
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic asymmetric [4+2] cycloadditions of unsaturated hydrocarbons by transition metal catalysis and photocatalysis
Jun-Xiong
He
and
Quan
Cai
 *
*
Department of Chemistry, Research Center for Molecular Recognition and Synthesis, State Key Laboratory of Green Chemical Synthesis and Conversion, Fudan University, Shanghai 200433, China. E-mail: quan_cai@fudan.edu.cn
First published on 13th April 2026
Abstract
The enantioselective Diels–Alder reaction is arguably one of the most efficient and straightforward approaches for the construction of chiral six-membered carbocycles. However, the generality of this conventional reaction is limited by restrictive electronic requirements and substitution patterns according to the classic Hoffmann–Woodward rules. To circumvent these limitations, several innovative approaches have emerged in recent years. Using these approaches, various types of electronic mismatched [4+2] cycloadditions of unsaturated carbons, such as alkenes, alkynes, and allenes, have been realized. This review will summarize these advancements in this rapidly growing area within the past three decades, including the transition metal-catalyzed asymmetric [4+2] cycloadditions via the oxidative cyclometallation and reductive elimination pathway, catalytic asymmetric [4+2] cycloadditions via the LUMO activation by π-acids and the HOMO activation by π-bases, as well as catalytic asymmetric radical-mediated [4+2] cycloadditions under light irradiation.
 Jun-Xiong He | Jun-Xiong He received his BSc degree from Jiangxi Normal University in 2018 and MSc from East China Normal University in 2021 under the supervision of Prof. Jian Zhou. Then, he earned his PhD degree in chemistry from Fudan University in 2025 under the supervision of Prof. Quan Cai. He is now working as a postdoctoral fellow in the lab of Prof. Shu-Li You at the Shanghai Institute of Organic Chemistry. |
 Quan Cai | Quan Cai received his BSc degree from the East China University of Science and Technology (2007) and his PhD degree from the Shanghai Institute of Organic Chemistry with Prof. Shu-Li You (2012). After postdoctoral work with Prof. K. C. Nicolaou at the Scripps Research Institute and Rice University (2012–2016) and with Prof. Nathanael Gray at Dana-Farber Cancer Institute, Harvard Medical School (2016–2017), he joined the Department of Chemistry at Fudan University as a tenure-track professor. In 2022, he was promoted to a full professor. His current research focuses on asymmetric catalysis, transition-metal catalysis, and total synthesis of natural products. |
1. Introduction
Six-membered ring structures are widely distributed in natural products, pharmaceuticals, and organic materials.1–4 Consequently, the development of methodologies for the synthesis of six-membered rings is a central theme in organic synthesis. From the synthetic perspective, the Diels–Alder reaction, which was first discovered by Otto Diels and Kurt Alder in 1928,5 is unambiguously one of the most efficient methods for the construction of six-membered rings.6–13 Using readily available 1,3-dienes and alkene/alkyne dienophiles as reaction components, this reaction could construct a new six-membered ring by forming two new bonds with up to four stereogenic centers in a single step. The popularity of this reaction in synthesis is not only credited to its ability to construct complex ring systems but also to the predictable regioselectivity and diastereoselectivity, thereby highly reinforcing its reliability in targeted synthesis.In general, Diels–Alder reactions are classified into three types: NEDDA reactions, IEDDA reactions, and electron-neutral Diels–Alder reactions (Scheme 1).14–16 According to the classic FMO (frontier molecular orbital) theory,17–19 NEDDA reactions are dominated by the energy gap between the HOMO of electron-rich dienes and the LUMO of electron-deficient dienophiles, while IEDDA reactions are controlled by the LUMO/HOMO interaction between electron-deficient dienes and electron-rich dienophiles. Compared with electron-neutral reactions, both NEDDA reactions and IEDDA reactions are facile to occur owing to the narrowed energy gap. Based on these modes, an appropriate chiral catalyst could be utilized to increase the HOMO energy or decrease the LUMO energy of reaction components, thus enhancing the reaction rate and controlling the stereoselectivity.
 | ||
| Scheme 1 Classification of Diels–Alder reactions. NEDDA reactions, normal-electron-demand Diels–Alder reactions; IEDDA reactions, inverse-electron-demand Diels–Alder reactions; HOMO, highest occupied molecular orbital; LUMO, lowest unoccupied molecular orbital; EDG, electron-donating group; and EWG, electron-withdrawing group. | ||
Since the first aluminum-catalyzed asymmetric Diels–Alder reaction discovered by Koga in 1979 (Scheme 2A),20,21 the synthetic community has witnessed great progress in the development of efficient catalytic systems to facilitate enantioselective Diels–Alder reactions, including the use of Lewis acids, biocatalysts, chiral Brønsted acids, chiral amines, and other organocatalytic systems.22–37 Despite significant advancements that have been achieved, the applicability of asymmetric Diels–Alder reactions in organic synthesis is severely limited by the requirement of restrictive electronic complementarity between dienes and dienophiles. In addition, the reaction partners need to be equipped with polar functional groups to provide binding sites for catalysts, thus further narrowing the application scope (Scheme 2B).
 | ||
| Scheme 2 Conventional catalytic asymmetric Diels–Alder reactions. | ||
To circumvent these limitations, several innovative approaches have been developed. Notably, transition metal-catalyzed cycloadditions of alkenes, alkynes, or allenes via the oxidative cyclometallation followed by the reductive elimination reaction pathway could overcome certain limitations of pericyclic cycloadditions.38–43 This reaction mode enables unactivated unsaturated hydrocarbons to participate in [4+2] cycloadditions that otherwise need to occur under harsh reaction conditions (Scheme 3A). On the other hand, by the coordination with a π-acid such as the gold(I) complex, the LUMO energy of C–C double or triple bonds can be lowered,44–50 thus facilitating efficient [4+2] cycloadditions with complementary reaction partners (Scheme 3B). More recently, a novel π-Lewis base catalytic strategy has been devised.51 It has been demonstrated that low-valent transition metal complexes could serve as π-Lewis bases to raise the HOMO energy of 1,3-dienes by π-backdonation upon η2 coordination, thereby promoting [4+2] cycloadditions with electron-deficient diene components (Scheme 3C). Particularly noteworthy are photocycloaddition reactions, which have gained significant attention owing to their ability to mediate reactions between simple olefins under mild conditions.52–57 In this rapidly growing field, catalytic asymmetric photo-induced [4+2] cycloadditions through radical reaction pathways have also emerged (Scheme 3D).
 | ||
| Scheme 3 New approaches for catalytic asymmetric [4+2] cycloadditions of alkenes, alkynes and allenes. | ||
In recent years, numerous excellent review articles on [4+2] cycloadditions have been reported. However, most of them have been predominantly devoted to conventional catalytic asymmetric Diels–Alder reactions.22–35 Given the fact that great progress in transition metal catalysis and photocatalysis has been realized in the past three decades, it is a good occasion to summarize the exciting advancements in the development of catalytic asymmetric [4+2] cycloadditions of unsaturated hydrocarbons (alkenes, alkynes and allenes) through these novel approaches. The aim of this review is to provide a concise overview of this research field to readers. According to the above discussion (Scheme 3), this review is organized into four sections based on catalytic activation modes: (1) catalytic asymmetric [4+2] cycloadditions via the oxidative cyclometallation and reductive elimination pathways, (2) enantioselective [4+2] cycloadditions via the LUMO activation of π-acids, (3) enantioselective [4+2] cycloadditions via the HOMO activation of π-bases, and (4) catalytic asymmetric radical-mediated [4+2] cycloadditions.
2. Catalytic asymmetric [4+2] cycloadditions via the oxidative cyclometallation/reductive elimination pathway
The Diels–Alder reaction plays a pivotal role in the construction of six-membered rings, rendering it one of the most extensively employed methodologies in organic synthesis. According to FMO rules, this reaction is facile to occur within electronically complementary components. Actually, the inherent reluctance of electronically similar reactants to undergo cyclization under ambient conditions still represents one of the most significant limitations for this remarkable reaction. In this regard, transition metal catalysts can accelerate [4+2] cycloadditions between electronically similar reaction components via oxidative cyclometallation and subsequent reductive elimination. Notably, great progress in catalytic asymmetric [4+2] cycloadditions of electronically unbiased hydrocarbons has been achieved by a diverse range of transition metals such as Rh, Pd, Ru, Ni, Fe, and Co. In this section, we will discuss the advancements in this area according to transition metals.2.1. Rh-catalyzed [4+2] cycloadditions
In 1990, Livinghouse et al. reported the Rh(I)-catalyzed intramolecular [4+2] cycloadditions of unactivated diene-ynes 1 (Scheme 4).58 By the catalysis of commercially available Rh(I) complexes [(PPh3)3RhCl] or [(i-C3HF6O)3P]2RhOTf, several types of 5/6 bicyclic ring scaffolds 2 were efficiently constructed with high yields under mild conditions. Moreover, terminal triene substrates also demonstrated excellent compatibility, affording the corresponding bicyclic adducts with moderate to high yields. In this study, a reasonable mechanism was suggested. The catalytic cycle was initiated by the coordination of substrate 1 and the Rh(I) complex, which then formed the allylic η3-coordinated Rh(III) complex 3 by oxidative cyclometallation. Subsequently, this intermediate underwent allylic isomerization to yield Rh(III) species 4, which ultimately proceeded reductive elimination to furnish the final product 2. | ||
| Scheme 4 Rhodium(I)-catalyzed intramolecular [4+2] cycloadditions of diene-ynes and trienes. | ||
Based on this study, the same group realized the first Rh(I)-catalyzed intramolecular asymmetric [4+2] cycloadditions in 1994.59 Using DIOP-type bisphosphines L1 as chiral ligands, 5/6 bicyclic products with diverse substitution patterns were obtained enantioselectively from unactivated triene and diene-yne substrates 5 in good yields with up to 87% ee (Scheme 5). Although only moderate enantioselectivities were observed in some examples, this seminal work laid the foundation for the subsequent exploration of transition metal-mediated stereoselective annulations.
 | ||
| Scheme 5 Rhodium(I)-catalyzed asymmetric intramolecular [4+2] cycloadditions of diene-ynes and trienes. | ||
It has been found that the counterion of the rhodium catalyst had a significant influence on the reactivity and stereocontrol in [4+2] cycloadditions. Using Rh(I) catalysts bearing hexafluoroantimonate (SbF6−) as the counterion, the Gilbertson group investigated the performance of various chiral biphosphine ligands in [4+2] cycloadditions.60,61 The utilization of DUPHOS L2 as the ligand was found to be effective in intramolecular asymmetric [4+2] cycloadditions of diene-yne substrates 7, affording products 8 in high yields (76–85%) and high enantioselectivities (88–95% ee) (Scheme 6). Remarkably, the utilization of BINAP L3 enabled the [4+2] cycloaddition of triene 9 with comparable efficiency, delivering cycloadduct 10 in 64% yield and 98% ee. This study demonstrated the critical role of chiral phosphines in achieving good reactivity and enantiocontrol in transition metal-catalyzed [4+2] cycloadditions.
 | ||
| Scheme 6 Asymmetric intramolecular [4+2] cycloadditions catalyzed by a rhodium(I)/DUPHOS complex or rhodium(I)/BINAP complex. | ||
In 2001, Livinghouse and coworkers synthesized a series of P-chirogenic diphosphine ligands and evaluated them in the cationic Rh(I)-catalyzed intramolecular [4+2] cycloaddition of nitrogen-tethered triene 11.62 The phosphine linking element showed a great influence on the enantioselectivity. When diisopropylsila-bridged P-chirogenic diphosphine ligand L4 was employed, the desired hexahydroisoindole product 12 was obtained in 85% yield and 91% ee (Scheme 7).
 | ||
| Scheme 7 Rhodium(I)-catalyzed asymmetric intramolecular [4+2] cycloaddition of the nitrogen-tethered triene. | ||
The Schmalz group developed a cationic rhodium-catalyzed enantioselective intramolecular [4+2] cycloaddition using TADDOL-derived phosphine–phosphite L5 bearing a phenyl group adjacent to the phosphite moiety as the chiral ligand (Scheme 8).63 By this method, 5/6-bicyclic products 14 were obtained as single diastereoisomers in good to excellent enantioselectivities (up to 93% ee) from diene-enes 13. The key to realize high enantioselectivities was the preparation of the “aged” catalyst by microwave irradiation of the pre-catalyst from [Rh(NBD)Cl]2, AgSbF6, and L5 (0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1.6). Mechanistic studies including 31P NMR and ESI-MS measurements suggested that the [Rh(L5)2]+ species was a more selective catalyst.
:1:1.6). Mechanistic studies including 31P NMR and ESI-MS measurements suggested that the [Rh(L5)2]+ species was a more selective catalyst.
 | ||
| Scheme 8 Asymmetric intramolecular [4+2] cycloadditions of unactivated trienes catalyzed by a rhodium(I)/TADDOL-derived phosphine–phosphite ligand complex. | ||
In previous studies, the Rh(I) catalysts were typically generated in situ from achiral Rh(I)/diene complexes and chiral phosphine ligands. However, the effect of diene ligands in reactions had been ignored for many years. In 2006, the Mikami group disclosed the asymmetric synergistic effect between the chiral diene and diphosphine ligands (Scheme 9).64 By the combination of [Rh(L7)Cl]2, DUPHOS ligand L6 and AgSbF6, 5/6-bicyclic products 16 were synthesized with high yields and enantioselectivities (up to 99% yield, 98% ee) from unactivated diene-ynes via intramolecular [4+2] cycloadditions. It should be noted that the sole utilization of diphosphine ligand L6 or diene ligand L7 led to the desired cycloadducts in low enantioselectivities. In a plausible mechanism, the Rh(I)/L6/L7 complex was proposed as the active catalyst. The catalytic cycle was initiated by the coordination of 17 with diene-yne 15. The resulting intermediate 18 was further transformed into 19 by oxidative cyclometallation. Subsequently, this intermediate underwent allylic rhodium(III) isomerization to yield intermediate 20. From 20, the reductive elimination led to the final product 16 and regenerated the active Rh(I) catalyst 17. In the structure of the key intermediate 18, the diene ligand was proposed to coordinate with the metal center via η2 coordination, while the biphosphine ligand complexed with the metal center via η4 coordination.
 | ||
| Scheme 9 Asymmetric intramolecular [4+2] cycloadditions catalyzed by a rhodium(I)/DUPHOS/diene complex. | ||
Chung et al. demonstrated that the phosphine-free rhodium(I)/diene complex [Rh(naphthalene)(cod)]BF4 could act as an effective catalyst to promote intermolecular and intramolecular [4+2] cycloadditions between unactivated 1,3-dienes and alkynes.65 However, only racemic products were afforded by this achiral catalytic system. In 2007, Shintani and Hayashi et al. realized the cationic Rh(I)-catalyzed asymmetric intramolecular [4+2] cycloaddition of diene-yne by the sole utilization of diene (S,S)-Ph-bicyclo[2.2.2]octadiene (bod*) L8 (Scheme 10).66 By this catalytic system, [4+2] cycloadditions of both aryl- and alkyl-terminated diene-ynes 21 proceeded smoothly, affording the desired cycloadducts 22 in high yields (up to 96%) with excellent enantioselectivities (up to 99% ee). Notably, the intermolecular [4+2] cycloaddition between 1,3-dienes 23 and electron-deficient alkyne 24 was also compatible, giving 1,4-cyclohexadienes 25 in good yields and enantioselectivities. By direct comparison, the diene ligand showed much higher reactivity than phosphine ligands in this Rh(I)-catalyzed [4+2] cycloaddition.
 | ||
| Scheme 10 Rh(I)/chiral diene-catalyzed asymmetric intramolecular and intermolecular [4+2] cycloadditions. | ||
In 2008, the Shibata group reported the intermolecular asymmetric [4+2] cycloadditions of 1,3-dienes with dimethyl acetylenedicarbonate (DMAD) 24. Using [Rh(cod)L3]BF4 as the chiral catalyst, a variety of 1,4-cyclohexadienes were obtained in moderate yields and good enantioselectivities (up to 66% yield, 94% ee) (Scheme 11).67 Notably, 1,1-disubstituted 1,3-butadiene was compatible, giving the corresponding 1,4-cyclohexadiene in 47% yield and 87% ee. 1,2-Disubstituted 1,3-butadiene was also viable, and the bicyclic product was produced in 65% yield and 78% ee. In a proposed mechanism, the Rh(I)/L3 catalyst initially coordinated with 1,3-dienes 26 and DMAD. Since the 3,4-position of the 1-substituted-1,3-diene was less sterically hindered than the 1,2-position, the subsequent oxidative cyclometallation occurred at the 3,4-position of 26 with the alkyne moiety of DMAD. Due to the steric repulsion between the methoxycarbonyl and alkenyl groups in intermediate 30, metallacyclopentene 29 was therefore preferentially formed, which then underwent 1,3-allylic rearrangement and reductive elimination to afford the final product 27.
 | ||
| Scheme 11 Rh(I)/BINAP-catalyzed asymmetric intermolecular [4+2] cycloadditions of 1,3-dienes and DMAD. | ||
In 2012, the Tanaka group reported an intermolecular [2+2+2] trimerization/asymmetric intramolecular [4+2] cycloaddition of 5-alkynals 32 and two aryl ethynyl ethers 33. Using [Rh(cod)2]BF4/L9 as the chiral catalyst, a wide range of annulated 1,4-cyclohexadienes 34 were obtained regioselectively and diastereoselectively with up to 67% yield, >99% ee, and >99:1 dr (Scheme 12).68 A plausible mechanism is depicted in Scheme 12. By the catalysis of Rh/L9, two molecules of aryl ethynyl ether 33 underwent the oxidative cyclometallation, generating the rhodacyclopentadiene intermediate 35. Subsequently, insertion of the formyl group of 5-alkynal 32 into 35 afforded rhodacycle 36, which then underwent reductive elimination to give pyran derivative 37. From 37, the electrocyclic ring-opening delivered dienyne 38, which then afforded rhodacyclopentene 39 with the rhodium catalyst. The 1,3-allylic migration of 39 followed by the reductive elimination afforded 34 as the final product.
 | ||
| Scheme 12 Rh(I)/(R)-H8-BINAP-catalyzed intermolecular [2+2+2] trimerization/asymmetric intramolecular [4+2] cycloaddition. | ||
In 2020, Shi et al. identified a highly efficient Rh(I) catalytic system to realize the intermolecular [4+2] cycloadditions of 1-substituted-1,3-dienes and DMADs with high enantioselectivities (Scheme 13).69 Using phosphoramidite L10 as the chiral ligand, a wide range of cyclohexa-1,4-dienes 43 were obtained with up to 96% yield and >99% ee under mild reaction conditions. A similar mechanism involving the oxidative cyclometallation, 1,3-allylic rearrangement, and reductive elimination was proposed.
 | ||
| Scheme 13 Rh(I)/phosphomidite-catalyzed asymmetric intermolecular [4+2] cycloadditions of 1,3-dienes and DMADs. | ||
The first Rh(I)-catalyzed intramolecular [4+2] cycloadditions of allene-1,3-dienes were reported by the Wender group in 1995.70,71 By the utilization of phosphites as ligands, [4+2] cycloadditions of 1,3-disubstituted allene-1,3-dienes 44 allowed the construction of 6/5 and 6/6-fused bicyclic ring systems 45 with high yields under mild reaction conditions. Intriguingly, when P[OCH(CF3)2]2 was used as the ligand, the cycloaddition of 46 occurred regioselectively on the external double bond of the allene moiety, giving 6/7-fused bicyclic product 47 in 89% yield (Scheme 14).
 | ||
| Scheme 14 Rh(I)/phosphite-catalyzed intramolecular [4+2] cycloaddition of 1,1-disubstituted allene-1,3-dienes. | ||
In 2018, the Ma group realized the RhCl(PPh3)3-catalyzed intramolecular [4+2] cycloadditions of racemic 1,3-disubstituted allene-1,3-dienes with axial chirality.72 By the catalysis of RhCl(PPh3)3 (2.0 mol%), cis-fused bicyclic products 49 were produced with up to 87% yields (Scheme 15). The cycloaddition process occurred regioselectively on the internal double bond of the allene moiety. Notably, the configuration of the non-bridging carbon center in the six-membered ring was governed by the configuration of the double bond of the 1,3-diene moiety. When optically active allene-1,3-diene substrate 50 reacted under standard conditions, the axial-to-central chirality transfer process occurred, giving 51 as a single diastereoisomer in 75% ee.
 | ||
| Scheme 15 RhCl(PPh3)3-catalyzed intramolecular [4+2] cycloadditions of 1,3-disubstituted allene-1,3-dienes. | ||
To improve the efficiency of the axial-to-central chirality transfer process, Ma et al. reexamined the Rh(I)-catalyzed intramolecular [4+2] cycloadditions of optically active 1,3-disubstituted allene-1,3-dienes.73 It was found that when Rh(PPh3)3SbF6 was used as the catalyst in ethanol, a complete chirality transfer was observed; the corresponding cis-5/6-fused bicyclic adducts 53 were obtained with high yields and retained enantioselectivities (up to 99% ee) (Scheme 16). A series of control experiments indicated that the axial chirality of the 1,3-disubstituted allene moiety governed the absolute configurations of in situ generated chiral centers by chirality transfer. In a proposed mechanism, both intermediate 54 and its enantiomeric isomer ent-54 could be formed via ligand exchange between allene-1,3-dienes 52 and the in situ generated [Rh(PPh3)3]SbF6 catalyst. Owing to the significant steric hindrance between the R1 substituent and the catalyst framework in ent-54, intermediate 54 was energetically more favored. From 54, the subsequent cyclometallation and allylic isomerization steps generated rhodabicyclic intermediate 56, in which the three tertiary hydrogen atoms were positioned in a cis-configuration. Finally, reductive elimination followed by ligand exchange with allene-1,3-dienes 52 gave product 53 and regenerated the active catalytic species 54.
 | ||
| Scheme 16 Rh(PPh3)3SbF6-catalyzed intramolecular [4+2] cycloadditions of optical active allene-1,3-dienes. | ||
In 2021, the Gilbertson group also investigated the Rh(I)-catalyzed intramolecular [4+2] cycloadditions of chiral allene-1,3-dienes (Scheme 17).74 It was found that employing [Rh(cod)Cl]2/L11 as the catalyst in toluene at 80 °C, optically active allene-1,3-dienes 58 (80–98% ee) could be transformed into 59 with excellent yields and retained enantioselectivities (up to 96% yield, 98% ee). In most cases, the diastereoselectivities ranged from 99:1 to 90:10. The utilization of phosphine-phosphine oxide ligand L11 [1,2-bis(diphenylphosphino)ethane oxide, dppeO] was the key for the success of this transformation. It should be noted that optically active allene-1,3-dienes were prepared by the CuBr2-catalyzed EATA (enantioselective allenation of terminal alkynes) reaction developed by Ma and coworkers.75,76
 | ||
| Scheme 17 Rh(I)/dppeO-catalyzed intramolecular [4+2] cycloadditions of optically active allenyl 1,3-dienes. | ||
In 2021, Ma and coworkers reported the first Rh-catalyzed kinetic resolution-based enantioselective [4+2] cycloaddition-isomerization of 1,3-disubstituted allene-1,3-dienes (Scheme 18).77 By the catalysis of [Rh(C2H4)2Cl]2 and AgSbF6 with (R,R)-Ph-BPE L12 in EtOH at 40 °C, a wide range of aza-[4.3.0]bicyclic compounds 61 bearing various functionalities were synthesized in satisfactory yields (up to 40%) and enantioselectivities (up to >99% ee) from racemic 1,3-disubstituted allene-1,3-dienes. Intriguingly, these reactions exhibited a substrate-controlled divergence. While the (Sa)-enantiomer of substrate 60 participated in cyclization, the residual (Ra)-enantiomer was exclusively transformed into 1,3-diene derivative 62via Rh/L12-catalyzed double bond isomerization.
 | ||
| Scheme 18 Rh(I)/Ph-BPE-catalyzed kinetic resolution of racemic 1,3-disubstituted allene-1,3-dienes. | ||
Based on a series of control experiments, a plausible mechanism was proposed. Initially, [Rh(C2H4)2Cl]2 reacted with AgSbF6 and L12 to generate the cationic catalyst [RhL12]SbF6, which then coordinated with (Sa)-60 to generate intermediate 63. The subsequent cyclometallation formed intermediate 63, which then underwent allylic isomerization to produce rhodiabicycloheptene 65. Reductive elimination delivered the [4+2] cycloadduct 66 and regenerated [RhL11]+ (Path a). From 66, the in situ formed [Rh]–H species promoted isomerization of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond via sequential hydrometallation and β-H elimination to afford 61 as the final product. Meanwhile, the enantiomeric (Ra)-60 coordinated with [RhL11]+ and underwent oxidative addition to generate allyl-Rh-hydride intermediate 69. The reductive elimination of 69 and ligand dissociation then produced tetraene products 62 (path b). It should be noted that (Sa)-60 could also proceed through path b slowly to give 62 in a reduced yield, thus revealing the delicate balance between cyclization and isomerization processes.
C bond via sequential hydrometallation and β-H elimination to afford 61 as the final product. Meanwhile, the enantiomeric (Ra)-60 coordinated with [RhL11]+ and underwent oxidative addition to generate allyl-Rh-hydride intermediate 69. The reductive elimination of 69 and ligand dissociation then produced tetraene products 62 (path b). It should be noted that (Sa)-60 could also proceed through path b slowly to give 62 in a reduced yield, thus revealing the delicate balance between cyclization and isomerization processes.
Based on the above achievements, the Ma group realized the first Rh-catalyzed dynamic kinetic resolution (DKR) of racemic axially chiral allene-1,3-dienes through the intramolecular [4+2] cycloaddition paradigm, establishing a powerful approach for the construction of enantiopure 5/6-bicyclic scaffolds (Scheme 19).78 By the catalysis of [Rh(coe)2Cl]2/AgOTs/L12 in isopropanol at 80 °C, a wide range of cis-5,6-fused bicyclic products 72 were obtained with high yields and excellent enantioselectivities (up to 84% yield and >99% ee) directly from racemic allene-1,3-dienes 71. The key for the success of this transformation was the judicious selection of Malphos L13 as the chiral ligand. Combined mechanistic experiments and DFT calculations revealed that the Rh/L13 complex could facilitate the isomerization of (Sa)-71 to (Ra)-71 and then stereoselectively promoted the subsequent [4+2] cycloaddition of (Ra)-71. It was proposed that the racemization of 71 was not caused by the in situ generated rhodium hydride species, but by the coordination with Rh/L13 followed by the subsequent C–H oxidative addition and reductive elimination. Compared with the ability of (R,R)-Ph-BPE L12 to promote the isomerization of allenes to 1,3-dienes, the distinctive reactivity of L13 demonstrated the striking ligand effect.
 | ||
| Scheme 19 Rh(I)/Malphos-catalyzed dynamic kinetic resolution (DKR) of racemic allene-1,3-dienes via the intramolecular [4+2] cycloaddition. | ||
2.2. Pd-catalyzed [4+2] cycloadditions
Despite remarkable achievements realized by Rh(I) complexes in intramolecular [4+2] cycloadditions of allene-1,3-dienes, the intermolecular version of these transformations remained elusive. In 1997, Murakami and Ito et al. developed the palladium-catalyzed intermolecular [4+2] cycloadditions of 1,3-dienes and vinylallenes (Scheme 20).79 Using Pd(PPh3)4 as the catalyst, vinylallenes reacted as diene components to contribute a four-carbon unit in [4+2] cycloadditions with 1,3-dienes at room temperature, delivering substituted cyclohexene derivatives with high yields and diastereoselectivities. Notably, the reaction between isoprene and vinylallene afforded a pair of isomers in 97% yield with 71:29 regioselectivity. In a proposed mechanism, the reaction began with the [4+1] type of oxidative cyclometallation between vinylallenes 73 and Pd(PPh3)4, generating the five-membered bent palladacycle intermediate 76 in an s-trans coordination geometry. Subsequently, the π-allylpalladium intermediate 77 was formed via migratory insertion of the 1,3-diene into the palladacycle, leading to 77, which subsequently underwent reductive elimination to give 75. This study significantly advanced the development of transition metal-catalyzed [4+2] cycloadditions, providing an unconventional approach for constructing structurally diverse six-membered carbocycles.
 | ||
| Scheme 20 Pd(PPh3)4-catalyzed intermolecular [4+2] cycloadditions of vinylallenes and 1,3-dienes. | ||
In 2000, the Ito group achieved a breakthrough in the development of a catalytic asymmetric variant of this transformation (Scheme 21).80 After evaluation of various ferrocene-derived monophosphine ligands, the combination of Pd2(dba)3·CHCl3 with L14 was found to be effective, facilitating the intermolecular cycloadditions of vinylallenes 78 and 1,3-dienes 74 in dichloromethane under mild reaction conditions. In these reactions, vinylallenes bearing ester, phenyl, and alkyl groups all reacted smoothly with 1,3-butadienes to afford the desired products in high yields, albeit with moderate enantioselectivities (up to 82% ee).
 | ||
| Scheme 21 Pd(0)-catalyzed asymmetric intermolecular [4+2] cycloadditions of vinylallenes and 1,3-dienes. | ||
2-Carboalkoxy-1,3-butadienes (2-CBDs) 80 are electron-deficient dienes which are facile to self-dimerization under mild conditions.81 Recently, the Cai group employed readily available Morita–Baylis–Hillman (MBH) carbonates 82 as precursors of 2-CBDs. Through the Pd(0)-catalyzed oxidative addition followed by the β-H elimination, 2-CBD intermediates were generated in situ and their dimerization was suppressed by coordination with Pd(0) (Scheme 22).82 Using L15 as the chiral ligand, the asymmetric [4+2] cycloaddition between MBH carbonates 84 and 1,3-enynes 85 went on smoothly, giving 1,4-cyclohexadiene derivatives 86 bearing aryl- and alkyl-substituents with excellent yields and high enantioselectivities (up to 98% yield, 96% ee). By DFT calculations, it was proposed that the in situ generated 2-CBD coordinated with Pd/L15, giving intermediate 87, which then underwent [4+4+1] oxidative cyclometallation with 1,3-enyne 85 to afford Pd(II) species 88. Owing to the trans-configuration of the olefin moiety in 88, allylic isomerization and rotation steps should occur before reductive elimination, thus delivering intermediate 90. From 90, the reductive elimination afforded the final product 86 in a chemo-, regio-, and enantioselective manner.
 | ||
| Scheme 22 Pd(0)-catalyzed asymmetric [4+2] cycloadditions of Morita–Baylis–Hillman carbonates and 1,3-enynes. | ||
2.3. Ir-catalyzed [4+2] cycloadditions
In 2002, the Shibata group demonstrated that the iridium complex had sufficient catalytic activity to promote [4+2] cycloadditions of unactivated diene-ynes (Scheme 23).83 The catalytic enantioselective intramolecular [4+2] cycloadditions of diene-ynes 91 were realized by [Ir(cod)Cl]2 and BDPP L16, enabling the synthesis of chiral cyclohexa-1,4-dienes 92 with high yields and enantioselectivities (up to 73% yield, 98% ee). This study illustrated the versatility of iridium catalysis in asymmetric cycloadditions. However, only aryl-substituted diene-yne substrates were evaluated, leaving alkyl-substituted variants unexplored. | ||
| Scheme 23 Ir(I)/BDPP-catalyzed asymmetric intramolecular [4+2] cycloaddition of diene-ynes. | ||
2.4. Ru-catalyzed [4+2] cycloadditions
In 2000, the Trost group realized the RuCp(MeCN)3FP6-catalyzed intramolecular [4+2] cycloadditions of yne-enones, giving bicyclic 4H-pyrans with high yields. According to the proposed mechanism, three vacant coordination sites were required on the Ru(II) center, thus making the use of a chiral cyclopentadienyl (Cp) ligand a promising strategy to achieve good enantioselectivity control.84 In 2015, the Cramer group developed an efficient route for the synthesis of various cationic CpRu(II) complexes bearing chiral ligands and then evaluated these complexes in intramolecular [4+2] cycloadditions of yne-enones.85,86 Using 94 as the catalyst at −20 °C, the intramolecular formal [4+2] cycloadditions of yne-enones 93 proceeded efficiently to deliver 2H-pyran derivatives 95 with high yields and excellent enantioselectivities (up to 95% yield, 98% ee) (Scheme 24). In a proposed mechanism, the CpRu(II) complex 94 initially coordinated with yne-enone 93 to generate intermediate 96, which then underwent oxidative cyclometallation, allylic isomerization, and reductive elimination to afford 95 as the final product. Notably, the enantioselectivity was determined in the oxidative cyclometallation step. | ||
| Scheme 24 Ruthenium (II)-catalyzed asymmetric intramolecular [4+2] cycloadditions of yne-enones. | ||
2.5. Ni-catalyzed [4+2] cycloadditions
The Ni(0)-catalyzed cycloadditions of 1,3-dienes represent one of the earliest and most important transition metal-catalyzed C–C bond-forming reactions.87–89 Since the early 1960s, the Wilke group systematically investigated the cyclodimerization of 1,3-butadiene.90–92 In 1974, the Garrat group developed the intermolecular [4+2] cycloaddition between two different 1.3-dienes.93 In the presence of a nickel catalyst [Ni(acac)2, Et3Al, PPh3], the cross-[4+2] cycloaddition of 1,3-butadiene 99 with methyl sorbate 100 gave a pair of diastereomeric formal inverse-electron-demand [4+2] cycloadducts 102 and 102′ in 90% yield with 2:1 dr. It should be noted that under thermal conditions, methyl sorbate 100 acted as the diene component, while 1,3-butadiene 99 served as the dienophile to afford normal-electron-demand cycloadduct 101 exclusively. A plausible mechanism for the Ni(0)-catalyzed [4+2] cycloaddition is illustrated in Scheme 25. The oxidative cyclometallation of 99 and 100 with the Ni(0)/PPh3 catalyst afforded the key bis(η3-allyl)nickel(II) intermediate 103, which then underwent reductive elimination to produce the cycloaddition product. These results clearly indicated that the transition metal catalysis could not only accelerate the reaction rate but also change the inherent periselectivity in [4+2] cycloadditions of unsaturated hydrocarbons.
 | ||
| Scheme 25 Ni(0)/PPh3-catalyzed cross-[4+2] cycloadditions of 1,3-butadiene with methyl sorbate. | ||
The first asymmetric Ni(0)-catalyzed intermolecular [4+2] cycloaddition of 1,3-dienes was reported by the Mortreux group in 1994.94 By the catalysis of Ni(cod)2 and bis(aminophosphine) ligand L17 in toluene at 80 °C, 1,3-butadiene 99 underwent [4+2] dimerization to generate 4-vinylcyclohexene 104 in 80% yield but with only 6% ee. In this reaction, 1,5-cyclooctadiene 105 by [4+4] co-dimerization of 99 was obtained as the main side product in 20% yield (Scheme 26). In 1995, the same group reported the Ni(0)-catalyzed cross-[4+2] cycloaddition of methyl penta-2,4-dienoate 95 with 1,3-butadiene 99.95 Using Ni(cod)2/(R)-BINAP L3 as the catalyst, the cross-[4+2] cycloadduct 107 was produced in 15% yield and 5% ee, and the co-dimerization product 108 from 106 was generated in 22% yield (Scheme 26). The poor results in these two reactions demonstrated the substantial challenge in controlling both the chemoselectivity and enantioselectivity concurrently in Ni(0)-catalyzed asymmetric [4+2] cycloadditions of 1,3-dienes.
 | ||
| Scheme 26 Ni(0)-catalyzed asymmetric [4+2] cycloadditions of 1,3-dienes. | ||
A breakthrough in this research field was achieved by the Chen group in 2022.96 Using Ni(cod)2 as the catalyst and chiral N-heterocyclic carbene (NHC) L18 as the ligand, a wide range of unnatural cyclic monoterpene derivatives bearing quaternary carbon stereocenters were obtained in high yields and enantioselectivities (up to 98% yield, 97% ee) from isoprene and heterocycles (Scheme 27). By this method, various heterocycles, such as purines, adenines, and imidazoles, could be incorporated into the terpene skeleton. Mechanistic studies and DFT calculations revealed that this atom-economic reaction proceeded through the enantioselective [4+2] dimerization of isoprene followed by a sequential C–H alkylation of the heterocycle pathway. As shown in Scheme 27, the oxidative cyclometallation of isoprene with the Ni(0) catalyst proceeded through intermediate 111, giving the five-membered nickelacycle intermediate 112. By coordination with another isoprene, intermediate 113 was generated and underwent migratory insertion through transition state 114, thus affording seven-membered nickelacycle 115. Subsequently, reductive elimination of the Ni(II) species 115 produced the [4+2] cycloadduct 116 bearing a quaternary carbon center. Parallelly, the nickel–hydride intermediate 118 was formed from the heterocycle substrate via C–H activation with the Ni(0) catalyst. From 116 and 118, the sequential coordination followed by the migratory insertion and reductive elimination produced the final product 110 and regenerated the Ni(0) catalyst. Notably, the key for the success of this transformation was the judicious selection of a bulky C2-symmetric NHC L18 to realize the chemo-, regio-, and enantioselectivity.
 | ||
| Scheme 27 Ni(0)/NHC-catalyzed asymmetric heteroarylative cyclotelomerization of isoprene. | ||
In addition to the Pd(0)-catalyzed [4+2] cycloadditions of MBH carbonates with 1,3-enynes, the Cai group also reported the asymmetric formal inverse-electron-demand [4+2] cycloaddition of MBH carbonates 121 with 1,3-dienes 122 by the catalysis of Ni(0)/L19 (Scheme 28).82 A wide range of para-substituted cyclohexenes 123 were obtained in high yields with excellent chemo-, regio- and stereoselectivities (up to 94% yield, 99% ee, >20:1 rr). Intriguingly, this catalytic system could be applied to [4+2] cycloadditions between two electron-deficient dienes, which was an unsolved reaction mode in organic synthesis before this study. In analogous to conventional Diels–Alder reactions, these reactions demonstrated remarkable stereospecificity. When internal (E,Z)-1,3-dienes were used, the reaction occurred on the double bond adjacent to the alkyl group, affording cis-cycloadducts. (E,E)-1,3-diene was also compatible, and the trans-cycloadduct was produced. Owing to the mild and redox-neutral conditions, a wide range of conjugated trienes and air/light-sensitive tetraenes were tolerated. The utilization of these reactions in complex molecule synthesis was demonstrated by the three-step synthesis of (+)-juvabione and (+)-epijuvabione.82 By DFT calculations, it was proposed that the [4+2] cycloaddition went through a pathway involving [4+4+1] oxidative cyclometallation, allylic isomerization, and reductive elimination steps.
 | ||
| Scheme 28 Ni(0)-catalyzed [4+2] cycloadditions of Morita–Baylis–Hillman carbonates with 1,3-dienes. | ||
2.6. Fe-catalyzed [4+2] cycloadditions
The applications of iron catalysts have attracted increasing attention in recent years, owing to their abundance, low price, and unique reactivities to promote synthetic transformations that are otherwise challenging to be achieved by other catalytic systems.97–100 In 2020, the Cramer group realized a catalytic asymmetric cross-[4+4] cycloaddition between two different unactivated 1,3-dienes to form substituted cyclooctadienes by a chiral α-diimine iron complex.101 Based on this study, they developed a new chiral α-diimine iron catalyst that enabled the cross-[4+2] cycloadditions between unactivated 1-substituted 1,3-dienes and 2-substituted 1,3-dienes in a regioselective and enantioselective manner (Scheme 29).102 Using the iron complex 130 or 131 as the catalyst, a series of alkyl- and aryl-substituted meta-substituted vinyl cyclohexene derivatives 129 were obtained in high yields with excellent enantioselectivities (up to 99% yield, 94% ee). It should be noted that by conventional Diels–Alder reactions, only para- and ortho-substituted cyclohexene adducts could be produced, thus demonstrating the complementary advantage of the iron catalysis. | ||
| Scheme 29 Fe(I)/α-diimine-catalyzed asymmetric cross-[4+2] cycloadditions of unactivated 1,3-dienes. | ||
In a proposed reaction mechanism, the Fe(II) complex was initially reduced by MgBu2 to generate the Fe(I) catalyst, which then coordinated with 1,3-diene 74 to form the catalytically active species 132. Then, the oxidative cyclometallation between intermediate 132 and 1-substituted 1,3-diene 128 led to the formation of seven-membered ferracycle 133 regioselectively. Finally, a reductive elimination formed 1,3-substituted vinyl cyclohexene 129 and regenerated 132. This study unambiguously showcased that the main skeletons of α-diimine ligands played an important role in controlling the periselectivity in cycloaddition.
2.7. Co-catalyzed [4+2] cycloadditions
As an important type of earth-abundant transition metal in organic synthesis, the cobalt catalysts demonstrated sufficient ability to promote cycloadditions of unsaturated hydrocarbons.103–108 The Hilt group has systematically investigated the Co(I)-catalyzed [4+2] cycloadditions between unactivated 1,3-dienes and alkynes.109–111 In these reactions, various functionalities were compatible on the 1,3-diene and alkyne moieties with various substitution patterns.103–105 However, catalytic asymmetric variants of these transformations were ignored for many years. In 2023, the Rajanbabu group reported the first cobalt-catalyzed enantioselective [4+2] cycloadditions between 1,3-dienes and internal alkynes (Scheme 30).112 In the presence of catalytic amounts of CoBr2/BenzP*(L20) and zinc powder, highly functionalized 1,4-cyclohexadienes 135 were obtained in high yields and enantioselectivities (up to 82% yield, 99% ee) from 1-substituted 1,3-dienes 128 and electron-deficient alkynes 134. Notably, unsymmetric 1,3-dienes and alkynes were compatible in these reactions, leading to excellent regioselectivities in most cases. In control experiments, when the isolated [Co(I)BrL20]2 complex was used as the catalyst, no [4+2] cycloadduct was observed. In contrast, the combination of the same complex with NaBARF led to the formation of the desired [4+2] product with 100% conversion, indicating the key role of the cationic properties of the situ generated [CoL20]+[BARF]− catalyst. | ||
| Scheme 30 Co(I)/BenzP*-catalyzed asymmetric [4+2] cycloadditions of 1,3-dienes and alkynes. | ||
In a possible mechanism, the Co(I)/L20 complex was initially generated from CoBr2 and L20 by reduction with zinc. Then, it coordinated with the 1,3-diene and alkyne substrates to form intermediate 137, which underwent the turn-over limiting oxidative cyclometallation to generate [Co(III)]-metallacycle 139. The subsequent rapid reductive elimination formed the second C–C bond, yielding the final product 135 and regenerating the active catalyst. Remarkably, the utilization of the (R)-tBu-PHOX ligand instead of L20 enabled asymmetric [2+2] cycloadditions between 1,3-dienes and alkynes, further highlighting the crucial role of chiral ligands in controlling both the regioselectivity and chemoselectivity.
Recently, the Rajanbabu group reported the asymmetric cobalt-catalyzed intramolecular [4+2]-cycloadditions of unactivated diene-ynes 141 and trienes 143 (Scheme 31).113 In the presence of catalytic amounts of CoBr2/(S)-H8-BINAP (L21) and zinc powder, a series of 5/6-bicyclic products 142 were obtained as single diastereoisomers with excellent yields and enantioselectivities (up to 96% yield, 93% ee) from diene-ynes 141. When the CoBr2/L22 complex was used, trienes 143 underwent highly selective [4+2] cycloaddition, affording the fused 5,6-bicyclic products 144 rather than the intramolecular [2+2] cycloaddition products. Notably, the appropriate ligand was proved to be crucial for controlling the periselectivity of the [2+2] cycloaddition reaction.
 | ||
| Scheme 31 Co(I)-catalyzed intramolecular [4+2] cycloaddition reactions of unactivated diene-ynes and trienes. | ||
3. Enantioselective [4+2] cycloadditions via the LUMO activation of π-acids
In the past two decades, homogenous gold(I) catalysis has showcased its powerful ability in the construction of carbon–carbon bonds and carbon–heteroatom bonds.44–50,114–117 According to the Dewar–Chatt–Duncanson (DCD) bonding model, the Au(I) metal can coordinate with the alkene or alkyne ligand, forming a strong dative σ bond by overlap of the π orbital of the ligand with the low-lying empty orbital of the Au(I) metal center. Meanwhile, the Au(I) center backdonates the electron density from its filled d orbital into the π* anti-bonding orbital of the alkene/alkyne ligand. Since this back-donation is relatively weak, the interaction between the metal center and the ligand is dominated by the ligand-to-metal σ electron-donation, therefore making the alkene or alkyne ligand electrophilic.42 Based on this unique π-acid activation mode, significant advancements have been achieved in Au(I)-catalyzed formal [4+2] cycloadditions.In 2009, the Toste group reported a striking example of ligand-controlled gold(I)-catalyzed intramolecular [4+2] or [4+3] cycloadditions of allene-1,3-dienes with high selectivity (Scheme 32).118 Using di-tert-butylbiphenylphosphine L23 as the ligand, the allene-1,3-diene substrates underwent intramolecular [4+3] cyclization, affording 5/7 bicyclic adducts 146 exclusively. Intriguingly, changing the ligand from L23 to triarylphosphite ligand L24 altered the reaction pathway completely, leading to [4+2] trans-adducts 147 with up to 92% yield. In a proposed mechanism, the Au(I) catalyst coordinated with the allene moiety and then induced the intramolecular cyclization with the 1,3-diene part by the π-acid activation mode. It was hypothesized that the electron-rich σ-donor ligand L23 preferentially stabilized intermediates 148 and 149, thereby favoring [4+3] cycloadduct formation over path a. In contrast, the π-acceptor ligand L24 preferentially stabilized intermediates 150 and 151, thereby promoting the reaction to proceed through path b to form the [4+2] cycloadduct. This study established a fundamental framework to control the chemoselectivity in Au(I)-catalyzed cycloaddition reactions by ligand variations.
 | ||
| Scheme 32 Au(I)-catalyzed intramolecular [4+3] and [4+2] cycloadditions of allene-dienes. | ||
Almost at the same time, the asymmetric gold(I)-catalyzed intramolecular asymmetric cycloaddition of allene-1,3-dienes was reported by the Mascareñas group (Scheme 33).119 Using phosphoramidite L25 with C3/C3’ substituents on the BINOL skeleton as the chiral ligand, the intramolecular [4+2] cycloaddition of nitrogen-tethered allene-1,3-dienes 152 enabled the synthesis of trans-5,6- and 6,6-fused bicyclic products 153 with up to 93% yield and 97% ee. By experimental investigations and DFT calculations, a plausible mechanism was proposed. Initially, the Au(I)/L25 catalyst coordinated and activated the allene unit by π-acid activation to form gold-allyl intermediate 155, which then underwent intramolecular and concerted exo-like [4+3] cycloaddition with the 1,3-diene moiety to generate cycloheptenyl Au-carbene intermediate 156. Subsequently, the ring contraction of 156via the 1,2-alkyl shift delivered the final [4+2] cycloadduct 157 and regenerated the Au(I) catalyst. According to this mechanism, both the formal [4+3] and [4+2] cycloaddition reaction pathways shown in Scheme 32 shared the same [4C(4π) + 3C(2π)] cycloaddition step between the diene moiety and the Au(I)-allyl cation intermediate.
 | ||
| Scheme 33 Au(I)/phosphoramidite-catalyzed asymmetric intramolecular [4+2] cycloadditions of nitrogen-tethered allene-dienes. | ||
In 2010, the Toste group also reported an enantioselective Au(I)-catalyzed intramolecular [4+2] cycloaddition of carbon-tethered allene-1,3-dienes (Scheme 34).120 Using the C3-symmetric monodentate phosphite L26 as the chiral ligand, a series of enantioenriched trans-hexahydroindenes were selectively accessible from allene-1,3-dienes by gold(I) catalysis. This study together with Mascareñas's discovery demonstrated that chiral monodentate phosphorus-based ligands were suitable in asymmetric Au catalysis despite the linear geometry of Au(I) complexes.
 | ||
| Scheme 34 Au(I)/C3-symmetric phosphite-catalyzed asymmetric intramolecular [4+2] cycloadditions of allene-dienes. | ||
The first Au(I)-catalyzed asymmetric intermolecular [4+2] cycloadditions were reported by the Mascareñas group in 2012.121 In the presence of catalytic amounts of Au(I) complex 163 and AgNTf2, the intermolecular [4+2] cycloadditions between allenamides and 1,3-dienes went on smoothly, affording optical active cyclohexene products with excellent yields and enantioselectivities (up to 88% yield, >99% ee, Scheme 35). The key for the success of these reactions was the utilization of an axially chiral N-heterocyclic carbene (NHC) ligand featuring a triazole scaffold. The X-ray structure demonstrated that Au(I) complex 163 had a high percentage of buried volume value (Vbur = 46.2%) around the Au(I) center, indicating the significant steric hinderance of the NHC ligand.
 | ||
| Scheme 35 Au(I)/NHC-catalyzed asymmetric intermolecular [4+2] cycloadditions of allenamides and 1,3-dienes. | ||
In 2015, the Zhang group disclosed the enantioselective [4+2] cycloadditions of 3-styrylindoles and N-allenamides catalyzed by an Au(I)/chiral phosphoramidite complex (Scheme 36).122 By this method, a wide range of optically active tetrahydrocarbazoles were obtained with high yields and enantioselectivities (up to 99% yield, 97% ee). DFT calculations revealed that an Au(I)-allyl cation species was generated from the N-allenamide substrate by the activation of the Au(I) catalyst. Then, a stepwise [4+2] cycloaddition occurred with the 3-styrylindole substrate, in which the C2-position of the indole moiety served as the nucleophilic site due to the electron-withdrawing character of the N-CO2Et group.
 | ||
| Scheme 36 Au(I)/phosphoramidite-catalyzed asymmetric intermolecular [4+2] cycloadditions of 3-styrylindoles and N-allenamides. | ||
In 2017, the Rossi group reported the Au(I)-catalyzed asymmetric intermolecular [4+2] cycloadditions between 3-substituted 2-vinylindoles and N-allenamides (Scheme 37).123 Using DTBM-Segphos L27 as the chiral ligand, dearomative tetrahydrocarbazoles bearing a quaternary stereogenic center at the C3-position were obtained in good yields and enantioselectivities (up to 90% yield, 94% ee). Encouraged by these results, isomeric 2-methyl-3-vinylindoles were also evaluated, and the corresponding tetrahydrocarbazoles bearing a quaternary stereogenic center at the C2-position were produced with excellent yields and enantioselectivities (up to 97% yield, 98% ee, >20:1 E/Z).
 | ||
| Scheme 37 Au(I)/DTBM-Segphos-catalyzed asymmetric dearomative [4+2] cycloadditions of 3/2-substituted 2/3-vinylindoles and N-allenamides. | ||
In a proposed mechanism (Scheme 38), these transformations proceeded via a stepwise pathway, commencing with nucleophilic attack of the indole's C2/C3 position on the terminal allene carbon to generate dearomatized cationic intermediates 174/175. The regioselectivity on the allene substrates was governed by the electron-withdrawing group on the nitrogen. The second bond was formed by intramolecular cyclization on the C–Au bond, thus affording the final products and regenerating the Au(I) catalyst.
 | ||
| Scheme 38 Proposed mechanism for the Au(I)-catalyzed [4+2] cycloadditions of substituted vinylindoles and N-allenamides. | ||
In 2017, the Mikami group realized the first gold(I)-catalyzed asymmetric intermolecular [4+2] cycloadditions between 1,3-dienes and ynones.124 Using pyrrolidyl phosphoramidite L28 as the chiral ligand, bicyclo[2.2.2]octadiene derivatives 178 were obtained in high yields and enantioselectivities from cyclohexadiene 176 and ynones 177 (Scheme 39). In control experiments, when substituted ynones were utilized as substrates, the desired products could not be afforded or generated in very low yields, thus indicating the key role of terminal alkynes in this transformation. Systematic investigations, including NMR studies and non-linear effect experiments, suggested the existence of the gem-digold ynone complex. In a proposed catalytic cycle, gem-digold species 179 was initially generated. Then, the Diels–Alder reaction of 179 with the cyclohexadiene occurred efficiently in a stepwise or concerted reaction pathway.
 | ||
| Scheme 39 Au(I)/phosphoramidite-catalyzed asymmetric intermolecular [4+2] cycloadditions of ynones and cyclohexa-1,3-dienes. | ||
The resulting gem-digold intermediate 181 was protonated to release the chiral bicyclo[2.2.2]octadiene product and regenerate the cationic Au(I) catalyst.
4. Enantioselective [4+2] cycloadditions via the HOMO activation of π-bases
In contrast to electrophilic Au(I) complexes, which demonstrated a weak π-backdonation ability, some low-valent metals exhibited stronger π-backdonation than σ-donation when complexed with alkenes or alkynes, thus serving as π-Lewis bases to increase the electron density of unsaturated hydrocarbons. Based on this mode, Harman and others developed various impressive transformations of inert arenes and 1,3-dienes by the π-base activation of Os(II), Rh(I), Mo(0), and W(0) complexation.125–131 Very recently, a breakthrough in this research field was made by the Chen group, who identified that Pd(0) complexes could serve as π-Lewis base catalysts.51,132–136 By increasing the HOMO energy of unsaturated hydrocarbons, many types of reactions, including some unprecedented [4+2] cycloadditions, were realized elegantly.Cyclopentadienones were privileged electron-deficient dienes in Diels–Alder reactions, which were widely used in the synthesis of polycyclic aromatic hydrocarbons (PAHs).137–139 However, the parent cyclopentadienone without substituents was rarely utilized in organic synthesis owing to its high tendency to undergo self-dimerization.140–142 In 2021, the Chen group disclosed that 4-hydroxy-2-cyclopentadienone carbonates could be used as precursors of cyclopentadienones (Scheme 40).143 In the presence of a Pd(0) catalyst, the oxidative addition of the 4-hydroxy-2-cyclopentenone carbonate produced the key π-allylpalladium intermediate 183, which then underwent α-deprotonation and isomerization (β-H elimination) to afford the cyclopentadienone intermediate. By forming η2-complex 185 with the regenerated Pd(0) catalyst, the cyclopentadienone was stabilized, thus avoiding the facile dimerization. Remarkably, the HOMO energy of 185 was raised to −5.26 eV (vs. −7.03 eV for free cyclopentadienone) by the π-backbonding from the Pd(0) center as the π-Lewis base, thus enabling the uncoordinated alkene in cyclopentadienone as an electron-rich dienophile in inverse-electron-demand Diels–Alder (IEDDA) reactions.
 | ||
| Scheme 40 Pd(0)-catalyzed umpolung asymmetric inverse-electron-demand Diels–Alder reactions of 4-hydroxy-2-cyclopentenone carbonates. | ||
Using the Pd2(dba)3/L29 complex as the π-Lewis base catalyst, the umpolung aza-type IEDDA reactions between 4-hydroxy-2-cyclo-pentenone derivatives 187 and 1-azadienes 188 were realized. Notably, these reactions demonstrated excellent functional group tolerance toward 1-azadienes 188, affording chiral nitrogen atom-fused cyclic products 189 in high yields and outstanding enantioselectivities (up to 98% yield, 99% ee, >19:1 dr, Scheme 40). Additionally, employing L30 as the chiral ligand, 9-nitroanthracene 190 could also be used effectively as a diene partner, yielding the bridged [4+2] cycloadduct 191 with 95% yield and >99% ee. Intriguingly, cycloheptatrienone 192 was also an effective electron-rich 2π component in aza-Diels–Alder cyclization with 1-azadiene 193. Notably, the regioselectivity was controlled by ligands. Regiodivergent cycloadducts 194 and 195 were afforded in moderate yields and high enantioselectivities by the utilization of L30 and L31, respectively.
Based on the same π-Lewis base activation strategy, the Chen group developed the Pd(0)-catalyzed asymmetric inverse-electron-demand oxa-Diels–Alder reactions between 4-hydroxy-2-cyclopentenone carbonates 187 and α,β-unsaturated ketones 196 (Scheme 41).144 By this method, a series of chiral fused dihydropyrans 197 with versatile structural and functional diversity, generally with high stereoselectivity, could be constructed by the utilization of bifunctional aminoalcohol-derived monophosphine ligand L30 or proline-derived bisphosphine ligand L32. Remarkably, a ligand-dependent stereodivergence was observed. Employing L30 as the chiral ligand produced exo-cycloadducts exclusively (up to >19:1 dr), while the utilization of L32 furnished endo-cycloadducts with high diastereoselectivities (up to >19:1 dr). DFT calculations revealed that the [4+2] cycloaddition exhibited a concerted reaction pathway between the in situ generated η2-Pd(0)-cyclopentadienone complex and α,β-unsaturated ketone. With L30 as the ligand, a hydrogen-bonding interaction was observed between the N–H group of the ligand and the O-atom of cyclopentadienone in the [4+2] cycloaddition transition state, thus forcing the cycloaddition to proceed in an exo-fashion.
 | ||
| Scheme 41 Pd(0)-catalyzed umpolung asymmetric inverse-electron-demand oxa-Diels–Alder reactions of 4-hydroxy-2-cyclopentenone carbonates and α-cyano chalcones. | ||
To expand the generality of the Pd(0)-catalyzed asymmetric IEDDA reactions, the Chen group disclosed that dienones generated from Morita–Baylis–Hillman (MBH) carbonates 198 could be umpolunged to serve as electron-rich dienophile components (Scheme 42).145 By regioselective coordination of the Pd(0) catalyst on the exo-double bond, η2-Pd(0)-dienone complexes 202 were formed, with raised HOMO energy of the endo-double bond through π-Lewis base activation. By the utilization of Pd2(dba)3/L33 and 200 as co-catalysts, enantioselective inverse-electron-demand oxa-Diels–Alder reactions between MBH carbonates 198 and α-cyano chalcones 199 were realized. Based on this method, a variety of cis-pyran derivatives 201 with fused architectures were produced efficiently with high yields, high enantioselectivities, and high diastereoselectivities (up to 92% yields, 96% ee, >19:1 dr).
 | ||
| Scheme 42 Pd(0)-catalyzed asymmetric inverse-electron-demand oxa-Diels–Alder reactions of MBH carbonates and α-cyano chalcones. | ||
In 2023, the same group reported asymmetric 2,4-dienylation/intramolecular [4+2] cycloaddition cascade reactions by tandem Pd(0) catalysis (Scheme 43).146 The reaction commenced with the intermolecular allylation between ortho-functionalized aryl enones 203 and 2,4-dienyl carbonates 204 to give the dienyl enone intermediate 206. Then, the HOMO energy of the diene moiety of 207 was raised by π-Lewis base activation of the Pd(0) catalyst, thus facilitating the enantioselective intramolecular 1,4-addition. After the π–σ–π allylic isomerization, the resulting η3-complex underwent the intramolecular allylic alkylation to produce the final product. Using L34 as the chiral ligand, various ortho-functionalized aryl enones and 2,4-dienyl carbonates bearing aryl, alkyl, and alkynyl substituents were compatible in this cascade reaction, affording a wide range of enantioenriched spirocyclic architectures 205 with good yields and excellent ee (up to 97% yield, 97% ee).
 | ||
| Scheme 43 Pd(0)-catalyzed tandem asymmetric Tsuji–Trost allylation and intramolecular [4+2] cycloaddition reactions. | ||
It has been reported that in uncatalyzed IEDDA reactions of aurone-derived 1-azadienes 209 and 1,3-cyclohexadiene 210, the endo-selectivity was often observed, giving cis-fused polycyclic product exclusively. In 2023, the Chen group demonstrated that the diastereoselectivity of this reaction could precisely be reversed by the ligand. In the presence of catalytic amounts of Pd2(dba)3 and L35, [4+2] cycloadditions of 209 and 210 proceeded in an exo fashion, giving cis-fused tetrahydropyridine cycloadduct 211 in high yields and enantioselectivities (up to 99% yield, 98% ee) (Scheme 44).147 It was proposed that the coordination of the Pd(0) catalyst with 210 generated HOMO-energy-elevated η2-Pd(0) complex 212, which then underwent 1,4-addition with 209. From the resulting allylic Pd(II) species, the subsequent intramolecular allylic amination afforded the final product and regenerated the Pd(0) catalyst. By judicious selection of chiral ligand L36, the diastereoselectivity of the [4+2] annulation was switched, leading to the construction of trans-fused tetrahydropyridines 211 as main products (up to 98% yield, 98% ee, >19:1 dr). Remarkably, 1,3-cyclopentadiene, 1,3-cycloheptadiene, and acyclic 1,3-dienes were also compatible in the reaction, thus highly expanding the reaction generality.
 | ||
| Scheme 44 Pd(0)-catalyzed formal inverse-electron-demand aza-Diels–Alder reactions between 1,3-cyclohexadiene and aurone-derived 1-azadienes. | ||
In 2024, the Chen group reported a relay catalytic strategy involving Au-catalyzed cycloisomerization and Pd(0)/CPA co-catalyzed asymmetric [4+2] cycloaddition between enynamides 214 and 4-hydroxy-2-cyclopentenone carbonate derivatives 182 (Scheme 45).148 The reaction sequence commenced with the Ph3PAuNTf2-catalyzed intramolecular 5-endo-dig cyclization of enynamides 214, which efficiently generated dihydrofuran-fused azadiene intermediates 216. Subsequently, a cooperative catalytic system comprising Pd2(dba)3/L37 and chiral phosphoric acid 217 enabled the asymmetric formal [4+2] cycloadditions between in situ formed dihydrofuran-fused azadiene intermediates 216 and 4-hydroxyl-2-cyclopentanone carbonates 182. In line with previous studies (Scheme 40), carbonates 182 were proposed to convert to the corresponding η2-Pd(0)-cyclopentadienone intermediates via sequential oxidative addition, α-deprotonation and isomerization. Owing to the strong backbonding effect of the Pd(0) catalyst, the cyclopentadienone could serve as an electron-rich dienophile to react with 216. Remarkably, this cascade process demonstrated a wide reaction scope, affording various 6,7-dihydro-5H-cyclopenta[b]furo[3,2-e] pyridine scaffolds with excellent yields and enantioselectivities (up to 99% yield, 99% ee).
 | ||
| Scheme 45 Asymmetric synthesis of furo[2,3-b]pyridines by sequential Au(I) and Pd(0)/chiral phosphoric acid catalysis. | ||
An enantioselective formal aza-[4+2] cycloaddition between 1,3-dienes and N-cyano imines was realized by Pd(0)–π-Lewis base catalysis (Scheme 46).149 By the combination of Pd2(dba)3, monophosphine ligand L38, and chiral squaramide 221, the [4+2] cycloadditions of 218 and 219 went on smoothly to afford multifunctional piperidine derivatives with high yields and moderate enantioselectivities (up to 93% yield, 82% ee). In a proposed mechanism, 1,3-dienes 218 were activated by the Pd(0) catalysis with elevated HOMO energy, thereby promoting the vinylogous 1,2-addition with electron-deficient N-cyano imines 219. The resulting allylic Pd(II) intermediate 223 underwent the subsequent intramolecular allylic amination to give the final product and regenerate the Pd(0) catalyst. It should be noted that the enantioselectivity was controlled by chiral squaramide 221, which served as a Brønsted acid to increase the electrophilicity of 219.
 | ||
| Scheme 46 Pd(0)-catalyzed asymmetric aza-[4+2] cycloadditions of acyclic 1,3-dienes with N-cyano imines. | ||
The cross-Diels–Alder reaction between two different 1,3-dienes often gave a complex mixture, owing to the challenge in controlling the periselectivity, regioselectivity, and stereoselectivity simultaneously. As we mentioned before, Cramer and Braconi realized the enantioselective cross-[4+2] cycloaddition between two different unactivated 1,3-dienes by iron catalysis (Scheme 29).102 In 2024, the Cai group reported a cooperative Pd(0)/chiral phosphoric acid catalytic system, enabling the first chemo-, regio- and enantioselective sequential cross-[4+2] cycloaddition/decarboxylation of 2-pyrones150–152225 with unactivated acyclic 1,3-dienes 226 (Scheme 47).153 By this method, a wide range of chiral vinyl-1,3-cyclohexadiene derivatives 227, which were challenging to be accessed by other methods, were obtained under mild reaction conditions with excellent yields and enantioselectivities (up to 92% yield, 99% ee).
 | ||
| Scheme 47 Pd(0)/chiral phosphoric acid co-catalyzed periselective and enantioselective cross-[4+2] cycloaddition and decarboxylation reactions. | ||
Based on systematic experimental investigations and DFT calculations, a reasonable mechanism was proposed (Scheme 47). The catalytic cycle of the [4+2] cycloaddition was initiated with the coordination of 1,3-diene 226 with the Pd(0)/L39 complex, forming the η2-complex 229 with increased nucleophilicity by π-backbonding from the Pd(0) center interaction. Concurrently, the electrophilicity of 2-pyrone 225 was elevated by hydrogen-bonding activation with the sterically hindered chiral phosphoric acid 228. This synergistic catalytic system enabled the stereoselective 1,6-addition of complex 229 to 2-pyrone 225, generating the zwitterionic intermediate 231. Regioselective intramolecular palladium-catalyzed allylic substitution then occurred at the C3 position of 225, giving the exo-cycloadduct 232 as the key intermediate. In the decarboxylation cycle, the lactone group of 233 was activated by 228, thus promoting the oxidative addition with the Pd(0)/L39 complex from the back of the lactone moiety. Since the palladium center was far away from the nascent carboxylic group in the resulting intermediate 235, the decarboxylation occurred directly without the assistance of Pd(II). This step furnished the final 1,3-cyclohexadiene product 227 while regenerated both the Pd(0) catalyst and chiral phosphoric acid 228 to complete the catalytic cycle. Notably, the key for the success of this transformation was the utilization of an achiral NHC ligand, which highly increased the electron density of the palladium center.
In 2024, the Chen group reported the regioselective and enantioselective cross inverse-electron-demand [4+2] cycloadditions between 2,4-dienyl carbonyls and 1-heterodienes or allylodenemalononitriles by the Pd(0)–π-Lewis base catalysis (Scheme 48).154 In these reactions, the reactivity of the electron-deficient dienes 236 was reversed by regioselective γ,δ-η2 coordination with the Pd(0) catalyst, which donated its d-electron by π-backbonding, thus rendering 236 as electron-rich dienophiles in [4+2] cycloadditions with 237. By the combination of catalytic amounts of Pd2(dba)3 and the chiral ligand L40, various types of 1-heterodienes or allylodenemalononitriles were compatible in this reaction, affording a wide range of multifunctional six-membered cyclic compounds 238 with excellent yields and enantioselectivities (up to 99% yield and 99% ee).
 | ||
| Scheme 48 Pd(0)-catalyzed asymmetric cross inverse-electron-demand [4+2] cycloadditions between 2,4-dienyl carbonyls and 1-heterodienes/allylidenemalononitriles. | ||
5. Catalytic asymmetric radical-mediated [4+2] cycloadditions
Free radical reactions play a central role in organic synthesis owing to their high reactivity and efficiency.155–160 In recent years, radical-mediated [4+2] cycloadditions have received widespread interest from the synthetic community.161,162 Compared with the classic ionic Diels–Alder reaction, which typically occurs between an electron-rich component and an electron-deficient partner, the radical-mediated [4+2] cycloaddition could utilize simple and electronically mismatched unsaturated hydrocarbon substrates, thus making this reaction more general.51–56 However, due to the lack of catalytic modes to manipulate the stereochemistry of highly reactive radical species, the development of catalytic asymmetric radical-mediated [4+2] cycloaddition remains a challenging topic in organic synthesis. It is not until very recently, a few breakthroughs have emerged, which are deemed to be inspiring for more research work in this field. In this section, we will discuss these advancements according to the mechanistic reaction pathways.5.1. Catalytic asymmetric radical cation [4+2] cycloadditions
Electronically mismatched Diels–Alder reactions between two electron-rich components are regarded as one of the most challenging reaction modes, which often require harsh reaction conditions and a longer reaction time. In this regard, the radical cation Diels–Alder reaction is highly attractive. In the presence of an oxidative electron transfer catalyst, the reaction proceeds through one-electron oxidation of the electron-rich olefins to form a radical cation intermediate, followed by the subsequent cycloaddition with electron-rich dienes, thereby increasing the reaction rate by several orders of magnitude. Since the pioneering work of Bauld and co-workers, the radical cation Diels–Alder reaction has been extensively investigated by the development of one-electron oxidative catalysts, such as ground-state aminium salts, Fe(III) salts, and photoinitiated electron transfer with organic photosensitizers.163–171 Despite these great achievements, catalytic asymmetric radical cation [4+2] cycloadditions remain elusive, and only two examples have been reported to date.The first catalytic asymmetric radical cation [4+2] cycloaddition was reported by the Nicewicz group in 2018 (Scheme 49).172 By the development of a novel class of photoredox catalytic system comprising an oxidizing triaryl pyrylium (TP) salt bearing a chiral N-triflyl phosphoramide counterion, the intramolecular [4+2] cycloadditions of unactivated diene-enes 239 were achieved successfully, albeit with moderate enantioselectivities (up to 50% ee). In a proposed mechanism, the reaction was initiated by single-electron oxidation of the olefin moiety in diene-ene 239via the excited-state catalyst 241, thus generating radical cation intermediate 242. Subsequent intramolecular cycloaddition of 220 gave radical cation intermediate 243, which then underwent single-electron reduction to furnish the final trans-5,6-fused bicycle products 240, while simultaneously regenerating the catalyst. Although only moderate enantioselectivities were obtained for the desired products, this study made an important proof-of-concept that ion pairing between radical cation intermediates and chiral anions was capable of inducing enantiocontrol in radical cation [4+2] cycloadditions.
 | ||
| Scheme 49 Cationic oxopyrylium-catalyzed asymmetric radical cation intramolecular [4+2] cycloadditions of unactivated diene-enes. | ||
A breakthrough in this research field was made by the Ishihara group very recently (Scheme 50).173 In 2023, they reported a judicious design of a chiral Fe(III) photoredox catalyst generated in situ from FeCl3 and a chiral silver(I) phosphate salt. In the presence of catalytic amounts of FeCl3 (5 mol%) and silver phosphoramide 247 (16.5 mol%), the intermolecular [4+2] cycloadditions between chalcone derivatives 245 and isoprene 244 went on smoothly to provide the corresponding cycloadducts 246 in good yields with high enantioselectivities (up to 81% yield, 94% ee). It should be noted that the methyl group of 246 was at the meta-position of the carbonyl group, which exhibited the opposite regioselectivity compared with Lewis acid-mediated Diels–Alder reactions based on the Woodward–Hoffmann rule.
 | ||
| Scheme 50 Asymmetric radical cation [4+2] cycloadditions catalyzed by the combination of FeCl3 and silver N-triflylphosphoramide. | ||
In a proposed mechanism, FeCl3 underwent anion exchange with three equivalents of 247 to generate the chiral FeX3 salt in situ. Upon irradiation with blue LEDs, this Fe(III) complex was excited and subsequently quenched with the chalcone derivative 245 through one-electron oxidation to afford FeX2 and radical cation intermediate 248 paired with the chiral phosphoramide anion (X−). The stereochemistry of the subsequent radical cation-induced [4+2] cycloaddition of 248 was controlled by the chiral anion to produce the radical cation intermediate 249. Finally, the one-electron reduction of 250 with FeX2 or chalcone derivative 245 delivered the cyclohexene product 246 with good enantioselectivities. Notably, this catalytic system was also applicable to the intermolecular asymmetric [2+2] cycloadditions of chalcone derivatives and styrenes, thus demonstrating the generality of this chiral counteranion strategy.
According to the above proposed mechanism, only one chiral phosphoramide anion (X−) was involved in the enantio-determining step. Consequently, the Ishihara group then developed a new chiral Fe(III) photoredox catalyst, which consisted of a chiral phosphoramide anion and an achiral bidentate ligand (Scheme 51).174 By the catalysis of the in situ generated 253 from FeCl3, achiral disilver salt 254, and silver phosphoramide 247, the intermolecular [4+2] cycloadditions between chalcone derivatives 251 and 1,3-butadiene derivatives 252 proceeded enantioselectively, leading to the corresponding cycloadducts with high yields and ee. By this strategy, only one equivalent of the chiral silver salt was needed. Notably, this new catalysis design also benefitted from the tunability of the achiral ligand in the disilver salt to achieve good stereocontrol.
 | ||
| Scheme 51 A new Fe(III) photocatalyst consisting of a chiral phosphoramide anion and an achiral bidentate ligand. | ||
5.2. Catalytic asymmetric triplet-state [4+2] cycloadditions
Besides the single-electron-transfer (SET) reaction pathway, the photocatalysis through the energy transfer process has also emerged as a powerful approach to facilitate [4+2] cycloadditions that are hard to be achieved by traditional methods.53 However, the high reactivity of radical species and excited-state intermediates poses an impetus to achieving high enantiocontrol by chiral catalysts. In 1990, the Schuster group reported the first enantioselective photochemical [4+2] cycloaddition.175 By the catalysis of enantioenriched (–)-1,1′-bis(2,4-dicyanonaphthalene) [BDCN] 259 (2.5 mol%, 70% ee) under irradiation (350 nm) in toluene, the [4+2] cycloaddition between 1,3-cyclohexadiene (CHD) 256 and trans-β-methylstyrene 257 gave the corresponding cycloadduct 258 in 15% ee (Scheme 52). Mechanistically, it was proposed that photosensitizer 259 initially formed a pair of diastereomeric exciplexes with prochiral dienophile 257 by different facial orientations, which then reacted with CHD 256 to form a triplex, thereby enabling the subsequent [4+2] cycloaddition. The observed enantioselectivity originated from the selective capture of the diastereomeric exciplexes with CHD 256, establishing the stereochemical basis for the asymmetric induction in this transformation. | ||
| Scheme 52 The BDCN-catalyzed photosensitized [4+2] cycloaddition of trans-β-methylstyrene with 1,3-cyclohexadiene. | ||
The first catalytic asymmetric photochemical [4+2] cycloadditions with high enantiocontrol were reported by the Jiang group in 2024 (Scheme 53).176 By the cooperative catalysis of photosensitizer dicyanopyrazine (DPZ) 263 and chiral phosphoric acid 264, the dearomative [4+2] cycloadditions between anthracenes 260 and alkenyl-azaarenes 256 went on smoothly under irradiation of visible light. By this method, a series of high-value cycloadducts incorporating the privileged azaarene motif were obtained in excellent yields with high enantioselectivities and diastereoselectivities (up to 99% yield, 99% ee, and >20:1 dr). The synthetic utility was unambiguously demonstrated by the construction of all-carbon quaternary stereocenters bearing multiple adjacent stereocenters. In a plausible mechanism, the DPZ sensitizer underwent intersystem crossing (ISC) to generate the triplet excited state 3DPZ. Subsequent triplet–triplet energy transfer of 3DPZ with 255 gave the triplet diradical species 260. Meanwhile, alkenyl-azaarene 261 complexed with chiral Brønsted acid 264via the hydrogen-bonding interaction to establish a chiral environment for the subsequent radical addition with 266. The resulting diradical intermediate 267 underwent ISC followed by the diradical coupling, affording final [4+2] cycloadduct 262. It should be noted that the pyrene groups on the chiral phosphoric acid were essential for the good enantioselectivity control, which were proposed to have π–π stacking interactions with substrates.
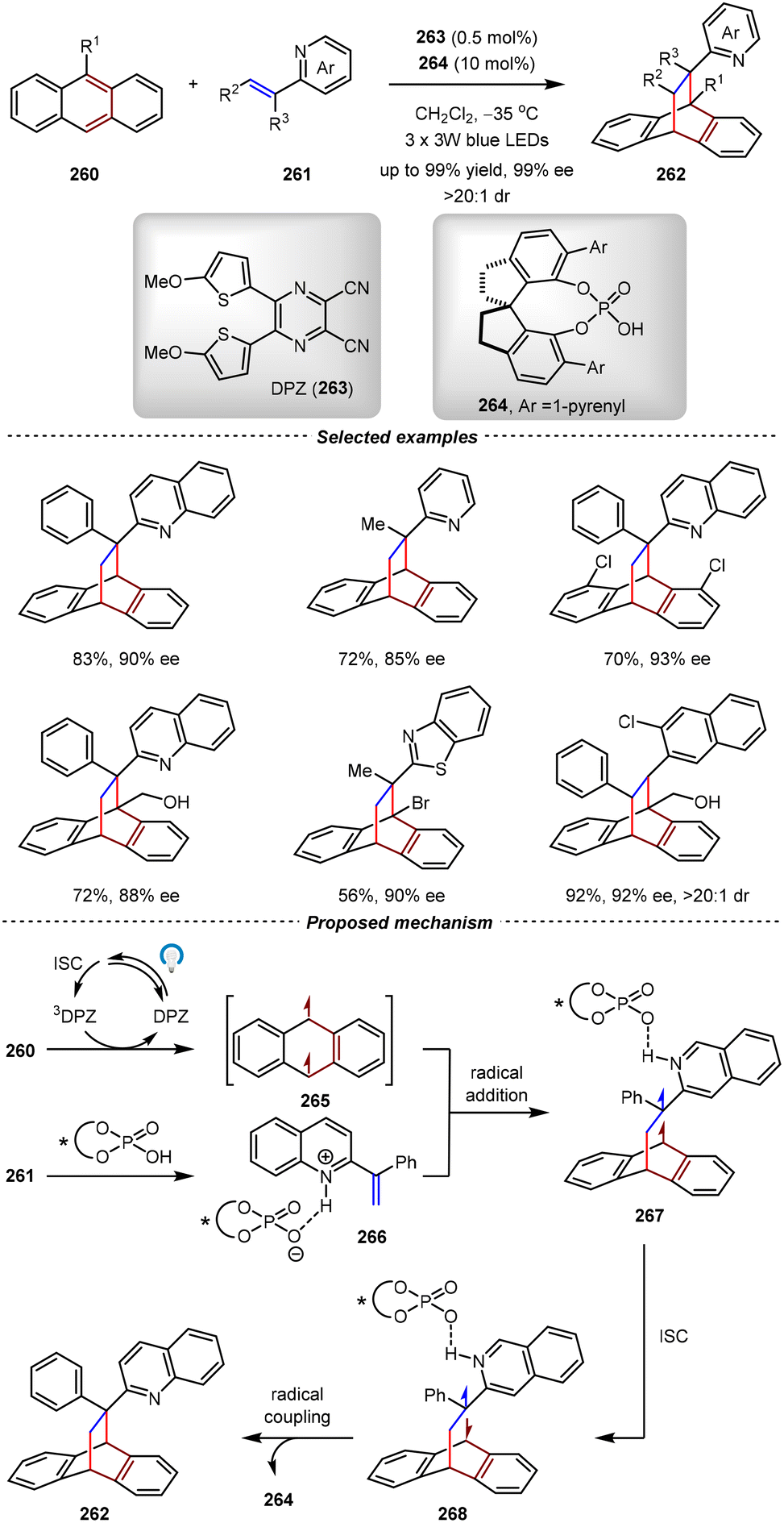 | ||
| Scheme 53 Asymmetric [4+2] dearomative photocycloadditions of anthracene with alkenylazaarenes. | ||
Almost at the same time, the You group reported the Lewis acid catalyzed asymmetric dearomative (CADA) [4+2] cycloadditions of naphthalene derivatives (Scheme 54).177 By the catalysis of Gd(OTf)3 and PyBox L41 under visible light irradiation, the dearomative [4+2] cycloadditions of naphthalenes and styrenes delivered a diverse range of bridged polycyclic products with excellent yields and enantioselectivities (up to 96% yield, >99% ee, >20:1 dr, and >20:1 rr). Notably, phenyl allene also showed good reactivity, giving the corresponding diene product in 61% yield and 93% ee. By extensive mechanistic investigations, it was found that the Gd(III)/L41 complex served dually as both the excited-state catalyst and the stereocontrol modulator. Initially, the coordination of Gd(III)/L41 with 269 produced ground-state intermediate 272, which then generated singlet excited-state 272* under blue LED irradiation. From 272*, triplet-state intermediate 273 was afforded via ISC. This reactive diradical species then mediated the stereoselective Si-face addition with 270, producing a new biradical intermediate 274. The subsequent ISC process enabled the intramolecular radical recombination to give intermediate 275, which then underwent ligand exchange with 269, thus simultaneously releasing the desired product 271 and regenerating intermediate 272 to complete the catalytic cycle. This effective photocatalytic dearomatization strategy opened a new gate for the development of future photochemical asymmetric [4+2] cycloaddition reactions.
 | ||
| Scheme 54 Gd(III)-catalyzed asymmetric [4+2] photocycloadditions of naphthalenes and styrenes. | ||
6. Conclusions
Over the past three decades, the field of organic synthesis has witnessed remarkable progress in catalytic asymmetric [4+2] cycloadditions involving alkenes, alkynes, and allenes, extending well beyond the conventional Diels–Alder reaction. Transition metal catalysis, in particular, enables these transformations by coordinating with unsaturated C–C bonds in reactants and facilitating subsequent oxidative cyclometallation and reductive elimination to afford [4+2] cycloadducts. Through careful selection of transition metals and ligands, high levels of chemo-, regio-, and stereoselectivity of [4+2] cycloadditions can be achieved. On another front, π-acidic Au(I) catalysts activate allenes to generate highly reactive Au-allyl cation species, which readily undergo intramolecular or intermolecular [4+2] cycloadditions with 1,3-dienes. More recently, low-valent Pd(0) complexes have emerged as efficient π-Lewis base catalysts that raise the HOMO energy of unsaturated hydrocarbons via η2 coordination, enabling previously inaccessible [4+2] cycloadditions. Notably, photo-induced catalytic asymmetric [4+2] cycloadditions proceeding via the radical cation or triplet-state pathway have introduced unique and powerful reaction modes. By these innovative strategies, [4+2] cycloadditions have been significantly extended to unactivated unsaturated hydrocarbons, which are typically unreactive substrates under conventional Diels–Alder reactions.Despite the notable achievements realized to date, the reaction scope and patterns of [4+2] cycloadditions beyond conventional Diels–Alder reactions remain limited. In transition metal-catalyzed [4+2] cycloadditions following the oxidative cyclometallation/reductive elimination pathway, current studies are predominantly focused on intramolecular reactions. In contrast, intermolecular [4+2] cycloadditions are rare and often suffer from low reactivity, along with poor control over stereoselectivity and regioselectivity. Regarding π-acid and π-base activation modes, only Au(I) and Pd(0) complexes have been employed as catalysts, leaving other transition metals largely unexplored. Notably, catalytic asymmetric radical-mediated [4+2] cycloadditions are still in infancy, especially when compared to the rapidly advancing field of photoinduced enantioselective [2+2] cycloadditions. To overcome these challenges, continued development of innovative catalysts and ligands, as well as new reaction modes is of great importance.
Author contributions
Q. C. conceptualized the concept, and J.-X. H. and Q. C. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review.Acknowledgements
Financial support from the National Natural Science Foundation of China (grant no. 22471043 and 22222104), the Shanghai Pilot Program for Basic Research, and Fudan University is gratefully acknowledged.Notes and references
- P. M. Dewick, Medicinal natural products: a biosynthetic approach, Wiley, Chichester, UK, 2009 Search PubMed.
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- S. Vaz Jr, Sustainable agrochemistry: a compendium of technologies, Springer, Cham, Switzerland, 2019 Search PubMed.
- J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed.
- O. Diels and K. Alder, Justus Liebigs Ann. Chem., 1928, 460, 98–122 CrossRef CAS.
- G. Brieger and J. N. Bennett, Chem. Rev., 1980, 80, 63–97 CrossRef CAS.
- W. Oppolzer, Angew. Chem., Int. Ed. Engl., 1984, 23, 876–889 CrossRef.
- J. D. Winkler, Chem. Rev., 1996, 96, 167–176 CrossRef CAS PubMed.
- K. C. Nicolaou, S. A. Snyder, T. M. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS.
- K. Takao, R. Munakata and K. Tadano, Chem. Rev., 2005, 105, 4779–4807 CrossRef CAS PubMed.
- Q. Cai, Chin. J. Chem., 2019, 37, 946–976 CrossRef CAS.
- J. Huang, R. Yin and T. Cao, Acta Chim. Sin., 2025, 83, DOI:10.6023/A25040131.
- Y. Ma, Y. Wang, F. Wang, S. Lu and X. Chen, Chin. Chem. Lett., 2025, 36, 110546 CrossRef CAS.
- R. Hoffmann and R. B. Woodward, J. Am. Chem. Soc., 1965, 87, 2046–2048 CrossRef CAS.
- R. B. Woodward and R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1969, 8, 781–853 CrossRef CAS.
- R. Hoffmann and R. B. Woodward, Acc. Chem. Res., 1968, 1, 17–22 CrossRef CAS.
- K. Fukui, T. Yonezawa and H. Shingu, J. Chem. Phys., 1952, 20, 722–725 CrossRef CAS.
- K. Fukui, in A Simple Quantum-Theoretical Interpretation of the Chemical Reactivity of Organic Compounds, Molecular Orbitals in Chemistry, Physics, and Biology, Academic Press, New York, 1964, pp. 513–537 Search PubMed.
- K. N. Houk, Acc. Chem. Res., 1975, 8, 361–369 CrossRef CAS.
- S. Hashimoto, N. Komeshima and K. Koga, J. Chem. Soc., Chem. Commun., 1979, 437–438 RSC.
- H. Takemura, N. Komeshima, I. Takahashi, S. Hashimoto, N. Ikota, K. Tomioka and K. Koga, Tetrahedron Lett., 1987, 46, 5687–5690 CrossRef.
- H. B. Kagan and O. Riant, Chem. Rev., 1992, 92, 1007–1019 CrossRef CAS.
- U. Pindur, G. Lutz and C. Otto, Chem. Rev., 1993, 93, 741–761 CrossRef CAS.
- E. J. Corey, Angew. Chem., Int. Ed., 2002, 41, 1650–1667 CrossRef CAS PubMed.
- W. Notz, F. Tanaka and C. F. Barbas, Acc. Chem. Res., 2004, 37, 580–591 CrossRef CAS PubMed.
- G. Desimoni, G. Faita and K. A. Jørgensen, Chem. Rev., 2006, 106, 3561–3651 CrossRef CAS PubMed.
- K. Ishihara, M. Fushimi and M. Akakura, Acc. Chem. Res., 2007, 40, 1049–1055 CrossRef CAS PubMed.
- P. Merino, E. Marqués-López, T. Tejero and R. P. Herrera, Synthesis, 2010, 1–26 CrossRef CAS.
- R. Wang and X. Jiang, Chem. Rev., 2013, 113, 5515–5546 CrossRef PubMed.
- X.-H. Liu, H. Zheng, Y. Xia, L. Lin and X. Feng, Acc. Chem. Res., 2017, 50, 2621–2631 CrossRef CAS PubMed.
- B. R. Lichman, S. E. O’Connor and H. Kries, Chem. – Eur. J., 2019, 17, 6864–6877 CrossRef.
- V. Laina-Martín, J. A. Fernández-Salas and J. Alemán, Chem. – Eur. J., 2021, 27, 12509–12520 CrossRef PubMed.
- G. Zhao, S. A. Blaszczy and J. Wang, Green Synth. Catal., 2021, 2, 198–215 Search PubMed.
- M.-M. Xu and Q. Cai, Chin. J. Org. Chem., 2022, 42, 698–713 CrossRef CAS.
- T. Bauer, Molecules, 2025, 30, 1978 CrossRef CAS PubMed.
- L. Dell’Amico, A. Vega-Peñaloza, S. Cuadros and P. Melchiorre, Angew. Chem., Int. Ed., 2016, 55, 3313–3317 CrossRef PubMed.
- K. N. Flesch, A. Q. Cusumano, P.-J. Chen, C. S. Strong, S. R. Sardini, Y. E. Du, M. D. Bartberger, W. A. Goddard III and B. M. Stoltz, J. Am. Chem. Soc., 2023, 145, 11301–11310 CrossRef CAS PubMed.
- M. Lautens, W. Klute and W. Tam, Transition Metal-Mediated Cycloaddition Reactions, Chem. Rev., 1996, 96, 49–92 CrossRef CAS PubMed.
- S. Saito and Y. Yamamoto, Chem. Rev., 2000, 100, 2901–2915 CrossRef CAS PubMed.
- C. Auvert, L. Fensterbank, P. Garcia, M. Malacria and A. Simonneau, Chem. Rev., 2011, 111, 1954–1993 CrossRef PubMed.
- P. A. Inglesby and P. A. Evans, Chem. Soc. Rev., 2010, 39, 1954–1993 RSC.
- L.-N. Wang and Z.-X. Yu, Chin. J. Org. Chem., 2020, 40, 3536–3558 CrossRef CAS.
- Y. Zhang, Y. Tang and Y.-Y. Zhou, Chin. J. Org. Chem., 2025, 45, 1–21 CrossRef CAS.
- A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410–3449 CrossRef PubMed.
- A. Fürstner, Acc. Chem. Res., 2014, 47, 925–938 CrossRef PubMed.
- E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326–3350 CrossRef PubMed.
- C. Aubert, L. Fensterbank, P. Garcia, M. Malacria and A. Simonneau, Chem. Rev., 2011, 111, 1954–1993 CrossRef CAS PubMed.
- A. Marinetti, H. Jullien and A. Voituriez, Chem. Soc. Rev., 2021, 41, 4884–4908 RSC.
- D. Campeau, D. F. León Rayo, A. Mansour, K. Muratov and F. Gagosz, Chem. Rev., 2021, 121, 8756–8867 CrossRef CAS PubMed.
- Y. Que, W. Lei, Y. Fang, S. He and Y. Chen, Green Synth. Catal., 2024, 5, 270–276 Search PubMed.
- Z.-C. Chen, Q. Ouyang, W. Du and Y.-C. Chen, J. Am. Chem. Soc., 2024, 146, 6422 CrossRef CAS PubMed.
- Y. Inoue, Chem. Rev., 1992, 92, 741–770 CrossRef CAS.
- S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS PubMed.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- J. Großkopf, T. Kratz, T. Rigotti and T. Bach, Chem. Rev., 2022, 122, 1626–1653 CrossRef PubMed.
- M. Zhu, X. Zhang, C. Zheng and S.-L. You, Acc. Chem. Res., 2022, 55, 2510–2525 CrossRef CAS PubMed.
- Y. Yin, M. You, X. Li and Z. Jiang, Chem. Soc. Rev., 2025, 54, 2246–2274 RSC.
- R. S. Jolly, G. Luedtke, D. Sheehan and T. Livinghouse, J. Am. Chem. Soc., 1990, 112, 4965–4966 CrossRef CAS.
- L. McKinstry and T. Livinghouse, Tetrahedron, 1994, 50, 6145–6154 CrossRef CAS.
- S. R. Gilbertson and G. S. Hoge, Tetrahedron Lett., 1998, 39, 2075–2078 CrossRef CAS.
- S. R. Gilbertson, G. S. Hoge and D. G. Genov, J. Org. Chem., 1998, 63, 10077–10080 CrossRef CAS.
- H. Heath, B. Wolfe, T. Livinghouse and S. K. Bae, Synthesis, 2001, 2341–2347 CrossRef CAS.
- A. Falk, L. Fiebig, J.-M. Neudörfl, A. Adler and H.-G. Schmalz, Adv. Syn. Catal., 2011, 353, 3357–3362 CrossRef CAS.
- K. Aikawa, S. Akutagawa and K. Mikami, J. Am. Chem. Soc., 2006, 128, 12648–12649 CrossRef CAS PubMed.
- S.-J. Paik, U. K. Son and Y. K. Chung, Org. Lett., 1999, 1, 2045–2047 CrossRef CAS.
- R. Shintani, Y. Sannohe, T. Tsuji and T. Hayashi, Angew. Chem., Int. Ed., 2007, 46, 7277–7280 CrossRef CAS PubMed.
- T. Shibata, D. Fujiwara and K. Endo, Org. Biomol. Chem., 2008, 6, 464–467 RSC.
- Y. Miyauchi, K. Noguchi and K. Tanaka, Org. Lett., 2012, 14, 5856–5859 CrossRef CAS PubMed.
- R. L.-Y. Bao, J. Yin, L. Shi and L. Zheng, Org. Biomol. Chem., 2020, 18, 2956–2961 RSC.
- P. A. Wender, T. E. Jenkins and S. Suzuki, J. Am. Chem. Soc., 1995, 117, 1843–1844 CrossRef CAS.
- B. M. Trost, D. R. Fandrick and D. C. Dinh, J. Am. Chem. Soc., 2005, 127, 14186–14187 CrossRef CAS PubMed.
- Y. Han and S. Ma, Org. Chem. Front., 2018, 5, 2680–2684 RSC.
- Y. Han, A. Qin and S. Ma, Chin. J. Chem., 2019, 37, 486–496 CrossRef CAS.
- J. Li and S. R. Gilbertson, Org. Lett., 2021, 23, 2911–2914 CrossRef CAS PubMed.
- J. Ye, S. Li, B. Chen, W. Fan, J. Kuang, J. Liu, Y. Liu, B. Miao, B. Wan, Y. Wang, X. Xie, Q. Yu, W. Yuan and S. Ma, Org. Lett., 2012, 14, 1346–1349 CrossRef CAS PubMed.
- X. Huang, T. Cao, Y. Han, X. Jiang, W. Lin, J. Zhang and S. Ma, Chem. Commun., 2015, 51, 6956–6959 RSC.
- A. Qin, Q. Zhang, H. Qian, Y. Han and S. Ma, Chin. J. Chem., 2021, 39, 559–565 CrossRef CAS.
- Y. Han, A. Qin, Q. Zhang, X. Zhang, H. Qian and S. Ma, Angew. Chem., Int. Ed., 2022, 61, e202211635 CrossRef CAS PubMed.
- M. Murakami, K. Itami and Y. Ito, J. Am. Chem. Soc., 1997, 119, 7163–7164 CrossRef CAS.
- M. Murakami, R. Minamida, K. Itami, M. Sawamura and Y. Ito, Chem. Commun., 2000, 2293–2294 RSC.
- C. Spino, J. Crawford, Y. Cui and M. Gugelchuk, J. Chem. Soc., Perkin Trans. 2, 1998, 1499–1506 RSC.
- J.-X. He, Q.-T. Lu, T. Zhang, R.-Y. Gao, Y.-S. Cui, Y. Lan and Q. Cai, Nat. Catal., 2025, 8, 1348–1360 CrossRef CAS.
- T. Shibata, K. Takasaku, Y. Takesue, N. Hirata and K. Takagi, Synlett, 2002, 1681–1682 CrossRef CAS.
- B. M. Trost, R. E. Brown and F. D. Toste, J. Am. Chem. Soc., 2000, 122, 5877–5878 CrossRef CAS.
- D. Kosser and N. Cramer, J. Am. Chem. Soc., 2015, 137, 12478–12481 CrossRef PubMed.
- D. Kosser and N. Cramer, Chima, 2017, 71, 186–189 CrossRef PubMed.
- Y. Tamaru, Modern organonickel chemistry, Wiley-VCH, Weinheim, German, 2005 Search PubMed.
- P. A. Wender and I. C. Nathan, J. Am. Chem. Soc., 1986, 108, 4678–4679 CrossRef CAS.
- P. A. Wender and T. E. Jenkins, J. Am. Chem. Soc., 1989, 111, 6432–6434 CrossRef CAS.
- W. Brenner, P. Heimbach, H. Hey, E. W. MüIler and G. Wilke, Liebigs Ann. Chem., 1969, 727, 161–182 CrossRef CAS.
- P. Heimbach and H. Schenkhdm, Top. Curr. Chem., 1980, 92, 45–108 CrossRef CAS.
- G. Wilke, Angew. Chem., Int. Ed. Engl., 1988, 27, 185–206 CrossRef.
- P. J. Garratt and M. Wyat, J. Chem. Soc., Chem. Commun., 1974, 251 RSC.
- I. Suisse, H. Bricout and A. Mortreux, Tetrahedron Lett., 1994, 35, 413–416 CrossRef CAS.
- A. EIAmrani, I. Suisse, N. Knouzi and A. Mortreux, Tetrahedron Lett., 1995, 36, 5011–5014 CrossRef.
- G. Zhang, C.-Y. Zhao, X.-T. Min, Y. Li, X.-X. Zhang, H. Liu, D.-W. Ji, Y.-C. Hu and Q.-A. Chen, Nat. Catal., 2022, 5, 708–715 CrossRef CAS.
- C. Bolm, J. Legros, J. L. Paih and L. Zani, Chem. Rev., 2004, 104, 6217–6254 CrossRef CAS PubMed.
- A. Correa, O. G. Mancheño and C. Bolm, Chem. Soc. Rev., 2008, 37, 1108–1117 RSC.
- I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed.
- P. J. Chirik, Angew. Chem., Int. Ed., 2022, 61, 5170–5181 Search PubMed.
- E. Braconi, A. C. Götzinger and N. Cramer, J. Am. Chem. Soc., 2020, 142, 19819–19824 CrossRef CAS PubMed.
- E. Braconi and N. Cramer, Angew. Chem., Int. Ed., 2017, 56, e202112148 Search PubMed.
- G. Hilt, Synlett, 2023, 23–28 CrossRef CAS.
- P. Röse and G. Hilt, Synthesis, 2016, 463–492 Search PubMed.
- W. Hess, J. Treutwein and G. Hilt, Synthesis, 2008, 3537–3562 CAS.
- S. Biswas, M. M. Parsutkar, S. M. Jing, V. V. Pagar, J. H. Herbot and T. V. Rajanbabu, Acc. Chem. Res., 2021, 54, 4545–4564 CrossRef CAS PubMed.
- V. V. Pagar and T. V. Rajanbabu, Science, 2018, 361, 68–72 CrossRef CAS PubMed.
- M. M. Parsutkar, V. V. Pagar and T. V. Rajanbabu, J. Am. Chem. Soc., 2019, 141, 15367–15377 CrossRef CAS PubMed.
- G. Hilt and F.-X. du Mesnil, Tetrahedron Lett., 2000, 41, 6757–6761 CrossRef CAS.
- G. Hilt and K. I. Smolko, Angew. Chem., Int. Ed., 2003, 42, 2795–2797 CrossRef CAS PubMed.
- G. Hilt, J. Janikowski and W. Hess, Angew. Chem., Int. Ed., 2006, 45, 5204–5206 CrossRef CAS PubMed.
- D. Singh and T. V. RajanBabu, Angew. Chem., Int. Ed., 2023, 62, e202216000 CrossRef CAS PubMed.
- K. K. Ghosh, R. Chowdhury, J. P. Gordon and T. V. RajanBabu, Angew. Chem., Int. Ed., 2025, 64, e202515154 CrossRef CAS PubMed.
- D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2018, 108, 3351–3378 CrossRef PubMed.
- A. Fürstner, Chem. Soc. Rev., 2009, 38, 3203–3221 RSC.
- A. S. K. Hashimi, Angew. Chem., Int. Ed., 2010, 49, 5232–5241 CrossRef PubMed.
- E. Jiménez-Núñez and A. M. Echavarren, Chem. Commun., 2007, 333–346 RSC.
- P. Mauleó, R. M. Zeldin, A. Z. González and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 6348–6349 CrossRef PubMed.
- I. Alonso, B. Trillo, F. López, S. Montserrat, G. Ujaque, L. Castedo, A. Lledós and J. L. Mascareñas, J. Am. Chem. Soc., 2009, 131, 13020–13030 CrossRef CAS PubMed.
- A. Z. González and F. D. Toste, Org. Lett., 2010, 12, 200–203 CrossRef PubMed.
- J. Francos, F. Grande-Carmona, H. Faustino, J. Iglesias-Sigüenza, E. Díez, I. Alonso, R. Fernádez, J. M. Lassaletta, F. López and J. L. Mascareñas, J. Am. Chem. Soc., 2012, 134, 14322–14325 CrossRef CAS PubMed.
- Y. Wang, P. Zhang, Y. Liu, F. Xia and J. Zhang, Chem. Sci., 2015, 6, 5564–5570 RSC.
- V. Pirovano, M. Borri, G. Abbiati, S. Rizzato and E. Rossi, Adv. Synth. Catal., 2017, 359, 1912–1918 CrossRef CAS.
- M. Nanko, S. Shibuya, Y. Inaba, S. Ono, S. Ito and K. Mikami, Org. Lett., 2018, 20, 7353–7357 CrossRef CAS PubMed.
- J. M. Keane and W. D. Harman, Organometallics, 2005, 24, 1786–1798 CrossRef CAS.
- B. K. Liebov and W. D. Harman, Chem. Rev., 2017, 117, 13721–13755 CrossRef CAS PubMed.
- J. A. Smith, K. B. Wilson, R. E. Sonstrom, P. J. Kelleher, K. D. Welch, E. K. Pert, K. S. Westendorff, D. A. Dickie, X. P. Wang, B. H. Pate and W. D. Harman, Nature, 2020, 581, 288–293 CrossRef CAS PubMed.
- K. B. Wilson, J. T. Myers, H. S. Nedzbala, L. A. Combee, M. Sabat and W. D. Harman, J. Am. Chem. Soc., 2017, 139, 11401–11412 CrossRef CAS PubMed.
- A. W. Lankenau, D. A. Iovan, J. A. Pienkos, R. J. Salomon, S. S. Wang, D. P. Harrison, W. H. Myers and W. D. Harman, J. Am. Chem. Soc., 2015, 137, 3649–3655 CrossRef CAS PubMed.
- W. Liu, F. You, C. J. Mocella and W. D. Harman, J. Am. Chem. Soc., 2006, 128, 1426–1427 CrossRef CAS PubMed.
- M. L. Spera, R. M. Chin, M. D. Winemiller, K. W. Lopez, M. Sabat and W. D. Harman, Organometallics, 1996, 15, 5447–5449 CrossRef CAS.
- B.-X. Xiao, B. Jiang, R.-J. Yan, J.-X. Zhu, K. Xie, X.-Y. Gao, Q. Ouyang, W. Du and Y.-C. Chen, J. Am. Chem. Soc., 2021, 143, 4809–4816 CrossRef CAS PubMed.
- J.-X. Zhu, Z.-C. Chen, W. Du and Y.-C. Chen, Angew. Chem., Int. Ed., 2022, 61, e202200880 CrossRef CAS PubMed.
- S.-Z. Tan, P. Chen, L. Zhu, M.-Q. Gan, Q. Ouyang, W. Du and Y.-C. Chen, J. Am. Chem. Soc., 2022, 144, 22689–22697 CrossRef CAS PubMed.
- P. Chen, S.-Z. Tan, L. Zhu, Q. Ouyang, Z.-J. Jia, W. Du and Y.-C. Chen, Angew. Chem., Int. Ed., 2023, 62, e202301519 CrossRef CAS PubMed.
- Y.-F. Li, W.-T. Gui, F. Pi, Z. Chen, L. Zhu, Q. Ouyang, W. Du and Y.-C. Chen, Angew. Chem., Int. Ed., 2024, 63, e202407682 CrossRef CAS PubMed.
- M. A. Ogliaruso, M. G. Romanelli and E. I. Becker, Chem. Rev., 1965, 65, 261–367 CrossRef CAS.
- A. Narita, X.-Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC.
- X.-Y. Wang, X. Yao and K. Müllen, Sci. China Chem., 2019, 62, 1099–1144 CrossRef CAS.
- F. Gavina, A. M. Costero, P. Gil, B. Palazon and S. V. Luis, J. Am. Chem. Soc., 1981, 103, 1797–1798 CrossRef CAS.
- P. G. Baraldi, A. Barco, S. Benetti, G. P. Pollini, E. Polo and D. Simoni, J. Chem. Soc., Chem. Commun., 1984, 1049–1050 RSC.
- S. Dong, T. Qin, E. Hamel, J. A. Beutler and J. A. Porco, J. Am. Chem. Soc., 2012, 134, 19782–19787 CrossRef CAS PubMed.
- X.-X. Yang, R.-J. Yan, G.-Y. Ran, C. Chen, J.-F. Yue, X. Yan, Q. Ouyang, W. Du and Y.-C. Chen, Angew. Chem., Int. Ed., 2021, 60, 26762–26768 CrossRef CAS PubMed.
- X.-X. Yang, X.-L. Zhao, Q. Ouyang, W. Du and Y.-C. Chen, Org. Chem. Front., 2022, 9, 1364–1369 RSC.
- J.-L. Cheng, F. Liu, X. Song, Z. Zhao, W. Du and Y.-C. Chen, J. Org. Chem., 2023, 88, 7800–7809 CrossRef PubMed.
- J.-X. Zhu, F. Pi, T. Sun, W.-Y. Huang, L. Gao, Z.-C. Chen, W. Du and Y.-C. Chen, Org. Lett., 2023, 25, 3682–3686 CrossRef CAS PubMed.
- Y. Hu, J.-Y. Huang, R.-J. Yan, Z.-C. Chen, Q. Ouyang, W. Du and Y.-C. Chen, Chem. Sci., 2023, 14, 1896–1901 RSC.
- K. Xie, G.-Q. Zhang, B.-R. Kong, Z.-J. Jia and Z.-C. Chen, Org. Chem. Front., 2024, 11, 3900–3905 RSC.
- J. Zhang, Y. Hu, T.-Y. Zhang, Z.-C. Chen, W. Du and Y.-C. Chen, Org. Lett., 2023, 25, 8133–8138 CrossRef CAS PubMed.
- M.-M. Xu and Q. Cai, Chin. J. Org. Chem., 2022, 42, 698–713 CrossRef CAS.
- X. Song, Y. Zhang, P. Ji, F. Zeng, F. Bi and W. Wang, Green Synth. Catal., 2020, 1, 66–69 Search PubMed.
- Y. Zhou, Z. Zhou, W. Du and Y. Chen, Acta Chim. Sinica, 2018, 76, 382–386 CrossRef CAS.
- M.-M. Xu, P.-P. Xie, J.-X. He, Y.-Z. Zhang, C. Zheng and Q. Cai, J. Am. Chem. Soc., 2024, 146, 6936–6946 CrossRef CAS PubMed.
- K. Xie, B.-R. Kong, C.-Q. Wang, Z.-C. Chen, W. Du and Y.-C. Chen, Org. Lett., 2025, 27, 7058–7063 CrossRef CAS PubMed.
- L. F. Tietze, Chem. Rev., 1996, 96, 115–136 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- M. Yan, J. C. Lo and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS PubMed.
- P. Xiong and H. C. Xu, Acc. Chem. Res., 2019, 52, 3339–3350 CrossRef CAS PubMed.
- J. Huang, R. Ying and T. Cao, Acta Chim. Sin., 2025, 83, 1424–1434 CrossRef CAS.
- S. Perveen, L. Qin, M. Zhao, Z. Ding, Y. Wang, Z. Nie and P. Li, Chin. Chem. Lett., 2026, 37, 111886 CrossRef CAS.
- D. J. Bellville, D. W. Wirth and N. L. Bauld, J. Am. Chem. Soc., 1981, 103, 718–720 CrossRef CAS.
- N. L. Bauld, D. J. Bellville, R. Pabon, R. Chelsky and G. Green, J. Am. Chem. Soc., 1983, 105, 2378–2382 CrossRef CAS.
- D. J. Beliviile, N. L. Bauld, R. Pabon and S. A. Gardner, J. Am. Chem. Soc., 1983, 105, 3584–3588 CrossRef.
- R. A. Pabon, D. J. Bellville and N. L. Bauld, J. Am. Chem. Soc., 1983, 105, 5158–5159 CrossRef CAS.
- G. C. Calhoun and G. B. Schuster, J. Am. Chem. Soc., 1984, 106, 6870–6871 CrossRef CAS.
- J. Mlcoch and E. Steckhan, Angew. Chem., Int. Ed. Engl., 1985, 24, 412–414 CrossRef.
- A. Gieseler, E. Steckhan, O. Wiest and F. Enoch, J. Org. Chem., 1991, 56, 1405–1411 CrossRef CAS.
- T. Horibe, S. Ohama and K. Ishihara, J. Am. Chem. Soc., 2019, 141, 1877–1881 CrossRef CAS PubMed.
- Y. Yu, Y. Fu and F. Zhong, Green Chem., 2018, 20, 1743–1747 RSC.
- P. D. Morse, T. M. Nguyen, C. L. Cruz and D. A. Nicewicz, Tetrahedron, 2018, 74, 3266–3272 CrossRef CAS PubMed.
- S. Ohamura, K. Kitagiri, H. Kato, T. Horibe, S. Miyakawa, J. Hasegawa and K. Ishihara, J. Am. Chem. Soc., 2023, 28, 15054–15060 CrossRef PubMed.
- H. Akao, S. Ohmura and K. Ishihara, J. Am. Chem. Soc., 2026, 148, 4867–4872 CrossRef CAS PubMed.
- J.-I. Kim and G. B. Schuster, J. Am. Chem. Soc., 1990, 112, 9635–9637 CrossRef CAS.
- D. Tian, W. Shi, X. Sun, X. Zhao, Y. Yin and Z. Jiang, Nat. Commun., 2024, 15, 4563 CrossRef CAS PubMed.
- M. Li, X.-L. Huang, Z.-Y. Zhang, Z. Wang, Z. Wu, H. Yang, W.-J. Shen, Y.-Z. Cheng and S.-L. You, J. Am. Chem. Soc., 2024, 146, 16982–16989 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |