
Lysosome-specific chemical platforms for precision oncology: from structural design to biological applications
Xing
Wang
 ab,
Yuqi
Tang
*ab and
Quan
Li
*abc
ab,
Yuqi
Tang
*ab and
Quan
Li
*abc
aInstitute of Advanced Materials and School of Chemistry and Chemical Engineering, Southeast University, Nanjing 211189, China. E-mail: yqtang@seu.edu.cn; quanli3273@gmail.com
bSchool of Intelligent Science and Engineering, Southeast University, Wuxi 214026, China
cMaterials Science Graduate Program, Kent State University, Kent, OH 44242, USA
First published on 6th March 2026
Abstract
The complex mechanisms of tumorigenesis and the inherent limitations of conventional therapies have severely restricted the clinical efficacy of tumor treatment. Owing to their pivotal roles in macromolecular degradation, energy metabolism, autophagy, and signal transduction, lysosomes are increasingly recognized as important targets for precision oncology. In this review, we summarize advances made over the past five years in the development of lysosome-specific chemical platforms for tumor therapy, covering molecular, material, and biomimetic platforms. Through comprehensive assessment of their structural designs, mechanisms of action, and representative applications, this review highlights the advantages and challenges of these platforms in drug delivery, photodynamic/photothermal therapy, immunotherapy, biomimetic strategies, and targeting imaging. Moreover, the unique acidic microenvironment and membrane permeability of lysosomes provide favorable conditions for lysosomal escape, offering another potential and critical mechanism to target as a new tumor treatment strategy. In addition to lysosomal escape, lysosome-specific platforms can target membrane permeability and remodeling of the immune microenvironment in enhancing therapeutic efficacy and overcoming drug resistance. Finally, we highlight the future directions and clinical translation prospects of lysosome-specific self-assembling peptide platforms, coacervates, chimeras, and near-infrared small-molecule probes, emphasizing the critical role of interdisciplinary integration in advancing precision tumor theranostics.
 Yuqi Tang | Yuqi Tang is a postdoctoral fellow at Institute of Advanced Materials and School of Chemistry and Chemical Engineering, Southeast University under the supervision of Prof. Quan Li. She received her PhD from Southeast University in June 2024, and she won the “NIMS Internship Program Award” from the National Institute for Materials Science (NIMS) in Japan. Her current research interests focus on stimuli-responsive smart materials, biomedical materials, and their applications. |
 Quan Li | Quan Li is Distinguished Chair Professor and Director of Institute of Advanced Materials at Southeast University. He held appointments in USA, Germany, and France. Li received his PhD in Organic Chemistry from the Chinese Academy of Sciences in Shanghai, where he was promoted to the youngest Full Professor in February 1998. He has been elected as a member of the European Academy of Sciences and the European Academy of Sciences and Arts. His current research interest spans from stimuli-responsive smart soft matter, advanced photonics, and optoelectronic materials for energy harvesting and energy saving to functional biocompatible materials, biomedical materials, and nanoparticles to nanoengineering and device fabrication. |
1. Introduction
The rising global incidence of cancer, which remains the second leading cause of death worldwide, has become a serious public health challenge.1–5 Despite progress, therapeutic efficacy against tumors is constrained owing to the complex etiology of the disease, together with insufficient drug accumulation, multidrug resistance, dynamic genetic mutations, and barriers within the tumor microenvironment (TME), often leading to treatment failure.6–12 For instance, chemotherapy remains a clinical mainstay in cancer treatment, but its efficacy is limited by low specificity and systemic toxicity. In recent decades, numerous novel strategies have emerged to overcome these challenges, including immunotherapy, targeted therapy, nanomedicine, phototherapy, RNA-based therapeutics, and gene editing, which are often combined as multimodal approaches to improve the precision of tumor therapy and overcome resistance.13–19 Moreover, advanced delivery systems, stimulus-responsive platforms, and targeted prodrugs have been investigated to address existing limitations; nevertheless, there remain important challenges to overcome with these systems, such as low loading, premature leakage, and insufficient drug release.20–22 Therefore, a single strategy is unlikely to fully overcome these obstacles. More promising strategies for enhancing precision oncology have been developed in recent years by integrating molecular structure modification, supramolecular chemistry, nanomedicine, biomimetic design, and tumor pharmacology through a multidisciplinary approach.23 Against this background, a systematic summary of strategies becomes particularly crucial.At the time of their identification by the Nobel laureate Christian de Duve first in the 1950s, lysosomes were viewed as the primary cellular degradation centers for macromolecules.24 Specifically, lysosomes mediate autophagy, a key process that recycles damaged organelles and proteins to maintain homeostasis, survival, and adaptation under stress; accordingly, dysregulation in these lysosome-mediated processes is linked to tumor development and neurodegeneration.25–27 Since their discovery, the functions of lysosomes have been expanded beyond the primary role in degradation. Lysosomes regulate metabolism, inter-organelle communication, host defense, and signaling, establishing these structures as central regulators of cellular function.28–30 Lysosomal abundance is often elevated in tumors, particularly in aggressive breast and gastric tumors, supporting proliferation, invasion, and stress adaptation.31–33 Since lysosomes recycle nutrients, regulate apoptosis, and manage oxidative stress, their destabilization may lead to cytosolic damage and inflammation of tumor cells.34,35 Owing to these multifaceted roles, lysosomes are increasingly recognized as promising therapeutic targets for tumor treatment.
Given the central role of lysosomes in tumor metabolism and autophagy, lysosome-targeted chemical platforms hold potential to achieve precise drug delivery and effectively induce tumor cell death, which may further help overcome chemoresistance. Integrating these strategies with other treatment modalities such as imaging, photothermal and photodynamic therapy (PDT), and immunotherapy could offer more effective and personalized approaches to tumor therapy. Recent efforts toward such integration include the design of lysosome-specific molecular, material-based, and biomimetic platforms, emphasizing the link between structural modifications and therapeutic precision. For example, Borkowska et al. developed mixed-charge nanoparticles that selectively aggregate in tumor lysosomes, and Diao et al. created a fluorescent probe for the real-time imaging of mitochondria–lysosome interactions.36,37 Genentech reported potent, selective VPS34 kinase inhibitors that modulate autophagy and show promise as lysosome-targeted antitumor agents.38 Collectively, these advances highlight the growing potential of lysosome-based strategies in oncology.
In this review, we summarize the recent advances (over the past five years) in the development of lysosome-targeted chemical platforms for precision oncology, providing a comprehensive perspective on their future development prospects. Fig. 1 provides a schematic of the three representative lysosome-specific chemical platforms highlighted in this review. First, we introduce molecular platforms, including organic small molecules, metal complexes, and supramolecular assemblies, along with their synthetic strategies. We focus on their advantages, limitations, and mechanisms of action as identified in experiments with mouse tumor models. Subsequently, we examine the chemical and biological properties of lysosome-specific material-based and biomimetic platforms derived from molecular platforms, with a focus on their design principles, synthetic routes, preparation methods, synergistic mechanisms, and clinical translational potential. Moreover, this review provides an in-depth analysis and forward-looking discussion of lysosome-mediated dual-organelle targeting. Beyond exploring the mechanistic role of lysosomes in these platforms, this review aims to expand the current scope of organelle-targeted therapeutics, highlighting the significance of membrane-less organelles as potential therapeutic targets. Overall, summarizing the relationship between lysosomal function and tumor progression offers a comprehensive perspective for understanding the role of lysosome-targeted platforms in biomedical research and clinical applications.
 | ||
| Fig. 1 Lysosome-targeted theranostic platforms for precision oncology, designed based on the biological features of lysosomes, have been developed, including molecular platforms, material-based platforms, and biomimetic platforms. | ||
2. Lysosomal function and therapeutic targets
Lysosomes are approximately 1-µm-diameter spherical organelles that play a pivotal role in cellular digestion, degradation, and redox homeostasis. Lysosomes are commonly known as the “suicide bags” of eukaryotic cells given their ability to trigger cell death upon membrane disruption, thereby representing a promising yet underexplored target for tumor therapy.39,40 Their acidic lumen (pH ∼ 4.5–5), containing V-ATPases, ion channels, and transport proteins, provides an optimal environment for hydrolytic enzymes that mediate macromolecular breakdown.40–42 Lysosomal hydrolytic enzymes degrade and recycle extracellular materials via endocytosis and phagocytosis, whereas intracellular components are degraded through autophagy; collectively, these processes maintain cellular quality control by breaking down damaged macromolecules such as polysaccharides, proteins, and nucleic acids.43 These catabolic processes also regenerate essential biomolecules and nutrients, supporting cellular growth, proliferation, and homeostasis.Autophagy exhibits a dual role in apoptosis, also influencing tumor progression and metastasis at different stages, although the underlying mechanisms remain incompletely understood.44 Targeting lysosomal autophagy has therefore emerged as a promising strategy to dissect these regulatory networks and develop precise antitumor therapies.45 Notably, relative to their normal counterparts, tumor cell lysosomes are larger, more fragile, and exhibit higher protease activity, making them more susceptible to stimuli that disrupt lysosomal membrane permeability (LMP) and trigger lysosome-mediated death.46 Accordingly, the lysosomal membrane structure, internal microenvironment, and metabolic pathways constitute the central targets for lysosome-mediated precision tumor therapy.
3. Endosomal and lysosomal escape
Autophagy is classified into microautophagy, chaperone-mediated autophagy, and macroautophagy; the latter form is the most widely studied and is often referred to simply as “autophagy”.47–55 In microautophagy, cellular components are directly engulfed via lysosomal membrane invagination; chaperone-mediated autophagy involves the selective delivery of proteins marked by specific motifs through heat shock proteins;55–59 and macroautophagy involves the formation of double-membrane autophagosomes from various sources, including the endoplasmic reticulum (ER), Golgi, mitochondria, and plasma membrane, which enclose cargo and fuse with lysosomes for degradation.56,60 Endosomes, formed via endocytosis, sort and process internalized materials and can fuse with lysosomes. The acidic environment of endosomes combined with their hydrolytic enzymes can induce the degradation of nanodrugs. Endosomal escape represents an early stage, and a prerequisite, of lysosomal escape; therefore, certain antitumor drugs that escape at the endosomal stage may bypass the lysosomal stage and avoid degradation. Therefore, achieving efficient endosomal and/or lysosomal escape is crucial for responsive drug release, precise targeting, and enhanced bioavailability.57–70Many lysosome-targeted drugs, including small molecules, metal complexes, and supramolecular drugs, directly exert antitumor effects by disrupting lysosomal function. The rational design of material carriers can greatly improve the targeting accuracy of molecular drug delivery, thereby markedly amplifying the overall therapeutic efficacy. Liu et al. developed deformable gallium (Ga) particle-based nanorobots that transform from a spherical to cactus-like shape at low temperatures. Membrane-coated Ga-based particles (GaPs or Ga/MPs) physically puncture lysosomes during this phase transition, enabling endosomal escape. The co-encapsulated anti-tumor drug paclitaxel (PTX) could then be released into the cytosol upon endosomal disruption, exerting cytotoxic effects with the aid of hydrolytic enzymes (Fig. 2a).69 Moreover, owing to their excellent radiopacity, injectability, and targetability, these Ga-based particles enable high-resolution computed tomography imaging of blood vessels and tumors (Fig. 2b), significantly enhancing the integration of diagnosis and therapy. Similarly, Fan et al. grafted cationic photosensitizers (NB-Br) onto PLK1 small interfering RNA (siRNA) to form amphiphilic conjugates (siPLK1-NB) that self-assemble into nanoparticles (siPLK1-NB-NPs). In lysosomes, the reactive oxygen species (ROS) released from siPLK1-NB-NPs disrupt the membrane structure, enabling the release of siRNA into the cytosol to downregulate PLK1 expression and effectively inhibit tumor growth.71
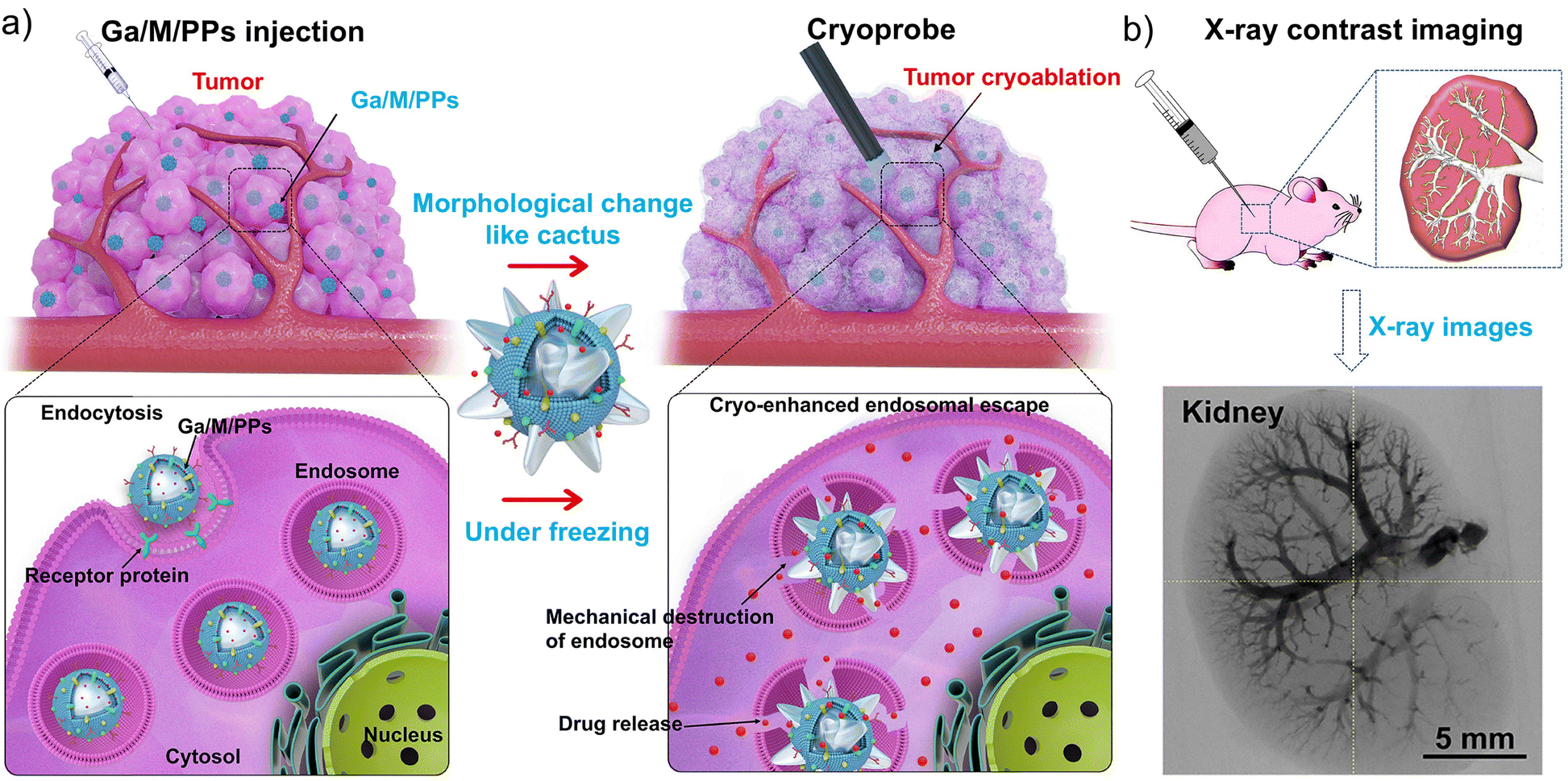 | ||
| Fig. 2 Cryo-treatment-driven liquid metal transformer to promote intracellular therapy. (a) Schematic illustration of cryo-facilitated liquid-metal particle transformation for endosomal escape. (b) Illustrated scheme of the injection process of gallium (Ga) particles solution into animal body and X-ray images of the kidney. Reproduced with permission from ref. 69. Copyright 2022, Elsevier Ltd. | ||
Overall, these studies demonstrate that the therapeutic efficacy of nanodrugs can be significantly enhanced by promoting endosomal and lysosomal escape, improving the delivery of drugs or nucleic acids to target organelles, and reducing off-target effects. Therefore, rationally designing drugs capable of endosomal and lysosomal escape offers distinct advantages in improving intracellular delivery efficiency and antitumor activity, while further serving as a general reference for the development of diverse lysosome-targeted chemical platforms.
4. Lysosome-specific molecular platforms
Small molecules or metal complexes bearing lysosome-targeting moieties can be efficiently delivered to lysosomes where they can directly exert their pharmacological effects. As shown in Fig. 3, morpholine rings (pKa ≈ 8–8.5), N,N-diethyl tertiary amines (pKa ≈ 10–11), tertiary amine moieties (pKa ≈ 9–11), piperazine rings (pKa ≈ 9.7 and 5.3), and polyamine chemical moieties (pKa ≈ 8.5–11) are commonly used for lysosomal targeting as these molecules are characterized by weak basicity to facilitate their selective accumulation within lysosomes, thereby maximizing local efficacy.72 While benzoindole quaternary ammoniums, as examples of permanent cations, target lysosomes via electrostatic interactions. Specifically, these groups can interact with lysosomal membrane structures, promoting fusion into the lysosomal lumen and enabling effective lysosomal targeting. Importantly, weakly basic moieties can increase the protonation of molecules within the lysosome, enhancing interactions with the negatively charged inner lysosomal membrane and prolonging their retention in the lysosomal lumen.73–75 Some lysosome-specific chemical molecules now serve as important tools for precise diagnosis and treatment of tumors. In recent years, the design of lysosome-targeted small molecules has shifted toward exploiting intrinsic lysosomal components to achieve precise targeting efficiency by recognizing lysosomal contents, including β-galactosidase, sulfatase, and lysosomal iron. Representative examples include DCM-Mor-Gal, YSO4F, and cLip-1, which have been successively developed and play an important role in advancing research on tumor therapy.76–78 The design of molecular platforms that interfere with the lysosomal phagocytosis-metabolism process is another key research focus. Hydrophobic molecules can readily embed into lysosomal membranes and interact with membrane structures or proteins through hydrophobic interactions. For example, tetrandrine binds directly to lysosomal integral membrane protein 2 (LIMP-2) and accumulates on the lysosome surface via endocytic pathways. In addition, the novel long-acting lysosome-targeting anticancer agent LLAD covalently binds to the lysosomal membrane protein SLC38A9, prolonging inhibition of this transmembrane transporter to induce lysosomal alkalization.79,80 Host–guest supramolecular platforms, constructed on small-molecule scaffolds, integrate multiple lysosome-targeting strategies, thereby further enhancing the intelligence, precision, and controllability of tumor diagnosis and therapy. Peptide-based targeting approaches, such as TP10, H5WYG, and Lauryl-P3GKS (GalNAc) peptides, have also been proposed to increase the precision of lysosomal localization.81,82 Overall, through in-depth mechanistic studies of lysosome specificity, the recent development of novel lysosome-targeting molecules has significantly advanced precision oncology (Fig. 3).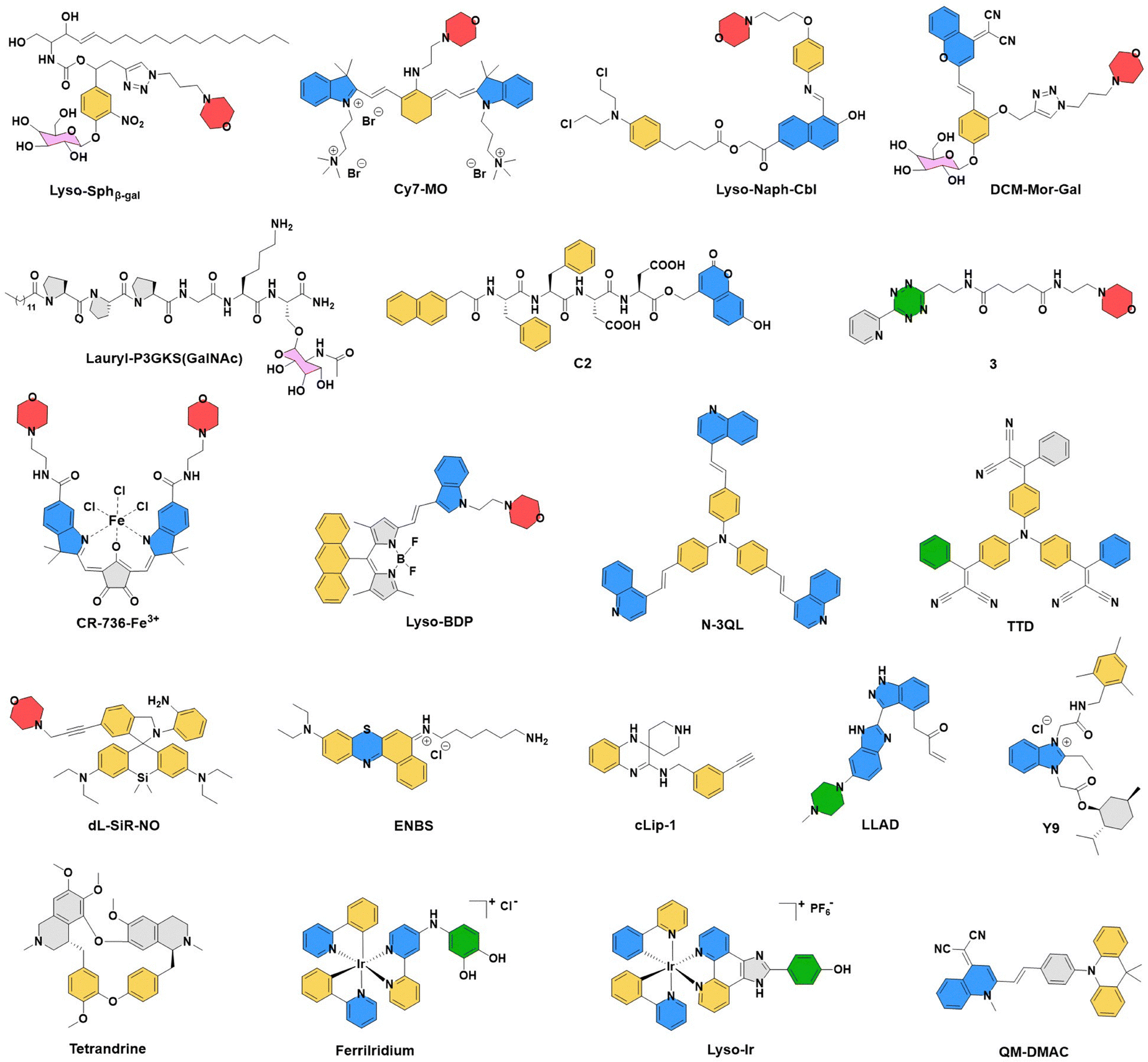 | ||
| Fig. 3 Recently reported classic single-molecule chemical structures targeting lysosomal metabolic mechanisms or biochemical structures, including the acidic lysosomal microenvironment, negative charge characteristics, membrane interactions, enzyme-triggered mechanisms, and metabolism. | ||
4.1. Organic small-molecule platforms
The degradation and recycling of biomacromolecules are key processes involved in the initiation, proliferation, and metastasis of non-small cell lung cancer (NSCLC).83 In addition to playing a role in these processes, lysosomal damage is a direct means of activating autophagy, a key mechanism through which NSCLC acquires drug resistance. Therefore, inducing lysosome-mediated lethal damage or inhibiting autophagy is a potential strategy for the effective treatment of NSCLC and/or the reversal of its drug resistance.84 Zhang et al. designed and synthesized a Gboxin analogue, designated as Y9, through chemical structure optimization (Fig. 4a).85 Gboxin is a small molecule that specifically inhibits the growth of primary glioblastoma cells by targeting mitochondrial adenosine triphosphate synthase without affecting the growth of normal cells. Unlike Gboxin, the basic cationic amphiphilic structure of Y9 enables specific binding to lysosomes, followed by their acidification to induce lysosomal dysfunction. As shown in Fig. 4b–d, Y9 inhibited tumor growth in a dose-dependent manner in a xenograft mouse model, not only suppressing NSCLC progression but also inducing cellular damage and apoptosis. In addition, fluorescence changes of Galectin-3 and mCherry-GFP-LC3B confirmed that Y9 not only disrupts lysosomal integrity but also impairs the maturation of autophagic lysosomes, thereby promoting tumor apoptosis via activation of the P38-JNK pathway (Fig. 4e and f). Therefore, Y9 represents a distinct Gboxin analogue that surpasses its prototype by inducing lysosomal dysfunction and promoting apoptosis, highlighting its potential as a novel lead compound for NSCLC therapy.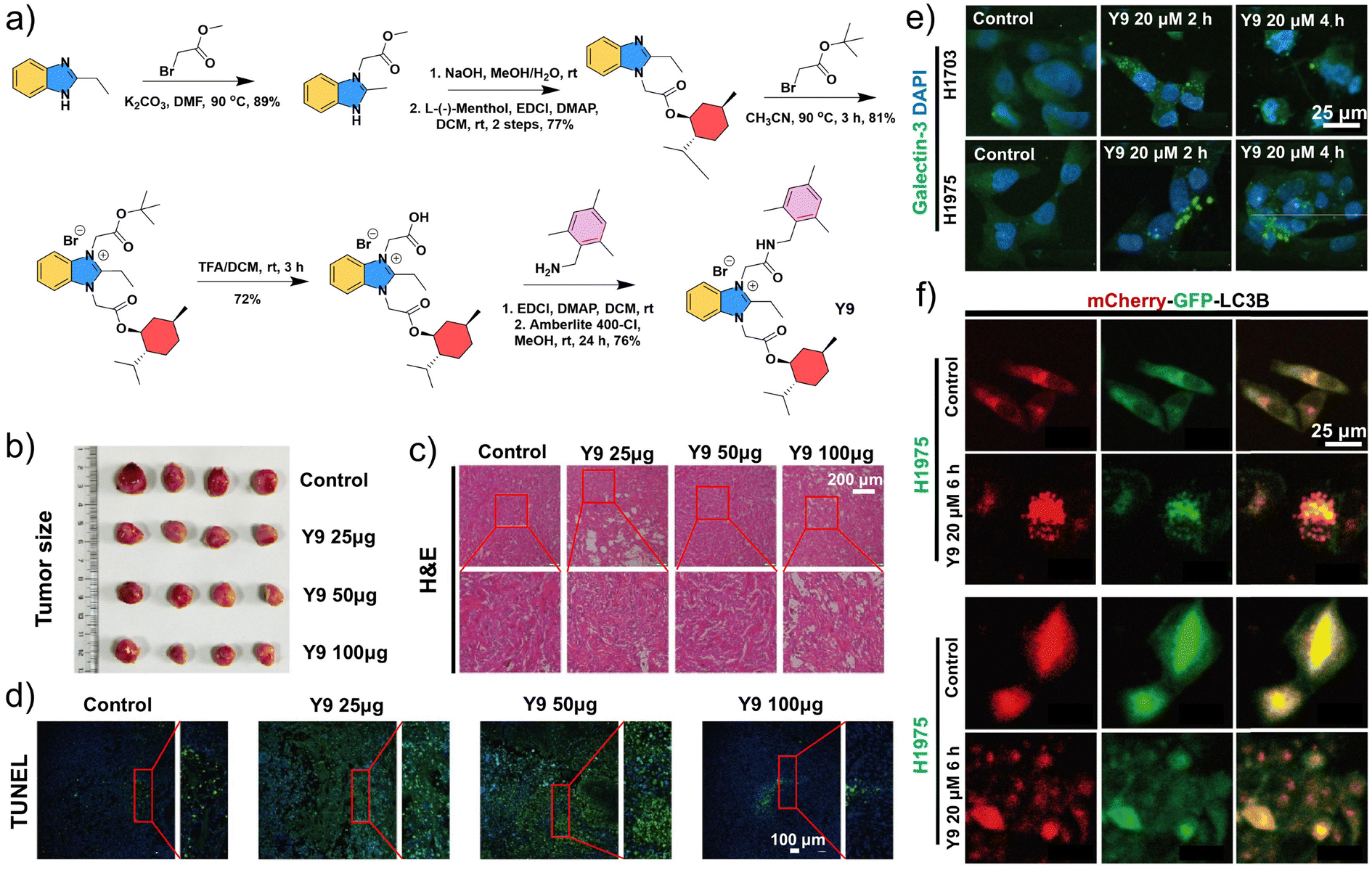 | ||
| Fig. 4 The structure and antitumor mechanism of the lysosome-targeted small molecule Y9. (a) Synthesis route of Y9. (b) NSCLC subcutaneous tumors treated with different doses of Y9. Images of (c) H&E and (d) TUNEL staining of tumors in mice treated with different doses of Y9. (e) Representative images of immunofluorescence staining of different tumor cells. (f) Representative images of mCherry or GFP fluorescence in NSCLC cells pre-infected with Ad-mCherry-GFP-LC3B for 48 h, followed by treatment with Y9 for 6 h. (a–f) Reproduced with permission from ref. 85. Copyright 2024, Elsevier Ltd. | ||
Wu et al. designed a nitrogen-containing heterocyclic cationic amphiphilic compound, designated as ENBS (Fig. 5a), which exerts antitumor effects specifically triggered by characteristics of the TME. Specifically, the acidic TME markedly enhances the protonation of ENBS. Under near-infrared (NIR) light activation, ENBS generates superoxide anions (O2−˙) to overcome the lethal hypoxia characteristics of the TME, effectively inducing necroptosis and triggering antitumor immunity.86 Necroptosis is a pro-inflammatory form of programmed cell death that relies on membrane pore formation by gasdermin N-terminal fragments, ultimately releasing large amounts of cellular contents and causing effective immunogenic cell death (ICD).87–89 Tumor-specific lysosomal targeting enabled ENBS to efficiently accumulate around the tumor within 90 min post-administration, achieving spatiotemporally controlled and precise PDT (Fig. 5b).
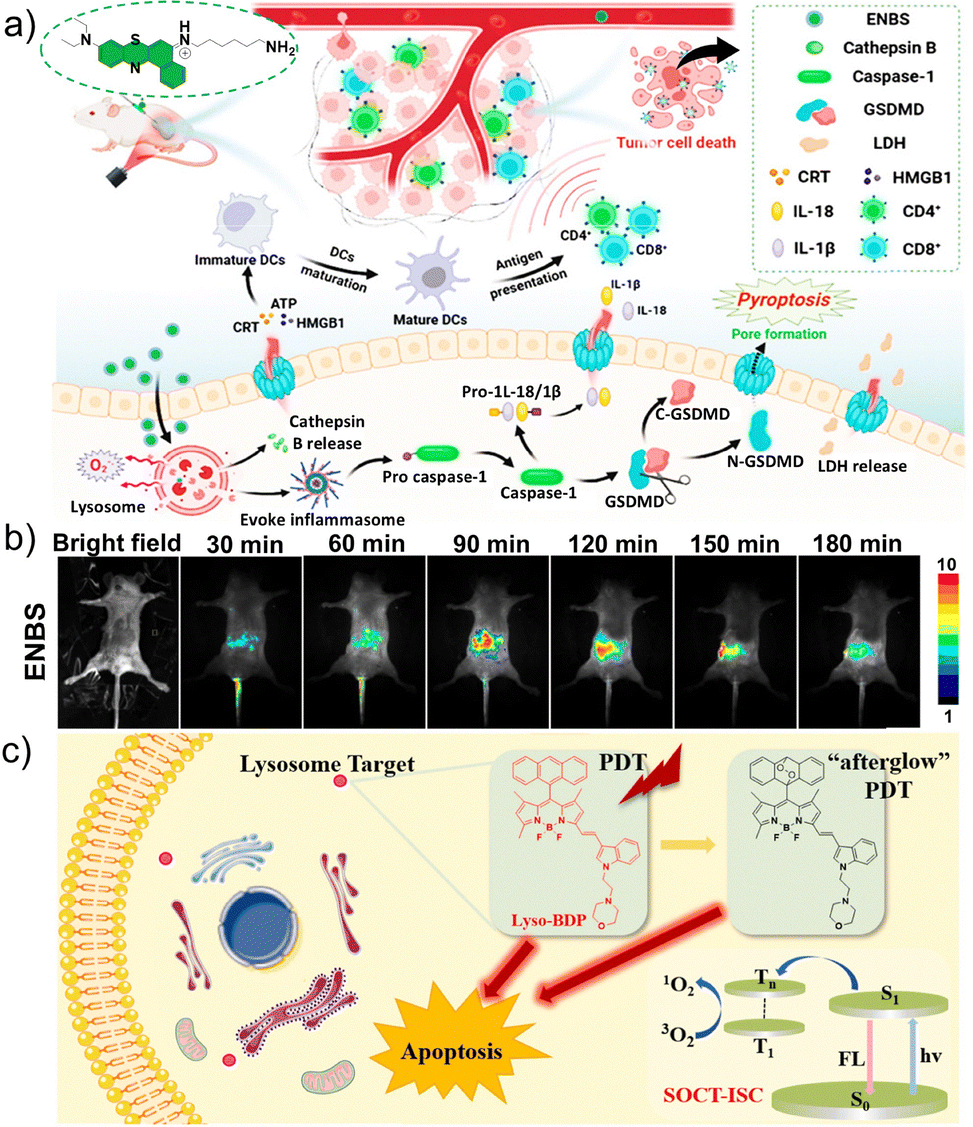 | ||
| Fig. 5 Lysosome-targeted photosensitizers for imaging-guided photodynamic therapy (PDT). (a) Schematic diagram of the lysosome-anchored type I photosensitizer ENBS, which can induce pyroptosis and an antitumor immune response. (b) Intravital fluorescence images of the tumor site acquired at various time points after intravenous injection of ENBS. (a and b) Reproduced with permission from ref. 86. Copyright 2024, American Chemical Society. (c) Schematic illustration of the PDT mechanism of the novel non-heavy-atom, lysosome-targeted BODIPY probe Lyso-BDP. Reproduced with permission from ref. 101. Copyright 2023, Elsevier Ltd. | ||
Inspired by these emerging lysosomal-targeting strategies, Xu et al. developed a series of novel phenothiazine- and pyridine-based photosensitizers that can be rapidly synthesized and exhibit potent antitumor activity.90 These compounds feature simple molecular structures, are easy to synthesize, and selectively accumulate in lysosomes. Notably, compound 5 exhibited a cellular ROS generation rate approximately 40-fold higher than that of conventional photosensitizers such as methylene blue, resulting in markedly enhanced PDT efficacy, with the phototoxicity index reaching 53.8. Lysosome-targeted, light-responsive compounds exhibit excellent theranostic properties, providing valuable insights for the further development of controllable and effective antitumor agents.91–93
The morpholine ring is also a classic moiety for lysosomal targeting.94–96 Heavy non-metals such as bromine and iodine are often incorporated into conventional photosensitizers to enhance PDT efficiency through the heavy-atom effect, which facilitates spin–orbit coupling and subsequently promotes intersystem crossing. However, the incorporation of heavy atoms often leads to intrinsic dark cytotoxicity, thereby limiting the clinical translation of these photosensitizers.97–100 To overcome this limitation, Zhao et al. designed and synthesized a novel heavy-atom-free, lysosome-targeted triplet photosensitizer, named Lyso-BDP.101 Upon laser irradiation, Lyso-BDP efficiently generates triplet states through spin–orbit charge transfer-induced intersystem crossing (Fig. 5c). Moreover, the anthracene moiety reinforces the triplet excited state while forming partially stable endoperoxide intermediates under irradiation, which are capable of gradually releasing singlet oxygen (1O2) in the absence of light. This unique “afterglow” 1O2 release further amplifies the PDT effect.
Yuan et al. designed a theranostic probe, Cy7-MO, with heptamethylphthalocyanine as the core, which was modified with quaternary ammonium groups on both sides and had a morpholine group introduced at the chlorinated atom.102 Confocal laser scanning microscopy imaging confirmed the lysosomal targeting of Cy7-MO, indicating a strong affinity between the morpholine group in Cy7-MO and lysosomes. Notably, the Cy7-MO probe exhibits excellent cellular tolerance and biocompatibility. Leeuwen et al. focused on the distinct role of lysosomes in the antigen presentation process. The examination of differences among antigen presentation pathways led to the development of a bio-orthogonal click-release chemical tool for lysosomal targeting in antigen-presenting cells.103 Specifically, the (2-aminoethyl)-morpholine group was combined with a triazine molecule for chemical de-caging to yield compound 3, which can specifically accumulate in lysosomes. Theoretically, if ligand molecules pass through the lysosomes, they will undergo a click-release chemical reaction with compound 3, thereby activating downstream T cells. This work therefore provides an important tool for studying the role of T cell behavior in antitumor immunity.
The precise localization of lysosomes can be achieved by specific binding to lysosomal proteins. As a classic example of lysosomal targeting achieved through the lysosomal uptake-metabolism pathway, Ryu et al. designed and synthesized an amphiphilic probe using lysosome-targeting peptides as targeting groups, confirming that binding to specific proteins on the lysosomal surface can enhance the targeting accuracy.104 Focusing on the same mechanism, Liu et al. leveraged the metabolic feature changes of lysosomes in senescent cells to design a small-molecule prodrug, SA-β-Gal, enabling the selective clearance of cells at different stages of senescence.105 This strategy first exploits the increased lysosomal content in senescent cells, allowing the prodrug to preferentially accumulate in their lysosomes via lysosomal-targeting groups. Second, leveraging the fragility of the lysosomal membrane, a sphingosine derivative with lysosomal affinity is used to selectively disrupt the lysosomal membrane upon activation, triggering lysosome-mediated apoptosis. This strategy achieved the selective clearance of senescent cells, thereby providing a proof-of-concept for tumor therapy through targeting senescent cells. Additionally, He et al. discovered that the natural product polyphyllin D (PD) targets enlarged lysosomes in hepatocellular carcinoma (HCC) cells by binding to the sphingomyelinase SMPD1, triggering lysosome-related cell death. Furthermore, PD enhances the release of sorafenib taken up by lysosomes, thereby strengthening the combined antitumor effect.106 These findings clarified the mechanism of PD in antitumor activity and in overcoming sorafenib resistance, providing new treatment options and preclinical data for HCC. Altogether, these studies provide important research directions for targeted lysosomal therapy in precision medicine for tumor treatment.
In summary, the design of recently reported lysosome-targeted organic small molecules primarily relies on several strategies. First, basic or cationic groups (such as piperidine rings or tertiary amines) are introduced into the molecular structure, and their weak basicity promotes their accumulation in lysosomes and specific subcellular regions. Second, selective accumulation and functional activation can be achieved through specific interactions with intrinsic lysosomal enzymes such as β-galactosidase and sphingomyelinase SMPD1. Third, targeting lysosomal membrane proteins such as SLC38A9 and LIMP-2, as well as relevant transport pathways, enables precise lysosomal localization and cross-talk with metabolic functions. Furthermore, in lysosome-targeted precision oncology, photochemical and responsive chemical strategies can enhance the antitumor activity of small molecules through photodynamic or controlled-release mechanisms while achieving spatiotemporal precision. Consequently, these organic small molecules offer significant advantages in precision tumor diagnosis and therapy, enabling accurate localization, enhancing local efficacy, and inducing specific cell death. Moreover, in certain cases, these strategies can help to overcome drug resistance or improve the efficacy of combination therapies.
Despite the rapid emergence in new lysosome-specific small-molecule antitumor agents, challenges remain to be addressed, such as their potential toxicity to normal tissues, limited targeting selectivity, and insufficient stability in complex in vivo environments. By contrast, metal complexes demonstrate excellent in vivo stability and, by enhancing their tumor-targeting capabilities, they can effectively minimize toxicity to healthy tissues. This approach represents a highly promising strategy for the development of novel tumor theranostic platforms.
4.2. Metal complex platforms
The TME is closely associated with tumor growth, immune evasion, the acquisition of drug resistance, and tumor inflammation. Identifying recognition probes or antitumor prodrugs targeting the TME, along with their integration into diagnostic and therapeutic reagents, holds great promise for applications in tumor diagnosis and precision treatment.107–112 Cancer can be characterized by the dysregulation of iron homeostasis, reflecting the close relationship between iron and intracellular oxidative cycles, along with free radical generation. However, there are relatively few reports on antitumor theranostic agents targeting the labile iron pool (LIP) in tumor cells.113–116 Chao et al. reported a gastric tumor theranostic agent based on an iridium(III) complex, known as FerriIridium.117 FerriIridium can be specifically activated by the LIP in the lysosomes of tumor cells, generating secondary products that possess both antitumor activity and phosphorescence (Fig. 6a). In vitro experiments demonstrated that FerriIridium can consume Fe3+ and convert it into Fe2+, showing high specificity for the catalytic process (Fig. 6b–d). Electron spin resonance analysis indicated that FerriIridium can catalyze the generation of O2−˙ and hydroxyl radicals (OH˙) in vitro, providing a foundation for O2−˙-mediated intratumoral catalysis and efficient tumor chemodynamic therapy (CDT) (Fig. 6d). Further research showed that as low-toxicity FerriIridium enters gastric cancer cells via endocytosis, its catechol group chelates lysosomal Fe3+ and is oxidized to intermediates. Under acidic lysosomal conditions, these intermediates hydrolyze into hydroquinone compounds and mitochondrial-targeting Ir–NH2 complexes. Altogether, they target lysosomes and mitochondria to exert synergistic antitumor effects via respiratory chain disruption and ROS generation. FerriIridium was activated in the tumor within 8 h and reached peak luminescence in 24 h (Fig. 6e). In vivo assays in mice showed that the emission intensity in AGS tumors was 3-fold higher than that in A549 tumors, further highlighting the potential of FerriIridium as a theranostic agent.117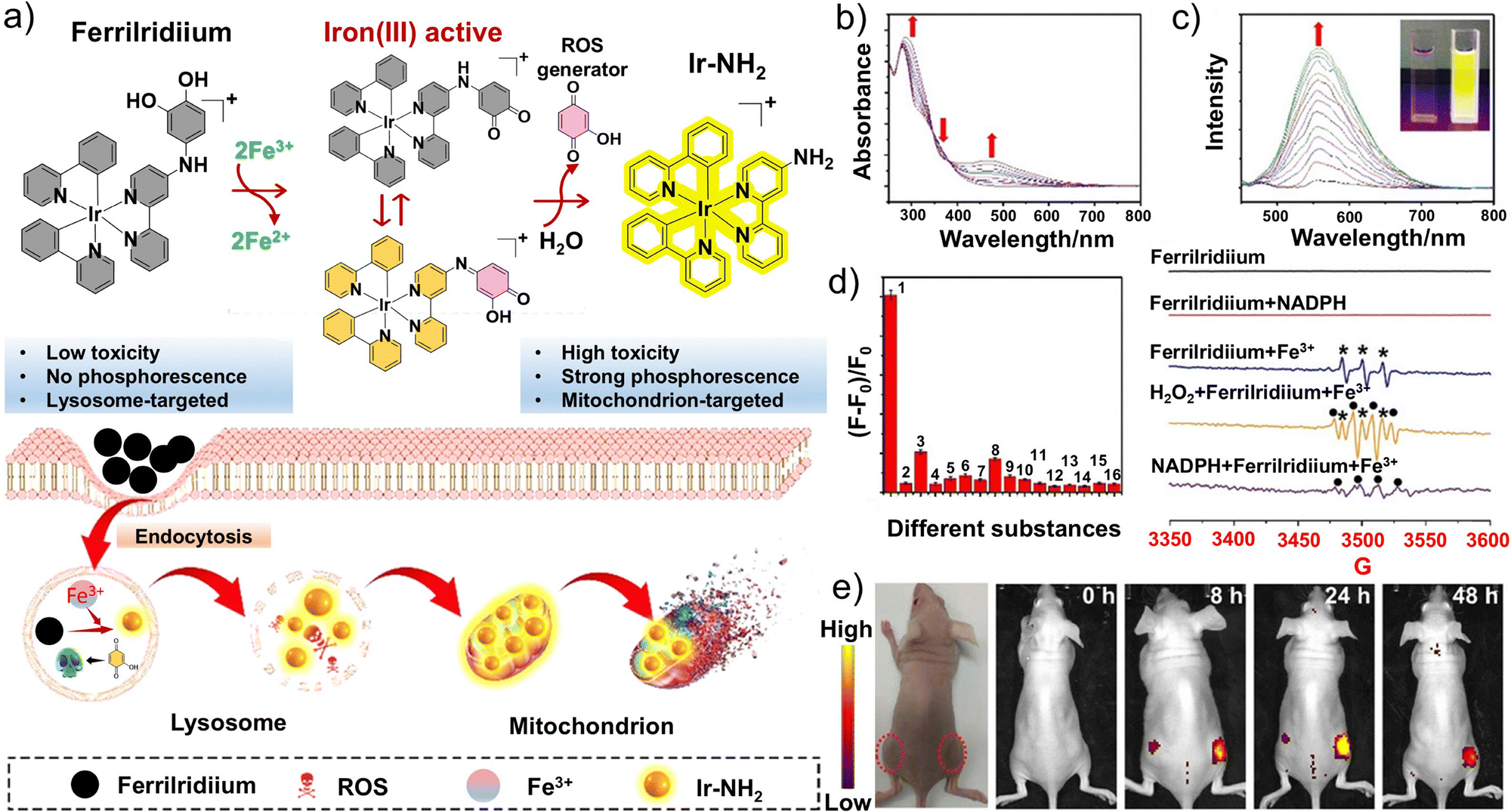 | ||
| Fig. 6 Antitumor mechanisms and tumor imaging of the lysosome-targeted metal complex FerriIridiium. (a) Schematic illustration of the iron(III) activation of FerriIridium and the subsequent cellular toxicity mechanism. (b) Absorption and (c) emission spectra of FerriIridium in the presence of Fe3+ from 0 to 4 equivalents. (d) Ion selectivity (1, Fe3+; 2, Fe2+; 3, Cu2+; 4, Cu+; 5, Zn2+; 6, Al3+; 7, Ni2+; 8, Mn2+; 9, Pb2+; 10, Co2+; 11, Cd2+; 12, Hg2+; 13, K+; 14, Na+; 15, Ca2+; 16, Mg2+) and electron spin resonance spectra of FerriIridium under different conditions. (e) FerriIridium-treated A549 (left hip) and AGS (right hip) tumors in nude mice after different times. (a–e) Reproduced with permission from ref. 117. Copyright 2021, Wiley-VCH. | ||
With the advancement of metal immunology, the expanded potential of platinum-based drugs in tumor immunotherapy has garnered widespread attention. Despite the significant clinical success of immunochemotherapy combining platinum drugs with immune checkpoint blockade (ICB) (such as PD-1/PD-L1), this combination therapy often brings new challenges, including dosage control, complex drug regimens, high costs, increased risk of side effects, and the development of drug resistance.118–121 These challenges have promoted the development of novel platinum-based drugs with both chemotherapeutic and ICB activities. Mao et al. developed a series of platinum–metformin conjugates as a promising alternative to antibody-based PD-L1 inhibitors. They confirmed that the cyclometalized platinum(II) group in Pt-2 significantly improved the transport of metformin into cells while facilitating its selective transport to lysosomes (Fig. 7a). Therefore, these conjugates offer advantages in theranostic applications with excellent photophysical properties for cellular imaging while delivering good antitumor activity for chemotherapy.122 Compared to cisplatin, Pt-2 effectively activates antitumor immunity in vivo by downregulating PD-L1 levels, thereby modulating macrophage polarization, promoting the maturation of dendritic cells (DCs), and enhancing lymphocyte infiltration within tumor tissues (Fig. 7b). Further mechanistic investigations revealed that Pt-2 selectively associates with lysosomes and induces lysosomal autophagy, while promoting lysosome-dependent PD-L1 degradation through inhibition of PD-L1 expression (Fig. 7c and d). Consequently, Pt-2 exhibits superior antitumor efficacy with minimal side effects (Fig. 7e). Since the primary action target of platinum-based drugs in tumor treatment is nuclear DNA, any factor that interferes with the binding of platinum to DNA may result in drug inactivation or the development of resistance. For example, the acidification caused by lysosomal phagocytosis or binding to glutathione (GSH) ultimately leads to the extrusion of platinum from cells, rendering it inactive.123–126 Therefore, the release of platinum-based drugs following lysosomal disruption can potentially maximize therapeutic efficacy. To overcome these inactivation mechanisms, rather than directly targeting lysosomes, He et al. developed a photosensitizing platinum complex with “light-activated” lysosomal escape functionality, realizing selective damage to solid tumors through precise and controllable light.127 The Pt-BDPA complex enters cells via energy-dependent phagocytosis and remains confined within lysosomes, exhibiting negligible dark toxicity due to its inability to bind nuclear DNA. Upon light activation, the compound generates ROS that rupture lysosomes, releasing the platinum complex to allow it to bind DNA and induce marked cytotoxicity to tumor cells. The light-induced ROS also deplete intracellular GSH, further enhancing nuclear platinum accumulation and antitumor efficacy. This study thus offered a proof of concept for developing escape-type phototherapeutic metal complexes.
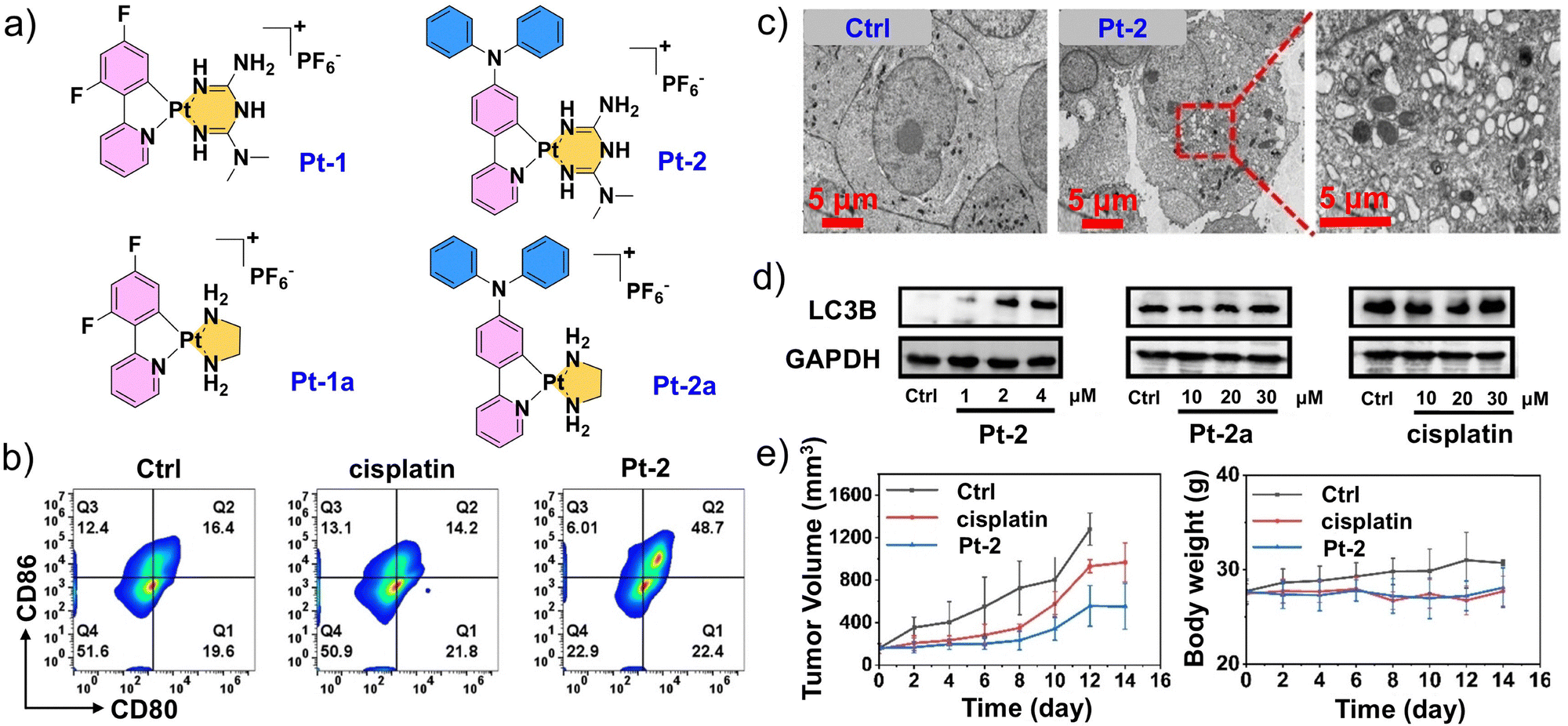 | ||
| Fig. 7 Design and performance evaluation of platinum (II)-metformin complex Pt-2. (a) Chemical structures of platinum(II)–metformin complexes (Pt-1 and Pt-2) and control compounds (Pt-1a and Pt-2a). (b) Flow cytometry images of CD80+CD86+ dendritic cells (DCs) among tumor cells after different treatments. (c) Transmission electron microscopy (TEM) images of A549 cells after Pt-2 treatment (2 µM, 24 h). (d) Western blot analysis of LC3B expression in A549 cells upon 12 h treatment with Pt-2, Pt-2a, and cisplatin at the indicated concentrations. (e) Tumor volume and weight change curves of mice in different treatment groups. (a–e) Reproduced with permission from ref. 122. Copyright 2024, Wiley-VCH. | ||
Gold (Au) complexes are considered potential antitumor agents due to their inherent inhibition of thioredoxin reductase.128 Tang et al. developed TBP-Au, an Au(I)-based aggregation-induced emission (AIE) luminogen that enables high-resolution two-photon tumor imaging and lysosome-targeted tumor PDT.129 Lo et al. designed a rhenium(I) complex targeting lysosomes, where one ligand bears a triazole fragment with a quenching effect, resulting in a very low overall quantum yield for the complex.130 Upon trans-cyclooct-4-enol-triggered cleavage, the 1O2 yield is markedly enhanced, so that the triazole fragment functions as a molecular switch for photosensitization. Sadler et al. reported a bimetallic platinum(IV)-iridium(III) complex with charge transfer leading to the accumulation of intracellular platinum. The platinum then induces nuclear damage upon irradiation, generating large amounts of ROS within only 1 h after administration. Simultaneously, the iridium localizes to lysosomal vesicles, suggesting that the complex undergoes cleavage and excretion via recycling vesicles.131 Based on the studies mentioned above, metal complexes offer good potential in controllable phototherapy due to their inherent heavy-atom effect, rich excited-state properties, and tunable photochemical activity.
However, lysosome-targeted optical metal complexes still face several significant limitations. First, these complexes generally exhibit relatively high dark toxicity and have comparatively short absorption wavelengths, which restrict their broad application in precise tumor therapy. Moreover, in many metal-based probes or targeting systems, complexes based on iridium, ruthenium, or platinum inherently generate large amounts of 1O2 under light irradiation, with an average diffusion length of only ∼10 µm. This confines the phototherapeutic effect to a very small spatial region, thereby significantly limiting their long-term therapeutic potential. Therefore, these intrinsic limitations must be carefully considered in the subsequent design and application of lysosome-targeted metal-based photosensitizers. In contrast, supramolecular assemblies demonstrate greater adaptability than single molecules through structural modulation, enhanced targeting, and multifunctional integration, offering advantages with respect to safety, therapeutic efficacy, and clinical feasibility.
4.3. Supramolecular platforms
Compared to other traditional therapies, PDT has several unique advantages such as spatial selectivity, efficiency, lack of resistance, and non-invasiveness.132–137 Nevertheless, there are unavoidable limitations that restrict its progress in both preclinical and clinical studies. Conventional PDT suffers from drawbacks such as the “always-on” state and poor tumor specificity, which force patients to remain in darkness for prolonged periods following therapy. To overcome these shortcomings, adaptive supramolecular photosensitizers have been proposed with switchable photosensitive effects. Huang et al. reported a supramolecular modification based on the host–guest interaction of pillar aromatic hydrocarbons.138 Specifically, they designed and synthesized a supramolecular ROS switch based on an anionic water-soluble pillar[5]arene (WP5) as the host and a tetraphenylethylene-based photosensitizer (G) as the guest. This supramolecular switch exhibits minimal fluorescence and ROS generation under neutral conditions, but emits bright red fluorescence and induces pronounced ROS production in acidic environments (Fig. 8a). Specifically, the AIE molecule G displays negligible fluorescence in water owing to its high solubility. However, under acidic conditions (pH ≤ 5.0), the protonation of WP5 triggers its dissociation from G, leading to aggregation of the amphiphilic photosensitizer (G) and marked enhancement in fluorescence emission (Fig. 8b). pH titration confirmed that the system exhibits a sharp fluorescence increase at pH below 6.0, highlighting the potential of this acid-triggered switch for tumor imaging and therapy (Fig. 8c).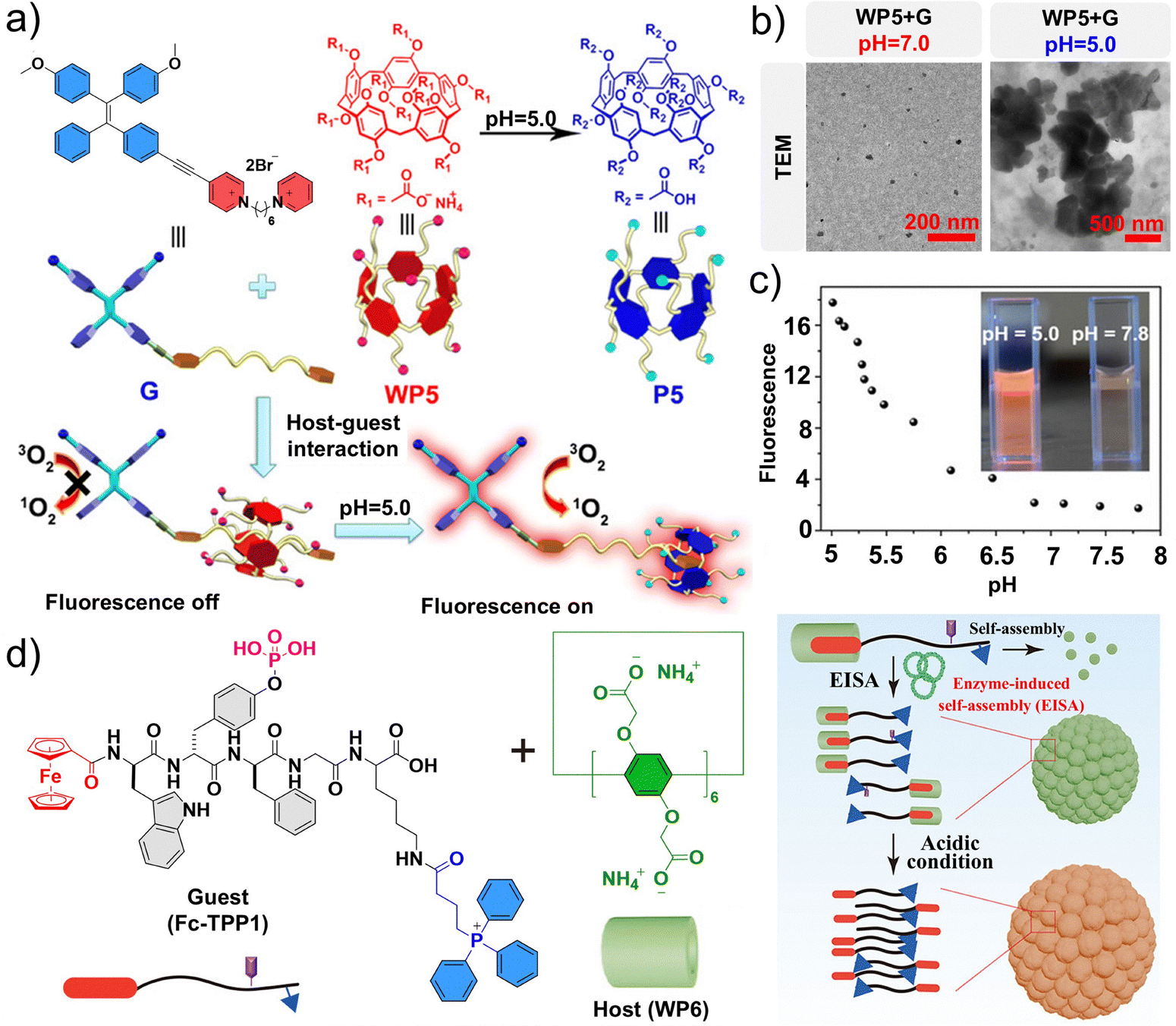 | ||
| Fig. 8 Assembly principle and structural characterization of lysosome-targeted supramolecular assemblies. (a) Chemical structures and cartoon representations of WP5, P5, and G. (b) Transmission electron microscopy (TEM) images of WP5 + G at different pH values. (c) Solution pH dependence of the fluorescence intensity of WP5 + G in aqueous solution at 620 nm. (a–c) Reproduced with permission from ref. 138. Copyright 2020, Wiley-VCH. (d) Chemical structures of Fc-TPP1 and pillar[6]arene (WP6), and schematic illustration of assemblies formed from the host–guest complexes between the molecules. Reproduced with permission from ref. 152. Copyright 2022, American Chemical Society. | ||
Other extensive studies have also demonstrated the potential of supramolecular ROS switches as precise antitumor therapeutics.139–142 Enzyme-driven self-assembly is an effective strategy for controlling molecular assembly dynamics, capable of mimicking most natural biological processes.143–145 Enzyme-induced self-assembly (EISA) is a strategy to control behaviors around or within cells through the overexpression of enzymes, offering an alternative to traditional tumor therapy methods as an emerging research hotspot.123,146–151 Wang et al. utilized host–guest complexes to regulate the formation dynamics of EISAs in tumor cells through lysosomal targeting and escape, leading to the selective induction of tumor cell death.152 In this system, a pillar[6]arene (WP6) serves as the host and Fc-TPP1 serves as the guest to form a supramolecular assembly, and their assembly behavior can be further driven by alkaline phosphatase (ALP) and an acidic microenvironment (Fig. 8d). After entering tumor cells, Fc-TPP1/WP6 escapes from lysosomes and subsequently targets mitochondria, resulting in the reduction of mitochondrial membrane potential and elevated production of ROS. This process triggers apoptosis in tumor cells, which is associated with ferroptosis.153–155 Overall, this work presents a versatile strategy for regulating enzyme-driven self-assembly in living cells to selectively program tumor cell death.
The effectiveness of the immune response induced by ICD has both direct and indirect associations with the generation of ROS; yet, many existing ICD inducers suffer from inherent limitations,156–163 including the potential for adverse reactions and challenges in inducing immune regulatory responses for deeply located solid tumors.163–165 Platinum(II) and ruthenium(II) metallacycles have shown good performance in biomedical imaging, chemotherapy, and PDT.166–171 Sun et al. developed a new NIR-emitting ruthenium(II) metallacycle named Ru1105, with a maximum emission wavelength (λem) of 1105 nm, which acts as a superb ICD inducer for chemical phototherapy of deep tumors while minimizing adverse reactions.172 The researchers introduced julolidinyl and anisole electron-donating units, along with a binuclear aromatic ruthenium coordination unit, into the Aza-BODIPY receptor framework to form the NIR-II–emitting ligand, L (Fig. 9a and b). Subsequently, L and the 0° ruthenium(II) receptor A were reacted at room temperature to yield the metallacycle Ru1105, which could serve as the supramolecular host for the construction of assemblies. Additionally, the geometry of Ru1105 was optimized using Gaussian 09, and its successful synthesis was confirmed by mass and nuclear magnetic resonance spectra (Fig. 9c–e). In vitro, Ru1105 exhibited remarkable tissue penetration capacity, entering tumor cells via clathrin-mediated endocytosis and preferentially localizing in lysosomes, highlighting its potential as a lysosome-targeted drug (Fig. 9f). In vivo, vaccination experiments in CT26 tumor-bearing BALB/c mice demonstrated that under 808 nm laser irradiation, Ru1105 effectively induced ICD by activating CD8+ T cells while sparing Foxp3+ T cells, resulting in significant tumor regression and eradication. Overall, Ru1105 serves as a case study for the development of an NIR light-activated supramolecular platform to induce ICD, providing a foundation for developing host–guest supramolecular antitumor agents.
 | ||
| Fig. 9 Characterization and performance evaluation of Ru1105. (a) The ligand L and the acceptor A self-assemble to form the ruthenium(II) metallacycle Ru1105. (b) Normalized absorption and the corresponding emission spectra (λex = 808 nm) of Ru1105 and L in N,N-dimethylformamide. (c) Partial 1H nuclear magnetic resonance spectra of the metallacycle Ru1105. (d) Calculated (blue) and experimental (red) mass spectra of Ru1105. (e) Molecular model of Ru1105 optimized by the B3LYP molecular orbital approach. (f) Reactive oxygen species (ROS) generation for Ru1105 in deep tissues under 808 nm laser irradiation for 3 min, and fluorescence images of Ru(bpy)3Cl2(I) and Ru1105(II) encapsulated in capillaries and immersed at varied depths in 1% intralipid. (a–f) Reproduced with permission from ref. 172. Copyright 2022, American Chemical Society. | ||
Overall, supramolecular platforms integrate multiple functional advantages, enabling structural tunability, controllable assembly, and multifunctional integration through noncovalent interactions. Accordingly, this type of platform can ultimately achieve a balance among targeting capability, responsiveness, and drug delivery efficiency. Compared with the relatively simple small-molecule or metal complex molecular platforms described above, supramolecular platforms not only enhance tumor selectivity and drug stability based on their structural design but also achieve stimuli-responsive release in response to specific characteristics of the TME (such as pH and ROS), thereby improving therapeutic efficacy while minimizing toxicity to normal tissues. Their high tunability and multifunctionality render supramolecular platforms particularly promising agents for precise tumor therapy. Nevertheless, these formulations face several challenges for application in vivo, including limited stability, short circulation times, off-target effects, and toxicity to normal tissues caused by premature drug release. To address these challenges, lysosome-specific material platforms can enhance drug stability and prolong circulation, achieve more precise tumor targeting through surface functionalization, and enable controlled release by exploiting the unique features of the TME.
5. Lysosome-specific material platforms
In addition to molecular platforms that achieve precise drug delivery through classical lysosome-targeted groups, exploiting the phagocytosis function of lysosomes to achieve precise delivery of biomaterials has long been an important area of research in precision oncology. Importantly, phenomena involving multi-organelle targeting are often observed after lysosomal escape, which can further enhance the efficiency of tumor treatment.173,174 The development of a series of lysosome-specific material platforms, including self-assembled peptides, nanomicelles, nanoparticles, liposomes, nanogels, inorganic nanodrugs, and coacervates, provides new approaches for precision tumor therapy. These platforms have been rapidly emerging in recent years, with preclinical studies demonstrating the great promise of lysosomes as effective targets for tumor treatment.5.1. Self-assembling peptide platforms
The self-assembly of peptides combines designability, biodegradability, and functional diversity, while also forming controllable nanostructures. Therefore, this approach holds substantial research value in drug delivery, tissue engineering, immunotherapy, and fundamental molecular science. Some candidate drugs for NSCLC can induce LMP to directly trigger tumor cell death. Lam et al. developed a deformable self-assembling peptide platform to induce lysosomal LMP in NSCLC.175 The nanomaterial (designated as CPTNP-FF) was constructed from amphiphilic peptides that self-assemble into nanoparticles and readily fuse with lysosomes (Fig. 10a and b). In vitro, CPTNP-FF undergoes an acid-triggered morphological transition from nanoparticles to nanofibers, and this process was effectively recapitulated within the lysosomes of A549 cells (Fig. 10c and d). This conformational transformation perturbs lysosomal pH homeostasis, leading to sustained lysosomal destabilization, consequently accelerating tumor cell death and enhancing cisplatin sensitivity. In xenograft models, CPTNP-FF therapy resulted in a punctate distribution of galectin-1, in contrast to the uniform staining observed in the tumors of the control, phosphate-buffered saline.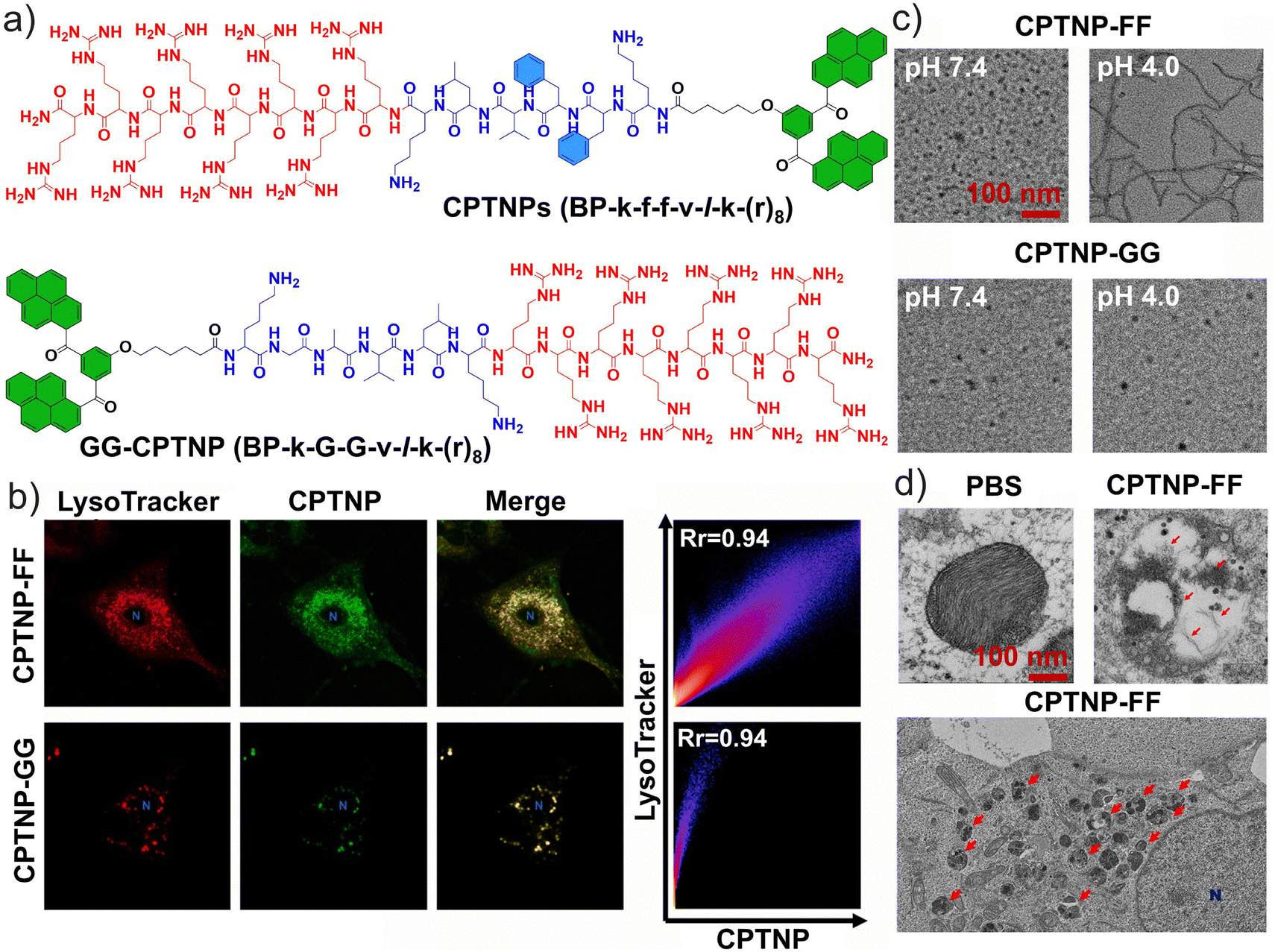 | ||
| Fig. 10 Self-assembly and targeting of peptides. (a) Chemical structures of CPTNPs, BP-k-f-f-v-l-k-(r)8 peptide, and the negative control GG-CPTNP, BP-k-G-G-v-l-k-(r)8 peptide. (b) A single A549 cell treated with CPTNP-FF and CPTNP-GG stained with LysoTraker Red; the nucleolus is represented by a blue N. The intensity heat map (represented by the pixel intensity distribution) shows a cell imaged after treatment with CPTNP-FF and CPTNP-GG with a Pearson correlation coefficient of 0.94 in each case. (c) Transmission electron microscopy (TEM) images of CPTNPs incubated at pH 7.4 and 4.0. (d) Low-magnification TEM image of the same sample; lysosomes are labeled with red arrows while the nucleolus is labeled with a blue N. (a–d) Reproduced with permission from ref. 175. Copyright 2021, Elsevier Ltd. | ||
Inspired by natural bioconjugates, Wang et al. constructed a pH-responsive peptide self-assembling hydrogel in live cell lysosomes, which was explored as a potential agent for tumor therapy.176 Specifically, this strategy addresses the acquisition of drug resistance in tumor cells caused by the tendency of chemotherapeutic agents to become trapped in lysosomes during tumor treatment.
The designed peptide molecules can adopt different morphologies under varying pH conditions, as demonstrated using peptide sequences derived from human insulin (Fig. 11a). TEM images confirmed that D-LTP exhibits pH-dependent morphological changes distinct from those of L-LTP; at pH values similar to those in the TME, D-LTP more readily transforms into nanofibers (Fig. 11b). Furthermore, by exploiting the acidic environment of tumor cell lysosomes to regulate the assembly behavior of D-LTP peptides, the nanofibers underwent a phase transition from solution to hydrogel within lysosomes. The resulting hydrogel expanded the lysosomal volume and increased lysosomal membrane permeability. Finally, intracellular hydrogelation in tumor cells enhanced the accumulation of doxorubicin (Dox), enabling its release from lysosomes and delivery to the nucleus (Fig. 11c). This strategy provides a novel approach for the precise formation of functional peptide aggregates in living cells with potential to overcome tumor drug resistance.
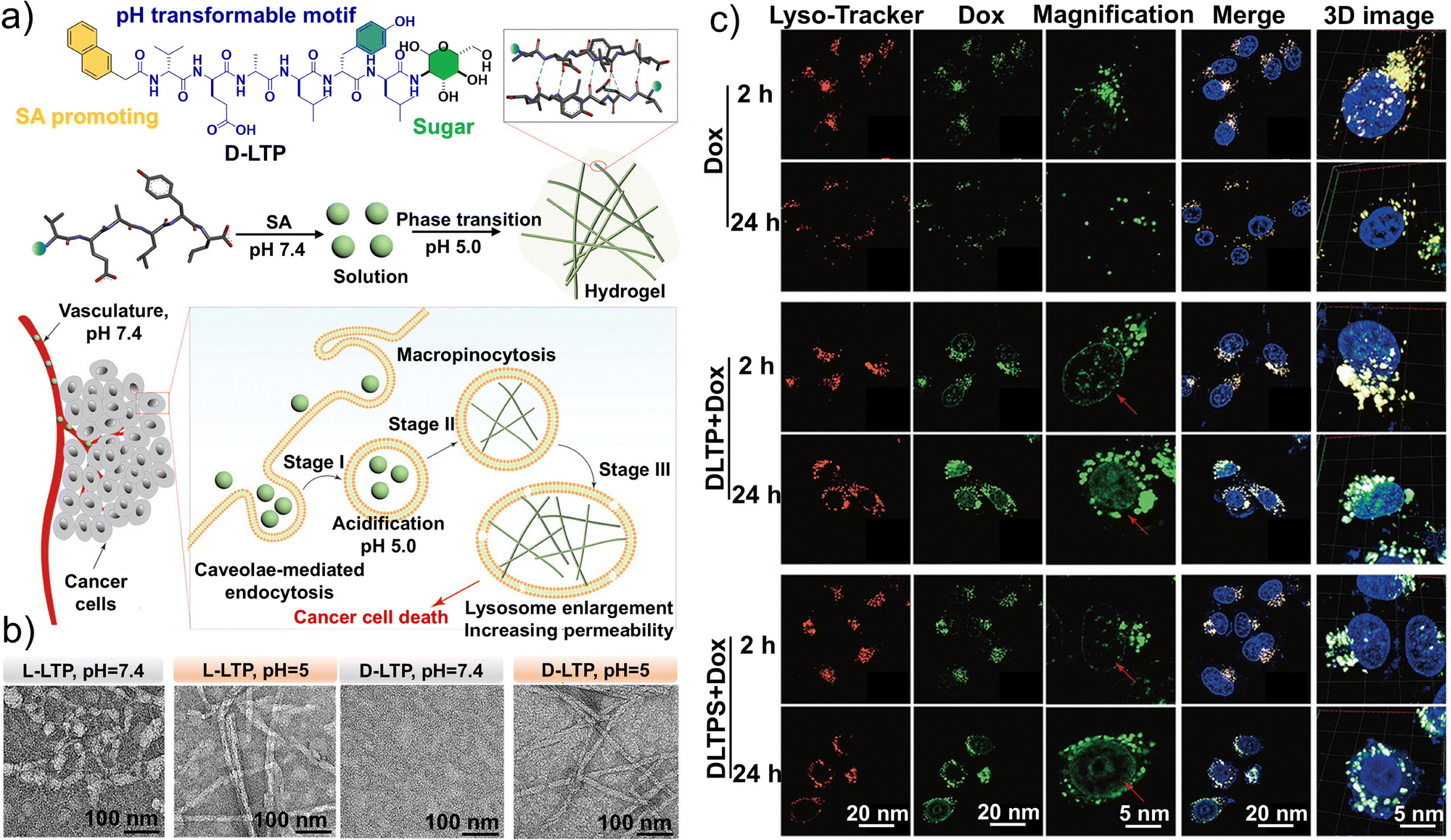 | ||
| Fig. 11 Structural design, self-assembly effect, and targeting properties of D-LTP. (a) Schematic illustration of pH-responsive transformable peptides and the self-assembly process in vitro and in vivo. (b) Transmission electron microscopy (TEM) images of L-LTP and D-LTP at a concentration of 500 × 10−6 M with pH 7.4 and 5.0. (c) Confocal laser-scanning microscopy (CLSM) images of HeLa cells treated with 5 × 10−6 M Dox and 500 × 10−6 M D-LTP or D-LTPS for 2 and 24 h. All cell images in a given column share the same scale bar. (a–c) Reproduced with permission from ref. 176. Copyright 2021, Wiley-VCH. | ||
Although inducing LMP and lysosome-associated enzyme-controlled self-assembly are effective approaches for spatiotemporally precise tumor therapy, studies combining lysosomal targeting with enzyme-controlled self-assembly strategies to selectively kill tumor cells remain limited. Liang et al. designed a pyrene-peptide conjugate, Py-Yp-Lyso, which integrates lysosomal targeting with ALP-regulated self-assembly. This molecule exhibited the highest selectivity toward HeLa cells and the strongest cytotoxic effect.177 In mice, Py-Yp-Lyso was specifically activated via overexpressed ALP on the tumor cell surface as well as within lysosomes, enabling concurrent diagnosis and therapy. Ding et al. achieved lysosome-targeted accumulation by modulating the charges of nanoassemblies, inducing ICD and transforming tumors from “cold” to “hot” via LMP and lysosomal membrane rupture (LMR), which subsequently triggered autophagy and apoptosis.178 Through molecular structure regulation, Ding et al. combined the AIE molecule TPE-Py, self-assembling peptides pYKpY, and a triphenylphosphonium cation to construct a water-soluble probe, named TPE-Py-pYK(TPP)pY, featuring responsive fluorescence and 1O2-quenching capability. Upon activation by ALP, the probe self-assembles into positively charged nanostructures, switching on fluorescence and efficiently generating 1O2, with acidic conditions (pH 5.5) further boosting the production of ROS. In ALP-overexpressing HeLa cells, the probe initially assembles on the cell membrane before accumulating in lysosomes; increasing concentrations of the probe triggered LMP, LMR, and tumor cell death. Similar outcomes were observed in ALP-overexpressing 4T1 cells. This ALP-activated self-assembling peptide induces lysosomal autophagy and apoptosis, establishing a direct link between lysosome-mediated cell death and tumor immunotherapy.
Owing to their water solubility, amphiphilic nature, and intrinsic charge, peptides can efficiently enter cells via endocytosis and preferentially accumulate in lysosomes. Within the acidic lysosomal environment or upon enzymatic activation, these peptides undergo self-assembly to form nanofibers, nanohydrogels, or charged nanoparticles. Such self-assembly may be directly triggered by acidic conditions or indirectly regulated by lysosomal enzymes such as ALP or cathepsin B (CTSB). Table 1 summarizes the mechanisms of direct or enzyme-mediated self-assembly and corresponding antitumor effects of recently reported self-assembling peptides, which can induce lysosomal damage (including LMP), lysosomal membrane swelling (LMS), and LMR, thereby impairing lysosomal function.65,105,175–180 These processes thus facilitate the controlled release of drugs or functional molecules from lysosomes, enabling precise intracellular delivery, while further triggering programmed cell death (e.g., apoptosis, pyroptosis, and necrosis) or activating ICD effects through regulated assembly, thereby enhancing antitumor efficacy and overcoming drug resistance.
| Name | Self-assembly | Trigger | Driving force | Tumor cell line | Lysosomal damage | Ref. | |
|---|---|---|---|---|---|---|---|
| a Alkaline phosphatase. b Cathepsin B. c Lysosomal membrane permeabilization. d Lysosomal membrane swelling. e Lysosomal membrane rupture. | |||||||
| Direct self-assembly | CPTNP-FF | Nanofibers | Lysosomal acidic microenvironment (pH ∼ 4.5–5.0) | Protonation of weakly basic functional groups | A549 | LMPc | 175 |
| D-LTP | Multidrug-resistant cell line | LMSd | 176 | ||||
| L-LTP | |||||||
| NTV2 | Nanosheets | B16F10-OVA | LMP, LMRe | 65 | |||
| Indirect self-assembly | Py-Yp-Lyso | Nanoparticles | ALPa | Non-covalent interactions | HeLa | LMP | 177 |
| TPE-Py-pYK(TPP)pY | HeLa, 4T1 | LMR | 178 | ||||
| C1 | Nanonetwork | MuM-2B, HeLa, HepG2 | LMP | 179 | |||
| DL-MN | Nanofibers | CTSBb | HeLa, U87MG | LMP, LMR | 180 | ||
| NDI-Lyso-RGD | HeLa, MCF7 | LMP, LMS, LMR | 105 | ||||
Collectively, this mechanism integrates peptide lysosomal accumulation, acid- or enzyme-triggered self-assembly, and modulation of lysosomal function, offering a robust strategy for spatiotemporally precise tumor therapy. Despite the promise of lysosome enzyme-triggered self-assembling peptide platforms, several challenges remain. The heterogeneous expression of lysosomal enzymes across different tumor types, coupled with their presence in normal tissues, limits both the specificity and safety of these approaches. Moreover, peptides often suffer from poor stability and suboptimal in vivo delivery, with activation lacking precise spatiotemporal control. Potential immunogenicity and metabolic safety issues also warrant further investigation. Therefore, future efforts should prioritize the optimization of selectivity, delivery efficiency, and controllable activation to fully realize the potential of these platforms in precision oncology.
5.2. Nanomicelle platforms
Compared with peptides, nanomicelles are more easily tunable in terms of surface area, size and shape, targeting ability, and controlled release, enabling more precise tumor therapy.181 Phototherapy has emerged as an effective, non-invasive alternative to conventional treatment for nasopharyngeal carcinoma (NPC), overcoming challenges such as incomplete surgery and severe chemotherapy/radiotherapy side effects.182–187 Yang et al. developed a lysosome-targeted and pH-responsive nano-phototherapy diagnostic agent for NIR-II fluorescence imaging-guided PDT and photothermal therapy (PTT) for NPC.188 Specifically, they synthesized an organic solubilizing group-donor–acceptor–donor-solubilizing group (S-D–A–D-S) type molecule, named IRFEM, through an existing chemical modification method with the introduction of morpholine rings to significantly enhance tumor-targeting precision (Fig. 12a).189 They subsequently encapsulated IRFEM with an acid-sensitive amphiphilic copolymer, DSPE-Hyd-PEG2k, to prepare the nanomicelle IRFEM@DHP (Fig. 12b). IRFEM@DHP demonstrated a lysosomal binding probability up to 90.6%, which is highly favorable for applications in lysosome-mediated precision tumor diagnosis and therapy (Fig. 12c). Owing to its multifunctional properties, including fluorescence, photothermal conversion, and ROS generation, IRFEM represents an ideal candidate for NIR-II imaging-guided combined PTT/PDT therapy. Moreover, IRFEM@DHP can preferentially accumulate within NPC cells via the enhanced permeability and retention effect, subsequently localizing in the acidic lysosomes where IRFEM is released. Upon release, the abundant morpholine groups enable IRFEM to specifically target lysosomes. Meanwhile, the light irradiation triggers robust photothermal and ROS production, thereby inducing lysosome-mediated cell death. This strategy was confirmed with in vivo experiments as IRFEM@DHP + NIR treatment markedly inhibited tumor growth in animal models (Fig. 12d). In addition, the morpholine rings confer excellent long-term NIR-II imaging capability, enabling precise tumor localization up to 48 h post-administration (Fig. 12e). The broad and intense NIR-II emission facilitates deep-tissue imaging and precise phototherapy, while the modular design of S-D–A–D-S molecules allows the fine-tuning of photophysical processes, generating a synergistic effect between PTT and PDT to enhance tumor eradication. The lysosome-targeting function further prolongs the retention of IRFEM@DHP, improving the efficiency of combined PDT/PTT in eliminating tumor cells.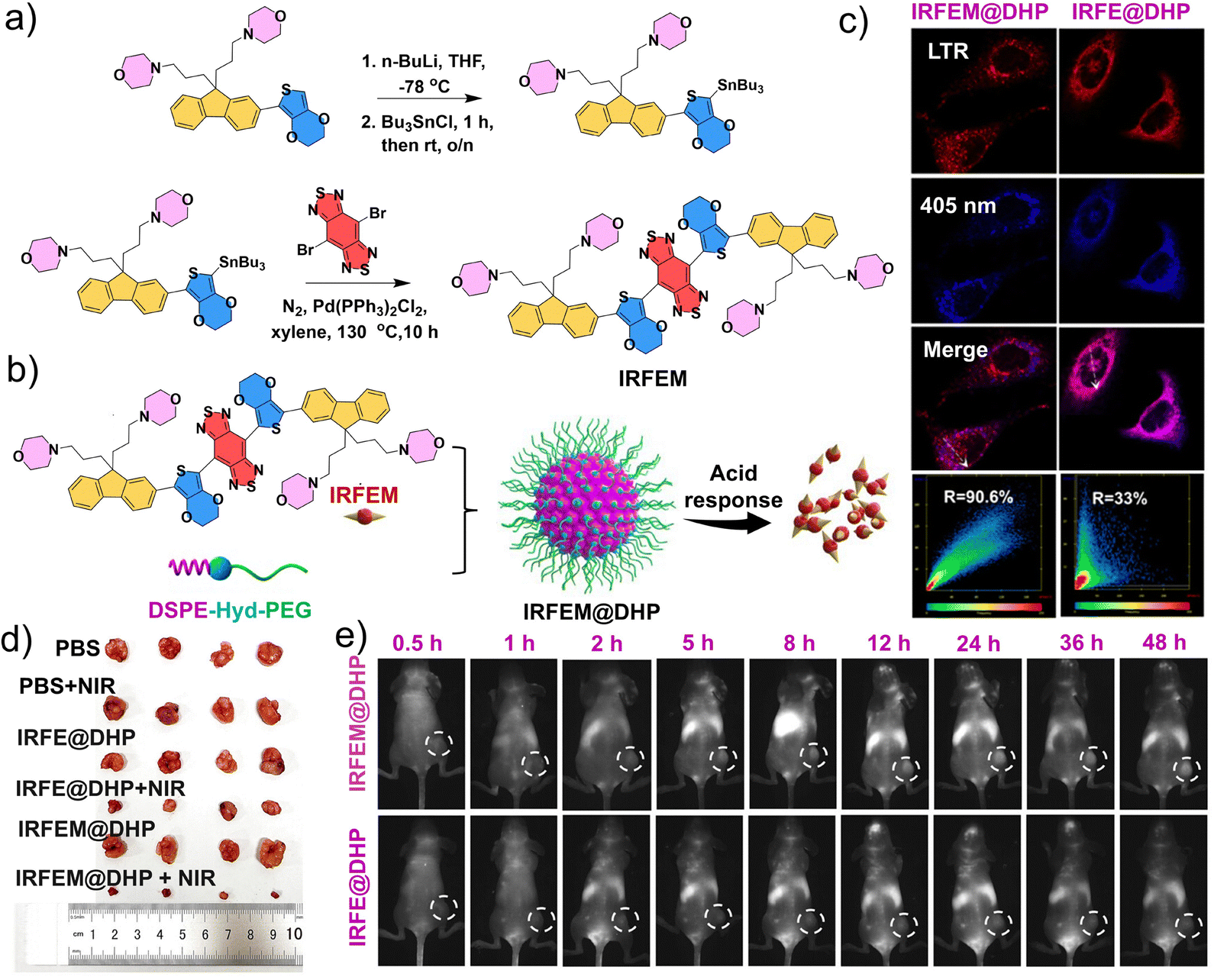 | ||
| Fig. 12 Preparation and application of the IRFEM@DHP nanomicelles. (a) Synthetic route of IRFEM. (b) Preparation and responsive behavior of IRFEM@DHP. (c) Confocal microscopy images of 5–8F cells following incubation with IRFEM@DHP and LysoTracker Red (LTR). (d) Photographs of tumors from mice following various treatments. (e) Real-time fluorescence images of 5–8F tumor-bearing mice following treatment with IRFE@DHP or IRFEM@DHP at different times. (a–e) Reproduced with permission from ref. 188. Copyright 2024, American Chemical Society. | ||
Inspired by the close relationships among lysosomal function, pyroptosis, and autophagy, Yin et al. synthesized a BODIPY dimer, BDPd, containing two lysosome-targeting morpholine groups, which was employed as an NIR photosensitizer for combined PDT/PTT (Fig. 13a).190 The amphiphilic triblock copolymer Pluronic F127 was then used as the building block to prepare BDPd nanomicelles (Fig. 13b). In vitro assays demonstrated that the intracellular uptake of BDPd was enhanced upon 660 nm laser irradiation, thereby increasing its cytotoxicity against 4T1 mouse breast tumor cells. Mechanistic investigations further revealed that PTT/PDT mediated by the BDPd nanomicelles induced pronounced lysosomal and mitochondrial damage, exposed ICD markers, and activated pyroptotic pathways via NLRP3/gasdermin D (GSDMD) and caspase-3/gasdermin E (GSDME) signaling, ultimately promoting the maturation of DCs. The induced lysosomal dysfunction also impaired autophagic degradation, thereby mitigating its cytoprotective effects. Both BDPd and BDPd nanomicelles efficiently localized to lysosomes in 4T1 cells. Following intratumoral administration, the temperature in the tumor region increased markedly (Fig. 13c–f). PTT/PDT mediated by BDPd nanomicelles significantly suppressed the growth of established 4T1 mammary tumors in vivo and elicited a robust local and systemic immune response, thereby preventing tumor recurrence (Fig. 13g). Overall, this study highlights a promising nanomicelle-based strategy that combines PTT/PDT with subcellular targeting to enhance programmed cell death in tumor cells, ultimately improving therapeutic outcomes.
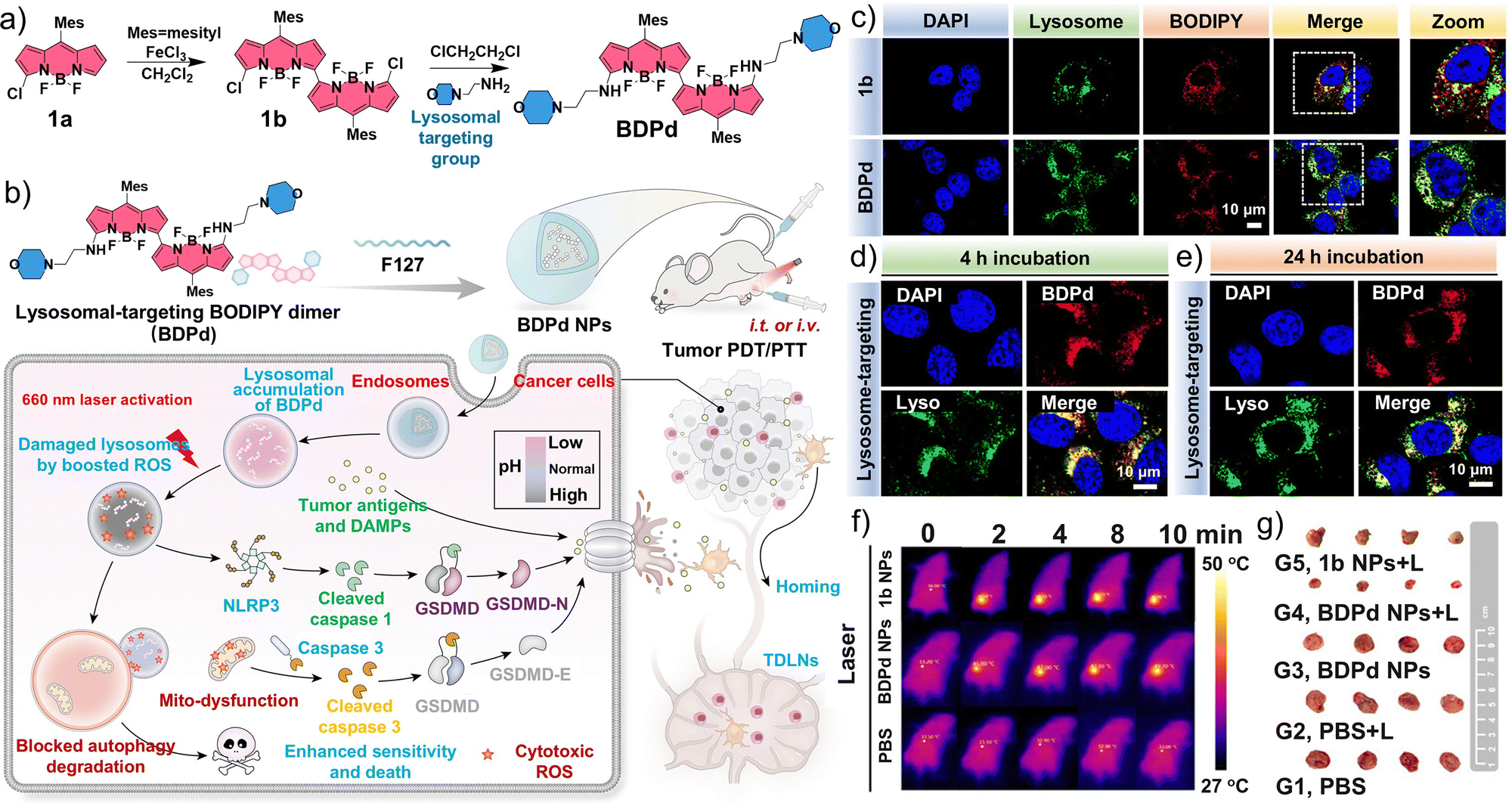 | ||
| Fig. 13 Preparation and application of BDPd nanomicelles (BDPd NPs). (a) Synthesis route of BDPd. (b) Schematic illustration of the construction of BDPd NPs and their treatment in a mouse breast cancer model with PTT/PDT. (c) Confocal laser-scanning microscopy (CLSM) images of the colocalization of BODIPY dimer 1b and BDPd with the lysosome. CLSM images of the colocalization of BDPd NPs with the lysosome at (d) 4 h and (e) 24 h after incubation. (f) In vivo thermal images of tumor-bearing mice treated with PBS, 1b NPs, or BDPd NPs and laser irradiation (660 nm, 200 mW cm−2, 10 min). (g) Representative photographs of collected tumors 14 days after treatment. (a–g) Reproduced with permission from ref. 190. Copyright 2024, American Chemical Society. | ||
Ferroptosis is a unique form of programmed cell death with distinct biological characteristics and mechanisms from apoptosis, necrosis, and autophagy.191–193 However, achieving spatiotemporal control of intracellular Fenton reactions to regulate tumor ferroptosis remains challenging. Zhang et al. reported a novel oxime-based activatable nanomicelle, PTO-Biotin NPs, capable of triggering lysosomal dysfunction-mediated Fenton pathways to induce ferroptosis with NIR laser-mediated spatiotemporal precision.194 Additionally, Zhou et al. screened out diketopyrrole red dyes containing two indole groups that exhibited favorable NIR fluorescence/photoacoustic imaging and photothermal therapy properties; they further incorporated two morpholine rings into the dyes to generate the lysosome-targeting and Fe3+-regulatory agent CR-736.195 A CR-736-Fe3+ nanomicelle platform was then developed to deliver these agents precisely to breast tumor sites, releasing CR-736 and Fe3+ in the acidic lysosomal microenvironment to activate multiple pH-responsive functions. The synergistic effect of PTT, ferroptosis, and CDT was maximized in the acidic tumor lysosomal microenvironment and these effects were validated both in vitro and in murine tumor models.
Supramolecular nanomicelles are primarily assembled via non-covalent interactions, such as hydrophobic interactions, hydrogen bonding, π–π stacking, electrostatic interactions, and metal coordination. Their highly modular molecular architecture enables rational design for disease targeting, microenvironment responsiveness, imaging, and the integration of multimodal therapies. However, nanomicelles formed via single-chain polymer folding exhibit enhanced structural stability, improved in vivo tolerance, and precise functional integration, particularly excelling in multifunctional incorporation, increased drug loading, prolonged blood circulation, and controlled nanostructure morphology. As summarized in Table 2, recent studies have reported a series of lysosome-targeted single-chain polymeric nanomicelles (SCPNs) that achieve synchronized structural and functional regulation via microenvironment-triggered intramolecular folding and self-assembly.194,196–201 Representative examples include TAENmi, which undergoes conformational contraction in the acidic TME to facilitate combined phototherapy and immunotherapy; ALR-PLA-SS-PEG, which releases functional modules at elevated GSH levels to mediate protein degradation therapy; PCL-PSDMA-PTX, which is characterized by a dual pH responsiveness mechanism for efficient intracellular paclitaxel delivery; PGN4.9, which precisely modulates lysosomal acidity and enzymatic activity to reprogram immune phenotypes; and HCPT-SCNP, which targets lysosomes to overcome multidrug resistance. Overall, these studies indicate that single-chain polymeric nanomicelles exhibit high stability and pronounced microenvironment responsiveness, while inducing lysosomal stress (LS) to exacerbate lysosomal damage, thereby synergistically mediating phototherapy, chemotherapy, immunotherapy, ferroptosis, and other multimodal antitumor mechanisms. Despite the distinct advantages of nanomicelle platforms for lysosome-targeted precision tumor therapy, there are some limitations to address. Although these platforms can effectively maintain cargo stability, their intrinsic stability in the blood circulation remains limited, and premature disassembly or drug release may compromise targeting efficiency. Of particular concern is their restricted drug-loading capacity, which is especially critical for combination therapies as this can markedly diminish any potential synergistic effects. Finally, achieving highly uniform nanomicelles with consistent stability and reproducibility at an industrial scale remains challenging.
| Name | Self-assembly driving force | Trigger | Function | Lysosomal escape | Lysosomal damage | Antitumor mechanism | Ref. |
|---|---|---|---|---|---|---|---|
| a Glutathione. b Lysosomal membrane permeabilization. c Lysosomal stress. d Lysosomal membrane rupture. e Paclitaxel. | |||||||
| TAENmi | Noncovalent interactions | Acidic microenvironment (pH < 6.5) | Phototherapy andimmunotherapy | No | LMPb/LSc | Pyroptosis-mediated immune response | 196 |
| PTO-biotin NPs | Acidic tumor microenvironment | Phototherapy and ferroptosis | LMP/LMRd | Phototherapy-enhanced ferroptosis | 194 | ||
| ALR–PLA–SS–PEG | Overexpressed GSHa in tumor | Targeted therapy | LMP/LMR | Protein degradation | 197 | ||
| PCL–PSDMA–PTX | Acidic extracellular environment (pH ∼ 6.8) | Chemotherapy and immunotherapy | Yes | LMP | Efficient delivery of PTXe | 198 | |
| Acidic lysosomal microenvironment (pH ∼ 4.5–5.0) | |||||||
| BDP NPs | Acidic lysosomal microenvironment (pH ∼ 4.5–5.0) | Phototherapy and immunotherapy | LMP/LMR | PDTf-mediated immune response | 199 | ||
| PGN4.9 | Immunotherapy | pH and enzyme activity interference | Lysosomal function interference and immune phenotype reprogramming | 200 | |||
| HCPT-SCNP | Acidic microenvironment (pH = 5.0) | Chemotherapy | No | LMR | Lysosome-targeted blockade of drug resistance | 201 | |
5.3. Organic nanoparticle platforms
 | ||
| Fig. 14 Preparation and functional evaluation of the lysosome-targeted spherical nanoparticle LTANP based on positive and negative charge interactions. (a) Schematic illustration for detachment of the polyethylene glycol (PEG) shell in the acidic tumor microenvironment (TME; pH 6.5) and protein nanocapsule aggregation in the lysosomal microenvironment (pH 4.5). (b) Schematic illustration of LTANP-targeted aggregation in lysosomes to induce lysosomal membrane permeabilization (LMP) and immunogenic cell death (ICD) for tumor immunotherapy through a biomimetic strategy. (c) Quantitative analysis of the CD8+ T lymphocyte population in rechallenged tumors based on flow cytometry; effector memory T cells (CD3+CD8+CD44+CD62L−) and central memory T cells (CD3+CD8+CD44+CD62L+) in the spleens of the mice after different treatments; and lung metastatic nodules in tumor-rechallenged mice after different treatments. (d) Representative images and H&E staining analysis of lung sections in tumor-rechallenged mice after different treatments. (a–d) Reproduced with permission from ref. 212. Copyright 2024, Wiley-VCH. | ||
Anionic nanoparticles are internalized slowly by adherent cells, whereas cationic nanoparticles interact with the cell membrane via strong electrostatic forces; although this increases membrane permeability, it comes at a cost of non-selective cytotoxicity. To address this issue, Grzybowski et al. developed mixed-charge spherical nanoparticles ([±] NPs) coated with varying ratios of anionic and cationic ligands. These nanoparticles selectively kill tumor cells, exhibit good tolerability to normal cells, and undergo precipitation or crystallization under different pH conditions (Fig. 15a and b).36 Leveraging the pH differences between tumor and normal tissues, the pH-dependent aggregation of [±] NPs enables selective lysosomal targeting within tumor cells (Fig. 15c). Assays with the 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 [±] NPs showed the formation of intermediate clusters in the presence of the lysosomal protease cathepsin D, followed by their further aggregation into larger nanosupercrystals (∼260 nm) within lysosomes. Subsequent studies demonstrated that the selective assembly of nanosupercrystals in tumor lysosomes disrupts lysosomal function, inducing lysosome-dependent cell death (Fig. 15d and e). This approach can potentially be broadly applied as a general pH-lysosome-targeted nanodrug delivery strategy, although further validation in animal models is necessary.
:20 [±] NPs showed the formation of intermediate clusters in the presence of the lysosomal protease cathepsin D, followed by their further aggregation into larger nanosupercrystals (∼260 nm) within lysosomes. Subsequent studies demonstrated that the selective assembly of nanosupercrystals in tumor lysosomes disrupts lysosomal function, inducing lysosome-dependent cell death (Fig. 15d and e). This approach can potentially be broadly applied as a general pH-lysosome-targeted nanodrug delivery strategy, although further validation in animal models is necessary.
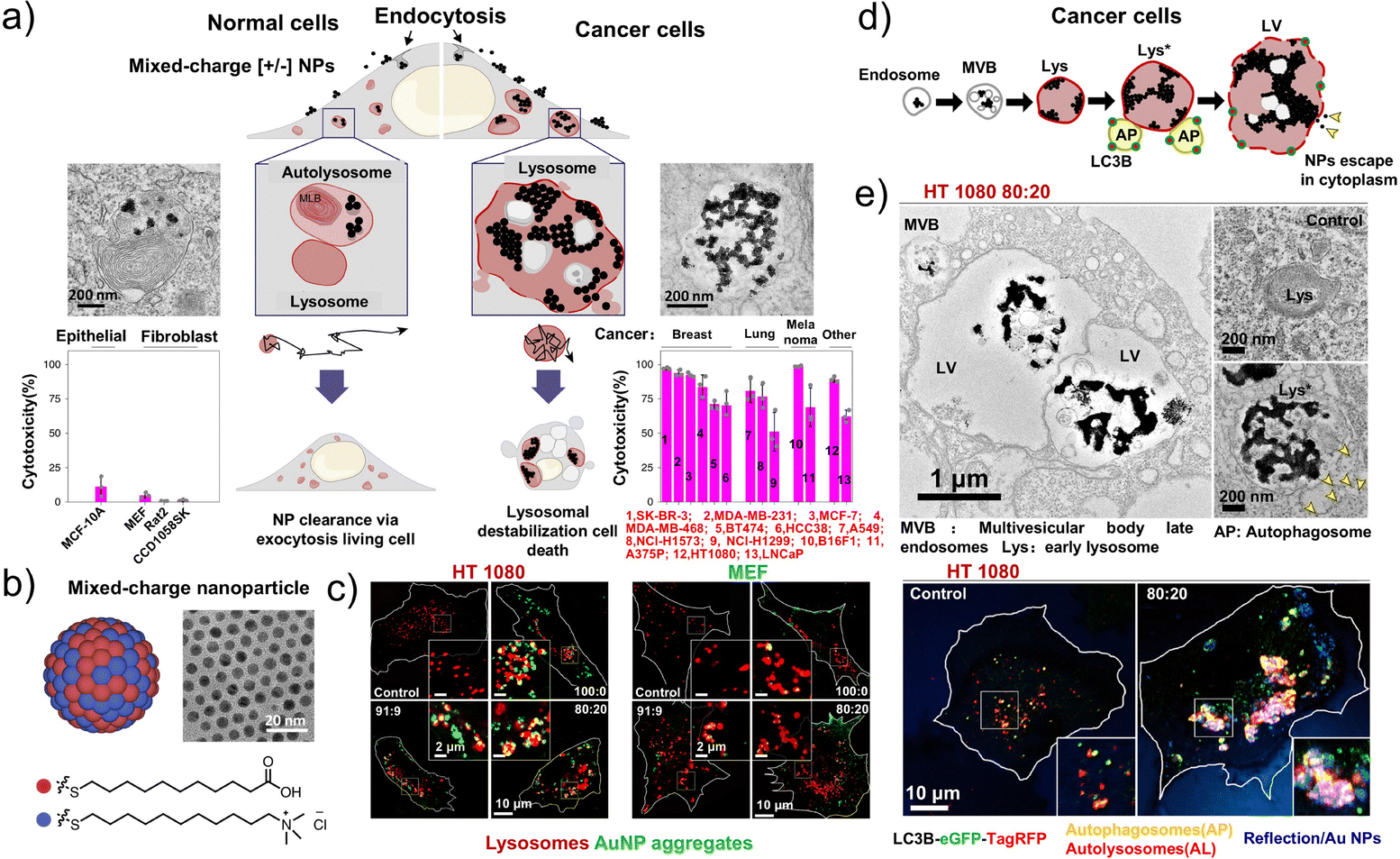 | ||
| Fig. 15 Schematic diagram of the mechanism underlying the lysosome targeting of spherical nanoparticles (NPs) based on the interaction of positive and negative charges. (a) The crystallization of mixed-charge NPs in tumor lysosomes leads to the selective killing of tumor cells. (b) Schematic and representative transmission electron microscopy (TEM) image of gold NPs. (c) HT1080 and MEF cells treated with the indicated NPs (50nM) for 6h. (d) Schematic of the proposed aggregation pathways for tumor cells. (e) Representative TEM images of HT1080 cells treated with 80:20 NPs for 24h; APs and ALs were marked with an eGFP-TagRFP-LC3B autophagy sensor, followed by cell incubation with 80:20 [±] NPs (50nM) for the indicated times. (a–e) Reproduced with permission from ref. 36. Copyright 2020, Springer Nature. | ||
Additionally, protein–polymer conjugates represent hybrid biomaterials in which the properties of proteins can be modulated or enhanced by synthetic polymers. With the widespread application of polyethylene glycol (PEGylation), proteins have been modified into various hydrophilic, hydrophobic, and reactive polymers.214–217 Poly(DPA), a pH-sensitive quaternary ammonium polymer, releases its payload upon quaternization and has therefore been employed to develop nanocarriers for controlled intracellular drug delivery; owing to its biocompatibility and stability, bovine serum albumin (BSA) is widely used in such carriers. For example, Velonia et al. loaded fluorescent dyes into BSA-poly(DPA) nanoparticles to track cellular uptake and achieve lysosomal bio-orthogonality, enabling precise drug delivery.218 With key advantages of enhanced stability and excellent controlled-release capabilities, the development of spherical nanoparticles is increasingly advancing from basic laboratory studies to preclinical evaluation of their therapeutic capacities.
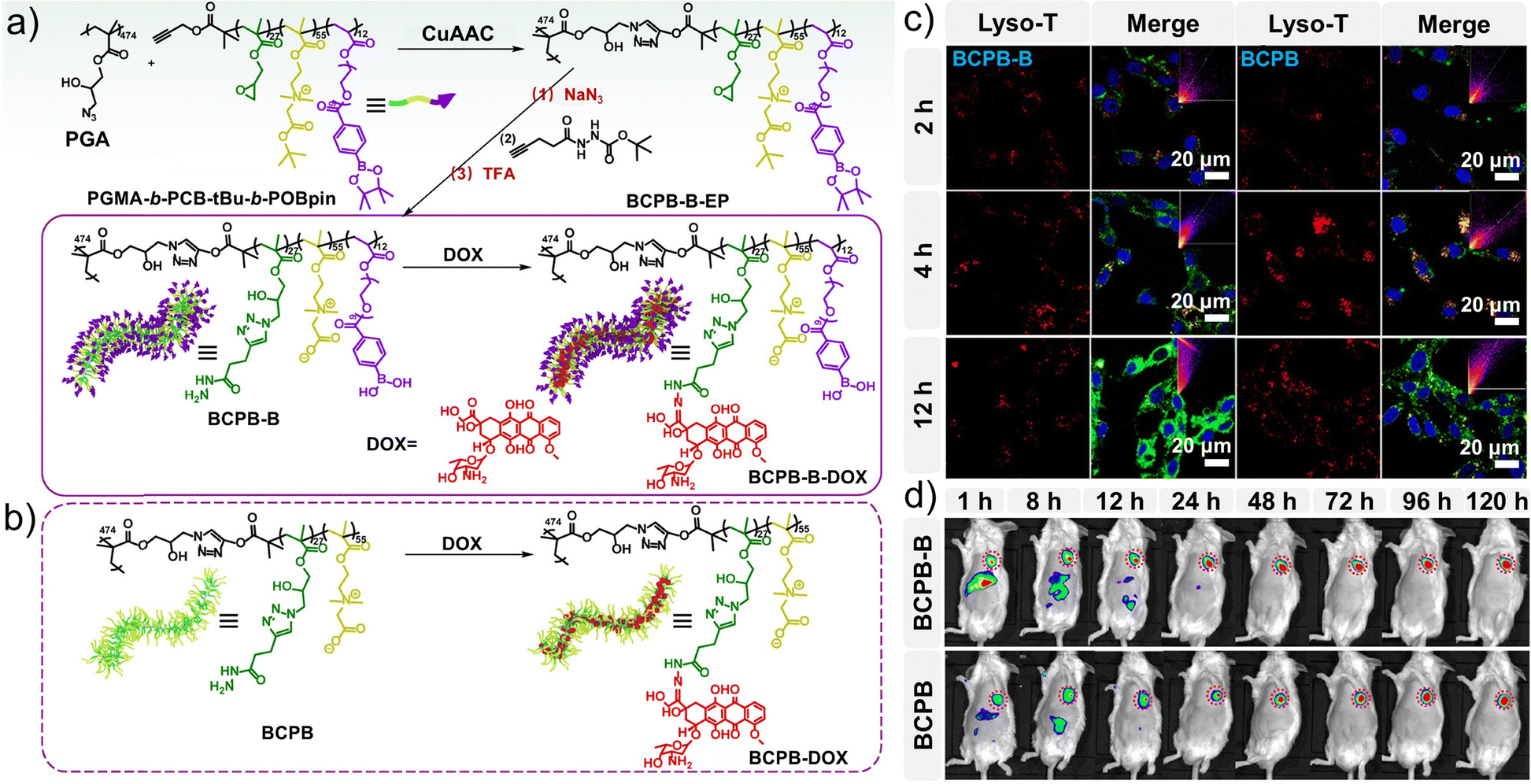 | ||
| Fig. 16 Preparation of BCPB derivatives and evaluation of their targeting properties. (a) Synthesis and drug loading of BCPB-B-DOX. (b) Synthesis and drug loading of BCPB-DOX. (c) Colocalization observation by confocal laser-scanning microscopy (CLSM) of FITC-labeled brushes (green) and Lyso-Tracker (red) in CT26 cells. (d) Near-infrared (NIR) fluorescence images of H22 tumor-bearing mice at different time points after tail vein injection of NIR-797-labeled BCPB-B-DOX and BCPB-DOX, respectively. (a–d) Reproduced with permission from ref. 223. Copyright 2021, American Chemical Society. | ||
Low-generation dendritic polymers have attracted considerable attention owing to their unique architectures and capacity for therapeutic delivery. However, the limited stability of high-generation dendritic macromolecules prepared ex vivo has hindered the development of dendritic nanoparticle platforms; thus, their in situ construction within tumor cells has emerged as a promising alternative strategy.224–227 Luo et al. developed a GSH-activated low-generation peptide dendritic polymer, SPr-G2, which exploits bio-orthogonal polymerization within tumor cells to generate high-generation dendritic polymeric nanoparticles (SPr-NPs) (Fig. 17a). This approach aims to perturb lysosomal function, enhance MHC-I expression, and induce ICD.228 Furthermore, SPr-G2 can be conjugated with the chemotherapeutic agent camptothecin to form SPr-CPT@PEG nanoparticles, promoting drug accumulation and release in tumor tissues in response to the acidic TME to ultimately improve chemotherapy efficacy (Fig. 17b). SPr-CPT further enhances antitumor immunity by promoting cytotoxic T cell activation and the maturation of DCs in mouse models (Fig. 17c–e). This study provided new insights into the design of peptide dendritic polymers for tumor therapy, highlighting their potential to augment chemotherapeutic efficacy while simultaneously activating antitumor immune responses.
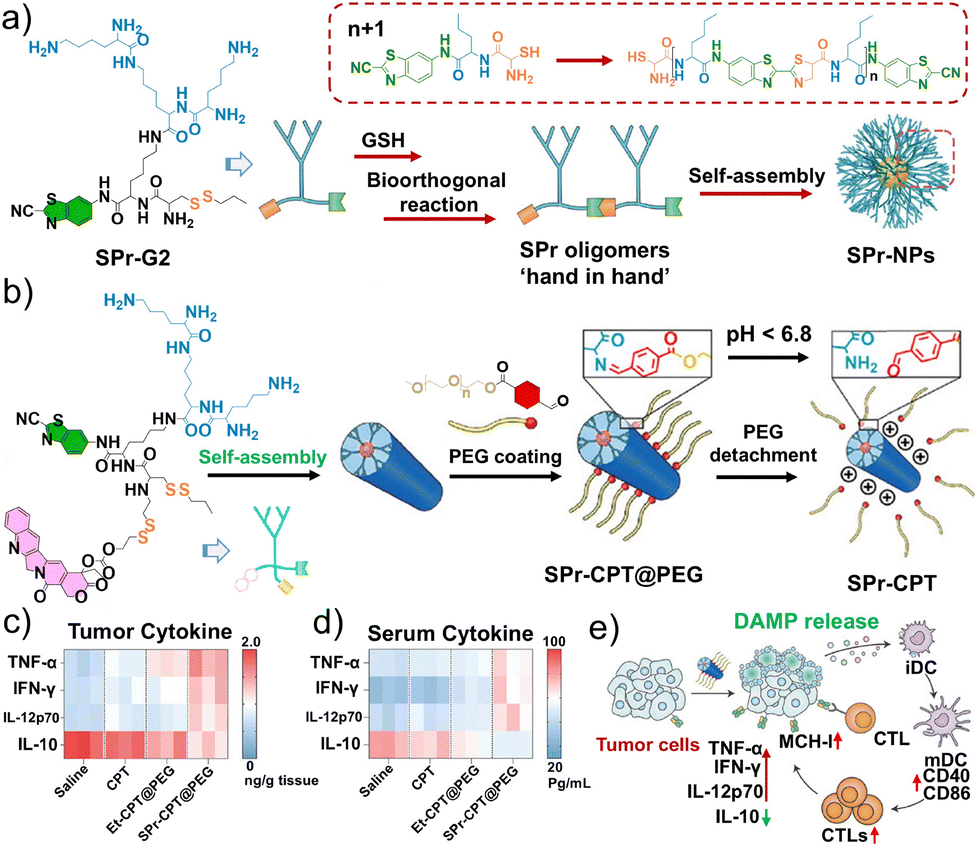 | ||
| Fig. 17 Schematic diagram of the mechanism of lysosome-targeting non-spherical nanoparticles. (a) Schematic illustration of the chemical structure of the SPr-G2 dendrimer and the formation of aggregates after GSH-triggered cleavage of disulfides and intermolecular condensation of SPr-G2. (b) Schematic of the preparation of PEG-coated SPr-CPT nanoparticles, SPr-CPT@PEG. Heatmap of the TNF-α, IFN-γ, IL-12p70, and IL-10 expression levels in (c) tumor tissue and (d) serum of 4T1 tumor-bearing model mice after various treatments. (e) Schematic illustration of the mechanism of SPr-CPT@PEG-induced MHC-I expression upregulation in tumor cells along with activation of CD8+ T cells. (a–e) Reproduced with permission from ref. 228. Copyright 2024, Wiley-VCH. | ||
The TME, composed of tumor cells, immune cells, stromal cells, and the extracellular matrix, plays a central role in tumor initiation, progression, and metastasis. The dynamic interactions among these components shape the biological behavior of tumors. Consequently, remodeling the TME and promoting immune cell infiltration and activity represent key strategies for enhancing the efficacy of immunotherapy.229–231 In addition to their use in lysosome-targeted tumor therapy, peptides are frequently conjugated with various organic small molecules or polymers to construct organic molecule–peptide conjugates, enabling the development of responsive amorphous nanoparticles. Based on developments in the fields of tumor immunotherapy, discovery of cell death mechanisms, nanotechnology, and TME remodeling, Ye et al. designed a new nanoparticle (NP-NH-D5) that is specifically responsive to the TME. Through precise lysosomal targeting, NP-NH-D5 transforms from a polymeric nanosphere into a polymeric nanofiber, achieving accurate tumor cell targeting and inducing necroptosis.232 This advance not only provides novel strategies for tumor immunotherapy but also highlights the enormous potential of nanotechnology in biomedicine.
Although amorphous lysosome-targeted antitumor nanoparticles show promise for precise drug delivery and induction of tumor cell death, their application still faces numerous challenges. These include insufficient structural stability leading to in vivo morphological changes and loss of function, limited targeting precision and selectivity resulting in off-target effects, uncertainties in drug loading and release, as well as potential immune responses and long-term biocompatibility issues. In addition, their preparation process poses difficulties in batch-to-batch consistency, creating obstacles for large-scale production and clinical translation. Future research in this field should focus on addressing these challenges to advance the development of amorphous nanomedicine materials and facilitate their clinical translation.
5.4. Liposome platforms
Necroptosis is a pro-inflammatory form of programmed cell death mediated by pore formation in the plasma membrane from the N-terminal fragment of gasdermin. This process leads to the release of substantial intracellular contents that in turn elicits robust immune responses, providing a strong basis for precise tumor therapy in combination treatment strategies.81–83 Shuai et al. constructed a novel nanoliposome, GM@LR, to co-deliver a plasmid expressing GSDME and carbonyl manganese (MnCO) to triple-negative breast cancer (TNBC) cells (Fig. 18a).233 The internalization of GM@LR encapsulating YOYO-1-labeled pGSDME (green) was tracked, revealing that the internalized GYOYO-1M@LR nanoparticles were initially transported to lysosomes (Fig. 18b). Further studies indicated that pGSDME was transported to the cell nucleus. In the presence of H2O2, MnCO generates Mn2+ and carbon monoxide (CO). CO activates caspase-3, which cleaves the expressed GSDME, transforming an apoptosis process into necroptosis. Additionally, Mn2+ promotes the maturation of DCs by activating the STING signaling pathway (Fig. 18c). In tumor-bearing mice, the proportion of mature DCs increased within the tumor, leading to significant infiltration of cytotoxic lymphocytes and a robust immune response that effectively inhibited tumor growth. Furthermore, GM@LR-induced necroptosis caused a strong local immune response at the tumor site, resulting in significant tumor regression. GM@LR could also stimulate antitumor immune memory, effectively preventing tumor metastasis in 4T1 lung metastasis models (Fig. 18d and e). Overall, this study offers a liposomal strategy targeting lysosomes that induces antitumor responses through necroptosis and cGAS-STING signal activation, which can be combined with immunotherapy for the efficient treatment of TNBC.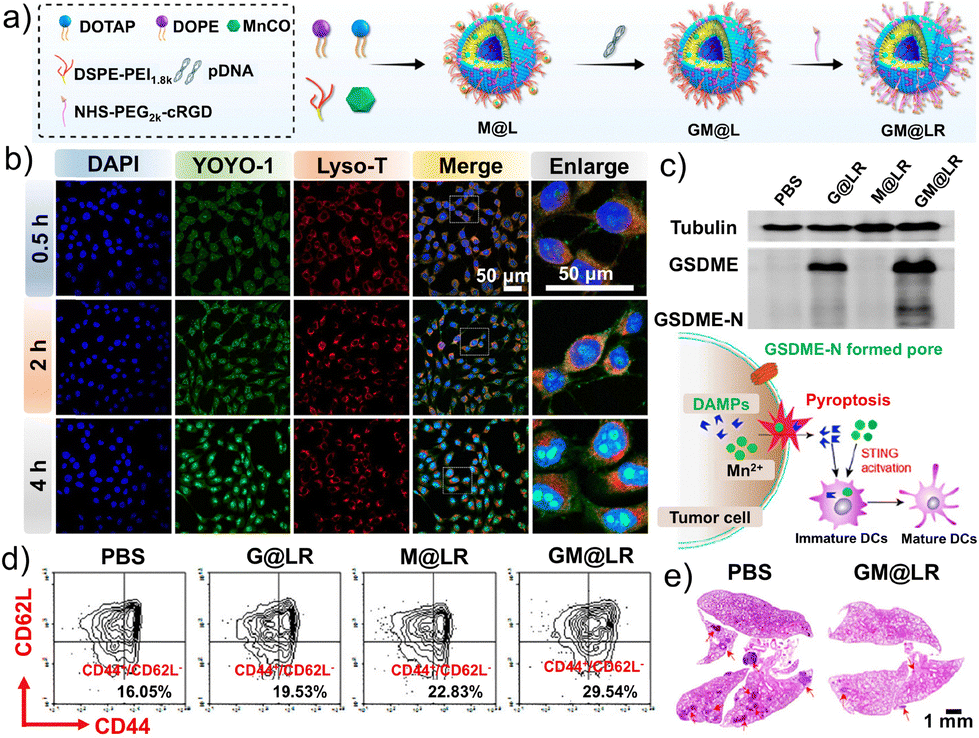 | ||
| Fig. 18 Schematic diagram of the preparation and mechanism of action of the lysosome-targeted liposome GM@LR. (a) Schematic illustration of the preparation of MnCO and pGSDME as a co-delivered nanodrug for tumor immunotherapy. (b) Confocal laser-scanning microscopy (CLSM) images of 4T1 cells incubated with pGSDME loaded-nanodrugs decorated with cRGD for various time periods. (c) Western blot assay indicating the expression of GSDME in 4T1 cells treated with PBS, G@LR, M@LR, or GM@LR, along with a schematic illustration of the nanodrugs inducing cell pyroptosis and stimulating the maturation of DCs. (d) Quantification of effector memory T cells (CD44+CD62L−, lower right) in 4T1 tumors detected by flow cytometry. (e) H&E staining of lung tissues 10 days after intravenous injection of 5 × 105 4T1 cells. (a–e) Reproduced with permission from ref. 223. Copyright 2023, American Chemical Society. | ||
Furthermore, PDT-induced ICD is a potential strategy to transform tumor cells into in situ vaccines, enabling the activation and maintenance of tumor immune cycles.234–236 Liu et al. developed an immune-enhancing polymeric liposome, IERL, with a stable nanostructure and bioactive surface (Fig. 19a).237 Confocal images (Fig. 19b) indicated that IERLs were phagocytosed by lysosomes and then escaped in vitro. Simultaneously, IERLs loaded with chlorin e6 and catalase (CAT) capture generated tumor-associated antigens (TAAs), which are transported to DCs via calreticulin-mediated phagocytosis. Upon uptake by DCs, IERL-Ps facilitate the lysosomal escape of TAAs, leading to significantly enhanced cross-presentation of TAAs and activation of cytotoxic T cells, thereby boosting the ICD-related antitumor immune response. By loading photosensitizers, IERLs can elicit robust antitumor immunity and long-term immune memory upon local irradiation, thereby inhibiting the growth of both primary and distant metastatic tumors. Furthermore, given the broad applicability of liposomal carriers, the photosensitizers in IERLs can be readily replaced with photothermal agents, radiosensitizers, or their combinations, providing a versatile liposomal nanoplatform for the rapid development of combination tumor immunotherapies. Given the inherent sensitivity of lung cancer cells to ferroptosis, therapeutic strategies that exploit ferroptosis mechanisms hold considerable promise for novel treatments.238–240 Huang et al. constructed an inhalable biomineralized liposome, LDM, co-loaded with dihydroartemisinin (DHA) and calcium phosphate (CaP), achieving effective accumulation in the lungs through nebulization. This approach further enhances ferroptosis therapy via an ER stress process centered on Ca2+ release.241 The mechanism is outlined in Fig. 19c, in which DHA is encapsulated within the bilayer of liposomes that are further coated with a pH-responsive biomineralized CaP shell. Structurally, DHA resides in the liposomal bilayer, while the CaP shell confers stability and enables controlled Ca2+ release. Lysosomal escape is ultimately achieved via the proton sponge effect of CaP, resulting in potent cytotoxicity and significant reduction in cell viability. Notably, A549 cells treated with LD appeared spherical due to DHA-induced cytotoxicity, whereas LDM treatment led to cell enlargement accompanied by abundant cellular debris, indicating disruption of membrane integrity and increased surface roughness (Fig. 19d and e). Upon delivery to lung cancer cells, the CaP shell decomposes in the acidic TME, triggering ER stress, lysosomal escape, and enhanced ferroptosis. The nebulized LDM format achieved approximately 6.8-fold higher tumor accumulation and stronger co-localization with A549 cells compared to intravenous administration, resulting in substantial tumor cell killing through synergistic ferroptosis and apoptosis mechanisms. In vivo, nebulized LDM suppressed tumor growth by ∼79.3% in lung cancer models and markedly increased pulmonary Ca2+ levels. Overall, LDM can overcome the limitations of poor drug accumulation and insufficient ROS generation, offering a potent ferroptosis-based strategy for lung cancer therapy.
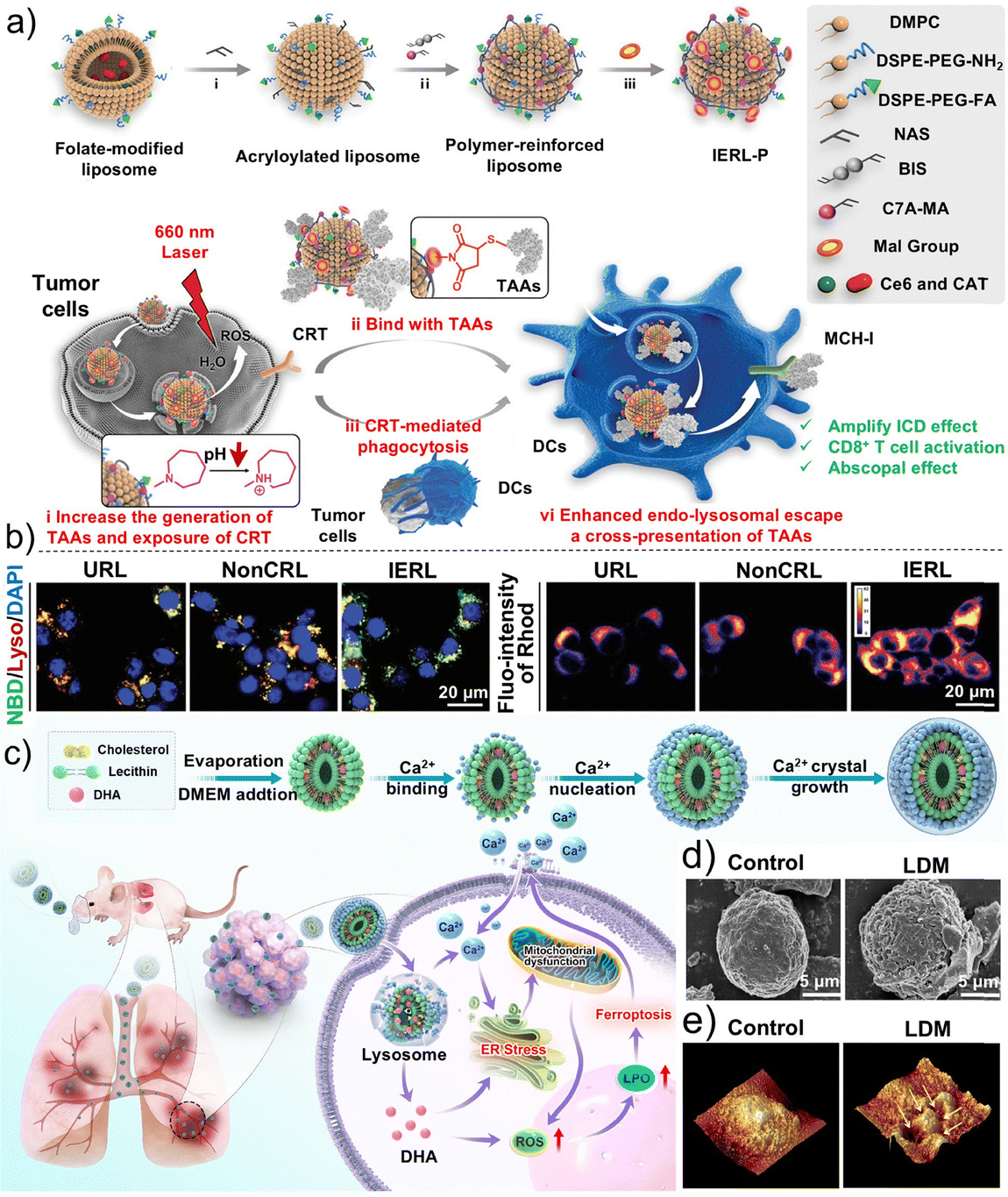 | ||
| Fig. 19 Schematic diagram of the preparation and action mechanism of the lysosome-targeted liposome IERL-P. (a) Design and preparation of IERL-Ps, and schematic illustration of IERL-Ps to amplify the immunogenic cell death (ICD)-associated antitumor efficiency and systemically induce T cell-dependent antitumor responses after photodynamic therapy (PDT). (b) Confocal laser-scanning microscopy (CLSM) images demonstrating the endo-lysosomal escape of URLs, NonCRLs, and IERLs in 4T1 tumor cells after 6 h and 12 h incubation. Reproduced with permission from ref. 237. Copyright 2022, Wiley-VCH. (c) Schematic illustration of a cyclic Ca2+-burst-centered ER stress process to enhance lung cancer therapy via ferroptosis. (d) Scanning electron microscopy (SEM) image of the morphology of A549 cells with different treatments. (e) Single-cell three-dimensional surface topography images of A549 cells with different treatments analyzed by atomic force microscopy. (c–e) Reproduced with permission from ref. 241. Copyright 2023, American Chemical Society. | ||
Additionally, Li et al. designed dual ligand-modified liposomes incorporating LyP-1 peptide and chondroitin sulfate, encapsulating paclitaxel and cryptotanshinone within the phospholipid bilayer to generate a dual-targeting, co-loaded, lysosome-specific liposome (CS/LyP-1-PC Lip).242 Following intravenous administration, these liposomes accumulated in tumor tissues via p32/CD44 dual receptor-mediated active targeting and efficiently penetrated 4T1 tumor cells. The released paclitaxel then exerts chemotherapeutic effects, induces apoptosis, and triggers ICD, consequently promoting the maturation of DCs. Simultaneously, cryptotanshinone downregulates STAT3 expression, reducing the secretion of immunosuppressive factors and cells, thus reversing tumor immunosuppression. These two mechanisms synergistically modulate the TME, activate effector T cells, and ultimately suppress the proliferation and metastasis of TNBC. Li et al. further developed lysosome-targeted liposomes for the co-delivery of docetaxel and sorafenib in the treatment of advanced hepatocellular carcinoma metastasis; this design significantly enhanced antitumor efficacy, providing novel insights for the clinical management of malignant liver tumors.243 Huang et al. designed an ultrasound-sensitive lysosome-targeted liposome system (MLipsiBcl-2) for the co-delivery of sonosensitizers and siRNA in hepatocellular carcinoma therapy.244 Upon ultrasound stimulation, MLipsiBcl-2 generates ROS, inducing liposome rupture, drug release, lysosomal membrane disruption, and downregulation of anti-apoptotic proteins. The development of this ultrasound-sensitive targeted gene delivery platform underscores its potential as a user-friendly multifunctional therapeutic system, offering prospects for combinatorial treatment strategies across diverse tumor types.
Owing to their excellent biocompatibility and controllable release properties, liposomes enable precise intracellular drug delivery to tumor cells. Lysosome-targeted liposome-based chemical platforms represent one of the most clinically translatable strategies for combination tumor immunotherapy and multimodal treatment.
5.5. Nanogels
Compared with self-assembled peptides, nanomicelles, nanoparticles, and liposomes, nanogels consist of a three-dimensional, highly hydrated, cross-linked network capable of accommodating diverse therapeutic agents. Nanogels further enable lysosome-responsive release via programmable mechanisms, including swelling, degradation, and diffusion.245–247 Their soft and deformable architecture facilitates cellular internalization and lysosome-specific delivery, endowing nanogels with pronounced structural and functional advantages for lysosome-targeted therapeutics.In contrast to the extensive research on solid tumor therapy, material-based drug delivery systems for hematological malignancies remain relatively underexplored. Hematological tumors, which originate from the blood and lymphatic systems, are characterized by diffuse distribution, diagnosis reliant on blood and bone marrow analyses, and treatment predominantly based on systemic chemotherapy and stem cell transplantation.248,249 Acute myeloid leukemia (AML) is a highly aggressive hematological malignancy characterized by the clonal expansion of poorly differentiated myeloid cells, Although differentiation-inducing therapies can convert leukemic cells into non-malignant phenotypes, their efficacy remains limited across multiple AML subtypes, and the disease remains characterized by high relapse rates and poor outcomes owing to chemoresistance. Accumulating evidence indicates that CD44 is overexpressed in various leukemias, functioning both as a key mediator of drug resistance and a poor prognosis factor, making it an attractive target for drug delivery. As a natural ligand of CD44, hyaluronic acid (HA)-based nanomedicines have been developed with significantly enhanced targeting efficiency. Typically, CD44-mediated endocytosis transports drugs into endolysosomes, opening the door for lysosomal regulation to play a pivotal role in AML therapy.250–252
Xu et al. synthesized an HA-based nanogel (HA/Cis/Dau) co-loaded with cisplatin and daunorubicin to achieve the differentiation-inducing therapy of refractory AML via the disruption of lysosomal homeostasis (Fig. 20a).253 Elemental mapping and dynamic light scattering (DLS) confirmed the nanogel structure and efficient co-loading of the two chemotherapeutics, which further demonstrated that HA/Cis/Dau was readily internalized by recurrent/refractory AML (rrAML) cells and delivered to lysosomes (Fig. 20a–d). Once localized in the lysosomes, HA/Cis/Dau induced LMP and intracellular ROS accumulation, which subsequently triggered the metabolic reprogramming-mediated differentiation of rrAML cells. Meanwhile, the released cisplatin and daunorubicin diffused into the nucleus to directly kill rrAML cells. Owing to the remarkable CD44-targeting ability and differentiation-induction effect, HA/Cis/Dau substantially reduced the systemic toxicity of chemotherapeutic agents (Fig. 20e and f). These findings underscore the promise of HA/Cis/Dau nanogels in enhancing targeted chemotherapy and facilitating the differentiation of rrAML cells.
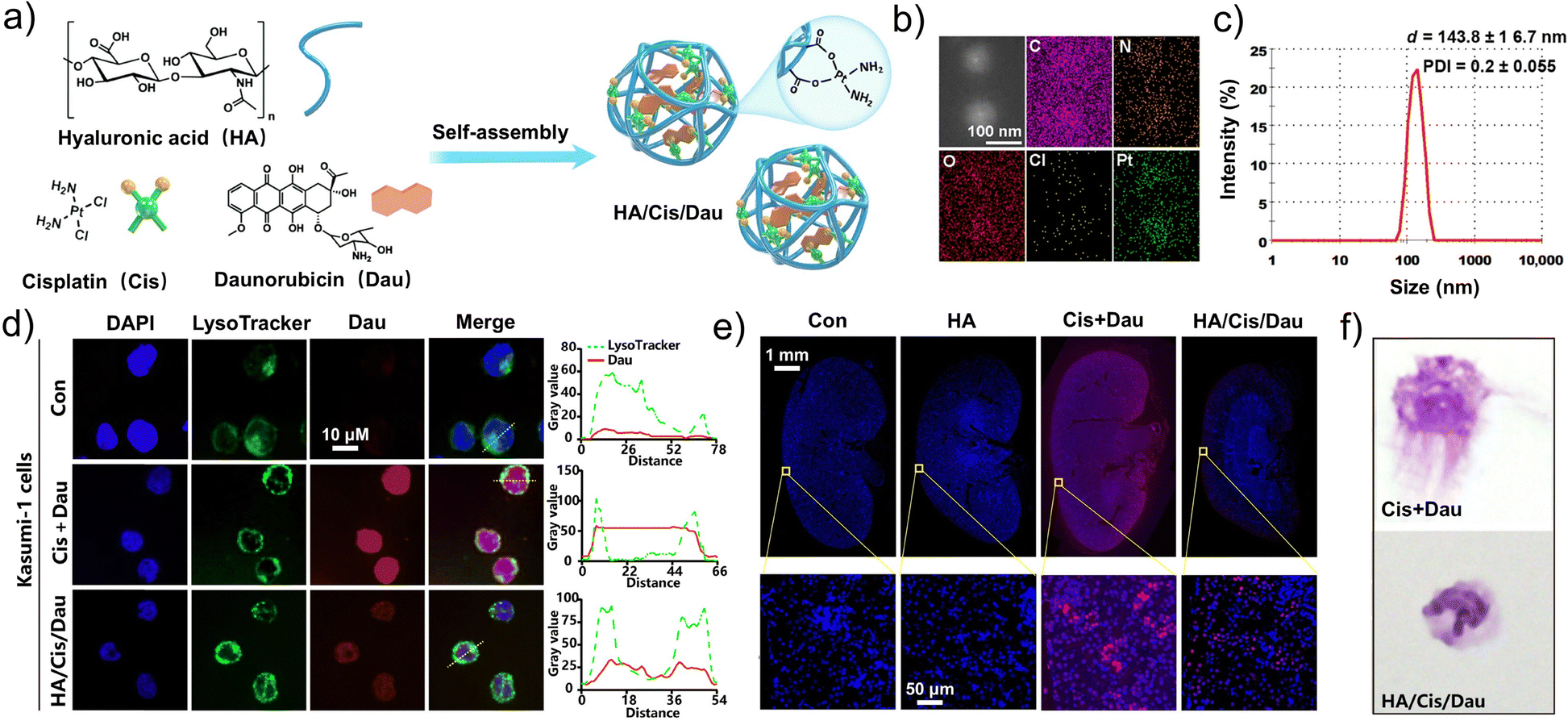 | ||
| Fig. 20 Preparation of an HA-based nanogel loaded with dual chemotherapeutic drugs and their applications in leukemia therapy. (a) Schematic illustration of HA/Cis/Dau self-assembly. (b) Elemental mapping of HA/Cis/Dau. (c) Hydrodynamic size and polydispersity index (PDI) of HA/Cis/Dau. (d) Kasumi-1 cells stained with 4′,6-diamidino-2-phenylindole (DAPI) and LysoTracker to visualize the nucleus and lysosomes, respectively; the colocalization of LysoTracker and daunorubicin fluorescence along the dashed lines was analyzed using ImageJ. (e) Cis–DNA staining of the kidney in healthy mice; the red fluorescence indicates the platinated DNA (Cis–DNA adducts). (f) Wright–Giemsa staining of primary AML progenitor cells from patients after HA/Cis/Dau treatment. (a–f) Reproduced with permission from ref. 253. Copyright 2025, American Association for the Advancement of Science. | ||
The highly hydrated, cross-linkable networks of nanogels enable the efficient encapsulation of small-molecule chemotherapeutics, proteins, and nucleic acids, allowing precise release in response to tumor-associated cues, including pH, enzymes, and redox conditions. This programmable release enhances the local drug concentration, mitigates systemic toxicity, and helps to overcome drug resistance. Compared with conventional nanocarriers, nanogels provide greater flexibility in size modulation, surface functionalization, and multi-drug co-delivery, rendering them suitable for deep tumor penetration and controlled release in solid tumors, as well as bone marrow and circulatory targeting in hematological cancers. Collectively, nanogels are driving the field of next-generation antitumor drug delivery toward high selectivity, safety, and multifunctionality, holding considerable potential for clinical translation and industrial applications.
5.6. Inorganic nanodrug platforms
Inorganic nanodrugs have advantages of stability and low immunogenicity. These properties allow them to be readily taken up by tumor cells and accumulate via the endosome–lysosome pathway, enabling efficient lysosome-targeted delivery. Accordingly, inorganic nanodrugs play an indispensable role in lysosome-targeted platforms for precise tumor therapy.254–256 As highlighted above, targeted therapy for ferroptosis has also garnered significant attention given its potential to eradicate iron-dependent tumor cells.257–259 Increasing the levels of unstable intracellular iron pools through small molecules and iron-containing nanomaterials is an effective method to induce ferroptosis. However, challenges such as rapid drug metabolism and toxicity to normal tissues often lead to insufficient therapeutic efficacy of such strategies. Therefore, it is crucial to develop a long-lasting and selective approach to modulate ferroptosis in tumor treatment. Cheng et al. proposed a lysosome-targeted magnetic nanoparticle torque generator, T7-MNT, as a dynamic tool for inducing endogenous ferroptosis in breast tumors.260 As illustrated in Fig. 21a, T7-MNTs are primarily localized in the lysosomes of MCF-7 and MDA-MB-231 cells, indicating that lysosome targeting occurs via transferrin receptor-mediated endocytosis. Under a programmed rotational magnetic field, T7-MNTs generate torque through the disruption of lysosomal membranes, triggering the release of endogenous Fe2+ and effectively initiating ferroptosis (Fig. 21b). This dynamic targeting strategy can be combined with existing ferroptosis inducers to enhance therapeutic efficacy. Most notably, the ability to specifically induce ferroptosis in tumor cells while minimizing effects on normal cells could represent a significant advancement in tumor therapy.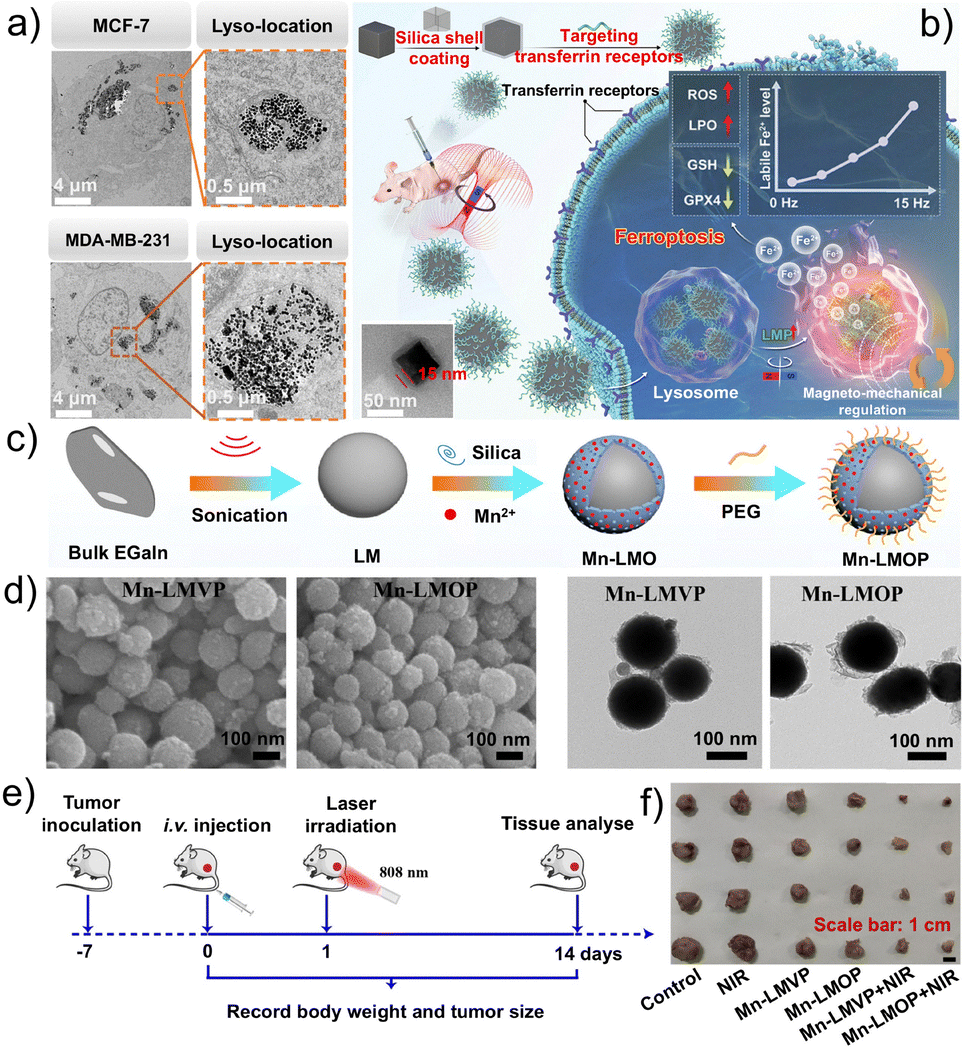 | ||
| Fig. 21 Preparation, mechanism of action, and in vivo antitumor activity of lysosome-targeted inorganic nanoplatforms. (a) Transmission electron microscopy (TEM) images of T7-MNTs located in lysosomes of MCF-7 and MDA-MB-231 cells. (b) Lysosome-targeted magnetic nanotorquers dynamically trigger the endogenous Fe2+ pool outbreak to evoke ferroptosis in a programmable manner by locally disrupting the lysosomal membrane under a magnetic field. (a and b) Reproduced with permission.260 Copyright 2023, Wiley-VCH. (c) Schematic illustration of the synthesis of Mn-LMOP for photothermal therapy (PTT) and chemodynamic therapy (CDT) combination tumor treatment. (d) Scanning and transmission electron microscopy images of Mn-LMVP/Mn-LMOP. (e) Scheme illustrating the experimental schedule. (f) Photographs of dissected tumors. (c–f) Reproduced with permission from ref. 261. Copyright 2025, Elsevier Ltd. | ||
Lv et al. developed an inorganic manganese-doped liquid metal (LM) nanomaterial to achieve a synergistic combination of PTT and CDT.261 The spherical nanomaterials Mn-LMO and Mn-LMOP were coated with silica on the surface of the photothermal agent LM surrounded by PEG, exhibiting uniform particle sizes (Fig. 21c and d). The tumor inhibition rates in mice treated with Mn-LMO and Mn-LMOP were 85.49% and 92.37%, respectively, resulting from the effective accumulation of Mn-LMO and Mn-LMOP at the tumor site through enhanced cellular uptake and lysosomal escape (Fig. 21e and f). Upon uptake by HepG2 tumor cells, these inorganic nanoparticles deplete GSH, which induces the manganese-based Fenton-like reaction to enhance CDT. Additionally, owing to the high photothermal conversion efficiency of LM, the temperature at the tumor site increases significantly, further enhancing CDT. Moreover, this combined CDT/PTT strategy can activate the gas-sting pathway, in turn enhancing the antitumor immune response.
Analogously, Gao et al. developed a novel inorganic nanocage (PFH@Ag@Ch-I), representing a new type of tumor treatment designated as a “nano-bomb,” composed of silver nanocages loaded with perfluorohexane (PFH), chitosan, and indocyanine green.35 Once PFH@Ag@Ch-I enters tumor cell lysosomes, the silver nanocages trigger a photothermal effect under NIR light, raising the temperature to 50 °C. This rapid heating causes PFH to undergo a phase transition, releasing a large amount of oxygen, thereby increasing the internal pressure within the lysosomes. The consequent lysosomal dysfunction results in enhanced exosome secretion. The exosome-encapsulated “nano-bomb” can be further delivered deep into the tumors. Meanwhile, the silver nanoparticles are fragmented into many smaller silver nanoparticles that catalyze Fenton-like reactions to generate OH˙ for CDT.
In summary, novel inorganic nanomaterials hold great promise for precise lysosomal targeting in tumor therapy. With excellent biocompatibility and stability, they can be functionalized for specific recognition and targeted delivery to tumor cells. Leveraging the acidic or enzymatic environment within lysosomes, these nanomaterials enable controlled drug release, enhancing therapeutic efficacy while sparing normal cells. Combined with imaging technologies, these nanomaterials also enable the real-time monitoring of drug distribution and release, supporting personalized treatment strategies. Advances in nanotechnology continue to expand the potential of inorganic nanomaterials for effective, tumor-targeted therapy. However, the potential biotoxicity, in vivo metabolism and clearance issues, poor degradability, complex manufacturing processes, and high costs of inorganic nanodrug platforms need to be addressed to further advance their applications in precise tumor therapy.
5.7. Coacervates
As a type of disordered self-assembled system, a coacervate is driven by non-specific interactions. Coacervates are characterized by loosely arranged molecules and highly dynamic structures capable of rapidly adapting to complex biological environments. Their structural flexibility and plasticity enable efficient binding with a variety of drugs or biomacromolecules, forming densely packed drug reservoirs while maintaining excellent adaptability and permeability at cell membrane and subcellular organelle interfaces.262–265 Compared with the highly ordered self-assembled systems described in the previous sections, including self-assembled peptides, nanomicelles, nanoparticles, and liposomes, disordered coacervates offer distinct advantages as therapeutic platforms, including rapid formation, combinatorial flexibility, and multi-drug co-loading, enabling fast-response drug release or environment-sensitive delivery. Furthermore, their dynamic and reconfigurable nature provides opportunities to overcome complex in vivo barriers, enhance the local drug concentration, and adapt to heterogeneous TMEs. Collectively, these properties render disordered coacervates multifunctional and versatile platforms for drug delivery.Yu et al. developed an enzyme-responsive, phase-separating peptide, YSO4F, capable of generating in situ liquid–liquid phase-separated coacervates with selective targeting of membraneless organelles, thereby achieving effective tumor therapy. The peptide features an “adhesive-spacer-adhesive” motif and is further functionalized with a virus-derived FGDF domain to specifically target the stress granule core protein G3BP2.77 Upon catalysis by lysosomal sulfatase, YSO4F is converted into the phase-separating peptide YF, forming coacervates that bind G3BP2 and subsequently fuse with stress granules. This process blocks the sequestration of RACK1 and activates pro-apoptotic signaling pathways, markedly enhancing the antitumor efficacy of sorafenib (Fig. 22a). As shown in Fig. 22b, YSO4F gradually loses its sulfate groups under sulfatase catalysis, converting into the hydrophobic peptide YF, which undergoes liquid–liquid phase separation to form spherical coacervate droplets. Fluorescence recovery after photobleaching (FRAP) and droplet fusion assays confirmed that these droplets exhibit the typical dynamic properties of coacervates, with a half-recovery time of 23.4 s, indicating rapid molecular exchange between small and larger coacervate droplets (Fig. 22c). For in vivo confirmation of the antitumor effects, Yu et al. administered m-YSO4F-LSG combined with intraperitoneal sorafenib intratumorally to mice. This combination therapy achieved marked tumor volume inhibition, surpassing the effects of monotherapy and demonstrating a robust synergistic antitumor effect (Fig. 22d and e). Immunofluorescence analysis further revealed that the functional coacervates formed in tumor tissues exhibited a high colocalization coefficient of 0.77 with the stress granule protein G3BP2 directly confirming their efficient targeting of stress granules. Western blot analysis indicated activation of the caspase-3-dependent apoptotic pathway in the combination therapy group. Furthermore, H&E staining of major organs demonstrated that the m-YSO4F-LSG system possesses excellent biocompatibility (Fig. 22f–h).
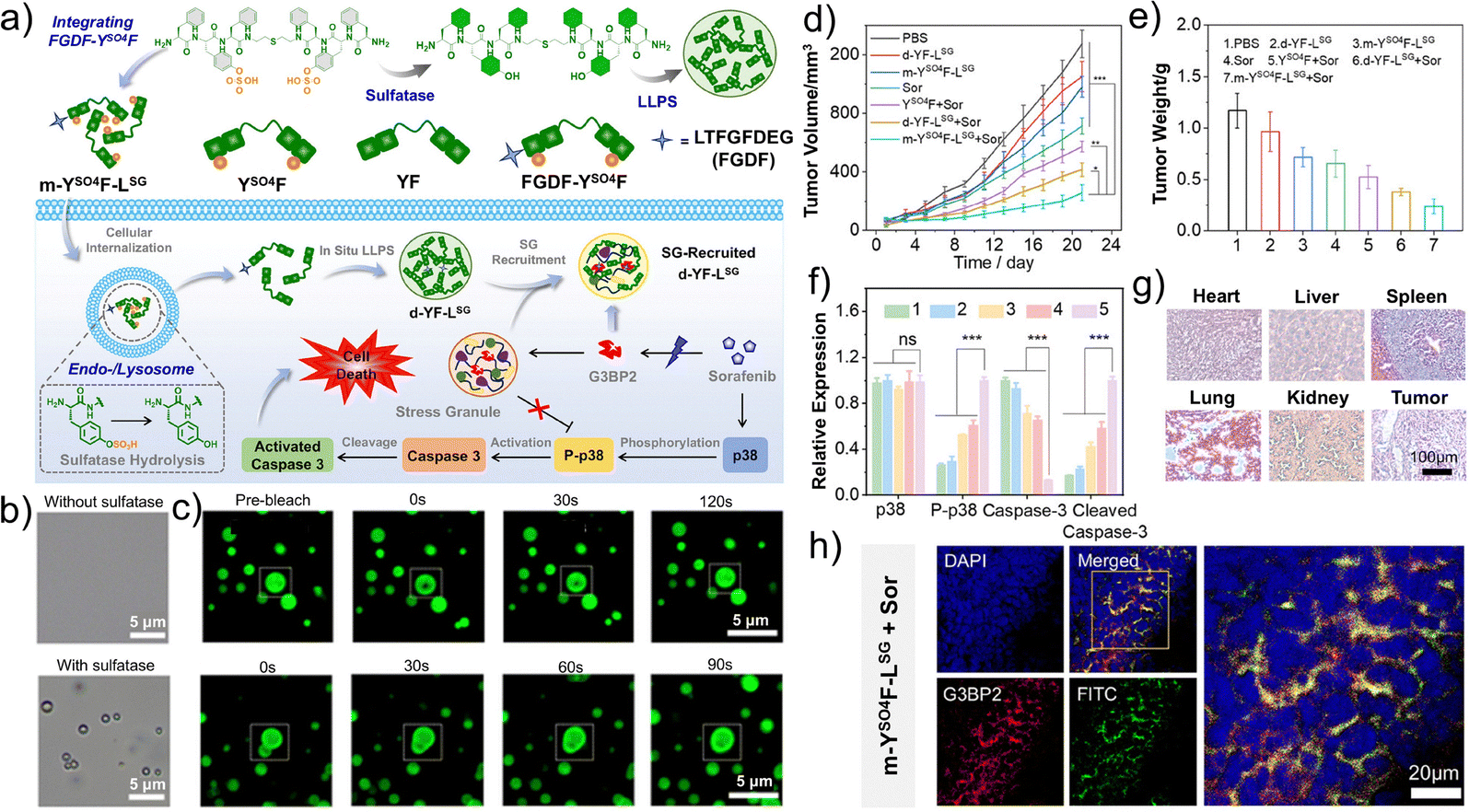 | ||
| Fig. 22 Lysosome-mediated formation of coacervates and their mechanism of action. (a) Schematic illustration of in situ phase separation of peptides into coacervate droplets in living cells targeting membraneless organelle stress granules (SGs) for cancer chemotherapy combined with sorafenib. (b) Optical microscopy images of peptide YSO4F in the solution prior to and after incubation with sulfatase for 24 h. (c) Confocal laser-scanning microscopy (CLSM) images of the droplets formed by sulfatase-treated YSO4F to illustrate the FRAP process, and CLSM images of the fusion of the droplets formed by sulfatase-treated YSO4F. (d) Tumor growth curves and (e) weight of tumor tissues dissected from mice treated with PBS, d-YF-LSG, m-YSO4F-LSG, Sor, YSO4F + Sor, d-YF-LSG + Sor, or m-YSO4F-LSG + Sor. (f) Protein levels of p38, P-p38, caspase-3, and cleaved caspase-3 in the tumor tissues from mice administered with PBS (1), m-YSO4F-LSG (2), Sor (3), d-YF-LSG + Sor (4), or m-YSO4F-LSG + Sor (5). (g) H&E-stained images of the tumor tissues and major organs from the mice after m-YSO4-LSG + Sor administration. (h) CLSM images of frozen sections of tumor tissues from the mice treated with m-YSO4F-LSG + Sor. (a–h) Reproduced with permission from ref. 77. Copyright 2025, Wiley-VCH. | ||
Peng et al. developed a QCS/GA/I/DOX-based coacervate chemoembolization agent that exhibits excellent fluidity, water immiscibility, and low viscosity, positioning it as a promising microvascular embolic platform for tumor chemotherapy.20 Owing to abundant hydrogen-bonding sites, the coacervate can incorporate contrast agents to enhance X-ray visibility, while its electrostatic interactions with Dox enable high-capacity loading and sustained intratumoral release. Building on the versatility of intracellular phase separation, Gu et al. further engineered a biomolecular coacervate system integrated with bio-orthogonal chemistry, establishing a new intracellular coacervate-based modality for drug delivery.266 This system readily forms intracellular drug reservoirs, markedly increasing the local drug concentration and prolonging intracellular drug retention, ultimately reversing chemoresistance in tumor cells. Building on condensate biology, Zhang et al. targeted tumor-associated FOXM1 protein condensates and designed a phospho-mimetic peptide, FIP4, to disrupt FOXM1-driven phase separation. The FIP4-enriched nanoparticles substantially inhibited tumor metastasis, enhanced tumor immunogenicity, and improved the therapeutic efficacy of antitumor immunotherapy, highlighting their considerable potential for combinatorial cancer treatment.267
Whether operating through lysosomal escape-mediated drug release (i.e., “departure-type” coacervates), leveraging in vitro-constructed coacervates for sustained release and organelle targeting (i.e., “arrival-type” coacervates), or engaging natural and artificial biomolecular coacervates within tumor cells, these strategies collectively underscore the unique advantages of disordered coacervates in drug delivery. Further systematic investigation into aggregate formation, dynamic reorganization, and drug loading-release behavior will provide essential mechanistic insights and design principles to advance the rational development of multifunctional material-based chemical platforms. The high structural flexibility, rapid responsiveness, multivalent drug-loading capacity, and strong adaptability to heterogeneous TMEs render coacervates particularly advantageous for enhancing drug targeting, improving therapeutic outcomes, and minimizing systemic toxicity. With continued elucidation of their formation mechanisms and dynamic behaviors, these disordered self-assembled systems are poised to evolve into versatile platforms for precision tumor therapy, combination drug delivery, and biological function modulation, offering an antitumor chemical platform with greater selectivity, safety, and functional integration.
6. Biomimetic platforms
6.1. Lysosome-targeted chimera
In recent years, targeted protein degradation (TPD) has emerged as an innovative therapeutic approach that exploits the cellular degradation system to selectively eliminate challenging drug targets. However, the effects of traditional proteolysis-targeting chimeras (PROTACs) are limited to TPD on intracellular proteins with cytoplasmic domains, leaving this strategy useless against non-cytoplasmic proteins (representing over 40% of human proteins) that play key roles in disease development.70,268–270 Lysosome-targeted chimeras (LYTACs) have been developed to selectively bind lysosomal receptors on the surface of tumor cells, thereby facilitating the trafficking and degradation of extracellular and membrane-associated proteins. However, LYTACs also face challenges such as poor tumor accumulation, non-specific activation, suboptimal pharmacokinetics, and limitations in combination with other therapies, hindering their clinical translation.271–273 Nanotechnology offers a promising solution to overcome these issues, as nanomaterials can improve the pharmacokinetics and biodistribution of drugs while further promoting ligand loading. Therefore, nanoparticle surface engineering can activate LYTAC-mediated TPD and act synergistically with other therapies to enhance antitumor efficacy.Dai et al. developed an excellent LYTAC system based on nanospheres that exploits intrinsic lysosomal degradation pathways, providing a biomimetic platform for targeted liver tumor therapy. This approach enables the simultaneous degradation of membrane or extracellular proteins while delivering therapeutic drugs, resulting in enhanced antitumor effects.81 Amphiphilic peptides modified with N-acetylgalactosamine (GalNAc) bind to HepG2 cells via ASGPR and self-assemble into approximately 200 nm nanospheres, enabling protein transport and degradation. Upon loading glucose oxidase (GOx), GOx-LYTACs are formed, which specifically target CD24, achieving over 80% tumor suppression in vitro and promoting CD24 degradation in vivo. These effects induce tumor-associated macrophages to polarize toward the M1 phenotype to realize targeted degradation, drug delivery, and tumor therapy simultaneously.
However, compared with the significant progress achieved by molecular glue- and PROTAC-mediated intracellular protein degradation, the limited availability of lysosomal surface binding sites hinders the identification of optimal targets for LYTAC development. As a result, lysosome-targeted platforms for membrane and extracellular proteins remain at the preclinical stage, with no LYTAC-based therapeutics approved for routine clinical application to date. Nevertheless, this has become an active research field. Geng et al. employed Glut1 as a lysosomal-targeting receptor, designing Glut1-facilitated lysosomal degradation for PD-L1-overexpressing TNBC.274 Ge et al. reported a recombinant IGF2 fusion protein strategy for extracellular and membrane protein degradation, offering a modular platform to enhance antibody drugs and reduce resistance.275 Tan et al. developed a multivalent aptamer-based LYTAC (AptLYTACs) platform, in which trivalent single-target AptLYTACs efficiently degrade membrane proteins, providing a proof-of-concept for single or dual target degradation.276 In addition to the studies mentioned above, as shown in Table 3, continuous efforts and advances in the development of LYTACs have been made, based on novel lysosomal membrane proteins, novel proteins of interest, new technologies, and new functionalities, greatly advancing the development of innovative, efficient, and low-toxicity TPD technologies.273,277–286
| Type | Name | Lysosomal membrane protein | Protein of interest | Ref. |
|---|---|---|---|---|
| Supra-LYTACs | Py-FFK-Az | Cl-M6PR | CAIX | 277 |
| Py-FFK-BMS | PD-L1 | |||
| HerTACs | LP | Her2 | PD-L1 | 278 |
| QP | ||||
| RP | ||||
| IFLDs | BMS-L1-RGD | αvβ3 | PD-L1 | 279 |
| BMS-L2-RGD | ||||
| BMS-L3-RGD | ||||
| PT-LYTAC | PBAC | Cl-M6PR | PTK7 | 280 |
| DR-TACs | TF-L5-B | CXCR4, FOLR1 | PD-L1 | 281 |
| CF-L5-G | EGFR | |||
| CF-L5-B | PD-L1 | |||
| CF-L5-B | CPCR4, FOLR1 | PD-L1 | ||
| Pep-TAC | Pep-1 | TFRC | PD-L1 | 273 |
| HGTACs | H1 + G1 | Cl-M6PR | NS-650 | 282 |
| H1 + G2 | EGFR | |||
| H1 + G3 | Her2 | |||
| KB-TACs | KB-CTX | Ketoboronate | EGFR | 283 |
| KB-TTZ | Her2 | |||
| KB-Bev | VEGFA | |||
| TDA-MLYTACs | TDN-1-3 | Cl-M6PR | PTK7 | 284 |
| TDN-2-2 | ||||
| TDN-3-1 | ||||
| IMTACs | EGFR IMTAC | Lysosomal targeted receptors | EGFR | 285 |
| PDL1 IMTAC | PD-L1 | |||
| EGFR/PDL1 IMTAC | EGFR, PDL1 | |||
| Small molecule LYMTACs | LYMTAC-1 | RNF152 | EPHA2, etc. | 286 |
| LYMTAC-2 | KRASG12D | |||
| LYMTAC-4 | KRASG12D, KRASG13D |
Liu et al. developed a novel bifunctional LYTAC nanoplatform, NLTC, which achieves the targeted degradation of extracellular or membrane-associated proteins by the in situ polymerization of various functional ligands on the surface of protein nanocapsules.287 By precisely modulating Ac-M6P, Ac-DNP, and Ac-BMS on the protein surface, effective targeting of lysosomes through Ac-M6P is achieved, further activating the TPD process and resulting in the efficient degradation of α-DNP protein and PD-L1 protein in lysosomes (Fig. 23a). Moreover, this synthesis method is applicable to various proteins and enzymes, providing broad possibilities for subsequent functional expansion in contrast to the relatively singular design model of traditional LYTACs. NLTC-BMS was used to target PD-L1 for antitumor studies, which effectively inhibited the growth of B16F10 tumors and activated T cell-mediated antitumor immunity upon tumor injection (Fig. 23b). Specifically, a significant increase in the levels of CD8+ T cells was observed in tumors treated with NLTC-BMS, reaching 31.4%, compared to only 11.7% in the PBS group, indicating enhanced infiltration of CD8+ T cells within the tumors. Additionally, NLTC-BMS significantly improved the activity and proliferation of CD8+ T cells. These findings highlight the potential of NLTC-BMS in restoring the activity and proliferation of CD8+ T cells. Overall, these results indicate that NLTC-BMS can effectively degrade PD-L1 in vivo, thus activating T cell-mediated antitumor immune responses and suppressing tumor growth. Despite the effectiveness of NLTC-BMS in degrading PD-L1, its antitumor efficacy remains limited due to the influence of an immunosuppressive TME. The researchers further encapsulated CAT within NLTC-BMS to form NLTC-BMS/CAT, followed by modification with a pH-responsive removable PEG shell to obtain NLTC-BMS/CAT-PDA. This design not only enables the nanoplatform to effectively degrade PD-L1 on the tumor cell surface but also utilizes CAT to catalyze the decomposition of H2O2 in the tumor tissue to generate O2, thereby alleviating the immunosuppressive inflammation and hypoxic environment within the tumor. This effect synergizes with blockade of the PD-1/PD-L1 pathway, enhancing the overall antitumor immune memory response (Fig. 23c). This combined strategy resulted in significant inhibition of tumor growth, recurrence, and metastasis in tumor-bearing mice, which is an effect that traditional single-treatment methods struggle to achieve.
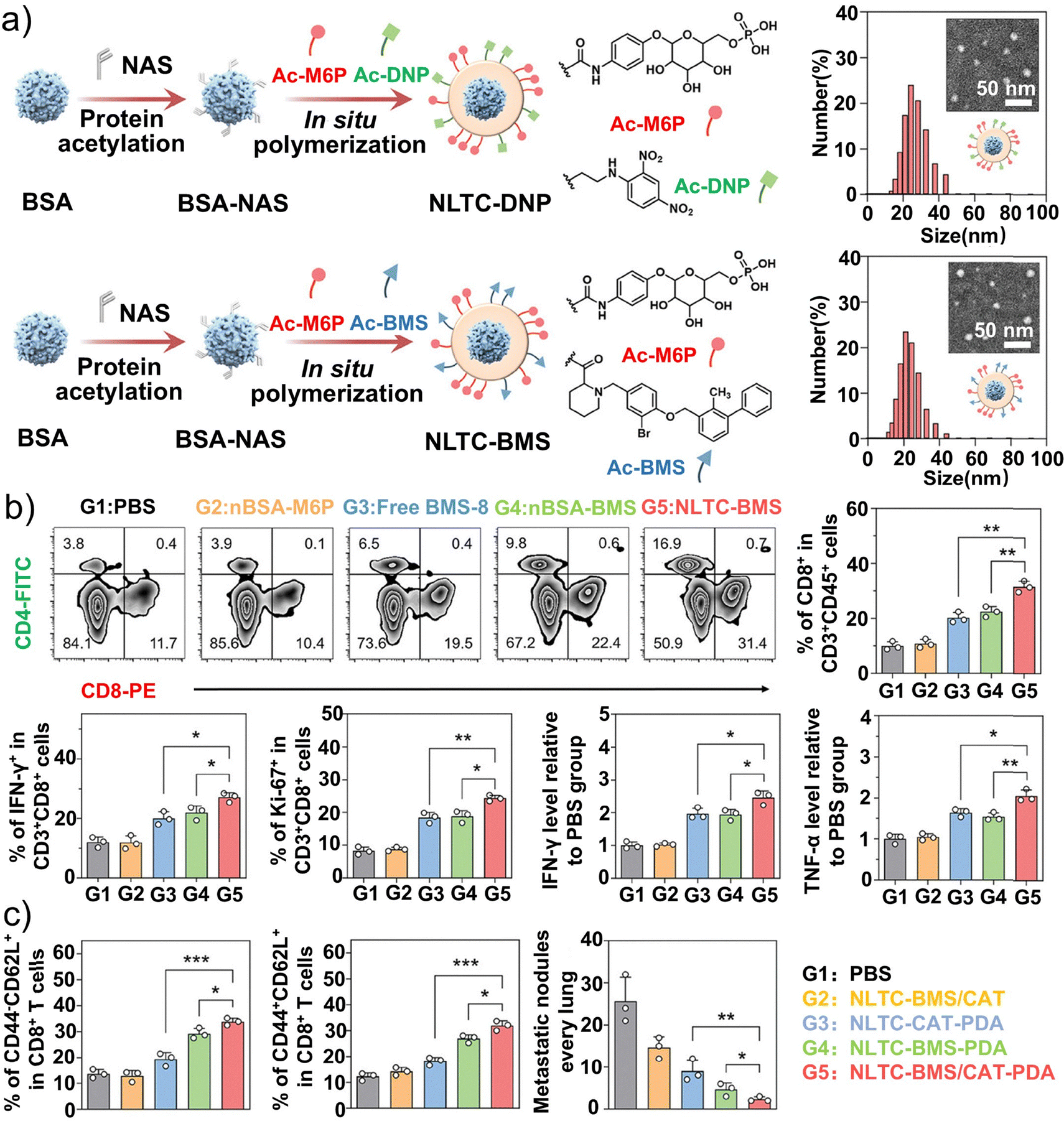 | ||
| Fig. 23 Structural schematic diagram and antitumor mechanism of lysosome-targeted chimeras. (a) Schematic illustration of the fabrication of NLTC-DNP and NLTC-BNS; particle size measurement and transmission electron microscopy (TEM) images of the chimeras; and their effects on extracellular α-DNP/membrane protein (PD-L1) degradation. (b) Representative flow cytometric plots and quantitative analysis of tumor-infiltrating CD8+ T cells, tumor-infiltrating IFN-γ+CD8+ T cells, and Ki-67+CD8+ T cells, along with relative expression levels of IFN-γ and TNF-α in tumor tissues after different treatments. (c) Quantitative analysis of effector memory and central memory T cells in the spleens of mice after different treatments, and quantitative analysis of lung metastatic nodules in tumor-rechallenged mice. (a–c) Reproduced with permission from ref. 287. Copyright 2025, Wiley-VCH. | ||
Inspired by the LYTAC design, Ding et al. developed novel semiconductor polymer nanoparticles (SP3 NPs) with a bioinspired protein secondary structure on the surface, enabling self-cooperative tumor immunotherapy by combining ICD with ICB therapy.288 Through a rational fluorination reaction, SP3 NPs demonstrated excellent photodynamic properties that can significantly induce ICD. Moreover, to optimize the preparation process, the researchers introduced peptide antagonists that self-assemble on the surface of the photodynamic nanoparticles to form SP3 NPs-DFDFGDPPA with β-sheet protein secondary structures, facilitating the effective LYTAC-mediated degradation of PD-L1 in lysosomes (Fig. 24a). In vivo experiments indicated that SP3 NPs-DFDFGDPPA not only induced a strong antitumor immune response, inhibiting both primary and distant tumors, but also led to long-term immune memory against tumor recurrence. The immune memory effect further prolonged the survival rate of mice (Fig. 24b–f). Combining light-sensitive agent-based ICD with protein secondary structure-mediated multivalent PD-L1 blockade can create a novel, potent immunotherapeutic agent that is analogous to LYTAC.
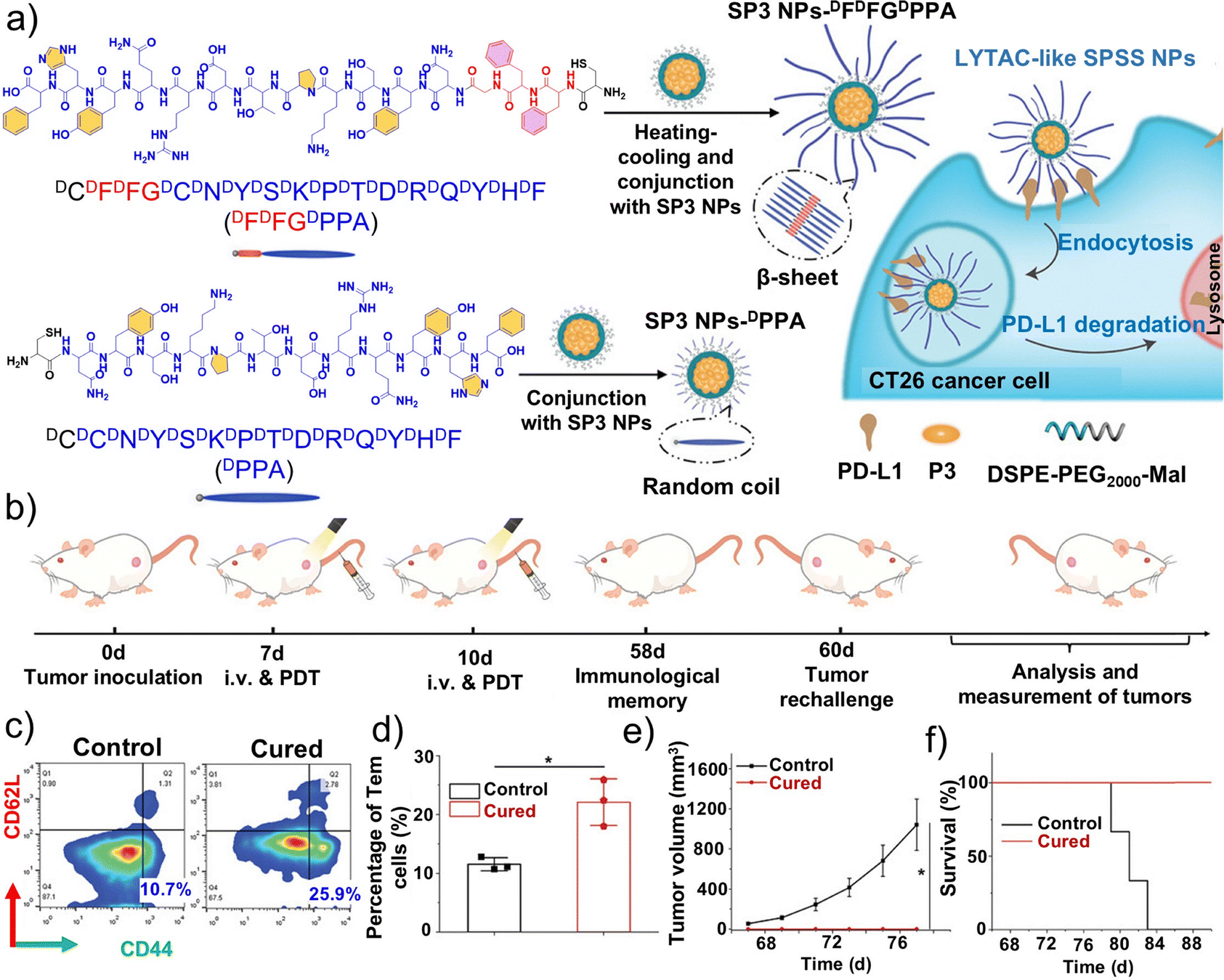 | ||
| Fig. 24 Preparation and in vivo evaluation of lysosome-targeted chimeras (SPSS NPs) for self-synergistic tumor immunotherapy. (a) Chemical structures of the DFDFGDPPA peptide and the DPPA peptide and schematic illustration of the preparation procedure of SP3 NPs-DFDFGDPPA. (b) Schematic illustration of the in vivo tumor rechallenge experimental schedule. Representative (c) flow cytometry data and (d) quantitative analysis of effector memory T cells in the peripheral blood of healthy and cured mice. (e) Volume of rechallenged CT26 tumors inoculated on healthy or cured mice. (f) Survival rate of the healthy and cured mice after tumor rechallenge. (a–f) Reproduced with permission from ref. 288. Copyright 2022, Wiley-VCH. | ||
Overall, LYTAC and its derivative technologies have achieved significant progress in the selective degradation of membrane and extracellular proteins. The continuous development of LYTAC-like biomimetic platforms, including small molecules, aptamers, nanomaterials, and bioinspired protein secondary structures, has not only expanded the potential to realize the degradability of previously undruggable targets but also enabled synergistic integration with ICD, ICB, photodynamic therapy, and immune modulation, yielding enhanced antitumor effects. However, research in this field remains largely at the preclinical stage, with translation constrained by limited lysosomal binding sites, restricted target expandability, incomplete pharmacokinetic and safety evaluations, and the influence of the TME on degradation efficiency. Future efforts should focus on identifying more engineerable lysosomal receptors, improving system generalizability, enhancing targeting via nanoplatforms or protein secondary structures, and further integrating these approaches with multimodal cancer therapies to facilitate clinical translation.
6.2. Cell membrane-camouflaged nanodrugs
The emergence of antibiotic resistance has rendered traditional antibacterial strategies less effective, highlighting an urgent need for the development of new alternative therapies. However, achieving selective eradication of infectious bacteria remains highly challenging.289–291 Liu et al. utilized the self-directed capture ability of macrophages for infectious bacteria to develop a precise in vivo antibacterial photodynamic therapy (APDT) strategy via the adoptive transfer of photosensitizer-loaded macrophages.292 As shown in Fig. 25a, they first synthesized TTD, which can generate ROS and exhibits bright fluorescence, and then constructed TTDm NPs to achieve lysosomal targeting. Subsequently, they prepared TTD-loaded macrophages (TLMs) by directly incubating TTDm NPs with macrophages. The TTD localized in lysosomes, where it encountered the captured bacteria within phagocytosed lysosomes. Encouraged by these in vitro results, the researchers further tested whether APDT based on adoptive TLMs could eliminate invading bacteria in vivo. As depicted in Fig. 25b and c, the fluorescence intensity of the TLM-treated skin slightly decreased over time, but the signal area gradually expanded 15 h post-injection. Furthermore, confocal imaging revealed numerous red fluorescence spots spreading within the infected tissues of mice treated with TLMs, indicating the presence of TTDm NPs in the areas infected by Staphylococcus aureus. The number of overlapping fluorescence spots found in TLM-treated tissues was greater than that found in TTDm NPs-treated tissues, suggesting a higher degree of fusion between TTDm NPs of TLM and Staphylococcus aureus in macrophage therapy. Following TLM treatment, the infected skin was exposed to white-light irradiation, and the colony-forming unit count in the TLM group was markedly lower than that of the control group on the second day post-treatment, demonstrating the superior antibacterial efficacy of TLMs against Staphylococcus aureus (Fig. 25d). H&E staining further revealed that skin sequentially treated with TLM and APDT exhibited minimal inflammatory cell infiltration and had a normal tissue morphology compared to all other groups (Fig. 25e). Collectively, these findings indicate that TLMs can selectively capture and eliminate bacteria while activated under light irradiation to adopt an inflammatory, antibacterial M1 phenotype. Upon subcutaneous administration, TLMs effectively inhibit bacteria in infected tissues via APDT, promoting tissue recovery following severe bacterial infection. This engineered cell-based therapeutic strategy thus holds considerable potential for treating severe bacterial infections, along with tumors associated with bacterial colonization, promoting precision tumor therapy using lysosome-targeted biomimetic platforms. | ||
| Fig. 25 Macrophage membrane-camouflaged biomimetic platform, TTD-loaded macrophages (TLM), and evaluation of its antibacterial properties. (a) Chemical structure of TTD, schematic mechanism of TTD to produce reactive oxygen species (ROS), TTD NPs with surface-modified lysosome-targeting morpholine groups, and the APDT process. Upon injection into the bacterial infection site, the TLMs are triggered to actively capture the infectious bacteria precisely. (b) In vivo fluorescence images of an S. aureus-infected mouse after subcutaneous injection of TLMs. (c) Confocal images of the TLMs injected into the infected skin tissue of mice. (d) Antibacterial activity of TLMs compared to TTD NPs and saline in the S. aureus-infected mice under white-light irradiation (400–780 nm, 300 mW cm−2) for 10 min after continuous treatment for two days. (e) Representative images of H&E-stained skin slices following different treatments. (a–e) Reproduced with permission from ref. 292. Copyright 2023, American Chemical Society. | ||
Traditional adoptive T cell transfer faces the same limitations described above, as ex vivo-expanded or genetically modified T cells show only modest improvements in tumor recognition and killing; accordingly, several strategies have been proposed to enhance T cell activity.293–295 Zhang et al. constructed a lysosome-targeted nanodrug, LYS-NPs, to modify T cells for harvesting adoptive T cell vectors (ATVs).296 Specifically, tumor antigens were presented to CD8+ T cells via DCs, and granzyme B and perforin encapsulated in the calcium carbonate-mineralized zeolitic imidazolate framework were endocytosed by the pre-activated T cells. LYS-NPs, modified with CD63 aptamers, targeted lysosomes to release granzyme B and perforin in a stable and activated form. The ATVs remained stable until T cell receptors were activated by tumor cells, triggering lysosomal release at the immunological synapse (Fig. 26a). In co-culture experiments, unlike the T cells in the control group, ATVs densely attached to 4T1 tumor cells. Upon tumor activation, the ATVs released perforin, granzyme B, and Ca2+, enabling effective tumor recognition and significantly improving antitumor efficacy (Fig. 26b).
 | ||
| Fig. 26 Schematic diagram of the mechanism of lysosome-targeted biomembrane camouflage nanoparticles. (a) Schematic diagram of the design and synthesis of LYS-NPs and their function. (b) Representative images of CD8+ T cells or adoptive T cell vectors (ATVs) interacting with tumor cells at 200× magnification; the bar graph on the right shows the quantification of T cell numbers around the tumor cells. (a and b) Reproduced with permission from ref. 296. Copyright 2021, Wiley-VCH. (c) Conceptual schematic diagram of the preparation process and intracellular mechanism of nanoparticles. Reproduced with permission from ref. 302. Copyright 2023, Wiley-VCH. | ||
Oral squamous cell carcinoma (OSCC) is commonly treated using systemic chemotherapy, which often causes multidrug resistance and adverse effects.297–299 Cobalt–ferrocene (Co–Fc) metal–organic frameworks are nanoparticles that catalyze a strong Fenton reaction, producing OH˙ to kill tumor cells. However, tumor cells can eliminate exogenous substances via autophagy to maintain homeostasis.300,301 To address this problem, Zhou et al. developed Co–Fc nanoparticles loaded with the autophagy inhibitor hydroxychloroquine (HCQ), forming Co–Fc@HCQ.302 As shown in Fig. 26c, ferrocene catalyzes endogenous ROS generation, while HCQ inhibits autophagy by blocking autophagosome-lysosome fusion, reducing ROS clearance. To further enhance targeting, tumor cell membranes from CAL-27 cells were used to construct CM@Co–Fc@HCQ nanoparticles, providing features of homologous targeting and immune evasion. The nanoparticles demonstrated excellent tumor lysosome targeting, biosafety, and therapeutic efficacy. Overall, CM@Co–Fc@HCQ offers an intelligent approach for localized chemotherapeutic delivery, enhancing ROS via autophagy inhibition and presenting a promising approach for effective OSCC therapy.
Compared with traditional drug delivery systems, biomembrane-camouflaged nanoparticles can better evade recognition by the immune system, thereby prolonging the circulation time of drugs in the body. Furthermore, these systems can achieve precision treatment by targeting specific receptors on tumor cells, enhancing the efficacy of antitumor therapies. Consequently, biomembrane-camouflaged nanoparticles are expected to play an increasingly critical role in tumor therapy, advancing the development of personalized medicine.
6.3. Molecular imprinting degraders
There have been significant clinical advancements in ICB therapy for various tumor types, particularly with strategies targeting the PD-1/PD-L1 axis. However, existing methods that inhibit the PD-1/PD-L1 signaling pathway show poor efficacy and pose a risk of inducing autoimmune diseases.303,304 Liu et al. reported a molecular imprinting lysosomal nanoparticle degrader (MILND) targeting tumor cells aimed at enhancing ICB-based immunotherapy.305 MILND is designed with the N-terminal epitope of PD-L1 as the imprint template (Fig. 27a), so that it specifically targets PD-L1 on tumor cells, facilitating cellular uptake. This process further induces the transport of PD-L1 to lysosomes for degradation, ultimately resulting in the downregulation of PD-L1 expression on tumor cells (Fig. 27b–d). By blocking the PD-1/PD-L1 signaling pathway, MILND activates T cell-mediated immune responses in vivo, triggering durable antitumor efficacy. In vivo experiments demonstrated that MILND effectively accumulates at tumor sites and exhibits strong tumor growth inhibition in xenograft tumor models, with no significant side effects observed. | ||
| Fig. 27 Chemical biology of a molecular imprinting platform and proton sponge platform. (a) Schematic illustration of boosting tumor immunotherapy via reversing PD-L1-mediated immunosuppression by MILND. (b) Relative fluorescence intensity of PD-L1 on 4T1 cells after different treatments. (c) Immunoblotting analysis of PD-L1 expressed on 4T1 cells after treatment with MILND and NILND, respectively, for 24 h. (d) Confocal laser-scanning microscopy (CLSM) images of 4T1 cells after 24 h of treatment with MILND and NILND, respectively. (a–d) Reproduced with permission from ref. 305. Copyright 2024, American Chemical Society. (e) Scheme showing the chemical structure and formation of the proton sponge nano-assembly from PSNA monomers. Reproduced with permission from ref. 309. Copyright 2024, Wiley-VCH. | ||
Unlike LYTACs, MILND is centered on the construction of a “nano-mold” that specifically recognizes the N-terminal epitope of PD-L1. By employing a short-peptide molecular imprinting template on the nanoparticle surface, MILND achieves highly selective recognition of the target epitope, which is then delivered to lysosomes for degradation. Compared with LYTACs, MILND offers enhanced precision in epitope recognition and increased multifunctionality of the nanoparticle carrier, enabling more accurate PD-L1 degradation and effective reversal of immunosuppression. Another important distinction from LYTACs is that MILND does not rely on lysosomal-targeting receptors; once internalized, it is naturally trafficked to the lysosomes. Thus, MILND represents a novel lysosome-specific biomimetic antitumor platform that can potentially be extended to degrade a wider range of key proteins, further providing a strategy to simplify LYTAC preparation and improve therapeutic efficiency.
6.4. Proton sponge nanoplatforms
In addition to constructing lysosome-specific molecular or material platforms by exploiting the acidic lysosomal microenvironment or facilitating lysosomal escape, modulation of lysosomal protonation levels represents an alternative and important approach to induce lysosomal cell death. Proton sponges such as high-molecular-weight branched polyethyleneimine (PEI) are highly effective in disrupting lysosomes. However, their fixed charge density and limited understanding of associated cell death mechanisms restrict achieving controllable cytotoxicity, thereby hindering their therapeutic applications.306–308 A proton sponge platform is commonly used to absorb protons in the lysosome, simulating the TME, in lysosome-targeted tumor therapy. Jokers et al. created a series of proton sponge nanoplatforms, PSNAs, with a self-assembled controllable surface charge density and cytotoxicity.309 These PSNAs were constructed through the covalent linkage of low-molecular-weight branched PEI and a self-assembling peptide carrying tetraphenylethylene pyridinium (PyTPE), a fluorophore based on aggregation-induced emission. The assembly of the peptide-PyTPE with PEI led to the construction of PSNAs with enhanced positive surface charge and cellular cytotoxicity (Fig. 27e).By adjusting the hydrophilic and hydrophobic components within the peptide, the self-assembly tendency of PSNAs was further optimized, achieving maximum fluorescence, positive surface charge density, cellular uptake, and cytotoxicity toward tumor cells. Comprehensive studies revealed that LMR-driven pyroptosis and necroptosis are the two key causes of the observed cell death. The ICD effects induced in tumors by PSNA therapy activate immune cells, highlighting the strong potential of PSNAs to elicit antitumor immune responses. Future research on these platforms should focus on developing biomimetic proton-regulating nanoplatforms with higher controllability. By tuning the charge density, self-assembly structures, and hydrophilic–hydrophobic properties, these platforms can achieve precise regulation of LMR and cell death. Moreover, this strategy can be combined with immune activation, drug delivery, or photodynamic therapy to enhance the overall therapeutic efficacy. Lysosome-targeted biomimetic platforms achieve efficient intracellular drug delivery and site-specific release by mimicking biological membranes or specific receptors, thereby enhancing antitumor or anti-infective efficacy while enabling imaging and immunomodulatory functions, with excellent biocompatibility. However, their complex fabrication, limited in vivo stability, restricted target selection, and potential toxicity remain major challenges. Future research should focus on optimizing their targeting efficiency and controllable release, improving in vivo stability, integrating multifunctional applications, evaluating the long-term safety, and exploring their potential applications in the treatment of lysosome-related diseases beyond tumors.
7. Conclusions
Lysosome-targeted chemical platforms represent a rapidly evolving frontier in precision oncology, offering innovative strategies to overcome traditional limitations of tumor therapy such as drug resistance, poor targeting specificity, and systemic toxicity. Over the past five years, advances in the design of lysosome-targeted tumor therapeutic platforms have demonstrated remarkable preclinical efficacy. The structural and functional characteristics of tumor lysosomal membranes highlight LMP and autophagy as important targets for lysosome-mediated precision oncology. Autophagy can promote tumor survival by sequestering drugs, but it can also enhance tumor elimination when combined with chemotherapy, immunotherapy, ferroptosis, necrosis, PDT, PTT, and biotherapy. This review summarizes the latest advances in the design and applications of lysosome-specific molecules, materials, and biomimetic platforms reported in the past five years, with a focus on their structural design, mechanisms of action, therapeutic efficacy, and translational potential. Specifically, key steps contributing to the progress in lysosome-targeted chemical platforms include the development of small molecules, metal complexes, and supramolecular assemblies that exploit the acidic lysosomal microenvironment for selective activation and drug release, as well as material-based platforms capable of controlling drug delivery and enhancing endosomal/lysosomal escape. Furthermore, lysosome-targeted biomimetic platforms such as LYTACs, cell membrane-camouflaged nanoparticles, molecularly imprinted degraders, and proton sponge platforms have shown superior synergistic effects by integrating lysosomal targeting with immune modulation, ferroptosis, and phototherapy mechanisms. Despite these advances, several challenges remain, including improving the stability and biocompatibility of delivery systems, enhancing targeting precision to minimize off-target effects, and addressing the complexity of large-scale manufacturing and clinical validation.8. Perspectives
Despite the immense potential of lysosome-specific chemical platforms in precision oncology, several critical challenges remain to be addressed. A deeper understanding of lysosomal biology across different tumor types, including its connections with the TME, immune regulation, and drug resistance, is needed. Such knowledge is crucial for the development of peptide self-assembly material platforms, as it will provide precise design guidance for peptide modules. By leveraging variations in lysosomal membrane composition, acidity, enzymatic activity, and endocytic–metabolic pathways, researchers can optimize peptide sequences, assembly-driving forces, and stimulus-responsive mechanisms. Such optimization will enable peptide self-assembly platforms to achieve controlled and selective accumulation in lysosomal compartments, maintain stable conformations, and undergo programmed assembly–disassembly. With consequent enhancement of delivery efficiency, immune modulation can be more accurately targeted, and release behaviors become more predictable, ultimately advancing material platforms aimed at overcoming drug resistance.Second, the drug-loading capacity, lysosomal targeting specificity, and intelligent responsiveness of these chemical platforms need to be further enhanced to achieve higher accumulation levels and more controlled release properties. The liquid-like nature of coacervates, combined with their high loading capacity, acid-triggered behavior, and intrinsic organelle-interaction mechanisms that favor lysosomal affinity, makes them ideal platforms for designing lysosome-specific drug delivery systems. Such systems enable precise, efficient, and intelligently regulated therapeutic delivery within tumor lysosomes. By modulating the liquid–liquid phase separation behavior, interfacial tension, and material composition of coacervates, their residence time in tumor lysosomes, cargo-protection capability, and controllable disassembly can be further improved, thereby synergistically enhancing both targeting specificity and stimulus responsiveness. However, the in vivo applications of coacervates still require continuous attention with respect to their stability, controllability, and targeting efficiency, and addressing these issues will directly determine their actual efficacy in precise drug delivery and cancer therapy. Currently, both the construction of coacervates for drug delivery and the development of tumor therapeutic platforms targeting biomolecular coacervates represent major directions in precision oncology and are expected to remain at the forefront of the field in the coming years.
Third, efforts should focus on integrating lysosome-specific biomimetic platforms with immunotherapy, chemotherapy, radiotherapy, targeted therapy, and gene therapy to maximize multimodal therapeutic effects. Functionalized lysosome-targeting chimeras, endowed with capabilities such as immune modulation, metabolic intervention, TME regulation, or multi-drug co-delivery, can achieve more efficient degradation of membrane and extracellular proteins, enhance antigen presentation, amplify immune responses, and increase the sensitivity of conventional chemotherapy or radiotherapy. Their modular chemical architectures further facilitate the combination of different payloads or the coupling of multiple functional units according to specific therapeutic regimens, thereby enabling truly multimodal synergistic targeted therapy.
Moreover, the identification of lysosome-associated biomarkers and the development of imaging-guided theranostic technologies are crucial for appropriate patient stratification and therapeutic monitoring. In this regard, lysosome-targeted NIR small-molecule probes play a particularly essential role. These molecular platforms enable high-sensitivity, real-time imaging in deep tissues by leveraging the lysosome's acidic environment, enzymatic reactions, or receptor-mediated endocytosis for precise localization, while simultaneously coupling diagnostic signal output with drug delivery or degradation processes to achieve an integrated “diagnosis-and-treatment” functionality. This strategy facilitates dynamic assessment of the in vivo drug distribution, lysosomal accumulation, and therapeutic progress, providing critical support for personalized treatment and real-time efficacy evaluation. Recent studies on pH-responsive europium(III) complexes have shown how targeted probes can monitor receptor internalization and lysosomal acidification in live cells.310,311 Building on these principles, commercially available reagents such as Revvity's pHSenseTM (2025) enable scalable, time-resolved assays for G protein-coupled receptor internalization, providing tools relevant to drug discovery for lysosome-related metabolic diseases including diabetes and obesity. Future efforts may focus on expanding chemical diversity, optimizing targeting, and integrating such lysosome-specific probes with theranostic platforms to bridge diagnostic imaging and functional response evaluation.
Equally important, the design and application of lysosome-targeted chemical platforms must incorporate rigorous evaluation of safety and long-term toxicity. Such considerations not only enhance standardized preparation and scalable production but, in combination with well-designed clinical trials and regulatory oversight, can further accelerate their clinical translation. In summary, with continued research in these directions, lysosome-targeted platforms hold considerable promise for advancing precision medicine in oncology and beyond.
Conflicts of interest
There is no conflicts of interest to declare.Data availability
Data sharing is not applicable to this article as no new data were created or analyzed in this study.Acknowledgements
The authors acknowledge financial support from the Jiangsu Innovation Team Program, the Fundamental Research Funds for the Central Universities, the National Natural Science Foundation of China (22505027), the Basic Research Program of Jiangsu (BK20251322), the Postdoctoral Fellowship Program of the CPSF (GZC20250793), the China Postdoctoral Science Foundation (2025M771164), and the Jiangsu Funding Program for Excellent Postdoctoral Talent (2025ZB618).Notes and references
- H. Nagai and Y. H. Kim, J. Thorac. Dis., 2017, 9, 448–451 CrossRef PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, CA Cancer J. Clin., 2020, 70, 7–30 Search PubMed.
- R. L. Siegel, A. N. Giaquinto and A. Jemal, CA Cancer J. Clin., 2024, 74, 12–49 Search PubMed.
- R. L. Siegel, T. B. Kratzer, A. N. Giaquinto, H. Sung and A. Jemal, CA Cancer J. Clin., 2025, 75, 10–45 Search PubMed.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, CA Cancer J. Clin., 2021, 71, 209–249 Search PubMed.
- S. T. Pan, Z. L. Li, Z. X. He, J. X. Qiu and S. F. Zhou, Clin. Exp. Pharmacol. Physiol., 2016, 43, 723–737 CrossRef CAS PubMed.
- A. Zhang, K. Miao, H. Sun and C. Deng, Int. J. Biol. Sci., 2022, 18, 3019–3033 CrossRef CAS PubMed.
- M. Attwaters, Nat. Rev. Gastroenterol. Hepatol., 2025, 22, 7 CrossRef PubMed.
- S. N. Aleksakhina, A. Kashyap and E. N. Imyanitov, Biochim. Biophys. Acta Rev. Cancer, 2019, 1872, 188310 CrossRef CAS PubMed.
- J. Ma, Y. Zhang, K. Tang, H. Zhang, X. Yin, Y. Li, P. Xu, Y. Sun, R. Ma, T. Ji, J. Chen, S. Zhang, T. Zhang, S. Luo, Y. Jin, X. Luo, C. Li, H. Gong, Z. Long, J. Lu, Z. Hu, X. Cao, N. Wang, X. Yang and B. Huang, Cell Res., 2016, 26, 713–727 CrossRef CAS PubMed.
- E. Lepeltier, P. Rijo, F. Rizzolio, R. Popovtzer, V. Petrikaite, Y. G. Assaraf and C. Passirani, Drug Resist. Update, 2020, 52, 100704 CrossRef PubMed.
- P. Wu, W. Gao, M. Su, E. C. Nice, W. Zhang, J. Lin and N. Xie, Front. Cell Dev. Biol., 2021, 9, 641469 CrossRef PubMed.
- O. Hiroki, H. Kawakubo, S. Mayanagi, K. Fukuda, O. Goto, R. Nakamura, K. Suda, N. Wada, N. Yahagi and Y. Kitagawa, J. Clin. Oncol., 2018, 36, 168 Search PubMed.
- Y. Tang, X. Wang, G. Zhu, Z. Liu, X. M. Chen, H. K. Bisoyi, X. Chen, X. Chen, Y. Xu, J. Li and Q. Li, Small, 2023, 19, 2205440 CrossRef CAS PubMed.
- X. Wang, Y. Tang, Y. Li and Z. Qi, Adv. Healthcare Mater., 2024, 13, 2401904 CrossRef CAS PubMed.
- X. Wang, Y. Li, K. Hasrat, L. Yang and Z. Qi, Small, 2023, 19, 2305101 CrossRef CAS PubMed.
- K. Morihiro, S. Morita, N. Harada, M. Baba, J. Yum, M. Naito, K. Miyata, G. Nagae and A. Okamoto, J. Am. Chem. Soc., 2025, 146, 1346–1355 Search PubMed.
- H. Luo, W. Ma, Q. Chen, Z. Yang and Y. Dai, Nano Today, 2023, 53, 102042 Search PubMed.
- S. Cornen and E. Vivier, Science, 2018, 362, 1355–1356 Search PubMed.
- M. Liu, Y. Sun, Y. Zhou, Y. Chen, M. Yu, L. Li, L. Yan, Y. Yuan, J. Chen, K. Zhou, H. Shan and X. Peng, Adv. Healthcare Mater., 2024, 13, 2304488 CrossRef CAS PubMed.
- X. Kong, Y. Qi, X. Wang, R. Jiang, J. Wang, Y. Fang, J. Gao and K. Chu Hwang, Prog. Mater. Sci., 2023, 134, 101070 CrossRef CAS.
- Y. Liu, B. Wang, J. Zhu, X. Xu, B. Zhou and Y. Yang, Adv. Mater., 2023, 35, 2208512 CrossRef CAS PubMed.
- M. Norouzi, M. Amerian, M. Amerian and F. Atyabi, Drug Discov. Today, 2020, 25, 107–125 CrossRef CAS PubMed.
- C. de Duve, B. C. Pressman, R. Gianetto, R. Wattiaux and F. Appelmans, Biochem. J., 1955, 60, 604–617 CrossRef CAS PubMed.
- C. Papadopoulos, B. Kravic and H. Meyer, J. Mol. Biol., 2020, 432, 231–239 CrossRef CAS PubMed.
- Y. Lei and D. J. Klionsky, Autophagy, 2024, 20, 1471–1472 CrossRef CAS PubMed.
- C. Papadopoulos and H. Meyer, Curr. Biol., 2017, 27, R1330–R1341 CrossRef CAS PubMed.
- A. M. Thelen and R. Zoncu, Trends Cell Biol., 2017, 27, 833–850 CrossRef CAS PubMed.
- X. Luo, C. Cheng, Z. Tan, N. Li, M. Tang, L. Yang and Y. Cao, Mol. Cancer, 2017, 16, 76 CrossRef PubMed.
- X. Yin, R. Xu, J. Song, R. Ruze, Y. Chen, C. Wang and Q. Xu, Cancer Commun., 2022, 42, 1234–1256 CrossRef PubMed.
- B. K. Kashyap, V. V. Singh, M. K. Solanki, A. Kumar, J. Ruokolainen and K. K. Kesari, ACS Omega, 2023, 8, 14290–14320 CrossRef CAS PubMed.
- N. Walweel and O. Aydin, ACS Omega, 2024, 9, 27832–27852 CrossRef CAS PubMed.
- N. Chauhan and B. S. Patro, Cancer Lett, 2024, 584, 216599 CrossRef CAS PubMed.
- C. Ding, C. Chen, X. Zeng, H. Chen and Y. Zhao, ACS Nano, 2022, 16, 13513–13553 CrossRef CAS PubMed.
- M. Yang, C. Cong, J. Bian, Z. Xu, X. Liu, L. Liu, X. Zhang, D. Wang and D. Gao, Appl. Mater. Today, 2021, 22, 100940 CrossRef.
- M. Borkowska, M. Siek, D. V. Kolygina, Y. I. Sobolev, S. Lach, S. Kumar, Y. K. Cho, K. Kandere-Grzybowska and B. A. Grzybowski, Nat. Nanotechnol., 2020, 15, 331–341 CrossRef CAS PubMed.
- Q. Chen, H. Fang, X. Shao, Z. Tian, S. Geng, Y. Zhang, H. Fan, P. Xiang, J. Zhang, X. Tian, K. Zhang, W. He, Z. Guo and J. Diao, Nat. Commun., 2020, 11, 6290 CrossRef CAS PubMed.
- D. X. Hu, S. Patel, H. Chen, S. Wang, S. T. Staben, Y. N. Dimitrova, H. A. Wallweber, J. Y. Lee, G. K. Y. Chan, C. J. Sneeringer, M. S. Prangley, J. G. Moffat, K. C. Wu, L. K. Schutt, L. Salphati, J. Pang, E. McNamara, H. Huang, Y. Chen, Y. Wang, W. Zhao, J. Lim, A. Murthy and M. Siu, J. Med. Chem., 2022, 65, 11500–11512 CrossRef CAS PubMed.
- M. T. Gyparaki and A. G. Papavassiliou, Trends Mol. Med., 2014, 20, 239–241 CrossRef CAS PubMed.
- R. M. Perera and R. Zoncu, Annu. Rev. Cell Dev. Biol., 2016, 32, 223–253 CrossRef CAS PubMed.
- X. Zhang, X. Cheng, L. Yu, J. Yang, R. Calvo, S. Patnaik, X. Hu, Q. Gao, M. Yang, M. Lawas, M. Delling, J. Marugan, M. Ferrer and H. Xu, Nat. Commun., 2016, 7, 12109 CrossRef CAS PubMed.
- M. Samie, X. Wang, X. Zhang, A. Goschka, X. Li, X. Cheng, E. Gregg, M. Azar, Y. Zhuo, A. G. Garrity, Q. Gao, S. Slaugenhaupt, J. Pickel, S. N. Zolov, L. S. Weisman, G. M. Lenk, S. Titus, M. Bryant-Genevier, N. Southall, M. Juan, M. Ferrer and H. Xu, Dev. Cell, 2013, 26, 511–524 CrossRef CAS PubMed.
- R. E. Lawrence and R. Zoncu, Nat. Cell Biol., 2019, 21, 133–142 CrossRef CAS PubMed.
- S. M. Davidson and M. G. Vander Heiden, Annu. Rev. Pharmacol. Toxicol., 2017, 57, 481–507 CrossRef CAS PubMed.
- R. Mathew, V. Karantza-Wadsworth and E. White, Nat. Rev. Cancer, 2007, 7, 961–967 CrossRef CAS PubMed.
- B. Levine and G. Kroemer, Cell, 2019, 176, 11–42 CrossRef CAS PubMed.
- A. Singh, M. Handa, M. Ruwali, S. J. S. Flora, R. Shukla and P. Kesharwani, Prog. Biochem., 2021, 109, 198–206 CrossRef CAS.
- L. M. D. Delbridge, K. M. Mellor, D. J. Taylor and R. A. Gottlieb, Nat. Rev. Cardiol., 2017, 14, 412–425 CrossRef CAS PubMed.
- A. Scrivo, M. Bourdenx, O. Pampliega and A. M. Cuervo, Lancet Neurol., 2018, 17, 802–815 CrossRef PubMed.
- Y. Feng, D. He, Z. Yao and D. J. Klionsky, Cell Res., 2014, 24, 24–41 CrossRef CAS PubMed.
- L. Galluzzi, J. M. Bravo-San Pedro, K. Blomgren and G. Kroemer, Nat. Rev. Neurosci., 2016, 17, 467–484 CrossRef CAS PubMed.
- S. Wang, H. Long, L. Hou, B. Feng, Z. Ma, Y. Wu, Y. Zeng, J. Cai, D. W. Zhang and G. Zhao, Signal Transduct. Target. Ther., 2023, 8, 304 CrossRef PubMed.
- X. Li, S. He and B. Ma, Mol. Cancer, 2020, 19, 12 CrossRef CAS PubMed.
- M. Raudenska, J. Balvan and M. Masarik, Mol. Cancer, 2021, 20, 140 CrossRef PubMed.
- Y. Kuchitsu and T. Taguchi, Trends Cell Biol., 2024, 34, 606–616 CrossRef CAS PubMed.
- S. Kaushik and A. M. Cuervo, Nat. Rev. Mol. Cell Biol., 2018, 19, 365–381 CrossRef CAS PubMed.
- L. Wang, D. J. Klionsky and H. M. Shen, Nat. Rev. Mol. Cell Biol., 2023, 24, 186–203 CrossRef CAS PubMed.
- K. Tekirdag and A. M. Cuervo, J. Biol. Chem., 2018, 293, 5414–5424 CrossRef CAS PubMed.
- J. Debnath, N. Gammoh and K. M. Ryan, Nat. Rev. Mol. Cell Biol., 2023, 24, 560–575 CrossRef CAS PubMed.
- F. M. Menzies, A. Fleming, A. Caricasole, C. F. Bento, S. P. Andrews, A. Ashkenazi, J. Füllgrabe, A. Jackson, M. Jimenez Sanchez, C. Karabiyik, F. Licitra, A. Lopez Ramirez, M. Pavel, C. Puri, M. Renna, T. Ricketts, L. Schlotawa, M. Vicinanza, H. Won, Y. Zhu, J. Skidmore and D. C. Rubinsztein, Neuron, 2017, 93, 1015–1034 CrossRef CAS PubMed.
- H. Kim, D. Lee, J. Kim, T. I. Kim and W. J. Kim, ACS Nano, 2013, 7, 6735–6746 CrossRef CAS PubMed.
- J. Shen, J. Chen, J. Ma, L. Fan, X. Zhang, T. Yue, Y. Yan and Y. Zhang, Asian J. Pharm. Sci., 2020, 15, 759–776 Search PubMed.
- L. Ye, H. Liu, X. Fei, D. Ma, X. He, Q. Tang, X. Zhao, H. Zou, X. Chen, X. Kong and P. Liu, Nano Res., 2022, 15, 1135–1144 CrossRef CAS.
- B. Shi, M. Zheng, W. Tao, R. Chung, D. Jin, D. Ghaffari and O. C. Farokhzad, Biomacromolecules, 2017, 18, 2231–2246 CrossRef CAS PubMed.
- N. Gong, Y. Zhang, X. Teng, Y. Wang, S. Huo, G. Qing, Q. Ni, X. Li, J. Wang, X. Ye, T. Zhang, S. Chen, Y. Wang, J. Yu, P. C. Wang, Y. Gan, J. Zhang, M. J. Mitchell, J. Li and X.-J. Liang, Nat. Nanotechnol., 2020, 15, 1053–1064 CrossRef CAS PubMed.
- X. Zhang, K. Wang, Z. Zhao, X. Shan, Y. Wang, Z. Feng, B. Li, C. Luo, X. Chen and J. Sun, ACS Nano, 2024, 18, 7136–7147 CrossRef CAS PubMed.
- J. Qiu and Y. Xia, Nat. Nanotechnol., 2020, 15, 252–253 CrossRef CAS PubMed.
- Y. Liu, L. Qiao, S. Zhang, G. Wan, B. Chen, P. Zhou, N. Zhang and Y. Wang, Acta Biomater., 2018, 66, 310–324 CrossRef CAS PubMed.
- X. Wang, X. Li, M. Duan, S. Shan, X. Zhu, Y. Chai, H. Wang, X. Sun, L. Sheng, G. Qing, W. Rao, L. Hu, J. Chen and J. Liu, Matter, 2022, 5, 219–236 CrossRef CAS.
- P. P. Chamberlain and L. G. Hamann, Nat. Chem. Biol., 2019, 15, 937–944 CrossRef CAS PubMed.
- Y. Yang, H. Ning, T. Xia, J. Du, W. Sun, J. Fan and X. Peng, Adv. Mater., 2023, 35, 2301409 CrossRef CAS PubMed.
- K. Qiu, H. Zhu, T. W. Rees, L. Ji, Q. Zhang and H. Chao, Coord. Chem. Rev., 2019, 398, 113010 CrossRef.
- A. Ballabio and J. S. Bonifacino, Nat. Rev. Mol. Cell Biol., 2020, 21, 101–108 CrossRef CAS PubMed.
- S. R. Bonam, F. Wang and S. Muller, Nat. Rev. Drug Discov., 2019, 18, 923–948 CrossRef CAS PubMed.
- S. M. Banik, K. Pedram, S. Wisnovsky, G. Ahn, N. M. Riley and C. R. Bertozzi, Nature, 2020, 584, 291–297 CrossRef CAS PubMed.
- Q. Yi, C. Pu, X. Tang, M. Liu, X. Lin, W. Lan, X. Zhang, M. Yang, M. Wang and J. Wang, Bioorg. Chem., 2025, 157, 108266 CrossRef CAS PubMed.
- W. Wang, H. Wang, Z. Zhang, X. Liu, B. Hu, F. Tian, Z. Ye, L. Shi and Z. Yu, Adv. Mater., 2025, 37, 2420399 CrossRef CAS PubMed.
- T. Caneque, L. Baron, S. Muller, A. Carmona, L. Colombeau, A. Versini, S. Solier, C. Gaillet, F. Sindikubwabo, J. L. Sampaio, M. Sabatier, E. Mishima, A. Picard-Bernes, L. Syx, N. Servant, B. Lombard, D. Loew, J. Zheng, B. Proneth, L. K. Thoidingjam, L. Grimaud, C. S. Fraser, K. J. Szylo, E. Der Kazarian, C. Bonnet, E. Charafe-Jauffret, C. Ginestier, P. Santofimia-Castano, M. Estaras, N. Dusetti, J. L. Iovanna, A. S. Cunha, G. Pittau, P. Hammel, D. Tzanis, S. Bonvalot, S. Watson, V. Gandon, A. Upadhyay, D. A. Pratt, F. P. Freitas, J. P. Friedmann Angeli, B. R. Stockwell, M. Conrad, J. M. Ubellacker and R. Rodriguez, Nature, 2025, 642, 492–500 CrossRef CAS PubMed.
- W. C. Chan, Q. Zhao, K. H. Wong, H. H. Tang, D. K. Mok, L. Pardeshi, Y. C. Ruan, Y. Yang, C. M. Wong, C. T. T. Wong, Y. J. Zhao, K. Y. Chan, Z. Yin, H. H. Leung, X. Ma, M. P. Chau, F. K. Leung, Y. Y. Lui, S. W. Au, K. Sun, C. Y. Mok, F. Wang, C. Ma, Y. H. Ho, X. Y. Ye, M. L. Yeung, Y. Zhang, Y. X. Zhao, J. Liu, K. H. Wong, L. M. C. Chow, X. D. Li, G. M. Tse, M. K. Wong, X. Shen, P. Chiu and B. C. B. Ko, Nat. Commun., 2025, 16, 6308 CrossRef CAS PubMed.
- S. Zhao, Q. Bai, G. Xue, J. Wang, L. Hu, X. Wang, Y. Li, S. Lu, Y. Sun, Z. Zhang, Y. Mu, Y. Zhi and Q. Chen, Acta Pharm. Sin. B, 2025, 15, 5867–5879 CrossRef CAS PubMed.
- K. Wang, A. Yu, K. Liu, C. Feng, Y. Hou, J. Chen, S. Ma, L. Huang and X. Dai, Adv. Sci., 2023, 10, 2300288 CrossRef CAS PubMed.
- Z. Li, J. Zou and X. Chen, Adv. Mater., 2023, 35, 2209529 CrossRef CAS PubMed.
- M. Cao, X. Luo, K. Wu and X. He, Signal Transduct. Target. Ther., 2021, 6, 379 CrossRef CAS PubMed.
- L. E. L. Hendriks and A. M. C. Dingemans, Expert. Opin. Investig. Drugs, 2017, 26, 541–550 CrossRef CAS PubMed.
- J. Yin, L. Ding, S. Yao, J. Huang, Y. Xiao, Y. Wang, B. Zhang, M. Rehmutulla, L. Gu, Q. Tong and Y. Zhang, Bioorg. Chem., 2024, 153, 107820 Search PubMed.
- G. Li, L. Gu, C. Yang, X. Kong, Y. Qin and L. Wu, ACS Mater. Lett., 2024, 6, 1820–1830 Search PubMed.
- Y. Tang, D. Xiang and Q. Li, Adv. Mater., 2025, 37, 2501184 CrossRef CAS PubMed.
- Z. Wang, Y. Tang and Q. Li, Light Sci. Appl., 2025, 14, 16 Search PubMed.
- Y. Tang, Z. Wang and Q. Li, Adv. Funct. Mater., 2024, 34, 2405367 CrossRef CAS.
- Q. Xiao, H. Lin, J. Wu, X. Pang, Q. Zhou, Y. Jiang, P. Wang, W. Leung, H. Lee, S. Jiang, S. Q. Yao, L. Gao, G. Liu and C. Xu, J. Med. Chem., 2020, 63, 4896–4907 Search PubMed.
- H. Zhang, Y. Liu and S. Qu, Responsive Mater., 2024, 2, e20240012 CrossRef CAS.
- Y. Tang, X. Wang, S. Chen and Q. Li, Responsive Mater., 2024, 2, e20240003 CrossRef CAS.
- C. Zhou, Q. Tang, S. Yang, G. Zhao, T. Wang, Z. Chen, Y. Zhang, P. Tan and X. Ma, Responsive Mater., 2025, 3, e20240043 CrossRef CAS.
- P. Liu, L. Wang, L. Chen, X. Su and X. Shi, Bioconjugate Chem., 2021, 32, 1117–1122 CrossRef CAS PubMed.
- J. Niu, Y.-H. Liu, W. Xu, W.-W. Xu, Y.-H. Song, J. Yu, Y.-M. Zhang and Y. Liu, Chem. Commun., 2023, 59, 4680–4683 RSC.
- Q. Feng, T. Yang, L. Ma, X. Li, H. Yuan, M. Zhang, Y. Zhang and L. Fan, ACS Appl. Mater. Interfaces, 2022, 14, 38594–38603 CrossRef CAS PubMed.
- J. Zhao, K. Xu, W. Yang, Z. Wang and F. Zhong, Chem. Soc. Rev., 2015, 44, 8904–8939 RSC.
- C. Zhang, J. Zhao, X. Cui and X. Wu, J. Org. Chem., 2015, 80, 5674–5686 CrossRef CAS PubMed.
- V.-N. Nguyen, Y. Yan, J. Zhao and J. Yoon, Acc. Chem. Res., 2021, 54, 207–220 CrossRef CAS PubMed.
- X. Kong, L. Di, Y. Fan, Z. Zhou, X. Feng, L. Gai, J. Tian and H. Lu, Chem. Commun., 2019, 55, 11567–11570 RSC.
- P. Zhao, Z. Wang, Y. Wang, Z. Wu, Y. Guo, C. Wang, X. Fang, Z. Qu, H. Wang and G. Zhao, Dyes Pigm., 2023, 214, 111214 CrossRef CAS.
- Y. Wu, Z. Chen, D. Shen, Z. He, J. Lv, H. Li, M. Yang, J. Tan, J. Yuan, J. Gao and Z. Yuan, ACS Omega, 2023, 8, 12481–12488 CrossRef CAS PubMed.
- N. A. M. Ligthart, M. A. R. de Geus, M. A. T. van de Plassche, D. Torres Garcia, M. M. E. Isendoorn, L. Reinalda, D. Ofman, T. van Leeuwen and S. I. van Kasteren, J. Am. Chem. Soc., 2023, 145, 12630–12640 CrossRef CAS PubMed.
- B. Jana, S. Jin, E. M. Go, Y. Cho, D. Kim, S. Kim, S. K. Kwak and J. H. Ryu, J. Am. Chem. Soc., 2023, 145, 18414–18431 CrossRef CAS PubMed.
- Y. Xia, J. Li, L. Wang, X. Luo, Y. Xie and Y. Liu, Angew. Chem., Int. Ed., 2022, 61, e202115764 CrossRef CAS PubMed.
- Y. Wang, Y. Y. Chen, G. B. Gao, Y. H. Zheng, N. N. Yu, L. Ouyang, X. Gao, N. Li, S. Y. Wen, S. Huang, Q. Zhao, L. Liu, M. Cao, S. Zhang, J. Zhang and Q. Y. He, Mol. Ther., 2023, 31, 2169–2187 CrossRef CAS PubMed.
- Y. Li, Y. Sun, J. Li, Q. Su, W. Yuan, Y. Dai, C. Han, Q. Wang, W. Feng and F. Li, J. Am. Chem. Soc., 2015, 137, 6407–6416 CrossRef CAS PubMed.
- M. Gao, F. Yu, C. Lv, J. Choo and L. Chen, Chem. Soc. Rev., 2017, 46, 2237–2271 RSC.
- Z. Wang, Z. Xu and G. Zhu, Angew. Chem., Int. Ed., 2016, 55, 15564–15568 CrossRef CAS PubMed.
- E.-J. Kim, S. Bhuniya, H. Lee, H. M. Kim, C. Cheong, S. Maiti, K. S. Hong and J. S. Kim, J. Am. Chem. Soc., 2014, 136, 13888–13894 CrossRef CAS PubMed.
- C. Liu, R. Zhang, W. Zhang, J. Liu, Y. L. Wang, Z. Du, B. Song, Z. P. Xu and J. Yuan, J. Am. Chem. Soc., 2019, 141, 8462–8472 CrossRef CAS PubMed.
- W. Piao, S. Tsuda, Y. Tanaka, S. Maeda, F. Liu, S. Takahashi, Y. Kushida, T. Komatsu, T. Ueno, T. Terai, T. Nakazawa, M. Uchiyama, K. Morokuma, T. Nagano and K. Hanaoka, Angew. Chem., Int. Ed., 2013, 52, 13028–13032 CrossRef CAS PubMed.
- T. T. Mai, A. Hamaï, A. Hienzsch, T. Cañeque, S. Müller, J. Wicinski, O. Cabaud, C. Leroy, A. David, V. Acevedo, A. Ryo, C. Ginestier, D. Birnbaum, E. Charafe-Jauffret, P. Codogno, M. Mehrpour and R. Rodriguez, Nat. Chem., 2017, 9, 1025–1033 CrossRef CAS PubMed.
- S. Fakih, M. Podinovskaia, X. Kong, H. L. Collins, U. E. Schaible and R. C. Hider, J. Med. Chem., 2008, 51, 4539–4552 CrossRef CAS PubMed.
- L. Qiu, C. Zhu, H. Chen, M. Hu, W. He and Z. Guo, Chem. Commun., 2014, 50, 4631–4634 RSC.
- Y. Yang, Q. Zhao, W. Feng and F. Li, Chem. Rev., 2013, 113, 192–270 CrossRef CAS PubMed.
- S. Kuang, X. Liao, X. Zhang, T. W. Rees, R. Guan, K. Xiong, Y. Chen, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2020, 59, 3315–3321 CrossRef CAS PubMed.
- H. Yao, J. Lan, C. Li, H. Shi, J. P. Brosseau, H. Wang, H. Lu, C. Fang, Y. Zhang, L. Liang, X. Zhou, C. Wang, Y. Xue, Y. Cui and J. Xu, Nat. Biomed. Eng., 2019, 3, 306–317 CrossRef CAS PubMed.
- M. de Miguel and E. Calvo, Cancer Cell, 2020, 38, 326–333 CrossRef CAS PubMed.
- L. Zhang, N. Montesdeoca, J. Karges and H. Xiao, Angew. Chem., Int. Ed., 2023, 62, e202300662 CrossRef CAS PubMed.
- S. Sen, M. Won, M. S. Levine, Y. Noh, A. C. Sedgwick, J. S. Kim, J. L. Sessler and J. F. Arambula, Chem. Soc. Rev., 2022, 51, 1212–1233 RSC.
- B. Liu, B. B. Liang, W. D. Cao, X. X. Su, Q. Cao and Z. W. Mao, Angew. Chem., Int. Ed., 2024, 63, e202410586 CrossRef CAS PubMed.
- X. Xue, C. Zhu, H. Chen, Y. Bai, X. Shi, Y. Jiao, Z. Chen, Y. Miao, W. He and Z. Guo, Inorg. Chem., 2017, 56, 3754–3762 CrossRef CAS PubMed.
- R. Safaei, B. J. Larson, T. C. Cheng, M. A. Gibson, S. Otani, W. Naerdemann and S. B. Howell, Mol. Cancer Ther, 2005, 4, 1595–1604 CrossRef CAS PubMed.
- S. S. Chauhan, X. J. Liang, A. W. Su, A. Pai-Panandiker, D. W. Shen, J. A. Hanover and M. M. Gottesman, Br. J. Cancer, 2003, 88, 1327–1334 CrossRef CAS PubMed.
- R. Safaei, K. Katano, B. J. Larson, G. Samimi, A. K. Holzer, W. Naerdemann, M. Tomioka, M. Goodman and S. B. Howell, Clin. Cancer Res, 2005, 11, 756–767 CrossRef CAS PubMed.
- X. Xue, C. Qian, H. Fang, H. K. Liu, H. Yuan, Z. Guo, Y. Bai and W. He, Angew. Chem., Int. Ed., 2019, 58, 12661–12666 CrossRef CAS PubMed.
- N. Feng, Z. Peng, X. Zhang, Y. Lin, L. Hu, L. Zheng, B. Z. Tang and J. Zhang, Nat. Commun., 2024, 15, 8187 CrossRef CAS PubMed.
- H. Zou, J. Zhang, C. Wu, B. He, Y. Hu, H. H. Y. Sung, R. T. K. Kwok, J. W. Y. Lam, L. Zheng and B. Z. Tang, ACS Nano, 2021, 15, 9176–9185 CrossRef CAS PubMed.
- G. X. Xu, L. C. Lee, P. K. Leung, E. C. Mak, J. Shum, K. Y. Zhang, Q. Zhao and K. K. Lo, Chem. Sci., 2023, 14, 13508–13517 RSC.
- H. Shi, O. W. L. Carter, F. Ponte, C. Imberti, M. A. Gomez-Gonzalez, F. Cacho-Nerin, P. D. Quinn, J. E. Parker, E. Sicilia, H. Huang and P. J. Sadler, Angew. Chem., Int. Ed., 2024, 63, e202400476 CrossRef CAS PubMed.
- G. Qi, F. Hu, Kenry, L. Shi, M. Wu and B. Liu, Angew. Chem., Int. Ed., 2019, 58, 16229–16235 CrossRef CAS PubMed.
- X. Wang, L. Yang, Y. Li, X. Wang and Z. Qi, Chem. Asian J., 2024, 19, e202400305 CrossRef CAS PubMed.
- J. He, Y. Wang, M. A. Missinato, E. Onuoha, L. A. Perkins, S. C. Watkins, C. M. St Croix, M. Tsang and M. P. Bruchez, Nat. Methods, 2016, 13, 263–268 CrossRef CAS PubMed.
- J. Gao, J. Li, W.-C. Geng, F.-Y. Chen, X. Duan, Z. Zheng, D. Ding and D.-S. Guo, J. Am. Chem. Soc., 2018, 140, 4945–4953 CrossRef CAS PubMed.
- Y. Wang, W. Wu, J. Liu, P. N. Manghnani, F. Hu, D. Ma, C. Teh, B. Wang and B. Liu, ACS Nano, 2019, 13, 6879–6890 CrossRef CAS PubMed.
- J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. S. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Chem. Rev., 2010, 110, 2795–2838 CrossRef CAS PubMed.
- L. Shao, Y. Pan, B. Hua, S. Xu, G. Yu, M. Wang, B. Liu and F. Huang, Angew. Chem., Int. Ed., 2020, 59, 11779–11783 CrossRef CAS PubMed.
- K. Zhou, Y. Wang, X. Huang, K. Luby-Phelps, B. D. Sumer and J. Gao, Angew. Chem., Int. Ed., 2011, 50, 6109–6114 CrossRef CAS PubMed.
- Z. Yang, W. Fan, W. Tang, Z. Shen, Y. Dai, J. Song, Z. Wang, Y. Liu, L. Lin, L. Shan, Y. Liu, O. Jacobson, P. Rong, W. Wang and X. Chen, Angew. Chem., Int. Ed., 2018, 57, 14101–14105 CrossRef CAS PubMed.
- D. Chen, Q. Tang, J. Zou, X. Yang, W. Huang, Q. Zhang, J. Shao and X. Dong, Adv. Healthcare Mater., 2018, 7, 1701272 CrossRef PubMed.
- F. Li, Y. Du, J. Liu, H. Sun, J. Wang, R. Li, D. Kim, T. Hyeon and D. Ling, Adv. Mater., 2018, 30, 1802808 CrossRef PubMed.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- H. He, W. Tan, J. Guo, M. Yi, A. N. Shy and B. Xu, Chem. Rev., 2020, 120, 9994 CrossRef CAS PubMed.
- H. Wu and M. Fuxreiter, Cell, 2016, 165, 1055 CrossRef CAS PubMed.
- K. Cheng, Y. Ding, Y. Zhao, S. Ye, X. Zhao, Y. Zhang, T. Ji, H. Wu, B. Wang, G. J. Anderson, L. Ren and G. Nie, Nano Lett., 2018, 18, 3250 CrossRef CAS PubMed.
- X. Yang, Z. Cao, H. Lu and H. Wang, Adv. Healthcare Mater., 2021, 10, 2100381 CrossRef CAS PubMed.
- M. T. Jeena, L. Palanikumar, E. M. Go, I. Kim, M. G. Kang, S. Lee, S. Park, H. Choi, C. Kim, S. M. Jin, S. C. Bae, H. W. Rhee, E. Lee, S. K. Kwak and J. H. Ryu, Nat. Commun., 2017, 8, 26 CrossRef CAS PubMed.
- J. Shi and J. P. Schneider, Angew. Chem., Int. Ed., 2019, 58, 13706 CrossRef CAS PubMed.
- A. Tanaka, Y. Fukuoka, Y. Morimoto, T. Honjo, D. Koda, M. Goto and T. Maruyama, J. Am. Chem. Soc., 2015, 137, 770–775 CrossRef CAS PubMed.
- M. Zhang, Y. Guan, Z. Dang, P. Zhang, Z. Zheng, L. Chen, W. Kuang, C. Wang and G. Liang, Sci. Adv., 2020, 6, eaba3190 CrossRef CAS PubMed.
- X. Yang, B. Wu, J. Zhou, H. Lu, H. Zhang, F. Huang and H. Wang, Nano Lett., 2022, 22, 7588–7596 CrossRef CAS PubMed.
- X. Chen, R. Kang, G. Kroemer and D. Tang, Nat. Rev. Clin. Oncol., 2021, 18, 280 CrossRef CAS PubMed.
- Z. Shen, J. Song, B. C. Yung, Z. Zhou, A. Wu and X. Chen, Adv. Mater., 2018, 30, 1704007 CrossRef PubMed.
- H. Wang, Y. Cheng, C. Mao, S. Liu, D. Xiao, J. Huang and Y. Tao, Mol. Ther., 2021, 29, 2185–2208 CrossRef CAS PubMed.
- D. V. Krysko, A. D. Garg, A. Kaczmarek, O. Krysko, P. Agostinis and P. Vandenabeele, Nat. Rev. Cancer, 2012, 12, 860–875 CrossRef CAS PubMed.
- L. Wang, R. Guan, L. Xie, X. Liao, K. Xiong, T. W. Rees, Y. Chen, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2021, 60, 4657–4665 CrossRef CAS PubMed.
- M. Sabbatini, I. Zanellato, M. Ravera, E. Gabano, E. Perin, B. Rangone and D. Osella, J. Med. Chem., 2019, 62, 3395–3406 CrossRef CAS PubMed.
- M. Li, J. Kim, H. Rha, S. Son, M. S. Levine, Y. Xu, J. L. Sessler and J. S. Kim, J. Am. Chem. Soc., 2023, 145, 6007–6023 CrossRef CAS PubMed.
- M. Zhang, X. Jin, M. Gao, Y. Zhang and B. Z. Tang, Adv. Mater., 2022, 34, 2205701 CrossRef CAS PubMed.
- B. Englinger, C. Pirker, P. Heffeter, A. Terenzi, C. R. Kowol, B. K. Keppler and W. Berger, Chem. Rev., 2019, 119, 1519–1624 CrossRef CAS PubMed.
- R. Kuai, W. Yuan, S. Son, J. Nam, Y. Xu, Y. Fan, A. Schwendeman and J. J. Moon, Sci. Adv., 2018, 4, eaao1736 CrossRef PubMed.
- H. Ma, Y. Lu, Z. Huang, S. Long, J. Cao, Z. Zhang, X. Zhou, C. Shi, W. Sun, J. Du, J. Fan and X. Peng, J. Am. Chem. Soc., 2022, 144, 3477 CrossRef CAS PubMed.
- N. Toupin, M. K. Herroon, R. P. Thummel, C. Turro, I. Podgorski, H. Gibson and J. J. Kodanko, Chem. – Eur. J., 2022, 28, e202104430 CrossRef CAS PubMed.
- Z. Gao, S. Jia, H. Ou, Y. Hong, K. Shan, X. Kong, Z. Wang, G. Feng and D. Ding, Angew. Chem., Int. Ed., 2022, 61, e202209793 CrossRef CAS PubMed.
- P. Zhang, Z. Zhou, W. Long, Y. Yan, Y. Li, T. Fu, Y. Liu, Z. Zhao, W. Tan and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2202255119 CrossRef CAS PubMed.
- H.-Y. Lin, Y.-T. Wang, X. Shi, H.-B. Yang and L. Xu, Chem. Soc. Rev., 2023, 52, 1129–1154 RSC.
- C. Li, Y. Pang, Y. Xu, M. Lu, L. Tu, Q. Li, A. Sharma, Z. Guo, X. Li and Y. Sun, Chem. Soc. Rev., 2023, 52, 5340–5342 RSC.
- K.-X. Teng, L.-Y. Niu, N. Xie and Q.-Z. Yang, Nat. Commun., 2022, 13, 6179 CrossRef CAS PubMed.
- P. Jeyakkumar, Y. Liang, M. Guo, S. Lu, D. Xu, X. Li, B. Guo, G. He, D. Chu and M. Zhang, Angew. Chem., Int. Ed., 2020, 59, 15199–15203 CrossRef CAS PubMed.
- A. López-Moreno, S. Ibáñez, S. Moreno-Da Silva, L. Ruiz-González, N. M. Sabanés, E. Peris and E. M. Pérez, Angew. Chem., Int. Ed., 2022, 61, e202208189 CrossRef PubMed.
- L. Tu, C. Li, Q. Ding, A. Sharma, M. Li, J. Li, J. S. Kim and Y. Sun, J. Am. Chem. Soc., 2024, 146, 8991 CrossRef CAS PubMed.
- X. Liu, Y. Zhou, W. Xie, S. Liu, Q. Zhao and W. Huang, Small Methods, 2020, 4, 2000566 CrossRef CAS.
- K. S. Allemailem, A. Almatroudi, F. Alrumaihi, S. A. Almatroodi, M. O. Alkurbi, G. T. Basfar, A. H. Rahmani and A. A. Khan, Int. J. Nanomedicine, 2021, 16, 5065–5098 CrossRef PubMed.
- C. M. Baehr, L. Zhang, Y. Wu, A. Domokos, W. Xiao, L. Wang and K. S. Lam, Biomaterials, 2021, 277, 121078 CrossRef CAS PubMed.
- J. Wang, L. Hu, H. Zhang, Y. Fang, T. Wang and H. Wang, Adv. Mater., 2022, 34, 2104704 CrossRef CAS PubMed.
- C. Wu, C. Wang, T. Zhang, G. Gao, M. Wei, Y. Chen, X. Li, F. Wang and G. Liang, Adv. Healthcare Mater., 2022, 11, 2101346 CrossRef CAS PubMed.
- S. Ji, J. Li, X. Duan, J. Zhang, Y. Zhang, M. Song, S. Li, H. Chen and D. Ding, Angew. Chem., Int. Ed., 2021, 60, 26994–27004 CrossRef CAS PubMed.
- W. Ji and J. L. Li, ACS Nano, 2025, 19, 12208–12221 CrossRef PubMed.
- L. Hu, Y. Li, X. Lin, Y. Huo, H. Zhang and H. Wang, Angew. Chem., Int. Ed., 2021, 60, 21807–21816 CrossRef CAS PubMed.
- H. Cabral, K. Miyata, K. Osada and K. Kataoka, Chem. Rev., 2018, 118, 6844–6892 CrossRef CAS PubMed.
- Y. P. Chen, A. T. C. Chan, Q. T. Le, P. Blanchard, Y. Sun and J. Ma, Lancet, 2019, 394, 64–80 CrossRef PubMed.
- R. Jiromaru, T. Nakagawa and R. Yasumatsu, Cancer Manag. Res., 2022, 14, 2681–2689 CrossRef CAS PubMed.
- K. C. W. Wong, E. P. Hui, K.-W. Lo, W. K. J. Lam, D. Johnson, L. Li, Q. Tao, K. C. A. Chan, K.-F. To, A. D. King, B. B. Y. Ma and A. T. C. Chan, Nat. Rev. Clin. Oncol., 2021, 18, 679–695 Search PubMed.
- H. Li, Y. Feng, Q. Luo, Z. Li, X. Li, H. Gan, Z. Gu, Q. Gong and K. Luo, Theranostics, 2023, 13, 5386–5417 CrossRef CAS PubMed.
- D. Jin, Y. Zhu, M. Liu, W. Yu, J. Yu, X. Zheng, L. Wang, Y. Wu, K. Wei, J. Cheng and Y. Liu, BME Front., 2023, 4, 0015 CrossRef CAS PubMed.
- C. Li, G. Jiang, J. Yu, W. Ji, L. Liu, P. Zhang, J. Du, C. Zhan, J. Wang and B. Z. Tang, Adv. Mater., 2023, 35, 2208229 CrossRef CAS PubMed.
- Z. Li, S. Yang, H. Xiao, Q. Kang, N. Li, G. L. Wu, S. Tan, W. Wang, Q. Fu, X. Tang, J. Zhou, Y. Huang, G. Chen, X. Tan and Q. Yang, Bioconjugate Chem., 2024, 35, 1015–1023 CrossRef CAS PubMed.
- G.-l Wu, B. Sun, Y. He, X. Tan, Q. Pan, S. Yang, N. Li, M. Wang, P. Wu, F. Liu, H. Xiao, L. Tang, S. Zhu and Q. Yang, Chem. Eng. J., 2023, 463, 142372 Search PubMed.
- Q. Sun, J. Yang, Q. Wu, W. Shen, Y. Yang and D. Yin, ACS Appl. Mater. Interfaces, 2024, 16, 127–141 Search PubMed.
- X. Jin, J. Tang, X. Qiu, X. Nie, S. Ou, G. Wu, R. Zhang and J. Zhu, Cell Death Discov., 2024, 10, 45 CrossRef PubMed.
- B. R. Stockwell, J. P. Friedmann Angeli, H. Bayir, A. I. Bush, M. Conrad, S. J. Dixon, S. Fulda, S. Gascón, S. K. Hatzios, V. E. Kagan, K. Noel, X. Jiang, A. Linkermann, M. E. Murphy, M. Overholtzer, A. Oyagi, G. C. Pagnussat, J. Park, Q. Ran, C. S. Rosenfeld, K. Salnikow, D. Tang, F. M. Torti, S. V. Torti, S. Toyokuni, K. A. Woerpel and D. D. Zhang, Cell, 2017, 171, 273–285 CrossRef CAS PubMed.
- X. Jiang, B. R. Stockwell and M. Conrad, Nat. Rev. Mol. Cell Biol., 2021, 22, 266–282 CrossRef PubMed.
- W. Li, S. Yin, Y. Shen, H. Li, L. Yuan and X. B. Zhang, J. Am. Chem. Soc., 2023, 145, 3736–3747 CrossRef CAS PubMed.
- P. You, F. Lu, C. Ouyang, J. Yu, J. Gonzalez-Garcia, J. Song, W. Ni, J. Wang, C. Yin and C. Q. Zhou, ACS Appl. Mater. Interfaces, 2024, 16, 46066–46078 CrossRef CAS PubMed.
- J. Liu, Y. Yan, Y. Zhang, X. Pan, H. Xia, J. Zhou, F. Wan, X. Huang, W. Zhang, Q. Zhang, B. Chen and Y. Wang, J. Am. Chem. Soc., 2024, 146, 34568–34582 CrossRef CAS PubMed.
- S. Lee, S. Kang and W. J. Kim, ACS Nano, 2025, 19, 7742–7754 CrossRef CAS PubMed.
- K. Zheng, X. Ouyang, J. Li, Y. Cao and S. Peng, J. Am. Chem. Soc., 2025, 147, 34742–34757 Search PubMed.
- S. Chen, B. Li, Y. Yue, Z. Li, L. Qiao, G. Qi, Y. Ping and B. Liu, Adv. Mater., 2024, 36, 2404296 Search PubMed.
- M. Tang, B. Chen, H. Xia, M. Pan, R. Zhao, J. Zhou, Q. Yin, F. Wan, Y. Yan, C. Fu, L. Zhong, Q. Zhang and Y. Wang, Nat. Commun., 2023, 14, 5888 Search PubMed.
- D.-Y. Hou, M.-D. Wang, N.-Y. Zhang, S. Xu, Z.-J. Wang, X.-J. Hu, G.-T. Lv, J.-Q. Wang, M.-Y. Lv, L. Yi, L. Wang, D.-B. Cheng, T. Sun, H. Wang and W. Xu, Nano Lett., 2022, 22, 3983–3992 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 Search PubMed.
- B. Li, A. Y. Jiang, I. Raji, C. Atyeo, T. M. Raimondo, A. G. R. Gordon, L. H. Rhym, T. Samad, C. MacIsaac, J. Witten, H. Mughal, T. M. Chicz, Y. Xu, R. P. McNamara, S. Bhatia, G. Alter, R. Langer and D. G. Anderson, Nat. Biomed. Eng., 2025, 9, 167–184 Search PubMed.
- N. Luo, J. K. Weber, S. Wang, B. Luan, H. Yue, X. Xi, J. Du, Z. Yang, W. Wei, R. Zhou and G. Ma, Nat. Commun., 2017, 8, 14537 Search PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discov., 2021, 20, 101–124 Search PubMed.
- T. Sun, Y. S. Zhang, B. Pang, D. C. Hyun, M. Yang and Y. Xia, Angew. Chem., Int. Ed., 2014, 53, 12320–12364 Search PubMed.
- E. P. Stater, A. Y. Sonay, C. Hart and J. Grimm, Nat. Nanotechnol., 2021, 16, 1180–1194 CrossRef CAS PubMed.
- X. Sun, Y. Zhang, J. Li, K. S. Park, K. Han, X. Zhou, Y. Xu, J. Nam, J. Xu, X. Shi, L. Wei, Y. L. Lei and J. J. Moon, Nat. Nanotechnol., 2021, 16, 1260–1270 Search PubMed.
- X. Wang, J. Wilhelm, W. Li, S. Li, Z. Wang, G. Huang, J. Wang, H. Tang, S. Khorsandi, Z. Sun, B. Evers and J. Gao, Nat. Commun., 2020, 11, 5828 Search PubMed.
- N. H. T. Petersen, O. D. Olsen, L. Groth-Pedersen, A. M. Ellegaard, M. Bilgin, S. Redmer, M. S. Ostenfeld, D. Ulanet, T. H. Dovmark, A. Lønborg, S. D. Vindeløv, D. Hanahan, C. Arenz, C. S. Ejsing, T. Kirkegaard, M. Rohde, J. Nylandsted and M. Jäättelä, Cancer Cell, 2013, 24, 379–393 CrossRef CAS PubMed.
- S.-y Zhu, R.-q Yao, Y.-x Li, P.-y Zhao, C. Ren, X.-h Du and Y.-m Yao, Cell Death Dis., 2020, 11, 817 Search PubMed.
- Y. Xing, J. Yang, A. Peng, Y. Qian, Y. Liu, P. Pan and Q. Liu, Adv. Mater., 2024, 36, 2412730 CrossRef CAS PubMed.
- M. Liu, M. Yang, X. Wan, Z. Tang, L. Jiang and S. Wang, Adv. Mater., 2023, 35, 2208995 Search PubMed.
- B. Kaupbayeva and A. J. Russell, Prog. Polym. Sci., 2020, 101, 101194 Search PubMed.
- Q. Zhang, M. Li, C. Zhu, G. Nurumbetov, Z. Li, P. Wilson, K. Kempe and D. M. Haddleton, J. Am. Chem. Soc., 2015, 137, 9344–9353 Search PubMed.
- D. Yadav and H. K. Dewangan, J. Biomater. Sci. Polym. Ed., 2021, 32, 266–280 CrossRef CAS PubMed.
- R. Falatach, C. McGlone, M. S. Al-Abdul-Wahid, S. Averick, R. C. Page, J. A. Berberich and D. Konkolewicz, Chem. Commun., 2015, 51, 5343–5346 Search PubMed.
- L. Papadimitriou, A. Theodorou, M. Papageorgiou, E. Voutyritsa, A. Papagiannaki, K. Velonia and A. Ranella, J. Drug Deliv. Sci. Technol., 2022, 75, 103591 Search PubMed.
- A. Ahmad, J. M. Khan and S. Haque, Biochimie, 2019, 160, 61–75 CrossRef CAS PubMed.
- D. Pei and M. Buyanova, Bioconjugate Chem., 2019, 30, 273–283 CrossRef CAS PubMed.
- L. I. Selby, C. M. Cortez-Jugo, G. K. Such and A. P. R. Johnston, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2017, 9, e1452 Search PubMed.
- A. K. Varkouhi, M. Scholte, G. Storm and H. J. Haisma, J. Control. Release, 2011, 151, 220–228 CrossRef CAS PubMed.
- R. Wang, C. Yin, C. Liu, Y. Sun, P. Xiao, J. Li, S. Yang, W. Wu and X. Jiang, J. Am. Chem. Soc., 2021, 143, 20927–20938 CrossRef CAS PubMed.
- Y. Li, Y. Wu, Z. Fang, Y. Zhang, H. Ding, L. Ren, L. Zhang, Q. Gong, Z. Gu and K. Luo, Adv. Mater., 2024, 36, 2307263 CrossRef CAS PubMed.
- S. Shao, Q. Zhou, J. Si, J. Tang, X. Liu, M. Wang, J. Gao, K. Wang, R. Xu and Y. Shen, Nat. Biomed. Eng., 2017, 1, 745–757 CrossRef CAS PubMed.
- Z. Wang, Y. Guo, Y. Fan, J. Chen, H. Wang, M. Shen and X. Shi, Adv. Mater., 2022, 34, 2107009 CrossRef CAS PubMed.
- K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062–5064 CrossRef CAS PubMed.
- D. Zhong, X. Hou, D. Pan, Z. Li, Q. Gong and K. Luo, Adv. Mater., 2024, 36, 2403588 CrossRef CAS PubMed.
- M. Mukhopadhyay, Nat. Methods, 2021, 18, 1274 CrossRef CAS PubMed.
- K. Cui, K. Wang and Z. Huang, J. Exp. Clin. Cancer Res., 2024, 43, 315 CrossRef PubMed.
- Z. Zhou and Z. R. Lu, Adv. Drug Deliv. Rev., 2017, 113, 24–48 CrossRef CAS PubMed.
- J. Zhang, Y. Hu, X. Wen, Z. Yang, Z. Wang, Z. Feng, H. Bai, Q. Xue, Y. Miao, T. Tian, P. Zheng, J. Zhang, J. Li, L. Qiu, J. J. Xu and D. Ye, Nat. Nanotechnol., 2025, 20, 563–574 CrossRef CAS PubMed.
- H. Zhong, G. Chen, T. Li, J. Huang, M. Lin, B. Li, Z. Xiao and X. Shuai, Nano Lett., 2023, 23, 5083–5091 CrossRef CAS PubMed.
- B. Ji, M. Wei and B. Yang, Theranostics, 2022, 12, 434–458 CrossRef CAS PubMed.
- Y. Zhang, Y. Liao, Q. Tang, J. Lin and P. Huang, Angew. Chem., Int. Ed., 2021, 60, 10647–10653 CrossRef CAS PubMed.
- M. Zha, G. Yang, Y. Li, C. Zhang, B. Li and K. Li, Adv. Healthcare Mater., 2021, 10, 2101066 CrossRef CAS PubMed.
- Y. Zhao, Z. Chen, Q. Li, X. Cao, Q. Huang, L. Shi and Y. Liu, Adv. Funct. Mater., 2022, 32, 2209711 CrossRef CAS.
- S. Wu, C. Zhu, D. Tang, Q. P. Dou, J. Shen and X. Chen, Biomark. Res., 2021, 9, 82 CrossRef PubMed.
- C.-H. Hsieh, H.-C. Hsieh, F.-H. Shih, P.-W. Wang, L.-X. Yang, D.-B. Shieh and Y.-C. Wang, Theranostics, 2021, 11, 7072–7091 CrossRef CAS PubMed.
- P. Koppula, G. Lei, Y. Zhang, Y. Yan, C. Mao, L. Kondiparthi, J. Shi, X. Liu, A. Horbath, M. Das, W. Li, M. V. Poyurovsky, K. Olszewski and B. Gan, Nat. Commun., 2022, 13, 2206 CrossRef CAS PubMed.
- F. Fu, W. Wang, L. Wu, W. Wang, Z. Huang, Y. Huang, C. Wu and X. Pan, ACS Nano, 2023, 17, 5486–5502 CrossRef CAS PubMed.
- K. Luo, L. Yang, C. Yan, Y. Zhao, Q. Li, X. Liu, L. Xie, Q. Sun and X. Li, Small, 2023, 19, 2302834 CrossRef CAS PubMed.
- Y. Yao, Y. Hu, X. Meng, F. Feng, F. Xu, G. Wang, H. Yu and J. Li, Pharm. Sci. Adv., 2024, 2, 100046 CrossRef CAS PubMed.
- G. Wang, H. Lu, Y. Pan, Y. Qi and Y. Huang, Small, 2024, 20, 2406182 CrossRef CAS PubMed.
- W. Shi, Y. Xi, X. Sheng, S. Das, D. He, K. Collins, Y. Hu and M. G. Finn, Adv. Funct. Mater., 2025, 35, 2409796 CrossRef CAS PubMed.
- S. Xie, Y. Li, W. Cao, J. Peng, K. Huang, J. Meng and X. Li, Adv. Healthcare Mater., 2024, 13, 2303671 CrossRef CAS PubMed.
- X. Zeng, M. Zuo, J. Yuan, G. Chen, S. Liu, C. Ou, Q. Chen, C. Wei, D. Yu and D. Cheng, Adv. Funct. Mater., 2025, 35, 2406122 CrossRef CAS.
- C. D. DiNardo, H. P. Erba, S. D. Freeman and A. H. Wei, Lancet, 2023, 401, 2073–2086 CrossRef CAS PubMed.
- J. Huuhtanen, D. Bhattacharya, T. Lonnberg, M. Kankainen, C. Kerr, J. Theodoropoulos, H. Rajala, C. Gurnari, T. Kasanen, T. Braun, A. Teramo, R. Zambello, M. Herling, F. Ishida, T. Kawakami, M. Salmi, T. Loughran, J. P. Maciejewski, H. Lahdesmaki, T. Kelkka and S. Mustjoki, Nat. Commun., 2022, 13, 1981 CrossRef CAS PubMed.
- M. Hullo, C. Mathe, N. Fonknechten, C. Lacrouts, G. Piton, J. Noireaux, S. Chevillard, A. Campalans and E. Bourneuf, ACS Nano, 2025, 19, 29160–29180 CrossRef CAS PubMed.
- Y. Liang, Y. Wang, L. Wang, Z. Liang, D. Li, X. Xu, Y. Chen, X. Yang, H. Zhang and H. Niu, Bioact. Mater., 2021, 6, 433–446 CAS.
- K. Y. Choi, H. S. Han, E. S. Lee, J. M. Shin, B. D. Almquist, D. S. Lee and J. H. Park, Adv. Mater., 2019, 31, 1803549 CrossRef PubMed.
- S. Xu, T. Wang, X. Hu, H. Deng, Y. Zhang, L. Xu, Y. Zeng, J. Yu, W. Zhang, L. Wang and H. Xu, Sci. Adv., 2025, 11, eado3923 CrossRef CAS PubMed.
- X. Shi, S. H. P. Sung, J. H. C. Chau, Y. Li, Z. Liu, R. T. K. Kwok, J. Liu, P. Xiao, J. Zhang, B. Liu, J. W. Y. Lam and B. Z. Tang, Small Methods, 2020, 4, 2000046 CrossRef CAS.
- K. Wu, J. Liu, X. Zhang, Z. Chao, Y. Fang, Y. Zhu, Y. Liu, X. Zhang, Q. Wang, H. Ju and Y. Liu, Biomaterials, 2025, 315, 122918 CrossRef CAS PubMed.
- X. Yang, X. Wang, X. Zhang, J. Zhang, J. W. Y. Lam, H. Sun, J. Yang, Y. Liang and B. Z. Tang, Adv. Mater., 2024, 36, 2402182 CrossRef CAS PubMed.
- M. A. Freitas-Cortez, F. Masrorpour, H. Jiang, I. Mahmud, Y. Lu, A. Huang, L. K. Duong, Q. Wang, T. A. Voss, C. S. Kettlun Leyton, B. Wei, W. K. Chan, K. Lin, J. Zhang, E. Tsouko, S. Ganjoo, H. B. Barsoumian, T. S. Riad, Y. Hu, C. Leuschner, N. Puebla-Osorio, J. Wang, J. Hu, M. A. Davies, V. K. Puduvalli, C. Billon, T. P. Burris, P. L. Lorenzi, B. Gan and J. W. Welsh, Mol. Cancer, 2025, 24, 40 CrossRef CAS PubMed.
- F. Zheng, Y. Wang, Q. Zhang, Q. Chen, C. L. Liang, H. Liu, F. Qiu, Y. Chen, H. Huang, W. Lu and Z. Dai, Front. Pharmacol., 2023, 14, 1145407 CrossRef CAS PubMed.
- B. Li, C. Ayala-Orozco, T. Si, L. Zhou, Z. Wang, A. A. Martí and J. M. Tour, Adv. Sci., 2024, 11, 2405965 CrossRef CAS PubMed.
- X. Wei, Y. Li, H. Chen, R. Gao, P. Ning, Y. Wang, W. Huang, E. Chen, L. Fang, X. Guo, C. Lv and Y. Cheng, Adv. Sci., 2024, 11, 2302093 CrossRef CAS PubMed.
- S. Wang, Y. Zou, L. Hu and Y. Lv, Acta. Biomater., 2025, 191, 369–385 CrossRef CAS PubMed.
- J. O. Law, C. M. Jones, T. Stevenson, T. A. Williamson, M. S. Turner, H. Kusumaatmaja and S. N. Grellscheid, Sci. Adv., 2023, 9, eadg0432 CrossRef CAS PubMed.
- C. Lim and W. C. Blocher McTigue, ACS Biomater. Sci. Eng., 2024, 10, 6766–6789 CrossRef CAS PubMed.
- C. Yin, C. Wu, X. Yu, Y. Zhu, B. Wu, Y. Wang and L. Tian, Adv. Sci., 2025, e18312 Search PubMed.
- P. Zhao, J. Guo, T. Jiang, X. Xu, S. Chen, Z. Li, J. Xu, G. Li and L. Bian, Mater. Today, 2023, 66, 26–35 Search PubMed.
- T. Liang, Y. Dong, I. Cheng, P. Wen, F. Li, F. Liu, Q. Wu, E. Ren, P. Liu, H. Li and Z. Gu, Nat. Biomed. Eng., 2024, 8, 1469–1482 Search PubMed.
- F. Xie, X. Zhou, Y. Ran, R. Li, J. Zou, S. Wan, P. Su, X. Meng, H. Yan, H. Lu, H. Ru, H. Hu, Z. Mao, B. Yang, F. Zhou and L. Zhang, Nature, 2025, 638, 1112–1121 CrossRef CAS PubMed.
- Y. Liu, X. Qian, C. Ran, L. Li, T. Fu, D. Su, S. Xie and W. Tan, ACS Nano, 2023, 17, 6150–6164 CrossRef CAS PubMed.
- Y. Chen, F. Liu, S. Pal and Q. Hu, Chem. Soc. Rev., 2024, 53, 9582–9608 Search PubMed.
- L. Zhao, J. Zhao, K. Zhong, A. Tong and D. Jia, Signal Transduct. Target. Ther., 2022, 7, 113 CrossRef CAS PubMed.
- J. Lin, J. Jin, Y. Shen, L. Zhang, G. Gong, H. Bian, H. Chen, D. G. Nagle, Y. Wu, W. Zhang and X. Luan, Theranostics, 2021, 11, 8337–8349 CrossRef CAS PubMed.
- M. Cui, D. Zhang, X. Zheng, H. Zhai, M. Xie, Q. Fan, L. Wang, C. Fan and J. Chao, J. Am. Chem. Soc., 2024, 146, 29609–29620 CrossRef CAS PubMed.
- Y. Xiao, Z. He, W. Li, D. Chen, X. Niu, X. Yang, W. Zeng, M. Wang, Y. Qian, Y. Su, F. Luo, G. Chen, J. Liu, X. Sui, X. Zhou and Y. Gao, Nat. Commun., 2025, 16, 1388 CrossRef CAS PubMed.
- J. Luo, Q. Gao, K. Tan, S. Zhang, W. Shi, L. Luo, Z. Li, G. E. Khedr, J. Chen, Y. Xu, M. Luo, Q. Xing and J. Geng, J. Am. Chem. Soc., 2024, 146, 17728–17737 Search PubMed.
- B. Zhang, R. K. Brahma, L. Zhu, J. Feng, S. Hu, L. Qian, S. Du, S. Q. Yao and J. Ge, J. Am. Chem. Soc., 2023, 145, 24272–24283 Search PubMed.
- Q. Duan, H.-R. Jia, W. Chen, C. Qin, K. Zhang, F. Jia, T. Fu, Y. Wei, M. Fan, Q. Wu and W. Tan, Adv. Sci., 2024, 11, 2308924 CrossRef CAS PubMed.
- D. Kim, G. Yang, C. Lim, G. Park, J. Lee, Y. Sim and J. H. Ryu, Adv. Sci., 2025, 12, 2503134 CrossRef CAS PubMed.
- S. He, W. Huang, Y. Zhu, F. Gao, Y. Fang, J. Zhu, Y. Bao, G. Dong and C. Sheng, Angew. Chem., Int. Ed., 2025, 64, e202511467 CrossRef CAS PubMed.
- J. Zheng, W. He, J. Li, X. Feng, Y. Li, B. Cheng, Y. Zhou, M. Li, K. Liu, X. Shao, J. Zhang, H. Li, L. Chen and L. Fang, J. Am. Chem. Soc., 2022, 144, 21831–21836 CrossRef CAS PubMed.
- R. Zhang, C. Yang, X. Gao, Z. He, D. K. Ji and W. Tan, J. Am. Chem. Soc., 2025, 147, 20989–21002 CrossRef PubMed.
- K. Wang, K. Wang, C. Wang, H. Yang, G. Zhou, P. Ji, X. Sun, G. Chen, X. Fan, K. Hu, J. Yi, H. Zhang and R. Wang, J. Am. Chem. Soc., 2025, 147, 40751–40762 CrossRef CAS PubMed.
- X. Chen, T. Wu, Y. Chen, H. Wu, W. Kang, N. Wang, Q. You, X. Guo and Z. Jiang, Angew. Chem., Int. Ed., 2025, 64, e202506618 CrossRef CAS PubMed.
- D. Pechalrieu, V. Nogueira, N. Qiu, B. Racioppo, D. Abegg, N. Hay and A. Adibekian, J. Am. Chem. Soc., 2025, 147, 43124–43138 CrossRef CAS PubMed.
- H. Zhu, G. Zhang, Y. Ning, Y. Zhang, Y. Wu, J. Li, H. Zhang, C. Yang and Z. Zhu, ACS Nano, 2025, 19, 24895–24903 Search PubMed.
- M. Cui, D. Zhang, X. Zheng, H. Zhai, M. Xie, Q. Fan, L. Wang, C. Fan and J. Chao, J. Am. Chem. Soc., 2025, 146, 29609–29620 CrossRef PubMed.
- D. A. Nalawansha, G. Mazis, G. Husemoen, K. S. Ashton, W. Deng, R. P. Wurz, A. T. Tran, B. A. Lanman, J. Xie, R. G. Guenette, S. Li, C. E. Smith, S. Archunan, M. K. Agnihotram, A. Sadhukhan, R. Kapoor, C. Wilde, S. Koirala, E. M. F. De Sousa and P. R. Potts, Nat. Commun., 2025, 16, 7812 CrossRef CAS PubMed.
- Y. Xing, J. Li, L. Wang, Z. Zhu, J. Yan, Y. Liu and Q. Liu, Adv. Mater., 2025, 37, 2417942 CrossRef CAS PubMed.
- J. Qi, S. Jia, X. Kang, X. Wu, Y. Hong, K. Shan, X. Kong, Z. Wang and D. Ding, Adv. Mater., 2022, 34, 2203309 Search PubMed.
- A. Frei, A. D. Verderosa, A. G. Elliott, J. Zuegg and M. A. T. Blaskovich, Nat. Rev. Chem., 2023, 7, 202–224 CrossRef CAS PubMed.
- M. Xie, M. Gao, Y. Yun, M. Malmsten, V. M. Rotello, R. Zboril, O. Akhavan, A. Kraskouski, J. Amalraj, X. Cai, J. Lu, H. Zheng and R. Li, Angew. Chem., Int. Ed., 2023, 62, 202217345 CrossRef PubMed.
- A. Upadhayay, J. Ling, D. Pal, Y. Xie, F. F. Ping and A. Kumar, Drug Resist. Update., 2023, 66, 100890 CrossRef CAS PubMed.
- P. Wang, B. Wu, M. Li, Y. Song, C. Chen, G. Feng, D. Mao, F. Hu and B. Liu, ACS Nano, 2023, 17, 10365–10375 CrossRef CAS PubMed.
- R. Zhou, H. Yu, T. Sheng, Y. Wu, Y. Chen, J. You, Y. Yang, B. Luo, S. Zhao, Y. Zheng, H. Li, Y. Zhang, Y. Guo, Z. Gu and J. Yu, Adv. Mater., 2024, 36, 2401667 Search PubMed.
- M. W. Braun, H. Abdelhakim, M. Li, S. Hyter, Z. Pessetto, D. C. Koestler, H. B. Pathak, N. Dunavin and A. K. Godwin, J. Immunother. Cancer, 2020, 8, e000706 Search PubMed.
- G. Parisi, J. D. Saco, F. B. Salazar, J. Tsoi, P. Krystofinski, C. Puig-Saus, R. Zhang, J. Zhou, G. C. Cheung-Lau, A. J. Garcia, C. S. Grasso, R. Tavaré, S. Hu-Lieskovan, S. Mackay, J. Zalevsky, C. Bernatchez, A. Diab, A. M. Wu, B. Comin-Anduix, D. Charych and A. Ribas, Nat. Commun., 2020, 11, 660 CrossRef CAS PubMed.
- Q. Zhao, Z. Gong, Z. Li, J. Wang, J. Zhang, Z. Zhao, P. Zhang, S. Zheng, R. J. Miron, Q. Yuan and Y. Zhang, Adv. Mater., 2021, 33, 2100616 Search PubMed.
- L. Gronbach, C. Wolff, K. Klinghammer, J. Stellmacher, P. Jurmeister, U. Alexiev, M. Schäfer-Korting, I. Tinhofer, U. Keilholz and C. Zoschke, Biomaterials, 2020, 258, 120277 CrossRef CAS PubMed.
- Z. Chang, Z. Liang, Y. Lan, J. Huang, L. Feng and J. Xu, ACS Appl. Mater. Interfaces, 2025, 17, 11688–11703 CrossRef CAS PubMed.
- L. Guan, D. K. Nambiar, H. Cao, V. Viswanathan, S. Kwok, A. B. Hui, Y. Hou, R. Hildebrand, R. von Eyben, B. J. Holmes, J. Zhao, C. S. Kong, N. Wamsley, W. Zhang, M. B. Major, S. W. Seol, J. B. Sunwoo, D. N. Hayes, M. Diehn and Q. T. Le, Cancer Res., 2023, 83, 861–874 Search PubMed.
- A. Picca, J. Faitg, J. Auwerx, L. Ferrucci and D. D’Amico, Nat. Metab., 2023, 5, 2047–2061 Search PubMed.
- P. Mavroeidi, F. Arvanitaki, M. Vetsi, S. Becker, D. Vlachakis, P. H. Jensen, L. Stefanis and M. Xilouri, Autophagy, 2022, 18, 2104–2133 Search PubMed.
- J. Chen, Z. Zhu, Q. Pan, Y. Bai, M. Yu and Y. Zhou, Adv. Funct. Mater., 2023, 33, 2300235 Search PubMed.
- G. C. Leonardi, J. F. Gainor, R. S. Azimi, J. Riess, H. Rizvi, M. D. Hellmann and M. M. Awad, J. Clin. Oncol., 2017, 35, 9081 Search PubMed.
- X. Lin, K. Kang, P. Chen, Z. Zeng, G. Li, W. Xiong, M. Yi and B. Xiang, Mol. Cancer, 2024, 23, 108 Search PubMed.
- H. Lu, P. Guan, S. Xu, Y. Han and Z. Liu, ACS Nano, 2024, 18, 23553–23565 Search PubMed.
- R. V. Benjaminsen, M. A. Mattebjerg, J. R. Henriksen, S. M. Moghimi and T. L. Andresen, Mol. Ther., 2013, 21, 149–157 Search PubMed.
- S. Roy, D. Zhu, W. J. Parak and N. Feliu, ACS Nano, 2020, 14, 8012–8023 Search PubMed.
- X. Yang, C. Zhang, M. Song, Z. Zhang, J. Zhou, H. Zhang and Y. Ding, ACS Nano, 2023, 17, 23568–23583 Search PubMed.
- T. He, J. Wen, W. Wang, Z. Hu, C. Ling, Z. Zhao, Y. Cheng, Y. C. Chang, M. Xu, Z. Jin, L. Amer, L. Sasi, L. Fu, N. F. Steinmetz, T. M. Rana, P. Wu and J. V. Jokerst, Adv. Mater., 2024, 36, 2307679 Search PubMed.
- M. Starck, J. D. Fradgley, R. Pal, J. M. Zwier, L. Lamarque and D. Parker, Chem. – Eur. J., 2021, 27, 766–777 Search PubMed.
- J. D. Fradgley, M. Starck, M. Laget, E. Bourrier, E. Dupuis, L. Lamarque, E. Trinquet, J. M. Zwier and D. Parker, Chem. Commun., 2021, 57, 5814–5817 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |