
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBand alignment and interfacial electrostatics: unraveling the dynamic space charge layer in all-solid-state batteries
Haoyuan
Lai
ab,
Jinli
Liu
c,
Qiqiang
Huang
ab,
Chenxi
Li
a,
Peng
Zhang
a,
Xiaofeng
Luo
a,
Lewei
Shi
a,
Zhibo
Han
a,
Wei
Peng
a,
Xingtai
Liu
a,
Xinman
Chen
b,
Languang
Lu
c,
Xuning
Feng
c,
Dongsheng
Ren
c,
Minggao
Ouyang
 *c and
Xiang
Liu
*a
*c and
Xiang
Liu
*a
aSchool of Materials Science and Engineering, Beihang University, 100191 Beijing, China. E-mail: xiangliu@buaa.edu.cn
bSchool of Electronic Science and Engineering (School of Microelectronics) South China Normal University, Foshan 528225, P. R. China
cSchool of Vehicle and Mobility, Tsinghua University, 100084 Beijing, China. E-mail: ouymg@tsinghua.edu.cn
First published on 24th February 2026
Abstract
All-solid-state batteries (ASSBs) are poised to transform electrochemical energy storage, yet their performance remains critically limited by high interfacial impedance. A central origin of this bottleneck is the space charge layer (SCL), an intrinsic electrostatic structure arising from electrochemical potential mismatch at solid–solid interfaces. Unlike the adaptive electric double layers in liquid electrolytes, SCLs in solids form rigid but dynamically evolving potential barriers that vary with state of charge and strongly regulate lithium-ion transport and interfacial stability. This review provides a critical and unified assessment of SCL physics in ASSBs by integrating defect chemistry, semiconductor band theory, and emerging operando characterization. We clarify SCL formation driven by Fermi level alignment, reconcile divergent views on its quantitative impact on interfacial resistance, and highlight recent experimental advances that directly visualize buried electrostatic fields. Importantly, we systematically compare two major classes of SCL regulation strategies—hierarchical band alignment engineering and interfacial field modulation—by analyzing their applicable electrolyte systems, processing complexity, scalability, and cost implications. While band alignment engineering via buffer or coating layers is particularly effective for oxide-based systems with severe electrostatic mismatch, field modulation strategies offer lower-cost and more scalable solutions for sulfide and composite electrolytes. By explicitly linking interfacial electrostatics to practical material selection and engineering constraints, this review establishes a physically grounded framework for tailoring SCL behavior and provides actionable guidance for the design of next-generation, high-performance ASSBs.
Broader contextAll-solid-state batteries (ASSBs) are widely viewed as a cornerstone of next-generation energy storage, offering improved safety and higher energy density compared to conventional liquid-based systems. However, their widespread application is critically limited by interfacial challenges, among which the space charge layer (SCL) plays a central, yet insufficiently understood role. Originating from Fermi level mismatch, the SCL governs lithium-ion redistribution, interfacial electric fields, and kinetic barriers, thus strongly influencing battery performance and degradation. In this work, we establish a comprehensive and multiscale framework to elucidate the formation and evolution of SCLs and their coupling with electrochemical and mechanical effects. By integrating theoretical modeling with advanced characterization techniques, we reveal how SCL-induced changes in activation energy, local current distribution, and ion transport pathways lead to increased impedance and structural instability, particularly under varying states of charge. Importantly, we highlight the dynamic and spatially heterogeneous nature of SCL effects, by bridging microscopic mechanisms with macroscopic performance. These advances provide critical insights into interfacial regulation strategies and offer general design principles for stabilizing solid–solid interfaces, and thereby accelerate the development of high-performance, durable ASSBs and advancing the broader field of electrochemical energy storage. |
1. Introduction
Amidst the global energy structure's transition towards decarbonization, high-energy-density lithium-ion batteries have emerged as the primary energy storage solution for electric vehicles and smart grids.1–7 While current mainstream lithium-ion batteries still rely on liquid electrolytes, these traditional systems exhibit limitations such as low ignition points and poor thermal stability, rendering them susceptible to fire or explosion incidents.8–10 Furthermore, advancements in technology have pushed the energy density of these batteries close to the theoretical limits of their constituent materials. This is increasingly insufficient to meet the demands for long-range operation and enhanced safety in electric vehicles.11–13 All-solid-state batteries (ASSBs) are regarded as a cornerstone for next-generation energy storage owing to their intrinsic safety, wide electrochemical stability window, and potential for high-voltage operation. Despite the rapid development of high-conductivity solid electrolytes (10−3–10−2 S·cm−1), the interfacial ionic resistance between solid electrolytes and electrodes typically ranges from 10 to 1000 Ω·cm−2, remaining two to four orders of magnitude higher than in liquid systems. This discrepancy constitutes one of the key bottlenecks impeding the practical implementation of ASSBs.14–19 While earlier studies mainly attributed this resistance to chemical incompatibility and mechanical detachment, such explanations fail to account for persistent interfacial polarization even in chemically stable systems (e.g., LiNbO3-coated LiCoO2/LiPON or oxide–oxide interfaces).20–26 This observation suggests the presence of an intrinsic electrostatic origin—the space charge layer (SCL)—resulting from the mismatch of Fermi levels, ion chemical potentials, or defect concentrations across the interface. Unlike liquid electrolytes where ions can freely rearrange to screen potentials, the immobile lattice in solid electrolytes facilitates the formation of extended depletion or accumulation zones, fundamentally altering transport kinetics.1,2,11,12,27,28The concept of the SCL in ionic solids has evolved over more than six decades, bridging classical defect chemistry, semiconductor physics, and modern battery science. A concise historical timeline (as shown in Fig. 1) can effectively illustrate this evolution: the idea of a space charge region was first introduced by W. Schottky in the context of semiconductor heterojunctions, where potential discontinuities were shown to redistribute charge carriers near the interface. Similar phenomena were later reported in ionic conductors, where heterointerfaces were found to induce local potential gradients and, in some cases, enhance ionic conductivity.29
 | ||
| Fig. 1 Historical evolution of the space charge layer concept in solid-state ionics and batteries.30 Copyright 2004, AIP Publishing. Reproduced with permission.31 Copyright 2014, American Chemical Society. Reproduced with permission.32 Copyright 2020, Elsevier. Reproduced under terms of the CC-BY license.33 Copyright 2022, Kazuhiro Hikima et al., published by Springer Nature. Reproduced with permission.34 Copyright 2025, Elsevier. | ||
In the early 21st century, Haruyama et al.31 employed density functional theory (DFT) calculations to demonstrate that inserting a suitable buffer layer could effectively mitigate the lithium-ion depletion or accumulation caused by SCL formation at solid–solid interfaces. This marked the beginning of rational interface design based on electronic structure. Subsequently, the magnitude of the SCL impact became a subject of debate. De Klerk et al.35 used atomistic simulations to estimate the SCL thickness to be less than 1 nm, suggesting that its impact on ionic transport might be negligible under certain conditions. In contrast, Cheng et al.32 experimentally tuned the interfacial electrochemical potential difference via in situ NMR measurements and cathode potential control, showing that the presence of an SCL significantly increases the Li+ migration barrier at the interface.
Recent advancements in characterization have provided deeper insights into the dynamic nature of these layers. Hikima et al.33 applied in situ XPS to monitor band structure evolution during cycling, revealing that the direction of band bending reverses at different states of charge. More recently, researchers have recognized that SCLs do not always hinder ion transport; under specific configurations, they can even facilitate interfacial Li+ conduction. For example, Ohta et al.34 reported that the LiCl/FeOCl heterointerface (LFH composite) exhibits higher ionic conductivity than either single-phase material, highlighting that a moderate interfacial potential offset may create a fast-ion migration pathway. This historical progression underscores that the SCL is not merely a side effect of interface reactions but a fundamental electrostatic phenomenon intrinsic to solid–solid contact in ionic conductors.
In ASSBs, the SCL dictates the local electrostatic potential, defect concentration, and effective ionic/electronic conductivity near interfaces. These changes, though confined within a few nanometers, can dramatically alter the overall cell impedance and electrochemical stability.36–41 Despite substantial progress, a comprehensive and quantitative understanding of SCLs in ASSBs remains elusive. Previous reviews often focused on chemical or mechanical aspects, leaving the electrostatic and band-structure perspective fragmented.
In this review, we aim to summarize the fundamental theories and experimental evidence of SCL formation in various solid-state battery systems, compare state-of-the-art characterization techniques capable of probing the SCL at atomic to mesoscale resolution, and discuss materials and interface-engineering strategies—such as band-alignment control and dielectric-layer coatings—to mitigate or exploit the SCL. Finally, we highlight emerging opportunities in multi-physics modeling and operando probes that could unify chemical, electrical, and mechanical effects at the solid–solid interface.
2. The formation of the SCL
In liquid electrolytes, mobile ions can rapidly rearrange to maintain electroneutrality at the electrode–electrolyte interface. Potential differences are screened within angstrom-scale distances by solvated ions, forming a compact electric double layer (EDL) with negligible internal field. However, in solid electrolytes, both ionic and electronic mobilities are drastically reduced. The redistribution of charged species becomes limited, preventing complete neutralization of interfacial charge imbalance. Consequently, an extended electrostatic potential gradient develops across the solid–solid interface, forming a SCL that cannot be efficiently screened as in liquid systems. This transition—from dynamic solvation equilibrium in liquids to electrostatic equilibrium in solids—marks a fundamental distinction that governs interfacial transport behavior in ASSBs.2.1 Origin of the space charge layer
When two solids with different Fermi levels and ionic chemical potentials come into contact, ions and/or electrons migrate across the interface until the electrochemical potential (![[small mu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e0.gif) = μ + z·eφ) of each species reaches equilibrium. This redistribution disrupts local charge neutrality and produces an electric potential gradient (dφ/dx) near the interface. The resulting regions of Li+ enrichment or depletion form the SCL, which modifies local ion concentration and conductivity. The potential profile within the SCL follows Poisson's equation:
= μ + z·eφ) of each species reaches equilibrium. This redistribution disrupts local charge neutrality and produces an electric potential gradient (dφ/dx) near the interface. The resulting regions of Li+ enrichment or depletion form the SCL, which modifies local ion concentration and conductivity. The potential profile within the SCL follows Poisson's equation:where ρ(x) is the charge density, ε0 the vacuum permittivity, and εr the dielectric constant of the electrolyte. The characteristic length over which the potential decays, known as the Debye length (λD), is given by:

where kB is the Boltzmann constant, T is the absolute temperature, n0 represents the effective concentration of mobile ionic charge carriers participating in electrostatic screening, and q is the elementary charge.
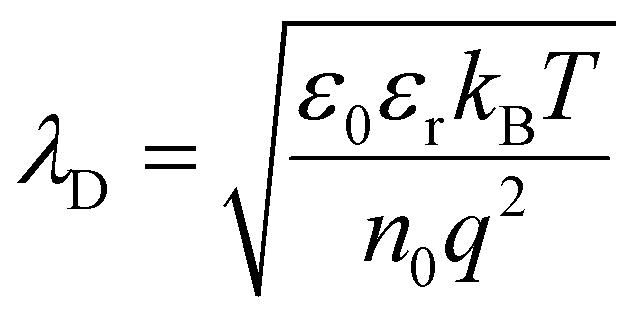
Using representative parameters reported for typical solid electrolytes at room temperature, sulfide electrolytes (e.g., LGPS- or argyrodite-type systems) are commonly characterized by relatively high dielectric constants (εr ≈ 20–40)42–45 and high concentrations of mobile Li+ carriers (n0 ≈ 1021 cm−3), reflecting their high ionic conductivity. Substituting these values into the Debye expression yields a characteristic screening length below 1 nm. In contrast, oxide electrolytes such as garnet-type or NASICON-type materials typically exhibit lower dielectric constants (εr ≈ 8–12)42–45 and lower effective mobile carrier concentrations (n0 ≈ 1019 cm−3), resulting in a Debye length on the order of several nanometers. From a purely electrostatic screening perspective, oxide electrolytes therefore intrinsically support spatially broader SCLs than sulfide electrolytes.35,46
However, the electrochemical impact of an SCL is not determined by its spatial extent alone, but by the combined effect of the screening length and the interfacial lithium chemical potential mismatch (ΔμLi) between the electrolyte and the electrode. For a given positive electrode material, sulfide electrolytes generally exhibit a larger lithium chemical potential difference than oxide electrolytes, owing to their higher-lying valence bands and reduced thermodynamic stability at high potentials. The characteristic interfacial electric field can be approximated as:

For oxide-based interfaces, ΔμLi is often limited to ∼0.1 V, distributed over a Debye length of several nanometers (∼2 nm), resulting in electric fields on the order of 107 V m−1. In contrast, sulfide-based electrolytes in contact with the same cathode material can experience a larger chemical potential mismatch (∼0.3–0.5 V), concentrated within a much shorter screening length (∼0.2 nm), giving rise to interfacial electric fields approaching or exceeding 108 V m−1. Therefore, although sulfide electrolytes possess a shorter intrinsic Debye length, the combination of a larger lithium chemical potential mismatch and a compressed screening region leads to a stronger and more abrupt interfacial electric field compared to oxide electrolytes. This explains why sulfide-based cathode interfaces often exhibit more pronounced space-charge-driven effects—such as severe lithium depletion or accumulation, enhanced electronic leakage, and accelerated interfacial degradation—even though the nominal SCL thickness is smaller.
This analysis highlights that SCL impact in ASSBs should be evaluated not solely in terms of spatial extent, but through the coupled consideration of electrostatic length scales and chemical potential driving forces.
2.2 The SCL formed at the cathode side of ASSB
The space charge effect is generally more pronounced at the cathode–electrolyte interface than at the anode side for several intrinsic reasons. First, layered oxide cathodes undergo substantial changes in lithium activity during delithiation, leading to a much stronger variation in their electrochemical potential compared with typical anode materials. Second, the potential difference between the cathode and solid electrolyte is intrinsically larger than that between the anode and electrolyte, resulting in a stronger driving force for charge redistribution and more significant band bending at the interface. Third, the cathode operates at much higher potentials, where parasitic reactions and electrolyte decomposition are more likely to occur, creating a chemically complex and dynamically evolving interfacial environment that further amplifies SCL formation.47 Unlike the subnanometer electric double layer formed in liquid electrolytes, the extended SCL in solid-state systems enables substantial variations in band structure and defect concentration, determined by the electronic character (e.g., n-type or p-type) and defect chemistry of the materials involved.Goodenough et al.48 demonstrated that the difference in the chemical potential of lithium between the positive and negative electrodes in lithium batteries is primarily dictated by the difference in the electrochemical potential of electrons (i.e., the Fermi level), which governs the open-circuit voltage of the battery. This observation underscores the importance of Fermi level research in battery science. Extending this concept to solid–solid interfaces, mismatched Fermi levels between the cathode and solid electrolyte drive electron and ion redistribution until thermodynamic equilibrium is achieved. This process generates band bending and establishes an internal electric field at the interface. Experimental studies have confirmed that such charge redistribution results in Li+ enrichment on the electrolyte side for various cathode–electrolyte combinations.35,49,50
However, this redistribution can also result in undesirable Li+-depleted regions (“Li-poor layers”), increased interfacial resistance, and accelerated performance degradation during prolonged cycling. These challenges remain major barriers to achieving high-rate and long-life all-solid-state batteries. Accordingly, interface engineering strategies—particularly the incorporation of electronically insulating buffer layers—have emerged as effective methods to moderate band bending, suppress excessive SCL formation, and stabilize the cathode–electrolyte interface.29,30,51 Moreover, with the continuous advancement of characterization techniques, we have gradually uncovered some properties of the SCL.
3. The SCL effect
Existing research on the space charge layer is typically categorized into three aspects: first, macroscopic manifestations, such as the increase in interfacial impedance, particularly at the cathode–solid electrolyte interface; second, microscopic characterization, focusing on the enrichment or depletion of lithium-ion or electron concentration at the interface; and third, predictions derived from theoretical calculations or simulation models. However, a definitive consensus on the precise impact of the space charge layer remains elusive. The prevailing view is that the space charge layer contributes to the increase in interfacial impedance. The elucidation of its underlying mechanisms is often confined to specific aspects, and a unified theory comprehensively explaining its impact is still lacking. Given the occasional equivalence of chemical potential and Fermi level under certain conditions, analyzing interface properties through the material's Fermi level offers a valuable perspective. The Fermi level's position dictates the formation and extent of the space charge layer. Ideally, consistent Fermi level alignment across the interface should prevent the generation of a built-in electric field, as depicted in Fig. 2a. With consistent Fermi levels, no chemical potential difference and consequently no band bending would occur. Conversely, Fig. 2b and d illustrate scenarios where the interface Fermi levels are misaligned. This Fermi level mismatch leads to band bending upon interfacial contact, thereby generating a built-in electric field and elevating the interfacial impedance. However, it is crucial to recognize that unlike semiconductor materials, batteries undergo lithium-ion and electron intercalation/deintercalation during charging and discharging. This process induces constant fluctuations in the cathode's Fermi level. Consequently, completely eliminating the space charge layer based solely on Fermi level matching might not be feasible.33 Nevertheless, its impact can be mitigated by, for example, identifying materials whose Fermi level shifts are less sensitive to lithium-ion intercalation or deintercalation, thereby alleviating the issues arising from Fermi level disparities. Alternatively, the use of an appropriate buffer layer can mitigate the SCL resulting from an excessive energy level difference. | ||
| Fig. 2 Schematic diagram of interface Fermi level matching. (a) A good interface state does not lead to band bending at the interface and does not produce a built-in electric field. (b–e) Mismatched Fermi levels, after the unification of Fermi levels, band bending occurs. (f) Lithium-ion aggregation phenomenon caused by different Fermi levels at the interface. Reproduced with permission.52 Copyright 2020, Elsevier. | ||
3.1 Impact of SCL on the interface
The impact of the space charge layer on the interface has been reported by numerous studies. Some researchers posit that the influence of the space charge layer on the interface can be negligible—for example, Haruta et al.'s (2015) study utilizing magnetron sputtering to fabricate all-solid-state thin-film batteries found interfacial resistance to be almost negligible.53 De Klerk and Wagemaker have presented theoretical calculations indicating that the space charge layer at the interface is very thin (less than 1 nanometer) and its impact can be disregarded.54 However, the majority of the scientific community still maintains that the space charge layer exerts a significant influence on the interface. The main effects attributed to the space charge layer on the interface are generally categorized into three types: alterations and imbalances in lithium-ion concentration; changes in interfacial activation energy; and increases in interfacial impedance. Fig. 3 summarizes the phenomena currently believed to be induced by the space charge layer. These phenomena ultimately manifest as elevated interfacial impedance, leading to a deterioration in the battery's rate and cycle performance.55–58 | ||
| Fig. 3 Impact of the space charge layer on the interface of all-solid-state batteries. | ||
 | ||
| Fig. 4 Changes and imbalances in lithium-ion concentration: (a) Electric potential distribution around the LiCoO2/electrolyte interface during the charge–discharge process. Reproduced under terms of the CC-BY license.61 Copyright 2010, John Wiley and Sons. (b) Lithium-ion distribution near grain boundary cores. Reproduced under terms of the CC-BY license.62 Copyright 2023, Zhenqi Gu et al., published by Springer Nature. (c) Trends of electric field at different voltages at the LCO and LPSCL interfaces. Reproduced under terms of the CC-BY license.50 Copyright 2020, Longlong Wang et al., published by Springer Nature. (d) Li-ion distribution acquired at Cu/LASGTP interface from SR-EELS. Reproduced with permission.49 Copyright 2019, John Wiley and Sons. | ||
 | ||
| Fig. 5 The effect of SCL at interfaces on activation energy and impedance: (a) Determination of the activation energy of Li-ion exchange and schematic of space charge layer effects on Li-ion transport. Reproduced with permission.32 Copyright 2020, Elsevier. (b,c) Arrhenius relationships of ohmic resistance (Rohm), composite cathode resistance (Rcc), and interface resistance (Rint) to the operation temperatures for (b) Ni90/LPSC/Li–In, reproduced with permission.66 Copyright 2024, American Chemical Society. (c) Ni90/LIC/LPSC/Li–In ASSBs. Reproduced with permission.66 Copyright 2024, American Chemical Society. | ||
 | ||
| Fig. 6 Relationship between interfacial impedance and SOC. Reproduced with permission.67 Copyright 2017, American Chemical Society. Reproduced with permission.20 Copyright 2019, John Wiley and Sons. | ||
Haruyama et al. employed EIS to investigate the impact of the space charge layer at the interface between oxide cathodes and sulfide electrolytes. Their findings indicated that the presence of the space charge layer significantly increased interfacial resistance, thereby limiting the lithium-ion transport efficiency.31,68 Brogioli et al. studied the interface between lithium–lanthanum–zirconate (LLZO) ceramic particles and PEO-based polymer electrolytes. They utilized theoretical calculations and experimental validation to demonstrate that the space charge layer at the LLZO/PEO interface made a substantial contribution to the interfacial resistance. By developing an electric double-layer model, they analyzed the charge distribution and potential difference at the interface and derived the relationship between interfacial capacitance and potential difference. The experimental results revealed that the interfacial resistance varied with the concentration of lithium salts and reached a minimum value at high concentrations.63
 | ||
| Fig. 7 The effect of ferroelectric materials on the interfacial SCL: (a) Schematic illustration of interface charge redistribution between oxide cathodes and sulfide SEs after different interface engineering approaches. Reproduced under terms of the CC-BY license.50 Copyright 2020, Longlong Wang et al., published by Springer Nature. (b) Schematic views of Li+ concentration profile in LiPON at open circuit conditions around a pristine LiPON/LNM interface. Reproduced with permission.69 Copyright 2014, John Wiley and Sons. (c) FEM simulations of the effects of BTO nanocrystals. Reproduced under terms of the CC-BY license.50 Copyright 2020, Longlong Wang et al., published by Springer Nature. | ||
3.2 SCL-dominated degradation mechanism
The formation of the space charge layer introduces complexities to interface issues. How, then, does the space charge layer accelerate interface degradation during the charge–discharge process of the battery? This section aims to explain the evolution of the space charge layer during the charge–discharge process from the perspective of band theory. Initially, upon interface formation, if the chemical potentials on both sides of the interface are identical, no space charge layer will form. This implies that the respective energy bands of the two materials at the interface will not bend, and no built-in electric field will be generated. However, when the chemical potentials of the two materials at the interface differ, the space charge layer will indeed form. Following the formation of the space charge layer, the interfacial activation energy increases, and the formation energy of lithium vacancies also increases, making it more energetically unfavorable for lithium ions to detach from one side of the interface and migrate to the other. This phenomenon is ultimately reflected in electrochemical measurements as increased interfacial impedance, leading to the degradation of battery performance.As the battery commences charging and discharging, lithium ions are continually intercalated and deintercalated within the cathode and anode, leading to a continuous shift in the Fermi level of the cathode.70 The Fermi level, representing the energy level with a 50% probability of electron occupation, also reflects the electron density of the material. During charging, as lithium ions are progressively extracted from the cathode, an equivalent number of electrons also leave, causing the Fermi level of the cathode to decrease correspondingly. This process induces a Fermi level mismatch across the interface. This mismatch results in the bending of the conduction band minimum and valence band maximum of both the cathode and the electrolyte and, consequently, to a dynamically evolving SCL (Fig. 8).
 | ||
| Fig. 8 SCL-dominated degradation mechanism. | ||
During electrochemical cycling, the relative dominance of chemical and mechanical degradation at solid–solid interfaces is strongly dependent on the state of charge (SOC), rather than being governed by a single universal failure mechanism. As lithium ions are continuously intercalated and deintercalated, the Fermi level of the cathode dynamically shifts, reflecting changes in both lithium content and electronic structure.70 This evolution leads to a SOC-dependent electrochemical potential mismatch across the cathode–electrolyte interface and, consequently, to a dynamically evolving SCL. At relatively low to moderate voltages, particularly for layered Ni-rich cathodes, the overall volumetric change of the cathode is comparatively limited, and interfacial mechanical contact can generally be maintained under typical stack pressures. In this regime, the electrochemical potential mismatch between the cathode and solid electrolyte remains significant, giving rise to pronounced band bending and a steep interfacial electric potential gradient. Under such conditions, the SCL plays a dominant role in interfacial degradation by locally redistributing lithium ions and charge carriers. The resulting strong internal electric fields can accelerate parasitic interfacial reactions, promote electrolyte decomposition, and induce the formation of lithium-rich or lithium-depleted regions. Importantly, the interphase products generated through these SCL-driven processes further modify the local chemical potential landscape and electric field distribution, thereby increasing interfacial heterogeneity and reinforcing chemical degradation pathways. As the SOC increases toward higher voltages, however, the degradation priority may shift. For Ni-rich cathodes, anisotropic lattice expansion and contraction become more pronounced at high degrees of delithiation. If the externally applied stack pressure is insufficient or spatially non-uniform, even a relatively narrow SOC window can trigger mechanical degradation, such as interfacial cracking, contact loss, or partial delamination. In this high-voltage regime, mechanical failure can locally disrupt ionic transport pathways and dominate interfacial degradation. Moreover, under certain SOC conditions, the electrochemical potentials across the interface may approach equilibrium, reducing the driving force for further SCL-induced charge redistribution. In such cases, SCL-related electrostatic effects no longer serve as the primary degradation origin, although they may still modulate local reaction kinetics at damaged regions. Hikima et al.'s in situ study on band structure changes revealed interface alterations in all-solid-state batteries.33 As depicted in Fig. 9b, significant band bending occurs at the cathode–electrolyte interface towards the end of discharge. More critically, the built-in electric field generated by the bending of the cathode's energy bands aligns with the external electric field applied during charging, potentially exacerbating band bending. The generated built-in electric field promotes the redistribution of lithium ions, leading to the formation of lithium-rich or lithium-poor regions. Zhang et al.20 studied the periodic evolution process of interfaces in all-solid-state batteries and established a model of the relationship between lithium-ion concentration at the interface and battery status. The model results show that during charging, lithium ions accumulate on the positive electrode side. During discharge, the distribution of lithium ions shows a trend opposite to that during charging. The blockage of lithium-ion transport channels can result in localized charge density inhomogeneities, rendering the interface more susceptible to physical or chemical transformations and consequently altering the interface composition. This, in turn, leads to further changes in the Fermi level. This cycle repeats continuously throughout the charge–discharge process, macroscopically manifesting as a gradual decline in battery performance, increased impedance, and interface degradation. Moreover, the volume changes in the Ni-rich cathode during cycling can cause the formation of mechanical contact failure at the cathode–electrolyte interface, potentially interrupting lithium-ion transport pathways and leading to an increase in local interfacial lithium-ion concentration. At high voltages, valence band electrons in the electrolyte may gain sufficient energy to transition to the conduction band, generating electron–hole pairs. Electrons excited to the conduction band, possessing higher energy, tend to migrate from the conduction band of the electrolyte to the conduction band of the cathode due to the cathode's higher conduction band energy position, as electrons move towards regions of higher potential. This electron migration causes the electrolyte to lose electrons and undergo oxidation reactions, further degrading the interface. Furthermore, the side reaction products formed due to the space charge layer during a charge–discharge cycle are often not fully reversible. Over multiple cycles, this leads to irreversible degradation of the interface and a corresponding decline in battery performance.71
 | ||
| Fig. 9 Band structure changes of all-solid-state batteries during cycling. (a) Band structure changes of all-solid-state batteries during cycling. Reproduced under terms of the CC-BY license.33 Copyright 2022, Kazuhiro Hikima et al., published by Springer Nature. (b) Band bending of the cathode side with additional assumptions represented by the relative energy level positions. Reproduced under terms of the CC-BY license.33 Copyright 2022, Kazuhiro Hikima et al., published by Springer Nature. (c and d) Schematic of the energy bands at the interface of LMO and LASGTP at both 5 V and 2 V. | ||
Overall, the space charge layer (SCL) at solid–solid interfaces in all-solid-state batteries should be understood as a dynamic and SOC-dependent interfacial phenomenon that continuously evolves during charge–discharge cycling. As lithium ions are repeatedly intercalated and deintercalated, the interfacial electrochemical potential mismatch and band alignment are periodically reconstructed, leading to the recurrent formation, relaxation, and redistribution of the SCL. This cyclic evolution gives rise to persistent lithium-ion accumulation or depletion at the interface, locally modifying ion transport pathways and electrostatic environments. Over extended cycling, such repetitive SCL reconstruction acts as a feedback loop that progressively amplifies interfacial heterogeneity, resulting in increased impedance and gradual performance degradation. Importantly, the relative contributions of chemical and mechanical degradation associated with SCL evolution are strongly dependent on the state of charge. At low to moderate SOC ranges, where volumetric changes of electrode materials are comparatively limited and interfacial contact is largely preserved, SCL-induced electrostatic effects tend to dominate. In this regime, steep interfacial potential gradients can hinder lithium-ion transport and promote parasitic chemical reactions, such as electrolyte decomposition or interphase growth, which further alter the local chemical potential and reinforce SCL evolution. By contrast, at high SOCs, anisotropic lattice strain and accumulated stress may trigger mechanical degradation, including contact loss or interfacial delamination, particularly under insufficient or non-uniform stack pressure. In such cases, mechanical failure can become the primary degradation origin, while the SCL plays an amplifying role by intensifying local electric fields and accelerating chemical reactions at damaged regions. Under certain SOC conditions, partial equilibration of interfacial electrochemical potentials may also reduce the direct driving force for further SCL growth, shifting the degradation priority away from electrostatic effects.
These considerations demonstrate that chemical and mechanical degradation processes at solid–solid interfaces are not independent phenomena but are inherently coupled through SOC-dependent interfacial electrostatics and structural evolution. While many existing studies focus on either chemical or mechanical degradation in isolation, such an approach is insufficient to fully capture the dynamic nature of SCL-governed interfaces. A comprehensive understanding of interfacial degradation in all-solid-state batteries therefore requires integrated electro–chemo–mechanical frameworks that explicitly incorporate SOC-dependent band alignment, space charge evolution, and stress–strain coupling. Developing such coupled models, together with advanced operando characterization techniques, will be essential for decoupling these intertwined mechanisms and for guiding rational interface design strategies.
Notably, emerging evidence suggests that under specific material combinations and interfacial architectures, the SCL can actively promote or facilitate lithium-ion transport. Wang et al.72 reported the formation of a fast ion-conducting interfacial region at the LYC–LZC interface, where the SCL was proposed to facilitate enhanced lithium-ion mobility. Similar enhancements in ionic conductivity have been observed in mixed-phase electrolyte systems reported by Ohta et al.,34 suggesting that appropriately engineered SCLs can function as ion-transport-promoting interfacial channels. These findings collectively indicate that the role played by the SCL in all-solid-state batteries is not intrinsically detrimental or beneficial but is governed by a complex interplay of interfacial electrostatics, materials chemistry, mechanical integrity, and operating conditions. A comprehensive understanding of SCL effects therefore requires advanced characterization techniques and physically informed modeling approaches capable of decoupling these intertwined factors across multiple time and length scales. More detailed discussions of characterization strategies and control methodologies are provided in the following sections.73–75
4. Characterization methods
A significant factor hindering the progress in studying the space charge layer is the lack of robust characterization methods. Given the internal location of the all-solid-state battery interface, conventional characterization and testing techniques often fall short of providing in-depth information regarding its microscopic morphology or distribution. Furthermore, the prevalent use of mixed composite cathodes in current designs further complicates the characterization of the space charge layer in these batteries.19,58,62,73,76–79 The primary challenges in characterizing the space charge layer can be summarized as follows:First, the microscopic-scale characteristics of the space charge layer pose a considerable difficulty. Typically existing at the atomic or nanoscale, its thickness can be limited to just a few nanometers or even less. But the majority of currently available characterization techniques are capable of providing only macroscopic or localized information, making it challenging to fully and quantitatively elucidate the microscopic structure and ion transport characteristics of the space charge layer. For instance, the widely used EIS can indirectly indicate changes in interfacial impedance but cannot directly visualize the microscopic structure of the space charge layer. X-ray photoelectron spectroscopy (XPS), while effective at analyzing the valence states and chemical composition of elements at the interface, lacks the ability to quantitatively describe the distribution of lithium ions. Existing characterization techniques face difficulties in directly observing and measuring the distribution and concentration changes of lithium ions at such minute scales. Conventional transmission electron microscopy (TEM), despite its high resolution, still encounters limitations in directly imaging the space charge layer and necessitates specialized sample preparation.
Second, the inherent complexity of the interface presents another major hurdle. The space charge layer forms at the solid–solid interface between the electrode and the electrolyte, where a multitude of intertwined physical and chemical phenomena may occur, including interface reactions, element diffusion, and interface side reactions. These interconnected processes make it difficult to isolate the specific impact of the space charge layer for individual analysis. Interface reactions, for example, can lead to the formation of new phases, further altering the electrochemical properties of the interface and potentially obscuring the intrinsic characteristics of the space charge layer. Moreover, the vast array of electrode and electrolyte materials utilized in all-solid-state batteries results in significant variations in the interface characteristics formed by different material combinations. The formation mechanism and influencing factors of the space charge layer are material-dependent, thus complicating the establishment of a universally applicable characterization model.
Third, the dynamic change characteristics of the space charge layer add another layer of complexity. The formation and evolution of the space charge layer are dynamic processes, and its properties fluctuate with the battery's charging and discharging cycles, as well as variations in temperature, pressure, and other operational conditions. The space charge layer within the battery may undergo continuous changes during operation, rendering it challenging to capture its complete behavior using static characterization techniques. Advanced in situ characterization methods are likely required to better understand its dynamic evolution.
Fourth, interface contact and stability issues further complicate the characterization efforts. The contact between the solid electrolyte and the electrode is often not perfectly intimate, potentially resulting in pores, cracks, and other defects at the interface. These imperfections can affect the formation and distribution of the space charge layer and also increase the difficulty of accurate characterization. Mechanical instability at the interface can lead to localized alterations in the space charge layer, potentially compromising the accuracy of the characterization results.
The challenge in characterizing the space charge layer in all-solid-state batteries arises from the synergistic effect of various factors, including its microscopic scale, interface complexity, dynamic nature, and interface contact and stability issues. These factors are intricately intertwined, contributing to the slow progress in the study of the space charge layer. However, the continuous advancement and application of sophisticated characterization techniques offer promising avenues to overcome this bottleneck.80 In particular, in situ characterization techniques hold significant potential as they can monitor the dynamic changes of the space charge layer in real time during battery operation, providing crucial insights into its formation mechanism and influencing factors. Takada's early work involved increasing the potential difference between the oxide cathode and the sulfide solid electrolyte to investigate the impact of the space charge layer effect on battery performance, demonstrating that the effect is primarily attributable to interface mismatch.31 However, finer characterization of the space charge layer remained limited by the available techniques at the time. Thanks to technological progress, researchers have since reported significant advancements in high-precision characterization instruments. For instance, Gu et al. successfully captured the charge distribution at the interface using high-resolution aberration-corrected electron microscopy, revealing a lithium-ion aggregation zone rather than the lithium-ion depletion zone previously reported in the literature.62 This finding underscores the indispensable role played by advanced characterization techniques in understanding interface evolution. Current research methodologies for studying the space charge layer predominantly involve simulation calculations and experimental characterization.
4.1 Simulation and calculation
First-principles calculations and density functional theory (DFT) are increasingly employed to provide atomic-level insights into defect formation energies, electronic band alignment, and potential distribution at the electrode–solid electrolyte interface (Fig. 10).52,79,81–85 These methods can predict the interfacial potential drop driven by electron transfer and the space charge layer of Li+, as well as how these potential drops vary with the state of charge (SOC) of the battery. More sophisticated interface models have been developed beyond simple equilibrium conditions, considering factors such as applied voltage, temperature, and specific properties of the electrode and electrolyte materials (e.g., dielectric constant) that influence the formation of the space charge layer and its impact on lithium-ion transport. These models reveal that the size of the space charge layer in solid electrolytes may be larger than that in liquid electrolytes and is strongly dependent on the dielectric properties of the materials. It has been recognized that the properties of the space charge layer (thickness, ion concentration, potential drop) are dynamic and depend on the SOC of the battery and the crystal orientation of the materials, which affect the defect formation energies and band alignment at the interface.86 | ||
| Fig. 10 Application of DFT and model simulations in the space charge layer. (a) Different sites on the contact surfaces of LCO, LPS, and LNO. Reproduced with permission.31 Copyright 2014, American Chemical Society. (b) The vacuum-aligned position of the electronic bands in lithium metal, LiPON, LiCoO2, and Li0.5CoO2 before contact. Occupied bands (VBM) are blue and unoccupied bands (CBM) are orange. Vacuum (dashed line) alignments and band gaps are indicated. Reproduced with permission.87 Copyright 2019, John Wiley and Sons. (c) Schematic representation of three domains in P2D model. Reproduced with permission.88 Copyright 2022, Elsevier. (d) Schematic of a metal/electrolyte/metal cell with the space charge layers at the interface and the proposed equivalent circuit model. Reproduced with permission.79 Copyright 2021, American Chemical Society. (e) Schematic of the electrostatic potential and valence bands at the interfaces between LiPON and LixCoO2 for x = 1 and x = 0.5. Reproduced with permission.87 Copyright 2019, John Wiley and Sons. | ||
For instance, in 2017, Fingerle et al. utilized DFT modeling to evaluate the defect concentration and Fermi level position at the interface between LiCoO2 and LiPON to elucidate the formation mechanism and evolution of the space charge layer.47 The modeling results showed consistency with the actual interface, indicating that the formation of the space charge layer arose from the equilibrium of the electrochemical potential of Li ions at the interface. This phenomenon persisted even after annealing, suggesting that the electrochemical potential gradient at the interface is intrinsic. Swift and Qi proposed a first-principles-based model to predict the potential distribution in all-solid-state lithium batteries (ASSLBs) and applied it to the Li/LiPON/LixCoO2 system.87,89 The model incorporated the interfacial potential drop resulting from lithium-ion transport and electron transfer and integrated defect formation energy calculations to predict the formation of the SCL, thereby forecasting the migration direction of lithium ions within the space charge layer. Their study demonstrated that the formation and interfacial potential energy of the space charge layer are dependent on the SOC of the cathode material, specifically the concentration of lithium ions in the cathode. De Klerk et al. employed a simplified model to evaluate the interfacial capacitance and resistance of the SCL in typical electrode–electrolyte combinations, applying it to the contact of LiCoO2 (LCO) and graphite electrodes with LLZO and LATP solid electrolytes.35,54 Their findings suggested that the thickness of the SCL is typically at the nanoscale, and the resistance to Li+ transport is negligible unless a completely depleted Li+ layer is formed in the solid electrolyte. This implies that the impact of the SCL on the performance of all-solid-state batteries is minor, and the primary causes of large interfacial resistance are poor contact between the electrode and the electrolyte, decomposition products, etc. The model's validity was supported by comparisons with experimental results, and the authors acknowledged some assumptions and uncertainties within the model, such as the simplified treatment of Coulomb interactions. Liu et al. proposed a novel equivalent circuit model that considers the interface to encompass not only capacitance but also space charge layer resistance and polarization resistance.79 The elements of the circuit model were quantified based on the improved Planck–Nernst–Poisson (MPNP) model. This new model offers a better explanation for the real impedance tail observed in the low-frequency region. The validity of the model was corroborated by comparisons with experimental results. Their study revealed that polarization resistance plays a significant role in the low-frequency region, particularly in solid electrolytes exhibiting non-ideal electron blocking. The model's accuracy was verified by comparison with the experimental impedance spectrum of LiPON, and the influence of temperature on ionic conductivity was discussed, further enhancing the understanding of solid electrolyte impedance. Moreover, Al-Ali et al. introduced a deep learning-based algorithm utilizing convolutional neural networks (CNN) and long short-term memory networks (LSTM) to identify the circuit model that best fits the measured impedance spectra.90 A two-stage optimization technique was also proposed to determine the optimal circuit model parameters. The efficacy of the proposed algorithm was validated using experimental impedance data from NiMH batteries and cherry tomato bioimpedance. The results indicated that the algorithm could successfully model and fit the given impedance data with low error, demonstrating its significant value for accurately identifying interfacial impedance. Regarding the dynamic evolution of the space charge layer, Katzenmeier et al. employed kinetic Monte Carlo (kMC) simulations to investigate the SCL phenomenon in solid electrolytes, aiming to predict the spatial extent of the SCL with a minimal number of input parameters and explore its temporal evolution.64,65,81 They constructed a kMC model to simulate the hopping mechanism of Li+ ions within the solid electrolyte, studied the formation process of the SCL, and validated the simulation results with experimental data. The kMC simulation results exhibited good agreement with the experimental findings, capturing the physical behavior of SCL formation and revealing the impact of material properties, such as Li+ ion concentration and dielectric constant, on the thickness and asymmetry of the SCL. The accuracy of the kMC model in predicting SCL thickness and capacitance was confirmed by comparison with experimental data.
The research conducted by these scholars underscores the considerable significance of model simulation and first-principles calculations in elucidating the formation and evolution mechanisms of the space charge layer. However, the accuracy of these models and calculation results necessitates experimental verification, highlighting the indispensable role played by experimental studies.88,90–98
4.2 Experimental characterization
In experimental validation, the synergistic application of advanced characterization techniques provides multidimensional data to support theoretical models (Fig. 11).100,104–112 X-ray photoelectron spectroscopy (XPS) can be employed for the quantitative analysis of the evolution of elemental chemical states at the interface. By comparing the shifts in the binding energies of Li 1s and transition metal 2p orbitals at varying depths, the direction of electron transfer predicted by DFT and the lithium-ion concentration gradient within the SCL can be verified. Transmission electron microscopy (TEM), often coupled with electron energy loss spectroscopy (EELS), enables nanoscale resolution of interface structure analysis. Notably, in situ observation of beam-sensitive regions via electron beam irradiation allows for the direct measurement of the actual thickness of the lithium-depleted layer, which can be statistically correlated with the extent of lattice distortion calculated by DFT. Furthermore, to overcome the spatial averaging limitations of conventional 2D TEM projections, 3D electron tomography has emerged as a cutting-edge approach. By reconstructing a series of 2D images acquired at different tilt angles, 3D electron tomography, often combined with EDS or EELS, provides a holistic view of the spatial heterogeneity of the SCL and its coupling with the cathode–electrolyte interphase (CEI). This 3D perspective is crucial for identifying non-uniform Li-ion percolation pathways in bulk-type composite electrodes where the SCL effects are influenced by complex local geometries. Synchrotron X-ray absorption spectroscopy (XAS) facilitates the dynamic tracking of changes in the oxidation states of transition metals at the interface during battery cycling, revealing the impact mechanism of the space charge layer effect on the structural stability of the cathode material. For example, Fingerle et al. integrated surface science methods and simulation calculations to investigate the reaction and space charge layer formation at the interface between LiCoO2 and lithium phosphorus oxynitride (LiPON).47 Utilizing XPS, synchrotron X-ray photoelectron spectroscopy (SXPS), and ultraviolet photoelectron spectroscopy (UPS), they meticulously detailed the chemical reactions and charge transfer processes occurring at the interface. XPS and SXPS can accurately monitor alterations in the valence states and chemical environment of elements at the interface, while UPS serves to measure the work function and valence band structure, thereby constructing a comprehensive band structure diagram. Yamamoto et al. employed quantitative electron holography (EH) to directly observe the potential distribution changes induced by lithium-ion diffusion in all-solid-state LIBs, revealing the lithium-ion potential distribution at the cathode/solid electrolyte interface during charge–discharge reactions.49,61 Complementing electron-based techniques, the combination of in situ Atomic Force Microscopy (AFM) and Kelvin Probe Force Microscopy (KPFM) offers a powerful platform for mapping the internal potential profiles of cross-sectioned solid-state cells. KPFM measures the contact potential difference (CPD) between a conductive tip and the sample surface with high spatial and voltage resolution, allowing for the direct visualization of the electric potential drop across the SCL. For instance, Masuda et al.100 utilized in situ KPFM to map the potential distribution within a charged all-solid-state LIB, successfully identifying the local overpotential at the cathode/electrolyte interface and quantifying the width of the resistive space-charge region under operative conditions. Electrochemical impedance spectroscopy serves as a critical verification tool. By establishing an equivalent circuit model that incorporates the impedance of the space charge layer (e.g., introducing a parallel combination of a Warburg diffusion element and interfacial capacitance), the contribution of the SCL to the lithium-ion migration barrier can be quantitatively separated. Yu et al. utilized EIS and solid-state nuclear magnetic resonance (NMR) techniques to study lithium-ion transport characteristics in Li7P3S11 sulfide-based solid electrolytes.60 EIS was used to measure the ionic conductivity of the electrolyte and to determine the activation energy for lithium-ion transport. Gu et al. combined high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), EELS, and ab initio molecular dynamics (AIMD) simulations to investigate the atomic configuration and ion transport behavior of the SCL in LLTO.62 Their findings indicated that the SCL in LLTO is actually a lithium-ion-rich region. The lithium ions within the SCL primarily occupy the 3c interstitial sites, and their ion transport efficiency is comparable to that of the bulk phase, suggesting that the SCL is not the primary cause of the large grain boundary resistance. On the contrary, the grain boundary core with lithium-ion depletion is the main bottleneck for the high grain boundary resistance in LLTO. Wang et al. utilized in situ differential phase contrast scanning transmission electron microscopy (DPC-STEM) to directly observe the net charge density distribution at the LiCoO2/Li6PS5Cl interface in sulfide-based all-solid-state batteries.50 DPC-STEM technology can monitor the accumulation of lithium ions at the interface in real time, thereby revealing the hindrance effect of the SCL on lithium-ion transport at the interface. Recent advances in 4D-STEM have further enhanced this capability, enabling the simultaneous mapping of local crystallinity, strain, and electric fields, which provides a more nuanced understanding of how structural degradation at the NMC/LGPS interface correlates with SCL formation.20,101 Isaac et al. employed EIS to investigate ion charge transport and transfer in multilayer model systems based on polyethylene oxide (PEO) polymer electrolytes and NaSICON-type ceramic electrolytes.99 Their study found that the total polarization resistance (Rp) of the multilayer system can be decomposed into the sum of the bulk electrolyte migration resistance (Rel), the interfacial charge transfer resistance (Rct), and the diffusion resistance (Rdif), i.e., Rp = Rel + Rct + Rdif. This indicates that EIS can quantitatively analyze changes in interfacial impedance by decomposing the raw data. Utilizing Li NMR technology, researchers can quantitatively analyze the diffusion coefficient and transport barrier of lithium ions in the solid electrolyte, as well as the lithium-ion transport characteristics at the electrode–electrolyte interface.32,101,113 Compared with conventional spectroscopic and electrochemical methods, these emerging techniques provide unprecedented spatial dimensionality and operando sensitivity, enabling direct correlation between electrostatic potential evolution, local lithium redistribution, and interfacial structural degradation. | ||
| Fig. 11 Currently commonly used methods for characterizing the space charge layer and their advantages on both spatial and temporal scales.20,32,49,50,54,60,61,71,74,76,77,79,80,82,88,98–104 (a) In situ charge-density-distribution characterization of the LCO/LPSCl interface. Reproduced under terms of the CC-BY license.50 Copyright 2020, Longlong Wang et al., published by Springer Nature. (b) Evolution of the four resistances obtained by fitting the impedance spectra. Reproduced with permission.67 Copyright 2017, American Chemical Society. (c) Electric potential distribution near Cu/LASGTP interface obtained from phase-shifting electron holography. Reproduced with permission.49 Copyright 2019, John Wiley and Sons. (d) Exchange phenomenon within LiV2O5 and Li2V2O5 bulk, 6Li 2D exchange NMR spectrum of LiV2O5-LAGP and Li2V2O5-LAGP at 363 K with 0.3 s mixing time. Reproduced with permission.32 Copyright 2019, Elsevier. | ||
Looking ahead, the development and application of more sophisticated in situ characterization techniques will hopefully lead to a more comprehensive understanding of the microscopic structure and mechanism of action of the space charge layer. This, in turn, will provide a robust theoretical foundation for the interface optimization and performance enhancement of all-solid-state batteries. It is precisely through the reciprocal interplay between theoretical modeling and experimental validation that the true impact of the space charge layer on the interface of all-solid-state batteries can be elucidated.
Table 1 shows the statistical data on methods for characterizing space charge layers.
| Ref. | Characterization techniques | Key advantages | Limitations | Application scenarios |
|---|---|---|---|---|
| Al-Ali et al.90 | EIS | Sensitive to interfacial resistance and capacitance changes; non-destructive; suitable for operando analysis. | Indirect probe; model-dependent interpretation; limited spatial resolution. | Identifying SCL-induced interfacial impedance growth; tracking dynamic interfacial evolution during cycling. |
| Isaac et al.99 | ||||
| Liu et al.79 | ||||
| Wang et al.50 | In situ DPC-STEM | Directly visualizes net-charge-density and Li-ion accumulation at atomic scale. | Highly sensitive to electron beam; requires precise FIB sample preparation. | Visualizing charge distribution at high-voltage LCO/Li6PS5Cl interfaces. |
| Cheng et al.32 | 2D exchange NMR | Non-invasive; quantifies spontaneous Li-ion exchange rates and activation energy barriers. | Low spatial resolution; requires specific isotopes (6Li/7Li) for signal clarity. | Quantifying Li-ion transport barriers across LixV2O5/LAGP or S-based interfaces. |
| Liu et al.102 | ||||
| Yu et al.60 | ||||
| Liu et al.103 | ||||
| Yu et al.54 | ||||
| Nomura et al.49 | Electron Holography (EH) | Direct observation of 2D electric potential maps and dynamic changes during (dis)charge. | Low signal-to-noise ratio; complex phase reconstruction; limited to thin-film models. | Mapping potential drops and SCL width in model all-solid-state thin-film batteries. |
| Yamamoto et al.61 | ||||
| Masuda et al.100 | In situ KPFM | High surface potential sensitivity; maps cross-sectional potential across the entire cell. | Surface-sensitive only; potential contamination from surface oxidation or humidity. | Analyzing potential distribution and local resistance in bulk-type solid-state cells. |
| Hikima et al.33 | Operando HAXPES/XPS | Probes electronic band structure and “band bending” to infer potential alignment. | Limited probing depth (nm scale); indirect evidence of SCL via energy level shifts. | Distinguishing between chemical decomposition layers and electrostatic SCLs. |
| Fingerle et al.47 | ||||
| Hakari et al.106 | ||||
| Sugiyama et al.98 | Low-energy muons (μSR) | Probes local magnetic fields and Li-ion dynamics at depth-tunable nanometer scales. | Requires synchrotron/accelerator facilities; extremely complex data interpretation. | Searching for Li-ion depletion layers in thin-film battery materials. |
| Feng et al.104 | EIS with DRT/TLM | Non-destructive; decouples overlapping processes (SCL, SEI, bulk) via relaxation times. | Model-dependent interpretation; lacks direct structural/spatial confirmation. | Monitoring the evolution of interfacial impedance during long-term cycling/aging. |
| Yu et al.111 | ||||
| Zhang et al.76 | ||||
| Isaac et al.99 | ||||
| Paranamana et al.101 | 4D-STEM/EDS | Provides structural and compositional maps of the interphase at the nanoscale. | Primarily captures chemical interphases (CEI) rather than pure electrostatic SCL. | Understanding the coupling between chemical reactions and charge distribution. |
| Zhang et al.20 | ||||
| Wang et al.50 | Multiphysics modeling (MPNP/DFT) | Predicts potential profiles, defect concentrations, and SCL thickness theoretically. | Simplified assumptions (e.g., ideal interfaces); requires accurate input parameters. | Theoretical validation and predictive design of buffer layers/coatings. |
| Swift & Qi87 | ||||
| Liu et al.103 | ||||
| Brogioli et al.63 |
5. Space charge layer control strategies
Following an in-depth analysis of the formation mechanism of the SCL, characterization methods, and its multifaceted impact on the performance of all-solid-state batteries, it is clear that the space charge layer represents a key bottleneck limiting the achievement of high-performance and long-cycle-life applications for all-solid-state Ni-rich batteries.114–117 Considering the substantial influence of the space charge layer on interfacial impedance, lithium-ion transport kinetics, chemical and mechanical stability, and overall battery performance, the exploration of effective control strategies to mitigate or overcome its detrimental effects is urgently warranted.77,118–123Fig. 12 summarizes the prevailing approaches, including surface coating, bulk/gradient doping, and dielectric engineering. Although traditionally treated as distinct categories, these strategies can be mechanistically reorganized into two unified frameworks: Hierarchical Band Alignment Engineering and Interfacial Field Modulation. This classification better reflects the underlying physics governing SCL formation and supports the rational design of next-generation interface engineering strategies.
 | ||
| Fig. 12 Currently used methods to improve the space charge layer effect: (a) Coating. Reproduced with permission.24 Copyright 2021, American Chemical Society. Reproduced with permission.123 Copyright 2021, John Wiley and Sons, (b) Element doping. Reproduced with permission.124 Copyright 2020, American Chemical Society. Reproduced with permission.125 Copyright 2021, American Chemical Society, (c) Coating dielectric materials. Reproduced under terms of the CC-BY license.50 Copyright 2020, Longlong Wang et al., published by Springer Nature. Reproduced with permission.69 Copyright 2014, John Wiley and Sons. | ||
5.1 Hierarchical band alignment engineering
Hierarchical Band Alignment Engineering refers to a multi-level electronic-structure design strategy in which both the interfacial and bulk energy levels are tuned to create a continuous, graded alignment across the cathode–electrolyte interface. This framework consolidates surface coating and bulk/gradient doping into a single concept, as both ultimately regulate the electrochemical potential landscape that governs SCL formation.Performance improvements associated with coatings have been widely demonstrated in Ni-rich batteries.125–135 For example, Takada and Zhang et al. reported that LiNbO3-coated NCM811 exhibits significantly lower interfacial resistance after cycling, confirming that appropriately selected coating layers can effectively weaken SCL-induced impedance growth.136 Similarly, Liu et al. identified that LiI coatings improve lithium-ion transport across the interface by reducing SCL-related resistance and stabilizing interfacial reactions.103 Despite the diversity of coating chemistries, their mechanistic influence converges on interfacial band modulation. Operando HAXPES studies by Hikima et al. directly revealed dynamic band bending at the Li2MnO3/LASGTP interface during charge–discharge processes.33 Upon charging to 5.0 V, the interfacial electronic structure evolved from a staggered (Type-II) configuration to a flat-band (Type-I) alignment. Such transitions underscore that coatings—particularly buffer layers—effectively adjust the interfacial Fermi level alignment and mitigate potential discontinuities that drive space charge accumulation. During discharge, partial reduction at the interface further highlights the sensitivity of band structures to interfacial environments. These insights reveal that the true function of coating layers lies in the tailored engineering of interfacial band alignment, rather than merely preventing side reactions. Coatings regulate interfacial electrostatics, lower the interfacial potential drop, and reduce the driving force for Li+ depletion or accumulation within the SCL. Consequently, coatings can be analyzed and compared through a unified descriptor—their capability to flatten or smooth the interfacial band landscape. Furthermore, this framework suggests a pathway for next-generation design: by rationally stacking coating layers with complementary band alignment characteristics, one may construct multi-step energy level transitions that impede electron leakage while enabling rapid Li+ transport. Such engineered, asymmetric band structures may surpass single-layer coatings and offer a more robust route for suppressing SCL effects.124,137–141
Gradient doping further enhances these effects, as the spatial variation in dopant concentration yields a graded internal potential landscape. This gradual band alignment enables a smoother Li+ diffusion transition from the bulk to the interface, effectively diminishing the formation intensity of the SCL. Lim et al.21 demonstrated this principle by incorporating Ta and W through precursor surface modification on LiNi0.6Co0.2Mn0.2O2, creating a doped surface layer that stabilizes the cathode–sulfide electrolyte interface. Beyond single-ion doping, multi-element doping has emerged as a powerful strategy for simultaneously enhancing structural integrity and interfacial energetics. Lee et al.125 introduced Al3+ and Nb5+ dual doping into Li[Ni0.92Co0.04Mn0.04]O2 (NCM92), achieving improved cycling stability by leveraging complementary mechanisms: Al3+ strengthened the host lattice, while Nb5+ refined primary particle morphology and modulated defect formation energies. This cooperative tuning of both structural stability and band energetics demonstrates the advantage of multicomponent doping in high-Ni cathodes.
Overall, element doping enriches the Hierarchical Band Alignment Engineering framework by offering bulk-level control over band structure, defect chemistry, and Li+ chemical potential, all of which converge to weaken the space charge layer and enhance the electrochemical stability of all-solid-state Ni-rich cathodes. Notably, doping and coating can be synergistically combined to create multiscale band modulation. Liu et al. achieved this by pairing La2Li0.5Al0.5O4 coating with Al3+ doping on single-crystalline LiNi0.8Co0.1Mn0.1O2.142 The dual modification reduced oxygen vacancy formation, suppressed internal cracking, and enhanced Li+ transport kinetics. These findings highlight that doping and coating are not competing strategies; instead, they influence SCL formation from distinct but complementary dimensions. Coatings regulate interfacial band alignment directly, while doping tunes intrinsic bulk band structures and defect populations that feed into the interfacial electrochemical potential landscape.31,33,77,143
5.2 Interfacial field modulation
While Hierarchical Band Alignment Engineering focuses on smoothing electronic energy-level discontinuities, a complementary and equally important strategy is to directly regulate the built-in electric field that drives SCL formation at solid–solid interfaces. The intensity of this internal electric field originates from the electrochemical potential mismatch between the cathode and solid electrolyte; thus, mitigating band bending through electrostatic compensation offers a fundamentally different pathway to suppress SCL formation. Dielectric engineering introduces high-permittivity or ferroelectric materials at the interface to generate polarization fields that counteract the intrinsic electric field. Wang et al.50 demonstrated this concept by integrating discontinuous nano-ferroelectric domains at the cathode–electrolyte interface, showing that their switchable dipoles can partially neutralize the interfacial electrostatic field and significantly suppress SCL-driven impedance growth. Yada et al.69 further confirmed the applicability of this approach using nano-ferrite domains, which enhanced cycling performance by mitigating polarization-induced Li+ migration barriers. Building upon these findings, their group employed pulsed laser deposition to coat LiCr0.05Ni0.45Mn1.5O4−δ with discontinuous BaTiO3 (BTO), achieving stable high-voltage cycling through effective suppression of band bending at the interface. In this framework, dielectric layers act as electric-field buffers and weaken the built-in potential responsible for SCL formation. Interfaces with high dielectric contrast can redistribute electric flux, attenuate the sharp potential drop, and weaken the electrostatic driving force for SCL formation. And the feasibility of this method has also been demonstrated in actual experiments. This makes the strategy particularly powerful when electronic bandgap engineering alone is insufficient—for example, when intrinsic band structures are difficult to manipulate without compromising cathode stability.In summary, the strategies discussed above—Hierarchical Band Alignment Engineering and Interface Electric-Field Modulation—offer two powerful, complementary frameworks for suppressing the detrimental effects of the space charge layer. Band alignment engineering reconstructs the electronic landscape at the electrode–electrolyte interface through coatings and dopants, creating graded pathways that minimize potential discontinuities and homogenize Li+ distribution. Dielectric and ferroelectric engineering, in contrast, directly compensates the built-in electric field that drives SCL formation, thereby weakening interfacial band bending and promoting smoother ion transport.144–149 However, the effectiveness of both strategies fundamentally depends on the intrinsic chemical and mechanical stability of the interface. Artificially constructed layers—whether band-alignment coatings, doped surface regions, or dielectric modifiers—can only function as intended when the interface maintains structural coherence and chemical integrity during long-term cycling. If interfacial reactions, lattice distortion, microcracking, or delamination continuously occur, the engineered interlayers will lose contact, degrade, or undergo unintended reactions, leading to new potential gradients that re-initiate SCL formation. Therefore, chemical and mechanical stabilization provides the foundational prerequisite for all SCL-mitigation strategies. Only when the underlying interface remains stable over extended cycling can the deliberately designed interlayers reliably regulate band alignment and electric-field distribution.
6. Summary and prospects
Understanding and controlling the space charge layer at solid–solid interfaces is central to unlocking the full potential of ASSBs. In this review, we systematically examined the origin, evolution, and consequences of SCL formation through the integrated lenses of defect chemistry, interfacial band alignment, and electrostatic field redistribution. We further clarified how the SCL dynamically evolves during cycling, influencing ion migration kinetics, interfacial activation energy, electronic leakage pathways, and long-term chemo-mechanical integrity. Beyond consolidating existing knowledge, this review establishes a unified physical framework that categorizes SCL-regulation strategies into Hierarchical Band Alignment Engineering and Interfacial Electric-Field Modulation, highlighting the electrostatic essence of the SCL and offering a coherent roadmap for rational interface design (Fig. 13). However, despite significant progress, the complexity of the SCL at the interface of all-solid-state batteries is far from being fully elucidated. Future research should be committed to the following key directions to fundamentally overcome the constraints imposed by the SCL on the performance of all-solid-state batteries. | ||
| Fig. 13 Summary and outlook. | ||
6.1 Multiphysics-coupled modeling of dynamic SCL evolution
Developing multi-physics, multi-scale theoretical frameworks is essential for describing the dynamic behavior of the SCL under realistic battery conditions. Existing models often oversimplify the strong coupling among electrochemical reactions, mechanical stress from volume changes, thermal gradients, and interfacial chemical evolution. High-fidelity, multi-scale computational approaches integrating density functional theory, molecular dynamics, phase-field modeling, and continuum electro-chemo-mechanics will be needed to accurately predict SCL evolution—its thickness, lithium-ion distribution, band bending, and internal fields—and to clarify its correlation with the structural evolution and oxygen release intrinsic to Ni-rich materials.6.2 High-performance advanced characterization techniques for interfaces
Breakthroughs in in situ/quasi-in situ characterization with high spatial and temporal resolution are critical for validating theory and guiding interface design. Advanced techniques—such as DPC-STEM, EELS, HRTEM, HAXPES, synchrotron XAS, and multi-dimensional solid-state NMR—should be further developed to directly visualize Li+ distribution, oxidation-state changes, lattice distortion, and local potential gradients within the SCL. Importantly, future tools must enable real-time observation of SCL evolution during charge–discharge cycling, thermal perturbation, and mechanical loading, allowing its effects to be decoupled from other interfacial processes and establishing causal links between electrostatic phenomena and performance degradation.6.3 Interface design based on electronic-structure precision engineering
Interface design guided by band-structure engineering is expected to become a central strategy for SCL control. Conventional coatings, though effective, often rely on empirical optimization without a mechanistic foundation. Moving forward, functional interlayers with tailored electronic structures, graded band alignment, epitaxially engineered interfaces, and interface layers designed through high-throughput computational screening could enable precise Fermi level matching and controlled interfacial potential landscapes. Such strategies may not only suppress detrimental SCL formation but also create low-impedance Li+ transport pathways and passivate reactive surfaces.From a practical and industrial perspective, the two major SCL-regulation strategies discussed above—Hierarchical Band Alignment Engineering and Interfacial Electric-Field Modulation—exhibit distinct advantages, limitations, and applicable scenarios, depending on electrolyte chemistry, processing constraints, and scalability requirements. Hierarchical Band Alignment Engineering, encompassing surface coatings and bulk or gradient doping, is broadly applicable to both oxide- and sulfide-based solid electrolytes. Coating-based approaches are particularly effective for sulfide electrolytes, where large interfacial chemical potential mismatch and narrow Debye lengths give rise to intense local electric fields; in such systems, thin buffer layers can efficiently smooth band discontinuities and suppress excessive space charge accumulation. Bulk or gradient doping, in contrast, is more readily integrated into oxide-based cathode materials and benefits from mature synthesis protocols, offering relatively high scalability and compositional tunability at moderate cost. However, both coating and doping strategies require precise control over thickness, composition, and interfacial uniformity, and excessive processing complexity may limit their large-scale manufacturability.
Interfacial Electric-Field Modulation strategies, such as dielectric or ferroelectric engineering, provide a fundamentally different route by directly compensating the built-in electric field that drives SCL formation. These approaches are particularly attractive for systems in which intrinsic band alignment is difficult to optimize without compromising structural stability, such as high-voltage cathodes or interfaces with severe electronic mismatch. Nevertheless, the fabrication of high-quality dielectric or ferroelectric interlayers often relies on advanced deposition techniques and stringent thickness control, which may increase processing cost and pose challenges for large-area or high-throughput production. Moreover, their effectiveness is highly sensitive to interfacial mechanical integrity, as polarization-induced field compensation requires stable and continuous contact during long-term cycling.
Taken together, Hierarchical Band Alignment Engineering offers greater materials universality and scalability, while Interfacial Electric-Field Modulation provides stronger electrostatic regulation capability in electronically mismatched systems but with higher processing complexity. For practical implementation, these strategies should not be viewed as mutually exclusive; rather, their judicious combination—guided by electrolyte type, operating voltage window, and manufacturing constraints—will be essential for balancing performance gains, process complexity, and cost. Such a comparative, application-oriented understanding provides a rational basis for selecting SCL-regulation strategies in industrially relevant all-solid-state battery designs.
Finally, realizing SCL control at the full-cell level requires integrating these strategies within a multi-scale interface engineering framework. The SCL does not exist in isolation but is closely coupled with particle microstructure, electrode mesoscale packing, macroscopic stress distribution, and battery assembly conditions. Therefore, particle-level optimizations (such as single-crystallization, controlled morphology, and densification) must be combined with mechanically adaptive solid electrolytes and rational stack-pressure designs to ensure robust, intimate contact that enables the engineered interfacial electrostatics to function reliably during long-term cycling. Importantly, although some studies argue that mechanical degradation dominates interfacial failure, this does not negate the role played by SCLs; rather, mechanical integrity is a prerequisite for SCL formation. Thus, SCL evolution and mechanical contact degradation are inherently coupled, and resolving both challenges will require integrated electro–chemo–mechanical strategies.
Conflicts of interest
The authors declare no conflict of interest.Data availability
No new experimental datasets were generated in this review. All data supporting the analysis, figures, and discussions presented here are derived from previously published literature, as cited throughout the manuscript. Additional information, including processed data files, schematics, is available from the corresponding authors upon reasonable request.Acknowledgements
This work was supported by the National Key R&D Program of China (2023YFB2503700).References
- M. Ahangari, B. Szalai, J. Lujan, M. Zhou and H. Luo, Materials, 2024, 17, 801 CrossRef CAS PubMed.
- U. Kim, J. Park, A. Aishova, R. M. Ribas, R. S. Monteiro, K. J. Griffith, C. S. Yoon and Y. Sun, Adv. Energy Mater., 2021, 11, 2100884 CrossRef CAS.
- W. Yuan, W. Peng, C. Wu, N. Liu, C. Shen, Z. Xiao, J. Liu, C. Li, Y. Guo, Q. Huang, P. Zhang, H. Pan, L. Wen, L. Shi, L. Lu, D. Ren, K. Wu, M. Ouyang and X. Liu, Adv. Energy Mater., 2025, 15, 2405907 CrossRef CAS.
- Y. Wu, W. Zhang, X. Rui, D. Ren, C. Xu, X. Liu, X. Feng, Z. Ma, L. Lu and M. Ouyang, Adv. Energy Mater., 2025, 15, 2405183 CrossRef CAS.
- H. Pan, B. Zhao, H. Lai, Q. Huang, Z. Xiao, H. Liu, X. Chen, D. Ren, L. Lu, X. Liu and M. Ouyang, Chem. Eng. J., 2025, 524, 169639 CrossRef CAS.
- R. Li, Y. Mao, N. Chen, Q. Liu, J. Shao, Z. Liao, K. Qiu, P. Wang, S. Hao, X. Liao, H. Wang, Y. Wei, C. Guo, X. Liu, G. Zhu, D. Ren, L. Lu and M. Ouyang, J. Energy Storage, 2025, 106, 114859 CrossRef CAS.
- W. Peng, J. Liu, W. Yuan, Q. Huang, P. Zhang, C. Li, Y. Guo, L. Wen, Z. Xiao, J. Liu, Y. Li, D. Ren, L. Lu, M. Ouyang and X. Liu, ACS Appl. Mater. Interfaces, 2025, 17, 47112 CrossRef CAS PubMed.
- D. Kitsche, F. Strauss, Y. Tang, N. Bartnick, A. Kim, Y. Ma, C. Kübel, J. Janek and T. Brezesinski, Batteries Supercaps, 2022, 5, e202100397 CrossRef CAS.
- X. Li, Q. Sun, Z. Wang, D. Song, H. Zhang, X. Shi, C. Li, L. Zhang and L. Zhu, J. Power Sources, 2020, 456, 227997 CrossRef CAS.
- C. Doerrer, I. Capone, S. Narayanan, J. Liu, C. R. M. Grovenor, M. Pasta and P. S. Grant, ACS Appl. Mater. Interfaces, 2021, 13, 37809 CrossRef CAS PubMed.
- J. Yang, X. Liang, H.-H. Ryu, C. S. Yoon and Y.-K. Sun, Energy Storage Mater., 2023, 63, 102969 CrossRef.
- S. S. Zhang, Energy Storage Mater., 2020, 24, 247 CrossRef.
- Q. Huang, J. Liu, X. Chen, P. Zhang, L. Lu, D. Ren, M. Ouyang and X. Liu, Adv. Mater., 2025, 37, 2410006 CrossRef CAS PubMed.
- S. Braun, C. Yada and A. Latz, J. Phys. Chem. C, 2015, 119, 22281 CrossRef CAS.
- X. Kang, H. Lin, G. Xu, J. Tao, Y. Chen, K. Zhong, J.-M. Zhang and Z. Huang, Phys. Rev. Mater., 2024, 8, 105402 CrossRef CAS.
- L. Jia, J. Zhu, X. Zhang, B. Guo, Y. Du and X. Zhuang, Electrochem. Energy Rev., 2024, 7, 12 CrossRef CAS.
- R. R. Gaddam, L. Katzenmeier, X. Lamprecht and A. S. Bandarenka, Phys. Chem. Chem. Phys., 2021, 23, 12926 RSC.
- Q. Zhang, Y. Kong, K. Gao, Y. Wen, Q. Zhang, H. Fang, C. Ma and Y. Du, Sci. China: Technol. Sci., 2022, 65, 2246 CrossRef CAS.
- Z. Xu, X. Wang, Z. Wang, X. Li, J. Liu, A. Bayaguud and L. Zhang, J. Power Sources, 2023, 571, 233079 CrossRef CAS.
- J. Zhang, C. Zheng, L. Li, Y. Xia, H. Huang, Y. Gan, C. Liang, X. He, X. Tao and W. Zhang, Adv. Energy Mater., 2020, 10, 1903311 CrossRef CAS.
- C. B. Lim and Y. J. Park, Sci. Rep., 2020, 10, 10501 CrossRef CAS PubMed.
- H.-J. Noh, S. Youn, C. S. Yoon and Y.-K. Sun, J. Power Sources, 2013, 233, 121 CrossRef CAS.
- R. Koerver, W. Zhang, L. De Biasi, S. Schweidler, A. O. Kondrakov, S. Kolling, T. Brezesinski, P. Hartmann, W. G. Zeier and J. Janek, Energy Environ. Sci., 2018, 11, 2142 RSC.
- D. Kitsche, Y. Tang, Y. Ma, D. Goonetilleke, J. Sann, F. Walther, M. Bianchini, J. Janek and T. Brezesinski, ACS Appl. Energy Mater., 2021, 4, 7338 CrossRef CAS.
- D. H. Jeon, Energy Storage Mater., 2019, 18, 139 CrossRef.
- H. Zhang, X. He, Z. Chen, Y. Yang, H. Xu, L. Wang and X. He, Adv. Energy Mater., 2022, 12, 2202022 CrossRef CAS.
- S.-T. Myung, F. Maglia, K.-J. Park, C. S. Yoon, P. Lamp, S.-J. Kim and Y.-K. Sun, ACS Energy Lett., 2017, 2, 196 CrossRef CAS.
- G. Xu, X. Liu, A. Daali, R. Amine, Z. Chen and K. Amine, Adv. Funct. Mater., 2020, 30, 2004748 CrossRef CAS.
- W. Schottky, Z. Phys., 1942, 118, 539 CrossRef CAS.
- K. Lehovec, J. Chem. Phys., 1953, 21, 1123 CrossRef CAS.
- J. Haruyama, K. Sodeyama, L. Han, K. Takada and Y. Tateyama, Chem. Mater., 2014, 26, 4248 CrossRef CAS.
- Z. Cheng, M. Liu, S. Ganapathy, C. Li, Z. Li, X. Zhang, P. He, H. Zhou and M. Wagemaker, Joule, 2020, 4, 1311 CrossRef CAS.
- K. Hikima, K. Shimizu, H. Kiuchi, Y. Hinuma, K. Suzuki, M. Hirayama, E. Matsubara and R. Kanno, Commun. Chem., 2022, 5, 52 CrossRef CAS PubMed.
- S. Ohta, N. Singh, R. K. Rai, H. Koh, Y. Zhang, W. Suk, M. J. Palmer, S.-J. Hwang, M. Jones, C. Wang, C. Ling, K. S. Masias, E. Stavitski, J. Sakamoto and E. A. Stach, Joule, 2025, 9, 101789 CrossRef CAS.
- N. J. J. De Klerk and M. Wagemaker, ACS Appl. Energy Mater., 2018, 1(10), 5609–5618 CAS.
- F. Zhang, Y. Guo, L. Zhang, P. Jia, X. Liu, P. Qiu, H. Zhang and J. Huang, eTransportation, 2023, 15, 100220 CrossRef.
- J. Gao, J. Hao, Y. Gao, X. Sun, Y. Zhang, D. Song, Q. Zhao, F. Zhao, W. Si, K. Wang, T. Ohsaka, F. Matsumoto, J. Wu and H. Xie, eTransportation, 2023, 17, 100252 CrossRef.
- D. Ren, L. Lu, R. Hua, G. Zhu, X. Liu, Y. Mao, X. Rui, S. Wang, B. Zhao, H. Cui, M. Yang, H. Shen, C.-Z. Zhao, L. Wang, X. He, S. Liu, Y. Hou, T. Tan, P. Wang, Y. Nitta and M. Ouyang, eTransportation, 2023, 18, 100272 CrossRef.
- L. Wang, Z. Chen, Y. Liu, Y. Li, H. Zhang and X. He, eTransportation, 2023, 16, 100239 CrossRef.
- X. Yang, K. Doyle-Davis, X. Gao and X. Sun, eTransportation, 2022, 11, 100152 CrossRef.
- Z. Sun, M. Li, B. Xiao, X. Liu, H. Lin, B. Jiang, H. Liu, M. Li, D.-L. Peng and Q. Zhang, eTransportation, 2022, 14, 100203 CrossRef.
- A. Kim, S. Woo, M. Kang, H. Park and B. Kang, Front. Chem., 2020, 8, 468 CrossRef CAS PubMed.
- C. Loho, R. Djenadic, M. Bruns, O. Clemens and H. Hahn, J. Electrochem. Soc., 2017, 164, A6131 CrossRef CAS.
- A.-K. Hatz, R. Calaminus, J. Feijoo, F. Treber, J. Blahusch, T. Lenz, M. Reichel, K. Karaghiosoff, N. M. Vargas-Barbosa and B. V. Lotsch, ACS Appl. Energy Mater., 2021, 4, 9932 CrossRef CAS.
- M. V. Reddy, C. M. Julien, A. Mauger and K. Zaghib, Nanomaterials, 2020, 10, 1606 CrossRef CAS PubMed.
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 16141 CrossRef.
- M. Fingerle, R. Buchheit, S. Sicolo, K. Albe and R. Hausbrand, Chem. Mater., 2017, 29, 7675 CrossRef CAS.
- K. Mizushima, P. C. Jones, P. J. Wiseman and J. B. Goodenough, Mater. Res. Bull., 1980, 15, 783 CrossRef CAS.
- Y. Nomura, K. Yamamoto, T. Hirayama, S. Ouchi, E. Igaki and K. Saitoh, Angew. Chem., 2019, 131, 5346 CrossRef.
- L. Wang, R. Xie, B. Chen, X. Yu, J. Ma, C. Li, Z. Hu, X. Sun, C. Xu, S. Dong, T.-S. Chan, J. Luo, G. Cui and L. Chen, Nat. Commun., 2020, 11, 5889 CrossRef CAS PubMed.
- J. Maier, J. Phys. Chem. Solids, 1985, 46, 309 CrossRef CAS.
- N. Togasaki, A. Nakao, T. Tanaka, U. Harada, H. Onish, H. Yasuda, S. Kobayashi, F. Maeda and T. Osaka, J. Electrochem. Soc., 2023, 170, 050519 CrossRef CAS.
- M. Haruta, S. Shiraki, T. Suzuki, A. Kumatani, T. Ohsawa, Y. Takagi, R. Shimizu and T. Hitosugi, Nano Lett., 2015, 15, 1498 CrossRef CAS PubMed.
- C. Yu, S. Ganapathy, E. R. H. Van Eck, L. Van Eijck, N. De Klerk, E. M. Kelder and M. Wagemaker, J. Energy Chem., 2019, 38, 1 CrossRef.
- Z. Moradi, A. Lanjan, R. Tyagi and S. Srinivasan, J. Energy Storage, 2023, 73, 109048 CrossRef.
- P. Ranque, E. Gonzalo, M. Armand and D. Shanmukaraj, Mater. Today Energy, 2023, 34, 101283 CrossRef CAS.
- H. Wang, J. Zhu, Y. Su, Z. Gong and Y. Yang, Sci. China: Chem., 2021, 64, 879 CAS.
- C. König, A. Ramanayagam, J. Kraus and B. Roling, Batteries Supercaps, 2024, 7, e202300578 CrossRef.
- K. Yamamoto, Y. Iriyama, T. Asaka, T. Hirayama, H. Fujita, C. A. J. Fisher, K. Nonaka, Y. Sugita and Z. Ogumi, Angew. Chem., Int. Ed., 2010, 49, 4414 CrossRef CAS PubMed.
- C. Yu, S. Ganapathy, E. R. H. V. Eck, H. Wang, S. Basak, Z. Li and M. Wagemaker, Nat. Commun., 2017, 8, 1086 CrossRef PubMed.
- K. Yamamoto, Y. Iriyama, T. Asaka, T. Hirayama, H. Fujita, C. A. J. Fisher, K. Nonaka, Y. Sugita and Z. Ogumi, Angew. Chem., 2010, 122, 4516 CrossRef.
- Z. Gu, J. Ma, F. Zhu, T. Liu, K. Wang, C.-W. Nan, Z. Li and C. Ma, Nat. Commun., 2023, 14, 1632 CrossRef CAS PubMed.
- D. Brogioli, F. Langer, R. Kun and F. La Mantia, ACS Appl. Mater. Interfaces, 2019, 11, 11999 CrossRef CAS PubMed.
- L. Katzenmeier, S. Helmer, S. Braxmeier, E. Knobbe and A. S. Bandarenka, ACS Appl. Mater. Interfaces, 2021, 13, 5853 CrossRef CAS PubMed.
- L. Katzenmeier, L. Carstensen and A. S. Bandarenka, ACS Appl. Mater. Interfaces, 2022, 14, 15811 CrossRef CAS PubMed.
- P. Lu, S. Gong, C. Wang, Z. Yu, Y. Huang, T. Ma, J. Lian, Z. Jiang, L. Chen, H. Li and F. Wu, ACS Nano, 2024, 18, 7334 CrossRef CAS PubMed.
- R. Koerver, I. Aygün, T. Leichtweiß, C. Dietrich, W. Zhang, J. O. Binder, P. Hartmann, W. G. Zeier and J. Janek, Chem. Mater., 2017, 29, 5574 CrossRef CAS.
- Y. Tateyama, B. Gao, R. Jalem and J. Haruyama, Curr. Opin. Electrochem., 2019, 17, 149 CrossRef CAS.
- C. Yada, A. Ohmori, K. Ide, H. Yamasaki, T. Kato, T. Saito, F. Sagane and Y. Iriyama, Adv. Energy Mater., 2014, 4, 1301416 CrossRef.
- C. Zhu, S. Kobayashi, Y. Sugisawa, F. Weber, K.-H. Lin, M. Kitamura, K. Horiba, H. Kumigashira, K. Nishio, R. Shimizu, D. Sekiba, T. Hitosugi and R. Berger, ACS Nano, 2025, 19, 39062 CrossRef CAS PubMed.
- Y.-F. Zhou, M.-Z. Yang, F.-Q. She, L. Gong, X.-Q. Zhang, J. Chen, S.-Q. Song and F.-Y. Xie, Application of X-ray photoelectron spectroscopy to study interfaces for solid-state lithium ion battery, Acta Phys. Sin., 2021, 70, 178801 CrossRef.
- B. Wang, M. S. R. Limon, Y. Zhou, K. Cho, Z. Ahmad and L. Su, ACS Energy Lett., 2025, 10, 1255 CrossRef CAS.
- T. Kim, K. Kim, S. Lee, G. Song, M. S. Jung and K. T. Lee, Chem. Mater., 2022, 34, 9159 CrossRef CAS.
- A. Neumann, S. Randau, K. Becker-Steinberger, T. Danner, S. Hein, Z. Ning, J. Marrow, F. H. Richter, J. Janek and A. Latz, ACS Appl. Mater. Interfaces, 2020, 12, 9277 CrossRef CAS PubMed.
- S. Huang, Y. Kimura, T. Nakamura, N. Ishiguro, O. Sekizawa, K. Nitta, T. Uruga, T. Takeuchi, T. Okumura, M. Tada, Y. Uchimoto and K. Amezawa, J. Phys. Chem. C, 2024, 128, 6213 CrossRef CAS.
- Y. Zhang, Y. Tian, Y. Xiao, L. J. Miara, Y. Aihara, T. Tsujimura, T. Shi, M. C. Scott and G. Ceder, Adv. Energy Mater., 2020, 10, 1903778 CrossRef CAS.
- Y. Chen, L. Huang, D. Zhou, X. Gao, T. Hu, Z. Zhang, Z. Zhen, X. Chen, L. Cui and G. Wang, Adv. Energy Mater., 2024, 14, 2304443 CrossRef CAS.
- Y. Han, S. H. Jung, H. Kwak, S. Jun, H. H. Kwak, J. H. Lee, S. Hong and Y. S. Jung, Adv. Energy Mater., 2021, 11, 2100126 CrossRef CAS.
- Y. Liu, Y. Bai, W. Jaegermann, R. Hausbrand and B.-X. Xu, ACS Appl. Mater. Interfaces, 2021, 13, 5895 CrossRef CAS PubMed.
- W. He, L. Zhou, M. K. Tufail, P. Zhai, P. Yu, R. Chen and W. Yang, Trans. Tianjin Univ., 2021, 27, 423 CrossRef CAS.
- L. Katzenmeier, M. Gößwein, A. Gagliardi and A. S. Bandarenka, J. Phys. Chem. C, 2022, 126, 10900 CrossRef CAS.
- R. Usiskin and J. Maier, Adv. Energy Mater., 2021, 11, 2001455 Search PubMed.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271 CrossRef CAS PubMed.
- Y. Wang, T. Liu, L. Estevez and J. Kumar, Energy Storage Mater., 2020, 27, 232 CrossRef.
- S. Sinzig, T. Hollweck, C. P. Schmidt and W. A. Wall, J. Electrochem. Soc., 2023, 170, 040513 CrossRef CAS.
- W.-L. Feng, F. Wang, X. Zhou, X. Ji, F.-D. Han and C.-S. Wang, Stability of interphase between solid state electrolyte and electrode, Acta Phys. Sin., 2020, 69, 228206, DOI:10.7498/aps.69.20201554.
- M. W. Swift and Y. Qi, Phys. Rev. Lett., 2019, 122, 167701 CrossRef PubMed.
- W. Jiang, Q. Zhou, F. Lu, Y. Chen and Z. Ma, J. Energy Storage, 2022, 55, 105655 CrossRef.
- M. W. Swift, H. Jagad, J. Park, Y. Qie, Y. Wu and Y. Qi, Curr. Opin. Solid State Mater. Sci., 2022, 26, 100990 CrossRef CAS.
- A. Al-Ali, B. Maundy, A. Allagui and A. Elwakil, J. Electroanal. Chem., 2022, 924, 116854 CrossRef CAS.
- K. Nishio, D. Imazeki, K. Kurushima, Y. Takeda, K. Edamura, R. Nakayama, R. Shimizu and T. Hitosugi, ACS Appl. Mater. Interfaces, 2022, 14, 34620 CrossRef CAS PubMed.
- S. Jayasubramaniyan, C. Lee and H.-W. Lee, J. Mater. Res., 2022, 37, 4017 CrossRef CAS.
- D. Wang, X. Yin, J. Wu, Y. Luo and S. Shi, Acta Phys.-Chim. Sin., 2024, 40, 2307029 CrossRef.
- S. Su, J. Ma, L. Zhao, K. Lin, Q. Li, S. Lv, F. Kang and Y. He, Carbon Energy, 2021, 3, 866 CrossRef CAS.
- Y. Huang, B. Shao and F. Han, Curr. Opin. Electrochem., 2022, 33, 100933 CrossRef CAS.
- A. Chakraborty, M. Dixit, D. Aurbach and D. T. Major, npj Comput. Mater., 2018, 4, 60 CrossRef.
- B.-Y. Chang, Electrochim. Acta, 2023, 462, 142741 CrossRef CAS.
- J. Sugiyama, E. Nocerino, O. K. Forslund, Y. Sassa, M. Månsson, S. Kobayashi, K. Nishio, T. Hitosugi, A. Suter and T. Prokscha, J. Phys.: Conf. Ser., 2023, 2462, 012046 CrossRef CAS.
- J. A. Isaac, L. R. Mangani, D. Devaux and R. Bouchet, ACS Appl. Mater. Interfaces, 2022, 14, 13158 CrossRef CAS PubMed.
- H. Masuda, N. Ishida, Y. Ogata, D. Ito and D. Fujita, Nanoscale, 2017, 9, 893 RSC.
- N. C. Paranamana, A. Werbrouck, A. K. Datta, X. He and M. J. Young, Adv. Energy Mater., 2024, 2403904 Search PubMed.
- M. Liu, S. Ganapathy and M. Wagemaker, Acc. Chem. Res., 2022, 55, 333 CrossRef CAS PubMed.
- M. Liu, C. Wang, C. Zhao, E. Van Der Maas, K. Lin, V. A. Arszelewska, B. Li, S. Ganapathy and M. Wagemaker, Nat. Commun., 2021, 12, 5943 CrossRef CAS PubMed.
- Z. Feng, X. Qiu, X. Chen, H. Wang and X. Guo, ACS Appl. Mater. Interfaces, 2024, 16, 42995 CrossRef CAS PubMed.
- T. Zhang, Z. Yang, H. Li, Q. Zhuang and Y. Cui, Acta Chim. Sin., 2019, 77, 525 CrossRef CAS.
- T. Hakari, M. Deguchi, K. Mitsuhara, T. Ohta, K. Saito, Y. Orikasa, Y. Uchimoto, Y. Kowada, A. Hayashi and M. Tatsumisago, Chem. Mater., 2017, 29, 4768 CrossRef CAS.
- Y.-H. Chen, S.-C. Lin, J.-A. Wang, S.-Y. Hsu and C.-C. M. Ma, J. Electrochem. Soc., 2018, 165, A3029 CrossRef CAS.
- B. Fan, Z. Guan, H. Wang, L. Wu, W. Li, S. Zhang and B. Xue, Solid State Ionics, 2021, 368, 115680 CrossRef CAS.
- H. Li, T. Zhang, Z. Yang, Y. Shi, Q. Zhuang and Y. Cui, Int. J. Electrochem. Sci., 2021, 16, 210229 CrossRef CAS.
- C. Wei, H. Xue, Z. Li, F. Zhao and F. Tang, J. Phys. D: Appl. Phys., 2022, 55, 105305 CrossRef CAS.
- C.-Y. Yu, J. Choi, J. Dunham, R. Ghahremani, K. Liu, P. Lindemann, Z. Garver, D. Barchiesi, R. Farahati and J.-H. Kim, J. Power Sources, 2024, 597, 234116 CrossRef CAS.
- Z. Tang, A. Morchhale, J. R. Sayre and J.-H. Kim, J. Energy Storage, 2025, 107, 114966 CrossRef CAS.
- R. Götz, M. Wagner, K. Song, L. Katzenmeier and A. S. Bandarenka, Batteries Supercaps, 2024, e202400570 Search PubMed.
- Q. Huang, K. Qiu, Z. Xiao, D. Li, H. Deng, L. Wen, W. Yuan, W. Peng, P. Zhang, J. Liu, D. Li, C. Zhan, X. Chen, L. Lu, J. Hua, Y. Wei, J. Shao, D. Ren, M. Ouyang and X. Liu, Adv. Funct. Mater., 2025, 35, 2422663 CrossRef CAS.
- Q. Huang, H. Pan, D. Wang, Z. Xiao, Y. Guo, S. Liu, J. Liu, G. Yuan, T. Cheng, L. Wen, C. Li, Y. Li, J. Liu, Y. Wei, X. Rui, J. Hua, X. Chen, J. Shao, D. Ren, B. Deng, S. Yin, L. Lu, X. Liu and M. Ouyang, Adv. Funct. Mater., 2025, e16819 Search PubMed.
- P. Zhang, J. Liu, Q. Huang, Y. Li, Y. Guo, Z. Xiao, C. Li, L. Wen, W. Peng, W. Yuan, G. Zhu, L. Yin, L. Fan, L. Zheng, J. Zhang, T. Tan, J. Hua, D. Ren, L. Lu and X. Liu, eTransportation, 2025, 26, 100493 CrossRef.
- J. Liu, C. Wang, J. Yu, Q. Huang, P. Zhang, Z. Xiao, S. Liu, W. Yuan, C. Li, W. Peng, X. Yan, Y. Zhang, L. Yin, M. Zhang, L. Zheng, J. Zhang, J. Xu, W. Yin, L. Lu, D. Ren, M. Ouyang and X. Liu, Nano Energy, 2025, 140, 111038 CrossRef CAS.
- W. Jiang, X. Zhu, R. Huang, S. Zhao, X. Fan, M. Ling, C. Liang and L. Wang, Adv. Energy Mater., 2022, 12, 2103473 CrossRef CAS.
- A. Masias, J. Marcicki and W. A. Paxton, ACS Energy Lett., 2021, 6, 621 CrossRef CAS.
- M. Jiang, D. L. Danilov, R. Eichel and P. H. L. Notten, Adv. Energy Mater., 2021, 11, 2103005 CrossRef CAS.
- S. H. Jung, U. Kim, J. Kim, S. Jun, C. S. Yoon, Y. S. Jung and Y. Sun, Adv. Energy Mater., 2020, 10, 1903360 CrossRef CAS.
- X. Liu, B. Zheng, J. Zhao, W. Zhao, Z. Liang, Y. Su, C. Xie, K. Zhou, Y. Xiang, J. Zhu, H. Wang, G. Zhong, Z. Gong, J. Huang and Y. Yang, Adv. Energy Mater., 2021, 11, 2003583 CrossRef CAS.
- C. Wang, S. Hwang, M. Jiang, J. Liang, Y. Sun, K. Adair, M. Zheng, S. Mukherjee, X. Li, R. Li, H. Huang, S. Zhao, L. Zhang, S. Lu, J. Wang, C. V. Singh, D. Su and X. Sun, Adv. Energy Mater., 2021, 11, 2100210 CrossRef CAS.
- Y. Huang, S. Cao, X. Xie, C. Wu, S. Jamil, Q. Zhao, B. Chang, Y. Wang and X. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 19483 CrossRef CAS PubMed.
- J. S. Lee and Y. J. Park, ACS Appl. Mater. Interfaces, 2021, 13, 38333 CrossRef CAS PubMed.
- U. Nisar, N. Muralidharan, R. Essehli, R. Amin and I. Belharouak, Energy Storage Mater., 2021, 38, 309 CrossRef.
- S. Kang, H. Kim, J. Y. Jung, K.-H. Park, K. Kim, J. H. Song, J.-S. Yu, Y.-J. Kim and W. Cho, ACS Appl. Mater. Interfaces, 2023, 15, 10744 CrossRef CAS PubMed.
- F. Walther, S. Randau, Y. Schneider, J. Sann, M. Rohnke, F. H. Richter, W. G. Zeier and J. Janek, Chem. Mater., 2020, 32, 6123 CrossRef CAS.
- S. Payandeh, D. Goonetilleke, M. Bianchini, J. Janek and T. Brezesinski, Curr. Opin. Electrochem., 2022, 31, 100877 CrossRef CAS.
- K. Jung and T. Yim, Rare Met., 2021, 40, 2793 CrossRef CAS.
- M. Akin, Y. Wang, X. Qiao, Z. Yan and X. Zhou, Electrochim. Acta, 2020, 355, 136779 CrossRef CAS.
- S. Payandeh, C. Njel, A. Mazilkin, J. H. Teo, Y. Ma, R. Zhang, A. Kondrakov, M. Bianchini and T. Brezesinski, Adv. Mater. Interfaces, 2023, 10, 2201806 CrossRef CAS.
- Y. Wang, E. Wang, X. Zhang and H. Yu, Energy Fuels, 2021, 35, 1918 CrossRef CAS.
- C. Wang, Y. Wu, S. Wang, E. Van Der Heide and X. Zhuang, Mater. Res. Bull., 2025, 181, 113078 CrossRef CAS.
- R. Ruess, S. Schweidler, H. Hemmelmann, G. Conforto, A. Bielefeld, D. A. Weber, J. Sann, M. T. Elm and J. Janek, J. Electrochem. Soc., 2020, 167, 100532 CrossRef CAS.
- N. Ohta, K. Takada, I. Sakaguchi, L. Zhang, R. Ma, K. Fukuda, M. Osada and T. Sasaki, Electrochem. Commun., 2007, 9, 1486 CrossRef CAS.
- R. Götz, E. Pugacheva, Z. Ahaliabadeh, P. S. Llanos, T. Kallio and A. S. Bandarenka, ChemSusChem, 2024, 17, e202401026 CrossRef PubMed.
- X. Li, L. Jin, D. Song, H. Zhang, X. Shi, Z. Wang, L. Zhang and L. Zhu, J. Energy Chem., 2020, 40, 39 CrossRef.
- B. Shen, L. Li, X. Yao and B. Huang, Ceram. Int., 2024, 50, 7150 CrossRef CAS.
- R. Zhang, Y. Ma, Y. Tang, D. Goonetilleke, T. Diemant, J. Janek, A. Kondrakov and T. Brezesinski, Chem. Mater., 2023, 35, 6835 CrossRef CAS.
- Y. Zou, K. Zhou, G. Liu, N. Xu, X. Zhang, Y. Yang, J. Zhang and J. Zheng, ACS Appl. Mater. Interfaces, 2021, 13, 16427 CrossRef CAS PubMed.
- Y. Liu, T. Zeng, G. Li, T. Wan, M. Li, X. Zhang, M. Li, M. Su, A. Dou, W. Zeng, Y. Zhou, R. Guo and D. Chu, Energy Storage Mater., 2022, 52, 534 CrossRef.
- K. Yang, Y. Sun, Q. Su, Y. Lu, K. Liu, Z. Li, H. Liu and L. Zhang, Chem. Eng. J., 2023, 471, 144405 CrossRef CAS.
- W. Liu, P. Oh, X. Liu, M. Lee, W. Cho, S. Chae, Y. Kim and J. Cho, Angew. Chem., Int. Ed., 2015, 54, 4440 CrossRef CAS PubMed.
- M. Liu, S. Zhang, E. R. H. Van Eck, C. Wang, S. Ganapathy and M. Wagemaker, Nat. Nanotechnol., 2022, 17, 959 CrossRef CAS PubMed.
- W. Li, M. Li, S. Wang, P.-H. Chien, J. Luo, J. Fu, X. Lin, G. King, R. Feng, J. Wang, J. Zhou, R. Li, J. Liu, Y. Mo, T.-K. Sham and X. Sun, Nat. Nanotechnol., 2025, 20, 265 CrossRef CAS PubMed.
- S. Deng, X. Li, Z. Ren, W. Li, J. Luo, J. Liang, J. Liang, M. N. Banis, M. Li, Y. Zhao, X. Li, C. Wang, Y. Sun, Q. Sun, R. Li, Y. Hu, H. Huang, L. Zhang, S. Lu, J. Luo and X. Sun, Energy Storage Mater., 2020, 27, 117 CrossRef.
- J. Liang, S. Hwang, S. Li, J. Luo, Y. Sun, Y. Zhao, Q. Sun, W. Li, M. Li, M. N. Banis, X. Li, R. Li, L. Zhang, S. Zhao, S. Lu, H. Huang, D. Su and X. Sun, Nano Energy, 2020, 78, 105107 CrossRef CAS.
- T.-T. Zuo, R. Rueß, R. Pan, F. Walther, M. Rohnke, S. Hori, R. Kanno, D. Schröder and J. Janek, Nat. Commun., 2021, 12, 6669 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |