
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemBERTa-3: an open source training framework for chemical foundation models
Riya Singh†
 a,
Aryan Amit Barsainyan†a,
Rida Irfana,
Connor Joseph Amorinb,
Stewart Heb,
Tony Davisa,
Arun Thiagarajan‡
a,
Shiva Sankarana,
Seyone Chithranandae,
Walid Ahmad‡a,
Derek Jonesb,
Kevin McLoughlinb,
Hyojin Kimc,
Anoushka Bhutanid,
Shreyas Vinaya Sathyanarayanaa,
Venkat Viswanathand,
Jonathan E. Allenb and
Bharath Ramsundar*a
a,
Aryan Amit Barsainyan†a,
Rida Irfana,
Connor Joseph Amorinb,
Stewart Heb,
Tony Davisa,
Arun Thiagarajan‡
a,
Shiva Sankarana,
Seyone Chithranandae,
Walid Ahmad‡a,
Derek Jonesb,
Kevin McLoughlinb,
Hyojin Kimc,
Anoushka Bhutanid,
Shreyas Vinaya Sathyanarayanaa,
Venkat Viswanathand,
Jonathan E. Allenb and
Bharath Ramsundar*a
aDeep Forest Sciences, USA. E-mail: bharath@deepforestsci.com
bGlobal Security Computing Applications Division, Lawrence Livermore National Laboratory, Livermore, CA, USA
cCenter for Applied Scientific Computing, Lawrence Livermore National Laboratory, Livermore, CA, USA
dUniversity of Michigan, USA
eUniversity of California, Berkeley, USA
First published on 19th January 2026
Abstract
The rapid advancement of machine learning in computational chemistry has opened new doors for designing molecules, predicting molecular properties, and discovering novel materials. However, building scalable and robust models for molecular machine learning remains a significant challenge due to the vast size and complexity of chemical space. Recent advances in chemical foundation models hold considerable promise for addressing these challenges, but such models remain difficult to train and are often fully or partially proprietary. For this reason, we introduce ChemBERTa-3, an open source training and benchmarking framework designed to train and fine-tune large-scale chemical foundation models. ChemBERTa-3 provides: (i) unified, reproducible infrastructure for model pretraining and fine-tuning, (ii) systematic benchmarking tooling to evaluate proposed chemical foundation model architectures on tasks from the MoleculeNet suite, and (iii) fully open release of model weights, training configurations, and deployment workflows. Our experiments demonstrate that although both graph-based and transformer-based architectures perform well at small scale, transformer-based models are considerably easier to scale. We also discuss how to overcome the numerous challenges that arise when attempting to reproducibly construct large chemical foundation models, ranging from subtle benchmarking issues to training instabilities. We test ChemBERTa-3 infrastructure in both an AWS-based Ray deployment and in an on-premise high-performance computing cluster to verify the reproducibility of the framework and results. We anticipate that ChemBERTa-3 will serve as a foundational building block for next-generation chemical foundation models and for the broader project of creating open source LLMs for scientific applications. In support of reproducible and extensible science, we have open sourced all ChemBERTa3 models and our Ray cluster configurations.
1 Introduction
Drug discovery is a complex and time-intensive process that involves identifying potential therapeutic compounds and evaluating their biological efficacy. Molecular property prediction models have had a significant impact in the drug discovery process, since predicted properties are central to evaluating, selecting, and generating candidate molecules.1 In recent years, deep learning has been widely used for molecular property prediction. A range of architectures including graph neural networks and transformer-based architectures, alongside methodologies such as pretraining, contrastive learning, multi-task learning, and transfer learning, have all been used to enhance predictive accuracy and generalization.1,2 Large pre-trained transformer architectures, also known as chemical foundation models, have risen to particular prominence in recent years due to their potential to learn basic chemistry directly from large unlabeled compound databases.3–9Despite rapid progress in the development of chemical foundation models, there has been little systematic comparison of how different pretraining methodologies perform across diverse model architectures. Most existing studies focus on individual model classes, offering only a limited perspective on their strengths. This results in a narrow or incomplete understanding of how different models perform and compare across various contexts. One contributing factor to this lack of benchmarking is the substantial infrastructure required to pretrain and evaluate chemical foundation models at scale. Even with the availability of large-scale chemical datasets,10 the process of pre-training these models remains computationally intensive, and requires both scalable hardware and software infrastructure. These challenges have made large-scale reproducible benchmarking difficult.
In previous work, we introduced and open-sourced ChemBERTa3 and ChemBERTa-2,4 BERT-like transformer models designed to learn molecular fingerprints through semi-supervised pretraining. ChemBERTa was trained on a dataset of 10 million compounds, leveraging masked-language modeling (MLM) to extract meaningful molecular representations.11 ChemBERTa-2 further explored the scaling hypothesis that pretraining on larger datasets enhances downstream performance by employing both MLM and multi-task regression (MTR) over a significantly larger corpus of 77 million SMILES strings. The associated pretrained models were open sourced and have been widely used12,13 (with over 700 citations for ChemBERTa and 200 citations for ChemBERTa-2 at time of writing). In past work, we have also introduced MoleculeNet,14 a widely used benchmarking framework for molecular property prediction (cited over three thousand times at time of writing). All these works (ChemBERTa, ChemBERTa-2, and MoleculeNet) have been released as part of the open source DeepChem ecosystem15 which has built a broadly used open-source framework (over six thousand stars on Github) for drug discovery, materials science, and biology.
Recent work, such as MegaMolBART6 and Chemformer,7 has introduced transformer-based models trained on significantly more data than ChemBERTa and ChemBERTa-2 for molecular property prediction. However, in some cases, models have not been fully open sourced and remain hard to use and reproduce. For example, while MoLFormer5 has demonstrated strong performance and conducted extensive benchmarking, the largest state-of-art MoLFormer models have not been open sourced. Furthermore, benchmarking was not performed in a unified fashion: comparisons between MoLFormer and other models relied primarily on results reported from prior studies and employed a differing dataset splitting strategy from past studies, making reported comparisons not fully accurate (as we will discuss in more detail later in this work).
To address these limitations, we introduce the ChemBERTa-3 framework, an open source extensible platform for training and benchmarking chemical foundation models. ChemBERTa-3 is fully integrated into the DeepChem library and ecosystem and is able to leverage the extensive collection of models and benchmarking infrastructure available in DeepChem.15 To scale data-parallel training to multiple GPUs, ChemBERTa-3 leverages Ray's distributed training infrastructure16 and provides tooling specifically designed for efficient pretraining and fine-tuning of large-scale chemical foundation models. This integration supports both transformer-based and graph-based pretraining, allowing users to seamlessly pretrain and fine-tune models within DeepChem's modular ecosystem. We also introduce benchmarking guidelines and scripts to benchmark proposed chemical foundation model architecture using datasets from the MoleculeNet suite.14
As our first core contribution, we leverage the ChemBERTa-3 framework to compare and contrast several model architectures and their associated pretraining methods by systematically benchmarking. In particular we investigate how transformer-based methods compare to graph-based methods. Our experiments indicate that while graph-based models and pretraining methodologies perform comparably to transformer-based models at small scale, transformer-based approaches are considerably easier to scale to large datasets. Our results suggest that further investment in scaling graph-based pretraining infrastructure may be worthwhile.
As our second core contribution, we use ChemBERTa-3 infrastructure to train fully open-source MoLFormer architecture models on the Zinc20 dataset. We find that reproducing reported past MoLFormer results is highly challenging due to several subtleties in both benchmarking and model training. In particular, we find that MoLFormer's scaffold splitting algorithm is not equivalent to the MoleculeNet/DeepChem scaffold splitting algorithm, making earlier reported comparisons between MoLFormer and ChemBERTa/ChemBERTa-2 models inaccurate. To prevent such issues from arising in future work, ChemBERTa-3 proposes a standard benchmarking process for chemical foundation models using the MoleculeNet suite, ensuring consistent evaluation protocols and enabling more reliable comparisons across different model architectures. This benchmarking infrastructure is easily extensible to new datasets, ensuring that the methodology can remain relevant over time.
To test reproducibility, we train two separate large MoLFormer models on Zinc20, using both an AWS-based Ray deployment and on-premise high-performance computing infrastructure. We find that both models are directly comparable, and demonstrate that ChemBERTa-3 infrastructure can be meaningfully deployed in very different computing contexts. In service of open science, we open source the AWS-trained models (along with other small models). We also open source all training code and configurations used for these experiments.
Finally, our last core contribution in this work is a series of improvements and extensions to the open source DeepChem library and ecosystem to facilitate foundation model development. We introduce a new class into DeepChem, ModularTorchModel, that streamlines the process of pretraining and fine-tuning models. We also integrate several new model architectures into DeepChem (discussed in section 3.2), along with support for training transformer models from the HuggingFace library. These updates make DeepChem significantly more useful for foundation model research. The released ChemBERTa-3 training and benchmarking framework is powered by these underlying improvements to the DeepChem library and ecosystem.
We anticipate that ChemBERTa-3 will provide foundational infrastructure for designing and training next-generation chemical foundation models by facilitating both pre-training and benchmarking of new large chemical foundation models. We also anticipate that the lessons shared here, alongside the open-source infrastructure, will serve as a basis for facilitating the construction of both scientific foundation models in other domains17,18 and for the construction of open source LLMs for scientific work.
2 Related work
The field of drug discovery has witnessed significant advancements through the application of machine learning techniques.19 Traditional approaches often relied on handcrafted features and shallow learning models, which limited their ability to capture complex relationships within molecular data. The advent of deep learning has transformed this landscape, enabling the development of more sophisticated models that leverage large datasets to learn sophisticated representations of the structure of chemical space. A broad range of different approaches have been proposed in recent years for molecular property prediction.2.1 Graph-based architectures
Graph Neural Networks learn directly from molecular graphs (atoms as nodes, bonds as edges) and naturally capture structural features of compounds. Message Passing Neural Networks (MPNNs)20 are a variant of graph convolutional networks that propagate messages between neighboring atoms across molecular graphs to construct informative graph representations. Directed MPNN (D-MPNN),21 a variant of MPNN popularized by the Chemprop library, differs primarily by associating messages with directed bonds rather than atoms, thereby preventing redundant cyclic message paths (totters) and leading to less noisy, more informative molecular embeddings.Researchers have explored several unsupervised pre-training methods for GNNs to improve generalization further, enabling these models to learn from unlabeled data before fine-tuning on labeled samples. Infograph22 maximizes the mutual information between the graph-level representation and the representations of substructures of different scales. Infomax3D23 improves GNNs for molecular property prediction by leveraging 3D molecular data during pre-training. It maximizes the mutual information (MI) between learned 3D representations and 2D molecular graphs, enabling GNNs to infer implicit 3D geometric information from 2D data.
2.2 Transformer architectures
While transformers are designed for efficient processing of large-scale NLP corpora, they also prove highly effective in capturing intricate structural and semantic patterns from large, unlabeled molecular datasets. Our preceding work introduced ChemBERTa3 and ChemBERTa-2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4 based on the RoBERTa24 transformer implementation in HuggingFace. Chemformer7 is based on BART25 architecture. However, these models were trained on relatively smaller datasets. Larger models like MegaMolBART from NVIDIA trained on approximately 1.45 Billion molecules, and MoLFormer,5 trained on 1.1 Billion chemicals,26 have recently become popular. While MoLFormer has shown promising results, only a model version pre-trained on a smaller dataset of 100M molecules has been open sourced. This dataset combines 10% of Zinc and 10% of PubChem molecules used for MoLFormer-XL training, the best model of the MoLFormer suite (This full model remains close sourced.)
4 based on the RoBERTa24 transformer implementation in HuggingFace. Chemformer7 is based on BART25 architecture. However, these models were trained on relatively smaller datasets. Larger models like MegaMolBART from NVIDIA trained on approximately 1.45 Billion molecules, and MoLFormer,5 trained on 1.1 Billion chemicals,26 have recently become popular. While MoLFormer has shown promising results, only a model version pre-trained on a smaller dataset of 100M molecules has been open sourced. This dataset combines 10% of Zinc and 10% of PubChem molecules used for MoLFormer-XL training, the best model of the MoLFormer suite (This full model remains close sourced.)
2.3 Graph transformer architectures
GROVER27 leverages self-supervised learning at the node, edge, and graph levels to capture structural and semantic information from unlabeled molecular data. Instead of predicting node or edge types in isolation, it masks local subgraphs and infers contextual properties, reducing ambiguity. GROVER integrates Message Passing Networks within a Transformer-style architecture to encode this complex information effectively.Efforts to combine GNNs and transformers aim to provide a comprehensive molecular representation, capturing both the molecular structure and the interactions and characteristics of individual atoms.28
2.4 Pretraining methodologies
In transformer-based models, the masked language modeling (MLM) pretraining task, commonly used for BERT-style architectures, is used to predict masked tokens in SMILES sequences.11 MLM masks 15% of the tokens in each input string and trains the model to correctly identify them. Multitask regression (MTR) pretraining is used to learn to predict multiple molecular properties simultaneously. For graph neural networks, learning to predict masked atom types or bond connections enables the GNN to capture structural patterns in a molecule's topology, much like MLM does for sequences.29 Mutual information maximization (e.g., InfoGraph and Infomax3D) aligns local substructure embeddings with global molecular embeddings, often leveraging 2D or 3D data to enrich the learned representation without explicit labels.222.5 Benchmarking frameworks
Evaluating different models and pre-training methodologies requires robust and standardized benchmarks to ensure meaningful comparisons and reproducibility. The MoleculeNet14 benchmark suite is a widely used collection of datasets for this purpose, aggregating data on properties like solubility, toxicity, and bioactivity from multiple public sources. While MoleculeNet has drawn criticism for data curation issues,30 it provides considerable ease-of-use and standardized reproducible protocols which make it a powerful tool for directly comparing different models and pretraining methodologies. Despite the variety of available models and pretraining methodologies, it remains unclear which approach is most effective across different model architectures. Recognizing this gap, our work evaluates multiple architectures and pretraining methodologies on MoleculeNet benchmarks.Beyond MoleculeNet, several modeling and benchmarking frameworks have pushed the field forward. Chemprop31 demonstrated the practical effectiveness of directed message-passing networks through an accessible platform supporting small-molecule property prediction with uncertainty estimation. Broader machine-learning ecosystems such as PyTDC32 and TorchDrug33 have further expanded benchmarking infrastructure by integrating multimodal biological data, geometric deep learning, generative models, and standardized evaluation pipelines. Despite these advances, recent large-scale studies have raised concerns about benchmarking rigor. For example, the comprehensive evaluations of pretrained molecular embedding models report minimal gains over classical fingerprints under controlled statistical testing,34 and ecotoxicology benchmarks show strong in-distribution performance but substantial drops when models are applied to new host species or unseen chemicals.35 These findings reinforce the need for standardized, architecture-agnostic, and reproducible evaluation frameworks. ChemBERTa-3 is designed to address this need by providing a unified and extensible benchmarking suite capable of systematically comparing graph, transformer, and hybrid chemical foundation models under consistent protocols.
3 Methods
3.1 Base framework: DeepChem
DeepChem15 is an open source Python library for machine learning and deep learning on molecular, biological and quantum datasets. It is built on top of PyTorch,36 and other popular ML frameworks such as Scikit-learn37 and XGBoost.38 It offers tools to streamline model development, training, and evaluation. DeepChem simplifies the application, benchmarking, and deployment of machine learning models by providing easy-to-use model export and deployment APIs, making it easier to use ML in both research and production environments. In addition to its core functionality, DeepChem has been extended and integrated into multiple larger frameworks, such as the ATOM Modeling Pipeline (AMPL),39 which combines a diverse array of ML and molecular featurization tools for drug discovery.DeepChem was expanded with a suite of new infrastructure to support the ChemBERTa-3 framework (Fig. 1 (a)). Central to these contributions is the ModularTorchModel, which enables flexible pretraining, and fine-tuning of graph-based architectures with support for intermediate-loss computation. A HuggingFace DeepChem Wrapper was added to seamlessly integrate models and tokenizers from the transformers ecosystem, while new featurizers such as the RDKitConformerFeaturizer and GroverFeaturizer introduced support for 3D conformers and GROVER's functional-group-aware graph representations. Multiple models were implemented using ModularTorchModel and HuggingFace DeepChem Wrapper including ChemBERTa, InfoGraph, GROVER, InfoMax3DModular, MoLFormer (Fig. 1 (b)), and DMPNN. Additional details can be found in appendix A.1.
 | ||
| Fig. 1 (a) ChemBERTa-3 Overview and (b) MolFormer model architecture used for open-sourced c3-MoLFormer models. | ||
3.2 The ChemBERTa-3 framework
The ChemBERTa-3 infrastructure is designed as a scalable and extensible framework integrated within the open source DeepChem ecosystem. It allows efficient pretraining and fine-tuning of large-scale chemical foundation models, leveraging DeepChem's extensive molecular machine learning utilities. To effectively handle large datasets and distributed training, we connected ChemBERTa-3 with Ray, an open source framework designed to simplify scaling AI and Python applications, particularly in machine learning.16 Ray provides a compute layer for parallel processing, enabling users to run distributed tasks. The ChemBERTa-3 Ray infrastructure is illustrated in Fig. 2. | ||
| Fig. 2 This figure illustrates the ChemBERTa-3 architecture deployed on Prithvi. The framework utilizes S3 storage service for data access and submits jobs to a Ray Cluster, managed by the Ray Cluster Nodes Controller. The cluster consists of multiple Ray Worker Nodes, each subdividing tasks into Ray Sub Workers for efficient parallel processing. This design leverages Ray's distributed computing capabilities to enable scalable training and evaluation of ChemBERTa-3 models. | ||
As part of this integration, we built a pipeline where RayDataset is implemented as a subclass of the DeepChem Dataset superclass, by combining Ray's ray.data.Dataset with DeepChem's data handling utilities. This allows datasets to be modified using DeepChem featurizers, stored efficiently as NPZ files using _RayDcDatasink, and iterated over using iterbatches() for training. This approach enables scalable data handling while maintaining compatibility with DeepChem's modeling APIs.
The distributed data parallel (DDP) strategy is employed to efficiently scale the LLM pretraining on multiple GPUs and machines. It synchronizes gradients and model parameters, ensuring that all processes remain in sync. Each process maintains its copy of the model and performs forward and backward passes independently. During backpropagation, DDP registers an ‘autograd hook’ that triggers gradient synchronization, ensuring consistency across all replicas before updating the model. This setup ensures efficient resource utilization and enhances scalability, making it easier to explore and optimize new molecules, materials, and designs.
The ChemBERTa-3 platform provides a unified benchmarking framework for evaluating various models, including MoLFormer, ChemBERTa, Infograph, Infomax3D, GROVER, DMPNN, Random Forest (RF), and Graph Convolutional Networks (GCN). It standardizes scaffold split analysis, model training, and evaluation, ensuring fair comparisons and reproducible results. By integrating diverse architectures within a consistent pipeline, our platform facilitates rigorous benchmarking, enabling researchers to assess model performance comprehensively and develop more effective molecular modeling approaches.
4 Datasets
4.1 Pre-training dataset
ZINC2040 is a chemical library containing 1.4 billion compounds, 1.3 billion of which are purchasable, sourced from 310 catalogs from 150 companies, specifically designed for virtual screening.
In our work, the model performance is benchmarked across ZINC data sets of varying sizes to understand the impact of the scale of the data on model accuracy and generalization. Additionally, MoLFormer-c3-550M and MoLFormer-c3-1.1B are pre-trained on a combination of (50% ZINC20 + 50% Pubchem) and (100% ZINC20 + 100% Pubchem) datasets, respectively. This evaluation highlights the importance of training on large-scale datasets, which tend to improve model performance on downstream tasks, but also provides insights into the diminishing returns of adding more data at certain points.
4.2 Fine-tuning datasets
The pre-trained models are fine-tuned on various regression and classification tasks from MoleculeNet,14 chosen to cover medicinal chemistry applications, including brain penetrability, toxicity, solubility, and on-target inhibition. The datasets used include BACE, Clearance, Delaney, Lipophilicity, BBBP, ClinTox, HIV, Tox21, and Sider.  |
5 Experiments
5.1 Pretraining data collection
For the pretraining experiments, GCN, RF, and DMPNN were trained on only the fine-tuning splits for baseline comparisons. InfoGraph, InfoMax3D, and GROVER were pre-trained on only a 250K ZINC dataset and fine-tuned. ChemBERTa and MolFormer models were pre-trained on progressively larger datasets and then finetuned on the finetuning splits. ChemBERTa-10M and ChemBERTa-100M were pretrained on 10M and 100M molecules from ZINC20,40 respectively. MolFormer-550M was pretrained on 500M ZINC molecules + 50M PubChem molecules, and MolFormer-1.1B used 1B ZINC molecules + 100M PubChem molecules before fine-tuning. Graph convolutional models were not pretrained on larger datasets due to the relative difficulty of scaling graph-based pretraining. Our results indicate below that transformer-based pretraining is broadly comparable to graph-based pretraining at small scales, but it remains for future-work to test graph-based pretraining at larger scales.5.2 Training MoLFormer on the 1.1B dataset on AWS
We utilized 40 T4 GPUs from AWS, which were spot instances chosen to reduce costs. However, these instances are susceptible to preemption, leading to occasional interruptions and necessitating multiple restarts. Each restart involved resuming from the latest checkpoint. Since synchronous training was used, all GPUs needed to resynchronize, further increasing costs in both time and money. To address these challenges, we are exploring the implementation of asynchronous training when restarting from checkpoints. Pre-training the model took approximately 10 days on 40 T4 GPUs.5.3 Experiment design
We fine-tuned several models including purely supervised baselines, graph-based pre-trained models, and transformer-based pre-trained models on several datasets from MoleculeNet.For graph-based pretraining, we chose to benchmark GROVER, InfoGraph, and InfoMax3D models. We chose GROVER as it bridges a gap between transformer models and graph models. We chose InfoGraph, to test its mutual-information based pretraining methodology, and InfoMax3D, an extension of InfoGraph captures 3D molecular information, to test the importance of spatial dependencies and conformational variations in pretraining. For transformer-based pretraining, we trained ChemBERTa and MoLFormer models using the Chemberta-3 harness, leveraging Ray during pre-training.
5.4 Hyperparameter optimization
We fine-tuned the models for up to 500 epochs with early stopping based on validation loss, exploring train-time hyperparameters like learning rate and batch size. For regression tasks, labels were normalized to zero mean and unit standard deviation.The MoLformer(1.1B)5 results are directly taken from the MoLFormer paper, trained on ≈ 1.1 B molecules (100% PubChem + 100% Zinc). Our MoLFormer model, trained using the ChemBERTa-3 infrastructure, performs comparably on the three classification datasets (BACE, BBBP, Tox21) but slightly underperforms on other classification and regression tasks, possibly due to insufficient hyperparameter optimization. Additional details for hyperparameter optimization can be found in section 11.3 in Appendix.
5.5 AWS deployment of ChemBERTa-3 on Prithvi
To efficiently pre-train and fine-tune ChemBERTa-3 models at scale, we leverage Prithvi, an open-core commercial suite built on top of DeepChem. Prithvi provides tools for fine-tuning and deploying scientific foundation models. In this work, we use it primarily as a testing environment for ChemBERTa-3 pretraining and evaluation.We run training on AWS spot instances to reduce computational costs. Although these instances can be preempted at any time, frequent checkpointing allows us to resume from the most recent stable state. Over multiple runs, the cost savings from spot instances typically outweigh the overhead of handling potential interruptions.
5.6 Deploying ChemBERTa-3 on HPC infrastructure
To further verify reproducibility, the software framework was tested on a local HPC cluster using 16 4th generation AMD EPYC nodes, with 4 AMD MI300A APUs per node. The AMPL39 software environment and the Ray multi-node multi-GPU training framework was used for training new models. The MoLFormer architecture was selected for comparison with 5 replicate training runs to measure variability between training runs. After training from scratch with identical conditions to the cloud-based training (including the same 1.1B chemical training set), foundation models were fine-tuned on the labeled tasks to report average accuracy and standard deviation. Each training run took approximately four days. This model is referred to as the Molfomer local HPC trained model (MoLFormer-LHPC).6 Results
This section presents the performance of ChemBERTa and MoLFormer models across a variety of molecular property prediction benchmarks. We first compare the models against established graph-based and fingerprint baselines (Tables 1 and 3), and then analyze how performance scales with pretraining dataset size (Tables 2 and 4). All classification tasks report ROC–AUC (↑ = better) and regression tasks report RMSE (↓ = better). Results are averaged across three runs unless otherwise noted; the “±” symbol indicates the range across runs. Detailed results are provided in Sections 11.4 and 11.5 in Appendix.| Dataset | BACE ↑ | BBBP ↑ | TOX21 ↑ | HIV ↑ | SIDER ↑ | CLINTOX ↑ |
|---|---|---|---|---|---|---|
| Tasks | 1 | 1 | 12 | 1 | 27 | 2 |
| Chemberta-MLM-10M | 0.849 ± 0.014 | 0.956 ± 0.005 | 0.797 ± 0.009 | 0.695 ± 0.018 | 0.611 ± 0.005 | 0.991 ± 0.001 |
| Chemberta-MLM-100M | 0.859 ± 0.009 | 0.961 ± 0.003 | 0.803 ± 0.002 | 0.789 ± 0.004 | 0.618 ± 0.018 | 0.992 ± 0.002 |
| c3-MoLFormer-10M | 0.829 ± 0.003 | 0.899 ± 0.006 | 0.829 ± 0.005 | 0.747 ± 0.019 | 0.617 ± 0.011 | 0.854 ± 0.035 |
| c3-MoLFormer-100M | 0.852 ± 0.013 | 0.899 ± 0.022 | 0.829 ± 0.006 | 0.793 ± 0.005 | 0.625 ± 0.030 | 0.836 ± 0.029 |
| c3-MoLFormer-550M | 0.844 ± 0.015 | 0.915 ± 0.012 | 0.840 ± 0.004 | 0.750 ± 0.062 | 0.610 ± 0.045 | 0.839 ± 0.010 |
| c3-MoLFormer-1.1B | 0.848 ± 0.015 | 0.900 ± 0.015 | 0.830 ± 0.004 | 0.715 ± 0.101 | 0.640 ± 0.008 | 0.846 ± 0.028 |
| MoLFomer (paper) | 0.882 | 0.937 | 0.847 | 0.822 | 0.690 | 0.948 |
6.1 Comparison with baseline models
Tables 1 and 3 compare transformer architectures (ChemBERTa, c3-MoLFormer, and MoLFormer-LHPC) with conventional and graph-based baselines (RF, GCN, DMPNN, Infograph, Infomax3D, Grover) under both the MoLFormer and DeepChem scaffold splits. 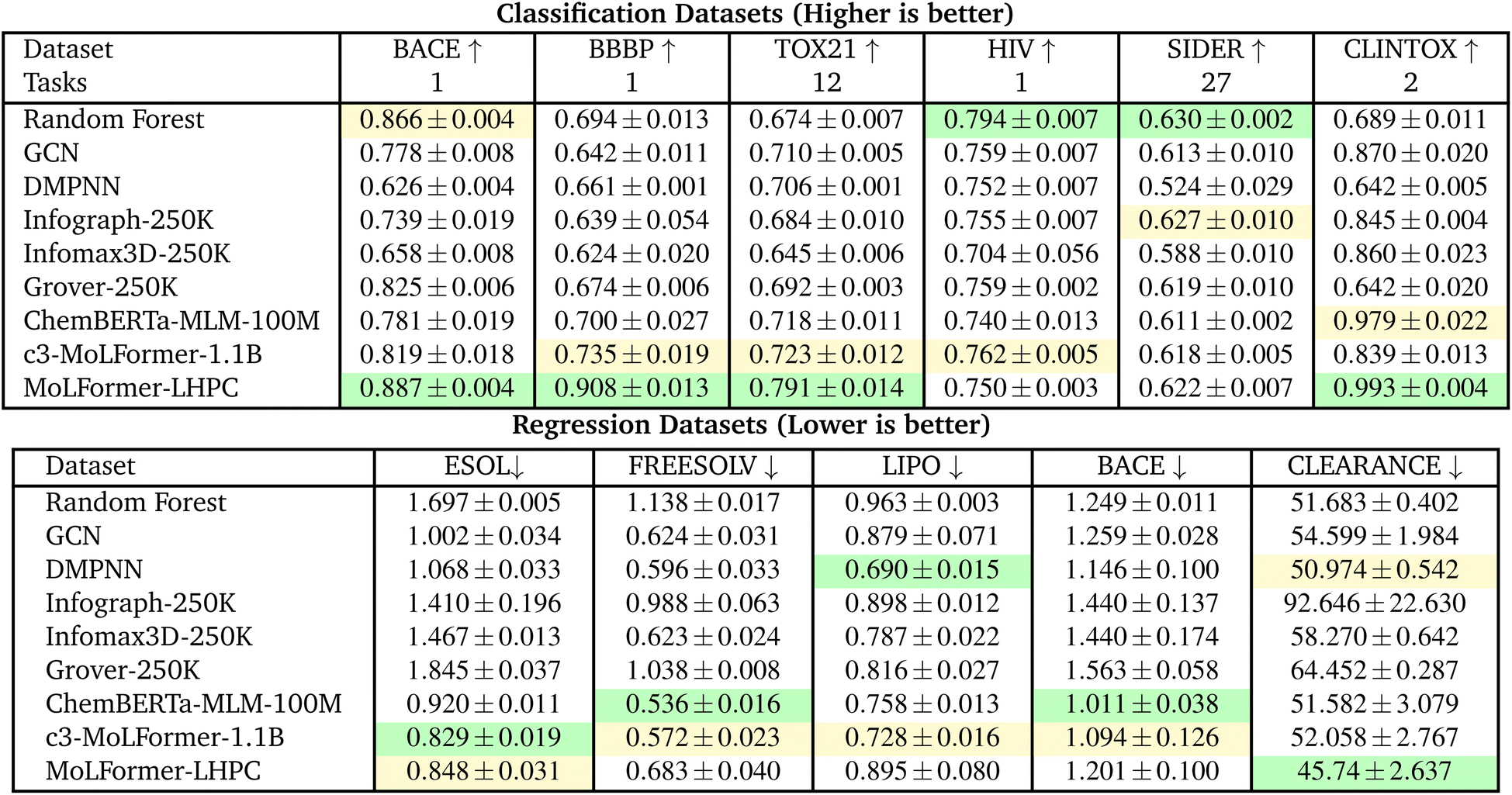 |
• The RF baseline is the top ranked model on three of the six classification tasks, BACE (0.884), HIV (0.803) and SIDER (0.711).
• Chemberta-MLM is top ranked on two classification tasks, BBBP (0.961) and CLINTOX (0.992).
• MoLFormer (reference-only) model is top ranked on only one classification task, TOX21 (0.847), but second ranked on four of the remaining five tasks.
• Surprisingly, c3-MoLFormer-1.1B under performs across all six tasks compared to the published MolFormer results, with second rank on one task only, Tox21 (0.830) and no top rank.
• There are no top ranked graph models and only one second ranked graph model: Grover-250 K, SIDER (0.699).
Under the DeepChem scaffold split (Table 3), absolute AUC values decrease due to stricter scaffold separation, but relative performance remains similar.
• Now, a transformer-based model is the first or second ranked model across all classification tasks except SIDER. With the MoLFormer-LHPC having the top rank in four tasks - BACE (0.887), BBBP (0.908), TOX21 (0.791) and CLINTOX (0.993).
• RF continues strong performance with top rank in the remaining two classification tasks, HIV (0.794) and SIDER (0.630).
• ChemBERTa-MLM-100M remains strong and stable (average 0.76 AUC) compared to graph-based baselines on average.
• On the MoLFormer splits, the MoLFormer (reference-only) model is the top ranked model across the four tasks and the open-source c3-MoLFormer-1.1B model is second ranked across all tasks but with a significantly higher RMSE.
• On the DeepChem splits, transformer models are the top ranked model in 4 of 5 tasks but DMPNN is the top-ranked model for LIPO and second ranked model for CLEARANCE presenting a better baseline than Random Forest for regression.
• For DeepChem splits the top transformer model is mixed, with ChemBERTa-MLM-100M being top ranked in two tasks (FREESOLV, BACE), c3-MoLFormer-1.1B top ranked in one task (ESOL) and second rank in three tasks (FREESOLV, LIPO and BACE) and MoLFormer-LHPC top ranked in one task (CLEARANCE) and second rank in one task (ESOL).
In summary, transformer architectures yield the most accurate and stable results across both scaffold split strategies. Among them, ChemBERTa-MLM-100M delivers high efficiency and strong performance without billion-scale pretraining, while MoLFormer-LHPC achieves the best overall accuracy when computational resources permit.
6.2 Effect of pretraining dataset scale
Tables 2 and 4 investigate how scaling the pretraining corpus influences downstream performance for ChemBERTa and MoLFormer models.| Dataset | BACE ↑ | BBBP ↑ | TOX21 ↑ | HIV ↑ | SIDER ↑ | CLINTOX ↑ |
|---|---|---|---|---|---|---|
| Tasks | 1 | 1 | 12 | 1 | 27 | 2 |
| ChemBERTa-MLM-10M | 0.773 ± 0.010 | 0.715 ± 0.006 | 0.713 ± 0.014 | 0.725 ± 0.017 | 0.616 ± 0.010 | 0.983 ± 0.010 |
| ChemBERTa-MLM-100M | 0.781 ± 0.019 | 0.700 ± 0.027 | 0.718 ± 0.011 | 0.747 ± 0.009 | 0.629 ± 0.023 | 0.979 ± 0.022 |
| c3-MoLFormer-10M | 0.776 ± 0.031 | 0.715 ± 0.021 | 0.718 ± 0.003 | 0.711 ± 0.014 | 0.618 ± 0.005 | 0.847 ± 0.024 |
| c3-MoLFormer-100M | 0.809 ± 0.019 | 0.730 ± 0.016 | 0.729 ± 0.005 | 0.747 ± 0.017 | 0.631 ± 0.009 | 0.854 ± 0.036 |
| c3-MoLFormer-550M | 0.812 ± 0.017 | 0.742 ± 0.020 | 0.726 ± 0.002 | 0.659 ± 0.140 | 0.594 ± 0.007 | 0.856 ± 0.020 |
| c3-MoLFormer-1.1B | 0.819 ± 0.018 | 0.735 ± 0.019 | 0.723 ± 0.012 | 0.762 ± 0.005 | 0.618 ± 0.005 | 0.839 ± 0.013 |
| MoLFormer-LHPC | 0.887 ± 0.004 | 0.908 ± 0.013 | 0.791 ± 0.014 | 0.750 ± 0.003 | 0.622 ± 0.007 | 0.993 ± 0.004 |
• ChemBERTa improves across most datasets (e.g., BACE 0.849 → 0.859, TOX21 0.797 → 0.803, HIV 0.695 → 0.789).
• c3-MoLFormer also benefits up to 100 M (BACE 0.829 → 0.852, HIV 0.747 → 0.793) but performance plateaus or slightly declines at larger scales (550 M–1.1 B).
• ChemBERTa-MLM-100M outperforms its 10M variant on BACE (0.773 → 0.781) and HIV (0.725 → 0.747) though the improvement now is less consistent.
• c3-MoLFormer exhibits improvements up to 100 M (e.g., BACE 0.776 → 0.809) before plateauing.
• The MoLFormer-LHPC generally surpassed all others (e.g., BACE 0.887, BBBP 0.908, CLINTOX 0.993).
7 Discussion
In the following sections, we discuss various takeaways from our experiments.7.1 Impact of dataset splitting on evaluation metrics
The choice of dataset splits is critical when benchmarking models. Scaffold splitting does not necessarily represent a single uniform algorithm; we find in particular that splits used by DeepChem and MoLFormer are not directly comparable. The discrepancy between different scaffold splitting methodologies represents a significant source of complexity and effort in benchmarking. We struggled for several months to reconcile ChemBERTa-3 results with MoLFormer results until we realized that MoLFormer scaffold splits differed from DeepChem's scaffold splits.To better understand how different splits affect evaluation, we compared the Minimum Tanimoto Distance (MTD) distributions produced by DeepChem's scaffold split with those produced by the MoLFormer-provided splits across multiple classification datasets. This analysis shows a consistent trend: DeepChem's scaffold splits lead to higher MTD values, which indicates that test molecules are structurally more dissimilar from the training set. In contrast, the MoLFormer splits produce lower MTD values, which reflects greater scaffold overlap between training, validation, and test sets.
This difference has clear implications for model evaluation. Because the MoLFormer splits contain more structurally similar compounds across partitions, models typically obtain higher ROC AUC scores under this setting. These higher scores likely reflect an easier prediction scenario rather than improved generalization. DeepChem's scaffold splits, in comparison, create a more challenging and often more realistic evaluation condition because models must generalize to compounds with novel scaffolds.
To illustrate this effect, Fig. 7 in the Appendix presents histograms of the MTD distributions for each dataset and split. These visualizations highlight how strongly the choice of splitting algorithm influences the structural separation between splits and therefore the difficulty of the evaluation task.
Additionally, in the original study,5 the MoLFormer model was evaluated on MoLFormer scaffold split, while baseline results were taken from prior literature which were based on different splits. By re-running all models under the same evaluation setup, we find that the performance differences between MoLFormer and baseline models such as GCN and DMPNN are more modest than those originally indicated. Directly comparing results across papers can lead to misleading comparisons, because different scaffold splits can cause large variations in scores. To mitigate these challenges and improve reproducibility, we recommend adopting DeepChem/ChemBERTa-3 as a standardized framework for future benchmarking and model development studies.
7.2 Graph versus transformer pretraining
We investigated various graph based pretraining approaches alongside transformer based pretraining approaches. In general, graph based approaches were considerably harder to scale, possibly due to the lower level of community investment in graph-pretraining infrastructure. For example, Grover featurization took up large amounts of disk space (100 GB for a 1M dataset) which made scaling difficult. Other graph models posed other scaling challenges. For this reason, we only pretrained models on graph based approaches on a 250 K subset of ZINC20. Broadly, graph-based approaches at this scale appeared broadly comparable to transformer-based approaches (see Table 1). Given the engineering challenges of scaling graph-based approaches, we chose to focus on transformer based approaches for larger pretraining scales in this work. However, the strong performance of graph-based approaches at smaller scales suggests that it may be worth investing in further graph-based training infrastructure since these models may exhibit strong performance at large scales comparable to that of transformer architectures.7.3 Scaling challenges
When scaling to larger datasets with a linear learning rate scheduler that includes a warmup phase, configuring the initial learning rate becomes challenging. Setting it too high before the warmup completes often leads to a spike in the loss, causing the model parameters to converge prematurely to a local minimum, as shown in Fig. 3. To address this, we opted to restart the training from the most recent stable checkpoint upon spikes, ensuring smoother convergence. We suggest this strategy as a useful stability trick for future efforts. | ||
| Fig. 3 This graph depicts the loss curve over training iterations, highlighting a sudden spike in loss that occurred during a training run. Such anomalies often arise from high learning rates or data irregularities, particularly when using a linear learning rate scheduler with a warmup phase. To mitigate this issue, checkpointing at regular intervals and restarting from the last stable checkpoint were used to recover from spikes. | ||
7.4 Lack of performance gains with increasing training dataset size
Scaling the pretraining data from 10M to 1.1B SMILES led to marginal improvements in downstream performance, showing diminishing returns. We hypothesize that the current pretraining objective does not fully capture the intrinsic chemical properties required for effective representation learning. More chemically informed objectives, such as those leveraging functional groups or 3D molecular representations, may be beneficial.Moreover, there are other areas within our pretraining setup that could be improved. One aspect may be the lack of SMILES canonicalization, which allows multiple syntactic variants of the same molecule to appear in the corpus. However, our experiments with canonicalization on a limited subset of the pretraining data did not yield any noticeable improvement. Additionally, MLM pretraining tends to favor prediction of frequent atom tokens (e.g., “C”), encouraging shortcut learning rather than chemically meaningful representations.42
7.5 Handling numerical issues in training
During the training process of the model, numerical instability was occasionally encountered, manifesting as NaN values in the loss. This issue was attributed to gradient explosion and sensitivity to the learning rate. To address this, a two-phase training strategy was implemented. Initially, the model was pre-trained on a smaller dataset with a very low learning rate to stabilize the initial weights and establish a robust starting point. Subsequently, training was conducted on the larger dataset with the actual learning rate, leveraging the pre-trained weights. Through this approach, instability was mitigated, convergence was improved, and resource utilization was made more efficient.7.6 Difficulty of hyperparameter optimization
We found that several models were very difficult to tune and required extensive hyperparameter optimization. For example, c3-MoLFormer required considerable tuning before it approached earlier reported MoLFormer results. Given the limitations of our compute budget, it is possible c3-MoLFormer and other models may perform better with additional optimization. We open source all our code and models in the hopes that community feedback and efforts will be able to correct any potential optimization issues despite our best-faith efforts. We list all best hyperparameters in Tables 7–14 and list costs for all model training in Table 6.8 Conclusion
In this paper, we introduced ChemBERTa-3, an open source training framework integrated into DeepChem. The framework supports training of transformer-based and graph-based models. We benchmarked multiple architectures including ChemBERTa, MoLFormer, GROVER, InfoGraph, and InfoMax3D on molecular property prediction tasks from the MoleculeNet dataset. We systematically evaluated several pretraining methodologies to compare their performance across different model types.Additionally, we contributed new tools to DeepChem, including the ModularTorchModel class and the HuggingFaceModel wrapper to improve model pretraining and fine-tuning. Our experiments identified transformer-based architectures as particularly scalable and effective, especially when trained on large-scale datasets. We also observed that scaffold split choices can cause substantial variability in reported performance, making cross-paper comparisons unreliable. To support more consistent and reproducible evaluation, we recommend adopting DeepChem/ChemBERTa-3 as a standardized benchmarking framework.
9 Future work
Due to the rapid growth in the chemical foundation model literature, we have not been able to benchmark every notable chemical foundation model release on the ChemBERTa-3 framework. Notably, ChemFormer and MegaMolBART remain to be added to benchmarks. We hope that our open release will incentivize broader community adoption, especially of our standardized benchmarking pipeline, and also incentivize open source community contributions for new chemical foundation models.10 Ethical statement
While these models are made openly available to advance research and innovation, it is important to acknowledge potential misuse. Specifically, the ability to design molecules with high precision could be exploited to create harmful or dangerous substances. Researchers are encouraged to adhere to ethical guidelines to mitigate such risks.Author contributions
RS – Main contributor on DeepChem implementations for ChemBERTa and MolFormer. Contributor on Grover implementation for DeepChem. Lead contributor to ChemBERTa3 repo. Contributor on Ray infrastructure in ChemBERTa3 repo. Led experimental execution. Contributed to writing and figures. RI – Literature review. Main contributor on writing and editing of manuscript. Design and iteration on figures. AAB – Contributor to Ray infrastructure in ChemBERTa3 repo. Main contributor on DeepChem DMPNN implementation. Contributed to experimental execution. Contributed to writing and figures. CJA – Conducted computational experiments on local HPC platforms. SH – Evaluated training data splits and experimental results. TD – Main contributor on the DeepChem implementations for ModularTorchModel, InfoGraph, Infomax3d, and RdkitConformerFeaturizer. AT – Main contributor to Grover implementation in DeepChem. Contributed to experiments and ray infrastructure in ChemBERTa3. SS – Contributed to HuggingFace infrastructure in DeepChem. Contributed to writing and editing. SC - Contributed to HuggingFace infrastructure in DeepChem. WA - Contributed to experimental design. Main contributor for dataset processing and gathering. DJ - Development of foundation model training and evaluation infrastructure on local HPC. KM – Helped plan experiments and prepared local HPC environment for testing and evaluation. HK - Evaluated experimental results. AB - Contributed to experimental evaluation and design. SVS - Contributed to experimental design. Contributed to writing and editing. VV – Co-designed study and evaluated computational experiments. JEA – Co-designed study, evaluated computational experiments and contributed to writing manuscript. BR – Co-designed study, designed and evaluated computational experiments, designed and evaluated visualizations, led DeepChem architecture design for new models, reviewed all DeepChem and ChemBERTa3 code and designed tests, and contributed to writing manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
All code for ChemBERTa-3 model pretraining and fine-tuning is available on GitHub at https://github.com/deepforestsci/chemberta3, with code, pretraining datasets (public S3 URLs) and fine-tuning datasets archived on Zenodo under DOI: 10.5281/zenodo.18235841. Pretrained MoLFormer and ChemBERTa models are released on Hugging Face through the DeepChem organization (https://huggingface.co/DeepChem).A. Appendices
A.1 Contributions to DeepChem in detail
We added several new pieces of infrastructure to DeepChem to support the ChemBERTa-3 framework and model pre-training.ModularTorchModel. The ModularTorchModel class is designed to simplify the process of building, pretraining, and fine-tuning both transformer and graph-based models. It allows users to define their model components as modular building blocks that can be easily connected to construct complex architectures. Unlike conventional DeepChem models such as TorchModel that compute loss solely from the final output, ModularTorchModel enables loss computation from intermediate network values, offering greater flexibility in optimization. While it integrates with HuggingFace for transformer-based pretraining, ModularTorchModel also fully supports custom graph pretraining implementations.
The existing TorchModel class in DeepChem provides an interface for training PyTorch models using DeepChem datasets. As shown in Fig. 4, the ModularTorchModel class is built upon the TorchModel class to provide flexibility in defining individual model components and their respective losses, which aids in fine-tuning specific components of the model for downstream tasks.
 | ||
| Fig. 4 Schematic representation of the ModularTorchModel framework in DeepChem, demonstrating its workflow for pretraining and fine-tuning tasks. The ModularTorchModel extends the existing TorchModel class. | ||
Fig. 5 shows an example usage that illustrates the build, pre-training, and fine-tuning of a model using ModularTorchModel and its raw code can be found in section 11.7.
 | ||
| Fig. 5 Example code illustrating the build, pre-training, and fine-tuning of a model using ModularTorchModel in DeepChem. | ||
HuggingFace Deepchem Wrapper. The HuggingFaceModel class in DeepChem acts as a wrapper to integrate HuggingFace43 models from the ‘transformers’ library into the DeepChem framework. This allows users to train, predict, and evaluate HuggingFace models using DeepChem's API, enabling direct comparisons between models from the two ecosystems. The wrapper also has a ‘tokenizer’ which tokenizes raw SMILES strings into tokens to be used by downstream models, leveraging the efficient tokenization and data handling utilities from the ‘transformers’ library, such as random masking of tokens for masked language model training.
RDKitConformer Featurizer. The RDKitConformerFeaturizer was added to DeepChem to generate 3D molecular representations for use in the InfoMax3DModular model. The conformer featurizer is an adaptation from RDKit,44 which featurizes an RDKit mol object as a GraphData object with 3D coordinates. The ETKDGv245 algorithm is used to generate 3D coordinates for the molecule. It is a conformation generation methodology that combines experimental torsion-angle preferences with knowledge-based terms and distance geometry to generate accurate 3D molecular structures.
ChemBERTa.3 The original ChemBERTa model was pretrained using Masked Language Modeling (MLM). ChemBERTa-2 extended this approach by adding multitask regression (MTR) pretraining on a larger dataset. At its core, Chemberta uses a byte-pair encoding (BPE) tokenizer, trained on the PubChem10M dataset using 60 k tokens.
For MTR, the RDKitDescriptorFeaturizer in DeepChem was used to compute a set of 200 molecular properties for each compound in our training dataset. Because these tasks have very different scales and ranges, the labels are mean-normalized for each task before training.
While ChemBERTa and ChemBERTa-2 primarily released pretrained models via HuggingFace, ChemBERTa-3 (discussed in detail in section 3.2) also has a released standalone github repository, fully integrated into the DeepChem ecosystem, that allows researchers to easily pretrain and fine-tune models themselves. ChemBERTa-3 also releases trained models on HuggingFace for ease of access.
InfoGraph.22 The DeepChem implementation of InfographModel learns graph representations through unsupervised contrastive learning by maximizing the mutual information between global graph embeddings and substructure embeddings. It is built upon the ModularTorchModel class to facilitate transfer learning. The model randomly samples pairs of graphs and substructures, and then maximizes their mutual information by minimizing their distance in a learned embedding space. The model can be used for downstream tasks such as graph classification and molecular property prediction. It utilizes the MolGraphConvFeaturizer in DeepChem for data preprocessing, and the pre-trained model can be fine-tuned on both regression and classification datasets.
GROVER.27 GROVER implementation in DeepChem utilizes the newly introduced GroverFeaturizer, which processes molecules from SMILES strings or RDKit objects to generate a molecular graph for message passing, functional group features for pretraining, and additional features for fine-tuning. Users can also specify an additional featurizer to extract extra molecular properties, enhancing transfer learning capabilities. As a ModularTorchModel, GROVERModel supports flexible fine-tuning and transfer learning.
Infomax3d.23InfoMax3DModular, implemented in DeepChem, is a ModularTorchModel that uses a 2D model (PNA) and a 3D model (Net3D) to maximize the mutual information between their representations, enabling the 2D model to be used for downstream tasks without requiring 3D coordinates. As mentioned before, it utilizes the RDKitConformerFeaturizer, which converts RDKit molecular structures into GraphData objects with 3D coordinates stored in the node_pos_features attribute. The ETKDGv246 algorithm is employed to generate these 3D conformers.
In our benchmark, we use InfoMax3DModular to compare the impact of incorporating 3D structural data versus relying solely on 2D representations, helping us evaluate the significance of 3D information in molecular property prediction tasks.
MoLFormer.5 The DeepChem implementation of MolFormer uses the HuggingFace DeepChem Wrapper to wrap the ‘ibm/MoLFormer-XL-both-10pct‘ pre-trained model readily available in the HuggingFace transformers library. It uses the ‘ibm/MoLFormer-XL-both-10pct‘ tokenizer.
DMPNN.21 DMPNN (Directed Message Passing Neural Network) implementation of DeepChem consists of a message-passing phase, where an encoder updates atom hidden states based on neighbor information, and a read-out phase, where a feed-forward network predicts molecular properties. The DMPNNFeaturizer in DeepChem extracts rich molecular representations for the DMPNN by encoding both atoms (nodes) and bonds (edges). Atom features (length 133) include properties like atomic number, degree, charge, chirality, hybridization, and aromaticity, while bond features (length 14) capture bond type, ring membership, conjugation, and stereo configuration. The DMPNN implementation in DeepChem is based on the Chemprop library but is adapted to interoperate with the DeepChem ecosystem.21
Fig. 6 illustrates overall architectures of each model. Table 5 provides a comparison of model architectures, feature/tokenization strategies, types of featurization, pretraining methods, and the corresponding DeepChem class implementations used in this study.
 | ||
| Fig. 6 The figure compares the key components and workflows of model architectures evaluated in this study: MoLFormer, ChemBERTa, InfoGraph, InfoMax3D, DMPNN and GROVER. | ||
| Models | Featurizer/Tokenizers | Featurization | Pretraining method | DeepChem classname |
|---|---|---|---|---|
| ChemBERTa-MLM | Roberta Tokenizer | SMILES | Masked language model |  |
| ChemBERTa-MTR | RDKitDescriptors | SMILES | Multi-task pretraining |  |
| Infograph | MolGraphConv | Graph | Mutual information |  |
| Maximization | ||||
| Grover | GroverFeaturizer | Graph | Self-supervised message |  |
| Passing transformer | ||||
| Infomax3D | RDKitConformer | Graph | Mutual information |  |
| Maximization | ||||
| MoLFormer | MoLFormer Tokenizer | SMILES | Masked language model |  |
| DMPNN | DMPNNFeaturizer | Graph | — |  |
A.2. Costs of model pre-training and benchmarking
Training and benchmarking large-scale model architectures, is both computationally expensive and time-consuming. These costs present a significant challenge to reproducibility, as rerunning experiments or comparing models under consistent settings requires substantial GPU resources. Even after pretraining, fine-tuning and evaluating across multiple tasks can be resource-intensive, making it difficult to perform large-scale, systematic benchmarks. Table 6 summarizes the costs of pretraining MolFormer models using AWS T4 GPU instances. Benchmarking took around 287 GPU hours. We have used T4 (1 GPU, g4dn.2xlarge) spot instance for benchmarking.| Models | Cost ($) | Date trained | Time taken (hours) | AWS region | Instance Configuration | Instance type |
|---|---|---|---|---|---|---|
| MoLFormer 1.1B | 4000 | 2025-01-16 | 260 | Us-east-2 | g4dn.12xlarge | Spot |
| MoLFormer 550M | 2400 | 2024-12-02 | 150 | Us-east-2 | g4dn.12xlarge | Spot |
| MoLFormer 250M | 1000 | 2024-11-18 | 70 | Us-east-2 | g4dn.12xlarge | Spot |
| Benchmarking | 150 | 2025-04-01 | 200 | Us-east-2 | g4dn.2xlarge | Dedicated |
A.3. Additional hyperparameter tuning details
Grid hyperparameter search is employed to tune pre-trained models on fine-tuning datasets. We use DeepChem's ‘GridHyperparamOpt‘ class, which performs an exhaustive search over a specified hyperparameter space. This approach iteratively evaluates all parameter combinations without parallelization, allowing flexible optimization of selected parameters.A hyperparameter sweep was performed for the fine-tuning strategy using limited grid search where we randomly picked certain number of variations for each task. The search space for each of the models are listed in the Tables 7–14. The best model with the lowest validation loss was picked for further analysis.
| Model | Dataset | Dataset type | No. of tasks | Best parameters | Score (RMSE/ROC AUC) | ||
|---|---|---|---|---|---|---|---|
| Learning rate | Batch size | Epochs | |||||
| c3-MolFormer | BACE | Class | 1 | 3.00 × 10−5 | 32 | 100 | 0.848 ± 0.015 |
| BBBP | Class | 1 | 3.00 × 10−5 | 32 | 150 | 0.900 ± 0.015 | |
| TOX21 | Class | 12 | 3.00 × 10−5 | 32 | 50 | 0.830 ± 0.004 | |
| HIV | Class | 1 | 3.00 × 10−5 | 32 | 50 | 0.715 ± 0.101 | |
| SIDER | Class | 27 | 1.00 × 10−6 | 16 | 213 | 0.640 ± 0.008 | |
| CLINTOX | Class | 2 | 2.00 × 10−5 | 32 | 100 | 0.846 ± 0.028 | |
| ESOL | Reg | 1 | 3.00 × 10−5 | 128 | 200 | 0.651 ± 0.034 | |
| FREESOLV | Reg | 1 | 3.00 × 10−5 | 128 | 150 | 1.052 ± 0.026 | |
| LIPO | Reg | 1 | 3.00 × 10−5 | 32 | 150 | 0.556 ± 0.004 | |
| Model | Dataset | Dataset type | No. of tasks | Best parameters | Score | |||
|---|---|---|---|---|---|---|---|---|
| Batch size | ffn_dropout_p | enc_dropout_p | Epochs | |||||
| DMPNN | BACE | Class | 1 | 128 | 0.2 | 0.2 | 100 | 0.878 ± 0.001 |
| BBBP | Class | 1 | 128 | 0.2 | 0.2 | 100 | 0.930 ± 0.002 | |
| TOX21 | Class | 12 | 128 | 0.2 | 0.2 | 100 | 0.824 ± 0.002 | |
| HIV | Class | 1 | 128 | 0.2 | 0.2 | 100 | 0.812 ± 0.020 | |
| SIDER | Class | 27 | 128 | 0.2 | 0.2 | 100 | 0.633 ± 0.009 | |
| CLINTOX | Class | 2 | 128 | 0.2 | 0.2 | 100 | 0.890 ± 0.001 | |
| ESOL | Reg | 1 | 64 | 0.2 | 0.2 | 100 | 0.699 ± 0.022 | |
| FREESOLV | Reg | 1 | 128 | 0.2 | 0.2 | 100 | 1.229 ± 0.044 | |
| LIPO | Reg | 1 | 128 | 0.2 | 0.2 | 100 | 0.577 ± 0.017 | |
| Model | Dataset | Dataset type | No. of tasks | Best parameters | Score (RMSE/ROC AUC) | |||
|---|---|---|---|---|---|---|---|---|
| Learning rate | Batch size | num_gc_layers | Epochs | |||||
| Infograph | BACE | Class | 1 | 0.001 | 128 | 4 | 100 | 0.840 ± 0.010 |
| BBBP | Class | 1 | 0.001 | 128 | 4 | 100 | 0.898 ± 0.013 | |
| TOX21 | Class | 12 | 0.001 | 128 | 4 | 100 | 0.793 ± 0.007 | |
| HIV | Class | 1 | 0.001 | 128 | 4 | 100 | 0.785 ± 0.001 | |
| SIDER | Class | 27 | 0.001 | 128 | 4 | 100 | 0.652 ± 0.016 | |
| CLINTOX | Class | 2 | 0.001 | 128 | 4 | 100 | 0.785 ± 0.044 | |
| ESOL | Reg | 1 | 0.001 | 128 | 4 | 100 | 0.792 ± 0.044 | |
| FREESOLV | Reg | 1 | 0.001 | 128 | 4 | 100 | 1.757 ± 0.363 | |
| LIPO | Reg | 1 | 0.001 | 128 | 4 | 100 | 0.697 ± 0.011 | |
| Model | Dataset | Dataset type | No. of tasks | Best parameters | Score (RMSE/ROC AUC) | ||
|---|---|---|---|---|---|---|---|
| Learning rate | Batch size | Epochs | |||||
| ChemBerta | BACE | Class | 1 | 3.00 × 10−5 | 32 | 100 | 0.859 ± 0.009 |
| BBBP | Class | 1 | 3.00 × 10−5 | 32 | 100 | 0.961 ± 0.003 | |
| TOX21 | Class | 12 | 3.00 × 10−5 | 32 | 100 | 0.803 ± 0.002 | |
| HIV | Class | 1 | 3.00 × 10−5 | 16 | 50 | 0.789 ± 0.004 | |
| SIDER | Class | 27 | 3.00 × 10−5 | 16 | 50 | 0.618 ± 0.018 | |
| CLINTOX | Class | 2 | 3.00 × 10−5 | 32 | 100 | 0.992 ± 0.002 | |
| ESOL | Reg | 1 | 3.00 × 10−5 | 32 | 100 | 0.682 ± 0.089 | |
| FREESOLV | Reg | 1 | 3.00 × 10−5 | 128 | 100 | 1.399 ± 0.051 | |
| LIPO | Reg | 1 | 3.00 × 10−5 | 128 | 100 | 0.615 ± 0.007 | |
| Model | Dataset | Dataset type | No. of tasks | Best parameters | Score | ||||
|---|---|---|---|---|---|---|---|---|---|
| Learning rate | Batch size | hidden_dim | target_dim | Epochs | |||||
| Infomax3D | BACE | Class | 1 | 0.001 | 64 | 64 | 10 | 100 | 0.787 ± 0.033 |
| BBBP | Class | 1 | 0.001 | 64 | 64 | 10 | 100 | 0.904 ± 0.012 | |
| TOX21 | Class | 12 | 0.001 | 64 | 64 | 10 | 100 | 0.781 ± 0.003 | |
| HIV | Class | 1 | 0.001 | 128 | 64 | 10 | 50 | 0.680 ± 0.023 | |
| SIDER | Class | 27 | 0.001 | 64 | 64 | 10 | 100 | 0.575 ± 0.005 | |
| CLINTOX | Class | 2 | 0.001 | 64 | 64 | 10 | 100 | 0.906 ± 0.006 | |
| ESOL | Reg | 1 | 0.001 | 32 | 64 | 10 | 500 | 0.767 ± 0.057 | |
| FREESOLV | Reg | 1 | 0.001 | 32 | 64 | 10 | 500 | 1.353 ± 0.041 | |
| LIPO | Reg | 1 | 0.001 | 32 | 64 | 10 | 500 | 0.569 ± 0.012 | |
| Model | Dataset | Dataset type | No. of tasks | Best parameters | Score(RMSE/ROC AUC) | ||
|---|---|---|---|---|---|---|---|
| Hidden size | Batch size | Epochs | |||||
| GROVER | BACE | Class | 1 | 128 | 128 | 100 | 0.652 ± 0.321 |
| BBBP | Class | 1 | 128 | 128 | 500 | 0.710 ± 0.322 | |
| TOX21 | Class | 12 | 128 | 100 | 100 | 0.789 ± 0.001 | |
| HIV | Class | 1 | 128 | 128 | 100 | 0.678 ± 0.243 | |
| SIDER | Class | 27 | 128 | 100 | 500 | 0.699 ± 0.007 | |
| CLINTOX | Class | 2 | 128 | 100 | 500 | 0.882 ± 0.013 | |
| ESOL | Reg | 1 | 128 | 128 | 100 | 3.761 ± 0.079 | |
| FREESOLV | Reg | 1 | 128 | 128 | 100 | 5.383 ± 0.028 | |
| LIPO | Reg | 1 | 128 | 128 | 500 | 1.082 ± 0.073 | |
| Model | Dataset | Dataset type | No. of tasks | Best parameters | Score (RMSE/ROC AUC) | ||
|---|---|---|---|---|---|---|---|
| Hidden size | Batch size | Epochs | |||||
| GCN | BACE | Class | 1 | 128 | 128 | 100 | 0.824 ± 0.004 |
| BBBP | Class | 1 | 128 | 128 | 100 | 0.898 ± 0.005 | |
| TOX21 | Class | 12 | 128 | 128 | 100 | 0.810 ± 0.004 | |
| HIV | Class | 1 | 128 | 128 | 100 | 0.768 ± 0.013 | |
| SIDER | Class | 27 | 128 | 128 | 100 | 0.603 ± 0.012 | |
| CLINTOX | Class | 2 | 128 | 128 | 100 | 0.838 ± 0.068 | |
| ESOL | Reg | 1 | 128 | 128 | 500 | 1.219 ± 0.094 | |
| FREESOLV | Reg | 1 | 128 | 128 | 500 | 4.368 ± 0.269 | |
| LIPO | Reg | 1 | 128 | 128 | 100 | 0.735 ± 0.005 | |
| Model | Dataset | Dataset Type | No.of tasks | Best parameters | Score (RMSE/ROC AUC) | |||
|---|---|---|---|---|---|---|---|---|
| n-estimators | min samples split | Criterion | Bootstrap | |||||
| RF | BACE | Class | 1 | 100 | 20 | Gini | True | 0.884 ± 0.004 |
| BBBP | Class | 1 | 100 | 20 | Gini | True | 0.926 ± 0.002 | |
| TOX21 | Class | 12 | 100 | 32 | Gini | False | 0.803 ± 0.004 | |
| HIV | Class | 1 | 100 | 20 | Gini | True | 0.829 ± 0.009 | |
| SIDER | Class | 27 | 100 | 32 | Gini | False | 0.711 ± 0.004 | |
| CLINTOX | Class | 2 | 100 | 16 | Entropy | False | 0.916 ± 0.011 | |
| ESOL | Reg | 1 | 100 | 2 | squared_error | True | 1.154 ± 0.008 | |
| FREESOLV | Reg | 1 | 100 | 2 | squared_error | True | 2.209 ± 0.028 | |
| LIPO | Reg | 1 | 100 | 2 | squared_error | True | 0.722 ± 0.001 | |
A.4. Expanded Benchmark Results for molformer scaffold splits
Tables 15–17 contain expanded benchmark results for molformer scaffold splits.| Classification datasets (higher is better) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Dataset tasks | BACE 1 ↑ | BBBP 1 ↑ | TOX21 12 ↑ | ||||||
| Run1 | Run2 | Run3 | Run1 | Run2 | Run3 | Run1 | Run2 | Run3 | |
| Random forest | 0.889 | 0.880 | 0.883 | 0.923 | 0.929 | 0.926 | 0.801 | 0.808 | 0.799 |
| GCN | 0.820 | 0.821 | 0.830 | 0.903 | 0.891 | 0.899 | 0.810 | 0.815 | 0.806 |
| DMPNN | 0.877 | 0.879 | 0.877 | 0.927 | 0.931 | 0.932 | 0.827 | 0.825 | 0.821 |
| Infograph-250K | 0.826 | 0.847 | 0.847 | 0.906 | 0.879 | 0.908 | 0.787 | 0.802 | 0.788 |
| Infomax3D-250K | 0.815 | 0.806 | 0.741 | 0.910 | 0.887 | 0.916 | 0.777 | 0.784 | 0.782 |
| Grover-250K | 0.883 | 0.880 | 0.878 | 0.937 | 0.936 | 0.939 | 0.788 | 0.791 | 0.788 |
| Chemberta-MLM-100M | 0.847 | 0.861 | 0.869 | 0.957 | 0.965 | 0.960 | 0.805 | 0.802 | 0.802 |
| c3-MoLFormer-1.1B | 0.869 | 0.833 | 0.843 | 0.917 | 0.902 | 0.881 | 0.831 | 0.824 | 0.835 |
| Classification datasets (higher is better) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Dataset tasks | HIV 1 ↑ | SIDER 27 ↑ | CLINTOX 2 ↑ | ||||||
| Run1 | Run2 | Run3 | Run1 | Run2 | Run3 | Run1 | Run2 | Run3 | |
| Random forest | 0.839 | 0.832 | 0.817 | 0.712 | 0.706 | 0.716 | 0.925 | 0.901 | 0.924 |
| GCN | 0.766 | 0.754 | 0.786 | 0.588 | 0.618 | 0.602 | 0.746 | 0.863 | 0.907 |
| DMPNN | 0.812 | 0.836 | 0.787 | 0.621 | 0.643 | 0.635 | 0.890 | 0.889 | 0.892 |
| Infograph-250K | 0.784 | 0.784 | 0.787 | 0.634 | 0.673 | 0.651 | 0.839 | 0.730 | 0.786 |
| Infomax3D-250K | 0.707 | 0.684 | 0.650 | 0.573 | 0.570 | 0.582 | 0.905 | 0.899 | 0.914 |
| Grover-250K | 0.849 | 0.851 | 0.852 | 0.690 | 0.707 | 0.699 | 0.883 | 0.865 | 0.897 |
| Chemberta-MLM-100M | 0.794 | 0.785 | 0.789 | 0.626 | 0.594 | 0.636 | 0.995 | 0.989 | 0.992 |
| c3-MoLFormer-1.1B | 0.573 | 0.773 | 0.799 | 0.629 | 0.640 | 0.650 | 0.824 | 0.828 | 0.886 |
| Regression datasets (lower is better) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Dataset tasks | ESOL 1 ↓ | FREESOLV 1 ↓ | LIPO 1 ↓ | ||||||
| Run1 | Run2 | Run3 | Run1 | Run2 | Run3 | Run1 | Run2 | Run3 | |
| Random forest | 1.165 | 1.147 | 1.149 | 2.247 | 2.183 | 2.196 | 0.721 | 0.719 | 0.723 |
| GCN | 1.103 | 1.221 | 1.333 | 4.463 | 4.000 | 4.641 | 0.741 | 0.736 | 0.729 |
| DMPNN | 0.669 | 0.707 | 0.721 | 1.213 | 1.184 | 1.289 | 0.568 | 0.601 | 0.562 |
| Infograph-250K | 0.767 | 0.756 | 0.855 | 1.766 | 1.308 | 2.198 | 0.692 | 0.688 | 0.712 |
| Infomax3D-250K | 0.847 | 0.725 | 0.728 | 1.304 | 1.404 | 1.351 | 0.552 | 0.574 | 0.581 |
| Grover-250K | 3.690 | 3.871 | 3.723 | 5.391 | 5.346 | 5.410 | 1.025 | 1.184 | 1.036 |
| Chemberta-MLM-100M | 0.610 | 0.808 | 0.628 | 1.364 | 1.472 | 1.363 | 0.614 | 0.606 | 0.625 |
| c3-MoLFormer-1.1B | 0.699 | 0.622 | 0.632 | 1.075 | 1.065 | 1.015 | 0.555 | 0.553 | 0.561 |
| Dataset tasks | BACE 1 ↑ | BBBP 1 ↑ | TOX21 12 ↑ | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Run1 | Run2 | Run3 | Run1 | Run2 | Run3 | Run1 | Run2 | Run3 | |
| Chemberta-MLM-10M | 0.869 | 0.839 | 0.841 | 0.963 | 0.952 | 0.954 | 0.806 | 0.785 | 0.800 |
| Chemberta-MLM-100M | 0.847 | 0.861 | 0.869 | 0.957 | 0.965 | 0.960 | 0.805 | 0.802 | 0.802 |
| c3-MoLFormer-10M | 0.829 | 0.824 | 0.832 | 0.907 | 0.892 | 0.899 | 0.829 | 0.824 | 0.836 |
| c3-MoLFormer-100M | 0.835 | 0.856 | 0.865 | 0.915 | 0.916 | 0.868 | 0.832 | 0.835 | 0.820 |
| c3-MoLFormer-550M | 0.843 | 0.826 | 0.863 | 0.899 | 0.927 | 0.918 | 0.838 | 0.838 | 0.846 |
| c3-MoLFormer-1.1B | 0.869 | 0.833 | 0.843 | 0.917 | 0.902 | 0.881 | 0.831 | 0.824 | 0.835 |
| Dataset tasks | HIV 1 ↑ | SIDER 27 ↑ | CLINTOX 2 ↑ | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Run1 | Run2 | Run3 | Run1 | Run2 | Run3 | Run1 | Run2 | Run3 | |
| Chemberta-MLM-10M | 0.719 | 0.690 | 0.676 | 0.607 | 0.607 | 0.618 | 0.991 | 0.989 | 0.992 |
| Chemberta-MLM-100M | 0.794 | 0.785 | 0.789 | 0.626 | 0.594 | 0.636 | 0.995 | 0.989 | 0.992 |
| c3-MoLFormer-10M | 0.754 | 0.766 | 0.721 | 0.605 | 0.632 | 0.613 | 0.807 | 0.868 | 0.889 |
| c3-MoLFormer-100M | 0.796 | 0.786 | 0.797 | 0.611 | 0.597 | 0.667 | 0.876 | 0.823 | 0.810 |
| c3-MoLFormer-550M | 0.663 | 0.782 | 0.805 | 0.642 | 0.643 | 0.546 | 0.826 | 0.845 | 0.849 |
| c3-MoLFormer-1.1B | 0.573 | 0.773 | 0.799 | 0.629 | 0.640 | 0.650 | 0.824 | 0.828 | 0.886 |
| Measure | MoLForm (paper) 1.1B | c3-MoLForm 1.1B | GCN | Infograph 250K | DMPNN | Infomax3D 250K | Grover250K | Chemberta 100M | RF |
|---|---|---|---|---|---|---|---|---|---|
| Alpha | 0.3327 | 0.6776 | 3.9494 | 0.6734 | 1.9431 | 2.4708 | 66.4236 | 1.2493 | 3.9187 |
| cv | 0.1447 | 0.6872 | 2.1584 | 0.5082 | 0.8730 | 0.9473 | 40.9454 | 0.3762 | 1.5656 |
| G | 0.3362 | 7.1345 | 17.1677 | 3.3537 | 11.3968 | 13.1874 | 506.3905 | 3.8939 | 18.4361 |
| Gap | 0.0038 | 0.0070 | 0.0125 | 0.0112 | 0.0066 | 0.0072 | 0.0829 | 0.00959 | 0.0079 |
| H | 0.2522 | 0.3900 | 256.2621 | 4.4229 | 11.1296 | 13.8275 | 453.8291 | 3.8955 | 18.3009 |
| Homo | 0.0029 | 0.0039 | 0.0085 | 0.0096 | 0.0048 | 0.0040 | 0.0101 | 0.0075 | 0.0066 |
| Lumo | 0.0027 | 0.0057 | 0.0086 | 0.0138 | 0.0047 | 0.0047 | 0.0898 | 0.00649 | 0.0071 |
| Mu | 0.3616 | 0.6231 | 0.5910 | 0.5950 | 0.5079 | 0.4893 | 0.8779 | 0.7633 | 0.5595 |
| r2 | 17.0620 | 26.2624 | 177.5807 | 83.6650 | 64.7802 | 81.4212 | 1210.7322 | 355.1189 | 98.4651 |
| u0 | 0.3211 | 5.1925 | 25.2800 | 9.1437 | 6.1491 | 13.6337 | 605.3215 | 54.9064 | 18.3840 |
| U | 0.2522 | 7.0372 | 28.764 | 6.3261 | 10.4231 | 13.3267 | 213.4233 | 2.9415 | 18.2680 |
| ZPVE | 0.0003 | 0.0017 | 0.0053 | 0.0048 | 0.0036 | 0.0028 | 0.2906 | 0.004 | 0.0100 |
A.5. Expanded Benchmark Results for DeepChem scaffold splits
Tables 18 and 19 contain expanded benchmark results for DeepChem scaffold splits.| Classification datasets (higher is better) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Dataset tasks | BACE 1 ↑ | BBBP 1 ↑ | TOX21 12 ↑ | ||||||
| Run1 | Run2 | Run3 | Run1 | Run2 | Run3 | Run1 | Run2 | Run3 | |
| ChemBERTa-MLM-10M | 0.763 | 0.770 | 0.787 | 0.709 | 0.724 | 0.712 | 0.733 | 0.704 | 0.703 |
| ChemBERTa-MLM-100M | 0.803 | 0.756 | 0.784 | 0.663 | 0.728 | 0.709 | 0.704 | 0.721 | 0.730 |
| c3-MoLFormer-10M | 0.738 | 0.778 | 0.814 | 0.701 | 0.744 | 0.699 | 0.722 | 0.716 | 0.715 |
| c3-MoLFormer-100M | 0.832 | 0.785 | 0.813 | 0.750 | 0.729 | 0.711 | 0.725 | 0.736 | 0.728 |
| c3-MoLFormer-550M | 0.791 | 0.812 | 0.832 | 0.769 | 0.721 | 0.739 | 0.727 | 0.729 | 0.723 |
| c3-MoLFormer-1.1B | 0.821 | 0.839 | 0.796 | 0.739 | 0.709 | 0.756 | 0.729 | 0.707 | 0.735 |
| Classification datasets (higher is better) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Dataset tasks | HIV 1 ↑ | SIDER 27 ↑ | CLINTOX 2 ↑ | ||||||
| Run1 | Run2 | Run3 | Run1 | Run2 | Run3 | Run1 | Run2 | Run3 | |
| ChemBERTa-MLM-10M | 0.713 | 0.713 | 0.749 | 0.617 | 0.626 | 0.603 | 0.970 | 0.993 | 0.986 |
| ChemBERTa-MLM-100M | 0.757 | 0.736 | 0.748 | 0.612 | 0.613 | 0.662 | 0.948 | 0.992 | 0.996 |
| c3-MoLFormer-10M | 0.717 | 0.692 | 0.725 | 0.623 | 0.611 | 0.621 | 0.880 | 0.824 | 0.837 |
| c3-MoLFormer-100M | 0.771 | 0.731 | 0.741 | 0.635 | 0.639 | 0.618 | 0.806 | 0.895 | 0.860 |
| c3-MoLFormer-550M | 0.742 | 0.748 | 0.461 | 0.603 | 0.592 | 0.588 | 0.835 | 0.853 | 0.882 |
| c3-MoLFormer-1.1B | 0.756 | 0.761 | 0.768 | 0.621 | 0.622 | 0.611 | 0.854 | 0.841 | 0.823 |
| Classification datasets (higher is better) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Dataset tasks | BACE 1 ↑ | BBBP 1 ↑ | TOX21 12 ↑ | ||||||
| Run1 | Run2 | Run3 | Run1 | Run2 | Run3 | Run1 | Run2 | Run3 | |
| Random forest | 0.870 | 0.866 | 0.861 | 0.702 | 0.676 | 0.704 | 0.675 | 0.666 | 0.682 |
| GCN | 0.789 | 0.773 | 0.771 | 0.639 | 0.657 | 0.630 | 0.710 | 0.716 | 0.704 |
| DMPNN | 0.627 | 0.631 | 0.621 | 0.661 | 0.661 | 0.663 | 0.705 | 0.706 | 0.706 |
| Infograph-250K | 0.739 | 0.762 | 0.716 | 0.705 | 0.572 | 0.640 | 0.692 | 0.669 | 0.691 |
| Infomax3D-250K | 0.648 | 0.666 | 0.660 | 0.653 | 0.612 | 0.608 | 0.653 | 0.645 | 0.639 |
| Grover-250K | 0.817 | 0.825 | 0.833 | 0.681 | 0.668 | 0.671 | 0.688 | 0.696 | 0.692 |
| ChemBERTa-MLM-100M | 0.803 | 0.756 | 0.784 | 0.663 | 0.728 | 0.709 | 0.704 | 0.721 | 0.730 |
| c3-MoLFormer-1.1B | 0.821 | 0.839 | 0.796 | 0.739 | 0.709 | 0.756 | 0.729 | 0.707 | 0.735 |
| MoLFormer-LHPC | 0.891 | 0.881 | 0.888 | 0.889 | 0.914 | 0.919 | 0.771 | 0.800 | 0.800 |
| Classification datasets (higher is better) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Dataset tasks | HIV 1 ↑ | SIDER 27 ↑ | CLINTOX 2 ↑ | ||||||
| Run1 | Run2 | Run3 | Run1 | Run2 | Run3 | Run1 | Run2 | Run3 | |
| Random forest | 0.803 | 0.793 | 0.785 | 0.632 | 0.628 | 0.631 | 0.699 | 0.674 | 0.694 |
| GCN | 0.752 | 0.756 | 0.769 | 0.617 | 0.623 | 0.600 | 0.866 | 0.896 | 0.848 |
| DMPNN | 0.759 | 0.742 | 0.756 | 0.539 | 0.549 | 0.484 | 0.648 | 0.643 | 0.635 |
| Infograph-250K | 0.746 | 0.763 | 0.755 | 0.614 | 0.639 | 0.628 | 0.839 | 0.849 | 0.846 |
| Infomax3D-250K | 0.742 | 0.746 | 0.625 | 0.596 | 0.573 | 0.594 | 0.892 | 0.848 | 0.841 |
| Grover-250K | 0.757 | 0.761 | 0.761 | 0.616 | 0.632 | 0.609 | 0.631 | 0.624 | 0.670 |
| ChemBERTa-MLM-100M | 0.758 | 0.736 | 0.727 | 0.612 | 0.613 | 0.609 | 0.948 | 0.992 | 0.996 |
| c3-MoLFormer-1.1B | 0.756 | 0.761 | 0.768 | 0.621 | 0.622 | 0.611 | 0.854 | 0.841 | 0.823 |
| MoLFormer-LHPC | 0.746 | 0.753 | 0.751 | 0.629 | 0.612 | 0.623 | 0.987 | 0.993 | 0.997 |
| Regression datasets (lower is better) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Dataset tasks | ESOL 1 ↓ | FREESOLV 1 ↓ | LIPO 1 ↓ | ||||||
| Run1 | Run2 | Run3 | Run1 | Run2 | Run3 | Run1 | Run2 | Run3 | |
| Random forest | 1.705 | 1.692 | 1.695 | 1.119 | 1.134 | 1.159 | 0.965 | 0.959 | 0.965 |
| GCN | 0.954 | 1.015 | 1.035 | 0.662 | 0.586 | 0.623 | 0.806 | 0.856 | 0.975 |
| DMPNN | 1.069 | 1.107 | 1.026 | 0.570 | 0.576 | 0.643 | 0.682 | 0.678 | 0.711 |
| Infograph-250K | 1.193 | 1.668 | 1.369 | 0.919 | 1.071 | 0.972 | 0.887 | 0.916 | 0.891 |
| Infomax3D-250K | 1.462 | 1.485 | 1.455 | 0.615 | 0.597 | 0.655 | 0.757 | 0.792 | 0.811 |
| Grover-250K | 1.796 | 1.885 | 1.853 | 1.049 | 1.038 | 1.028 | 0.798 | 0.796 | 0.855 |
| ChemBERTa-MLM-100M | 0.905 | 0.924 | 0.932 | 0.542 | 0.551 | 0.514 | 0.760 | 0.772 | 0.742 |
| c3-MoLFormer-1.1B | 0.439 | 0.446 | 0.392 | 0.552 | 0.559 | 0.604 | 0.594 | 0.587 | 0.597 |
| MoLFormer-LHPC | 0.804 | 0.871 | 0.869 | 0.627 | 0.700 | 0.720 | 0.967 | 0.783 | 0.933 |
| Regression datasets (lower is better) | ||||||
|---|---|---|---|---|---|---|
| Dataset tasks | BACE 1 ↓ | CLEARANCE 1 ↓ | ||||
| Run1 | Run2 | Run3 | Run1 | Run2 | Run3 | |
| Random forest | 1.234 | 1.251 | 1.262 | 51.123 | 51.876 | 52.049 |
| GCN | 1.225 | 1.295 | 1.256 | 57.149 | 52.311 | 54.336 |
| DMPNN | 1.073 | 1.287 | 1.078 | 50.438 | 51.717 | 50.768 |
| Infograph-250K | 1.426 | 1.281 | 1.615 | 124.305 | 72.759 | 80.873 |
| Infomax3D-250K | 1.685 | 1.335 | 1.301 | 57.658 | 57.997 | 59.158 |
| Grover-250K | 1.484 | 1.619 | 1.585 | 64.744 | 64.061 | 64.551 |
| ChemBERTa-MLM-100M | 1.037 | 0.958 | 1.039 | 47.618 | 52.002 | 55.126 |
| c3-MoLFormer-1.1B | 1.066 | 1.261 | 0.956 | 48.793 | 51.823 | 55.559 |
| MoLFormer-LHPC | 1.334 | 1.160 | 1.10 | 49.412 | 43.950 | 43.852 |
A.6. Extended comparison analysis of splits
Fig. 7 presents histograms of the Minimum Tanimoto Distance (MTD) distributions between the training split and the validation and test splits, for both DeepChem and MolFormer scaffold splitting strategies across several MoleculeNet classification datasets: BACE, SIDER, HIV, BBBP, ClinTox, and Tox21. | ||
| Fig. 7 Histograms of Minimum Tanimoto Distance (MTD) distributions comparing validation and test sets across multiple MoleculeNet classification datasets: BACE, SIDER, HIV, BBBP, CLINTOX and TOX21. | ||
A.7. Sample code for ModularTorchModel
Refer Fig. 8. | ||
| Fig. 8 Example Python code for DeepChem's ModularTorchModel class. | ||
Acknowledgements
Funding in part by DTRA project HDTRA13081-40035. All work performed at Lawrence Livermore National Laboratory is performed under the auspices of the U.S. Department of Energy under Contract DE-AC52-07NA27344.References
- J. Shen and C. A. Nicolaou, Drug Discovery Today: Technol., 2019, 32–33, 29–36 Search PubMed.
- C. Pang, H. Tong and L. Wei, Quant. Biol., 2023, 11(4), 359–473 Search PubMed.
- S. Chithrananda, G. Grand and B. Ramsundar, Clin. Orthop. Relat. Res., 2020, arXiv: arXiv:2010.09885, DOI:10.48550/arXiv.2010.09885.
- W. Ahmad, E. Simon, S. Chithrananda, G. Grand and B. Ramsundar, ChemBERTa-2: Towards Chemical Foundation Models, 2022, https://arxiv.org/abs/2209.01712 Search PubMed.
- J. Ross, B. Belgodere, V. Chenthamarakshan, I. Padhi, Y. Mroueh and P. Das, Nat. Mach. Intell., 2022, 4, 1256–1264 Search PubMed.
- NVIDIA, MegaMolBART: A deep learning model for small molecule drug discovery and cheminformatics based on SMILES, https://github.com/NVIDIA/MegaMolBART, 2021.
- R. Irwin, S. Dimitriadis, J. He and E. J. Bjerrum, Mach. learn.: sci. technol., 2022, 3, 015022 Search PubMed.
- D. Beaini, S. Huang, J. A. Cunha, Z. Li, G. Moisescu-Pareja, O. Dymov, S. Maddrell-Mander, C. McLean, F. Wenkel, L. Mülleret al., arXiv, 2023, preprint arXiv:2310.04292, DOI:10.48850/arXiv:2310.04292.
- A. M. Bran and P. Schwaller, Drug Development Supported by Informatics, Springer, 2024, pp. 143–163 Search PubMed.
- D. Beaini, S. Huang, J. A. Cunha, Z. Li, G. Moisescu-Pareja, O. Dymov, S. Maddrell-Mander, C. McLean, F. Wenkel, L. Müller, J. H. Mohamud, A. Parviz, M. Craig, M. Koziarski, J. Lu, Z. Zhu, C. Gabellini, K. Klaser, J. Dean, C. Wognum, M. Sypetkowski, G. Rabusseau, R. Rabbany, J. Tang, C. Morris, M. Ravanelli, G. Wolf, P. Tossou, H. Mary, T. Bois, A. W. Fitzgibbon, B. Banaszewski, C. Martin and D. Masters, The Twelfth International Conference on Learning Representations, 2024.
- J. Devlin, M.-W. Chang, K. Lee and K. Toutanova: BERT: Pre-Training of Deep Bidirectional Transformers for Language Understanding, 2019, pp. 4171–4186, https://aclanthology.org/N19-1423/ Search PubMed.
- D. Van Tilborg, A. Alenicheva and F. Grisoni, J. Chem. Inf. Model., 2022, 62, 5938–5951 Search PubMed.
- A. Yüksel, E. Ulusoy, A. Ünlü and T. Doğan, Mach. learn.: sci. technol., 2023, 4, 025035 Search PubMed.
- Z. Wu, B. Ramsundar, E. N. Feinberg, J. Gomes, C. Geniesse, A. S. Pappu, K. Leswing and V. S. Pande, arXiv, 2017, abs/1703.00564, Search PubMed.
- B. Ramsundar, P. Eastman, E. Feinberg, J. Gomes, K. Leswing, A. Pappu, M. Wu, and V. Pande., DeepChem: Democratizing Deep-Learning for Drug Discovery, Quantum Chemistry, Materials Science and Biology, 2016.
- P. Moritz, R. Nishihara, S. Wang, A. Tumanov, R. Liaw, E. Liang, W. Paul, M. I. Jordan and I. Stoica, Clin. Orthop. Relat. Res., 2017, 561–577 Search PubMed.
- T. Nguyen, J. Brandstetter, A. Kapoor, J. K. Gupta and A. Grover, arXiv, preprint arXiv:2301.10343, DOI:10.48550/arXiv:2301.10343, 2023.
- Y. Liu, J. Sun, X. He, G. Pinney, Z. Zhang and H. Schaeffer, arXiv, preprint arXiv:2409.09811, DOI:10.48550/arXiv:2409.09811, 2024.
- J. Vamathevan, D. Clark, P. Czodrowski, I. Dunham, E. Ferran, G. Lee, B. Li, A. Madabhushi, P. Shah, M. Spitzer and S. Zhao, Nat. Rev. Drug Discovery, 2019, 18, 463–477 CrossRef CAS PubMed.
- J. Gilmer, S. S. Schoenholz, P. F. Riley, O. Vinyals and G. E. Dahl, Proceedings of the 34th International Conference on Machine Learning, 2017, pp. 1263–1272 Search PubMed.
- K. Yang, K. Swanson, W. Jin, C. Coley, P. Eiden, H. Gao, A. Guzman-Perez, T. Hopper, B. Kelley, M. Mathea, A. Palmer, V. Settels, T. Jaakkola, K. Jensen and R. Barzilay, J. Chem. Inf. Model., 2019, 59, 3370–3388 Search PubMed.
- F.-Y. Sun, J. Hoffmann, V. Verma and J. Tang, arXiv, 2020, DOI:10.48550/arXiv:abs/1908.01000.
- H. Stärk, D. Beaini, G. Corso, P. Tossou, C. Dallago, S. Günnemann and P. Liò, Proceedings of the 39th International Conference on Machine Learning, PMLR 162:20479-20502, 2022 Search PubMed.
- Y. Liu, M. Ott, N. Goyal, J. Du, M. Joshi, D. Chen, O. Levy, M. Lewis, L. Zettlemoyer and V. Stoyanov, RoBERTa: A Robustly Optimized BERT Pretraining Approach, 2019 DOI:10.48550/arXiv.1907.11692.
- M. Lewis, Y. Liu, N. Goyal, M. Ghazvininejad, A. Mohamed, O. Levy, V. Stoyanov and L. Zettlemoyer, Clin. Orthop. Relat. Res., 2019, 7871–7880 Search PubMed.
- J. Ross, B. Belgodere, V. Chenthamarakshan, I. Padhi, Y. Mroueh and P. Das, Clin. Orthop. Relat. Res., 2021 Search PubMed.
- Y. Rong, Y. Bian, T. Xu, W. Xie, Y. Wei, W. Huang and J. Huang, arXiv, 2020, DOI:10.48550/arXiv:abs/2007.02835.
- M. V. Sai Prakash, N. Siddartha Reddy, G. Parab, V. Varun, V. Vaddina and S. Gopalakrishnan, Synergistic Fusion of Graph and Transformer Features for Enhanced Molecular Property Prediction, Cold Spring Harbor Laboratory, 2023, DOI:10.1101/2023.08.28.555089.
- W. Hu, B. Liu, J. Gomes, M. Zitnik, P. Liang, V. Pande and J. Leskovec, Strategies for Pre-training Graph Neural Networks, 2020 DOI:10.48550/arXiv.1905.12265.
- P. Walters, We Need Better Benchmarks for Machine Learning, https://practicalcheminformatics.blogspot.com/2023/08/we-need-better-benchmarks-for-machine.html, 2023, Blog post, accessed 21 Apr 2025.
- E. Heid, K. P. Greenman, Y. Chung, S.-C. Li, D. E. Graff, F. H. Vermeire, H. Wu, W. H. Green and C. J. McGill, J. Chem. Inf. Model., 2023, 64, 9–17 Search PubMed.
- A. Velez-Arce and M. Zitnik, arXiv preprint, 2025 Search PubMed.
- Z. Zhu, C. Shi, Z. Zhang, S. Liu, M. Xu, X. Yuan, Y. Zhang, J. Chen, H. Cai, J. Lu, C. Ma, R. Liu, L.-P. Xhonneux, M. Qu and J. Tang, arXiv preprint, 2022 Search PubMed.
- M. Praski, J. Adamczyk and W. Czech, arXiv preprint, 2025 Search PubMed.
- X. Lou, J. Cai, C.-w. Un and S. W. I. Siu, ACS Omega, 2025, 10, 37549–37560 CrossRef CAS PubMed.
- A. Paszke, S. Gross, F. Massa, A. Lerer, J. Bradbury, G. Chanan, T. Killeen, Z. Lin, N. Gimelshein, L. Antiga, A. Desmaison, A. Köpf, E. Z. Yang, Z. DeVito, M. Raison, A. Tejani, S. Chilamkurthy, B. Steiner, L. Fang, J. Bai and S. Chintala, PyTorch: An Imperative Style, High-Performance Deep Learning Library, 2019 DOI:10.48550/arXiv.1912.01703.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. VanderPlas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and E. Duchesnay, Scikit-learn: Machine Learning in Python, 2012 Search PubMed.
- T. Chen, C. Guestrin, Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, ACM, 2016, pp. 785–794 Search PubMed.
- A. J. Minnich, K. McLoughlin, M. Tse, J. Deng, A. Weber, N. Murad, B. D. Madej, B. Ramsundar, T. Rush, S. Calad-Thomson, J. Brase and J. E. Allen, J. Chem. Inf. Model., 2020, 60, 1955–1968 CrossRef CAS PubMed.
- J. J. Irwin, K. G. Tang, J. Young, C. Dandarchuluun, B. R. Wong, M. Khurelbaatar, Y. S. Moroz, J. Mayfield and R. A. Sayle, J. Chem. Inf. Model., 2020, 60, 6065–6073 CrossRef CAS PubMed.
- G. W. Bemis and M. A. Murcko, J. Med. Chem., 1996, 39, 2887–2893 Search PubMed.
- X. Zheng and Y. Tomiura, J. Cheminf., 2024, 16, 71 Search PubMed.
- T. Wolf, L. Debut, V. Sanh, J. Chaumond, C. Delangue, A. Moi, P. Cistac, T. Rault, R. Louf, M. Funtowicz, J. Brew, HuggingFace's Transformers: State-of-the-art Natural Language Processing, 2020. https://arxiv.org/abs/1910.03771 Search PubMed.
- G. Landrum, P. Tosco, B. Kelley, R. Rodriguez, D. Cosgrove, R. Vianello, sriniker, P. Gedeck, G. Jones, E. Kawashima, NadineSchneider, D. Nealschneider, A. Dalke, tadhurst-cdd, M. Swain, B. Cole, S. Turk, A. Savelev, N. Maeder and J. Lehtivarjo, RDKit, RDKit: Open-source cheminformatics, http://www.rdkit.org, 2021.
- S. Wang, J. Witek, G. A. Landrum and S. Riniker, J. Chem. Inf. Model., 2020, 60, 2044–2058 CrossRef CAS PubMed.
- rdkit.Chem.rdDistGeom.ETKDGv2 Documentation, https://rdkit.org/docs/source/rdkit.Chem.rdDistGeom.html#rdkit.Chem.rdDistGeom.ETKDGv2, 2024.
Footnotes |
| † Equal Contribution. |
| ‡ Work done during their time at Deep Forest Sciences. |
| This journal is © The Royal Society of Chemistry 2026 |