
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deep learning methods for 2D material electronic properties
Artem
Mishchenko
 *,
Anupam
Bhattacharya
*,
Xiangwen
Wang
,
Henry Kelbrick
Pentz
,
Yihao
Wei
and
Qian
Yang
*,
Anupam
Bhattacharya
*,
Xiangwen
Wang
,
Henry Kelbrick
Pentz
,
Yihao
Wei
and
Qian
Yang
Department of Physics and Astronomy, University of Manchester, Manchester, UK. E-mail: artem.mishchenko@manchester.ac.uk; anupam.bhattacharya@manchester.ac.uk
First published on 9th December 2025
Abstract
This review explores the impact of deep learning (DL) techniques on understanding and predicting electronic structures in two-dimensional (2D) materials. We highlight unique computational challenges posed by 2D materials and discuss how DL approaches – such as physics-aware models, generative AI, and inverse design – have significantly improved predictions of critical electronic properties, including band structures, density of states, and quantum transport phenomena. Through selected case studies, we illustrate how DL methods accelerate discoveries in emergent quantum phenomena, topology, superconductivity, and autonomous materials exploration. Finally, we outline promising future directions, stressing the need for robust data standardization and advocating for integrated frameworks that combine theoretical modeling, DL methods, and experimental validations.
1 Introduction
Two-dimensional (2D) materials offer diverse applications in (opto)electronics, energy storage, catalysis, sensing, and quantum technologies. Their reduced dimensionality leads to properties such as tunable electronic band structures, strong light–matter interactions, and high catalytic activity.1–3 For 2D materials to continue to drive innovation, accurate and efficient predictions of their electronic structures are paramount for both fundamental understanding and technological applications.Despite their conceptual simplicity compared to bulk materials, modeling the electronic structure of 2D systems presents unique challenges. Most computational approaches rely on 3D periodic boundary conditions, requiring a large vacuum along the out-of-plane direction to prevent artificial interactions between repeated layers. However, there is no widely established method designed specifically for 2D systems – that is, periodic within the plane but non-periodic out-of-plane. This limitation affects both accuracy and computational costs, particularly for multilayer 2D (hetero)structures, moiré superlattices, and strongly correlated systems.4 The growing shift toward automated, closed-loop materials discovery laboratories further underscores the need for computationally efficient methods, as rapid screening is critical for accelerating breakthroughs.5–7
Traditional electronic structure methods, such as tight-binding models and density functional theory (DFT), have long been used to study 2D materials. Tight binding models are computationally efficient and offer some analytical insights but fail to capture many-body interactions and other complex effects. DFT, while more robust, struggles with accurately modeling van der Waals (vdW) interactions – which are crucial in layered heterostructures8,9 – and remains computationally expensive, limiting its feasibility for large-scale high-throughput studies.
To overcome these limitations, artificial intelligence (AI), particularly deep learning (DL), has emerged as a powerful tool for electronic structure prediction. Unlike traditional methods, which rely on explicitly defined physical models, DL learns structure–property relationships directly from data, enabling it to approximate computationally demanding calculations at a fraction of the cost. A recent comprehensive perspective further emphasizes strategies for AI-driven research in chemistry and materials science.10 Indeed, recent studies have shown that DL models can improve predictive accuracy by learning from diverse datasets, including both high-fidelity simulations and experimental measurements.11–13
This review explores how DL is transforming electronic structure modeling in 2D materials, addressing key computational challenges and accelerating materials discovery. We begin with an overview of traditional computational and experimental approaches, followed by a discussion of databases and data representations used in training AI models. We then examine DL-driven predictions of electronic properties, such as band structures, density of states, and quantum transport phenomena, distinguishing between forward design, which predicts material properties from known structures, and inverse design, which identifies materials with target functionalities. We further differentiate between strictly inverse design (non-generative), which selects materials from existing databases, and generative inverse design, where AI models propose entirely novel structures using methods such as variational autoencoders (VAEs) and generative adversarial networks (GANs).
Beyond property prediction, we discuss how DL aids in discovering emergent quantum phenomena, including nontrivial topology, strongly correlated phases, and moiré superlattices. We also highlight the challenges in integrating DL with first-principles methods and experimental validation, emphasizing the need for improved interpretability, generalization, and data standardization. Finally, we outline future directions, including foundation models, AI-driven automation in experiments, and the integration of DL with quantum computing, providing a comprehensive perspective on how DL is shaping the future of 2D materials research.
We emphasize that this is a rapidly evolving field, with a continual influx of new publications. Consequently, it is not feasible to provide an exhaustive account of all developments. Instead, we highlight a selection of recent review articles that cover adjacent areas and complement the scope of this work. We recommend the review by Malica et al. for details of AI-assisted synthesis and interpretability of experiments of materials,14 Vital et al. for details of machine learning (ML) based interatomic potentials,15 the overview of numerical techniques for layered materials by Gray and Herbert,16 and new research avenues in electronic structure analysis with AI summarized by Kulik et al.17
2 Foundations for DL in 2D materials
2.1 Electronic band structures of 2D materials: key concepts and computational challenges
Electrons in materials are most commonly represented as wavefunctions that satisfy Schrödinger's equation, which can be solved to yield eigenvalues E and eigenvectors or wave functions Ψ. For materials with many electrons, solving this equation is practically impossible due to the complex nature of many-body electronic interactions. Various numerical techniques have been developed to address this challenge, including Hartree–Fock (HF), Density Functional Theory (DFT), Dynamic Mean-Field Theory (DMFT), Coupled Cluster (CC) theory, and Quantum Monte-Carlo (QMC) methods.18 DFT, the workhorse of computational materials science, handles many-body physics by mapping the complex many-electron problem onto a simpler system of non-interacting electrons moving in an effective potential, with electron–electron interactions approximated through exchange-correlation potentials that capture quantum mechanical effects.While these computational methods were originally developed for bulk 3D materials, they can be adapted to 2D materials by restricting periodic boundary conditions (PBC) to two dimensions. However, for multilayer 2D materials, accurately modeling van der Waals interactions between layers remains an active area of research.16
The electronic band structure of a material is typically represented through its dispersion relation – the relationship between energy eigenvalues and wave vectors. Conventionally, these energies are plotted as a function of wave vectors k (or momenta p = ℏk, where ℏ is the reduced Planck's constant) along high-symmetry lines in the reciprocal space, forming what is called the electronic band structure. For 2D materials, these high-symmetry lines lie in a plane, reflecting their reduced dimensionality.
Materials databases store this electronic structure information in various formats. Most commonly, dispersion relations along high-symmetry paths are stored as electronic band structures. Some databases also provide electronic dispersion on uniform k-grids, as a result, requiring significantly more storage space. Another common representation is the Density of States (DOS), which quantifies the occupation of electronic states at different energies and momenta, and is often presented alongside band structures. More detailed representations include electronic wave functions and charge density distributions (Ψ2), which would undoubtedly require substantially more storage capacity.
Complementing computational approaches are various experimental techniques that can directly probe the electronic structure of 2D materials. Spectroscopic methods such as X-ray Photoemission Spectroscopy (XPS), X-ray Absorption Spectroscopy (XAS), Ultraviolet Photoelectron spectroscopy (UPS), and Ultraviolet-visible-Near-Infrared spectroscopy (UV-Vis/NIR) provide valuable information about energies of electronic states in 2D materials.19 Techniques like Electron Energy Loss Spectroscopy (EELS)20 and Low Energy Electron Diffraction (LEED)21 have also proven instrumental in identifying electronic structures of 2D materials. Methods like Scanning Tunneling Microscopy (STM) allow direct visualization of local DOS over 2D surfaces with atomic resolution. Momentum-resolved techniques, particularly Angle-Resolved Photoemission Spectroscopy (ARPES) and Quasi-Particle Interference (QPI), offer unique capabilities to simultaneously measure electron energies and momenta, providing comprehensive mapping of band structures in reciprocal space.
So far, a wide range of computational and experimental methods are available to determine electronic structures of 2D materials, producing vast data stored in various formats across different platforms. In general, current computational datasets provide more structured information that could be systematically categorized. However, to predict properties of new materials from existing knowledge, AI approaches are required, as the volume of data and the complex interdependencies between variables far exceed the capabilities of conventional analytical methods.
The challenge intensifies when it comes to experimental data, which are largely unstructured and dispersed throughout scientific literature. AI techniques become even more critical for effective information extraction, data curation, standardization across sources, and accurate interpolation and extrapolation for prediction purposes. In the following subsections, we examine the existing databases, data representation formats, and computational tools for mining this wealth of electronic structure information.
2.2 Databases and data curation
The advancement of first-principles calculations has enabled the development of comprehensive computational databases, which have become indispensable tools for electronic structure exploration.22 These databases provide large-scale, standardized datasets that accelerate both materials discovery and machine learning model development. While major repositories such as the Materials Project23 and JARVIS-DFT24 were initially designed for 3D materials, they contain valuable information on 2D systems as well. Researchers can extract band structures and DOS, specific to monolayers or layered structures, by filtering database entries for reduced-dimensionality systems. This versatility extends the utility of these general-purpose databases to 2D materials research, yet challenges still exist in accurately identifying and integrating 2D-specific data, particularly when materials exhibit properties that differ between their bulk and 2D forms.To address the need for specialized 2D materials information, dedicated databases have emerged as focal points for the research community. The Computational 2D Materials Database (C2DB),25,26 Materials Cloud 2D Database (MC2D),27,28 and 2D Materials Encyclopedia (2DmatPedia)29 provide comprehensive catalogs of crystal structures, phonon dispersions, electronic band structures, and optical properties. These representations contain thousands of 2D materials with properties predicted using DFT and many-body perturbation theory methods.
Expanding beyond monolayers, computational studies have demonstrated the rich potential of stacking engineering in 2D materials.30,31 For instance, over 2500 (out of 8000) stable homo-bilayers structures with emergent properties distinct from their monolayer constituents are identified through high-throughput DFT,30 highlighting the extent which stacking engineering leads to novel physics and functionalities. Many homo-bilayers exhibit multiple stable stacking configurations, giving rise to sliding ferroelectricity – a phenomenon where the relative displacement of layers breaks the inversion symmetry and induces out-of-plane polarization. This effect has been demonstrated in various systems, including bilayer boron nitride, rhombohedral-stacked transition metal chalcogenides, and marginally twisted 2D materials.32 Recent developments in DFT-based high-throughput studies have extended this further to hetero-bilayers,33,34 offering insight into interfacial band alignment in van der Waals heterostructures. Computational materials databases, including those for 2D materials, are summarized in Table 1.
| Database | Description | # Of entries | URL | |
|---|---|---|---|---|
| 3D | Materials Project23 | A database of computed materials properties for research | 170![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000+ 000+ |
https://www.materialsproject.org |
| Automatic FLOW for materials discovery (AFLOW) | Largest 3D materials database based on VASP calculations | 3.5 million | https://www.aflowlib.org | |
| JARVIS-DFT24 | A repository of VASP-calculated properties for mostly 3D materials and 1000 2D materials | 80000+ |
https://jarvis.nist.gov | |
| Crystallography open database (COD) | Database of experimentally observed crystal structures | 500000+ |
https://www.crystallography.net/cod/ | |
| Open quantum materials database (OQMD)35 | VASP calculated material properties database | 300000+ |
https://www.oqmd.org | |
| Cambridge structural database (CSD) | Large experimental structure database including XRD, neutron diffraction | 1.25 million | https://www.ccdc.cam.ac.uk/structures/ | |
| GNoME dataset36 | AI predicted mosty 3D and some 2D stable materials | 2.2 million | https://github.com/google-deepmind/materials_discovery.git | |
| 2D | Computational 2D materials database (C2DB)25,26 | A database of properties for 2D materials calculated using DFT code GPAW | 4000: DFT 11600: AI | https://www.cmr.fysik.dtu.dk/c2db/c2db.html |
| Materials cloud 2D crystals database (MC2D)27,28 | 2D crystal structures exfoliated from 3D; DFT simulation with QE | 3077 | https://mc2d.materialscloud.org | |
| 2D materials encyclopedia (2DMatPedia)29 | 2D materials created with top–down and bottom–up approaches from materials project; DFT calculations with VASP | 6351 | https://www.2dmatpedia.org | |
| Virtual 2D materials database (V2DB)37 | A database of AI generated likely-stable 2D materials with key properties | 316505: AI |
https://doi.org/10.7910/DVN/SNCZF4 | |
| MatHub-2D38 | VASP and phonopy calculation results on high mobility semiconductors | 1900 | http://www.mathub2d.net/materials/matdb | |
| 2D octahereal materials database (aNANt)39 | Functional 2D materials simulated using VASP | 3099 | https://anant.mrc.iisc.ac.in/apps/2D | |
| Topological 2D materials database (2D-TQCDB)40 | DFT with VASP (with SOC) hosting band structures and detail topological properties | 8872 | https://topologicalquantumchemistry.com/topo2d/index.html | |
| Ferromagnetic 2D materials41 | VASP calculation identifying nonmagnetic to ferromagnetic transition via hole doping | 122 | SI of ref. 41 | |
| 2D topological insulators42 | DFT calculations with QE (with SOC) materials from MC2D | 1825 | https://www.materialscloud.org/discover/2dtopo/dashboard/ptable | |
| Experimental 2D materials43 | List of reported experimentally synthesized 2D materials | 300+ | https://zenodo.org/records/10887700 | |
| 2D materials platform44 | Experimental results such as XPS, RHEED, Raman, AFM on 2D materials | — | https://2dmat.chemdx.org/ | |
| Homo-bilayer | van der Waals bilayer database (BiDB)30 | A database of van der Waals bilayer material properties calculated with GPAW | 2586 | https://cmr.fysik.dtu.dk/bidb/bidb.html |
| Bilayer materials DataSet (BMDS)31 | Band structures calculated with SOC using VASP | 760 | BMDS | |
| Hetero-bilayers | InterMatch45 | Calculates interface properties like mismatch, charge transfer from bulk properties | — | https://contribs.materialsproject.org/projects/intermatch/ |
| Layered heterostructures on materials project46 | Active learning platform to compute electronic property of heterostructures | — | https://magics.usc.edu/data/ |
Despite the growing availability of databases for 2D materials, consolidating data for machine learning applications remains challenging. Researchers frequently need to extract and integrate data from multiple databases to assemble comprehensive training datasets. However, issues such as data duplication and inconsistencies intensify as these repositories rapidly expand. Different DFT functionals used across databases result in slight but evident variations in the predicted crystal structures and properties for nominally identical materials. Moreover, disparate representations of physical properties – such as differing conventions for elastic tensors – further compromise data compatibility and comparability. Without robust deduplication and harmonization strategies, repeated occurrences of similar or identical materials with varying properties may inadvertently introduce biases into machine learning models.
A notable example of addressing these challenges comes from Meng et al.,41 who developed a systematic approach to data integration. The authors collected 2D crystal structures from 2DmatPedia, C2DB, and Materials Cloud to identify non-magnetic 2D semiconductors with potential for hole-induced ferromagnetism. Their high-throughput screening process incorporated critical data cleaning steps: exclusion of magnetic metals, identification and removal of duplicates across databases, and filtering out materials with low thermodynamic stability. This methodical approach yielded a curated dataset of 3000 materials for further study, demonstrating a proper data cleaning workflow that strengthens materials discovery.
While computational databases provide structured insights, experimental data from the published literature constitute a critical complementary resource that offers essential real-world validation. Techniques such as ARPES, STM, and XPS could generate rich, realistic datasets capturing the true behavior of materials beyond idealized simulations. However, extracting and curating high-quality experimental data presents even greater challenges, including variability in experimental setups, inconsistencies in sample quality, non-uniform reporting standards, and the substantial costs and time required for such experiments.47 Addressing these challenges requires robust pre-processing pipelines and standardized data formats to assess measurement uncertainties, filtering out experimental noise, and assembling high-confidence datasets suitable for machine learning applications. The development of community-wide data standardization protocols would significantly accelerate the integration of experimental and computational data, representing a frontier opportunity to develop more robust and transferable predictive models.
2.3 Data representation
Data representation refers to the process of encoding the structural, electronic, or other properties of materials into mathematical forms that can be understood and processed by ML algorithms. Effective representation of data is central to the success of DL models in understanding and predicting the electronic structures of 2D materials. The choice of representation determines what information the model can learn, its ability to generalize, and the physical interpretability of its predictions. In this section, we discuss the key material representations used in DL workflows for 2D materials, categorized into structural, electronic, and hybrid representations.Graph-based representations have emerged as the predominant approach for encoding atomic structures, where atoms are represented as nodes and their interactions as edges. Isayev et al.48 pioneered this approach with property-labeled materials fragments (PLMFs), which use Voronoi tessellation and covalent radii cutoffs to partition crystal structures into meaningful subunits labeled with elemental properties such as valence electron count and electronegativity, creating a structural representation for predicting electronic properties, Fig. 1A. Almost simultaneously, neural message passing pioneered by Gilmer et al.,53 provided a natural, powerful, and flexible way to capture both local chemical environments and long-range atomic interactions by updating NNs through sending nodal information only along graph edges. This was one of the first use of graph neural networks (GNNs) in electronic structure prediction. Other early GNNs for materials, such as the crystal graph convolutional neural network (CGCNN),49 build a convolutional architecture on top of crystal graphs where atoms are nodes and bonds are edges, using iterative message passing to capture local chemical environments and predict material properties with DFT-level accuracy, including formation energy, band gap, or elastic modulus, Fig. 1B.
 | ||
| Fig. 1 Crystal structure representations. (A) Property-Labeled Materials Fragments (PLMFs), adapted from ref. 48 with permission from Springer Nature, Nat. Commun., 2017. (B) Crystal Graph Convolutional Neural Network (CGCNN), adapted from ref. 49 with permission from American Physical Society, Phys. Rev. Lett., 2018. (C) Atomistic Line Graph Neural Network (ALIGNN), reproduced from ref. 50 with permission from Springer Nature, npj Comput. Mater., 2021. (D) Materials Graph Network (MEGNet), adapted from ref. 51 with permission from American Chemical Society, Chem. Mater., 2019. (E) Simplified Line-Input Crystal-Encoding System (SLICES), adapted from ref. 52 with permission from Springer Nature, Nat. Commun., 2023. | ||
Building upon this foundation, more sophisticated graph architectures have been developed. The atomistic line graph neural network (ALIGNN)50 extends CGCNN by performing message passing on both the interatomic bond graph and its line graph corresponding to bond angles, explicitly incorporating angular information to improve prediction accuracy for diverse materials properties, Fig. 1C. MatErials Graph Network (MEGNet)51 further enriches graph representations by including global state attributes such as temperature, pressure, or entropy, alongside atom and bond features, allowing for more accurate prediction of materials properties at various thermodynamic conditions, Fig. 1D. AtomSets54 goes further by treating atoms and bonds as unordered sets rather than fixed graph elements, providing greater flexibility and making it better suited for diverse material representations without atomic ordering constraints. MatterGen, introduced by Zeni et al.,55 presents a diffusion-based generative model that encodes materials universally as a combination of atomic types, lattice vectors, and fractional atomic coordinates within the unit cell. This representation ensures invariance to permutations, translations, rotations, and supercell transformations, while achieving remarkable performance in generating stable, unique, and new inorganic materials across the periodic table with properties closely matching DFT predictions.
While graph-based representations are more intuitive for crystal structures, string-based representation allows researchers to take advantage of the extensive and rapidly evolving field of natural language processing. The Simplified Line-Input Crystal-Encoding System (SLICES),52 shown in Fig. 1E, is a string-based crystal representation analogous to SMILES for molecules,56 offering both invertibility and invariance to transformations. SLICES encodes compositional and topological data of crystal structures, successfully reconstructing 94.95% of over 40000 diverse crystal structures. This representation facilitates proof-of-concept inverse design studies in solid-state materials, for example, exploring candidates for direct narrow-gap semiconductors in optoelectronics, while the quantitative accuracy of such applications ultimately depends on the reliability of the underlying forward bandgap predictors.
Very recently, sequence models have also started using Wyckoff representation as input strings. Wyckoff representation of atomic coordinates is easy to put into a sequence for easy integration with a transformer. It also encodes the symmetries of the crystallographic sites catering to providing physical insight to the model.57–59
Topology-based methods, such as those described in Chen et al.60 and Jiang et al.,61 utilize persistent homology to encode atomic configurations and their interactions into simplified topological descriptors. These representations effectively capture both the in-plane structural relationships and reduced dimensionality, complementing graph-based approaches by providing a powerful framework for predicting electronic and physical properties with enhanced accuracy.
Physical property-based representations focus on encoding key electronic, vibrational, and optical properties of 2D materials. The Coulomb matrix is a widely used descriptor that encodes atomic interactions as the Coulomb potential between nuclei. However, its effectiveness is limited by its sensitivity to atomic ordering, leading to the development of several improved variants.62,63 Key approaches include the randomly sorted Coulomb matrix, which generates multiple permutations to improve prediction accuracy, and the Bag of Bonds (BoB) descriptor, which encodes atomic interactions through bond-type-specific vectors and maintains permutation invariance.64 For periodic systems, extensions such as the Sine matrix and Ewald-summation matrix further improve scalability and accuracy by incorporating lattice periodicity.65,66
The GW approximation calculates accurate quasiparticle energies by combining the single-particle Green's function (G) with the screened Coulomb interaction (W) to correct mean-field energies with many-body effects, but requires substantial computational resources. To bypass this, Knøsgaard et al. used DFT derived electronic structure descriptors as a starting point as DFT states encode materials structure well. They have designed two electronic descriptors viz. Energy Decomposed Operator Matrix Elements (ENDOME) and Radially Decomposed Projected Density of States (RAD-PDOS)67 to predict GW corrections from DFT calculations (see Fig. 2A). ENDOME first creates projections of position, momentum, Laplacian operators of a reference state with other states. Subsequently, it bins these projections based on energy differences of the states onto a Gaussian energy grid, producing a 6 × 50 fingerprint of energy-dependent features. Complementarily, RAD-PDOS constructs a correlation function in energy and radial distance, encoding DOS across different atomic orbitals into a 25 × 20 energy–distance grid that preserves orbital-specific electronic distributions. Thus, RAD-PDOS contains the information of environment of orbitals in Hilbert space. Finally, the concatenated fingerprints are passed into an XGBoost regression algorithm to train G0W0 energy corrections for each eigenvalues. Such physics-motivated approach enables accurate prediction of many-body effects at a fraction of the computational cost of traditional GW calculations, achieving mean absolute errors as low as 0.14 eV for electronic states in 2D materials.
 | ||
| Fig. 2 Electronic structure representations. (A) Energy Decomposed Operator Matrix Elements (ENDOME) and Radially Decomposed Projected Density of States (RAD-PDOS) fingerprints, adapted from ref. 67 with permission from Springer Nature,67 copyright 2025. (B) Segmentation of band structure images, adapted from ref. 68 with permission from Springer Nature,68 copyright 2025. (C) Convolutional autoencoder (CAE) elf, adapted from ref. 69 with permission from Springer Nature,69 copyright 2025. | ||
Bhattacharya et al.68 pioneered segmentation techniques for band structure images, by dividing them into energy strips (of 0.5 eV each) and along high-symmetry k-paths and applying a supervised convolutional neural network (CNN), to identify flat bands, Fig. 2B. This approach overcomes the limitations of parameterized band structures, which often miss important electronic features due to band crossings and complex dispersions. Their CNN achieved high accuracy in detecting flat bands from segmented images without relying on arbitrary bandwidth definitions. Building on this, Pentz et al.69 developed elf (electronic fingerprint), a convolutional autoencoder framework with RESNET architecture that encodes band structure images into 98-dimensional fingerprint vectors, Fig. 2C. By training the model to reproduce electronic band structures within ± 4 eV around the Fermi level (even when portions were artificially obscured during training), elf effectively captures essential electronic patterns and creates meaningful fingerprints that cluster materials with similar band structures, revealing chemical and electronic relationships that traditional analysis methods had overlooked. Specifically, elf was able to group chemical compounds with similar stoichiometry by using their similarity in band structures, and was also able to identify duplicate entries in the 2D materials encyclopedia database autonomously.
The relationship between structural and electronic representations is vital to 2D materials research. The crystal structure, which defines how atoms are arranged, directly influences the distribution of electronic states that govern material functionality. Combining these representations in ML has been shown to improve prediction accuracy. Wang et al.70 developed a feature engineering strategy that constructs seven element-specific feature matrices from 2D material structure graphs. By processing these matrices via mean-pooling, they can create adaptive descriptors and select property-specific matrices through performance ranking to capture both structural topology and elemental information.
Recent advancements have further expanded the hybrid representations by incorporating diverse data sources. These include graph-based features derived from crystal structures, physical property measurements, and text-derived insights mined from scientific literature. Such comprehensive descriptors could enable richer understanding of 2D materials. For example, MatSciBERT71 utilizes transformer architectures to distill electronic and structural insights from vast materials science literature, offering a scalable approach to knowledge extraction. Additionally, MatText72 provides a benchmarking framework designed to evaluate and enhance text-based representations, focusing on predicting numerical properties from textual inputs.
2.4 Tools for data processing and mining
General high-throughput tools for navigating and analyzing materials databases are essential for accelerating ML applications in materials science, addressing challenges like duplicate structure identification, feature extraction, and materials space navigation. Efficient data processing and organization are prerequisite for applying DL for large datasets in 2D materials research. A range of tools and methods have been developed to extract features, ensure data consistency, and enhance ML model training by filtering redundant or inconsistent information, Table 2.| Tools | Description | Link |
|---|---|---|
| Matminer | A python library for extracting and analyzing materials data, useful for feature engineering in machine learning studies of 2D materials | https://github.com/hackingmaterials/matminer |
| Pymatgen | A robust tool for materials analysis, enabling structure manipulation, property calculation, and simulation of 2D material systems | https://pymatgen.org/ |
| Matbench | A benchmarking platform for machine learning models, providing datasets and tasks relevant to predicting 2D material properties | https://matbench.materialsproject.org/ |
| Optimade | An API standard for querying materials databases, facilitating access to 2D materials data across repositories | https://www.optimade.org |
| MLMD | A machine learning framework for molecular dynamics, applicable to simulating and studying 2D material behaviors | https://github.com/Jiaxuan-Ma/MLMD |
| AlphaMat | A platform for computational materials design, offering tools to explore and optimize 2D material structures and properties | http://www.aimslab.cn |
| Constructor platform | A modular software suite for materials modeling, supporting simulations and workflows for 2D materials research | https://docs.constructor.tech/home/en-us/ |
Matminer automates the extraction of multiple descriptors – spanning electronic, structural, and thermodynamic properties – converting raw data into machine-readable formats.73 It supports high-throughput workflows by automatically fetching relevant descriptors and organizing them to input into models like CNNs and hybrid architectures. Pymatgen offers robust tools for retrieving and pre-processing data, including atomic positions, crystal structures, band gaps, etc., by interfacing with large databases like Materials Project.74 It is widely used to construct graph-based representations of 2D materials, which serve as inputs for GNNs. Matbench provides standardized datasets for tasks like band gap or formation energy prediction.75 It also hosts a dataset of experimentally observed band gaps. Apart from providing standardized datasets, Matbench-discovery76 also provides a leaderboard which show the current best machine learning potentials. Jarvis-leaderboard is another source of standardized datasets including accurate data from quantum calculations, experimental superconductivity transition temperature, and interatomic potentials.77 These benchmarking datasets are essential for model validation, allowing consistent comparisons across different architectures and algorithms. These well-curated benchmarks ensure reproducible and generalizable model performance, helping researchers optimize hyperparameters and assess algorithm robustness.78
In addition to these tools, high-throughput workflows require addressing data duplication and inconsistencies, which often arise from variations in cell parameters or small perturbations in atomic positions within the accuracy of DFT. Isometry-based comparisons have been developed to detect duplicates robustly, ensuring database integrity and improving ML model reliability by avoiding redundancy.79 Metrics like the Local Novelty Distance (LND) further quantify deviations in structure similarity using continuous descriptors, enabling efficient navigation of materials spaces. Advanced tools such as Predicted Fraction of Improved Candidates (PFIC) and Cumulative Maximum Likelihood of Improvement (CMLI) have also been developed to directly assess the quality of design spaces, helping researchers prioritize regions with higher discovery potential.80
The development of integrated workflows for seamless data exchange and collaboration across domains is also worth noting. A few recent examples include: OPTIMADE81 application programming interface provides standardized access across multiple databases, acting as an enabler for AI-driven materials discovery; MLMD platform82 is dedicated to the integration of experiments, computation, and design of novel materials; AlphaMat platform83 aims at uniting materials science and AI; and the Constructor Platform is designed to simplify and accelerate the scientific research lifecycle.84 Such integrated workflows promise to speed up materials discovery to meet our increasing technological requirements.
3 Forward design with DL for properties prediction
Forward design refers to predicting material properties from its known atomic structure. DL has transformed this process by providing data-driven insights for electronic structures and their derived properties. Unlike traditional computational approaches that rely on first-principles calculations, DL offers accelerated prediction pathways while maintaining comparable accuracy, enabling more efficient exploration of material design spaces.This forward design workflow can be systematically divided into three primary levels of prediction,85 also see Fig. 3 for details. First, DL can model fundamental electronic quantum mechanical properties by emulating underlying numerical methods, such as DFT or tight binding (TB). Here, DL models mimic DFT-like methods and use generative algorithms to predict the electronic structure or phonon frequencies on a discretized grid. Second, DL can predict derived single-point outputs – energy gaps, total energy, or interatomic forces – effectively replacing computationally expensive DFT calculations. These predictive models serve as machine learning force fields (MLFF), enabling rapid molecular dynamics simulations. Finally, DL can analyze outputs from these simulations to predict measurable material properties such as thermoelectric coefficients, superconducting transition temperature, or photoelectric efficiency, to name but a few. In some advanced implementations, DL frameworks can even directly correlate atomic configurations to these physical properties, bypassing intermediate calculations entirely.
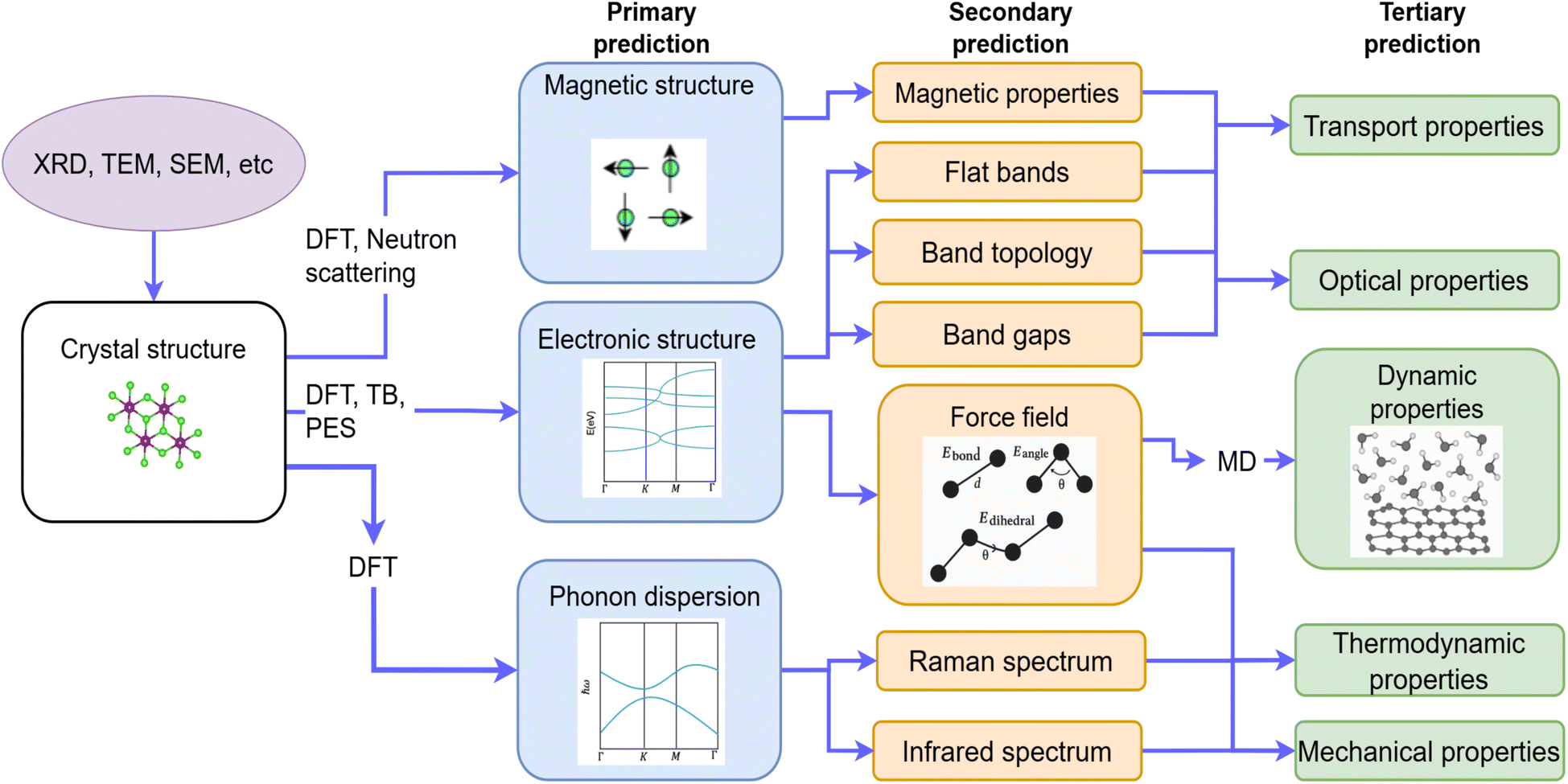 | ||
| Fig. 3 Forward design pathways for 2D material crystal structures to properties. Crystal structures are validated using experimental techniques like XRD, TEM etc. First principles simulations like DFT calculates electronic, phononic, and magnetic structure which in turn yields end properties like optical, transport, mechanical and dynamic properties. | ||
Beyond these general approaches to forward design, specialized DL architectures have been developed to incorporate physical knowledge and handle multiple objectives simultaneously. Physics-aware Neural Networks (PNNs)86 represent a specialized DL approach that explicitly embeds physical laws into neural network architectures. By incorporating known physical principles like symmetry considerations or conservation laws, PNNs excel in solving governing equations with sparse training data. Although their application to 2D materials' electronic structures remains limited, they show significant potential for modeling band structures or carrier dynamics in these systems.
Multi-Objective Optimization (MOO), enhanced by DL, enables the simultaneous optimization of multiple competing properties. MOO produces Pareto-optimal solutions representing the best possible trade-offs between different properties. When applied to 2D materials, this approach could optimize multiple properties simultaneously, such as electronic band gap and thermal conductivity, leading to more targeted material designs.
Although direct examples of PNNs and MOO applications in electronic structure prediction for 2D materials remain scarce87 – likely due to the complexity of quantum mechanical modeling and limited available datasets – the success of these methods in related fields suggests substantial untapped potential. This section explores the application of diverse DL approaches for forward design, examining methods for predicting fundamental electronic structures, applications for topological properties and strong correlations, prediction of flat bands and other quantum phenomena, and downstream property prediction critical to functional applications of 2D materials. Through these areas, we examine how DL is transforming our ability to predict and understand the complex electronic behaviors of 2D materials.
3.1 DL for predicting electronic structure
The prediction of electronic structures with DL can be approached in two fundamental ways. The first approach involves training neural networks to solve the underlying quantum many-body problems, specifically the electronic structure problem based on the many-electron Schrödinger equation or its approximations. In this physics-aware approach, the model outputs the electronic wavefunctions or electron density directly. The second approach focuses on directly predicting specific electronic properties – such as band dispersion, band gaps, or DOS – from crystal structures, effectively bypassing the computational complexity of quantum computations at the cost of reduced theoretical versatility. Detailed descriptions of these approaches are provided below and in Table 3.DFT handles many-body physics by approximating electron–electron interactions through exchange-correlation potentials. Several studies have demonstrated AI's effectiveness in modeling these exchange-correlation interactions,12,89–91 see Fig. 4A for details of one such architecture. This approach extends beyond standard DFT, enabling models to emulate advanced methods such as hybrid functionals and meta-generalized gradient approximation (meta-GGA) calculations, which provide a more accurate solution for exchange-correlation effects.18,92
 | ||
| Fig. 4 DL architectures (panel A, B and E) and techniques such as DL-assisted quantum embedding, message passing and equivariance for electronic structure prediction. (A) Solving Kohn–Sham equation with exchange-correlation energy estimated with DL, adapted from ref. 91 with permission from American Physical Society,91 copyright 2025. The left image show the iterative Kohn–Sham scheme, in which each iteration is divided into step shown in the right. The xc-energy term is modeled using the convolutional network described in the bottom right of panel A. (B) Architecture of Ferminet (adapted from ref. 95 with permission from arXiv,95 copyright 2025) shows L serial composite layers each made from a couple of MLP and convolution layers. (C) Comparison of the traditional quantum embedding path (left) with ML assisted calculation of embedding Hamiltonian in DL assisted quantum embedding calculations, adapted from ref. 108 with permission from American Physical Society,108 copyright 2025 (right) (D) Message passing (top) within a graph updates the nodes from neighboring node information. Equivariance (bottom) in GNN ensures the embedding transforms the same way as the input structure, adapted from ref. 113 with permission from Nordic Machine Intelligence,113 copyright 2025 (E) Graph based transformer network, Bandformer architecture graph encoder and graph2sequence modules. It is used for predicting band structure, adapted from ref. 114 with permission from arXiv,114 copyright 2025. | ||
Solving the complete many-electron Schrödinger equation presents a more formidable challenge, as it requires proper treatment of wavefunction anti-symmetry and complex electron–electron correlations. A particularly successful approach is using the Variational Monte Carlo method (VMC) technique which is a DL assisted Quantum Monte-Carlo method. In this probabilistic approach, a trial wavefunction respecting Pauli anti-symmetry and correlation is assumed and a variational method is used to reach the ground state. Hermann et al.93 introduced PauliNet, a DL framework using first-quantization representation that achieves nearly exact solutions for strongly correlated systems containing up to 30 electrons. In parallel, Pfau et al.94 developed FermiNet (see Fig. 4B for its architecture), another approach which uses first quantization to solve the many-electron Schrödinger equation. Both PauliNet and FermiNet use VMC with Slater–Jastrow–backflow (SJB) ansatz/representation for the wavefunction which consists of the Slater determinant for Pauli exclussion, Jastrow Factor for the short-range electron–electron correlation and backflow transformation to accurately incorporate correlation. More recently, von Glehn et al.95 introduced Psiformer, which replaces conventional neural networks in these models with transformer architecture, to better capture long-range electron interactions and improve convergence. While PauliNet, FermiNet (Fig. 4B), and Psiformer differ in their specific neural network architectures, they all employ VMC techniques to optimize electronic wavefunctions toward the ground state.96 Other ansatz/representations of quantum state wavefunctions which can be directly modeled as neural networks come under the umbrella of Neural Network Quantum States (NNQS) modelled with Restricted Boltzmann Machines (RBM)97 architecture. RBMs are probabilistic generative neural networks consisting of visible input and output layers and hidden layers for latent representation, and are trained to reconstruct the original distribution, thereby generative. An extension of RBMs called the deep Boltzmann machine98,99 was the first few architectures used as NNQS. Later, CNNs100 and autoregressive models101 were also used as neural networks modeling wavefunctions. Specifically, the attention mechanism within the autoregressive methods, e.g. transformer or Recursive neural networks, can automatically encode quantum correlations.102,103 All these networks are also optimized to reach the ground states using VMC.
Numerical techniques like DFT decompose the total energy of an electron to separate out the electron–electron interaction as exchange-correlation term, and solves the total energy by approximating this term. For strongly correlated systems, this assumption does not capture the interaction very well and we need special techniques104 in which we embed regions of strong correlations (where exact Hamiltonians need to be solved) within a region of weakly correlated systems where approximations works. Examples of such quantum embedding theories are Dynamic Mean Field Theory (DMFT), Density Matrix Embedding Theory (DMET), Quantum Defect Embedding Theory (QDET), Gutzwiller Approximation (GA) etc. DMFT uses a iterative frequency-dependent Green's function, which treats strong correlation as impurity within a dynamic bath of electrons, thus capturing dynamic effects e.g. phase changes. In contrast, DMET88 performs a computationally cheaper, frequency-independent self-consistency on the local density matrix, making it more efficient for ground-state properties e.g. energy. QDET105 is an embedding method using many-body perturbation theory to derive an effective Hamiltonian for localized defects, focusing on their ground and excited states. The Gutzwiller Approximation (GA),106 uses variational principles to provide a simplified wave function approach by suppressing double occupancy, offering a computationally inexpensive way to estimate correlation effects. Most of these approaches use expensive calculation of an embedding Hamiltonian making them almost an order of magnitude slower than traditional DFT. Almost a decade back, application of ML for replacing these computationally expensive steps were predicted to be feasible.107 However, application of DL in quantum embedding remains a handful. Rogers et al. have found an computational framework that replaces calculation of the expensive embedding Hamiltonian in a range of quantum embedding methodologies using a DL step, thereby reducing the computational cost to merely DFT level108 as shown in Fig. 4C. DL versions of these embedding techniques were also used for accurate interatomic force calculation in presence of string correlations. Suwa et al. have demonstrated a DL equivalent of a GA to carry out molecular dynamics showing 106 times performance improvement over traditional109 quantum calculations. Structural dynamic studies in f and d-electron correlated systems have been carried out with a combined quantum embedding technique and DL based interatomic potentials. The interatomic potential is trained on DFT data with GA.110 Lately, NNQS has also been used in combination with DMET to model strongly correlated systems.111
Zheng et al.112 developed AIQM1, an AI-enhanced quantum mechanical method that combines semi-empirical calculations with DL and dispersion corrections. This hybrid approach achieves coupled-cluster level accuracy for a range of properties, including ground-state energies for complex compounds like fullerenes, while maintaining computational efficiency comparable to semi-empirical methods. Such capabilities make AIQM1 particularly valuable for studying 2D materials with delocalized electrons and complex electronic structures.
Building on the graph-based representations introduced in Section 2.3.1, message-passing layers of GNNs have proven particularly effective for electronic structure predictions.53,115 They propagate information through crystal graphs as shown in Fig. 4E, by iteratively updating atomic features based on their local chemical environments, effectively capturing quantum–mechanical interactions between atoms. Recent advancements include MGNN (Moment Graph Neural Network), which uses moment representations to capture spatial relationships between atoms while maintaining rotational invariance.116 Unlike many equivariant models (see below) that process tensor information throughout the entire network, MGNN contracts moment tensors to scalars at the beginning of message passing, making it computationally efficient while accurately predicting properties like energies, forces, dipole moments, and polarizabilities. This approach allows MGNN to handle complex systems with accuracy approaching traditional electronic structure methods but much greater computational efficiency.
An increasingly popular approach among researchers is the adoption of equivariant neural networks (ENNs). These networks are designed such that their outputs transform predictably under symmetry operations – such as translation, rotation, or inversion – enabling them to inherently respect the underlying symmetries of the physical system (see Fig. 4D for example). By embedding these invariance properties, ENNs reduce model complexity, decrease the demand for extensive training data, and enhance both prediction accuracy and physical consistency. Notable applications include improved electronic density predictions.117,118
A tensor-based DL model, OrbNet-Equi, developed by Qiao et al.,119 incorporates geometric data by enforcing equivariance under symmetry operations. The model shows promising results for predicting electronic structures of complex molecules. In a complementary study, Tang et al.120 introduced DeepH-hybrid, an E(3)-equivariant neural network designed to learn hybrid functional Hamiltonians as a function of material structure. By bypassing the computationally intensive self-consistent field iterations of traditional methods, DeepH-hybrid achieves accuracy comparable to conventional hybrid functionals, demonstrating its effectiveness in predicting electronic structures for large-scale 2D moiré supercells, such as twisted bilayer graphene. Knøsgaard et al.67 developed a gradient boosting (GB) model using physics-aware ENDOME and RAD-PDOS fingerprints (as detailed in Section 2.3.2) to predict non-self-consistent or one-shot GW (G0W0) band structures for approximately 700 nonmagnetic 2D semiconductors from the C2DB database.26
The prediction of electronic band structures has evolved significantly with transformer-based architectures. For instance, the model Bandformer114 treats mapping of the structural graph of a crystal to its electronic band structure as a language translation task (as shown in Fig. 4F, it uses graph encoder and graph2seq modules to make sequences from structure graphs), encoding local atomic environments and high-symmetry k paths to predict the band centers (mean values) and dispersions (deviations from the mean) of the electronic bands near the Fermi level. Tested on the Materials Project database, Bandformer achieves mean absolute errors of 72 meV for band centers and 84 meV for band dispersions. As the first end-to-end approach for direct crystal structure to band structure prediction, these results are promising. While visual comparison between predicted and DFT-calculated band structures shows the model captures general electronic features, some fine details important for property prediction remain challenging to be reproduced accurately.
In the context of a more modest task of bandgap prediction, a material descriptor was developed for hybridized boron–nitrogen graphene with various supercell configurations, enabling DL models such as CNNs with transfer learning to capture the correlation between localized atomic clusters and the overall bandgap, achieving accurate bandgap prediction across different configuration scales.126
Zhang et al.127 evaluated four machine learning algorithms – support vector regression (SVR), multilayer perceptron (MLP), gradient boosting decision trees (GBDT), and random forest (RF) – for predicting bandgaps of 2D materials using C2DB. Their analysis revealed that GBDT and RF were the best for predicting bandgaps of 2D materials. Meanwhile, Rajan et al.39 focused on MXene materials, building a database of 7200 structures and applying LASSO (Least Absolute Shrinkage and Selection Operator) regularization to identify eight key features from an initial set of 47. Their Gaussian process regression model achieved exceptional accuracy with an RMSE (Root Mean Square Error) of 0.14 eV for bandgap prediction, enabling rapid screening of novel MXene compositions without requiring computationally intensive GW calculations.
Recently, transformer-based language models have been explored for predicting semiconductor band gaps by directly encoding material text descriptions. Yeh et al.128 demonstrated that the RoBERTa model can predict band gaps with high accuracy by processing textual representations of material properties, achieving a mean absolute error of approximately 0.33 eV. In a complementary approach, Lee et al.129 proposed CAST, a cross-attention based multimodal framework that fuses graph-encoded crystal structures with textual descriptions to predict material properties, showing improvements of up to 22.9% across multiple properties including band gap prediction.
In recent years, there have been concrete demonstrations of structure-to-band-structure prediction by machine learning models. Gong et al.114 introduced a graph-transformer framework that, given a crystal structure, predicts the full electronic band structure, including band gap, dispersion, and related features. Zhang et al.130 developed machine-learning models to predict the computed band gaps of double perovskite materials, illustrating progress in forward models for electronic properties. More recently, Wang et al.131 proposed a structure-informed framework for discovering flat-band two-dimensional materials, which combines a data-driven flatness score with multi-modal learning from atomic structures to identify topologically nontrivial flat-band candidates. These works underscore that while full accuracy remains a challenge, particularly for subtle features of band dispersion, AI models are increasingly capable of providing useful predictions beyond simple band gap estimates.
Beyond diffraction, other spectroscopic techniques have also benefited from DL-driven structure prediction. Vladyka et al.133 developed an MLP to analyze changes in X-ray emission spectra, focusing on Ge Kβ peaks at elevated pressures in amorphous GeO2. By encoding local atomic environments using Coulomb matrices, their model reliably predicts changes in coordination of a target atom from emission spectra, allowing for structural reconstruction from spectral moments.
Chen et al.134 trained their simple feedforward neural network with two hidden layers to predict ground-state electronic structures from core-loss spectroscopy. Using carbon K-edge ELNES/XANES spectra as input, their feedforward neural network with two hidden layers accurately reconstructed the carbon s- and p-orbital partial density of states (PDOS) for both occupied and unoccupied states. Their approach not only predicted electronic structures from experimental data but also demonstrated successful extrapolation to larger molecules, showing that noise-filtering preprocessing and careful model training enhance prediction performance for real experimental spectra.
| Model type | Title of study | Structure representation | Summary | Target materials |
|---|---|---|---|---|
| First principles | PauliNet93 | First-quantization, Slater–Jastrow-backflow wavefunction | Variational Monte Carlo (VMC) with DL ansatz, nearly exact for strongly correlated systems up to 30 electrons | Strongly correlated systems |
| FermiNet94 | First-quantization | Deep NN solves many-electron Schrödinger equation with antisymmetry, optimized by VMC | Strongly correlated systems | |
| Psiformer95 | Transformer in first-quantization wavefunctions | Replaces conventional NN with transformer, better captures long-range electron interactions, improves VMC convergence | Correlated molecular systems | |
| Neural network quantum states (NNQS)96,97,99 | RBM, DBM, CNN, autoregressive | Neural network wavefunctions trained with VMC; autoregressive transformers encode correlations efficiently | Generic quantum systems | |
| DL-assisted quantum embedding108,109 | Embedding Hamiltonian | DL replaces costly embedding Hamiltonian step in DMFT/DMET/QDET/GA, reduces scaling to DFT level | Strongly correlated f/d electron systems | |
| AIQM1 (ref. 112) | Hybrid semi-empirical + DL + dispersion corrections | Achieves coupled-cluster accuracy with semi-empirical cost, effective for delocalized electrons | Complex compounds, fullerenes | |
| Physics informed DL | MGNN (moment GNN)116 | Crystal graphs, moment tensors | Efficient and accurate prediction of energies, forces, dipoles, polarizabilities | General crystals |
| OrbNet-Equi119 | Tensor + equivariant geometry | Enforces equivariance, improves accuracy in electronic property predictions | Complex molecules | |
| DeepH-hybrid120 | E(3)-equivariant NN | Learns hybrid functional Hamiltonians without SCF with comparable accuracy | Moire supercells | |
| ENDOME/RAD-PDOS67 | Physics-aware electronic fingerprints ENDOME and RAD-PDOS | Predicts G0W0 band structures for ∼700 semiconductors | Nonmagnetic 2D semiconductors | |
| Data-based DL arroaches | Bandformer transformer114 | Graph-to-sequence (structural graph > band sequence) | Treats band prediction as language translation; MAE ∼72 meV for band centers | Materials project crystals |
| Basic CNN126 | Local cluster descriptors | Captures bandgap variations in B–N graphene supercells | Hybridized B–N, graphene | |
| Gaussian process regression + LASSO39 | Descriptors from materials properties | Predicts MXene bandgaps (RMSE 0.14 eV) after feature selection | MXenes | |
| RoBERTa (transformer, NLP)128 | Textual materials descriptions | Language model predicts bandgaps from text (MAE ∼0.33 eV) | Semiconductors | |
| CAST (cross-attention multimodal)129 | Graph + text fusion | Improves predictions (band gap + others) by combining structure + literature embeddings | General crystalline materials | |
| DL for experiments | PhAI (CNN + MLP)132 | Diffraction patterns + phase recycling | Reconstructs electron density maps, solves crystallography phase problem | 2D crystals |
| MLP (Coulomb matrices)133 | Encoded local environments | Reconstructs structure from X-ray emission spectra under pressure | Amorphous GeO2 | |
| Feedforward NN (2 hidden layers)134 | ELNES/XANES spectra | Reconstructs carbon PDOS from spectroscopy, generalizes to larger molecules | Carbon-based materials |
3.2 DL for interatomic potentials
Interatomic potentials capture empirical forms of interactions among species under various geometries. Traditional approaches fit forces and energies from first-principles simulations to predetermined functional forms, including simple empirical potentials (Lennard–Jones, Morse), bond-order potentials modeling directionality and variable bond strengths (Tersoff, REBO), and embedded-atom method potentials for metallic systems. These conventional fittings typically rely on least-squares methods or evolutionary algorithms such as genetic algorithms (GAs), which often struggle with complex structural configurations.DL has revolutionized this field by eliminating the need for predefined functional forms, instead learning the potential energy surfaces directly from data (Table 4). DL-based potentials can accurately model geometrical configurations, energy–distance relationships, and many-body interactions while reducing human bias in the fitting process.141 This approach allows for substantially improved accuracy and transferability across diverse atomic environments, particularly important for 2D materials with their unique bonding characteristics and surface effects. Various implementations have emerged, including DEEPMD135 (see Fig. 5A for its architecture), which uses deep neural networks to represent the many-body potential energy function, and ænet-PyTorch,142 which implements atom-centered neural networks with specialized symmetry functions. These ML potentials can achieve near-DFT accuracy at a fraction of the computational cost, enabling large-scale molecular dynamics simulations of 2D materials that would be prohibitively expensive with conventional ab initio methods.
 | ||
| Fig. 5 Various architectures used for generating interatomic potentials and identifying topological properties. (A) DPMD architecture, one of the first deep learning based interatomic potential, adapted from ref. 135 with permission from American Physical Society,135 copyright 2025. (B) Comparison of architectures of various high-dimensional NN potentials, adapted from ref. 136 with permission from American Chemical Society,136 copyright 2025. (C) SchNet architecture, a rotationally invariant interatomic potential, adapted from ref. 137 with permission from arXiv,137 copyright 2025. (D) PAINN architecture, one of the equivariant graph NN, adapted from ref. 138 with permission from arXiv,138 copyright 2025. (E) Topogivity pipeline, adapted from ref. 139 with permission from American Physical Society,139 copyright 2025. (F) Deep learning based identification of topological insulators, adapted from ref. 140 with permission from arXiv,140 copyright 2025. | ||
Unlike conventional DL approaches that rely purely on data-driven optimization or generative frameworks, physics-aware neural networks integrate explicit physical laws – such as governing differential equations, conservation laws, or symmetry constraints – directly into their architectures or training procedures. Physics-informed neural networks (PINNs) represent a specific subset of these approaches, typically enforcing physical constraints through additional terms in the loss function. More broadly, physics-aware DL models include various architectures and methods designed to respect and incorporate physical insights beyond just loss-function regularization. This integration of data-driven learning and physical knowledge provides enhanced interpretability, improved accuracy, and physical consistency. Consequently, physics-aware models, including PINNs, are especially beneficial in scenarios involving limited data, constrained computational resources, or when predictions must rigorously adhere to fundamental principles, as commonly encountered in modeling complex systems like 2D materials.
One of the earliest attempts to make physically aware potentials was by encoding the local environment in the model, allowing it to choose a reference geometry close to the training examples during prediction. For example, PINN potentials were proposed to enhance the transferability of machine-learning interatomic potentials by combining a physics-based analytical bond-order model with neural network regression, showcased through a general-purpose PINN potential for aluminum and tantalum.143–145 Recently, it was found that in case of 2D materials, separating interlayer and intralayer interactions while modeling interatomic potential can lead to almost an order of magnitude rise in accuracy over potentials that treat all interactions together. The authors also found that for Moire lattices, a physics-aware validating metric based on stacking configurations performs much better than traditional metrics like force and energy.146
A challenge for any interatomic potential is to remain transferable for any geometric configuration, especially for NN-based potentials as they do not work from first principles. Towards this, Behler and Parrinello147 designed high-dimensional NN potential which calculates potential energy surfaces as superposition of atomic contributions. This allowed complex geometries to be modeled precisely using NN potentials. This also encodes the atomic structures as Atomic Environment Vectors (AEVs) and embeds the associated symmetries with Behler–Parrinello symmetry functions or Justin Smith symmetry functions.136 Several extensions of these high-dimensional potentials are lately designed e.g. ANAKIN-ME (ANI),148 AIMNet,149 AIMNet-ME,150 ML-EHM.151Fig. 5B gives a comparative picture of their architectures. Recently, further iterations of these potentials were reported with improved long-range interaction models with dispersion correction, electrostatic interactions, etc152. Another challenge for interatomic potentials is to remain relevant for various chemical environments. In last five years, several such Universal ML Inter-atomic Potentials (UMLIPs) were developed opening the door for creating new materials which are chemically stable.153 Even though UMLIPs are typically trained on extensive datasets covering diverse chemical and coordination environments, they often struggle with out-of-distribution predictions154 – for instance, accurately predicting surface energies, since surfaces inherently break periodic boundary conditions by definition.155 These potentials have not only shown promises in predicting evolving dynamic simulations, they show accurate prediction for even collective phenomena like phonon behavior.156 The first UMLIP was MEGNet51 which used a graph architecture and was trained on the Materials Project database. Since then several UMLIPs were developed which mostly used graphs to encode structure information.15,157–160 Clearly, graph NNs play a pivotal role in encoding wide range of chemical and coordinate information helping build UMLIPs. Therefore, we are going to discuss the various graph-based ML potentials in the coming paragraphs.
Graph NNs have been the representation of choice for physics aware potential development. Some of the graphs include physical awareness by encoding invariance to rotations. Examples of such GNNs are ALIGNN-FF,161 SchNet137(Fig. 5C), MEGNet,51 M3GNet158 and so on.
Similar to electronic structure prediction, equivariance principle has also been introduced for designing interatomic potentials as well to explicitly respect symmetries such as translational, rotational, and permutation invariance. Several popular interatomic potentials were generated around this equivariance principle. NequIP is an interatomic potential which employs message-passing GNNs and E(3)-equivariant convolution operations on tensors, thus requiring much less data for training and achieving accuracy of ab initio simulations.162 Several interatomic potentials were then developed based on message passing GNNs, e.g. EGNN,163 E2GNN,164 PAINN138 (Fig. 5D), GemNet,165 SEGNN,166 NewtonNet167 and many more. Allegro is another interatomic potential which does not use atom-centered message passing but makes a many-body potential using a tensor product of equivariant representations.168 Another message passing equivariant NN potential is MACE, which uses higher order messages to reduce the number of message passing iterations.159
The development of accurate UMLIPs has greatly accelerated simulations and property predictions of 2D materials, though significant challenges remain in ensuring their reliability and transferability across diverse conditions.15
3.3 DL for topology and strong electronic correlations
Topological materials represent a frontier in condensed matter physics, characterized by electronic properties that remain robust against perturbations due to the underlying mathematical principles. Over the past decade, systematic approaches have been developed to discover and classify topological band structures in condensed matter systems by considering both local symmetries and crystalline symmetries.169–171 A key feature of these systems is the topological obstruction that prevents smooth transitions between electronic states with different topological characteristics, requiring the closing of energy gaps or other dramatic changes in the electronic structure.| Model architecture | Title of study | Structure representation | Summary | Target materials |
|---|---|---|---|---|
| Deep NN based models | DEEPMD135 | Atomic coordinates + environment descriptors | Deep NN represents many-body potential energy surface; achieves near-DFT accuracy for large-scale MD | 2D + 3D materials |
| ænet/ænet-PyTorch142 | Atom-centered symmetry functions and descriptors | Training of neural network interatomic potentials on both energies and forces, using force with energy improves performance | Metallic and ionic systems | |
| PINN potentials143,144 | Analytical bond-order + NN regression | Hybrid representation gives better transferability to unseen atomic configurations (defects, surfaces, compression, etc.); demonstrated for Al and Ta | General crystalline solids | |
| HDNNPs147 (e.g. ANI, AIMNet, AIMNet-ME, ML-EHM) | Atomic environment vectors (AEVs) + Behler-parrinello/Justin-Smith symmetry functions | High-dimensional NN potentials calculates energy as superposition of atomic contributions; handles complex geometries | Diverse chemical systems | |
| Graph NN based models | ALIGNN-FF50 | Graph + line graph (bond angles) | Physics-aware graph NN interatomic potential; explicitly includes bond angles | 2D/3D crystals |
| SchNet137 | Continuous-filter convolution on atomic environments | Rotationally invariant NN potential, widely used as benchmark | Molecular + solid-state systems | |
| M3GNet158 | Graph NN with 3-body interactions | Incorporates higher-order interactions, universal interatomic potential | Wide range including 2D materials | |
| Equivariant representations | NequIP162 | Message-passing GNN + E(3)-equivariant convolutions | Internal features that transform like tensors under rotation/translation. This requires far less training data to achieve ab initio accuracy | 2D and 3D systems |
| PaiNN138 | Equivariant message passing with scalar + vector features | PaiNN can predict tensorial molecular properties (like dipole moments, polarizability) and simulate molecular spectra (IR, Raman) | Molecular & crystalline | |
| GemNet165 | Equivariant graph-based encoding | Two-hop message passing to capture distances, angles, and dihedrals, invariant to translations, equivariant to permutations and rotations | Molecular & crystalline | |
| SEGNN166 | Equivariant graph-based encoding | Node and edge features include physical quantities like vectors or tensors (e.g. forces, velocities). Uses steerable MLPs and equivariant message passing | Molecular & crystalline | |
| MACE159 | High order E(3) equivariant message passing | Uses higher-order messages (up to four-body interactions) rather than only pairwise ones, reaches SOTA accuracy in low data regime | Molecular & crystalline | |
| Allegro168 | Tensor product equivariant reps | Builds many-body potential without atom-centered message passing | Molecular & crystalline |
The mathematical relationship between TB Hamiltonians and topological invariants has been rigorously established in condensed matter theory, providing a good foundation for ML applications. This precise correspondence has been explored by building supervised ML algorithms that learn mapping from TB parameters to topological properties without requiring explicit calculation of topological invariants.172–174
Complementing these supervised approaches, Scheurer and Slager175 demonstrated that unsupervised clustering techniques can also classify topologically distinct TB Hamiltonians. This approach is particularly powerful because it does not rely on specific parameterizations of the Hamiltonian. Taking a different direction, Peano et al.176 employed CNN to generate TB Hamiltonians directly from unit cell geometries, effectively capturing topological electronic features. This method leverages the NN to map arbitrary atomic structures to symmetry-enhanced TB models, enabling prediction of band structures and their topological properties, such as fragile topologies and Chern numbers, with high accuracy and computational efficiency. Extending this paradigm of leveraging ML for topological design, explicit topology optimization, utilizing the Moving Morphable Components (MMC) method as described by Du et al.,177 defines a structure descriptor, where a multitask learning (MTL) model concurrently predicts discrete-valued topological invariants and bandgaps for higher-order topological insulators.
As multiple DFT databases of 2D materials have been developed, ML tools mapping between realistic 2D material structures and topological structures were made possible. Schleder et al.178 employed the multi-task Sure Independence Screening and Sparsifying Operator (SISSO) method to engineer atomic feature-based descriptors from DFT databases, followed by the XGBoost tree algorithm to classify materials as topologically trivial or non-trivial with over 90% accuracy. This approach enabled the prediction of 56 novel topological materials, including 17 quantum spin Hall insulators, without requiring a priori structural knowledge, demonstrating significant advancement over traditional trial-and-error methods.
Building on this trend of ML applications in topological material discovery, Xu et al.139 introduced “topogivity”, a machine-learned chemical parameter that quantifies each element's tendency to form topological materials, enabling researchers to predict whether a material is topological based solely on its chemical formula with high accuracy (>80%). This DL architecture shown in Fig. 5E led to the discovery of new topological materials that could not be identified using traditional symmetry-based methods, demonstrating a simple and effective heuristic for materials discovery.
DL has proven to be valuable for phase classification in topological strongly correlated systems. For Fractional Quantum Hall (FQH) systems specifically, both supervised approaches, as demonstrated by Matty et al.179 or Li et al.140 (see Fig. 5F), and unsupervised methods, such as those employed by Jiang et al.,180 Jin and Wang,181 have been successfully applied to identify and characterize the complex phases in these systems. Zhang and Kim182 developed quantum loop topography, which constructs specialized input features for neural networks that successfully distinguish Chern insulators and fractional Chern insulators from trivial insulators. More recently, Teng et al.183 applied attention-based neural network-variational Monte Carlo methods to accurately predict wavefunctions in FQH systems, revealing microscopic features beyond traditional approximations. In a different approach, Noronha et al.184 demonstrated that neural networks can predict the Bott index (a topological invariant) in 2D topological superconductors with magnetic impurities by analyzing local DOS, providing an efficient method to identify topological phases from experimentally accessible measurements.
The presence of flat bands is an indicator of strong electronic correlations since suppression of kinetic energy enhances electron–electron interactions, leading to correlated quantum phenomena such as chiral plasmons,185 Chern insulators,186 and unconventional superconductivity,2 observed in twisted bilayer graphene and other 2D systems. Hence, high-throughput computational methods have been employed to identify flat bands in 2D materials.
Top-down data-driven searches leverage constraints such as bandwidth187–189 to screen materials using DFT calculations. These attempts suffer from arbitrariness in labeling band index due to band crossings. A CNN model was introduced to detect flat bands directly from images of electronic band structures, eliminating the dependency on band indexing.68 Through periodic table representations,190,191 recent studies employed CNN to predict occurrence of flat bands in 25 new Heusler alloys. Ma et al.192 have used a MLP to predict band gap in flat band system of twisted bilayer graphene (TBLG) with help of a physically interpretable descriptor designed with SISSO method. In a similar study on twisted bilayers dubbed DeepH, a DL model is used in predicting band gap and bandwidths of flat bands.193 Another study194 uses the DeepH model to explore MoSe2/WSe2 moiré lattices. Classification of flat band systems was achieved through an autoencoder based self-supervised model and subsequent clustering algorithms.69 In another research, a CNN is used to identify unique signatures of flat band states to distinguish them from conventional localized and extended states by training on wavefunctions from a molecular orbital representation.195
3.4 DL for other downstream tertiary properties
Beyond fundamental electronic structures, DL has successfully predicted numerous application-specific materials properties that directly inform practical applications. These downstream tertiary properties are crucial for identifying 2D materials that are suitable for specific technological needs.In thermoelectric applications, several approaches have shown promising results. Gan et al.196 combined high-throughput DFT calculations with neural networks to accurately predict maximum ZT (dimensionless thermoelectric figure of merit that quantifies the efficiency of a material in converting heat to electricity), and optimal doping types in layered semiconductors. Na et al.197 introduced DopNet, which explicitly models host materials and dopants separately, achieving 68% lower prediction errors for unseen materials. Ishiyama et al.198 demonstrated the usage of Bayesian optimization to enhance thermoelectric properties of III-V semiconductor thin films, achieving a three-fold ZT improvement in just six optimization cycles. Beyond thermoelectrics, Wang et al.199 developed a self-supervised probabilistic model for shape memory alloys that learns atomic representations directly from crystal structure data, enabling the discovery of novel shape memory alloy candidates. Magnetic properties have also been successfully predicted using GNNs. Minch et al.200 developed a graph-based DL algorithm using ALIGNN50 model to predict atomic magnetic moments of 2D materials based on a Cr2Ge2Te6 prototype.
The Hierarchical Correlation Learning for Multi-property Prediction (H-CLMP) framework, as presented by Kong et al.,201 addresses the challenge of predicting multiple material properties simultaneously. Their approach integrates three key components: (i) composition-based property prediction, (ii) the learning of correlations among target properties within a multi-target regression framework, and (iii) the use of transfer learning to leverage training data from related but distinct property domains. The model was demonstrated by predicting spectral optical absorption coefficients across a range of photon energies for complex metal oxides, using only their elemental compositions. The best performance was achieved with the transfer learning extension, where a GAN was pre-trained on computational DOS data – a tangential property domain – and then employed to augment the prediction of absorption coefficients. This work shows how extra data can improve predictions when direct training data is limited.
4 DL models for inverse design of 2D materials
While forward design maps material structures to their electronic properties as reviewed in the previous section, inverse design addresses the more challenging problem: identifying material structures that yield desired electronic properties. This reverse mapping presents significant challenges because the forward relation is not one-to-one (injective) – multiple material structures (polymorphs) or conditions (temperature, pressure) can produce similar properties, and small structural variations can lead to dramatically different electronic behavior.Data-driven inverse mapping from the property/functional space to the chemical space202 has evolved substantially in recent years, transforming from traditional search-based approaches to sophisticated generative AI methods. This evolution represents a paradigm shift in how we conceptualize materials discovery, moving from discrete sampling to continuous exploration of chemical space.
Inverse design approaches can be broadly categorized into two main frameworks: non-generative and generative methods. Non-generative approaches include: (a) high-throughput screening of discrete chemical space to locate the desired material candidate, (b) evolutionary algorithms such as genetic algorithms, particle swarm optimization, Monte Carlo tree-search, and random walk based materials design, and (c) iterative optimization techniques including Bayesian optimization (BO) and reinforcement learning (RL).13,203 The DL architectures behind these approaches are illustrated in Fig. 6. In these non-generative approaches, models identify optimal candidates with desired properties from an existing pool of materials.
 | ||
| Fig. 6 Inverse design using DL to identify material structures can start from either desired electronic structures or end properties of the materials. The DL architectures can be broadly classified into non-generative and generative categories. Examples of non-generative model architectures are dense or convolutional NNs or invertible NNs, whereas examples of generative architectures are GANs, VAEs, diffusion and autoregressive models. | ||
In contrast, generative architectures – including VAE, GAN, autoregressive models, and diffusion models, as shown in Fig. 6 – represent a fundamental shift, as they learn to generate entirely new material candidates within a continuous chemical space.202–205 Rather than searching existing databases, these methods learn the underlying distribution of valid materials and can generate novel structures that may not exist in the training datasets but possess target properties.
For 2D materials specifically, inverse design presents unique opportunities and challenges. The reduced dimensionality and distinctive quantum confinement effects of 2D systems create electronic properties highly sensitive to structural modifications, making them particularly suitable targets for AI-driven design. In this section, we examine both non-generative and generative DL approaches for inverse design of 2D materials, their implementation strategies, and the challenges they face.
4.1 Non-generative DL for inverse design of 2D materials
While traditional inverse material design approaches like evolutionary algorithms often operate without neural networks, modern implementations increasingly incorporate DL to enhance their efficiency and performance. This section focuses specifically on neural network-assisted inverse design methodologies, including decision tree frameworks, direct mapping networks, invertible neural networks, and optimization-enhanced approaches.A notable advancement in decision tree frameworks is CASTING206 (see Fig. 7A), which significantly enhances efficiency of decision making by introducing Monte Carlo Tree Search (MCTS) based on reinforcement learning. The framework has been used for predicting structures of representative examples of 2D materials – graphane and hexagonal boron nitride.
 | ||
| Fig. 7 Non-generative inverse deep learning. (A) CASTING: a Continuous Action Space Tree search framework for inverse design, adapted from ref. 206 with permission from Springer Nature, npj Comput. Mater., 2023. (B) Inverse design in quantum nanophotonics via LDOS-guided deep learning, reproduced from ref. 207 with permission from Walter de Gruyter/Science Wise Publishing, Nanophotonics, 2023. (C) Deep-learning inverse design model predicting compositions from the target DOS, adapted from ref. 208 with permission from American Chemical Society, J. Mater. Chem. A, 2024. (D) Inverse materials design workflow and parameter-space specification for MoS2, reproduced from ref. 209 with permission from Springer Nature, npj Comput. Mater., 2021. | ||
Combination of high-throughput experiments with active learning algorithm called Gaussian process BO have recently been used for designing quasi-2D halide perovskites by optimizing photoluminescence intensity and chemical stability.211
For more complex property–structure relationships, CNNs have proven particularly effective. Bang et al.208 used a CNN architecture to directly predict composition vectors (see. Fig. 7C) describing crystal structures from vector representations of DOS data, enabling the discovery of inorganic crystals optimized for catalysis and hydrogen storage applications. The CNN's ability to capture spatial hierarchies in data makes it well-suited for translating between electronic and structural representations.
In the domain of semiconductor heterostructures, Pimachev and Neogi212 developed a hybrid approach combining random forests and neural networks for forward prediction of electronic properties from crystal graph representations. For inverse design, they employed a CNN to map desired band structures back to corresponding heterostructure configurations, effectively establishing a bidirectional relationship between structure and properties.
The bijective nature of INNs allows them to be trained in the forward direction (structure to properties) and directly applied to reverse (properties to structure) without additional optimization steps. MatDesINNe,209 shown in Fig. 7D, demonstrated this capability for 2D materials by developing an INN that predicts electronic bandgaps of MoS2 under varying conditions of tensile strain and applied electric field. Once trained, their model could directly generate combinations of strain and field values that would yield a target bandgap, providing a computationally efficient pathway for property-based materials design.
4.2 Generative DL methods for inverse design
Unlike non-generative approaches that search existing material spaces, generative DL methods create entirely new material candidates by learning the underlying distribution of valid materials. These approaches offer unprecedented opportunities for exploring the vast chemical space of 2D materials by generating novel structures with targeted electronic properties.Generative AI models – including VAEs, GANs, diffusion models, and sequence-based models – have demonstrated significant impact in inverse materials design. As summarized in Table 5, these frameworks employ diverse architecture designs and structure embedding algorithms. We organize these approaches into five categories: (a) GAN-based approaches, (b) VAE-based approaches, (c) diffusion models, (d) RNN and transformer-based sequence models, and (e) hybrid approaches combining multiple generative AI models.
| Model architecture | Title of study | Structure representation | Summary | Target materials |
|---|---|---|---|---|
| Cross domain GAN | CrystalGAN214 (Fig. 8A) | Simple matrices formed of lattice vectors and fractional coordinates | Brings in cross domain learning in GAN instead of learning against noise. Model predicts novel, chemically stable ternary crystal structure | Hydride compounds |
| GAN | Kim et al.218 | Point cloud representation consisting of cell vectors and scaled coordinates | The method generates new crystal structures for Mg–Mn–O ternary materials and evaluates their properties via high-throughput virtual screening | Ternary system: Mg–Mn–O |
| GAN | CycleGAN240 | Image of surface showing atomic sites | Image to image generation between STM image and surface crystal structure and vice versa; uses discriminators between actual and generated STM image as well as surface atomic structures | Any surface |
| Graph attention transformer + GAN | EquiformerV2 (ref. 219) | Equivariant graphs | The model uses self-supervised learning of masked crystal structures representations, which are then fine-tuned for downstream tasks such as stability classification and regression | Inorganic crystals |
| GAN | GAN-DDLSF220 | Continuity vector matrix consisting of cell vectors and atomic coordinates | Optimize the latent space via data-driven fusion to mitigate mode collapse of GANs | Gallium nitride GaxNy compositions |
| VAE | FTCP (Fourier transformed crystal Properties)221 | Matrix with real and reciprocal-space features with element property matrix | Using a VAE with property-structured latent space (both input and latent space have combined lattice and property data), demonstrates generation of novel inorganic crystals at user-defined formation energy, bandgap, TE power factor | Inorganic crystals |
| Diffusion-VAE | CDVAE215,223 (Fig. 8B) | Direct coordinate representation with an equivariant graph network (node = atom, edges = bonds) | Trains the diffusion-VAE (GNN encoder, diffusion based decoder) on a database of 2D crystals, then generates new 2D materials | 2D materials |
| Diffusion-VAE | Con-CDVAE224 | Equivariant graph network | Extends CDVAE framework to allow target properties (band gap, formation energy, etc.). Implements a two-step training (first building property-aware latent space, then generating structures) | Inorganic crystals |
| Diffusion-VAE | Cond-CDVAE216 (Fig. 8C) | Matrix with atomic species, coordinates and lattice vectors | Trained on 670000 materials from Calypso dataset; enables user-defined composition and pressure to generate physically plausible, stable crystal candidates |
Inorganic crystals |
| VAE | WyCryst222 | Wyckoff position-based representation | Enforces space group symmetry via Wyckoff positions. Combines a VAE with DFT refinement to generate stable, symmetry-compliant structures | Inorganic crystals |
| Diffusion using a three-channel matrix representation | Supercon-Diffusion217 (Fig. 8E) | A three-channel matrix that encodes the stoichiometry (integer, first decimal, second decimal) of superconductors | The method accurately learns doping characteristics, achieving high doping effectiveness and electrical neutrality, and proposes 200 new potential high Tc superconductors | Doped high Tc superconductors (cuprates, iron-based, etc.) |
| Guided diffusion model | GaUDI241 | Molecules are represented using a graph-of-rings (GOR) representation that captures ring connectivity and geometry | Combines an equivariant graph neural network for property prediction with a diffusion denoising process | Organic molecules |
| Diffusion model with Riemannian manifold | CrystalGRW225 | EquiformerV2: equivariant GNN | Framework with geodesic random walks to denoise random noise into crystal structures; preserves crystallographic symmetry and enables conditional control (e.g., specifying point groups) to generate novel crystals with desired properties | Inorganic crystals |
| Diffusion | MatterGen55 (Fig. 8D) | Tuples of atomic species, coordinates and lattice vectors | The forward diffusion process corrupts an input structure. An equivariant score network does reverse denoising process with an adapter module that guides to target chemistry, symmetry, and scalar properties (e.g., band gap, bulk modulus) | Inorganic crystals |
| Diffusion | SymmCD226 | Space group, lattice parameters, asymmetric unit coordinates (fractional), site symmetries | A diffusion model that explicitly encodes crystallographic symmetry via asymmetric units and site symmetries, enabling diverse yet valid crystal generation | Inorganic crystals |
| Diffusion | WyckoffDiff227 | Protostructures: String representations with space group and Wyckoff positions | A discrete diffusion model that generates symmetry-constrained protostructures using Wyckoff positions, enabling fast generation of thermodynamically stable crystals | Inorganic crystals |
| Transformer | BLMM231 | Text representation for stoichiometry | A blank-filling LLM trained on ‘material grammars’ | Inorganic crystals |
| Transformer + UMLIP | Material transformer generator (MTG)232 | BLMM231 | Two transformer architectures simultaneously generate material compositions which are then relaxed with M3GNET UMLIP to predict structures | 2D materials |
| GPT2 | ATOMGPT233 | ALIGNN graph | Bidirectional prediction of structure-to-property and property-to-structure | Superconducting materials |
| RNN | Xiao et al.52 | SLICES: String-based crystal representation ensuring symmetry invariance and invertibility | Uses a transfer learning framework to train RNN on materials project dataset for learning SLICES representations. A transfer learned RNN then predicts SLICES for novel semiconductors | Direct gap crystalline semiconductors |
| Transformer | Wyformer58 | Wyckoff representation consisting of point group notations for each atom | A permutation-invariant transformer generates symmetry aware representations of novel materials for each space group | Inorganic crystals |
| Transformer | Matra-Genoa57 | Sequence containing composition, lattice, Wyckoff position tokens and atomic coordinates | An autoregressive transformer that conditions generation on target properties (e.g., energy above the convex hull) to produce stable crystal structures | Inorganic crystals |
| Transformer | CrystalFormer242 | Sequence that integrates space group numbers, Wyckoff letters, chemical species, fractional coordinates, and lattice parameters | An autoregressive transformer that exploits space group symmetry to reduce the degrees of freedom in crystal generation. It shows improved performance for symmetric structure initialization, element substitution, and property-guided design | Inorganic crystals |
| Wasserstein GAN + VAE | WGAN-VAE238 | Voxel-based representation capturing both atomic positions and lattice parameters using VAE | WGAN generates thermodynamically stable structures whereas VAE retains chemical validity | Vanadium oxide VxOy compositions |
| VAE + GAN + diffusion | VGD-CG237 | One hot encoding of composition and properties like band gap | A generator consisting of VAE, GAN and diffusion model (VGD-CG) generates compositions of target materials. A template-based structure prediction algorithm then predicts the crystal structures | Inorganic crystals |
| Diffusion + autoregressive token prediction | UniGenX59 | Text sequence consisting of chemical formula, lattice vectors and atomic coordinates | The diffusion model improves the precision of prediction, whereas attention based autoregression excels in predicting sequences | Inorganic crystals, organic compounds |
 | ||
| Fig. 8 Generative inverse deep learning methods. (A) CrystalGAN architecture, adapted from ref. 214, arXiv, preprint, 2018. (B) Workflow for generating 2D material candidates using the CDVAE generative model, reproduced from ref. 215 with permission from Springer Nature, npj Comput. Mater., 2022. (C) Architecture of Cond-CDVAE, adapted from ref. 216, arXiv, preprint, 2024. (D) Inorganic materials design with MatterGen, reproduced from ref. 55 with permission from Springer Nature, Nature, 2025. (E) Supercon-Diffusion architecture, adapted from ref. 217 with permission from Wiley, InfoMat, 2024. | ||
For example, CDVAE model215,223 and its extensions integrate a diffusion-based decoder within a VAE architecture, as discussed in Section 4.2.2. Recent studies have explored combining VAEs with Deep Kernel Learning (DKL), where the VAE's generative capabilities are aligned with target properties via Gaussian process regression in the latent space, facilitating generation of materials with specific properties.235,236
Further hybridization involving GANs, VAEs, and diffusion models has been demonstrated for the inverse design of target material compositions.237 Microsoft researchers have successfully combined the sequence modeling strengths of autoregressive transformers with the precision offered by diffusion models, enabling the generation of diverse organic and inorganic materials using Wyckoff representations for crystalline structures.59 Another notable approach involves integrating a Wasserstein GAN and VAE, with the GAN generating candidate structures and the VAE ensuring chemical validity, as demonstrated in the accelerated discovery of stable vanadium oxide compositions.238
Moreover, integration of generative large language models (LLMs) with high-throughput experimental data has been applied to the inverse design of doped perovskites. Here, a fine-tuned LLM trained on ferroelectric domain-specific knowledge constructs knowledge graphs linking structural phases, synthesis conditions, and desired properties, significantly enhancing targeted material discovery.239 These hybrid methodologies illustrate the growing potential of multi-modal approaches to overcome individual model limitations and accelerate advanced materials discovery.
4.3 Multi-objective optimization
MOO provides a powerful framework for designing 2D materials by balancing conflicting properties, yielding a Pareto front of optimal trade-off solutions. While early MOO efforts laid foundational groundwork, such as using genetic algorithms to optimize bandgap and mass in 2D phononic crystals,243 or differential evolution for inverse design,244 the integration of MOO with DL has significantly enhanced its capability, particularly for navigating through the complex design space of 2D materials.Recent advancements highlight the synergy between MOO and DL, enabling significant progress in material design and discovery. For instance, Krishnamoorthy A. et al. employed NSGA-III to parameterize interatomic potentials for MoSe2, simultaneously optimizing structural and thermal properties while quantifying thermal conductivity uncertainty.245 Similarly, Varasteanu & Kusko combined NSGA-II with the transfer matrix method to enhance the sensitivity and reflectivity of 2D materials, modified surface plasmon resonance sensors, achieving configurations like Ag–BaTiO3–graphene/WS2.246 Zhang et al. utilized a multi-objective generic algorithm to parameterize potentials for MoSe2, improving transferability for large deformation and fracture simulations.247 Additionally, Jablonka et al. introduced a bias-free active learning algorithm using Pareto dominance to reconstruct the Pareto front for polymer design, in principle adaptable to 2D systems, drastically reducing evaluation needs.248
Contemporary developments further emphasize the power of coupling DL with MOO. Roy et al. employed multi-objective Bayesian optimization (MOBO) with active learning to identify Pareto-optimal 2D material compositions, reducing the search space by up to 36%.249 Chen et al. optimized the optical properties of liquid-phase exfoliated MoS2 using a genetic algorithm-coupled artificial neural network, precisely tuning absorbance and bandgap.250
Although much recent work emphasizes optimizing structural, thermal, or optical properties, the potential of MOO for electronic structure design is also very promising. By simultaneously considering multiple electronic parameters – such as effective mass, bandgap size and band alignment – MOO can efficiently address inherent trade-offs critical to electronic applications. For example, incorporating MOO with a compound loss function into DL could accelerate the discovery of 2D materials with tailored electronic properties for next-generation electronics, where trade-offs between different electronic characteristics are often necessary. A recent study demonstrated this capability by leveraging Wyckoff position augmentation and transfer learning to optimize for targeted space group characteristics, bandgap, and formation energies, ultimately predicting several stable structures.251
As 2D material datasets continue to grow and computational resources expand, we anticipate that MOO coupled with DL will become increasingly central to electronic structure design, enabling researchers to navigate complex property spaces and identify optimal candidates for specific technological applications.
5 Opportunities and future directions
The exciting advances in deep learning for 2D materials research offer a wealth of opportunities. Yet, significant challenges remain. DL models have already shown great promise, but their ability to generalize beyond specific datasets still needs improvement.154,252–254 This is especially important for 2D materials, known for their diverse behavior and unique properties arising from intricate interactions between chemistry, structure, and physics. Improving model architectures and ensuring high-quality data will be key to unlocking broader generalization and driving further breakthroughs in this vibrant field.A key area for future advancement is improving the interpretability of deep learning models. As these models become increasingly complex, understanding their decision-making processes is essential for scientific validation and building trust. Promising approaches include physics-aware DL and explainable artificial intelligence (XAI), which integrate fundamental physical and chemical principles directly into model architectures.14,205,255 Such strategies not only enhance predictive accuracy but also offer valuable scientific insights, enabling researchers to better understand underlying mechanisms and develop hypotheses guided by model outputs.
Another promising direction involves improving data accessibility, comprehensiveness, and standardization. Major databases like Materials Project, 2DMatpedia, and C2DB have been instrumental in accelerating materials discovery; however, there remain significant opportunities to expand their scope, particularly by incorporating more comprehensive electronic structure data such as wavefunctions. Enriching these databases with detailed datasets would enable advanced computational analyses, especially for exploring novel quantum and topological phenomena. We suggest establishing clear and standardized reporting protocols, complemented by structured ontological frameworks (e.g., MatOnto,256 MatOWL,257 and others258,259) specifically designed for 2D materials. Such frameworks would significantly enhance data usability, making it easier for researchers to find, interpret, and apply relevant information. Furthermore, embracing semantic web technologies like JSON-LD260 or Splink261 could greatly improve data interoperability. This would facilitate automated reasoning and seamless integration across diverse datasets, enabling more reliable benchmarking and validation efforts. By comprehensively standardizing structural parameters, electronic properties, synthesis metadata, computational methodologies, uncertainty quantification, and validation procedures, the research community can substantially amplify the impact and efficiency of DL-driven autonomous experimentation in materials science.
Another direction of improvement is ensuring DL predictions are consistent with experiments. Initial data-based DL models used to be purely trained on data from high-throughput first-principles calculations. Their accuracy is therefore directly related to the accuracy of the DFT data. However, later on, with the rise of physics-aware DL models like equivariant GNNs and message-passing architectures, the DL models try to predict with the awareness of the physical structure, thus being more accurate over out-of-training regime. Furthermore, accurate datasets, which closely mimic experiments, e.g. coupled cluster methods, GW, hybrid methods, and dynamic mean field theory, are also being progressively incorporated in training to reach experimental level accuracy. However, large experimental datasets with electronic properties are still missing, preventing training DL models for realistic electronic structures. Some experimental datasets on band-gaps are available, e.g. Matbench_expt_gap,75 and the dataset created by Google DeepMind.262 Another way to reach experiment-level accuracy is through the use of self-driving labs in which AI plans experiments, executes them via robotics, analyzes results, and then plans new experiments to train DL models in real time. We discuss this topic further below.
The emergence of foundation models – large-scale pretrained AI systems capable of performing multiple tasks – offers exciting new opportunities for 2D materials discovery. Although this field is still developing, foundation models have immense potential to address some of the most challenging issues facing materials science today. These challenges include diverse representations of materials data, the need to handle physics across multiple length scales, varied computational approaches, and complex interdependencies between different material properties.263 AFLOW-ML48,264 offers early capabilities for property prediction across material classes. The NIST-JARVIS framework has evolved to support numerous materials informatics tools and property prediction modules. The MACE architecture265 demonstrates foundation-like capabilities for molecular dynamics simulations, phonon spectra prediction, and battery modeling. The MLMD platform82 provides an active learning system for multiple property predictions without requiring specialized coding. Recently developed transformer-based models like MatterGPT or AtomGPT have already demonstrated encouraging progress, showing their capability to generalize across a wide range of materials properties and tasks.233,266–268 Additionally, innovative methodologies such as symbolic regression present intriguing possibilities for creating transparent, interpretable models.269,270 These approaches not only help researchers build clearer connections between theory and experiment but also foster deeper scientific insights into the underlying physical phenomena.271–273
Transformer-based large language models have also shaped the materials discovery landscape in last few years using their incredible ability to utilize vast cross-domain knowledge. Text only transformer models like,71 and bigger models like Llama274 are enabling reliable extraction of synthesis conditions, substrate choices, and performance metrics for graphene derivatives, TMDs, MXenes, and other van der Waals systems. These information then can be fed into dataset creation, inverse-design loops and lab automation. Some big examples of LLMs are Concensus, Scite,275 Elicit276 which are trained on large amount of scientific literature and materials databases. Multimodal foundation models extend this by fusing text with images e.g. surface images from TEM/AFM, diffraction, and spectroscopy (Raman/PL/ARPES) to learn both atomic structure and electronic signatures for 2D materials. Gemini, GPT-5, NotebookLM are examples of such multi-modal models. Together, these text-based and multi-modal models are helping to build structured knowledge-base to revolutionize 2D materials design.
Combining advanced AI into fully autonomous materials discovery holds immense potential to revolutionize innovation in 2D materials.5,6 A self-driven laboratory with cutting-edge AI models could propose entirely new classes of 2D materials, carefully tailored for specific applications. Once a promising candidate is identified, robotic systems will carry out precise synthesis and characterization experiments automatically. Every piece of data collected – whether successful or not – feeds back into AI, adjusting their models and improving the quality of future proposals. This iterative, closed-loop process is already seen in early experimental systems which demonstrates the feasibility of these autonomous workflows. These pioneering examples showcase how continuous interaction between AI-driven prediction and robotic experimentation can rapidly refine materials design and discovery. Looking ahead, fully integrated autonomous labs could dramatically accelerate the identification and development of new materials, going far beyond human speed and efficiency. Ultimately, this integration promises to transform materials science – not merely accelerating current methods, but enabling entirely new approaches, materials, and scientific breakthroughs previously unimaginable.36,277
Finally, quantum computing represents a fascinating emerging frontier, poised to greatly influence the future of materials discovery. Current classical computational methods often face major bottlenecks – especially when simulating strongly correlated electronic systems common in many 2D materials.278 Hybrid quantum-classical algorithms279 and quantum embeddings methods280 offer promising new ways to tackle these longstanding challenges. As quantum technologies mature, combining them effectively with existing AI approaches may open doors to breakthroughs that were previously unattainable. This powerful integration could redefine our understanding of materials and accelerate the discovery of novel phenomena.
Together, these opportunities paint a compelling vision of a future where discovering, characterizing, and optimizing new 2D materials is dramatically faster and more effective. Powered by advanced agentic AI systems, autonomous experimentation platforms, and breakthroughs in quantum computing, researchers will rapidly unlock practical innovations across diverse technological fields – from electronics and energy to quantum technologies and healthcare.
Despite their promise, current DL approaches face important limitations. Most models lack robust uncertainty quantification, making it difficult to assess predictive reliability, particularly when extrapolating beyond the training data. They are also prone to biases inherited from limited or imbalanced datasets, and their interpretability remains a bottleneck for extracting physical insight rather than just numerical predictions. Additionally, the growing computational cost and associated carbon footprint of training large-scale models raise ethical and sustainability concerns in the long term. Addressing these challenges, through improved uncertainty-aware architectures, physics-informed learning, interpretable model design, and more energy-efficient training protocols, will be essential for ensuring that DL develops as a trustworthy and responsible tool for materials discovery.
6 Conclusions
This review highlights the transformative impact of deep learning in studying and predicting electronic properties of 2D materials. DL models have rapidly evolved into powerful tools. They not only accelerate computational predictions but also unveil subtle electronic phenomena often missed by traditional methods.Combining DL with physics-aware models and autonomous experimentation represents a significant advancement. This integrated approach offers deeper scientific insights, facilitating the discovery and development of novel materials tailored for specific applications. Furthermore, emerging technologies such as hybrid quantum-classical computing expand the potential of DL and related AI approaches. Such methods are particularly valuable for simulating complex electronic interactions in strongly correlated materials – an area traditionally challenging for classical computation alone.
Looking forward, dedicated efforts toward standardizing data, innovating new methodologies, and developing autonomous experimentation platforms will be critical. These advancements will ensure DL evolves from merely a computational aid into a fundamental aspect of research strategies, greatly enriching our scientific understanding and practical utilization of 2D materials.
Author contributions
Conceptualization: AM. writing – original Draft: AM, AB, XW, HKP, YW. writing – review & editing: AM, AB, XW, YW, QY. visualization: AB, XW, YW. supervision: AM, QY. project administration: AM. funding acquisition: AM, QY.Conflicts of interest
The authors have no conflict of interest to declare.Data availability
No primary research results, no software or code have been developed, and no new data were generated or analyzed as part of this review.Acknowledgements
This study was supported by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (Grant Agreement No. 865590) and the Research Council UK [BB/X003736/1]. Q. Y. acknowledges the funding from Royal Society University Research Fellowship URF\R1\221096 and UK Research and Innovation Grant [EP/X017575/1].Notes and references
- D. Voiry, J. Yang and M. Chhowalla, Adv. Mater., 2016, 28, 6197–6206 CrossRef PubMed.
- Y. Cao, V. Fatemi, S. Fang, K. Watanabe, T. Taniguchi, E. Kaxiras and P. Jarillo-Herrero, arXiv, 2018, preprint, arXiv: 1803.02342, DOI:10.48550/arXiv.1803.02342.
- L. Yin, R. Cheng, J. Ding, J. Jiang, Y. Hou, X. Feng, Y. Wen and J. He, ACS Nano, 2024, 18, 7739–7768 CrossRef PubMed.
- S. Carr, S. Fang, P. Jarillo-Herrero and E. Kaxiras, Nat. Rev. Mater., 2020, 5, 748–763 CrossRef.
- L. Hung, J. A. Yager, D. Monteverde, D. Baiocchi, H.-K. Kwon, S. Sun and S. Suram, Digital Discovery, 2024, 3, 1273–1279 RSC.
- N. J. Szymanski, B. Rendy, Y. Fei, R. E. Kumar, T. He, D. Milsted, M. J. McDermott, M. Gallant, E. D. Cubuk and A. Merchant, et al. , Nature, 2023, 624, 86–91 CrossRef PubMed.
- K. T. Butler, D. W. Davies, H. Cartwright, O. Isayev and A. Walsh, Nature, 2018, 559, 547–555 CrossRef PubMed.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef PubMed.
- T. Björkman, A. Gulans, A. V. Krasheninnikov and R. M. Nieminen, Phys. Rev. Lett., 2012, 108, 235502 CrossRef PubMed.
- A. H. Cheng, C. T. Ser, M. Skreta, A. Guzmán-Cordero, L. Thiede, A. Burger, A. Aldossary, S. X. Leong, S. Pablo-García, F. Strieth-Kalthoff and A. Aspuru-Guzik, Faraday Discuss., 2025, 256, 10–60 RSC.
- Y. Zhang, Q. Tang, Y. Zhang, J. Wang, U. Stimming and A. A. Lee, Nat. Commun., 2020, 11, 1706 CrossRef PubMed.
- S. Dick and M. Fernandez-Serra, Nat. Commun., 2020, 11, 3509 CrossRef PubMed.
- J. Xie, Y. Zhou, M. Faizan, Z. Li, T. Li, Y. Fu, X. Wang and L. Zhang, Nat. Comput. Sci., 2024, 1–12 Search PubMed.
- C. Malica, K. Novoselov, A. S. Barnard, S. V. Kalinin, S. R. Spurgeon, K. Reuter, M. Alducin, V. L. Deringer, G. Csanyi and N. Marzari, et al. , J. Phys.: Mater., 2025, 021001 Search PubMed.
- C. A. Vital, R. J. Armenta-Rico and H. E. Sauceda, Machine Learned Force Fields: Fundamentals, its reach, and challenges, 2025, https://arxiv.org/abs/2503.05845 Search PubMed.
- M. Gray, J. M. Herbert and J. M. Herbert, Elsevier, 2024, 20, 1–61 Search PubMed.
- H. J. Kulik, T. Hammerschmidt, J. Schmidt, S. Botti, M. A. Marques, M. Boley, M. Scheffler, M. Todorović, P. Rinke and C. Oses, et al. , Electron. Struct., 2022, 4, 023004 CrossRef.
- N. Marzari, A. Ferretti and C. Wolverton, Nat. Mater., 2021, 20, 736–749 CrossRef.
- M. Garrido, A. Naranjo and E. M. Pérez, Chem. Sci., 2024, 3428–3445 RSC.
- R. Yu and F. J. Garcia de Abajo, Sci. Adv., 2020, 6, eabb4713 CrossRef PubMed.
- W. Jin and R. M. Osgood, Adv. Phys.:X, 2019, 4, 1688187 Search PubMed.
- P. Xu, X. Ji, M. Li and W. Lu, npj Comput. Mater., 2023, 9, 42 CrossRef.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner and G. Ceder, et al. , APL Mater., 2013, 1, 011002 CrossRef.
- K. Choudhary, K. F. Garrity, A. C. Reid, B. DeCost, A. J. Biacchi, A. R. Hight Walker, Z. Trautt, J. Hattrick-Simpers, A. G. Kusne and A. Centrone, et al. , npj Comput. Mater., 2020, 6, 173 CrossRef.
- S. Haastrup, M. Strange, M. Pandey, T. Deilmann, P. S. Schmidt, N. F. Hinsche, M. N. Gjerding, D. Torelli, P. M. Larsen and A. C. Riis-Jensen, et al. , 2D Mater., 2018, 5, 042002 CrossRef.
- M. N. Gjerding, A. Taghizadeh, A. Rasmussen, S. Ali, F. Bertoldo, T. Deilmann, N. R. Knøsgaard, M. Kruse, A. H. Larsen and S. Manti, et al. , 2D Mater., 2021, 8, 044002 CrossRef.
- N. Mounet, M. Gibertini, P. Schwaller, D. Campi, A. Merkys, A. Marrazzo, T. Sohier, I. E. Castelli, A. Cepellotti and G. Pizzi, et al. , Nat. Nanotechnol., 2018, 13, 246–252 CrossRef PubMed.
- D. Campi, N. Mounet, M. Gibertini, G. Pizzi and N. Marzari, ACS Nano, 2023, 17, 11268–11278 CrossRef PubMed.
- J. Zhou, L. Shen, M. D. Costa, K. A. Persson, S. P. Ong, P. Huck, Y. Lu, X. Ma, Y. Chen and H. Tang, et al. , Sci. Data, 2019, 6, 86 CrossRef PubMed.
- S. Pakdel, A. Rasmussen, A. Taghizadeh, M. Kruse, T. Olsen and K. S. Thygesen, Nat. Commun., 2024, 15, 932 CrossRef PubMed.
- R. K. Barik and L. M. Woods, Sci. Data, 2023, 10, 232 CrossRef PubMed.
- F. Xue, Y. Ma, H. Wang, L. Luo, Y. Xu, T. D. Anthopoulos, M. Lanza, B. Yu and X. Zhang, Matter, 2022, 5, 1999–2014 CrossRef.
- Y.-J. Zhang, Y.-T. Ren, X.-H. Lv, X.-L. Zhao, R. Yang, N.-W. Wang, C.-D. Jin, H. Zhang, R.-Q. Lian and P.-L. Gong, et al. , Phys. Rev. B, 2023, 107, 235420 CrossRef.
- X. Li, T. Liu, L. Li, M. He, C. Shen, J. Li and C. Xia, Phys. Rev. B, 2022, 106, 125306 CrossRef.
- S. Kirklin, J. E. Saal, B. Meredig, A. Thompson, J. W. Doak, M. Aykol, S. Rühl and C. Wolverton, npj Comput. Mater., 2015, 1, 1–15 Search PubMed.
- A. Merchant, S. Batzner, S. S. Schoenholz, M. Aykol, G. Cheon and E. D. Cubuk, Nature, 2023, 624, 80–85 CrossRef.
- M. C. Sorkun, S. Astruc, J. V. A. Koelman and S. Er, npj Comput. Mater., 2020, 6, 106 CrossRef.
- M. Yao, J. Ji, X. Li, Z. Zhu, J.-Y. Ge, D. J. Singh, J. Xi, J. Yang and W. Zhang, Sci. China Mater., 2023, 66, 2768–2776 CrossRef.
- A. C. Rajan, A. Mishra, S. Satsangi, R. Vaish, H. Mizuseki, K.-R. Lee and A. K. Singh, Chem. Mater., 2018, 30, 4031–4038 CrossRef.
- U. Petralanda, Y. Jiang, B. A. Bernevig, N. Regnault and L. Elcoro, arXiv, 2024, preprint, arXiv:2411.08950, DOI:10.48550/arXiv.2411.08950.
- R. Meng, L. da Costa Pereira, J.-P. Locquet, V. Afanas’ ev, G. Pourtois and M. Houssa, npj Comput. Mater., 2022, 8, 230 CrossRef.
- A. Marrazzo, M. Gibertini, D. Campi, N. Mounet and N. Marzari, Nano Lett., 2019, 19, 8431–8440 CrossRef PubMed.
- J. Buha and S. Bellani, Database on available 2D materials, 2024 Search PubMed.
- J.-H. Yang, H. Kang, H. J. Kim, T. Kim, H. Ahn, T. G. Rhee, Y. G. Khim, B. K. Choi, M.-H. Jo and H. Chang, et al. , Digital Discovery, 2024, 3, 573–585 RSC.
- E. Gerber, S. B. Torrisi, S. Shabani, E. Seewald, J. Pack, J. E. Hoffman, C. R. Dean, A. N. Pasupathy and E.-A. Kim, Nat. Commun., 2023, 14, 7921 CrossRef PubMed.
- L. Bassman Oftelie, P. Rajak, R. K. Kalia, A. Nakano, F. Sha, J. Sun, D. J. Singh, M. Aykol, P. Huck and K. Persson, et al. , npj Comput. Mater., 2018, 4, 74 CrossRef.
- P. Kalhor, N. Jung, S. Bräse, C. Wöll, M. Tsotsalas and P. Friederich, Adv. Funct. Mater., 2024, 34, 2302630 CrossRef.
- O. Isayev, C. Oses, C. Toher, E. Gossett, S. Curtarolo and A. Tropsha, Nat. Commun., 2017, 8, 15679 CrossRef PubMed.
- T. Xie and J. C. Grossman, Phys. Rev. Lett., 2018, 120, 145301 CrossRef PubMed.
- K. Choudhary and B. DeCost, npj Comput. Mater., 2021, 7, 185 CrossRef.
- C. Chen, W. Ye, Y. Zuo, C. Zheng and S. P. Ong, Chem. Mater., 2019, 31, 3564–3572 CrossRef.
- H. Xiao, R. Li, X. Shi, Y. Chen, L. Zhu, X. Chen and L. Wang, Nat. Commun., 2023, 14, 7027 CrossRef CAS PubMed.
- J. Gilmer, S. S. Schoenholz, P. F. Riley, O. Vinyals and G. E. Dahl, Proceedings of the 34th International Conference on Machine Learning, 2017, pp. 1263–1272 Search PubMed.
- C. Chen and S. P. Ong, npj Comput. Mater., 2021, 7, 173 CrossRef.
- C. Zeni, R. Pinsler, D. Zügner, A. Fowler, M. Horton, X. Fu, Z. Wang, A. Shysheya, J. Crabbé and S. Ueda, et al. , Nature, 2025, 639, 624–632 CrossRef CAS PubMed.
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef CAS.
- P.-P. De Breuck, H. A. Piracha, G.-M. Rignanese and M. A. Marques, arXiv, 2025, preprint, arXiv:2501.16051, DOI:10.48550/arXiv.2501.16051.
- N. Kazeev, W. Nong, I. Romanov, R. Zhu, A. Ustyuzhanin, S. Yamazaki and K. Hippalgaonkar, arXiv, 2025, preprint, arXiv:2503.02407, DOI:10.48550/arXiv.2503.02407.
- G. Zhang, Y. Li, R. Luo, P. Hu, Z. Zhao, L. Li, G. Liu, Z. Wang, R. Bi, K. Gao andet al., arXiv, 2025, preprint, arXiv:2503.06687, DOI:10.48550/arXiv.2503.06687.
- X. Chen, D. Chen, M. Weng, Y. Jiang, G.-W. Wei and F. Pan, J. Phys. Chem. Lett., 2020, 11, 4392–4401 CrossRef CAS PubMed.
- Y. Jiang, D. Chen, X. Chen, T. Li, G.-W. Wei and F. Pan, npj Comput. Mater., 2021, 7, 28 CrossRef CAS PubMed.
- K. Hansen, G. Montavon, F. Biegler, S. Fazli, M. Rupp, M. Scheffler, O. A. Von Lilienfeld, A. Tkatchenko and K.-R. Muller, J. Chem. Theor. Comput., 2013, 9, 3404–3419 CrossRef CAS.
- G. Montavon, M. Rupp, V. Gobre, A. Vazquez-Mayagoitia, K. Hansen, A. Tkatchenko, K.-R. Müller and O. A. Von Lilienfeld, New J. Phys., 2013, 15, 095003 CrossRef CAS.
- K. Hansen, F. Biegler, R. Ramakrishnan, W. Pronobis, O. A. Von Lilienfeld, K.-R. Muller and A. Tkatchenko, J. Phys. Chem. Lett., 2015, 6, 2326–2331 CrossRef CAS PubMed.
- A. Y. Toukmaji and J. A. Board Jr, Comput. Phys. Commun., 1996, 95, 73–92 CrossRef CAS.
- F. Faber, A. Lindmaa, O. A. Von Lilienfeld and R. Armiento, Int. J. Quantum Chem., 2015, 115, 1094–1101 CrossRef CAS.
- N. R. Knøsgaard and K. S. Thygesen, Nat. Commun., 2022, 13, 468 CrossRef PubMed.
- A. Bhattacharya, I. Timokhin, R. Chatterjee, Q. Yang and A. Mishchenko, npj Comput. Mater., 2023, 9, 101 CrossRef.
- H. K. Pentz, T. Warford, I. Timokhin, H. Zhou, Q. Yang, A. Bhattacharya and A. Mishchenko, Commun. Phys., 2025, 8, 25 CrossRef PubMed.
- S. Lu, Q. Zhou, Y. Guo, Y. Zhang, Y. Wu and J. Wang, Adv. Mater., 2020, 32, 2002658 CrossRef CAS PubMed.
- T. Gupta, M. Zaki, N. A. Krishnan and Mausam, npj Comput. Mater., 2022, 8, 102 CrossRef.
- N. Alampara, S. Miret and K. M. Jablonka, arXiv, 2024, preprint arXiv:2406.17295, DOI:10.48550/arXiv.2406.17295.
- L. Ward, A. Dunn, A. Faghaninia, N. E. Zimmermann, S. Bajaj, Q. Wang, J. Montoya, J. Chen, K. Bystrom and M. Dylla, et al. , Comput. Mater. Sci., 2018, 152, 60–69 CrossRef.
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. L. Chevrier, K. A. Persson and G. Ceder, Comput. Mater. Sci., 2013, 68, 314–319 CrossRef CAS.
- A. Dunn, Q. Wang, A. Ganose, D. Dopp and A. Jain, npj Comput. Mater., 2020, 6, 138 CrossRef.
- J. Riebesell, R. E. Goodall, P. Benner, Y. Chiang, B. Deng, G. Ceder, M. Asta, A. A. Lee, A. Jain and K. A. Persson, Nat. Mach. Intell., 2025, 7, 836–847 CrossRef.
- K. Choudhary, D. Wines, K. Li, K. F. Garrity, V. Gupta, A. H. Romero, J. T. Krogel, K. Saritas, A. Fuhr and P. Ganesh, et al. , npj Comput. Mater., 2024, 10, 93 CrossRef.
- S. Gong, K. Yan, T. Xie, Y. Shao-Horn, R. Gomez-Bombarelli, S. Ji and J. C. Grossman, Sci. Adv., 2023, 9, eadi3245 CrossRef.
- D. E. Widdowson and V. A. Kurlin, arXiv, 2024, preprint, arXiv:2410.13796, DOI:10.48550/arXiv.2410.13796.
- Y. Kim, E. Kim, E. Antono, B. Meredig and J. Ling, npj Comput. Mater., 2020, 6, 131 CrossRef.
- C. W. Andersen, R. Armiento, E. Blokhin, G. J. Conduit, S. Dwaraknath, M. L. Evans, Á. Fekete, A. Gopakumar, S. Gražulis and A. Merkys, et al. , Sci. Data, 2021, 8, 217 CrossRef PubMed.
- J. Ma, B. Cao, S. Dong, Y. Tian, M. Wang, J. Xiong and S. Sun, npj Comput. Mater., 2024, 10, 59 CrossRef.
- Z. Wang, A. Chen, K. Tao, J. Cai, Y. Han, J. Gao, S. Ye, S. Wang, I. Ali and J. Li, npj Comput. Mater., 2023, 9, 130 CrossRef.
- Constructor Tech, https://docs.constructor.tech/home/en-us/.
- A. Chandrasekaran, D. Kamal, R. Batra, C. Kim, L. Chen and R. Ramprasad, npj Comput. Mater., 2019, 5, 22 CrossRef.
- L. G. Wright, T. Onodera, M. M. Stein, T. Wang, D. T. Schachter, Z. Hu and P. L. McMahon, Nature, 2022, 601, 549–555 CrossRef CAS PubMed.
- B. Lu, Y. Xia, Y. Ren, M. Xie, L. Zhou, G. Vinai, S. A. Morton, A. T. Wee, W. G. van der Wiel and W. Zhang, et al. , Advanced Science, 2024, 11, 2305277 CrossRef PubMed.
- M. H. Kalos, Monte Carlo methods in quantum problems, Springer Science & Business Media, 2012, vol. 125 Search PubMed.
- J. Schmidt, C. L. Benavides-Riveros and M. A. Marques, J. Phys. Chem. Lett., 2019, 10, 6425–6431 CrossRef CAS PubMed.
- M. F. Kasim and S. M. Vinko, Phys. Rev. Lett., 2021, 127, 126403 CrossRef CAS PubMed.
- L. Li, S. Hoyer, R. Pederson, R. Sun, E. D. Cubuk, P. Riley and K. Burke, Phys. Rev. Lett., 2021, 126, 036401 CrossRef CAS PubMed.
- J. Wu, S.-M. Pun, X. Zheng and G. Chen, J. Chem. Phys., 2023, 159, year Search PubMed.
- J. Hermann, Z. Schätzle and F. Noé, Nat. Chem., 2020, 12, 891–897 CrossRef CAS PubMed.
- D. Pfau, J. S. Spencer, A. G. Matthews and W. M. C. Foulkes, Phys. Rev. Res., 2020, 2, 033429 CrossRef CAS.
- I. von Glehn, J. S. Spencer and D. Pfau, arXiv, 2022, preprint, arXiv:2211.13672, DOI:10.48550/arXiv.2211.13672.
- J. Hermann, J. Spencer, K. Choo, A. Mezzacapo, W. M. C. Foulkes, D. Pfau, G. Carleo and F. Noé, Nat. Rev. Chem., 2023, 7, 692–709 CrossRef PubMed.
- G. Carleo and M. Troyer, Science, 2017, 355, 602–606 CrossRef CAS PubMed.
- X. Gao and L.-M. Duan, Nat. Commun., 2017, 8, 662 CrossRef PubMed.
- G. Carleo, Y. Nomura and M. Imada, Nat. Commun., 2018, 9, 5322 CrossRef CAS PubMed.
- J. Carrasquilla and G. Torlai, PRX Quantum, 2021, 2, 040201 CrossRef.
- H. Shang, C. Guo, Y. Wu, Z. Li and J. Yang, arXiv, 2023, preprint arXiv:2307.09343, DOI:10.48550/arXiv.2307.09343.
- Y. Wu, C. Guo, Y. Fan, P. Zhou and H. Shang, Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis, 2023, pp. 1–13 Search PubMed.
- M. Geier, K. Nazaryan, T. Zaklama and L. Fu, arXiv, 2025, preprint, arXiv:2502.05383, DOI:10.48550/arXiv.2502.05383.
- L. O. Jones, M. A. Mosquera, G. C. Schatz and M. A. Ratner, J. Am. Chem. Soc., 2020, 142, 3281–3295 CrossRef CAS PubMed.
- S. Chen, V. W.-z. Yu, Y. Jin, M. Govoni and G. Galli, J. Chem. Theory Comput., 2025, 21, 7797–7812 CrossRef CAS PubMed.
- Z. Ye, Y.-X. Yao, X. Zhao, C.-Z. Wang and K.-M. Ho, J. Phys.: Condens. Matter, 2019, 31, 335601 CrossRef CAS PubMed.
- L.-F. Arsenault, A. Lopez-Bezanilla, O. A. Von Lilienfeld and A. J. Millis, Phys. Rev. B, 2014, 90, 155136 CrossRef.
- J. Rogers, T.-H. Lee, S. Pakdel, W. Xu, V. Dobrosavljević, Y.-X. Yao, O. Christiansen and N. Lanatà, Phys. Rev. Res., 2021, 3, 013101 CrossRef CAS.
- H. Suwa, J. S. Smith, N. Lubbers, C. D. Batista, G.-W. Chern and K. Barros, Phys. Rev. B, 2019, 99, 161107 CrossRef CAS.
- R. Rao and L. Zhu, Phys. Rev. B, 2024, 110, 245111 CrossRef CAS.
- H. Ma, H. Shang and J. Yang, npj Comput. Mater., 2024, 10, 220 CrossRef CAS.
- P. Zheng, R. Zubatyuk, W. Wu, O. Isayev and P. O. Dral, Nat. Commun., 2021, 12, 7022 CrossRef CAS PubMed.
- H. Kneiding and D. Balcells, Nordic Machine Intelligence, 2023, vol 3, pp. 8–14 Search PubMed.
- W. Gong, T. Sun, H. Bai, J.-Y. Tsai, H. Ling and Q. Yan, arXiv, 2024, preprint, arXiv:2411.16483, DOI:10.48550/arXiv.2411.16483.
- P. Reiser, M. Neubert, A. Eberhard, L. Torresi, C. Zhou, C. Shao, H. Metni, C. van Hoesel, H. Schopmans and T. Sommer, et al. , Commun. Mater., 2022, 3, 93 CrossRef CAS PubMed.
- J. Chang and S. Zhu, npj Comput. Mater., 2025, 11, 55 CrossRef CAS.
- P. B. Jørgensen and A. Bhowmik, npj Comput. Mater., 2022, 8, 183 CrossRef.
- E. M. Sunshine, M. Shuaibi, Z. W. Ulissi and J. R. Kitchin, J. Phys. Chem. C, 2023, 127, 23459–23466 CrossRef CAS.
- Z. Qiao, A. S. Christensen, M. Welborn, F. R. Manby, A. Anandkumar and T. F. Miller III, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2205221119 CrossRef CAS PubMed.
- Z. Tang, H. Li, P. Lin, X. Gong, G. Jin, L. He, H. Jiang, X. Ren, W. Duan and Y. Xu, Nat. Commun., 2024, 15, 8815 CrossRef CAS PubMed.
- J. P. Janet and H. J. Kulik, Chem. Sci., 2017, 8, 5137–5152 RSC.
- K. T. Schütt, M. Gastegger, A. Tkatchenko, K.-R. Müller and R. J. Maurer, Nat. Commun., 2019, 10, 5024 CrossRef PubMed.
- L. Fiedler, N. A. Modine, S. Schmerler, D. J. Vogel, G. A. Popoola, A. P. Thompson, S. Rajamanickam and A. Cangi, npj Comput. Mater., 2023, 9, 115 CrossRef.
- L. Fiedler, N. A. Modine, K. D. Miller and A. Cangi, Phys. Rev. B, 2023, 108, 125146 CrossRef.
- G. A. Tritsaris, S. Carr and G. R. Schleder, Appl. Phys. Rev., 2021, 8, 031401 Search PubMed.
- Y. Dong, C. Wu, C. Zhang, Y. Liu, J. Cheng and J. Lin, npj Comput. Mater., 2019, 5, 26 CrossRef.
- Y. Zhang, W. Xu, G. Liu, Z. Zhang, J. Zhu and M. Li, PLoS One, 2021, 16, e0255637 CrossRef PubMed.
- Y. T. Yeh, J. Ock, A. Chandrasekhar, S. Maheshwari and A. B. Farimani, arXiv, 2025, preprint, arXiv:2501.03456, DOI:10.48550/arXiv.2501.03456.
- J. Lee, C. Park, H. Yang, S. Lim and S. Han, arXiv, 2025, preprint, arXiv:2502.06836, DOI:10.48550/arXiv.2502.06836.
- J. Zhang, Y. Li and X. Zhou, arXiv, 2023, preprint, arXiv:2301.03372, DOI:10.48550/arXiv.2301.03372.
- X. Wang, Y. Wei, A. Bhattacharya, Q. Yang and A. Mishchenko, arXiv, 2025, preprint, arXiv:2506.07518, DOI:10.48550/arXiv.2506.07518.
- A. S. Larsen, T. Rekis and A. Ø. Madsen, Science, 2024, 385, 522–528 CrossRef PubMed.
- A. Vladyka, C. J. Sahle and J. Niskanen, Phys. Chem. Chem. Phys., 2023, 25, 6707–6713 RSC.
- P.-Y. Chen, K. Shibata, K. Hagita, T. Miyata and T. Mizoguchi, J. Phys. Chem. Lett., 2023, 14, 4858–4865 CrossRef PubMed.
- L. Zhang, J. Han, H. Wang, R. Car and W. E, Phys. Rev. Lett., 2018, 120, 143001 CrossRef PubMed.
- T. Zubatiuk and O. Isayev, Acc. Chem. Res., 2021, 54, 1575–1585 CrossRef PubMed.
- K. Schütt, P.-J. Kindermans, H. E. Sauceda Felix, S. Chmiela, A. Tkatchenko and K.-R. Müller, arXiv, 2017, preprint, abs/1706.08566, DOI:10.48550/arXiv.1706.08566.
- K. T. Schütt, O. T. Unke and M. Gastegger, arXiv, 2021, preprint, abs/2102.03150, DOI:10.48550/arXiv.2102.03150.
- H. Xu, Y. Jiang, H. Wang and J. Wang, Phys. Rev. B, 2024, 109, 035122 CrossRef.
- X. Li, Y. Chen, B. Li, H. Chen, F. Wu, J. Chen and W. Ren, arXiv, 2025, preprint, arXiv:2503.11756, DOI:10.48550/arXiv.2503.11756.
- B. Mortazavi, X. Zhuang, T. Rabczuk and A. V. Shapeev, Mater. Horiz., 2023, 10, 1956–1968 RSC.
- J. López-Zorrilla, X. M. Aretxabaleta, I. W. Yeu, I. Etxebarria, H. Manzano and N. Artrith, J. Chem. Phys., 2023, 158, 164105 CrossRef PubMed.
- G. P. Pun, R. Batra, R. Ramprasad and Y. Mishin, Nat. Commun., 2019, 10, 2339 CrossRef PubMed.
- G. P. Pun, V. Yamakov, J. Hickman, E. Glaessgen and Y. Mishin, Phys. Rev. Mater., 2020, 4, 113807 CrossRef.
- Y.-S. Lin, G. P. P. Pun and Y. Mishin, Comput. Mater. Sci., 2022, 205, 111180 CrossRef.
- J. D. Georgaras, A. Ramdas, C. H. Shan, E. Halsted, T. Li, F. H. da Jornada and et al., arXiv, 2025, preprint, arXiv:2503.15432, DOI:10.48550/arXiv.2503.15432.
- J. Behler and M. Parrinello, Phys. Rev. Lett., 2007, 98, 146401 CrossRef PubMed.
- J. S. Smith, O. Isayev and A. E. Roitberg, Chem. Sci., 2017, 8, 3192–3203 RSC.
- R. Zubatyuk, J. S. Smith, J. Leszczynski and O. Isayev, Sci. Adv., 2019, 5, eaav6490 CrossRef CAS PubMed.
- R. Zubatyuk, J. S. Smith, B. T. Nebgen, S. Tretiak and O. Isayev, Nat. Commun., 2021, 12, 4870 CrossRef CAS.
- T. Zubatiuk, B. Nebgen, N. Lubbers, J. S. Smith, R. Zubatyuk, G. Zhou, C. Koh, K. Barros, O. Isayev and S. Tretiak, J. Chem. Phys., 2021, 154, 244108 CrossRef CAS PubMed.
- D. M. Anstine and O. Isayev, J. Phys. Chem., 2023, 127, 2417–2431 CrossRef CAS PubMed.
- M. Aykol, A. Merchant, S. Batzner, J. N. Wei and E. D. Cubuk, Nat. Comput. Sci., 2025, 5, 105–111 CrossRef PubMed.
- B. Deng, Y. Choi, P. Zhong, J. Riebesell, S. Anand, Z. Li, K. Jun, K. A. Persson and G. Ceder, npj Comput. Mater., 2025, 11, 1–9 CrossRef.
- B. Focassio, L. P. M. Freitas and G. R. Schleder, ACS Appl. Mater. Interfaces, 2024, 13111–13121 Search PubMed.
- A. Loew, D. Sun, H.-C. Wang, S. Botti and M. A. Marques, arXiv, 2024, preprint, arXiv:2412.16551, DOI:10.48550/arXiv.2412.16551.
- H. Yang, C. Hu, Y. Zhou, X. Liu, Y. Shi, J. Li, G. Li, Z. Chen, S. Chen, C. Zeni and et al., arXiv, 2024, preprint, arXiv:2405.04967, DOI:10.48550/arXiv.2405.04967.
- C. Chen and S. P. Ong, Nat. Comput. Sci., 2022, 2, 718–728 CrossRef PubMed.
- I. Batatia, D. P. Kovacs, G. Simm, C. Ortner and G. Csányi, Adv. Neural Inf. Process. Syst., 2022, 35, 11423–11436 Search PubMed.
- B. Deng, P. Zhong, K. Jun, J. Riebesell, K. Han, C. J. Bartel and G. Ceder, Nat. Mach. Intell., 2023, 5, 1031–1041 CrossRef.
- K. Choudhary, B. DeCost, L. Major, K. Butler, J. Thiyagalingam and F. Tavazza, Digital Discovery, 2023, 2, 346–355 RSC.
- S. Batzner, A. Musaelian, L. Sun, M. Geiger, J. P. Mailoa, M. Kornbluth, N. Molinari, T. E. Smidt and B. Kozinsky, Nat. Commun., 2022, 13, 2453 CrossRef CAS PubMed.
- V. G. Satorras, E. Hoogeboom and M. Welling, International conference on machine learning, 2021, pp. 9323–9332 Search PubMed.
- Z. Yang, X. Wang, Y. Li, Q. Lv, C. Y.-C. Chen and L. Shen, npj Comput. Mater., 2025, 11, 49 CrossRef CAS.
- J. Gasteiger, F. Becker and S. Günnemann, Adv. Neural Inf. Process. Syst., 2021, 34, 6790–6802 Search PubMed.
- J. Brandstetter, R. Hesselink, E. van der Pol, E. J. Bekkers and M. Welling, arXiv, 2021, preprint, arXiv:2110.02905, DOI:10.48550/arXiv.2110.02905.
- M. Haghighatlari, J. Li, X. Guan, O. Zhang, A. Das, C. J. Stein, F. Heidar-Zadeh, M. Liu, M. Head-Gordon and L. Bertels, et al. , Digital Discovery, 2022, 1, 333–343 RSC.
- A. Musaelian, S. Batzner, A. Johansson, L. Sun, C. J. Owen, M. Kornbluth and B. Kozinsky, Nat. Commun., 2023, 14, 579 CrossRef CAS PubMed.
- S. Ryu, A. P. Schnyder, A. Furusaki and A. W. Ludwig, New J. Phys., 2010, 12, 065010 CrossRef.
- L. Fu, Phys. Rev. Lett., 2011, 106, 106802 CrossRef PubMed.
- R.-J. Slager, A. Mesaros, V. Juričić and J. Zaanen, Nat. Phys., 2013, 9, 98–102 Search PubMed.
- P. Zhang, H. Shen and H. Zhai, Phys. Rev. Lett., 2018, 120, 066401 CrossRef CAS PubMed.
- L.-F. Zhang, L.-Z. Tang, Z.-H. Huang, G.-Q. Zhang, W. Huang and D.-W. Zhang, Phys. Rev. A, 2021, 103, 012419 CrossRef CAS.
- N. Sun, J. Yi, P. Zhang, H. Shen and H. Zhai, Phys. Rev. B, 2018, 98, 085402 CrossRef CAS.
- M. S. Scheurer and R.-J. Slager, Phys. Rev. Lett., 2020, 124, 226401 CrossRef CAS PubMed.
- V. Peano, F. Sapper and F. Marquardt, Phys. Rev. X, 2021, 11, 021052 CAS.
- Z. Du, J. Luo, Z. Xu, Z. Jiang, X. Ding, T. Cui and X. Guo, Int. J. Mech. Sci., 2023, 255, 108441 CrossRef.
- G. R. Schleder, B. Focassio and A. Fazzio, Appl. Phys. Rev., 2021, 8, 031409 CAS.
- M. Matty, Y. Zhang, Z. Papić and E.-A. Kim, Phys. Rev. B, 2019, 100, 155141 CrossRef CAS.
- N. Jiang, S. Ke, H. Ji, H. Wang, Z.-X. Hu and X. Wan, Phys. Rev. B, 2020, 102, 115140 CrossRef CAS.
- Q. Jin and H. Wang, Phys. Lett. A, 2022, 427, 127921 Search PubMed.
- Y. Zhang and E.-A. Kim, Phys. Rev. Lett., 2017, 118, 216401 Search PubMed.
- Y. Teng, D. D. Dai and L. Fu, arXiv, 2024, preprint, arXiv:2412.00618, DOI:10.48550/arXiv.2412.00618.
- F. Noronha, A. Canabarro, R. Chaves and R. G. Pereira, Phys. Rev. B, 2025, 111, 014501 CrossRef CAS.
- T. Huang, X. Tu, C. Shen, B. Zheng, J. Wang, H. Wang, K. Khaliji, S. H. Park, Z. Liu and T. Yang, et al. , Nature, 2022, 605, 63–68 CrossRef CAS PubMed.
- Y. Choi, H. Kim, Y. Peng, A. Thomson, C. Lewandowski, R. Polski, Y. Zhang, H. S. Arora, K. Watanabe and T. Taniguchi, et al. , Nature, 2021, 589, 536–541 CrossRef CAS PubMed.
- H. Liu, S. Meng and F. Liu, Phys. Rev. Mater., 2021, 5, 084203 CrossRef CAS.
- N. Regnault, Y. Xu, M.-R. Li, D.-S. Ma, M. Jovanovic, A. Yazdani, S. S. Parkin, C. Felser, L. M. Schoop and N. P. Ong, et al. , Nature, 2022, 603, 824–828 CrossRef CAS PubMed.
- J. Duan, D.-S. Ma, R.-W. Zhang, W. Jiang, Z. Zhang, C. Cui, Z.-M. Yu and Y. Yao, Adv. Funct. Mater., 2024, 34, 2313067 CrossRef CAS.
- X. Zhang, Y.-M. Zhao, Z. Song and L. Shen, Phys. Rev. Mater., 2023, 7, 064804 CrossRef CAS.
- X. Zhang, APS March Meeting Abstracts, 2023, pp. N53–013 Search PubMed.
- X. Ma, Y. Luo, M. Li, W. Jiao, H. Yuan, H. Liu and Y. Fang, Chin. Phys. B, 2023, 32, 057306 CrossRef CAS.
- Z.-X. Que, S.-Z. Li, B. Huang, Z.-X. Yang and W.-B. Zhang, J. Chem. Phys., 2024, 160 CrossRef CAS PubMed.
- S. Yang, J. Chen, C.-F. Liu and M. Chen, Phys. Rev. B, 2024, 110, 235410 Search PubMed.
- T. Kuroda, T. Mizoguchi, H. Araki and Y. Hatsugai, J. Phys. Soc. Jpn., 2022, 91, 044703 CrossRef.
- Y. Gan, G. Wang, J. Zhou and Z. Sun, npj Comput. Mater., 2021, 7, 176 CrossRef.
- G. S. Na, S. Jang and H. Chang, npj Comput. Mater., 2021, 7, 106 CrossRef.
- T. Ishiyama, K. Nozawa, T. Nishida, T. Suemasu and K. Toko, NPG Asia Mater., 2024, 16, 17 CrossRef.
- Y. Wang, T. Li, H. Zong, X. Ding, S. Xu, J. Sun and T. Lookman, npj Comput. Mater., 2024, 10, 185 CrossRef.
- P. Minch, R. Bhattarai, K. Choudhary and T. D. Rhone, Phys. Rev. Mater., 2024, 8, 114002 CrossRef.
- S. Kong, D. Guevarra, C. P. Gomes and J. M. Gregoire, Appl. Phys. Rev., 2021, 8 Search PubMed.
- B. Sanchez-Lengeling and A. Aspuru-Guzik, Science, 2018, 361, 360–365 CrossRef PubMed.
- M. Cheng, C.-L. Fu, R. Okabe, A. Chotrattanapituk, A. Boonkird, N. T. Hung and M. Li, arXiv, 2025, preprint, arXiv:2502.02905, DOI:10.48550/arXiv.2502.02905.
- H. Park, Z. Li and A. Walsh, Matter, 2024, 7, 2355–2367 CrossRef.
- P. Tiwary, L. Herron, R. John, S. Lee, D. Sanwal and R. Wang, arXiv, 2024, preprint, arXiv:2409.03118, DOI:10.48550/arXiv.2409.03118.
- S. Banik, T. Loefller, S. Manna, H. Chan, S. Srinivasan, P. Darancet, A. Hexemer and S. K. Sankaranarayanan, npj Comput. Mater., 2023, 9, 177 CrossRef.
- G.-X. Liu, J.-F. Liu, W.-J. Zhou, L.-Y. Li, C.-L. You, C.-W. Qiu and L. Wu, Nanophotonics, 2023, 12, 1943–1955 CrossRef PubMed.
- K. Bang, J. Kim, D. Hong, D. Kim and S. S. Han, J. Mater. Chem. A, 2024, 12, 6004–6013 RSC.
- V. Fung, J. Zhang, G. Hu, P. Ganesh and B. G. Sumpter, npj Comput. Mater., 2021, 7, 200 CrossRef.
- C. Aytekin, arXiv, 2022, preprint, arXiv:2210.05189, DOI:10.48550/arXiv.2210.05189.
- M. Um, S. L. Sanchez, H. Song, B. J. Lawrie, H. Ahn, S. V. Kalinin, Y. Liu, H. Choi, J. Yang and M. Ahmadi, Adv. Energy Mater., 2024, 2404655 Search PubMed.
- A. K. Pimachev and S. Neogi, arXiv, 2023, preprint, arXiv:2302.00261, DOI:10.48550/arXiv.2302.00261.
- L. Ardizzone, J. Kruse, S. Wirkert, D. Rahner, E. W. Pellegrini, R. S. Klessen, L. Maier-Hein, C. Rother and U. Köthe, arXiv, 2018, preprint, arXiv:1808.04730, DOI:10.48550/arXiv.1808.04730.
- A. Nouira, N. Sokolovska and J.-C. Crivello, arXiv, 2018, preprint, arXiv:1810.11203, DOI:10.48550/arXiv.1810.11203.
- P. Lyngby and K. S. Thygesen, npj Comput. Mater., 2022, 8, 232 Search PubMed.
- X. Luo, Z. Wang, P. Gao, J. Lv, Y. Wang, C. Chen and Y. Ma, arXiv, 2024, preprint, arXiv:2403.10846, DOI:10.48550/arXiv.2403.10846.
- C. Zhong, J. Zhang, Y. Wang, Y. Long, P. Zhu, J. Liu, K. Hu, J. Chen and X. Lin, InfoMat, 2024, 6, e12519 Search PubMed.
- S. Kim, J. Noh, G. H. Gu, A. Aspuru-Guzik and Y. Jung, ACS Cent. Sci., 2020, 6, 1412–1420 Search PubMed.
- F. Liu, Z. Chen, T. Liu, R. Song, Y. Lin, J. J. Turner and C. Jia, iScience, 2024, 27, 110672 Search PubMed.
- Z. Chen, H. Li, C. Zhang, H. Zhang, Y. Zhao, J. Cao, T. He, L. Xu, H. Xiao and Y. Li, et al. , J. Chem. Theory Comput., 2024, 20, 9627–9641 Search PubMed.
- Z. Ren, S. I. P. Tian, J. Noh, F. Oviedo, G. Xing, J. Li, Q. Liang, R. Zhu, A. G. Aberle and S. Sun, et al. , Matter, 2022, 5, 314–335 Search PubMed.
- R. Zhu, W. Nong, S. Yamazaki and K. Hippalgaonkar, Matter, 2024, 7, 3469–3488 CrossRef.
- P. M. Lyngby and K. S. Thygesen, 2D Mater., 2024 Search PubMed.
- C.-Y. Ye, H.-M. Weng and Q.-S. Wu, Comput. Mater. Today, 2024, 1, 100003 CrossRef.
- K. Tangsongcharoen, T. Pakornchote, C. Atthapak, N. Choomphon-anomakhun, A. Ektarawong, B. Alling, C. Sutton, T. Bovornratanaraks and T. Chotibut, arXiv, 2025, preprint, arXiv:2501.08998, DOI:10.48550/arXiv.2501.08998.
- D. Levy, S. S. Panigrahi, S.-O. Kaba, Q. Zhu, K. L. K. Lee, M. Galkin, S. Miret and S. Ravanbakhsh, arXiv, 2025, preprint, arXiv:2502.03638, DOI:10.48550/arXiv.2502.03638.
- F. E. Kelvinius, O. B. Andersson, A. S. Parackal, D. Qian, R. Armiento and F. Lindsten, arXiv, 2025, preprint, arXiv:2502.06485, DOI:10.48550/arXiv.2502.06485.
- M. Moret, L. Friedrich, F. Grisoni, D. Merk and G. Schneider, Nat. Mach. Intell., 2020, 2, 171–180 CrossRef.
- Q. Yuan, A. Santana-Bonilla, M. A. Zwijnenburg and K. E. Jelfs, Nanoscale, 2020, 12, 6744–6758 RSC.
- N. Fu, L. Wei, Y. Song, Q. Li, R. Xin, S. S. Omee, R. Dong, E. M. D. Siriwardane and J. Hu, Mach. Learn.: Sci. Technol., 2023, 4, 015001 Search PubMed.
- L. Wei, Q. Li, Y. Song, S. Stefanov, R. Dong, N. Fu, E. M. Siriwardane, F. Chen and J. Hu, Adv. Sci., 2024, 11, 2304305 CrossRef PubMed.
- R. Dong, Y. Song, E. M. Siriwardane and J. Hu, Adv. Intell. Syst., 2023, 5, 2300141 CrossRef.
- K. Choudhary, J. Phys. Chem. Lett., 2024, 15, 6909–6917 CrossRef PubMed.
- L. M. Antunes, K. T. Butler and R. Grau-Crespo, Nat. Commun., 2024, 15, 1–16 CrossRef PubMed.
- B. N. Slautin, U. Pratiush, D. C. Lupascu, M. A. Ziatdinov and S. V. Kalinin, arXiv, 2025, preprint, arXiv:2503.02978, DOI:10.48550/arXiv.2503.02978.
- A. Ghosh, M. Ziatdinov and S. V. Kalinin, arXiv, 2024, preprint, arXiv:2403.01234, DOI:10.48550/arXiv.2403.01234.
- C. Qin, J. Liu, S. Ma, J. Du, G. Jiang and L. Zhao, J. Mater. Chem. A, 2024, 12, 22689–22702 RSC.
- D. Ebrahimzadeh, S. S. Sharif and Y. M. Banad, arXiv, 2025, preprint, arXiv:2501.04604, DOI:10.48550/arXiv.2501.04604.
- K. Barakati, A. Molak, C. Nelson, X. Zhang, I. Takeuchi and S. V. Kalinin, arXiv, 2025, preprint, arXiv:2503.13833, DOI:10.48550/arXiv.2503.13833.
- Z. Zhu, J. Lu, S. Yuan, Y. He, F. Zheng, H. Jiang, Y. Yan and Q. Sun, J. Phys. Chem. Lett., 2024, 15, 1985–1992 CrossRef PubMed.
- T. Weiss, E. Mayo Yanes, S. Chakraborty, L. Cosmo, A. M. Bronstein and R. Gershoni-Poranne, Nat. Comput. Sci., 2023, 3, 873–882 CrossRef PubMed.
- Z. Cao, X. Luo, J. Lv and L. Wang, arXiv, 2024, preprint, arXiv:2403.15734, DOI:10.48550/arXiv.2403.15734.
- H.-W. Dong, X.-X. Su and Y.-S. Wang, J. Phys. D: Appl. Phys., 2014, 47, 155301 CrossRef.
- Y.-Y. Zhang, W. Gao, S. Chen, H. Xiang and X.-G. Gong, Comput. Mater. Sci., 2015, 98, 51–55 CrossRef.
- A. Krishnamoorthy, A. Mishra, N. Grabar, N. Baradwaj, R. K. Kalia, A. Nakano and P. Vashishta, Comput. Phys. Commun., 2020, 254, 107337 CrossRef.
- P. Varasteanu and M. Kusko, Appl. Sci., 2021, 11, 4353 CrossRef.
- X. Zhang, H. Nguyen, J. T. Paci, S. K. Sankaranarayanan, J. L. Mendoza-Cortes and H. D. Espinosa, npj Comput. Mater., 2021, 7, 113 CrossRef.
- K. M. Jablonka, G. M. Jothiappan, S. Wang, B. Smit and B. Yoo, Nat. Commun., 2021, 12, 2312 CrossRef PubMed.
- T. Park, E. Kim, J. Sun, M. Kim, E. Hong and K. Min, Mater. Today Commun., 2023, 37, 107245 CrossRef.
- J.-R. Jacobo, O. F. Olea-Mejía, A. L. Martínez-Hernández and V.-S. Carlos, FlatChem, 2024, 45, 100654 CrossRef.
- S. Yamazaki, W. Nong, R. Zhu, K. S. Novoselov, A. Ustyuzhanin and K. Hippalgaonkar, arXiv, 2025, preprint, arXiv:2503.16784, DOI:10.48550/arXiv.2503.16784.
- Y. Liu, Z. Yang, Z. Yu, Z. Liu, D. Liu, H. Lin, M. Li, S. Ma, M. Avdeev and S. Shi, J. Materiomics, 2023, 9, 798–816 CrossRef.
- J. Yang, K. Zhou, Y. Li and Z. Liu, arXiv, 2021, preprint, arXiv:2110.11334, DOI:10.48550/arXiv.2110.11334.
- H. Yu, J. Liu, X. Zhang, J. Wu and P. Cui, arXiv, 2024, preprint, arXiv:2403.01874, DOI:10.48550/arXiv.2403.01874.
- T. Liu and A. S. Barnard, Cell Rep. Phys. Sci., 2023, 4 Search PubMed.
- K. Cheung, J. Drennan and J. Hunter, AAAI Spring Symposium: Semantic Scientific Knowledge Integration, 2008, pp. 9–14 Search PubMed.
- X. Zhang, C. Hu and H. Li, Data Sci. J., 2009, 8, 1–17 CrossRef.
- Y. Ye, J. Ren, S. Wang, Y. Wan, I. Razzak, B. Hoex, H. Wang, T. Xie and W. Zhang, Adv. Neural Inf. Process. Syst., 2024, 37, 56878–56897 Search PubMed.
- P. Lambrix, R. Armiento, H. Li, O. Hartig, M. Abd Nikooie Pour and Y. Li, Semant. Web, 2024, 15, 481–515 Search PubMed.
- M. Lanthaler, Proceedings of the 22Nd international conference on world wide web, 2013, pp. 35–38 Search PubMed.
- R. Linacre, S. Lindsay, T. Manassis, Z. Slade, T. Hepworth, R. Kennedy and A. Bond, International Journal of Population Data Science, 2022, 7 Search PubMed.
- S. J. Yang, S. Li, S. Venugopalan, V. Tshitoyan, M. Aykol, A. Merchant, E. D. Cubuk and G. Cheon, arXiv, 2023, preprint, arXiv:2311.13778, DOI:10.48550/arXiv.2311.13778.
- S. Takeda, A. Kishimoto, L. Hamada, D. Nakano and J. R. Smith, Proc. AAAI Conf. Artif. Intell., 2023, 15376–15383 Search PubMed.
- V. Stanev, C. Oses, A. G. Kusne, E. Rodriguez, J. Paglione, S. Curtarolo and I. Takeuchi, npj Comput. Mater., 2018, 4, 29 CrossRef.
- I. Batatia, P. Benner, Y. Chiang, A. M. Elena, D. P. Kovács, J. Riebesell, X. R. Advincula, M. Asta, W. J. Baldwin, N. Bernstein, A. Bhowmik, S. M. Blau, V. Cărare, J. P. Darby, S. De, F. D. Pia, V. L. Deringer, R. Elijošius, Z. El-Machachi, E. Fako, A. C. Ferrari, A. Genreith-Schriever, J. George, R. E. A. Goodall, C. P. Grey, S. Han, W. Handley, H. H. Heenen, K. Hermansson, C. Holm, J. Jaafar, S. Hofmann, K. S. Jakob, H. Jung, V. Kapil, A. D. Kaplan, N. Karimitari, N. Kroupa, J. Kullgren, M. C. Kuner, D. Kuryla, G. Liepuoniute, J. T. Margraf, I.-B. Magdău, A. Michaelides, J. H. Moore, A. A. Naik, S. P. Niblett, S. W. Norwood, N. O'Neill, C. Ortner, K. A. Persson, K. Reuter, A. S. Rosen, L. L. Schaaf, C. Schran, E. Sivonxay, T. K. Stenczel, V. Svahn, C. Sutton, C. van der Oord, E. Varga-Umbrich, T. Vegge, M. Vondrák, Y. Wang, W. C. Witt, F. Zills and G. Csányi, arXiv, 2023, preprint, arXiv:2401.00096, DOI:10.48550/arXiv.2401.00096.
- T. Ma, H. Wang and L. J. Guo, Opto-Electron. Adv., 2024, 7, 240062–1 Search PubMed.
- Y. Chen, X. Wang, X. Deng, Y. Liu, X. Chen, Y. Zhang, L. Wang and H. Xiao, arXiv, 2024, preprint, arXiv:2408.07608, DOI:10.48550/arXiv.2408.07608.
- K. Choudhary, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-ztp85.
- S.-M. Udrescu and M. Tegmark, Sci. Adv., 2020, 6, eaay2631 CrossRef PubMed.
- M. Krenn, R. Pollice, S. Y. Guo, M. Aldeghi, A. Cervera-Lierta, P. Friederich, G. dos Passos Gomes, F. Häse, A. Jinich and A. Nigam, et al. , Nat. Rev. Phys., 2022, 4, 761–769 CrossRef PubMed.
- G. Wang, E. Wang, Z. Li, J. Zhou and Z. Sun, Interdiscip. Mater., 2024 Search PubMed.
- Y. Wang, N. Wagner and J. M. Rondinelli, MRS Commun., 2019, 9, 793–805 CrossRef.
- N. Mekras, E. Mekra and C. Georgiou, MATEC Web of Conferences, 2024, p. 14004 Search PubMed.
- H. Touvron, T. Lavril, G. Izacard, X. Martinet, M.-A. Lachaux, T. Lacroix, B. Rozière, N. Goyal, E. Hambro, F. Azhar, A. Rodriguez, A. Joulin, E. Grave and G. Lample, LLaMA: Open and Efficient Foundation Language Models, 2023, https://arxiv.org/abs/2302.13971 Search PubMed.
- J. M. Nicholson, M. Mordaunt, P. Lopez, A. Uppala, D. Rosati, N. P. Rodrigues, P. Grabitz and S. C. Rife, Quant. Sci. Stud., 2021, 2, 882–898 CrossRef.
- Elicit, Elicit: The AI Research Assistant, 2023, https://elicit.com Search PubMed.
- C. Lu, C. Lu, R. T. Lange, J. Foerster, J. Clune and D. Ha, arXiv, 2024, preprint, arXiv:2408.06292, DOI:10.48550/arXiv.2408.06292.
- K. R. Abidi and P. Koskinen, Phys. Rev. Mater., 2022, 6, 124004 CrossRef.
- D. Kim, P. Noh, H.-Y. Lee and E.-G. Moon, Phys. Rev. A, 2023, 108, L010401 CrossRef.
- C. Vorwerk, N. Sheng, M. Govoni, B. Huang and G. Galli, Nat. Comput. Sci., 2022, 2, 424–432 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2026 |