduce environmental sustainability.
In summary, EPSO functions as an efficient and environmentally friendly plasticizer that reinforces the mechanical, barrier, and thermal properties of TPS-based composites while preserving their biodegradability. The optimized formulations, specifically TPS-G20-E10-PBAT and TPS-G15-E15-PBAT, demonstrated their strong potential for use in sustainable packaging, mulch films, and other biodegradable applications.
6. Future outlook
The present study establishes the mechanical, thermal, barrier, and biodegradation performance of EPSO-modified TPS/PBAT composites, showing their suitability for sustainable packaging applications. However, for practical deployment, the long-term performance stability of these materials under environmental factors such as humidity, temperature fluctuations, and ultraviolet (UV) radiation requires further evaluation. Previous studies on TPS/PBAT systems have reported that prolonged exposure to such conditions can lead to plasticizer migration, oxidation, and gradual mechanical deterioration.36 In the present formulation, the incorporation of hydrophobic components such as epoxidized soybean oil (EPSO) and rice bran wax (RBW) is expected to mitigate these effects by reducing water uptake and providing a protective barrier against moisture and UV-induced degradation. The reactive epoxy functionality of EPSO can also promote limited crosslinking, improving structural cohesion and resistance to environmental aging. Future studies will therefore include accelerated aging and UV-resistance analyses to assess the retention of mechanical and barrier properties over extended storage periods.In addition to performance stability, the investigation into the long-term safety of these films for potential food-contact applications is essential. Epoxidized soybean oil (EPSO) is a bio-based and non-toxic additive that is listed by the U.S. Food and Drug Administration (FDA) for use as a plasticizer and stabilizer in food packaging materials. To ensure full regulatory compliance, overall and specific migration studies will be conducted in accordance with EU Regulation No. 10/2011. Previous reports15,16 have shown that EPSO exhibits very low migration levels because its reactive epoxy groups can partially crosslink with hydroxyl functionalities of starch, PBAT, and cellulose, thereby immobilizing it within the polymer matrix. This indicates that the developed biocomposite films are not only environmentally benign but also hold strong potential for use in compostable and food-safe packaging applications, pending confirmation through migration and aging validations.
Author contributions
Vineet Aniya: supervision, funding acquisition, conceptualization and overall execution. Chandramani Batsh: manuscript writing – original draft, experimentation and data curation. Chandan Kumar Munagala: experimentation and suggestions. Bitopan Boro: formal analysis. Devasish Chowdhury: suggestions and support for the experimental work. Harsha Nagar: suggestions and support for the experimental work.Conflicts of interest
There are no conflicts to declare.Data availability
The data will be made available from the corresponding author upon reasonable request.The supplementry information includes the elemental and rheological analysis of oils, mechanical properties of composite thin films, barrier properties of composite thin films. along with the 1H-NMR and 13C-NMR of oil. See DOI: https://doi.org/10.1039/d5lp00302d.
Acknowledgements
This research was funded by the CSIR – Fast Track Translation (FTT/FTC) Mode Project (MLP-0088) “Process for Cellulose-Derived Reinforced Thermoplastic Starch Blends for Biodegradable Carry Bag Application” under the CLP theme against the sanction reference no. 33/FTT/FTC/4thTr/CLP/2022-TMD. The authors thank the Director CSIR-IICT (Manuscript No. IICT/Pubs./2025/311) for providing all required facilities to carry out this work. Also, the authors acknowledges SAIC, IASST for the instrument facilities. BB Thanks CSIR for the fellowship.References
- I. D. Gómez and A. S. Escobar, The dilemma of plastic bags and their substitutes: A review on LCA studies, Sustain. Prod. Consum., 2022, 30, 107–116 CrossRef
.
- M. Dhairykar, S. Jawre and N. Rajput, Impact of plastic pollution on wildlife and its natural habitat, Pharm. Innov. J., 2022, 141–143 CAS
- M. Piccardo, F. Provenza, E. Grazioli, S. Anselmi, A. Terlizzi and M. Renzi, Impacts of plastic-made packaging on marine key species: Effects following water acidification and ecological implications, J. Mar. Sci. Eng., 2021, 9(4), 432 CrossRef
- M. S. Parvez, H. Ullah, O. Faruk, E. Simon and H. Czédli, Role of microplastics in global warming and climate change: A review, Water Air Soil Pollut., 2024, 235(3), 201 CrossRef CAS
- L. T. Helm, C. Venier-Cambron and P. H. Verburg, The potential land-use impacts of bio-based plastics and plastic alternatives, Nat. Sustain., 2025, 8(2), 190–201 CrossRef
- J. Thrän, G. Garcia-Garcia, C. Parra-López, A. Ufarte, C. García-García, S. Parra and S. Sayadi-Gmada, Environmental and economic assessment of biodegradable and compostable alternatives for plastic materials in greenhouses, Waste Manage., 2024, 175, 92–100 CrossRef
- J. Bai, H. Pei, X. Zhou and X. Xie, Reactive compatibilization and properties of low-cost and high-performance PBAT/thermoplastic starch blends, Eur. Polym. J., 2021, 143, 110198 CrossRef CAS
- N. Nomadolo, A. Mtibe, O. Ofosu, C. Mekoa, J. Letwaba and S. Muniyasamy, The effect of mechanical recycling on the thermal, mechanical, and chemical properties of poly (butylene adipate-co-terephthalate)(PBAT), poly (butylene succinate)(PBS), poly (lactic acid)(PLA), PBAT-PBS blend and PBAT-TPS biocomposite, J. Polym. Environ., 2024, 32(6), 2644–2659 CrossRef CAS
- M. Lackner, F. Ivanič, M. Kováčová and I. Chodák, Mechanical properties and structure of mixtures of poly (butylene-adipate-co-terephthalate)(PBAT) with thermoplastic starch (TPS), Int. J. Biobased Plast., 2021, 3(1), 126–138 CrossRef CAS
- S. Mohanty and S. K. Nayak, Starch based biodegradable PBAT nanocomposites: Effect of starch modification on mechanical, thermal, morphological and biodegradability behavior, Int. J. Plast. Technol., 2009, 13(2), 163–185 CrossRef CAS
- J. B. Olivato, C. M. Müller, F. Yamashita, M. V. Grossmann and M. M. Nobrega, Study of the compatibilizer effect in the properties of starch/polyester blends, Polímeros, 2013, 23, 346–351 CrossRef CAS
- Y. Fourati, Q. Tarrés, M. Delgado-Aguilar, P. Mutjé and S. Boufi, Cellulose nanofibrils reinforced PBAT/TPS blends: Mechanical and rheological properties, Int. J. Biol. Macromol., 2021, 183, 267–275 CrossRef CAS PubMed
- Z. Li, J. Shang, A. Abdurexit, R. Jamal, T. Abdiryim, E. Su and J. Wei, Improving the performance of polylactic acid/polypropylene/cotton stalk fiber composites with epoxidized soybean oil as a high efficiency plasticizer, Int. J. Biol. Macromol., 2024, 283, 137814 CrossRef CAS
- A. K. Kesari, A. M. Mulla, S. M. Razak, C. K. Munagala and V. Aniya, Cellulose nanocrystals engineered TPS/PBAT granulation through extrusion process and application for compostable carry bags, J. Ind. Eng. Chem., 2024, 136, 623–634 CrossRef
- H. H. Wang, S. J. Zhou, S. J. Xiong, Q. Liu, H. Tian, Y. S. Yuan and T. Q, High-performance thermoplastic starch/poly (butylene adipate-co-terephthalate) blends through synergistic plasticization of epoxidized soybean oil and glycerol, Int. J. Biol. Macromol., 2023, 242, 124716 CrossRef CAS PubMed
- Z. X. Fei, J. Sun, C. Cui, C. Yin, R. Zhan, L. Y. Shi, K. K. Yang and Y. Z. Wang, Highly enhanced mechanical strength and toughness of biodegradable PBAT plastics through a biobased multiple hydrogen bonding strategy, Macromolecules, 2024, 57(15), 7043–7051 CrossRef CAS
- H. Lang, X. Chen, J. Tian, J. Chen, M. Zhou, F. Lu and S. Qian, Effect of microcrystalline cellulose on the properties of PBAT/thermoplastic starch biodegradable film with chain extender, Polymers, 2022, 14(21), 4517 CrossRef CAS PubMed
- V. Gigante, L. Aliotta, I. Canesi, M. Sandroni, A. Lazzeri, M. B. Coltelli and P. Cinelli, Improvement of interfacial adhesion and thermomechanical properties of PLA based composites with wheat/rice bran, Polymers, 2022, 14(16), 3389 CrossRef CAS
- K. M. Dang and R. Yoksan, Thermoplastic starch blown films with improved mechanical and barrier properties, Int. J. Biol. Macromol., 2021, 188, 290–299 CrossRef CAS
- J. V. de Quadros Jr. and R. Giudici, Epoxidation of soybean oil at maximum heat removal and single addition of all reactants, Chem. Eng. Process., 2016, 100, 87–93 CrossRef CAS
- P. Pongmuksuwan, T. Jangmee and W. Kitisatorn, Microencapsulated epoxidized palm oil: A self-healing coating solution, Appl. Surf. Sci. Adv., 2023, 18, 100458 CrossRef
- R. A. Garalde, R. Thipmanee, P. Jariyasakoolroj and A. Sane, The effects of blend ratio and storage time on thermoplastic starch/poly (butylene adipate-co-terephthalate) films, Heliyon, 2019, 5(3), e01251 CrossRef PubMed
- S. Rahman, C. Batsh, S. Gurumayam, J. C. Borah and D. Chowdhury, Sodium alginate-nanocellulose-based active composite film for edible oils packaging applications, Mat. Adv., 2024, 5(23), 9314–9329 RSC
- M. P. Calderaro, C. I. de Luca Sarantopóulos, E. M. Sanchez and A. R. Morales, PBAT/hybrid nanofillers composites—Part 1: Oxygen and water vapor permeabilities, UV barrier and mechanical properties, J. Appl. Polym. Sci., 2020, 137(46), 49522 CrossRef CAS
- G. W. Thomson, The Antoine equation for vapor-pressure data, Chem. Rev., 1946, 38(1), 1–39 CrossRef CAS PubMed
- W. B. Garuma, T. K. Bedru, G. T. Gindaba, M. Jayakumar, I. T. Edae and A. G. Demesa, Optimization and characterization of ultrasonically extracted oil from watermelon seeds using response surface methodology: a pathway to biodiesel and epoxy oil production, Biomass Convers. Biorefin., 2025, 1–9 Search PubMed
- P. T. Wai, P. Jiang, M. Lu, Z. Cui, S. Feng and P. Zhang, An Easy and Promising Tool for the Determination of Iodine and Epoxy Values of Epoxidized Soybean Oil by 1H NMR Spectrometry, J. Anal. Chem., 2024, 79(2), 233–240 CrossRef CAS
- G. Z. Li, Q. Wang, C. Zhu, S. Zhang, F. Wang, L. Tao, Y. Jiang, Q. Zhang, W. Wang and R. Han, Synthesis and Performance of Epoxy-Terminated Hyperbranched Polymers Based on Epoxidized Soybean Oil, Molecules, 2025, 30(3), 583 CrossRef CAS
- S. Dalle Vacche, L. H. Esposito, D. Bugnotti, E. Callone, S. F. Orsini, M. D'Arienzo, L. Cipolla, S. Petroni, A. Vitale, R. Bongiovanni and S. Dirè, Modification of Epoxidized Soybean Oil for the Preparation of Amorphous, Nonretrogradable, and Hydrophobic Starch Films, Polysaccharides, 2025, 6(2), 40 CrossRef CAS
- D. Phothisarattana, P. Wongphan, K. Promhuad, J. Promsorn and N. Harnkarnsujarit, Blown film extrusion of PBAT/TPS/ZnO nanocomposites for shelf-life extension of meat packaging, Colloids Surf., B, 2022, 214, 112472 CrossRef CAS PubMed
- L. Lendvai, A. Apostolov and J. Karger-Kocsis, Characterization of layered silicate-reinforced blends of thermoplastic starch (TPS) and poly (butylene adipate-co-terephthalate), Carbohydr. Polym., 2017, 173, 566–572 CrossRef CAS
- K. Cai, X. Wang, C. Yu, J. Zhang, S. Tu and J. Feng, Enhancing the mechanical properties of PBAT/thermoplastic starch (TPS) biodegradable
composite films through a dynamic vulcanization process, ACS Sustainable Chem. Eng., 2024, 12(4), 1573–1583 CrossRef CAS
- P. Wongphan, C. Nerín and N. Harnkarnsujarit, Enhanced compatibility and functionality of thermoplastic cassava starch blended PBAT blown films with erythorbate and nitrite, Food Chem., 2023, 420, 136107 CrossRef CAS PubMed
- Q. Xu, J. Lin and G. Jiang, Synthesis, characterization and properties of soybean oil-based polyurethane, Polymers, 2022, 14(11), 2201 CrossRef CAS
- N. Kotov, P. A. Larsson, K. Jain, T. Abitbol, A. Cernescu, L. Wågberg and C. M. Johnson, Elucidating the fine-scale structural morphology of nanocellulose by nano infrared spectroscopy, Carbohydr. Polym., 2023, 302, 120320 CrossRef CAS
- Y. Laorenza and N. Harnkarnsujarit, Surface adhesion and physical properties of modified TPS and PBAT multilayer film, Food Packag. Shelf Life, 2024, 44, 101312 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2026 |