
Harnessing rare earth coordination chemistry for advanced photofunctional materials: from fundamental principles to emerging technologies
Fu-Jia
Song
a,
Pingru
Su
*a,
Xue
Li
a,
Chun-Hua
Yan
 ab,
Jun-Long
Zhang
*b and
Yu
Tang
*ac
ab,
Jun-Long
Zhang
*b and
Yu
Tang
*ac
aKey Laboratory of Nonferrous Metal Chemistry and Resources Utilization of Gansu Province, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou, 730000, P. R. China. E-mail: supr@lzu.edu.cn; tangyu@lzu.edu.cn
bBeijing National Laboratory for Molecular Sciences, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, P. R. China. E-mail: zhangjunlong@pku.edu.cn
cState Key Laboratory of Baiyunobo Rare Earth Resource Researches and Comprehensive Utilization, Baotou Research Institute of Rare Earths, Baotou 014030, P. R. China
First published on 28th April 2026
Abstract
Rare earth (RE) ions, characterized by unique 4f electronic configurations and shielded f–f transitions, serve as exceptional optical centers exhibiting narrow-band emission, long-lived luminescence, and rich energy-level structures. The construction of high-performance RE coordination-based photofunctional materials critically relies on the synergistic integration of the “antenna effect”, provided by meticulously designed organic ligands, and the distinctive excited-state properties of RE ions. This molecular engineering approach not only maximizes the intrinsic photophysical advantages of RE elements, but also enables the precise tailoring of materials for diverse cutting-edge applications. This review provides a systematic and comprehensive analysis of RE coordination-based photofunctional materials, spanning from fundamental design principles and controllable synthesis strategies to emerging applications. We delve into the structure–activity relationships across various categories, including molecular complexes, supramolecular assemblies, coordination polymers, metal–organic frameworks (MOFs), and RE-covalent-bonded organic frameworks (RE-COFs). Furthermore, we highlight their transformative roles in optoelectronics, advanced anti-counterfeiting, biomedical imaging/therapy, radiation detection (scintillators) and photochemical catalysis. Finally, we outline current challenges and future perspectives, aiming to inspire interdisciplinary innovation and accelerate the commercialization of next-generation RE molecular photonic materials.
 Fu-Jia Song | Fu-Jia Song is currently a PhD student under the supervision of Professor Yu Tang in the College of Chemistry and Chemical Engineering at Lanzhou University. He earned his BS degree in applied chemistry from Shandong University. His primary research interests lie in the construction of chiral supramolecular assemblies and the study of chiral transfer. |
 Pingru Su | Pingru Su is a Youth Researcher and Master's Supervisor at Lanzhou University. He earned his PhD in Inorganic Chemistry from Lanzhou University and conducted postdoctoral research at Shenzhen University. His current research interests focus on the self-assembly of rare earth complexes and their applications. |
 Xue Li | Xue Li is currently a PhD student under the supervision of Professor Yu Tang in the College of Chemistry and Chemical Engineering at Lanzhou University. Her primary research interest focuses on perovskite quantum dots and rare earth-based organic–inorganic hybrid materials. |
 Chun-Hua Yan | Chun-Hua Yan received his PhD in 1988 from Peking University, China. He was promoted to full professor in the Department of Chemistry at Peking University in 1992. He serves as Editor-in-Chief for J. Rare Earths (Elsevier). He was elected as a Member of Chinese Academy of Sciences (2011) and Fellow of the Academy of Sciences for the Developing World (TWAS, 2012). His research focuses on rare earth functional materials, rare earth separation processes, and their industrial applications. |
 Jun-Long Zhang | Jun-Long Zhang received his BSc from Sichuan University (1997), MSc from the Chengdu Institute of Organic Chemistry, Chinese Academy of Sciences (2000), and PhD from the University of Hong Kong. After postdoctoral research at the University of Illinois at Urbana-Champaign, he started his independent academic career at Peking University and now he is a full professor. Professor Zhang's research focuses on bioinorganic chemistry, metal medicines, and spin chemical biology. He has received several honors, including a Wiley-ACES Excellent Young Professor Award, a Distinguished Lectureship Award, being named an Emerging Investigator in Bioinorganic Chemistry (American Chemical Society), and a Chemistry Europe Early Career Award. He was the winner of the inaugural AsBIC James Hoeschele Award (2020). |
 Yu Tang | Yu Tang is a Changjiang Scholar Professor of Inorganic Chemistry and a PhD supervisor at the College of Chemistry and Chemical Engineering, Lanzhou University. She received her BS (1993), MS (1996) and PhD (1999) degrees in Inorganic Chemistry from Lanzhou University, where she also conducted her postdoctoral research. Her research focuses on rare earth coordination chemistry and functional materials, with an emphasis on smart luminescent systems and energy-related catalysis. She is a Fellow of the Chinese Chemical Society (FCCS) and the Royal Society of Chemistry (FRSC). She has been serving as an associate editor for Inorganic Chemistry (ACS) from 2021 to present. |
1. Introduction
Rare earth (RE) elements, distinguished by their unique [Xe]4fn electronic configurations, have become indispensable in modern science and technology owing to the exceptional photophysical properties of their trivalent ions (RE3+). The partially filled and shielded 4f orbitals give rise to sharp, line-like emission, long luminescence lifetimes, and a broad spectrum spanning from the ultraviolet to the near-infrared region.1 However, the same 4f–4f electronic transitions responsible for these superior optical characteristics are Laporte-forbidden, resulting in intrinsically weak light absorption and severely limiting direct excitation efficiency. This fundamental drawback is effectively circumvented by the “antenna effect”, wherein judiciously designed organic ligands serve as efficient light-harvesting units. First identified by Weissman in 19422 and systematically developed by Crosby and Whan in the 1960s,3 this mechanism involves ligand photoexcitation, intersystem crossing, and energy transfer to the RE3+ center, thereby sensitizing its characteristic luminescence.4,5 RE coordination-based photofunctional materials refer to a class of materials in which RE elements serve as the central optical units and are chemically bonded to organic ligands or other molecular components. By integrating the distinctive optical signatures of RE ions with the structural and functional tunability of molecular systems, these materials combine the intrinsic advantages of RE photophysics with the designability of coordination-based materials.6–8Beyond sensitization, the incorporation of organic components establishes a powerful molecular design paradigm. The structural diversity and synthetic flexibility of organic ligands enable precise regulation of the coordination environment, energy transfer pathways, and overall material architecture. This synergistic interplay allows for the rational engineering of luminescence efficiency, emission color, stability, and stimulus-responsive behavior, thereby forming the foundation of RE coordination-based photofunctional materials. Situated at the dynamic convergence of coordination chemistry, materials science, and photonics, rare earth coordination-based photofunctional materials are currently undergoing rapid expansion, as evidenced by the following statistics since 2020: approximately 4000 publications focusing on rare earth complex design and synthesis, 2400 on rare earth metal–organic frameworks and coordination polymers, 2700 on upconversion and downconversion nanomaterials, 1200 on organic–inorganic hybrid systems, and 900 on device fabrication and photonic integration. These materials are driving breakthroughs in a wide range of applications, including next-generation displays,9,10 near-infrared bioimaging,11,12 photon-upconversion solar cells,13,14 and high-security optical encryption,15–18 highlighting their broad scientific and technological significance. For the broader scientific community, a comprehensive understanding of the structure–property relationships in RE molecular photonic materials is essential. Such insight not only offers clear guidelines for rational material design but also effectively facilitates progress toward sustainable energy solutions, scalable biomedical theranostics, and advanced optoelectronic devices. Accordingly, this review targets a multidisciplinary audience, including chemists, materials scientists, physicists, and biomedical researchers, with the goal of fostering cross-disciplinary collaboration and bridging the gap between fundamental discovery and practical applications.
Although numerous reviews on RE luminescent materials have been published, the existing literature remains largely fragmented by material class or application focus. In the area of molecular RE complexes, foundational reviews have advanced the understanding of their construction, photophysical properties, and applications in areas such as bioimaging and biosensing19 and as emitters in devices like OLEDs.20 Other reviews concentrated on specific ligand platforms, such as RE porphyrinoids for phototheranostics21 and their supramolecular assemblies.22 For extended framework materials, comprehensive studies have focused on RE MOFs, highlighting their structural diversity, optical properties, and applications from sensing to biomedicine.23,24 Related framework systems, such as covalent organic frameworks (COFs) and coordination polymers, have also been reviewed independently.25,26 In the nanomaterials domain, separate reviews address RE-doped nanoparticles with an emphasis on sensitization mechanisms and applications,27 their hybrid systems with carbon dots,28 specific functions like nanothermometry,29 and their doping in inorganic matrices.30 Additional reviews are devoted to highly specialized applications, such as in neuroscience.31 Notwithstanding these valuable contributions, a unifying and conceptually integrated perspective that systematically links molecular design, synthetic strategies, property regulation, and emerging applications across the full spectrum of material architectures from discrete complexes to extended frameworks remains notably lacking. Addressing this gap is essential for advancing the field beyond empirical optimization toward predictive and programmable materials design.
In this review, we present a cohesive framework organized along the logical trajectory of “molecular design → controlled synthesis → property regulation → emerging applications”. By explicitly correlating ligand–RE3+ molecular engineering with macroscopic functional performance, this approach provides a unified roadmap for the field. Accordingly, this review is structured as follows (Fig. 1). We first discuss the coordination chemistry and molecular design principles underlying the antenna effect and ligand engineering. We then systematically examine synthesis strategies and performance-regulation approaches, including molecular complexes, crystalline frameworks (MOFs/COFs), supramolecular assemblies, and hybrid composites.
 | ||
| Fig. 1 Schematic diagram of RE coordination-based photofunctional materials. | ||
Subsequently, we highlight emerging applications in three frontier areas: optical anti-counterfeiting, biomedical theranostics, and radiation scintillators. Finally, we address current challenges and future perspectives, outlining pathways toward sustainable and innovative next-generation materials. By integrating molecular-level insight with materials-level functionality, this review aims to serve as a comprehensive and forward-looking reference for researchers across chemistry, materials science, photonics, and biomedicine, and to stimulate cross-disciplinary innovation in the rapidly evolving landscape of RE molecular photonics.
2. Ligand engineering for tunable photophysics
The exceptional luminescence of RE3+ originates from their rich 4fn electronic configurations, which generate a dense manifold of closely spaced energy levels and more than 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 possible transition pathways.32,33 As a consequence, RE3+ ions exhibit absorption and emission spanning a broad spectrum from the ultraviolet (UV) to the near-infrared (NIR) regions (Fig. 2).34 Notably, the parity-forbidden 4f–4f transitions produce sharp, fingerprint-like line emissions characterized by high color purity, large effective Stokes shifts, and long excited-state lifetimes (micro- to- milliseconds).35–37 These intrinsic photophysical properties, which are largely shielded from environmental perturbation by outer 5s25p6 electrons, establish RE3+ ions as ideal “privileged” centers for constructing high-performance luminescent materials. Despite these advantages, the practical exploitation of RE3+ luminescence is fundamentally limited by the inherently low absorption cross-sections of RE3+ ions. This challenge is overcome through the pivotal “antenna effect”, in which tailored organic chromophores serve as efficient light-harvesting units.38,39 Following photoexcitation, these ligands absorb incident photons, undergo intersystem crossing to a triplet state (T1) and subsequently transfer energy to the RE3+ ion, thereby sensitizing its characteristic emission. The efficiency of this energy transfer is governed by the energy gap (ΔE) between the ligand's T1 state and the accepting energy level of the RE3+ ion. Optimal ΔE values of approximately 2500–3500 cm−1 for Eu3+ and 2500–4000 cm−1 for Tb3+40 ensure efficient energy transfer while suppressing back-transfer and non-radiative losses. Accordingly, rational ligand design focuses on three core antenna parameters: high molar absorptivity (ε) for effective light harvesting, appropriately positioned excited-state energy levels for resonant energy transfer, and rigid coordination environments that minimize non-radiative quenching pathways.41 It should be noted, however, that the antenna effect is not limited to triplet-state sensitization. Experimental evidence confirms direct energy transfer from the ligand singlet state, including S1 → ILCT1 → Eu3+ pathways42 and ultrafast S1 → Ln3+ transfer rates reaching up to 1012 s−1 for various lanthanides.43 These findings demonstrate that direct energy transfer from the ligand singlet state, whether via an intraligand charge transfer intermediate or through ultrafast direct coupling, can also significantly contribute to lanthanide sensitization.
000 possible transition pathways.32,33 As a consequence, RE3+ ions exhibit absorption and emission spanning a broad spectrum from the ultraviolet (UV) to the near-infrared (NIR) regions (Fig. 2).34 Notably, the parity-forbidden 4f–4f transitions produce sharp, fingerprint-like line emissions characterized by high color purity, large effective Stokes shifts, and long excited-state lifetimes (micro- to- milliseconds).35–37 These intrinsic photophysical properties, which are largely shielded from environmental perturbation by outer 5s25p6 electrons, establish RE3+ ions as ideal “privileged” centers for constructing high-performance luminescent materials. Despite these advantages, the practical exploitation of RE3+ luminescence is fundamentally limited by the inherently low absorption cross-sections of RE3+ ions. This challenge is overcome through the pivotal “antenna effect”, in which tailored organic chromophores serve as efficient light-harvesting units.38,39 Following photoexcitation, these ligands absorb incident photons, undergo intersystem crossing to a triplet state (T1) and subsequently transfer energy to the RE3+ ion, thereby sensitizing its characteristic emission. The efficiency of this energy transfer is governed by the energy gap (ΔE) between the ligand's T1 state and the accepting energy level of the RE3+ ion. Optimal ΔE values of approximately 2500–3500 cm−1 for Eu3+ and 2500–4000 cm−1 for Tb3+40 ensure efficient energy transfer while suppressing back-transfer and non-radiative losses. Accordingly, rational ligand design focuses on three core antenna parameters: high molar absorptivity (ε) for effective light harvesting, appropriately positioned excited-state energy levels for resonant energy transfer, and rigid coordination environments that minimize non-radiative quenching pathways.41 It should be noted, however, that the antenna effect is not limited to triplet-state sensitization. Experimental evidence confirms direct energy transfer from the ligand singlet state, including S1 → ILCT1 → Eu3+ pathways42 and ultrafast S1 → Ln3+ transfer rates reaching up to 1012 s−1 for various lanthanides.43 These findings demonstrate that direct energy transfer from the ligand singlet state, whether via an intraligand charge transfer intermediate or through ultrafast direct coupling, can also significantly contribute to lanthanide sensitization.
 | ||
| Fig. 2 (a) Typical emission spectra of RE ions (visible to NIR). (b) Simplified energy-level diagram (Dieke diagram) showing major 4fn transitions of RE3+. Reprinted with permission from ref. 34. Copyright 2025, Elsevier. | ||
Guided by the hard and soft acid–base theory, RE ions as classical hard acids exhibit strong affinity toward oxygen- and nitrogen-donors. Combined with their high and flexible coordination numbers, this chemical versatility has enabled the development of an extensive library of ligands and corresponding advanced materials. These systems span from discrete mono-, di-, and polynuclear complexes to sophisticated higher-order architectures including luminescent cages, clusters, supramolecular assemblies, coordination polymers, and hybrid composite materials.44–47 Representative ligand families that effectively sensitize RE3+ ions include: the β-diketone ligand,48–53 porphyrin ligand,54–59 tacn ligand,60,61 pyclen ligand,62,63 cyclen ligand,64–66 Schiff-base ligand,67–72 pyridine ligand,73–77N-oxide/phosphoryl ligand,78–84 and emerging chiral ligand systems85–88 (Fig. 3).
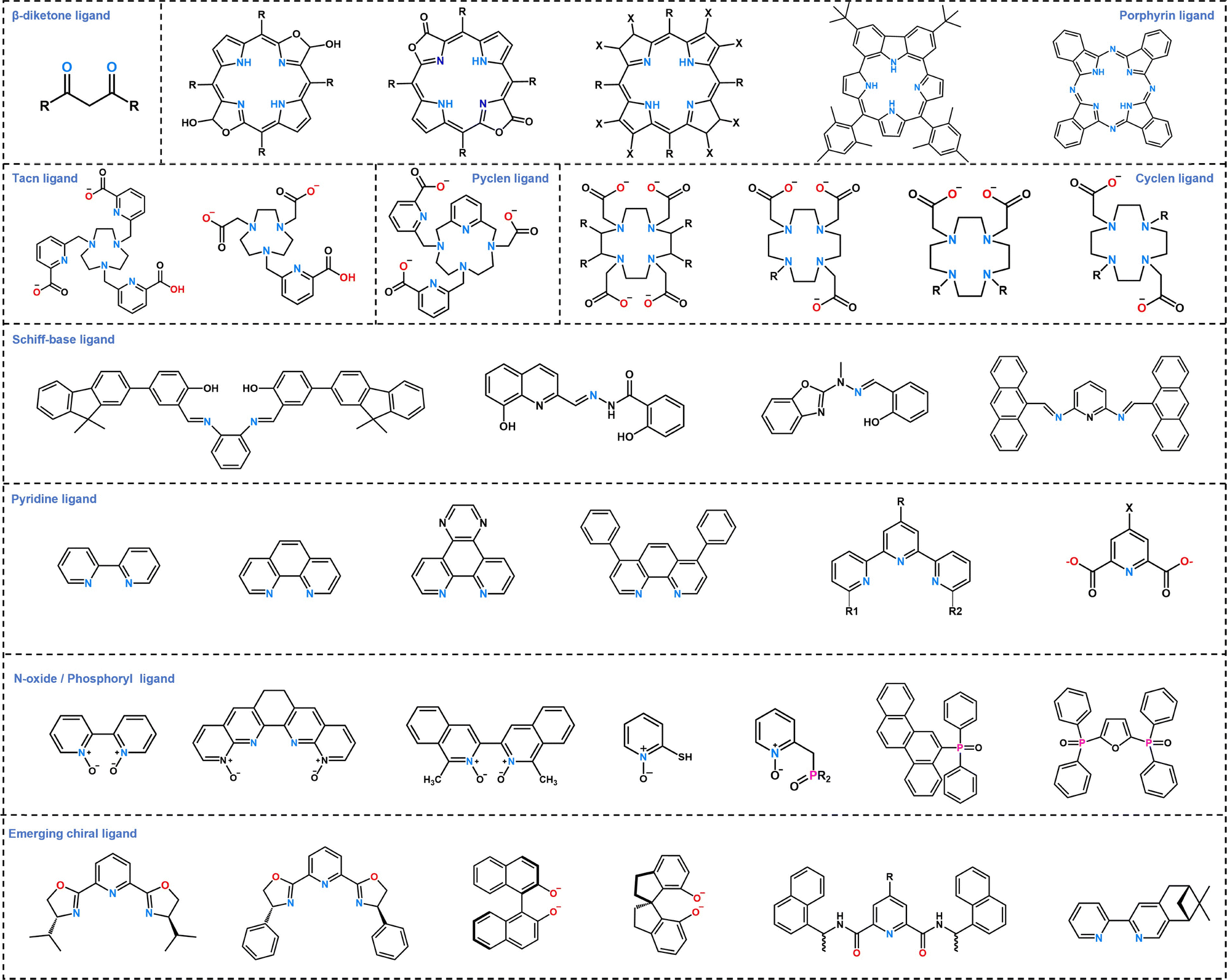 | ||
| Fig. 3 The structures of representative ligands for coordination with RE ions. | ||
Building upon this diverse ligand repertoire, ligand engineering emerges as a precise “art of molecular regulation”, enabling comprehensive and multidimensional control over RE photofunctional materials, spanning from photophysical input to functional output. Beyond the aforementioned ligands, ligand engineering has established a specialized category of antenna-metal complexes as mature molecular antenna systems for sensitizing lanthanide luminescence,89 where the core strategy employs transition metal centers, including Ru(II), Pt(II), and Zn(II), to construct efficient light-harvesting units. These units absorb visible light via metal-to-ligand charge transfer (MLCT) excited states and relay energy to lanthanide ions (e.g., Nd(III)/Yb(III)/Er(III)) through triplet energy transfer pathways. Key efficiency-enhancement approaches include: utilizing rigid bipyridine90/porphyrin ligands91 to enhance structural stability and suppress vibrational deactivation; precisely regulating donor triplet-to-acceptor excited-state energy gaps (e.g., matching the ∼6000 cm−1 gap for Ru(II) → Yb(III));92 intensifying superexchange interactions via cyanide-bridged supramolecular systems93 or reduced intermetallic distances (<10 Å)94 and mitigating solvent quenching through hydrophobic ligand encapsulation or solid-state matrices. These designs extend rare-earth complex excitation wavelengths into the visible spectrum while elevating near-infrared luminescence quantum yields to ∼10−2,95 thereby establishing a universal platform for novel bioprobes and optical communication materials.
At the level of light absorption and excitation, extending π-conjugation, covalently grafting highly absorbing organic dyes, or designing “nonlinear antenna” systems with large nonlinear absorption cross-sections allows the effective excitation window to be shifted from the UV into the visible or even NIR regions. Such strategies enable multi-photon excitation processes, overcoming the limitations of conventional UV excitation and opening new opportunities for deep-tissue bioimaging and optical communication.96–99
Beyond conventional ligand optimization strategies involving structural modifications (e.g., incorporation of As![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds for T1 state tuning),100 the enhancement of luminescence efficiency and quantum yield is achieved through optimized donor–acceptor energy transfer pathways and effective suppression of quenching effects within the acceptor's microenvironment. In addition to traditional triplet-state matching paradigms, the strategic integration of intraligand/ligand-to-ligand charge-transfer (ILCT/LLCT) states as efficient “energy relay stations” enables unconventional energy-transfer pathways. This approach circumvents the limitations of conventional S1 → T1 → Ln3+ models—specifically addressing the intersystem crossing bottleneck caused by large ΔEST gaps (>150 cm−1), by providing a direct conduit for excited-state energy migration from ligands to lanthanide ions (e.g., Eu3+), achieving a record Φ = 67% in model systems.101 Importantly, this experimental breakthrough validates the second operational mode of the antenna effect, where CT-mediated pathways bypass traditional Dexter energy-transfer constraints. The adoption of perfluorinated ligand design (–CH3 → –CF3 substitution) or fluoride-bridged architectures effectively mitigates C–H vibrational quenching (ν ∼ 2900 cm−1), resulting in a 19-fold enhancement of near-infrared quantum yield for the Er–Binol complex (Φ: 0.58% → 11%).102 Furthermore, the Tb-FC lanthanide cluster synthesized via a mixed-ligand strategy achieves exceptional emission efficiency, reaching Φ = 99% under optimized coordination conditions.103
O bonds for T1 state tuning),100 the enhancement of luminescence efficiency and quantum yield is achieved through optimized donor–acceptor energy transfer pathways and effective suppression of quenching effects within the acceptor's microenvironment. In addition to traditional triplet-state matching paradigms, the strategic integration of intraligand/ligand-to-ligand charge-transfer (ILCT/LLCT) states as efficient “energy relay stations” enables unconventional energy-transfer pathways. This approach circumvents the limitations of conventional S1 → T1 → Ln3+ models—specifically addressing the intersystem crossing bottleneck caused by large ΔEST gaps (>150 cm−1), by providing a direct conduit for excited-state energy migration from ligands to lanthanide ions (e.g., Eu3+), achieving a record Φ = 67% in model systems.101 Importantly, this experimental breakthrough validates the second operational mode of the antenna effect, where CT-mediated pathways bypass traditional Dexter energy-transfer constraints. The adoption of perfluorinated ligand design (–CH3 → –CF3 substitution) or fluoride-bridged architectures effectively mitigates C–H vibrational quenching (ν ∼ 2900 cm−1), resulting in a 19-fold enhancement of near-infrared quantum yield for the Er–Binol complex (Φ: 0.58% → 11%).102 Furthermore, the Tb-FC lanthanide cluster synthesized via a mixed-ligand strategy achieves exceptional emission efficiency, reaching Φ = 99% under optimized coordination conditions.103
Ligand engineering further enables dynamic programming of emission color and lifetime, with ligands functioning as both a “color palette” and a “time encoder”. Precise tuning of the ligand field or the deliberate construction of interionic energy transfer networks enables accurate adjustment from monochromatic purification to white-light emission.104–107 Meanwhile, controlling interionic distances and coupling strengths through ligand bridges allows luminescence lifetimes to be continuously programmed over hundreds of microseconds without altering emission, providing a powerful platform for time-resolved multiplexing, optical data storage, and information encryption.108–113 Finally, the integration of chiral motifs, photo- or chemo-responsive units, and catalytic functionalities into ligand frameworks enables the transition of RE materials from static emitters to dynamic, intelligent systems. Such designs have led to advanced functionalities such as circularly polarized luminescence,114 photo-switchable magneto-optical properties,115 and RE-based ultra-bright afterglow.116 This facilitates the transition from a static emitter to a dynamic “smart material”.117–123
In summary, ligand engineering provides a versatile “molecular toolbox” that enables the bottom-up construction and diversification of RE photofunctional material properties. By programming optical behavior at the molecular source, this approach offers unprecedented freedom in designing advanced luminescent systems, laying a robust foundation for future innovations in optoelectronics, information technology, and biotechnology.
3. Synthesis and performance modulation
Building upon the systematic elucidation of ligand engineering, which provides a fundamental framework for modulating RE photophysics through the rational control of frontier orbitals, spatial coordination environments, and vibrational landscapes to achieve “on-demand programming” of emission characteristics, we now confront the central challenge of translation: transforming these molecular-level design principles into practical, high-performance materials. Addressing this challenge requires the organized integration of discrete luminescent centers into well-defined architectures spanning multiple length scales and dimensionalities. Accordingly, the second part of this review is structured along the core progression of “molecular design → controlled synthesis → cross-scale performance regulation”. Our perspective expands from isolated molecule/ion pairs to hierarchical, ordered material systems, following the intrinsic structural hierarchy that governs their assembly and function.We will systematically demonstrate how ligand engineering principles are realized through precise synthetic and assembly strategies across ascending levels of architectural complexity. These range from discrete complexes (mononuclear/polynuclear species and molecular clusters and 1D coordination polymers) to supramolecular assemblies (constructed via non-covalent interactions like π–π stacking, hydrogen bonding, and electrostatics). We further extend this discussion to long-range ordered crystalline networks (including MOFs and COFs) and ultimately to organic–inorganic hybrid systems. This exposition maps a coherent translational pathway, progressing from well-defined molecular models with explicit structure–property relationships, through successive stages of assembly engineering (non-covalent driving), crystalline network engineering (coordination-directed), and interface engineering (heterogeneous coupling). Such a stepwise approach enables the construction of material systems with increasing structural complexity and functional integration, thereby bridging microscopic molecular attributes to macroscopic collective and intelligent functionalities.
3.1. Molecular complexes
Building upon the fundamental principles of ligand engineering established in Part 1, this section moves into the practical domain of synthesis, focusing on how discrete molecular and polynuclear complexes function as versatile platforms for both deciphering photophysical mechanisms and realizing advanced optical functions. We chart a progression from simplicity to complexity: beginning with mononuclear systems, where structure–property relationships can be unambiguously established; advancing to dinuclear and trinuclear architectures, in which deliberate intermetallic interactions give rise to cooperative and emergent behavior; and culminating in the rational design of complexes targeting advanced photonic phenomena such as upconversion and circularly polarized luminescence (CPL), wherein CPL is defined as the differential emission of left- and right-circularly polarized light from chiral luminescent systems, quantified by the dissymmetry factor (glum = 2ΔI/I, where ΔI = IL − IR).124 Throughout this discussion, a central theme that functional sophistication scales directly with synthetic precision in controlling molecular architecture emerges.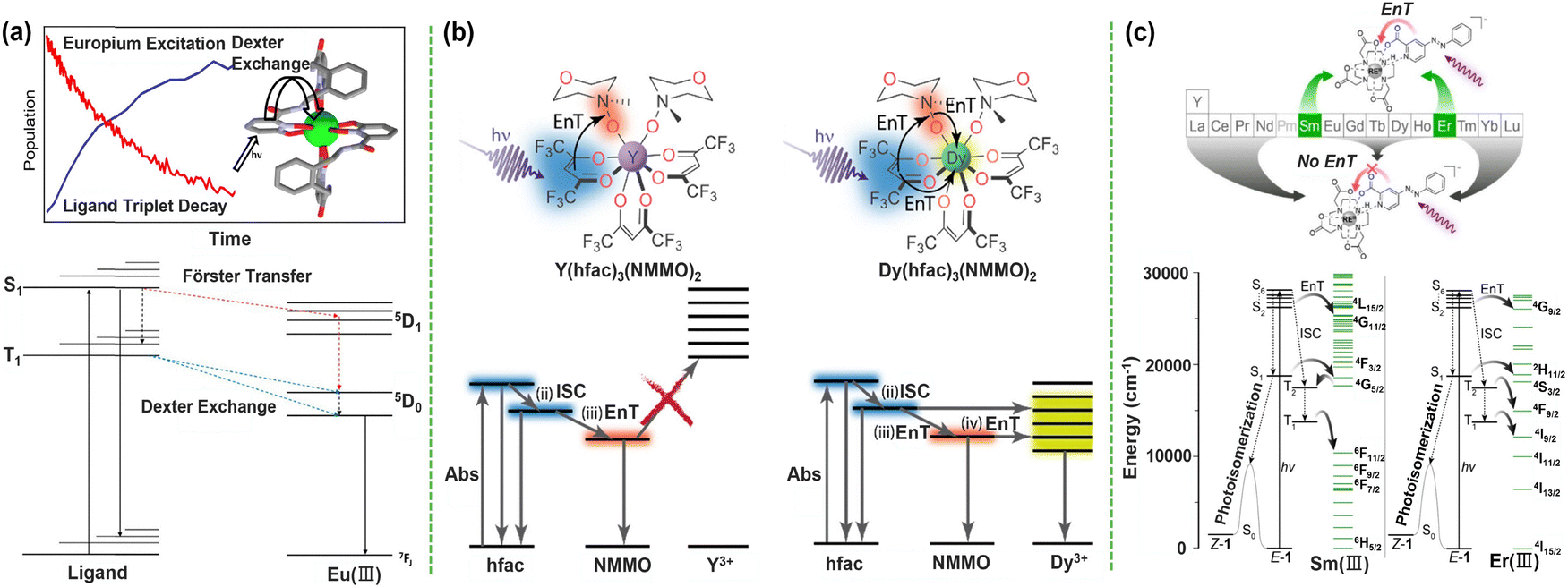 | ||
| Fig. 4 (a) Schematic illustration of energy transfer from the ligand to Eu3+ and the Jablonski diagram of the ligand and RE center following photoexcitation; Förster (red) and Dexter (blue) energy transfer pathways are also indicated. Reprinted with permission from ref. 125. Copyright 2019, American Chemical Society. (b) Jablonski-type diagrams showing the relative excited states of the ligand and metal, and the difference in energy transfer pathways after photoexcitation of Y(hfac)3(NMMO)2 and Dy(hfac)3(NMMO)2. Reprinted with permission from ref. 126. Copyright 2023, American Chemical Society. (c) Schematics and energy level diagram illustrating the proposed photoisomerization quenching of Sm3+ and Er3+via energy transfer. Reprinted with permission from ref. 128. Copyright 2025, American Chemical Society. | ||
Furthermore, it revealed that variations in the overall sensitization efficiency are determined almost entirely by differences in the ligand-centered intersystem crossing rates, thereby establishing definitive rules for achieving near-unity ET efficiency. Beyond ET dynamics, mononuclear complexes have emerged as powerful platforms for uncovering the intimate coupling between photophysics and reactivity. Schelter et al. demonstrated this paradigm using isostructural RE(hfac)3(NMMO)2 models (RE = Y, Dy), showing that the oxygen atom transfer reaction rate of the Dy3+ complex is two orders of magnitude slower than that of Y3+ (hfac = hexafluoroacetylacetone; NMMO = N-methylmorpholine-N-oxide). Ultrafast spectroscopic analysis revealed that accessible 4f levels of Dy3+ act as an “energy sink”, competitively quenching the ligand triplet states via ET and thereby suppressing photochemical reactivity. This work established that ET kinetics, rather than classical thermodynamic parameters such as ionic radius or Lewis acidity, can dictate reaction rates, offering a new strategy for differentiating REs (Fig. 4b).126 Extending this concept, studies on azobenzene-based photo-switchable RE–DO3A complexes127 (DO3A = 1,4,7,10-tetraazacyclododecane-1,4,7-tris-t-butyl acetate) showed that when ligand excited states are resonant with specific RE3+ 4f levels (Sm3+ and Er3+), efficient ET can fully quench the ligand photoisomerization, overriding ground-state coordination chemistry (Fig. 4c).128 Altogether, these examples show how mononuclear complexes serve as critical testbeds for validating theory and revealing new paradigms in which excited-state photophysics governs chemical outcomes.
The simplicity of mononuclear systems is also instrumental in addressing fundamental knowledge gaps across the entire RE series. This is illustrated by the study of promethium (Pm), the radioactive and elusive 61st element. By chelating 147Pm in aqueous solution with a diglycolamide ligand, researchers were able, through a combination of synchrotron X-ray absorption spectroscopy and computation, to determine its coordination geometry and bond distances for the first time (Fig. 5a–g).129 This achievement enabled the first complete experimental analysis of lanthanide contraction across an isostructural solution-phase series, capturing its non-linear trend and providing indispensable benchmarks for theory.
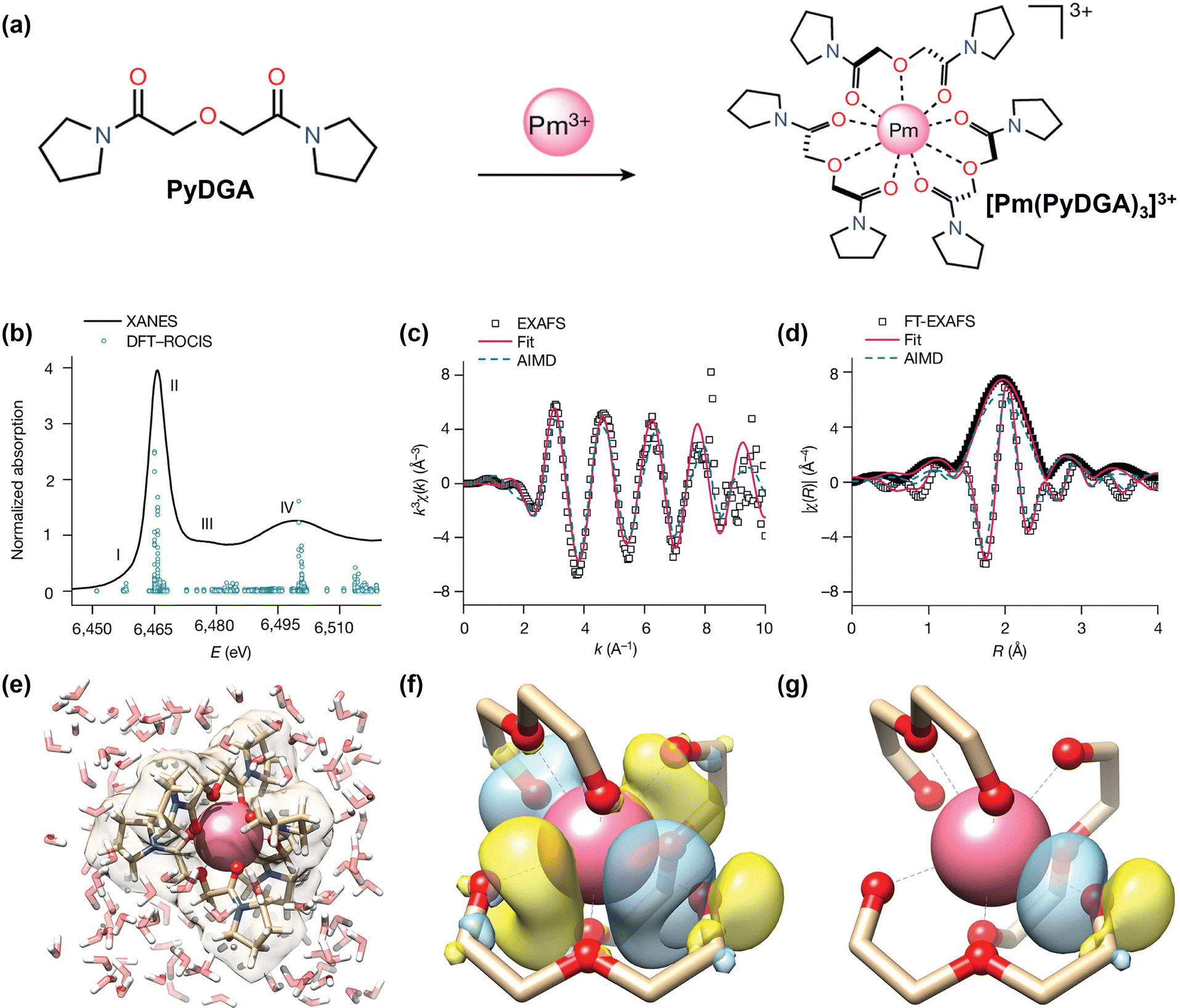 | ||
| Fig. 5 (a) Synthetic route to [Pm(PyDGA)3]3+. (b) Pm L3-edge XANES spectrum (line) and DFT–ROCIS fit (circles); E is the incident energy. (c) and (d) EXAFS data (squares), fit (pink), and AIMD simulation (turquoise). (e) AIMD snapshot of a solvated Pm complex. (f) Dative Pm–O bond showing overlap of the O lone pair and the Pm 5d orbital. (g) Resulting Pm–O bonding NBO (∼4% Pm character; Pm nodal feature below visualization threshold). Reprinted with permission from ref. 129, CC BY 4.0. | ||
The modular coordination chemistry of mononuclear RE complexes also allows for the precise customization of binding sites and optical responses, making them ideal platforms for constructing high-performance sensing systems. The core design logic integrates a stimulus-responsive motif into the ligand framework, which acts as a “molecular switch” to modulate the ET pathway between the sensitizing antenna and the RE center, enabling turn-on/off or ratiometric detection. For instance, M. Nazaré et al. developed a time-gated Tb3+ luminescent probe for nitroreductase detection in live bacteria (Fig. 6).130 Upon NTR-triggered reduction, a caged, non-sensitizing antenna precursor undergoes enzyme-triggered reduction and self-immolative activation to form an efficient carbostyril antenna, thereby switching on Tb3+ emission. In a complementary strategy, Faulkner et al. designed Eu3+ complexes based on Eu3+-phenacyl-DO3A derivatives for cyanide detection,131 where nucleophilic attack by cyanide disrupts the intramolecular ET pathway and quenches Eu3+ luminescence.
 | ||
| Fig. 6 Design rationale and mode of action of the carbostyril-based NTR activatable luminescent RE probe. Reprinted with permission from ref. 130, CC BY 4.0. | ||
Achieving strong CPL, especially in the near-infrared (NIR), with mononuclear systems demands exquisite control over chiral ligand design, coordination geometry, and the electronic coupling between the chiral environment and the RE emitter.34 T. Gunnlaugsson et al. demonstrated efficient chirality transfer from enantiopure pyridyl-diamide ligands to their Eu3+ complexes, which self-assemble into chiral crystalline structures. Using CPL laser scanning confocal microscopy (CPL-LSCM), they visually distinguished the handedness of emission at the single-crystal level, confirming the propagation of molecular chirality to the supramolecular architecture.132 To push the limits of glum and BCPL, G. Ung et al. employed rigid, axially chiral BINOL and helical chiral Spinol ligands. The [(BINOL)3ErNa3] complex achieved intense CPL at the strategic 1550 nm telecom window with a high glum of 0.47 (BINOL = 1,1′-bi-2-naphthol), marking the first observation of molecular CPL beyond 1200 nm (Fig. 7a and b).88 Subsequent spinol-based systems achieved record-high glum values up to 0.53 for Tb3+/Dy3+ and discovered NIR-CPL from Sm3+, while delivering an unprecedented BCPL of 3760 M−1 cm−1 (Fig. 7c and d).133 Further refining the classic chiral tetrakis(camphorato)europate system, Y. Hasegawa et al. introduced ammonium cations (e.g., TEA+) to replace Cs+. This strategic modification, coupled with ligand tuning, synergistically enhanced both the emission quantum yield and the glum, resulting in a record-breaking glum of −1.54, the largest magnitude reported for any chiral luminescent molecule, while also improving the overall CPL brightness (Fig. 7e–h).114 Collectively, these advances mark a transition from empirical discovery to symmetry-guided, rational design of chiroptical RE materials.
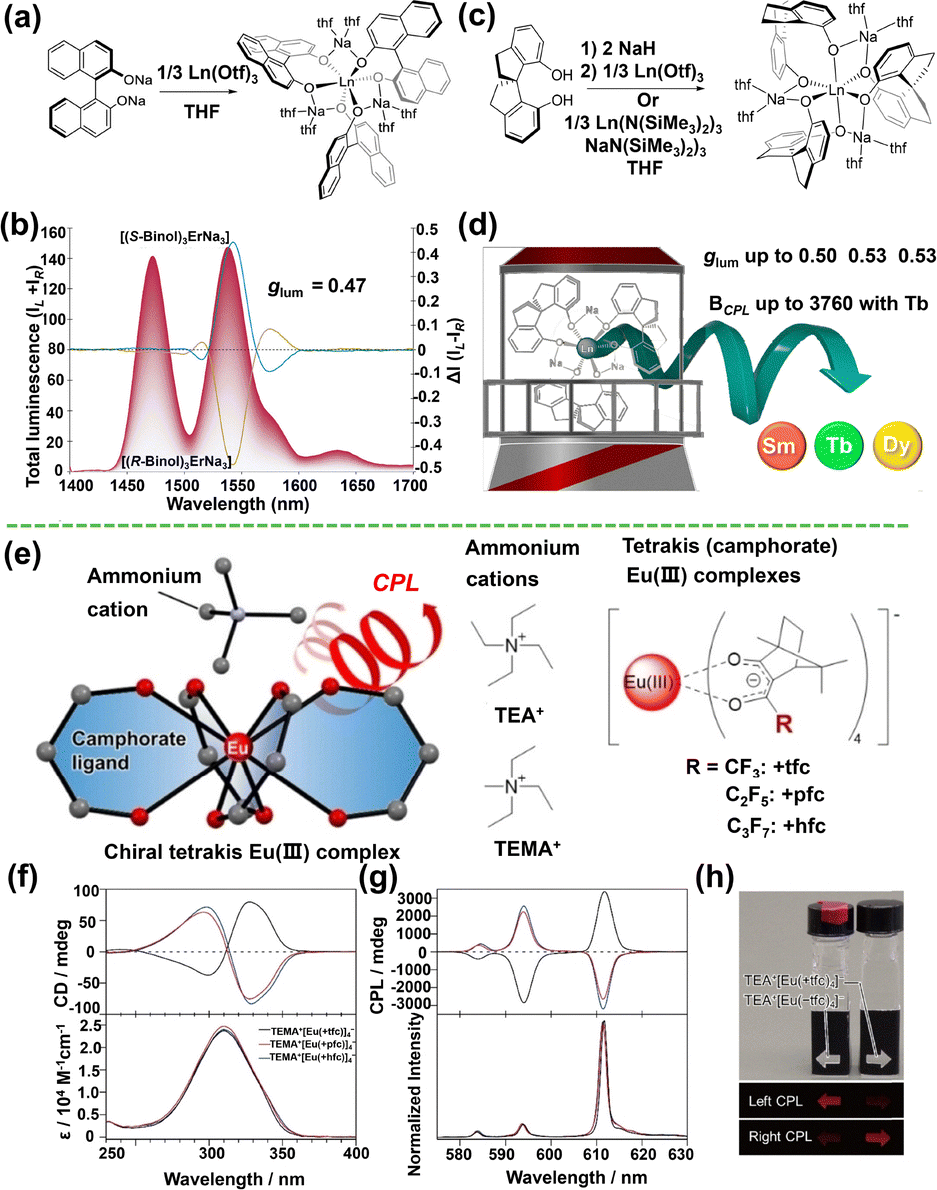 | ||
| Fig. 7 (a) Synthesis of [(R-Binol)3LnNa3(thf)6] (Ln = Er, Yb, Nd) from sodium R-binolate. Reprinted with permission from ref. 88. Copyright 2022, American Chemical Society. (b) Normalized CPL spectra of [(R/S-Binol)3ErNa3] in tetrahydrofuran solutions. (c) Synthesis of [(R-Spinol)3RENa3(thf)6] (RE = Sm, Eu, Tb, Dy). (d) Schematic illustration of CPL and corresponding glum values for the complexes in (c). Reprinted with permission from ref. 133. Copyright 2022, American Chemical Society. (e) Chemical structures of chiral Eu3+ complexes bearing ammonium cations. (f) and (g) CD and CPL spectra of TEA+[Eu(+tfc)4]−, TEA+ [Eu(+pfc)4]−, and TEA+[Eu(+hfc)4]− in chloroform. (h) Photographs of the emission from TEA+[Eu(+tfc)4]−and TEA+[Eu(−tfc)4]− in chloroform. Reprinted with permission from ref. 114. Copyright 2022, Wiley-VCH. | ||
Beyond classical antenna sensitization, recent efforts exploit “dark” triplet states and unconventional excitation pathways to unlock emergent luminescence phenomena like afterglow and radioluminescence (scintillation). Based on previous research foundations,134 F. Li et al. constructed a novel photochemical afterglow system by integrating a custom photo-energy cache unit (PCU) with a Eu3+ emitter via coordination bonds. Under NIR light initiation, a cascade involving ROS generation and chemiexcitation populates a ligand-based dark state (a TICT state), which then directly transfers energy to the Eu3+ center, yielding bright red afterglow with a record-high quantum yield of 27.5%. This system enabled ultra-sensitive in vivo imaging and was applied in lateral flow immunoassays, demonstrating a new pathway to convert chemical energy into long-lived, background-free luminescence (Fig. 8a).116 In parallel, X. Liu et al. developed ultra-bright molecular scintillators by optimizing ligand triplet states to efficiently capture “dark” triplet excitons generated by X-ray absorption. Through near-unity triplet exciton recycling to the RE center, they achieved radioluminescence enhancements of over three orders of magnitude compared to conventional organic scintillators, enabling high-resolution X-ray imaging and radiotherapy applications (Fig. 8b–e).135 These studies reflect a conceptual evolution of ligands from passive antennas to active “exciton managers”.
 | ||
| Fig. 8 (a) Schematic illustration of afterglow luminescence and the energy transfer process for the Eu3+ complex. Reprinted with permission from ref. 116. Copyright 2025, Wiley-VCH. (b) Schematic illustration of triplet exciton-mediated energy transfer from the ligand to the RE center. (c) Triplet exciton recycling in organolanthanide molecules. (d) Relative alignment of the ligand triplet states and the Eu3+ 5D0 emissive level. (e) Comparison of X-ray-excited radioluminescence intensity versus UV-excited photoluminescence intensity for various organoeuropium molecules. Reprinted with permission from ref. 135. Copyright 2024, Springer Nature. | ||
Pushing mononuclear design to its extreme enables the suppression of energetic disorder, unlocking properties essential for quantum information science, which capitalizes on RE ions' intrinsic shielded 4f electronic transitions to sustain long-coherence quantum states for fault-tolerant computing and communication.136 The pursuit of systems with ultra-narrow optical linewidths (long coherence times) is a prime example.
In solution, J. R. Caram and coworkers designed a ferrocene-supported Yb3+ complex, (thiolfan)YbCl(THF), [thiolfan = 1,1′-bis(2,4-di-tert-butyl-6-thiomethylenephenoxy) ferrocene], featuring a rigid ligand sheath that dramatically shields the Yb3+ center from environmental fluctuations. This resulted in an unprecedented room-temperature absorption linewidth of 0.625 meV for the f–f transition, with a homogeneous linewidth of 410 peV at 77 K. This “atomlike molecular sensor” (ALMS) demonstrated liquid-phase magnetic circular dichroism magnetometry capable of detecting Earth-scale magnetic fields, establishing a new paradigm for quantum sensing in condensed phases (Fig. 9a–c).137 In the solid state, P. Goldner et al. achieved optical homogeneous linewidths as narrow as 30 kHz in a molecular crystal of the mononuclear complex [Eu(BA)4(pip)], (BA = benzoylacetonate; pip = piperidin-1-ium), rivalling some of the best RE-doped inorganic crystals. This exceptional coherence enabled key quantum technology primitives: optical spin initialization with >95% efficiency, coherent optical storage via atomic frequency combs, and the observation of controllable ion–ion interactions, the basis for quantum gates (Fig. 9d–g).138 These breakthroughs highlight how meticulous ligand design in mononuclear complexes can create highly coherent spin-photon interfaces, combining the atomic-like properties of RE ions with the synthetic versatility and potential for integration offered by molecular materials. While extensive research focuses on RE(III) 4f–4f transition luminescence systems, divalent rare-earth elements (e.g., Eu(II)) exhibit unique advantages in broad-spectrum emission, high-efficiency luminescence, and stimuli-responsive behavior through their parity-allowed 5d–4f transitions, positioning them as emerging targets in optoelectronic materials and molecular sensing.139
 | ||
| Fig. 9 (a) Structure and X-ray crystal structure of (thiolfan)YbCl(THF). (b) Absorption spectrum with an expanded view of Yb-related features. (c) Normalized emission spectrum at room temperature. Reprinted with permission from ref. 137. Copyright 2024, AAAS. (d) X-ray crystal structure of the Eu3+ complex. (e) Photoluminescence spectrum of Eu3+ showing the characteristic 5D0 → 7FJ (J = 0–4) transitions (see the inset). (f) 7F0 → 5D0 absorption line recorded on a 500 µm thick powder sample. (g) Fluorescence decay of the 5D0 → 7FJ (J = 1–4) transitions. Reprinted with permission from ref. 138. Copyright 2022, Springer Nature. | ||
To address persistent challenges in air stability and device efficiency, Z. Liu et al. developed hydroxytris(pyrazolyl)borate complexes: fluorinated Eu-1 [Eu(htfpzb)3], [htfpzb = hydrotris(3-trifluoromethylpyrazolyl) borate)], achieved unprecedented 96% photoluminescence quantum yield (PLQY) with >2200-hour ambient stability. The methyl-substituted Eu-2 as an OLED emitter attained 6.5% external quantum efficiency (EQE), 30620 cd m−2 maximum brightness, and near-unity exciton utilization.140 Further optimized azacryptand-type Eu(II) complexes (EuX2-Nn) yielded record-breaking OLED performance with EuI2–N8 (99% PLQY, mechanochromic luminescence) delivering 25470 cd m−2 brightness and 17.7% EQE.141 Concurrently, M. Murugesu et al. pioneered sensing mechanism design: structural elucidation of the dinuclear organometallic complex [Cp*Eu(BH4)(THF)2]2 (THF = tetrahydrofuran) clarified 5d–4f transition pathways.142 In pseudo-C4v symmetric semi-sandwich architectures [CprEu(µ-BH4) (solv)n]m (solv = diglyme/toluene), stereoelectronic effects of cyclopentadienyl substituents, Eu(II)–ligand bond distances, and flexible pendant bonds were revealed to synergistically modulate luminescent thermometric properties, establishing new paradigms for molecular-level lanthanide sensing.143
In summary, mononuclear RE complexes benefit from the evolution of ligand engineering from achieving fundamental photophysical control and constructing responsive probes to empowering advanced biomedical theranostics and pioneering novel quantum and chiroptical functionalities. Across these diverse examples, a unifying principle that atomic-level precision in ligand design is essential for transforming the innate properties of RE ions into programmable, high-performance macroscopic functions emerges. The concepts and molecular blueprints established at this molecular level provide the essential foundation for construction of more sophisticated polynuclear and hierarchical architectures, as discussed in the following sections.
Achieving efficient UC in discrete molecular systems, where low-energy photons (typically NIR) are converted to higher-energy (visible) emission, represents a formidable challenge due to stringent requirements for minimizing non-radiative losses and maximizing interionic ET rates. A seminal strategy involves the covalent linkage of sensitizer (S) and activator (A) ions at fixed, optimal distances. For instance, the triple-stranded helicate [CrErCr(Lz)3]9+ [LZ = 2,6-bis(1-methyl-1H-benzimidazol-2-yl)pyridine] sandwiches an Er3+ activator between two strong-field Cr3+ sensitizers at distances of ∼8.9 Å. This design creates an exceptionally high local sensitizer concentration and ensures near-unity efficiency for the Cr3+(2E) → Er3+(4I11/2) ET step. Upon NIR excitation into the Cr3+ absorption, a sequential energy transfer upconversion mechanism populates the Er3+ 4S3/2 state, resulting in green emission.144 Further conceptual advances were achieved by L. J. Charbonnière et al.; they synthesized a tritopic ligand L1 (tris-heteropolytopic ligand), designed to hierarchically assemble hetero-polynuclear Tb/Yb complexes. The ligand features two strong triazacyclononane (tacn) binding sites for initial RE3+ coordination and a weaker polyether bridge for subsequent metalation, enabling the controlled formation of species like [Yb2L1] and ultimately [Tb(Yb2L1)]. Upon 980 nm excitation of Yb3+, this tetranuclear assembly exhibits cooperative sensitization upconversion (CS-UC), where two excited Yb3+ ions simultaneously transfer energy to a single Tb3+ ion, producing green Tb3+ emission even in water (Fig. 10a–c).60 This work demonstrated that encoding a fixed, short S⋯A distance (∼11 Å) within a molecular scaffold is crucial for overcoming the inherent inefficiency of cooperative processes, achieving a molecular UC quantum yield of 9.0 × 10−7 in D2O. The pursuit for room-temperature UC in solution has further motivated designs emphasizing ultra-short intermetallic contacts.
 | ||
| Fig. 10 (a) Synthesis of Tb(Yb2L1). (b) Emission spectra measured during titration with TbCl3·6H2O of a 1.01 mM solution of [Yb2L1] in D2O (λexc = 980 nm, P980 = 1.08 W, 10 scans). (c) Emission spectra measured for batches of a solution of [Yb2L1] in D2O with different amounts of TbCl3·6H2O, after 10 days at 60 °C (λexc = 980 nm, P980 = 1.08 W, 10 scans). Reprinted with permission from ref. 60. Copyright 2025, American Chemical Society. (d) Synthesis of [(ErL2)2F]+. (e) Energy level diagram showing the two upconversion mechanisms (ETU in blue and ESA in red) in Er3+. (f) Schematic comparison of the two mechanisms (vertical positions are not to scale). Reprinted with permission from ref. 145, CC BY 4.0. | ||
L. J. Charbonnière et al. exploited the synergistic binding of a single fluoride anion to assemble a supramolecular [(ErL2)2F]+ [L2 = 1,4,7-tris(1H-indazol-1-ylmethyl)-1,4,7-triazacyclononane-1,4-diacetic acid] dimer from macrocyclic [ErL2(H2O)]+ units. DFT calculations revealed an exceptionally short Er⋯Er distance of ∼4.5 Å, bridged by a linear Er–F–Er unit and stabilized by π-stacking and hydrogen bonds. This proximity dramatically enhances the ETU efficiency between the two Er3+ ions. Consequently, excitation at 980 nm in D2O at room temperature produces visible UC emission, with intensity maximized at a 0.5:1 F−:Er3+ ratio corresponding to the dimer's formation (Fig. 10d–f).145 Pushing this concept further, M.-H. Zeng et al. designed cationic [(Yb)2Er]+ clusters using a quinolinoxide ligand, achieving Yb⋯Er distances as short as ∼3.7 Å. The combination of ultra-short distances and fluoride-mediated quenching suppression enabled UC emission in non-deuterated solvents at room temperature under remarkably low excitation power densities (<0.3 W cm−2), marking a significant step toward practical molecular UC materials.146
Dinuclear d–f complexes provide an elegant platform for exploring the interplay between luminescence and molecular magnetism, particularly spin-crossover (SCO) behavior. The core strategy employs a luminescent RE3+ ion (e.g., Eu3+) as a reporter for the spin state of an adjacent transition-metal ion (e.g., Fe2+), modulated through temperature-dependent ET quenching. C. Piguet et al. carried out foundational studies using triple-helical [EuFe(Lk)3]5+ complexes (k = 4, 5, 6; Lk = didentate benzimidazole-2-yl-pyridine units). Ligands L4–L6 subtly modify the coordination environment of Fe2+ to tune its SCO properties. In [EuFe(L4)3]5+, Fe2+ remains high-spin (HS) at all temperatures, and Eu3+ emission is strongly yet incompletely quenched (ηET ∼ 98%). In contrast, in [EuFe(L5)3]5+, Fe2+ undergoes SCO near room temperature. Theoretical kinetic modeling revealed that ET from Eu3+ to low-spin (LS) Fe2+ is nearly quantitative (ηET ≥ 99.9%), leading to almost complete emission quenching whenever a significant LS fraction is present (Fig. 11a).147 This extreme sensitivity restricted detectable modulation to solid-state samples at higher temperatures where the HS form dominates, thereby proving the concept while highlighting a design challenge. To achieve a clear luminescent readout of SCO in solution, subsequent work focused on tuning both the SCO temperature and the spectral overlap integral. Replacing the terminal pyridine in the ligand with a pyrimidine unit (giving L2), [L2 = 1-methyl-2-(pyrimidin-2-yl)-1H-benzo[d]imidazole-based segmental ligand] weakened the ligand field at Fe2+, lowered the SCO transition temperature (T1/2 ≈ 317 K), and crucially reduced the intensity of the LS Fe2+ metal-to-ligand charge-transfer (MLCT) band. This decreased the spectral overlap with Eu3+ emission and thus reduced the ET rate constant from Eu3+ to LS Fe2+. In the resulting [EuFe(L2)3]5+, Eu3+ emission became detectable in acetonitrile solution across the SCO region (Fig. 11b).148 The integrated emission intensity exhibited a distinctive “wavy” temperature dependence: an initial decrease due to thermal quenching, followed by an increase as LS (a strong quencher) converts to HS (a weaker quencher), and a final decrease at higher temperatures.
 | ||
| Fig. 11 (a) Monitoring Fe2+ spin-state equilibria via Eu3+ luminescence in [EuFe(Lk)3]5+ complexes. Reprinted with permission from ref. 147. Copyright 2020, American Chemical Society. (b) Detecting Fe2+ spin-crossover by modulation of appended Eu3+ luminescence in [EuFe(L2)3]5+. Reprinted with permission from ref. 148. Copyright 2024, American Chemical Society. | ||
Collectively, studies on dinuclear and trinuclear RE systems reveal a landscape where function is exquisitely, yet delicately, dictated by synthetic control over molecular architecture. These complexes bridge mononuclear models and advanced functional materials, demonstrating that key photophysical processes, energy migration, cooperative sensitization, and exchange coupling are directly governed by precision in ion spacing, geometry, and the coordination microenvironment. However, this structural precision also exposes a fundamental optimization dilemma: strategies that shorten interionic distances to enhance energy transfer often simultaneously introduce new non-radiative decay pathways or exacerbate excited-state quenching, while ligand frameworks essential for structural integrity may themselves act as competitive energy sinks.
Heterodinuclear RE1–RE2 complexes, with fixed interionic distances and stoichiometric donor/acceptor ratios, represent the ideal model systems for developing precise ratiometric luminescent thermometers. The key is to engineer a thermally activated energy transfer (TA-ET) pathway from a donor (D) to an acceptor (A) RE3+ ion. A prime example is the heterodinuclear cycTb-phEu complex, in which Tb3+ and Eu3+ are site-specifically placed in distinct coordination environments separated by ∼10.6 Å. The TA-ET from Tb3+ (5D4) to Eu3+, proceeding primarily via the Eu3+ (7F1) level at room temperature, causes the Tb3+/Eu3+ emission intensity ratio to exhibit a linear and sensitive dependence on temperature over a broad range (50–298 K). As a stoichiometric molecular dyad, this system eliminates the uncertainties of doped materials (random distances, concentration gradients, and energy migration), enabling precise quantification of ET parameters.149
An alternative strategy to enhance thermal sensitivity focuses on managing ligand triplet states. In a dinuclear Tb–Nd complex featuring fluorinated β-diketonate (hfa) and bridging triphenylene ligands, the Tb3+(5D4) emission is quenched by back-ET to the nearby hfa triplet state (T1). Crucially, the T1 level of hfa is aligned with that of triphenylene, which efficiently transfers energy to the Nd3+ acceptor. This creates an “energy escape route” that shortens the effective lifetime of the hfa T1 state competing with Tb3+ emission. This ingenious design resulted in a record-high temperature sensitivity of 4.4% K−1 for the Tb T1 emission lifetime among Ln molecular thermometers, showcasing ligand-mediated exciton management for enhanced functionality.150 Beyond photon upconversion, dinuclear systems can leverage alternative excitation mechanisms. For instance, the Tb–DO3A complex (DO3A = 1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid) was used to demonstrate Cherenkov radiation energy transfer (CRET) (Fig. 12a). The Cherenkov radiation continuum generated by decaying radionuclides (18F and 89Zr), with maximum intensity in the UV/blue region, can efficiently excite the organic antenna of the Tb3+ complex. This strategy enables the optical imaging of discrete RE complexes without external light sources, opening avenues for radiation-guided photodynamic therapy or combined radiotherapy/imaging modalities (Fig. 12b).151 Notably, binuclear/trinuclear d–f systems exhibit remarkable potential in synergizing molecular nanomagnetism with luminescent functionality. For instance, S.-I. Ohkoshi et al. reported a trinuclear [YbCo2] supramolecular framework that concurrently exhibits slow magnetic relaxation (molecular nanomagnetism), NIR luminescence from Yb(III) (quantum yield >1%), high-sensitivity optical thermometry (>1% K−1), and proton conductivity within a single molecule – establishing a novel paradigm for multifunctional integration.152 Furthermore, their binuclear [HoIII(4-pyridone)4(H2O)2][MIII(CN)6] system innovatively exploited the luminescence re-absorption effect of Ho3+ to correlate single-molecule magnet behavior with ratiometric optical thermometry. By modulating [MIII(CN)6]3− (M = Co/Rh/Ir), they precisely tuned magnetic relaxation barriers and thermometric ranges (25–205 K), significantly advancing the field of d–f complexes in magneto-optical synergy in d–f complexes.153 While these pioneering studies demonstrate the remarkable multifunctionality achievable in d–f systems, their full potential for tailored photonic applications remains constrained by the empirical nature of current design approaches. To overcome this fundamental challenge, performance optimization must transcend simple structural adjustment; it is a multiparameter balancing act that requires concurrently managing energy transfer efficiency, excited-state lifetime, and overall photostability at the atomic scale.
 | ||
| Fig. 12 (a) Structure of RE complexes and 89Zr[Tb(L2)]− investigated in this work. (b) Schematic of CR-mediated excitation of a discrete, luminescent 89Zr[Tb(L2)]− complex. Reprinted with permission from ref. 151. Copyright 2018, Wiley-VCH. | ||
Collectively, studies on dinuclear and trinuclear RE systems reveal a landscape where function is exquisitely, yet delicately, dictated by synthetic control over molecular architecture. These complexes bridge mononuclear models and advanced functional materials, demonstrating that key photophysical processes, energy migration, cooperative sensitization, exchange coupling, are directly governed by precision in ion spacing, geometry, and the coordination microenvironment. However, this structural precision also exposes a fundamental optimization dilemma: strategies that shorten interionic distances to enhance energy transfer often simultaneously introduce new non-radiative decay pathways or exacerbate excited-state quenching, while ligand frameworks essential for structural integrity may themselves act as competitive energy sinks. Future progress will depend on a shift from qualitative design rules to quantitative prediction, enabled by: (i) synthetic methodologies capable of delivering precisely defined ion arrangements with tailored electronic structures and minimized energetic disorder and (ii) advanced spectroscopic and theoretical tools that directly correlate structural parameters with the dynamics, rates, and coherence of interionic interactions. These advances will be essential for transforming molecular intermetallic communication into a truly predictive and programmable foundation for next-generation RE photonic materials.
3.1.3.1. RE clusters. The central pursuit in luminescent RE cluster research is the assembly of dozens to hundreds of RE ions and their ligands into atomically precise nanoparticles that exhibit exceptional chemical stability, luminescence efficiencies approaching theoretical limits, and advanced biomedical functionalities. The evolution of this field clearly follows the logic of “solving stability bottlenecks → pursuing luminescence efficiency limits → pioneering biomedical other emerging applications”.156–158
Early generations of luminescent RE clusters were hampered by air/moisture sensitivity and quenching via high-energy vibrations (e.g., O–H oscillators). The breakthrough began with “phonon engineering” of the cluster core. The representative work on RE16(µ4-F)6(µ3-F)12(tBuCOO)18[N(CH2CH2O)3]4 (abbreviated as RE16F18) employed a clever “mixed-ligand” strategy. Using sterically bulky pivalate (tBuCOO−) and strongly chelating triethanolamine derivatives as peripheral protecting ligands, the researchers successfully introduced low-vibrational F− into the cluster core, replacing traditional hydroxo–bridges (Fig. 13a). This design yielded revolutionary improvements: the resulting Eu and Tb clusters were stable in air with thermal decomposition temperatures above 435 °C. Crucially, the F bridges drastically suppressed non-radiative decay caused by O–H vibrations, pushing the photoluminescence quantum yields (PLQYs) to 87.7% (Eu) and 99.0% (Tb), nearing theoretical limits (Fig. 13b and c).103
 | ||
| Fig. 13 (a) Structural representations of the [Eu16] cluster core: ball-and-stick model, bridging modes of fluoride ions (µ3-F and µ4-F), and space-filling model of a single molecule. (b) Photographic images of EuFC-16 and TbFC-16 single crystals under ambient light and upon 365 nm UV excitation (in glass vials). (c) Luminescence of THF solutions containing varying molar ratios of dissolved EuFC-16 and TbFC-16 crystals (in quartz cuvettes). Reprinted with permission from ref. 103 Copyright 2024, Wiley-VCH. | ||
Beyond delivering record-breaking performance, this work established a general design principle for luminescent clusters: a low-vibrational core combined with a rigid, hydrophobic shell. This paradigm removed a fundamental bottleneck for cluster integration into optoelectronic devices such as LEDs. With chemical robustness achieved, subsequent efforts focused on optimizing photophysical performance and integrating multiple functions. For instance, those based on the [Ln20] structure allows for precise tuning of energy transfer between different RE ions. This has enabled groundbreaking applications in molecular upconversion and the development of highly sensitive luminescent thermometers, pushing quantum yields in molecular systems to new heights.159 Moreover, advanced sensitization strategies like the “dual antenna effect”, achieved by incorporating metalloligands, can significantly enhance luminescence intensity while enabling complex functionalities, including multi-ion sensing and differentiated live-cell imaging. The pursuit for performance limits spurred heterometallic strategies.
X.-J. Kong et al. reported the first RE3+/Cu+ heterometallic cluster, [YbCu54O6C13(2-MeO-PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C)36] (ClO4)6 (Fig. 14a). Its structure resembles an “egg” with a [YbO6]9− unit as the “yolk” encapsulated by a two-layer copper “shell”. This architecture is exceptionally stable due to strong Cu+–alkynyl coordination and exhibits unique synergistic luminescence: the incorporation of Yb3+ significantly enhances the cluster's near-infrared-II (NIR-II, 986 nm) emission, achieving a solid-state quantum yield of 33.3%, a record for copper clusters (Fig. 14b and c).121 This work highlights how rationally engineered heterometallic core–shell architectures can generate “1 + 1 > 2” photophysical synergy, offering a novel route for developing new high-efficiency NIR emitters.
C)36] (ClO4)6 (Fig. 14a). Its structure resembles an “egg” with a [YbO6]9− unit as the “yolk” encapsulated by a two-layer copper “shell”. This architecture is exceptionally stable due to strong Cu+–alkynyl coordination and exhibits unique synergistic luminescence: the incorporation of Yb3+ significantly enhances the cluster's near-infrared-II (NIR-II, 986 nm) emission, achieving a solid-state quantum yield of 33.3%, a record for copper clusters (Fig. 14b and c).121 This work highlights how rationally engineered heterometallic core–shell architectures can generate “1 + 1 > 2” photophysical synergy, offering a novel route for developing new high-efficiency NIR emitters.
 | ||
| Fig. 14 (a) Synthesis route and crystal image of LnCu54. Temperature-dependent spectrum at (b) 100–300 K and (c) 293–413 K. Reprinted with permission from ref. 121. Copyright 2024, American Chemical Society. | ||
High nuclearity and atomic precision enable advanced functional integration. [Ln60L60(NO3)20(OAc)24(OH)12]·4OH (abbreviated as Ln60) is a milestone in this direction. Utilizing flexible Schiff base ligands, this work assembled one of the highest-nuclearity RE clusters known, featuring a key innovation: an internal cavity approximately 1.5 nm in diameter (Fig. 15).160 This cavity can efficiently load the anticancer drug doxorubicin (DOX), forming monodisperse drug-loaded nanoparticles (∼5 nm). Beyond therapeutics, the precise compositional control afforded by high-nuclearity clusters enables their use as optical barcodes with exceptionally high security levels, as well as tunable white-light emitters for display and anti-counterfeiting technologies.161,162
 | ||
| Fig. 15 (a) Hollow hexahedral structure of Eu60 viewed along the a-, b- or c-axis. (b) Basic structural unit (RE3L3(NO3)) templated by NO3−. (c) Three-dimensional channel structure of Ln60 viewed along the a-, b- or c-axis. (d) High-resolution TEM image and dynamic light scattering (DLS) analysis of Eu60. (e) TGA-DTG curves of Eu60. (f) RE luminescence spectra of Eu60 (λex = 395 nm) and (g) Tb60 (λex = 370 nm) in CH3CN. Reprinted with permission from ref. 160. Copyright 2024, Wiley-VCH. | ||
Beyond their biomedical applications, heterometallic lanthanide clusters have demonstrated remarkable potential in magneto-optical modulation. The [EuxTby] cluster developed by M. Murugesu et al. and the Ln2Ti7 cluster reported by X.-J. Kong et al. collectively show the synergistic enhancement mechanism arising from heterometallic component design in magneto-optical functionality. The former achieves programmable MCPL spectra through dynamic modulation of the gMCPL factor. Controlled energy transfer and magnetic coupling between Eu(III) and Tb(III),163 with the latter benefiting from the d-electron-free nature of Ti(IV) to eliminate energy quenching, enables Eu(III)/Sm(III) to simultaneously exhibit MCPL and near-infrared magnetic circular dichroism (MCD). This highlights the integrated advantages of 3d–4f systems in achieving multifunctional magneto-optical performance.164 In the domain of optical thermometry, the [Tb10Yb10] cluster introduces an innovative approach by utilizing the temperature-dependent excitation lifetime of the Yb(III) → Tb(III) upconversion process. This methodology effectively mitigates excitation source fluctuations inherent in traditional intensity-based thermometry through lifetime sensing, thereby significantly enhancing the reliability of biological temperature detection.165 These studies demonstrate that precise heterometallic design can directionally regulate energy transfer pathways, magneto-optical coupling effects, and luminescence kinetics, establishing novel paradigms for the development of multifunctional magneto-optical devices and high-precision sensing technologies.
Collectively, research on luminescent RE clusters has established a mature research paradigm: “structural stabilization, luminescence optimization and functional integration”. Looking forward, further advances will likely rely on the convergence of theoretical prediction, data-driven design, and automated synthesis to enable the precise construction of increasingly complex core–shell architectures and stimuli-responsive behaviors. Equally important will be systematic investigations into their interfacial behavior in biological environments and device architectures, which are essential for translating these sophisticated molecular clusters into practical technologies.
3.1.3.2. Coordination supramolecular cages/helicates. The scientific narrative of coordination supramolecular cages and helicates centers on the amplification of molecular-level chiral information and spatial information into discrete, higher-order architectures through coordination-directed self-assembly. By translating local stereochemical and geometric cues into well-defined cavities and topologies, these systems provide unparalleled control over chiroptical responses, host–guest chemistry, and adaptive, stimuli-responsive behavior.46
Effective chiral transfer and amplification are key to achieving strong CPL.166 Two complementary design strategies have emerged: (i) bottom-up construction using predesigned chiral ligands to enforce global stereochemical order and (ii) top-down chiral induction and resolution of racemic assemblies. A paradigmatic example of the bottom-up approach is the octanuclear circular helicate [(R)- or (S)-iPr-Pybox]8RE3+8(THP)8. In this system, chiral bis(oxazolinyl)pyridine (iPr-Pybox) ligands, which, together with tridentate β-diketonate ligands (THP) and RE3+ ions, undergo one-step thermodynamic assembly to form an octanuclear circular helicate with D4 symmetry. Stereochemical interactions between ligands force all eight RE3+ centers to adopt the same chiral configuration (Δ8 or Λ8) (Fig. 16a), achieving perfect chiral transfer from the ligand to the entire superstructure, resulting in a remarkably high glum of 1.25 at the Eu3+ magnetic dipole transition (592 nm) (Fig. 16b–f).167 In contrast, an alternative top-down strategy is exemplified by the Δ6/Λ6-Ln6L8 octahedral cages and their arginine-induced derivatives. Here, racemic hexanuclear cages are first assembled from achiral tridentate β-diketonate ligands. Subsequently, a natural chiral source (e.g., L-arginine) was used to selectively induce the crystallization of one enantiomer via non-covalent interactions, achieving chiral resolution. The resulting Eu6 cage maintains a high quantum yield of 61% alongside a glum of 0.53, demonstrating excellent overall “CPL brightness” (quantum yield × glum).168 Altogether, these two strategies illustrate complementary pathways that prioritize maximal stereochemical precision or practical post-assembly resolution, respectively. Beyond conventional luminescence, supramolecular assembly offers unique advantages for realizing highly challenging photonic functions such as molecular photon upconversion (UC). Traditional UC mechanisms primarily rely on direct ET between metal ions, limiting the designability of structures and properties. Recent research, utilizing well-designed multicomponent RE-organic assemblies, has revealed a novel upconversion pathway mediated by excited-multimer states. In the homometallic Yb8(L2R/S)12 assembly (Fig. 17a–c), upconverted multimer green fluorescence was observed under 980 nm excitation via a cooperative sensitization process. More importantly, in the heterometallic (Yb/Eu)8L12 assembly, upconverted red emission from Eu3+ was achieved for the first time through an excited-multimer-mediated energy relay, i.e., via the pathway (Yb* + Yb) → excited-multimer → Eu (Fig. 17d and e).169 This mechanism fundamentally deviates from classical interionic energy transfer by placing ligand-based excited multimer states at the core of the upconversion process, thereby introducing a new degree of freedom for UC material design. This concept has been further extended to integrate chirality with UC. Q.-F. Sun et al. have successfully extended this strategy to synergistically integrate chiral signals with the upconversion process. By assembling C2-symmetric chiral ligands with RE3+ ions, they constructed enantiopure tetrahedral Ln4(LR/S)6 cages with ΔΔΔΔ or ΛΛΛΛ metal stereochemistry. Under 980 nm excitation, the heterometallic assemblies (Yb/Eu)4(LR/S)6 and (Yb/Sm)4(LR/S)6 achieved upconversion emissions from Eu3+ and Sm3+ centers in solution at room temperature via a multimeric triplet-mediated cooperative sensitization pathway (Fig. 17f). Remarkably, this system delivers the first example of upconverted circularly polarized luminescence (UCCPL) in a RE supramolecular assembly, yielding glum on the order of 10−2 (Fig. 17g and h).170 This achievement demonstrates the feasibility of merging nonlinear photonic processes with intrinsic supramolecular chirality.
 | ||
| Fig. 16 (a) Reaction scheme of D4-symmetrical circular Ln3+ helicates of [(R)- or (S)-iPr-Pybox]8(LnIII)8(THP)8. (b) and (c) Absorption spectra and CD spectra of [(R)- or (S)-iPr-Pybox]8(LnIII)8(THP)8. Normalized CPL spectra of (d) [(R)- or (S)-iPr-Pybox]8Eu(III)8(THP)8 and (e) [(R)- or (S)-iPr-Pybox]8TbIII8(THP)8 in chloroform at 298 K. (f) Visible Eu3+ luminescence images of R and S isomers in chloroform (top) and PMMA films (bottom) with a bandpass filter (592 nm) and left- and right-circularly polarized filter. Reprinted with permission from ref. 167. Copyright 2020, American Chemical Society. | ||
 | ||
| Fig. 17 (a) Self-assembly of octanuclear Ln8L12-type assemblies by ligands L1 and L2R/S. X-ray crystal structures of Eu8(L1)12 (b) and Eu8(L2S)12 (c). (d) CSU from Ln to ligand's multimer; and (e) excited-multimer mediated CSU. Reprinted with permission from ref. 169. Copyright 2023, American Chemical Society. (f) Crystal structure of Eu4(LS)6. (g) Proposed multimer triplet-mediated CSU process. (h) UCCPL emission spectra of (Yb/Eu)4(LR/S)6 in CD3CN/MeOD (4.0 mM, λex = 980 nm, r.t.). Reprinted with permission from ref. 170, CC BY 4.0. | ||
The cavity, arguably the defining feature of coordination cages or helicates, is undergoing a functional transition from static encapsulation to dynamic intelligence.171,172 Defect engineering provides a key insight into this evolution. For instance, by tuning the steric hindrance of chiral ligands, flexible tetrahedral and cubic cages with missing edges (Ln4(L2)5 and Ln8(L1)10, can be synthesized.173 These “defects” confer valuable conformational flexibility to the cavity, enabling it to “breathe” and adapt to encapsulate guests of different sizes (e.g., C60, perylene, and BODIPY dyes), achieving a leap from “rigid size-matching” to “adaptive encapsulation”. This characteristic is further highlighted in guest-driven self-assembly. Studies show that the presence of polyaromatic guests can drive ligand assembly toward the formation of otherwise inaccessible Ln4L4-type (L: tris-tridentate ligand) tetrahedral cages (Fig. 18a and b). Crystal structures reveal that this cage possesses a remarkably guest-adaptive cavity, with volumes variable between 420 and 779 Å3 (Fig. 18c).174 Noncovalent confinement of pyrene within the Eu4L4 cage not only suppresses its excimer emission but also opens a new channel for efficient energy transfer from the guest to the host, sensitizing the RE luminescence of the cage. Furthermore, stereoselective self-assembly of either Λ4- or Δ4-type Eu4L4 cages can be achieved using chiral templates like R/S-BINOL, yielding high glum values of up to ±0.125, providing a new platform for chiral recognition and sensing.
 | ||
| Fig. 18 (a) Schematic of guest-driven assembly/chiral induction for Eu4L4 tetrahedral cages. (b) Achiral and chiral guest molecules used as templates in cage formation. (c) Crystal structures of the corresponding host–guest complexes. Reprinted with permission from ref. 174. Copyright 2022, American Chemical Society. | ||
Coordination supramolecular cages and helicates have reached a high level of maturity in chiral amplification and cavity engineering. The next frontier involves the deep integration of dynamic covalent chemistry and stimuli-responsive units with chiral cavities to develop intelligent integrated systems capable of enantioselective recognition, feedback-controlled drug release, or catalytic cycles, marking a transition from “precision structures” to “smart systems”. Such architectures hold promise for enantioselective recognition, controlled guest release, and catalytic cycles with built-in optical readout, marking a transition from exquisitely precise structures to multifunctional, adaptive photonic systems.
3.2. One-dimensional coordination polymers
Starting from rare-earth mononuclear complexes, the evolution through dinuclear and trinuclear systems toward higher-nuclearity states enables the assembly of discrete cage architectures and polynuclear clusters; further dimensionality expansion via ligand-mediated bridging enables the formation of one-dimensional chain-like coordination polymers (1D CPs), whose scientific appeal stems from the anisotropic physicochemical properties that emerge from structurally directed, one-dimensionally extended backbones. Positioned between discrete complexes and fully extended 3D solids, 1D RE-CPs provide a uniquely tractable platform for investigating directional energy transfer, polarized emission, and dimensionality-dependent excited-state dynamics.175–177 Their photophysical performance is governed by ligand field design and supramolecular assembly, which collectively dictate the local coordination environment, interchain coupling, and the balance between radiative and non-radiative decay.Mixed-ligand strategies are particularly effective because they enable simultaneous optimization of light harvesting, coordination rigidity, and vibrational quenching suppression. For instance, X. Liu et al. employed a combination of trifluoromethyl-substituted pyridyl diketone (tfpd) and 1,10-phenanthroline (phen) or 2,2′-bipyridine (bipy) ligands to construct helical Eu3+ CPs [Eu(tfpd)3(phen)]n (1) and [Eu(tfpd)3(bipy)]n (2), which exhibit photoluminescence quantum yields of 68.55% and 63.81%, respectively. Notably, these materials also demonstrate efficient mechanoluminescence activity along with an unusual anti-thermal quenching effect, revealing complex stimulus-responsive luminescence mechanisms (Fig. 19).178 In a complementary supramolecular design, O. Guillou et al. used flexible glutarate alongside rigid 1,10-phenanthroline ligands to assemble dimer-based 1D chains. The resultant strong interchain π–π stacking interactions provide critical supramolecular coupling pathways that underpin their high luminescence efficiencies.179 Additionally, multinuclear RE clusters were employed as precursors to react with halogenated benzoic acids, yielding topologically regular 1D chains. Although halogen substituents do not alter chain topology, they significantly modulate Tb3+ → Eu3+ energy transfer efficiency. This represents a “molecular alloying” approach for fine-tuning luminescence properties through subtle electronic and vibrational perturbations.180
 | ||
| Fig. 19 (a) Synthetic routes of CPs 1 and 2, as well as their crystal structures. (b) Photoluminescence (PL) and mechanoluminescence emissions (ML) of CP 1. Inset: Emission photograph of CP 1. (c) Schematic diagram of the ML luminescence mechanism. (d) ML spectra of 1 under different mechanical forces. Inset: Linear fitting of ML intensities under different forces. (e) ML spectra of CP 1 at different temperatures under 20 N. Inset: Comparison of ML intensities of CP 1 at different temperatures. Reprinted with permission from ref. 178. Copyright 2025, Springer Nature. | ||
One of the most captivating attributes of 1D CPs is their ability to amplify molecular chirality into macroscopic, often micron-scale, chiral morphologies, revealing the decisive role of morphological engineering in dictating optical activity. L.-M. Zheng et al. systematically elucidated this phenomenon through their study on Tb-based CPs. They demonstrated that homochiral Tb-CPs, formulated as [Tb (R- or S-pempH)3]·2H2O [pempH2 = (1-phenylethylamino)methylphosphonic acid], could self-assemble into either well-defined block-like single crystals or macroscopic helical bundles simply by modulating the pH of the reaction system. The origin to this morphological divergence lies in the protonation state of the chiral phosphonate ligand, which dictates the geometric compatibility between different inorganic chains ([Tb–O–P–]n) in the crystal lattice. The macroscopic chirality is visually confirmed by scanning electron microscopy (SEM), with R-1 and S-1 forming distinct left- and right-handed helical bundles, respectively (Fig. 20a). Their mirror-image circular dichroism (CD) spectra further corroborate the handedness of these assemblies at the optical level (Fig. 20b). At the molecular level, crystal structure analysis reveals that the crystalline phases (R-2, R-2′, and R-3) consist of chiral chains containing left-handed triple –Tb–O–P–O– helical strands. Under specific pH conditions, the coexistence and packing of two geometrically incompatible chains induce a persistent twist at the supramolecular level (Fig. 20c and d).181 This geometric mismatch drives a hierarchical self-assembly pathway, ultimately transcribing the molecular chirality (R or S configuration) into visually distinct macroscopic helices. Extending this concept, the same group achieved Eu3+ superhelical structures with identical chemical compositions and chain architectures but distinct morphologies, hollow versus solid helices. The hollow superhelices exhibit pronounced optical microcavity effects, which dramatically enhance the circularly polarized glum to 1.60 × 10−3.182
 | ||
| Fig. 20 (a) Molecular structures of chiral ligands (R/S)-pempH2 and their helical Tb3+ assemblies (R/S)-1. (b) Circular dichroism (CD) spectra of R-1 and S-1. (c) Crystal structures of complexes R-2, R-2′, and R-3, highlighting their chain formation along screw axes (simplified; N/C atoms omitted). (d) Side and top views of the left-handed triple helical chains in R-2, R-2′ and R-3, indicating pitch and diameter. Reprinted with permission from ref. 181, CC BY 4.0. | ||
The 1D topology is also conducive to enhancing magnetic anisotropy and facilitating the integration of optical and magnetic functionalities.183 A paradigm-setting work by J. Crassous et al. reported the helicene-based one-dimensional coordination polymer P-/M-[H6(py)2Yb(hfac)3]n (Fig. 21a–c). This material achieves a tripartite functional synergy within a single chiral system: (1) ligand-sensitized near-infrared emission from Yb3+, accompanied by observable circularly polarized luminescence; (2) field-induced single-molecule magnet behavior resulting from the strong magnetic anisotropy induced by the one-dimensional ligand field; and (3) a significant room-temperature magneto-chiral dichroism effect arising from the interplay between chiral structure and magnetism (Fig. 21d and e).184 Notably, compared to its mononuclear analogue, the polymer exhibits markedly enhanced magnetic anisotropy, demonstrating that magnetic anisotropy, rather than chirality alone, is the predominant factor governing its strong MChD response. This work represents a seminal advance toward genuine magneto-optic coupling in molecular materials, laying a foundation for applications in chiral spintronics and quantum information science.
 | ||
| Fig. 21 (a) Synthesis of a coordination polymer (2). (b) Crystal structure of M-2 viewed along the c axis, with the H6(Py)2 ligand shown in capped-stick style and the Yb(hfac)3 units in space-filling representation. (c) Supramolecular packing of the two enantiomers along the a axis. (d) Temperature-dependent MChD spectra (920–1005 nm) of oriented single crystals of P-2 and M-2 under B = ±1.0 T. (e) Magnetic field dependence of the MChD spectra for P-2 and M-2 recorded at T = 4 K. Reprinted with permission from ref. 184. Copyright 2022, Wiley-VCH. | ||
In conclusion, high-nuclearity clusters, coordination supramolecular cages/helicates, and one-dimensional coordination polymers systematically expand the frontiers of RE photofunctional materials from three distinct dimensions: “supramolecular precision assembly”, “dimensional anisotropy”. and “nanoparticle engineering”, Their common core lies in achieving precise and ultimate control over photophysical processes and material functions through rational design at the atomic and molecular scales. Future developments will trend towards: (1) a shift in synthetic methodology from “art” to “science”, leveraging artificial intelligence and automated platforms for the predictive and precise synthesis of complex structures; (2) an evolution of functionality from “integration” to “synergy and emergence”, exploring novel phenomena arising from the coupling of multiple functions (e.g., magneto-chromic effects and photoelectro-cooperative catalysis); (3) an advancement in applications from “materials” to “devices and systems”, focusing on addressing processability, stability, and interface issues of materials in real devices. Polynuclear RE systems, with their rich structural chemistry and fascinating photophysics, continue to propel the development of next-generation photonic and quantum technologies.
The directional nature of 1D structures creates ideal pathways for managing complex photophysical events. This is exemplified in UC processes, where the 1D arrangement serves as a directional “energy migration highway”. In [Eu1-x(pfbz)2phen)Cl]n, (pfbz = pentafluorobenzoate; phen = 1,10-phenanthroline) the rigid 1D backbone suppresses vibrational quenching, while the linear array of Yb3+ ions facilitates efficient energy migration, enabling a cooperative sensitization process that results in bright, visually detectable Eu3+ red emission upon 980 nm excitation, a rare achievement in molecular-based systems.185 Pushing the boundaries further, a landmark study on [Y2−2xYb2x(FCA)6]n (FCA = 9-fluorenone-2-carboxylate) demonstrated that the harvested NIR energy from Yb3+ ions can be transferred directly to the organic ligand (Fig. 22a), generating bright ligand-centered green upconversion fluorescence with an absolute quantum yield of ∼0.003% (Fig. 22b–d).186 This work establishes a distinct molecular-level “cooperative sensitization” UC mechanism at the molecular level and decisively expands the functionality of 1D CPs beyond metal-centered emission.
 | ||
| Fig. 22 (a) One-dimensional chain structure of [Y2−2xYb2x(FCA)6]n and its UC luminescence photograph under 980 nm laser excitation. (b) Energy-level diagram illustrating the CSU mechanism. (c) Temperature-dependent UC luminescence spectra of [Y2−2xYb2x(FCA)6]n (260–390 K). (d) Cycling stability test of UC luminescence intensity over five heating–cooling cycles between 293 and 390 K. Reprinted with permission from ref. 186. Copyright 2025, Wiley-VCH. | ||
Looking ahead, the rational design of 1D RE CPs will benefit greatly from deeper integration of predictive computational modelling and in situ spectroscopic techniques to unravel the subtle interplay between ligand dynamics, supramolecular interactions, and external stimuli. Expanding the ligand toolbox to incorporate stimuli-responsive and multifunctional moieties could enable dynamic control over anisotropic emission and chirality at multiple scales. Furthermore, coupling these 1D CPs with nanoscale photonic architectures or incorporating them into hybrid composite materials may unlock unprecedented applications in chiral sensing, spin photonics, and quantum information processing. Harnessing the unique synergy between structural simplicity, tunable anisotropy, and hierarchical chirality promises to elevate 1D RE CPs from fundamental models to versatile platforms for next-generation optoelectronic and photonic technologies.
3.3. RE luminescent crystalline porous materials
Building upon the inherent anisotropy of 1D coordination polymers, dimensional expansion through crystalline engineering strategies facilitates the construction of advanced porous architectures with collective photonic functionalities. RE luminescent crystalline porous materials represent an evolutionary step from discrete molecular emitters toward collective and programmable photonic functionalities, achieved by precisely arranging RE ions within ordered porous lattices. Such architectures integrate the “genetic” photophysical characteristics of RE ions (narrow linewidth, long lifetime, and large Stokes shift) with the intrinsic advantages of crystalline porous frameworks (structural order, anisotropy, porosity, and tunability). Based on structural dimensionality and chemical bonding modes, research in this field primarily focuses on two interrelated systems: RE metal–organic frameworks (RE-MOFs)187 and RE-modified covalent organic frameworks (RE-COFs).188,189 Altogether, these systems illustrate an evolution from linear chains to 3D porous architectures and finally to fully organic platforms for functional integration. The core scientific premise is the use of crystal engineering to gain precise, mesoscale control over the entire sequence of energy absorption, transfer, emission, and response.3.3.1.1. Metal node engineering. Metal nodes are the origin of the luminescent properties in RE-MOFs. Their identity, spatial arrangement, and interactions directly determine the energy migration pathways, excited-state dynamics, and ultimately macroscopic optical output.197
Employing a single luminescent RE ion (e.g., Eu3+ and Tb3+) as a node enables construction of MOFs with well-defined structures and uniform luminescence, forming the basis for high-precision sensing. For instance, a single-Eu3+ node MOF reported by L. D. Carlos's group utilized the small energy gap (ΔE = 553 cm−1) between the ligand triplet state and the Eu3+ excited state to achieve temperature-dependent back-energy transfer, serving as a paradigm for high-sensitivity single-ion ratiometric thermometry.198 B. Chen et al. first proposed and validated the strategy of utilizing mixed-RE MOFs to construct a dual-emission ratiometric luminescent thermometer. The authors designed and synthesized a series of isostructural MOFs (Fig. 23a and b), EuxTb1−x-DMBDC (DMBDC = 2,5-dimethoxy-1,4-benzenedicarboxylate), among which the optimal material, Eu0.0069Tb0.9931-DMBDC, exhibited unique thermo-responsive behavior that, within the temperature range of 10–300 K, the emission intensity of Tb3+ (545 nm) decreased due to energy transfer, while that of Eu3+ (613 nm) increased as the temperature rose (Fig. 23c).199 This opposing trend originated from the temperature-dependent Tb3+ → Eu3+ energy transfer.
 | ||
| Fig. 23 (a) Schematic illustration of the synthesis and energy transfer in Eu/Tb-MOFs. (b) Crystal structure of Eu/Tb-DMBDC. (c) The luminescent Eu0.0069Tb0.9931-DMBDC (10–300 K). Reprinted with permission from ref. 199. Copyright 2012, American Chemical Society. (d) The schematic diagram of the selected Ln-BPTC material (Tb0.95Eu0.05-BPTC) with a tunable temperature-dependent XEL color. (e) The XEL spectra of Tb0.95Eu0.05-BPTC at different X-ray dose rates. (f) The temperature-dependent XEL spectra of Tb0.95Eu0.05-BPTC (303–368 K). Reprinted with permission from ref. 200. Copyright 2024, Wiley-VCH. | ||
Building upon this concept, the same research group achieved a significant leap in the application scenario by successfully introducing this mechanism into the field of X-ray excited luminescence (XEL) (Fig. 23d), developing a novel intelligent scintillator material. They proposed a design strategy employing radio-luminescent functional building units (RBUs) and constructed the TbxEu1−x-BPTC series of MOFs [BPTC = (1,1′-biphenyl)-3,3′,5,5′-tetracarboxylic acid]. While inheriting the thermally enhanced Tb3+ → Eu3+ energy transfer mechanism (with energy transfer efficiency increasing from 26.5% to 71.1%), this material achieved the first example of ratiometric thermometry under X-ray excitation in a MOF, with an impressive absolute sensitivity of 6.74% K−1 (Fig. 23e, f).200 Conventional RE MOF thermometers are typically limited to operation within a single temperature region. In contrast, T. Lazarides et al. designed and synthesized low-doped RE metal–organic frameworks based on mixed ligands of 1,4-benzenedicarboxylic acid (BDC) and 2-amino-1,4-benzenedicarboxylic acid (ABDC). By exploiting the exceptionally long phosphorescence lifetime of ABDC (0.33 s) and the temperature-dependent ligand-to-RE3+ (Eu3+, Tb3+, Dy3+, and Sm3+) energy transfer mechanism, they achieved, for the first time, highly sensitive ratiometric luminescence thermometry in two distinct temperature regions: cryogenic (10–110 K) and ambient (70–330 K), with maximum relative sensitivities of 11.1% K−1 and 2.2% K−1, respectively. Notably, the low doping levels of emissive centers (0.25–10 mol%) effectively circumvented concentration quenching effects.201
Incorporating optically inert ions (e.g., Gd3+, Y3+, and Lu3+) serves as a strategic means of “structural dilution” or “crystal-field modulation”, precisely tuning luminescent ion spacing and local symmetry to suppress concentration quenching and regulate energy-transfer efficiency.202 Building upon the aforementioned work, which demonstrated the feasibility of multi-dimensional encoding, optically inert Gd3+ was introduced as a “spacer”. This enabled a systematic investigation of the energy transfer network among Eu3+ (visible emitter), Yb3+ (NIR emitter), and Gd3+. Through precise control of their ratios and spatial distribution, fine and continuous modulation of fluorescence lifetime (on the order of hundreds of microseconds) independent of the emission spectrum was achieved for the first time in a MOF. This work revealed that the distance-dependent intra-cluster energy transfer is the key to controlling lifetime, a mechanism further supported by theoretical calculations(Fig. 24a and b).203
 | ||
| Fig. 24 (a) Molecular structure of the tetratopic organic linker TCPB and the resulting porous framework constructed from nonanuclear RE clusters. (b) Pathways of complex energy transfer within the framework, including ligand-to-metal charge transfer (LMCT) and metal-to-metal charge transfer among the emissive RE ions (e.g., Eu3+, Yb3+, and Nd3+). Reprinted with permission from ref. 203. Copyright 2020, Wiley-VCH. (c) Mechanism of emission lifetime modulation via optically inert ion doping. (d) Orthogonal encoding enabled by composition engineering. Reprinted with permission from ref. 113, CC BY 4.0. | ||
Complementary work by D. F. Sava Gallis et al. employed nonanuclear RE clusters as nodes and a tetratopic organic linker TCPB to synthesize a series of heterometallic MOFs incorporating Eu3+, Yb3+, and Nd3+. The study selected three RE ions with complementary luminescent properties: Eu3+ (providing characteristic orange-red visible emission) and Yb3+ and Nd3+ (providing distinct emissions in the near-infrared region). By systematically varying the ratios of these three ions within the MOF framework, their photophysical outputs were systematically regulated (Fig. 24c and d).113 Given the favorable structural characteristics of MOFs which allow modulation to finely optimize the photoluminescence performance of lanthanide ions, there has been growing interest in designing and synthesizing porous MOF materials with intrinsic upconversion luminescence since the early 2000s, as evidenced by early studies.204,205 Beyond Stokes emission, metal node engineering has been extended to achieve anti-Stokes upconversion luminescence (UCL), enabling functionalities excited in the near-infrared region.206 Building upon these foundational studies, L. Sun et al. recently propelled this direction to new frontiers by discovering cooperative luminescence between Yb3+ ion pairs in a 1D Yb-BTC MOF, producing blue-green emission under 980 nm excitation. By further doping with various activators (Er3+, Ho3+, Tb3+, Eu3+, etc.), they systematically mapped diverse Yb3+-mediated ET pathways within a single MOF lattice, including energy transfer upconversion (to Er3+/Ho3+) and cooperative sensitization (to Tb3+/Eu3+) (Fig. 25a–d).207 This work established a foundational library of upconversion mechanisms achievable through precise heterometallic node integration in MOFs.
 | ||
| Fig. 25 (a) Schematic of cooperative luminescence between Yb3+ pairs and upconversion emission from various activators. (b)–(d) Modulation of cooperative luminescence through doping: (b) diluting with inert Y3+ increases the Yb–Yb distance; (c) doping with Tb3+/Eu3+ leads to cooperative sensitization; (d) doping with resonant ions (e.g., Er3+ and Ho3+) enables efficient energy transfer upconversion and quenches the Yb3+ signal. Reprinted with permission from ref. 207. Copyright 2022, Wiley-VCH. (e) Schematic of the energy transfer process in a 2D Ho/Tb,Yb-PMA MOF. (f) Schematic of the analogous process in a 2D Tb, Yb-PMA MOF, yielding Ho3+ upconversion emission at 660 nm. (g) Illustration of the analogous process in a 2D Tb,Yb-PMA MOF, yielding green upconversion at 545 nm. Reprinted with permission from ref. 208. Copyright 2025, Wiley-VCH. | ||
Building on this mechanistic understanding, later research advanced the platform from 1D to 2D layered MOFs (e.g., Yb-PMA). In this 2D system, efficient UCL was achieved with Ho3+ and Tb3+ activators. More innovatively, by exfoliating and restacking monolayers of pure Yb-MOF and pure Ln-MOF (Ln = Ho, Tb), upconversion was realized via interfacial ET, a novel physical assembly strategy distinct from traditional lattice doping (Fig. 25e–g).208 This progression, from discovering fundamental UCL mechanisms in 1D MOFs to engineering sophisticated energy transfer across van der Waals interfaces in 2D heterostructures, exemplifies the evolution of metal node engineering from controlling simple emission to programming complex, spatially resolved energy transfer networks. Extending these principles of lanthanide-mediated energy transfer, V. P. Fedin et al. designed and synthesized mixed-lanthanide metal–organic frameworks (Ln-MOFs) incorporating Yb3+, Tb3+, and Eu3+ ions utilizing the ether-containing organic linker 5,5′-(2,6-pyridinediylbis(oxy))diisophthalic acid, thereby achieving tunable multicolor upconversion luminescence under NIR-I (980 nm) excitation through Yb3+/Tb3+/Eu3+ ion-mediated stepwise energy transfer pathways, while also remarkably observing for the first time a four-photon upconversion process in the NIR-III (1960 nm) window.209
In the aforementioned studies, the types of RE nodes are typically limited to one or two species for modulating the properties of RE MOFs. Notably, breaking from the conventional RE MOF designs, C. Yang et al. constructed high-entropy RE-MOFs (HE-RE-MOFs) via concurrent integration of five lanthanides (La, Ce, Eu, Dy, and Er). This strategy yields multifunctional porous scintillators that simultaneously achieve: high-efficiency X-ray/γ-ray detection with ∼17000 photons per MeV light yield and 302 nGyair s−1 detection limit; self-calibrating temperature sensing; and record uranium adsorption (1532 mg g−1) enabling dual-mode on-site monitoring through integrated photoluminescence-scintillation mechanisms.210 RE-MOFs also provide an ideal platform for integrating sensitizers and activators to achieve advanced photonic functions. A groundbreaking paradigm is the introduction of a second metal ion as an extremely efficient “metallic antenna”. A seminal study demonstrated this by constructing a heterobimetallic uranyl-europium (UO22+/Eu3+) organic framework. In this architecture, the uranyl ion, with its strong, broad absorption in the 350–500 nm range, acts as a superior sensitizer. Theoretical and experimental evidence confirmed a near-unity energy transfer efficiency (∼100%) from UO22+ to Eu3+, attributed to their well-matched excited-state energy levels. This exceptional process led to a record-breaking photoluminescence quantum yield (PLQY) of 92.68% among all reported Eu-MOFs (Fig. 26).211 Moreover, the high atomic number of uranium endowed the framework with an intrinsically strong X-ray stopping power. As a scintillator, it achieved an ultralow detection limit of 1.243 µGyair s−1, significantly outperforming commercial benchmarks and meeting the stringent requirements for medical X-ray diagnostics. This work establishes a novel “metal-to-metal” sensitization pathway, transcending the limitations of organic linkers and opening a new dimension for designing ultra-bright and highly sensitive Ln-MOFs for radiation detection and solid-state lighting. Furthermore, RE-MOFs rich in heavy atoms show unique potential in high-energy radiation detection. M.-J. Lin et al. constructed a high-brightness, highly stable Eu-MOF scintillator via a “dual-antenna ligand” strategy.212
 | ||
| Fig. 26 (a) Design principle for fabricating EuMOFs with high luminescence efficiency by introducing UO22+ into the structure. (b) Infinite layer of SCU-UEu-2 along the [010] plane. (c) Three-dimensional structure of SCU-UEu-2 along the [001] plane. (d) Energy transfer pathway among BTEC, UO22+ and Eu3+. (e) Simplified model (PhCOO)2UO2Eu(COOPhCOO)45− complex. (f) Predicted UV-Vis-NIR electronic absorption spectra of model (PhCOO)2UO2Eu(COOPhCOO)45− and Eu(COOPhCOO)45− complexes by TDDFT/PBE0 calculations. (g) Energy levels of ground-state and low-lying excited states corresponding to Eu 4f → Eu 4f transitions in Eu(COOPhCOO)45− and singlet/triplet excited states of the ligand part in Eu(COOPhCOO)45− as well as those of the UO22+ part in (PhCOO)2UO2Eu(COOPhCOO)45−. Reprinted with permission from ref. 211. Copyright 2023, American Chemical Society. | ||
The node engineering strategy has been extended beyond homometallic rare-earth nodes. Pioneering work by S.-I. Ohkoshi et al. applied this concept to 3D cyanido-bridged Ln(III)–Co(III) heterometallic frameworks (e.g., [Dy(H2O)2][Co(CN)6]·nH2O).213 Through dehydration, they transformed eight-coordinate hydrated phases into six-coordinate, high-symmetry dehydrated phases, precisely modulating the Ln3+ coordination environment. This geometric engineering of the Ln3+ node generated a strong axial crystal field, which not only activated slow magnetic relaxation (SMM behavior) but also significantly tuned luminescence properties (e.g., shifting Dy-Co emission from near-white to deep yellow). Importantly, in the Tb-Co analogue, this approach enabled the synergistic integration of SMM properties and self-monitoring temperature sensing within a single-phase material: the dehydrated phase exhibited sensitive thermoluminescence over the 6–100 K range, with its operational range highly overlapping the SMM regime (<42 K).214 This work demonstrates that precise control of the Ln3+ node environment is a powerful strategy for achieving magneto-optical synergy and advancing smart materials.
3.3.1.2. Organic linker engineering. In RE-MOFs, organic linkers serve not only as the material's backbone but also act as “molecular programmers”, precisely regulating the luminescence behavior of RE ions through the “antenna effect”. Rational design of the ligand structure dictates the emission color, intensity, lifetime, and response mode of RE-MOFs to external stimuli, forming the central pillar of their functionalization.215–217
The essence of ligand design lies in matching its energy levels with the target RE ion. For instance, G. Qian et al. designed a fluorinated ligand with an appropriate triplet energy level to effectively sensitize Tb3+, resulting in an excellent low-temperature ratiometric thermometer.218 W. Liu et al. designed a V-shaped ligand incorporating naphthyl groups. The energy gap between its triplet state and the 5D0 level of Eu3+ was tailored to be at the edge of efficient energy transfer, achieving an incomplete “antenna effect” and constructing a dual-emission system.219
Moving beyond static energy-level matching, cutting-edge research has begun to harness the dynamic conformational responses of ligands themselves to external stimuli, endowing MOFs with unprecedented intelligent luminescence switching capabilities. A representative study employed the molecular rotor ligand TPA-COOH (4,4′,4″-triphenylamine tricarboxylate), known for its aggregation-induced emission (AIE) properties, to assemble with various RE3+ ions (Sm, Eu, Gd, Tb, Dy, etc.), constructing a series of layered RE-MOFs (Fig. 27a). This work revealed that the TPA-COOH ligand exhibits a “dynamic adaptive antenna effect”: the dihedral angles between its three benzene rings change systematically with increasing temperature (e.g., from 68.74°/60.01°/56.56° to 70.38°/59.80°/60.34°), leading to a corresponding decrease in its lowest triplet state (T1) energy level (Fig. 27b). This dynamically tunable energy level functions like an intelligent, adjustable antenna, enabling it to adaptively select and optimally match the energy levels of different RE ions based on the ambient temperature. The intelligent matching is perfectly demonstrated in co-doped EuxTb1−x-MOFs. Research found that at low temperatures (e.g., 80 K), the T1 level of TPA-COOH favors energy transfer to the 5D4 level of Tb3+, resulting in the material primarily emitting the characteristic green light of Tb3+. As the temperature rises, increased conformational twisting of the ligand lowers its T1 level, significantly improving its match with the 5Do level of Eu3+(Fig. 27c and d). Consequently, the energy transfer pathway intelligently switches from Tb3+ to Eu3+, and the material's luminescence gradually becomes dominated by the characteristic red light of Eu3+ (Fig. 27e).220 This reversible, temperature-driven transition enables multicolor (green-yellow-red) luminescence switching with excellent cyclability. Furthermore, by pre-installing specific functional groups on the ligand backbone or constructing a tailored pore chemical environment, high-selectivity recognition of target analytes can be achieved, subsequently triggering advanced optical responses. For example, Y.-Y. Wang et al. constructed RE-MOFs using linkers bearing uncoordinated triazole groups, which served as Lewis basic sites for the specific recognition of nitroaromatic explosives.221 Through ingenious ligand engineering, Q. Shi et al. synthesized a V-shaped dicarboxylate ligand featuring rigid bipyridine sites. During assembly with Yb3+, the RE ions preferentially coordinated with carboxylate oxygen atoms, leaving the bipyridine nitrogen atoms as free Lewis basic sites within the framework channels, thereby exhibiting strong specific binding affinity for Cu2+.222
 | ||
| Fig. 27 (a) Schematic of the TPA-COOH ligand–RE ion assembly to form layered Ln-MOFs. (b) Changes in the twisting angle of the TPA-COOH ligand in the Eu0.01Tb0.99-MOF structure at different temperatures. (c) and (d) Temperature-dependent emission spectra of Eu0.01Tb0.99-MOF at 80–400 K and the relationship between the ratio of emission peaks and temperature. (e) Relationship between the emission peak intensity of EuxTb(1−x)-MOFs and the contents of Tb3+ and Eu3+ ions, respectively. Reprinted with permission from ref. 220. Copyright 2025, Wiley-VCH. | ||
Recently, Y. Tang et al. employed a strategy of “pre-programming” pore chemistry through ligand design, which not only enables static binding but also facilitates the construction of dynamic, stimulus-responsive intelligent systems. A series of isostructural RE-MOFs (RE-TCPP) were constructed using a tetraphenylpyrazine-derived ligand (H4TCPP). The ligand framework, rich in nitrogen atoms, forms a three-dimensional porous structure containing exchangeable [(CH3)2NH2]+ cations. The study revealed for the first time that when Ag+ ions enter the channels of Eu-TCPP via ion exchange and replace the [(CH3)2NH2]+ cations, they significantly enhance the red luminescence intensity of Eu3+ while quenching the intrinsic emission of the ligand, thereby achieving an efficient ratiometric fluorescence response (Fig. 28a–d).223 More advanced ligand design focuses on dynamically modulating excited-state processes to confer ratiometric fluorescent response capabilities to chemical stimuli. B. Li et al. constructed a sensing platform with dual emission channels using a rigid, large π-conjugated pyrazinoquinazoline tetracarboxylic acid ligand.224 When biamine molecules entered the ligand-constituted pores via hydrogen bonding, they acted as electron donors to dynamically modulate the ligand's electron distribution. This altered the balance between ligand-to-ligand charge transfer (LLCT) and ligand-to-metal charge transfer (LMCT) processes. This ligand-mediated dynamic switching of charge transfer pathways resulted in remarkable ratiometric fluorescence changes and visible color transitions, enabling real-time visual monitoring of food freshness. Post-synthetic modification of ligands is a precise strategy for functionalizing MOF materials. This approach first involves constructing a parent framework with stable structure, ideal porosity, and predefined active sites, such as free amino groups, hydroxyl groups, or unsaturated metal sites, through coordination-driven self-assembly. Subsequently, under mild conditions, a range of well-established organic reactions, including amidation, Schiff base formation, click chemistry, and ligand exchange, are employed to carry out site-specific chemical modifications on the organic linkers or inorganic nodes within the framework.
 | ||
| Fig. 28 (a) The crystal structures of Ln-TCPP. (b) Schematic illustration of the replacement of [(CH3)2NH2]+ cations with Ag+ within the pores of Eu-TCPP. (c) 1H-NMR spectra (400 MHz, D2O, 298 K) after immersing the Eu-TCPP in the solution containing Ag+ ions and in pure water, respectively. (d) Fluorescence intensity ratio (I616/I416) of Eu0.52Tb0.48-TCPP suspension versus the concentration of Ag+. Reprinted with permission from ref. 223. Copyright 2025, Springer Nature. | ||
This enables the “grafting” or “transformation” of specific functional units into predetermined positions of the framework. This methodology offers several key advantages: (1) it allows for the introduction of complex or sensitive functional units, such as biomolecules, fluorescent dyes, catalytic centers, or stimulus-responsive modules, into the confined pores while largely preserving the integrity of the crystalline host structure, thereby effectively expanding the functional scope of the material. (2) Modification not only imparts new functions to the material but can also induce synergistic properties that are absent in both the parent material and the modifying unit alone. (3)Through sequential or selective multi-step modifications, multiple functions, such as sensing, catalysis, and drug loading, can be integrated into a single MOF platform, enabling the construction of integrated systems capable of intelligent responses and even logic operations. For instance, B. Chen et al. used post-synthetic modification to covalently graft the pH-sensitive dye FITC onto an amino-rich Eu-MOF, constructing the EuMOF-FITC probe. The probe exhibits a dual-signal ratiometric response to biogenic amines, weakly basic compounds produced during food spoilage. The amines induce a local pH increase, triggering enhanced FITC fluorescence and diminished Eu3+ emission, leading to a distinct color change from orange-red to green (Fig. 29a).225 Similarly, Z. Li et al. employed FITC to modify an Eu-MOF post-synthetically, developing the EuTPTC-NH-FITC [TPTC = 2′-amino-(1,1′:4′,1″-terphenyl)-3,3″,5,5″-tetracarboxylic acid] sensor for the specific detection of the neurotransmitter serotonin in biomedical applications. The response mechanism combines competitive absorption (quenching Eu3+ emission) and pH sensitivity (enhancing FITC emission), achieving ultrafast response (30 seconds) and high sensitivity. The pronounced color difference allows for visual or smartphone-assisted instant diagnosis (Fig. 29b).226 They also introduced another pH indicator, methyl red, into an Eu-MOF to prepare MR@EuMOF, which similarly shows a dual fluorescence response to biogenic amines such as histamine.227 At the frontier of linker modification, N. L. Rosi et al. adopted a “ship-in-a-bottle” post-synthetic strategy, where small-molecule precursors react within the pores of the Yb-MOF to generate densely packed conjugated structures (Fig. 29c), enabling direct modification of single crystals (Fig. 29d). This method creatively endows the material with a collective near-infrared excitation capability (up to 800 nm) (Fig. 29e and f),96 efficiently sensitizing Yb3+ emission, a property not present in the unmodified MOF or the free molecular components.
 | ||
| Fig. 29 (a) Schematic illustration of the preparation of the EuMOF-FITC probe. Reprinted with permission from ref. 225. Copyright 2021, Wiley-VCH (b) Schematic diagram showing the synthesis of EuTPTC-NH-FITC and its ratiometric detection of serotonin (5-HT). Reprinted with permission from ref. 226. Copyright 2025, Elsevier. (c) Illustration of the “ship-in-a-bottle” strategy for constructing long-wavelength molecular antennae within Ln-MOFs. (d) Bright-field optical microscopy images of MOF-1114(Yb) crystals before and after post-synthetic modification (PSM) with methyl propiolate (MP) (scale bar = 500 µm). (e) and (f) Photoluminescence properties of miniaturized MOF-1114(Yb) dispersed in aqueous solution containing 0.5% Tween-20: (e) Yb3+ emission spectra (λem = 980 nm) under excitation at λex = 380 nm and λex = 500 nm; (f) excitation spectra monitored at λem = 980 nm. Reprinted with permission from ref. 96. Copyright 2020, American Chemical Society. | ||
In summary, organic linker engineering transforms linkers in RE-MOFs from static “energy antennas” into optically responsive, intelligent functional units. This is achieved through energy-level matching, dynamic conformational control, and post-synthetic modification. Key advances include dynamic ligands with an adaptive antenna effect, selective sensing via pre-installed recognition sites, and ratiometric probes from covalently grafted dyes, enabling sensitive detection of temperature, metal ions, biogenic amines, and neurotransmitters. Furthermore, advanced strategies like the “ship-in-a-bottle” method generate synergistic luminescence within frameworks, opening new pathways for applications such as near-infrared imaging. Looking forward, organic linker engineering will propel “intelligent responsive units” toward a higher level of sophistication. Efforts will focus on developing ligand systems with logical decision-making capabilities, enabling on-demand programming of energy-transfer pathways via external fields, and integrating closed-loop “sensing-feedback-action” functionalities within a single framework. These advances will elevate RE MOFs beyond conventional sensing, transforming them into genuine smart materials for applications in point-of-care diagnostics and adaptive devices.
3.3.1.3. Pore environment engineering. The inherent porosity of MOFs provides a uniquely programmable nanospace, offering a powerful platform for pore environment engineering. By selectively encapsulating functional guest species (e.g., organic dyes, perovskite nanocrystals, or metal ions) or engineering intrinsic pore-wall functionalities (e.g., free basic sites and molecular rotors), the photophysical properties of RE-MOFs can be precisely tailored. This approach enables a spectrum of functionalities, from static functionalization for enhanced or tunable emission, to primary dynamic responses for sensing, and further to advanced dynamic intelligent systems with adaptive, multi-stimuli responsive behaviors for high-level information security. This approach enables functionalities spanning from static emission enhancement and color tuning to dynamic sensing responses and ultimately to advanced intelligent systems capable of adaptive, multi-stimuli-responsive behavior for information security and logic operations.228–230 The direct application of pore engineering is the static encapsulation of luminescent guests to achieve predictable and enhanced optical outputs, forming composite materials with new or optimized properties. G. Qian et al. demonstrated this by encapsulating a blue-emitting perylene dye into a porous Eu-MOF (ZJU-88) (Fig. 30a) (ligand: 1,1′:4′,1″:4″,1‴-quaterphenyl-3,3‴,5,5‴-tetracarboxylic acid). The resulting composite ZJU-88⊃perylene exhibited dual emission from the dye (∼473 nm) and Eu3+ (∼615 nm). Crucially, the energy transfer efficiency from the dye to Eu3+ was highly temperature-dependent (Fig. 30b). This created a perfect self-referencing ratiometric thermometer for the physiological range (20–80 °C) (Fig. 30c),231 achieving a high sensitivity of 1.28% °C−1, superior to several reported mixed-RE MOF thermometers. By encapsulating functional dye molecules as “antenna” guests within the pores of Ln-MOFs, the inherent weak photon-absorption ability of RE ions can be efficiently overcome, leading to a significant enhancement of their luminescence. For instance, to develop highly luminescent MOFs for NIR-II bioimaging, Z. Liu et al. proposed a strategy integrating the cyanine dye IR-3C, which possesses strong NIR photon-harvesting capability, with RE3+ ions (RE = Yb, Nd, Er) into the same framework (Fig. 30d). Via a resonance energy transfer pathway, IR-3C transfers the absorbed energy to RE3+, thereby boosting the emission intensity of Er3+, Yb3+ and Nd3+ emission by several orders of magnitude (Fig. 30e and f).232 The rigid porous skeleton of MOFs not only sensitizes the luminescence of the guest species but also provides a protective microenvironment that shields them from quenchers such as water molecules, which is crucial for applications in aqueous or biological environments. Beyond organic guests, MOF pores can immobilize metal ions to create new luminescent composites. N. L. Rosi et al. utilized the channels of an anionic MOF (bio-MOF-1) to encapsulate RE ions like Tb3+, Sm3+ and Eu3+ through a straightforward cation exchange process (Fig. 30g). The MOF framework effectively functioned as an antenna, sensitizing the emission of these ions in both the visible and NIR regions (Fig. 30h–j).233 Beyond encapsulating guests, the pore walls themselves can be functionalized to act as responsive sites. Wang et al. constructed a water-stable Eu-MOF (JXUST-29) featuring accessible, uncoordinated N atoms from the thiadiazole group on the pore surface. These sites act as proton receptors, enabling the MOF to function as a highly sensitive pH sensor, with luminescence intensity enhancing ∼54-fold from pH 2 to 5. Moreover, these pores selectively recognized basic amino acids arginine (Arg) and lysine (Lys) in aqueous solution and even in living cells, via fluorescence enhancement and blue-shift, with ultra-low detection limits.234
 | ||
| Fig. 30 (a) Design of a dual-emitting ZJU-88⊃perylene composite (EnT: energy transfer, Em: emission). (b) Emission spectra of ZJU-88⊃perylene recorded from 20 to 80 °C, excited at 388 nm. (c) Temperature-dependent intensity ratio of Eu3+ (615 nm) to perylene (473 nm) and the fitted curve for ZJU-88⊃perylene. Reprinted with permission from ref. 231. Copyright 2015, Wiley-VCH. (d) Schematic illustration of the energy transfer cascade in RE-BTC-IR. (e) and (f) Schematic illustration of the energy transfer cascade in Er-BTC-IR, Yb-BTC-IR and Nd-BTC-IR excited by 808-nm laser. Reprinted with permission from ref. 232, CC BY 4.0. (g) Schematic illustration of Ln3+ incorporation into bio-MOF-1 and subsequent Ln3+ sensitization by the framework. Excitation and emission spectra of Sm3+@bio-MOF-1 (h), Tb3+@bio-MOF-1 (i), and Eu3+@bio-MOF-1 (j). Reprinted with permission from ref. 233. Copyright 2011, American Chemical Society. | ||
Through deliberate host–guest design and pore environment engineering, RE-MOFs can serve not only as static “nano-containers” to immobilize and enhance guest functionalities, but also as dynamic “intelligent platforms” enabling precise control over chiral transfer, energy transfer, and multi-stimuli-responsive behaviors.235,236 A representative example involves encapsulating achiral MAPbBr3 perovskite nanocrystals (NCs) into a chiral Eu-MOF with 1D channels (Fig. 31a). The confined NCs inherit chirality from the host framework via host–guest coordination (Eu⋯Br and Pb⋯O), resulting in intense circularly polarized luminescence (CPL). This chiral host–guest composite exhibits multiple reversible CPL switching behaviors in response to chemical stimuli (H2O/CH3NH3Br solution) and temperature (Fig. 31b–h),237 based on which complex chiroptical logic gates and multi-level information encryption systems have been successfully designed. On the other hand, research has also demonstrated the feasibility of a reverse strategy: encapsulating chiral CsPbBr3 nanoparticles (NPs) into achiral RE-MOFs (e.g., Eu-MOF) can induce CPL in the originally achiral host (Fig. 31i and j).238 Through π–π interactions between the host and guest, both chirality and energy are transferred from the guest NPs to the host MOF, achieving a high luminescence dissymmetry factor (glum = ∼1.3 × 10−2). This composite material possesses two distinct chiral emission centers (originating from the NPs and the sensitized RE3+, respectively). By varying the excitation wavelength, wavelength-programmable CPL colors can be obtained, thereby constructing an optical stimulus-responsive system that provides an ideal platform for high-security anti-counterfeiting applications. The precisely engineered pore channels in RE-MOFs not only serve as spatial platforms for molecular recognition but also as critical structural units for regulating electron transfer, protonation/hydrogen bonding interactions, and conformational dynamics. Through strategic design and functionalization of their components, metal nodes, organic linkers, and pore environments, multi-dimensional control over luminescence and sensing responses can be achieved. The field has thus advanced from simple color tuning to sophisticated manipulation of energy transfer, excited-state dynamics, and stimuli-responsive behavior, establishing RE-MOFs as versatile “solid-state molecular photonics platforms”. This progress underpins their expanding applications in environmental monitoring, biosensing, and smart information materials. Future development hinges on tackling key challenges: enhancing quantum yields, upconversion efficiency, and stability through deeper insights into excited-state processes; designing robust yet complex host–guest systems; integrating multiple dynamic units to realize adaptive and logic-responsive materials; and advancing device integration via micro/nano-fabrication for practical sensors, displays, and memory devices. Ultimately, the multifunctional integration within the confined pores of MOFs not only advances fundamental photonic science but also paves the way for next-generation optical technologies, driving interdisciplinary innovation that bridges the theory and applications.
 | ||
| Fig. 31 (a) Schematic diagram illustrating the synthesis and CPL mechanism of chiral MOFs encapsulating achiral MAPbX3 perovskite nanocrystals (NCs). (b) Excitation-dependent CPL spectral switching of (P)-(+)/(M)-(−)-EuMOF⊃MAPbBr3 in ethanol dispersion, achieved by alternating 365 nm and 294 nm excitation. (c) Reversible CPL on/off switching of the (M)-(−)-EuMOF⊃MAPbBr3 powder sample monitored at 530 nm, triggered by alternating treatment with water (black dots) or a CH3NH3Br (MABr) ethanol solution (green dots). (d) Time-resolved photoluminescence (PL) decay curves of bulk MAPbBr3 powder (top) and the (M)-(−)-EuMOF⊃MAPbBr3 composite (bottom). (e) Normalized Eu L3-edge X-ray absorption near-edge structure (XANES) spectra of (M)-(−)-EuMOF⊃MAPbBr3, EuBr3, and Eu2O3. (f) Fourier-transform of the k2-weighted Eu L3-edge extended X-ray absorption fine structure (EXAFS) spectra for (M)-(−)-EuMOF⊃MAPbBr3, EuBr3, and Eu2O3. (g) Structural model depicting MAPbBr3 NCs confined within enantiomeric MOFs, highlighting key host–guest interactions (Eu⋯Br bonds, green dashed lines; Pb⋯O bonds, purple dashed lines). (h) Schematic illustration of the mechanism for chirality induction in perovskite NCs based on the chiral space of the host MOF. Reprinted with permission from ref. 237. Copyright 2022, Wiley-VCH. (i) Schematic of the preparation process for chiral R/S-CsPbBr3 nanoparticle (NP)@achiral Eu-MOF composites (R/S-CsPbBr3@Eu-MOF). (j) CPL spectra of the R/S-CsPbBr3@Eu-MOF composite under 365 nm and 254 nm excitation. Reprinted with permission from ref. 238, CC BY 4.0. | ||
Reversible physicochemical interactions between the pore surface and external stimuli (gas molecules, ions, vapors, temperature, etc.) enable real-time, reversible switching of luminescence signals, constituting the core mechanism for MOFs in sensing and dynamic anti-counterfeiting. The pores not only provide spatial sites for molecular recognition but also modulate intermolecular interactions and electron/energy transfer pathways, thereby governing the material's responsive mechanisms.239–241 The pore channels within RE-MOFs can create well-defined donor–acceptor interfaces that effectively regulate electron transfer processes. For instance, E.-Q. Gao et al. designed MOFs featuring electron-deficient terpyridine linkers, where the pore surfaces act as strong electron acceptors. Upon exposure to amine vapors, electron transfer occurs at the interface, leading to quenching of Eu3+ luminescence and a visible color change. This demonstrates the precise control of electron transfer enabled by the pore environment, offering a novel approach for highly sensitive gas sensing.242 Functional groups within the pore channels dynamically regulate selective recognition and optical responses through reversible protonation and hydrogen-bond networks. In the JXUST-29 MOF reported by S. J Liu et al., uncoordinated nitrogen atoms inside the pores undergo reversible protonation, significantly modulating the luminescence of the linker and enabling selective detection of amino acids such as arginine and lysine.243 Moreover, common stimuli like water molecules form adjustable hydrogen-bond networks with amino groups inside the pores, altering the linker's energy levels and enhancing back energy transfer. This drives reversible luminescence switching between red and blue emissions.244
In summary, precisely engineered pore channels in RE-MOFs serve as platforms for molecular recognition and regulate electron transfer, protonation, hydrogen bonding, and conformational dynamics. Strategic design of metal nodes, organic linkers, and pore environments enables multi-dimensional control of luminescence and sensing. The field has advanced from simple color tuning to sophisticated manipulation of energy transfer, excited-state dynamics, and stimuli-responsive behavior, establishing RE-MOFs as versatile photonics platforms. This progress underpins expanding applications in environmental monitoring, biosensing, and smart information materials. Future development hinges on enhancing quantum yields, upconversion efficiency, and stability; designing robust host–guest systems; integrating dynamic units for adaptive materials; and advancing device integration via micro/nano-fabrication for practical sensors, displays, and memory devices. Ultimately, multifunctional integration in RE-MOFs not only advances fundamental photonic science but also paves the way for next-generation optical technologies, bridging theory and applications.
 | ||
| Fig. 32 Schematic of synthesis strategies for RE-modified COFs: (a) direct synthesis; (b) sub-component self-assembly; (c) post-synthetic metalation. Reprinted with permission from ref. 189. Copyright 2020, Wiley-VCH. | ||
To address this issue and simplify the synthetic process, a subcomponent self-assembly strategy can be adopted, which combines dynamic covalent bonds (e.g., imine bonds) with coordinative bonds for the preparation of RE-COFs (Fig. 32b). This method has been widely used to construct various supramolecular coordination cage systems. It is noteworthy that this strategy typically requires dynamic and reversible characteristics during bond formation, which may lead to metal-to-metal exchange within the framework. Moreover, the assembly driven by RE ions may further improve the crystallization behavior of RE-COFs. When target RE-COFs cannot be achieved via direct synthesis due to differences in the reactivity of RE precursors, post-synthetic metalation serves as an alternative approach: modifying target RE ions into the existing framework through metal–ligand coordination (Fig. 32c), thereby providing a reliable preparation route for RE-COFs tailored for specific applications. For example, RE-COFs demonstrate unique advantages and broad potential in the field of luminescent ratiometric temperature sensing, fully embodying the design philosophy that “the pre-designable framework ensures efficient and stable immobilization of RE ions, while the ordered pore channels promote rapid diffusion and selective recognition of analytes”. A. M. Kaczmarek et al. confirmed that grafting RE ions (e.g., Eu3+ and Tb3+) and their complexes onto COF skeletons (e.g., TpBpy-COF) can successfully construct novel luminescent thermometers, which exhibit excellent sensing performance over a wide temperature range (e.g., 10–360 K), with thermal sensitivity as high as 1.403% K−1 and a temperature uncertainty of δT < 1 K (Fig. 33a).245 Furthermore, the same group constructed a multifunctional platform integrating catalysis and temperature sensing by co-modifying COFs (e.g., TTA-DFP-COF) with both RE ions (Eu3+/Tb3+) and transition metal ions (e.g., Cu2+). This platform not only serves as a nanothermometer but also enables in situ, non-invasive monitoring of local temperature gradients and variations during catalytic reactions, such as the Glaser coupling reaction (Fig. 33b).117
 | ||
| Fig. 33 (a) Synthesis of the TpBpy COF and TpBpy-Ln_acac materials. Reprinted with permission from ref. 245. Copyright 2020, Wiley-VCH. (b) Schematic representation of the TTA-DFP COF and grafting of the RE ions and d-metal into the framework. Reprinted with permission from ref.117. Copyright 2020, Wiley-VCH. | ||
More broadly, functionalizing RE ions on COF platforms, researchers have successfully constructed a series of high-performance optical sensors targeting environmental pollutants (TC and UO22+), biomarkers (AA), pharmaceuticals (SMZ/TMP), and food components (organic acids).246–254 These works collectively demonstrate the design advantages of such hybrid materials: the precise pre-designability and modifiability of the framework ensure efficient and stable immobilization of RE3+, while its ordered nanoporous channels promote rapid diffusion and selective recognition of analytes. Specifically, the dual fluorescence response mechanisms–enhancement and quenching–as depicted in (Fig. 34), provide the fundamental basis for such selective sensing. In the enhancement pathway, the analyte facilitates energy transfer from the COF ligand to the RE3+ center, thereby boosting luminescence (Fig. 34a). Conversely, in the quenching mechanism, the analyte competes for coordination or interacts directly with the RE3+, resulting in its diminished emission. This tunable response is key to achieving high sensitivity and specificity (Fig. 34b).
 | ||
| Fig. 34 Schematic illustration of the detection mechanism based on fluorescent RE3+-COFs. (a) Fluorescence enhancement mechanism of RE3+-COFs induced by the analyte. (b) Fluorescence quenching mechanism of RE3+-COFs induced by the analyte. | ||
The field of RE luminescent crystalline materials has fully transitioned from a structure-oriented “construction” phase to a function-oriented “refinement” era. Through insights gained from anisotropy and chirality transfer in one-dimensional coordination polymers, synergistic programming of metal nodes, organic linkers, and pore environments in three-dimensional MOFs, and the precise design of modular COF/HOF sensing platforms, researchers can now achieve extreme control over photophysical properties and integrate complex intelligent responsive functions within these ordered crystalline systems. Looking ahead, future developments in this field will focus on several frontier directions: (1) data- and AI-assisted materials design, enabling predictive discovery of RE crystalline materials with higher luminescence efficiency, longer coherence or exciton lifetimes, and enhanced chiroptical activity; (2) intelligent and adaptive crystals capable of logic operations and sequential responses to multiple stimuli, mimicking feedback mechanisms found in biological systems; (3) hybrid heterostructures, integrating RE-based crystalline materials with 2D materials, semiconductors, or photonic architectures to achieve controllable interfacial energy and charge transfer for advanced optoelectronic devices.; (4) scalable and sustainable synthesis, including mild and green preparation methods, alongside integration into flexible films and miniaturized chips for real-world deployment.
3.4. Self-assemblies of RE complexes
Following the discussion on extended crystalline frameworks (MOFs/CPs) in Section 3.2, which are constructed through strong and highly directional coordination bonds, we now turn to a distinct paradigm in hierarchical material design: supramolecular assembly. This section focuses on the creation of functional architectures through the organized aggregation of pre-formed, luminescent RE complexes (the building blocks from Sections 3.1 and 3.2) via directional non-covalent interactions. The core objective of supramolecular engineering in this context is to transcend the properties of individual molecular units. By precisely controlling the spatial organization of these building blocks, we can amplify their intrinsic photophysical traits (e.g., via environmental rigidification), integrate multiple functions within a single entity, and emerge with new collective properties such as energy transfer networks, chiral amplification, and dynamic stimulus-responsiveness that are inaccessible to isolated complexes. In this review, “RE luminescent supramolecular assemblies” are defined as ordered, nanoscale to microscale architectures in which well-defined RE complexes serve as the primary structural and functional units, integrated through programmed, reversible non-covalent interactions such as π–π stacking, hydrogen bonding, electrostatic interaction, and hydrophobic effects. This definition intentionally distinguishes them from: (1) coordination polymers/MOFs (Section 3.2), which are extended crystalline networks formed by strong, often irreversible coordination bonds defining a fixed, periodic lattice and (2) hybrid materials (Section 3.4), which primarily involve the heterogeneous combination of chemically distinct phases (e.g., nanoparticles in a polymer), with molecular hybrids constituting a supplementary category where homogeneous crystalline materials form through covalent/ionic bonding between molecular precursors (e.g., metal–organic salts that may exhibit coupled ferroelectric-photoluminescent functionalities). In contrast, the assemblies discussed here are characterized by their dynamic, processable, and solution-based formation, leading to structural hierarchies that are environment-dependent. The design logic follows a coherent “Building Block → Driving Force → Architecture → Emergent Function” sequence. First, the molecular complex is engineered not only for photophysical performance but also to encode interaction motifs (e.g., aromatic surfaces, H-bond donors/acceptors, charges, and hydrophobic tails). Second, the choice of solvent and conditions selectively activates one or more of these motifs, driving the self-assembly process. Third, this process yields an architecture with a specific dimensionality and internal packing. Finally, and most crucially, this specific spatial arrangement dictates the collective optical output. For instance, dense packing can suppress vibrational quenching, precise chromophore alignment can enable efficient energy migration, and helical stacking can induce strong CPL. In the following subsections, we categorize and analyze the major classes of RE supramolecular assemblies according to their dominant morphologies and interaction modes, demonstrating how the interplay of weak forces dictates structure and, consequently, function.The self-assembly-induced luminescence (SAIL) strategy, developed by Y. Tang et al., is a typical representative of this direction. They demonstrated that Eu3+ β-diketonate complexes can self-assemble into uniform Eu-NPs in mixed solvents (Fig. 35a). By measuring the luminescence intensity of the assembly in systems with varying water content, it was observed that at lower water content (<30%), the luminescence of Eu3+ is quenched due to O–H vibrational deactivation. However, as the water content increases further, the fluorescence intensity gradually rises, exhibiting an assembly-induced luminescence phenomenon (Fig. 35b). Comparative experiments using H2O and D2O confirmed that the enhancement in luminescence is primarily due to the reduction in the number of water molecules surrounding the luminescent centers and the physical as well as spatial constraints imposed by the assembly, which lower the probability of non-radiative transitions in the Eu3+ complexes. (Fig. 35c and d).119
 | ||
| Fig. 35 (a) Schematic diagram of the synthetic route for self-assembly-induced luminescence (SAIL) in the Eu–complex. (b) Photoluminescence spectra of the Eu-complex in acetone/water mixed solvents. (c) Emission lifetime at 613 nm for the Eu-complex in different ratios of acetone/D2O (black) and acetone/H2O (red). (d) Overall luminescence quantum yield (Φoverall) and ligand-to-Eu3+ energy-transfer efficiency (Φsen) of the Eu-complex in various acetone/water mixtures. Reprinted with permission from ref. 119, CC BY 4.0. (e) Hierarchical self-assembly of a hexameric RE–organic capsule from triple-helical building blocks. (f) Aggregation-induced emission enhancement during the hexamerization of RE triple helicates. Reprinted with permission from ref. 118. Copyright 2021, American Chemical Society. (g) Chemical structure of the exchangeable RE antenna PAnt and schematic of its self-exchange mechanism for circumventing photobleaching in PLIM cellular imaging. (h) Time-gated phosphorescence spectral changes of PAnt upon titration with increasing Eu3+ concentrations. Reprinted with permission from ref. 259, CC BY 4.0. | ||
This confinement concept can be extended to integrate multiple emission mechanisms within a single nanostructure. For example, co-assembly of an aggregation-induced emission (AIE)-active unit with a Tb3+ complex produced TPE-Tb NPs, yielding a dual-emissive system suitable for ratiometric sensing.258 Such systems highlight how confinement assembly not only enhances luminescence but also enables functional integration. Q.-F. Sun et al. demonstrated a hierarchical approach by simulating the formation mechanism of insulin hexamers: coordination first formed triple helices, which were then templated by anions via hydrogen bonds into a discrete Eu6 capsule (Fig. 35e). Helical aggregation imposes constraints on the rotational motion of ligands on the hexameric capsule and reduces non-radiative losses, thereby enhancing luminescence intensity and quantum yield (Fig. 35f).118 This secondary assembly resulted in significant luminescence enhancement, illustrating how cascaded interactions create complex architectures with superior properties. The PAnt-Eu/Tb self-assembly system developed by J. A. González-Vera et al. achieves a similar effect through a different approach by precisely matching coordination dynamics reversibility with self-assembly pathways (Fig. 35g). With RE ions effectively isolated from quenching agents like water molecules within the assembly, while ligand absorption capacity is enhanced by ordered stacking, the system exhibits characteristic RE ion emission and outstanding photostability (Fig. 35h).259 Furthermore, fresh ligands dynamically replace photobleached ligands, enabling the system to exhibit significantly superior photobleaching resistance under continuous excitation compared to traditional complexes (e.g., ATBTA-Eu3+). This property facilitates outstanding applications in PL lifetime imaging microscopy (PLIM).
Extending beyond mere enhancement, supramolecular assembly enables the creation of adaptive and multifunctional nanoparticles. A quintessential example is the spontaneous formation of amorphous nanoparticles from nucleotides and RE ions. Nucleotides (e.g., AMP and GMP) act as multitasking ligands, coordinating to RE3+via phosphate groups and nucleobases. The high coordination flexibility of RE3+ prevents crystallization, yielding dynamic networks with remarkable adaptive inclusion properties. These NPs can encapsulate guests such as dyes (preventing aggregation and yielding high quantum yields), metal nanoparticles (forming core–shell structures), and proteins for NPs@guest by self-assembly (Fig. 36a). They also exhibit intrinsic functions like sensitized Tb3+ luminescence (via energy transfer from nucleobase) and superior performance as MRI contrast agents (due to increased rotational correlation time of assembled Gd3+) (Fig. 36b–d).260 This adaptive platform finds advanced applications in therapy and immunomodulation. A template-induced self-assembly strategy using nanomicelles enables precise size control of RE nucleotide nanoparticles (LNNPs) from sub-5 nm to over 100 nm (Fig. 36e–g).261 These ultrasmall LNNPs efficiently load drugs like doxorubicin, enable tumor-microenvironment-responsive release, and combine effective tumor retention with renal clearance, addressing long-term toxicity concerns. Furthermore, nucleotide/RE3+ coordination NPs can be designed as the stimulator of interferon genes (STING agonists). For instance, Eu-GAMP-NPs activate the STING pathway, promote dendritic cell maturation, and serve as potent nanovaccine adjuvants, eliciting strong humoral and cellular immunity to inhibit tumor growth (Fig. 36h).262
 | ||
| Fig. 36 (a) A schematic illustration of NPs@Guest formation through the self-assembly of 5′-AMP, guest and Ln3+ ions. (b) Photographs of aqueous dispersions of 5′-AMP/Tb3+, 5′-GMP/Tb3+, 5′-UMP/Tb3+, and 5′-CMP/Tb3+ NPs (λex = 254 nm). (c) T1-weighted MR images of Magnevist, 5′-GMP/Gd3+ NPs and 5′-AMP/Gd3+ NPs in buffer. (d) Determination of longitudinal relaxivity (r1) values for magnevist, 5′-GMP/Gd3+ NPs and 5′-AMP/Gd3+ NPs. Reprinted with permission from ref. 260. Copyright 2009, American Chemical Society. (e) Strategy for probing the coordination environment of Ln3+ in LNNPs by employing Eu3+ as the spectroscopic probe. (f) Schematic of the size-tunable synthesis of LNNPs, spanning from sub-5 nm to 105 nm. (g) TEM images of LNNPs synthesized with different Ln3+ ions (Ln = Ce, Nd, Eu, Gd, Tb, Er) (scale bar = 100 nm) and STEM images and the corresponding elemental mappings of ATP-Eu LNNPs, ATP-Gd LNNPs, and ATP-Er LNNPs (scale bar = 10 nm). Reprinted with permission from ref. 261. Copyright 2022, Wiley-VCH. (h) Schematic illustration of RE-nucleotide coordination nanoparticles for STING activation. Reprinted with permission from ref. 262. Copyright 2022, American Chemical Society. | ||
In summary, constructing a rigid nanoenvironment through confined self-assembly is an effective strategy to overcome luminescence quenching of RE complexes in aqueous phases. Different systems consistently indicate that physical isolation of quenchers and restriction of molecular motion are the core mechanisms for enhancing luminescence. Importantly, dynamically reversible assembly processes can further confer advanced properties such as anti-photobleaching and tunable lifetimes. These insights establish confinement assembly as a powerful design principle for next-generation RE-based optical materials, bioimaging probes, and theranostic nanoplatforms.
Translating chiral luminescent RE assemblies from the solution state to highly ordered two-dimensional interfaces is a crucial step toward practical device applications and imposes higher demands for achieving uniform and stable macroscopic chiral optical signals. Stable Langmuir–Blodgett (LB) films can be constructed by designing amphiphilic ligands that combine strong coordination capacity, chiral luminescent antennas, and long alkyl chains. For instance, Eu3+ complexes based on chiral naphthylamine antennas and hexadecyl chains can self-assemble into stable monolayers at the air–water interface, which can be completely transferred onto solid substrates (Fig. 37a). The resulting LB films not only preserve the characteristic red emission and long lifetime of Eu3+ but, more importantly, represent the first observation of clear CPL signals within a RE-based LB film system (Fig. 37b).266 The interfacial confinement effect forces molecules into a highly ordered, close-packed arrangement, effectively transferring the chiral environment of the ligands to the central Eu3+ ions and giving significant circularly polarized luminescence in the macroscopic film.
 | ||
| Fig. 37 (a) Schematic illustration of the Eu3+-directed synthesis of chiral amphiphilic self-assembled complexes and the formation of stable LB films on quartz substrates. (b) Total luminescence (black) and circularly polarized luminescence (CPL, blue) spectra of the 13·Eu monolayer, along with the CPL spectrum (red) of the 23·Eu monolayer (inset: comparison of Eu3+-centered emission spectra of 13Eu·LB films after one and two immersion-deposition cycles). Reprinted with permission from ref. 266. Copyright 2011, Wiley-VCH. | ||
High CPL performance depends on constructing assemblies with well-defined helical chirality. J. H. Jung et al. were among the first to investigate the direct correlation between helical structural parameters and luminescence properties within a supramolecular gel system. Initially, they designed and synthesized ligands featuring chiral alanine linkers and terpyridine coordinating units (R/S-L1) (Fig. 38a). Upon introducing Tb3+ or Eu3+ ions, coordination-driven assembly R-L1 yielded right-handed helical nanofibers. Atomic force microscopy (AFM) studies revealed a strong dependence of the helical pitch length on the concentration of the RE ions (Fig. 38b).267 Importantly, the luminescence intensity exhibited a linear correlation with the pitch: a smaller pitch corresponded to a larger intermolecular tilt angle. This effectively suppressed the strong face-to-face π–π stacking that could lead to quenching, resulting in an approximately threefold enhancement in luminescence efficiency. To achieve higher precision in controlling helical structures necessitates moving beyond thermodynamically dominated random growth. Inspired by the concept of “living” supramolecular polymerization, the same group subsequently developed a seed-induced living polymerization strategy based on RE complexes to prepare structurally well-defined supramolecular multiblock copolymers. This strategy cleverly exploits the difference between the kinetic and thermodynamic products of Eu3+/Tb3+ complexes. Mononuclear complexes act as kinetic products, forming spherical structures, while dinuclear complexes assemble into fibers as the thermodynamic product. Using the thermodynamically stable fibers as “seeds” can initiate the chain-growth polymerization of the kinetic monomers, yielding polymers with uniform length.
 | ||
| Fig. 38 (a) Chemical structures of chiral terpyridine-based ligands R-L1 and S-L1. (b) AFM images of gel-Tb prepared at different Tb3+ concentrations, alongside 3D models illustrating the internal and external right-handed helical structures (top: helical pitch; bottom: tilt angle between two ligand molecules coordinated to a RE ion). Reprinted with permission from ref. 267. Copyright 2017, American Chemical Society. (c) Schematic of seeded living supramolecular polymerization for preparing homomultiblock (right) and heteromultiblock (left) copolymers. Reprinted with permission from ref. 268. Copyright 2024, American Chemical Society. | ||
Furthermore, through sequential monomer addition, they successfully synthesized Eu/Tb heterometallic block copolymers with A-B-A and B-A-B-A-B sequences (Fig. 38c).268 This “living” control enables precise programming of both the size and sequence of the assemblies, laying the groundwork for constructing sophisticated hierarchical structures featuring alternating helical domains and integrated multicolor luminescence.
The formation of helical chirality fundamentally depends on the precise design of molecular building blocks. Research centered on amino acid-modified benzene-1,3,5-tricarboxamide (BTA) vividly demonstrates how minor structural variations can dictate entirely distinct self-assembly pathways and final morphologies. When the linked amino acid is glycine, the system forms gel fibers via one-dimensional supramolecular polymerization. In contrast, substitution with alanine, phenylalanine, or leucine leads to the self-assembly of monodisperse, solid microspheres (Fig. 39a). These orthogonal assembly pathways allow for the independent formation of microspheres and gel fibers within the same medium. More intriguingly, the post-synthetic introduction of Eu3+ or Tb3+ ions can trigger further hierarchical assembly through coordination bonds: gel fibers can transform into microspheres (Fig. 39b), while microspheres can be crosslinked into higher-order superstructures (Fig. 39c). Circular dichroism (CD) and CPL studies confirmed that the molecular chirality of the BTA core is successfully transferred and amplified throughout the entire assembly, determining the absolute configuration around the RE ions, producing strong chiral luminescence (Fig. 39d).269
 | ||
| Fig. 39 (a) Schematic representation of the BTA scaffold before and after stereogenic encoding with selected amino acids, yielding compounds 2–5. (b) Preparation scheme of crosslinked microparticles and corresponding SEM images of the microparticles in both uncrosslinked and crosslinked states. (c) Schematic of the morphology transition from a fibrous gel network of 2 to spherical nanoparticles upon Eu3+ diffusion, accompanied by SEM images of both structures. (d) Emission and CPL spectra of Tb3+-(S)-3 microspheres (green) and Eu3+-(S)-3 microspheres (red). Reprinted with permission from ref. 269, CC BY 4.0. | ||
Beyond the static architectures, recent advances emphasize intrinsically dynamic chiral assemblies whose luminescence and chiroptical properties are stimulus-dependent. By coordinating Eu3+ with chiral ligands featuring “molecular rotor” structures, aggregation-induced emission (AIE)-active dynamic chiral Eu3+ complexes have been successfully constructed (Fig. 40a and b). This system is almost non-emissive in its molecularly dispersed state. However, in the aggregated state, the restriction of intramolecular rotation and vibration not only significantly enhances the characteristic emission of the Eu3+ ion (Fig. 40c and d), but also synchronously achieves a dramatic amplification of the chiral optical signal. This is evidenced by a glum as high as 0.64 and a circularly polarized luminescence brightness (BCPL) reaching 2429 M−1cm−1 (Fig. 40e and f),270 representing a major breakthrough in the “dual enhancement” of core CPL performance parameters.
 | ||
| Fig. 40 (a, b) Schematic illustration of the one-pot construction of dynamic RE complexes exhibiting RIR (a) and RIV (b) properties using ligands containing molecular rotors and vibrational units. (c) and (d) Emission spectra of R-Eu-Et-1 (c) and R-Eu-Et-2 (d) in glycerol/DMSO mixtures under 365 nm excitation; insets show corresponding luminescence photographs of R/S-Eu-Et-1 and R/S-Eu-Et-2 in 0% and 99% glycerol/DMSO under UV light. (e) and (f) CPL spectra and corresponding glum of R/S-Eu-Et-1 (e) and R/S-Eu-Et-2 (f). Reprinted with permission from ref. 270, CC BY 4.0. | ||
 | ||
| Fig. 41 (a) Schematic of a tunable, panchromatic luminescent assembly based on RE supramolecular cascade energy transfer. Reprinted with permission from ref. 271. Copyright 2025, Springer Nature. (b) Synthesis of metal-chelating ligand end-capped poly(butadiene) films with varied contents of tetrathiol cross-linker and photoinitiator. Reprinted with permission from ref. 272. Copyright 2011, American Chemical Society. (c) and (d) Synthesis of photoresponsive luminescent hybrid hydrogels via hierarchical self-assembly of trimeric α-CD-Ln complexes and Guazo units with SPLN, enabling remote-controlled reversible sol ↔ gel transition mediated by photoswitchable host–guest inclusion. Reprinted with permission from ref. 273. Copyright 2018, Wiley-VCH. | ||
The convergence of supramolecular assembly with multi-stimuli responsiveness paves the way for intelligent systems capable of complex information processing and encryption. A sophisticated platform is demonstrated by Eu3+-based nanoassemblies (Eu-NPs) that exhibit triply orthogonal, reversible responses (Fig. 42a). The confined assembly enhances luminescence and enables: (1) pH-switching between red Eu3+ emission and green ligand-centered aggregation-induced emission (AIE) (Fig. 42b); (2) linear thermal quenching for ratiometric thermometry(Fig. 42c); (3) UV/vis-light-modulated FRET with a photochromic dithienylethene (DTE) for emission ON/OFF switching (Fig. 42d).274 Co-assembling with DTE (Eu/DTE-NPs) integrates these responses, enabling the creation of dynamic anti-counterfeiting labels and physically unclonable functions (PUFs) with dual-key (intensity/lifetime) authentication.
 | ||
| Fig. 42 (a) Schematic illustration of Eu-NP assemblies and Eu/DTE-NP co-assemblies. (b) pH-responsive properties of Eu-NPs. (c) Temperature-responsive characteristics of Eu-NPs. (d) Schematic of the photo-triggered luminescence switching behavior of Eu/O-DTE NPs under alternating UV and vis light irradiation. Reprinted with permission from ref. 274. Copyright 2025, Wiley-VCH. | ||
Drawing inspiration from biological metamorphosis, the concept of post-synthetic structure-editing can be implemented in hydrogel actuators. Using a stepwise “inside-out growth” strategy, an initial Eu3+-containing hydrogel can sequentially grow additional hydrogel layers with distinct temperature- and pH-responsive fluorescence properties. This postnatal editing creates an actuator that can display different 3D shapes and fluorescence patterns in response to specific environmental “keys” (OH−, temperature, and pH), mimicking adaptive biological systems for environment-interactive information encryption (Fig. 43).275 Furthermore, certain specific assemblies also exhibit multi-stimuli responsiveness towards circularly polarized luminescence (CPL). Innovatively adopting a “pre-assembly followed by coordination” strategy, Y. Tang et al. implanted Eu3+ ions or Eu(L)3 complexes into preformed helical frameworks. This approach effectively propagates chirality to the lanthanide luminescent centers via coordination bonds, achieving intense CPL with |glum| ≈ 0.022 and a high CPL brightness of 147.90. Crucially, orthogonal stimuli-responsiveness was realized through coordination mode modulation: directly coordinated HNRs-Eu demonstrated humidity sensitivity with thermal stability, while β-diketone-protected HNRs-Eu(L)3 exhibited thermoresponsiveness with moisture resistance.276 This enabled the construction of an advanced anti-counterfeiting platform based on multistage information encryption.
 | ||
| Fig. 43 Bio-inspired structure-editing process of a hydrogel actuator. Mimicking morphogenesis, APS guides the sequential growth of T-Bgel and pH-Cgel on the Eu-gel, yielding adaptive shapes for environment-triggered visual communication (fluorescent ink: red carbon dots). Reprinted with permission from ref. 275. Copyright 2023, Wiley-VCH. | ||
This section has detailed the evolution of RE luminescent supramolecular assemblies from simple confinement structures for emission enhancement to complex, adaptive, and intelligent systems. Through the strategic use of dynamic non-covalent interactions, including coordination networks, electrostatic binding, host–guest inclusion, and hydrophobic forces, these assemblies achieve emergent properties such as adaptive encapsulation, cascade energy transfer, multi-stimuli responsiveness, and post-synthetic structural editing. They bridge molecular functionality with macroscopic applications, offering versatile platforms for biomedical theranostics, sensing, anti-counterfeiting, and next-generation adaptive materials.
3.5. Interface engineering and organic–inorganic hybrid materials
In the preceding sections, we discuss RE coordination-based photofunctional materials, from discrete complexes to extended crystalline frameworks. The core logic lies in achieving the ultimate regulation of photophysical properties through precise molecular and supramolecular engineering within homogeneous or periodic structures. However, extending the functionality of RE luminescent centers to more complex application scenarios, such as in vivo bioimaging, solid-state light-emitting devices, or dynamic responsive systems, often requires materials to possess heterogeneous structures, environmental stability, and effective coupling with external systems. This drives the in-depth research on RE-based organic–inorganic hybrid materials. This section no longer treats hybrid materials as an independent category but rather understands them as an extension and application of molecular engineering principles to the new dimension of heterogeneous interfaces. The core scientific question is: How can we achieve precise control over energy/charge transfer across scales and phases, enhance stability, and integrate functionalities through rational design of the interface between organic components and inorganic components (especially RE-doped nanocrystals and RE-NPs)? We discuss this around three interrelated themes: “interfacial energy management”, “interfacial stability and functionalization”, and “interface engineering for device integration”, revealing how interface engineering serves as a key nexus connecting molecular attributes to macroscopic performance.Grafting organic dye molecules with high light absorption efficiency onto the surface of RE-NPs via covalent or coordinative bonding is a classic strategy to broaden their excitation spectrum and significantly enhance luminescence intensity. This process can be viewed as an extension of the “antenna effect” from discrete complexes to heterogeneous interfaces. Its efficiency is governed by three key factors: spectral overlap, donor–acceptor distance, and the excited-state dynamics of the dye (involving competition between singlet and triplet channels).277–283 The early foundational work of J. C. Hummelen et al. combined the NIR dye IR-806 with β-NaYF4:Yb,Er nanoparticles. Energy transfer from the dye to the Yb3+ sensitizer within the particle via Förster resonance energy transfer (FRET) led to an upconversion luminescence enhancement of up to ∼3300-fold (Fig. 44a–c).284 This example demonstrated the immense potential of interfacial energy transfer (IET). Subsequent studies successfully expanded the effective excitation window from the visible to the near-infrared region (∼700–900 nm) by using dyes with broader absorption profiles (e.g., indocyanine green, ICG) or constructing multi-dye sensitization systems.285,286
 | ||
| Fig. 44 (a) Schematic illustration of the dye-sensitized upconversion nanoparticle. (b) Experimental upconversion excitation spectra of β-NaYF4:Yb,Er nanoparticles with and without IR-806 dye sensitization. (c) Steady-state emission spectra of β-NaYF4:Yb,Er nanoparticles, β-NaYF4:Yb,Er nanoparticles/IR-780, β-NaYF4:Yb,Er nanoparticles and IR-806. Reprinted with permission from ref. 284. Copyright 2012, Springer Nature. | ||
In-depth mechanistic investigations revealed the complexity of interfacial photophysics.287,288 Q. Liu et al. achieved a 2413-fold upconversion enhancement by shortening the distance between the dye chromophore and the nanoparticle surface (using conjugated linkers), highlighting the extreme sensitivity of the distance parameter (Fig. 45a and b).289 A. Rao et al., by systematically varying the linker length and employing pump–probe spectroscopy, directly resolved the dynamics of singlet energy transfer (SET) and triplet energy transfer (TET) from organic ligands to RE-NPs (Fig. 45c and d). They found that TET follows a Dexter mechanism with the rate exponentially decaying with the distance (Fig. 45e and f),290 providing quantitative guidance for interface design. Beyond distance effects, the composition of the inorganic matrix itself can modulate interfacial energy transfer efficiency. For example, Chen et al. discovered that the heavy-atom effect (from Cs+ and Lu3+) in CsLu2F7:Yb/Er nanocrystals significantly promotes the intersystem crossing efficiency of the surface-bound dye IR808 (up to 99.3%), thereby sensitizing Er3+via efficient triplet energy transfer, resulting in a 1309-fold UCL enhancement.291 These studies collectively indicate that interfacial energy transfer has evolved from simple “antenna grafting” into a sophisticated “interfacial photophysics engineering” endeavor, involving the synergistic control of multiple parameters such as distance, energy levels, spin states, and matrix effects.
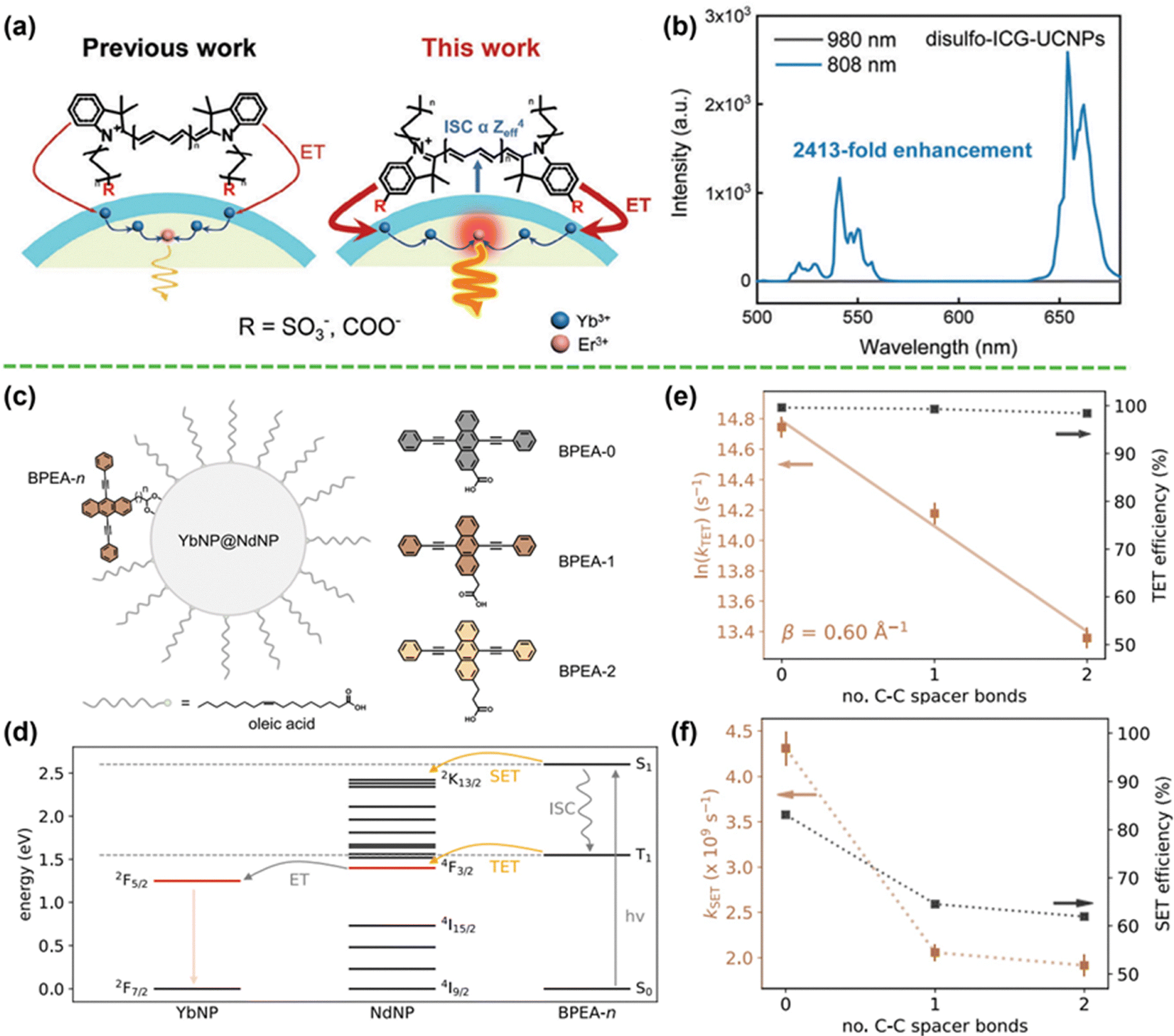 | ||
| Fig. 45 (a) Dye's chemical structure scheme and mechanism of energy transfer in previous and this work. (b) The UCL spectra of UCNPs upon excitation at 980 nm and dye-UCNPs upon excitation with 808 nm laser at the optimal concentrations of disulfo-ICG. Reprinted with permission from ref. 289. Copyright 2023, Wiley-VCH. (c) Molecular structures of the three BPEA carboxylic acid derivatives and a schematic illustration of the architecture of the YbNP@NdNP@BPEA nanohybrids. (d) Energy level diagram describing the energy transfer processes in YbNP@NdNP@BPEA nanohybrids. (e) Triplet energy transfer rates (kTET, brown squares) as extracted from the Tn ← T1 photoinduced absorption decay kinetics. (f) Singlet energy transfer rates (kSET) and singlet energy transfer efficiencies versus the number of C–C bonds, i.e., n in BPEA-n, of the aliphatic spacer. Reprinted with permission from ref. 290, CC BY 4.0. | ||
To overcome the distance limitation and efficiency bottleneck of traditional FRET, cutting-edge research focuses on constructing multi-step “energy relay” and cascade transfer pathways at hybrid interfaces. For instance, Han et al. modified core–shell-shell nanoparticles with organic ligands, realizing a multi-step energy relay: “Tm3+ → ligand (FRET) → ligand triplet state (ISC) → surface Tb3+/Eu3+ (TET)”, transforming conventional downconversion into efficient upconversion emission (Fig. 46a–c).292 R. Deng et al. utilized NaGdF4:Nd3+ nanocrystals and introduced a porphyrin molecule (TCPP) as an energy relay to construct a cascade pathway: “Nd3+ → TCPP (fTET) → annihilator triplet state (TTET) → upconversion emission (TTA)”, achieving efficient sensitization of triplet–triplet annihilation upconversion in multiple organic annihilators under single-wavelength NIR excitation (Fig. 46d–f).293 These examples highlight how interfacial energy relays can bridge the ladder-like energy levels of RE ions with organic photophysics, enabling entirely new emission mechanisms.
 | ||
| Fig. 46 (a) Schematic of the core–shell-shell nanoparticle (NaYF4@NaYbF4: Tm@NaYF4: Tb) and conjugated ligands. (b) Proposed energy transfer mechanism for photon upconversion in ligand-modified nanoparticles. (c) UCL spectra of CPPOA-modified nanocrystals (red) and cation-exchanged NaGdF4: Yb/Tm@NaGdF4: Tb nanoparticles (black) under 980 nm excitation. Reprinted with permission from ref. 292, CC BY 4.0. (d) Proposed cascade process for RE nanocrystal (RENC)-sensitized triplet–triplet annihilation (TTA) upconversion: 4f-triplet energy transfer (fTET) from Nd3+ to a relay molecule, triplet–triplet energy transfer (TTET) to an annihilator, and TTA between annihilators. (e) Schematic of RENC-sensitized TTA upconversion of organic annihilators mediated by a TCPP relay molecule. (f) Corresponding photographs of upconversion luminescence from annihilators: TIPSAn, DPBF, BPEA, and Rub. Reprinted with permission from ref. 293. Copyright 2025, Wiley-VCH. | ||
In addition to distance and matrix composition, the geometric orientation of organic molecules at the interface is a subtle yet critical factor governing the dynamics of interfacial energy transfer. A. Rao et al. quantitatively elucidated the influence of molecular orientation on the full dynamics of triplet excitons by studying the coupling of three anthracene carboxylic acid isomers (1-ACA, 2-ACA, and 9-ACA), differing in the position of the carboxylic acid group, with RE-doped nanoparticles (Fig. 47).294 While the three isomers exhibit similar properties in their free state, their distinct binding geometries on the nanoparticle surface, resulting from the different carboxylate positions, significantly alter their photophysical behaviors: 1-ACA demonstrates the highest triplet generation rate (up to 86%) and yield, whereas 9-ACA exhibits the fastest triplet energy transfer rate to RE3+ ions (up to 1.1 × 108 s−1). This study, for the first time in a RE hybrid system, establishes a quantitative relationship between molecular orientation, interfacial coupling strength, triplet exciton dynamics, and energy transfer efficiency. This work established a quantitative relationship between molecular orientation, interfacial coupling strength, and energy-transfer efficiency, underscoring spatial configuration as a crucial design dimension. Energy transfer can also proceed in the reverse direction, using luminescent RE-NPs as donors and surface-modified organic molecules (or another luminescent center) as acceptors. This strategy not only leverages the photophysical properties of organic molecules (e.g., emission wavelength and environmental sensitivity) but also modulates the luminescence intensity and lifetime of the RE-NPs themselves by controlling the energy transfer efficiency, providing a powerful tool for constructing ratiometric fluorescent probes and smart responsive systems. The mechanisms mainly include FRET, Dexter energy transfer, and reabsorption.
 | ||
| Fig. 47 Tuning triplet exciton pathways via molecular geometry: divergent ISC and TET rates in positional isomers on RE nanoparticles. Reprinted with permission from ref. 294, CC BY 4.0. | ||
F. Zhang et al. developed a nanosensor based on hydrophobic interactions between a cyanine dye MY-1057 and Nd3+-doped downconversion nanoparticles (Fig. 48a and b). Benefiting from the strong spectral overlap between Nd3+ emission (1060 nm) and dye absorption, efficient FRET occurs. In the tumor microenvironment, excessive ONOO− degrades the dye molecule, blocking the FRET pathway and thus restoring the Nd3+ luminescence intensity and lifetime, enabling an “OFF–ON” lifetime-based sensing mode (Fig. 48c–f).295 The same team further constructed a dual-emission ratiometric NIR-II sensor by electrostatically assembling J-aggregated cyanine dye (FD-J) with core–shell–shell–shell nanoparticles (Fig. 48g). The selective absorption of FD-J quenched the emission of the spectrally overlapping Nd3+ channel (1330 nm), while leaving the Er3+ channel (1550 nm) unaffected, establishing an initial ratiometric signal (Fig. 48h–k).296 In inflammatory microenvironments, overexpressed ClO− disrupts the FD-J structure, suppressing energy transfer and restoring Nd3+ luminescence, enabling dynamic changes in the ratiometric responses.
 | ||
| Fig. 48 (a) Scheme of the ONOO−-responsive nanosensor DSNP@MY-1057-GPC-3. (b) In the presence of ONOO−, the structure of the energy acceptor MY-1057 degrades sensitively. (c) Overlap of the MY-1057 absorption and DSNP luminescence emission spectra. (d) Absorption of MY-1057 as a function of ONOO− (0–1/4 equiv.). (e) Luminescence emission intensity and (f) lifetime response of DSNP@MY-1057-GPC-3 at 1060 nm as a function ONOO− concentration. Reprinted with permission from ref. 295. Copyright 2020, Wiley-VCH. (g) Schematic illustration of emission spectral response of the DSNP@FD-J nanosensor to ClO−. (h) Overlap of the FD-J absorbance and DSNP fluorescence emission spectra. (i) Emission spectra of DSNP@FD-J with varied concentrations of FD-J (0–72 µM) conjugation. (j) Fluorescence decay of DSNP and DSNP@FD-J at 1330 nm. (k) Plot of fluorescence ratio changes as a function of FD-J concentration. Reprinted with permission from ref. 296, CC BY 4.0. | ||
X. Liu et al. found that coordinating bidentate phosphonate ligands to NaGdF4:Yb/Tm upconversion nanoparticles induced reconstruction of the surface RE sublattice, reducing the energy mismatch between surface and core Yb3+ ions, thereby suppressing surface quenching and enabling an 11000-fold enhancement of four-photon upconversion luminescence.297 F. Wang et al. demonstrated that a rigid organic shell not only improved stability but also effectively suppressed thermal quenching, allowing upconversion nanoparticles to maintain high brightness at elevated temperatures.298 Novel surface modification strategies can confer exceptional environmental tolerance to nanoparticles. Z. Nie et al. proposed using polymers containing N-heterocyclic carbenes (NHCs) for nanoparticle surface modification.299 The modified nanoparticles maintained excellent monodispersity across a wide pH range (0–14), a broad temperature span (–78 °C to 100 °C), and at high salt concentrations (up to 1000 mM NaCl), significantly outperforming conventional thiol-polyethylene glycol (PEG) modification approaches.
Beyond stability, surface engineering is key to endowing nanoparticles with supramolecular interaction capabilities, enabling controlled assembly and intelligent behavior. By modifying surfaces with specific functional molecules, nanoparticles can be promoted to undergo controlled aggregation via covalent linkage, host–guest recognition, or electrostatic interactions. F. Zhang et al. modified ultrasmall RE nanoparticles with glutathione (GSH). The oxidation-induced cross-linking of its thiol groups under reactive oxygen species (ROS) conditions enabled in situ aggregation and size regulation at target sites, significantly improving imaging signal enrichment efficiency (Fig. 49a).300 Introducing photo-responsive host–guest recognition systems enables high spatiotemporal precision and remote control over nanoparticle interactions. Based on this, they proposed a NIR light-regulated supramolecular assembly/disassembly strategy (Fig. 49b). β-Cyclodextrin-modified downconversion nanoprobes and azobenzene-modified upconversion nanoparticles were sequentially injected into tumor-bearing mice. Host–guest interaction enabled specific assembly at the tumor site, enhancing probe accumulation. Simultaneously, 980 nm NIR light triggered upconversion luminescence, inducing azobenzene isomerization and enabling controllable disassembly (Fig. 49c),301 accelerating clearance to reduce background and toxicity. This design concept based on host–guest recognition and photo-responsiveness has been further extended to the construction of macroscopic, dynamically reversible smart luminescent materials and functional hydrogels.302,303
 | ||
| Fig. 49 (a) Illustration of bioimaging for acute local epidermal inflammation in mice utilizing ultra-small DCNP@GSH nanoprobes. Reprinted with permission from ref. 300. Copyright 2019, Wiley-VCH. (b) Fabrication of azobenzene (Azo) modified NaGdF4:10%Y,25%Yb,0.5%Tm@NaGdF4 UCNP@Azo and β-CD modified NaGdF4:5%Nd@NaGdF4 DCNP@β-CD. (c) Supramolecular recognition-induced assembly and 980 nm NIR-regulated disassembly of nanoparticles. Reprinted with permission from ref.301. Copyright 2018, Wiley-VCH. | ||
 | ||
| Fig. 50 (a) Schematic illustration of the device architecture of LnLEDs with a close-up schematic of LnNP@9-ACA nanohybrids. (b) Simplified schematic showing electron and hole injection through organic molecules to turn on RE ions in an insulating host lattice. Reprinted with permission from ref. 306, CC BY 4.0. (c) Schematic of the synthetic procedure for 4-nm NaGdF4:Tb/Eu nanocrystals capped with ArPPOA ligands. (d) Energy-level diagram of NaGd0.6F4: Tb0.4@ArPPOA illustrating the energy-transfer mechanism. Reprinted with permission from ref. 307. Copyright 2025, Springer Nature. | ||
Complementary to the preceding discussion, it is noteworthy that organic–inorganic interfacial engineering in perovskite systems has evolved into molecular hybridization engineering, wherein atomic-scale coupling between organic cations and inorganic frameworks enables the synergistic manipulation of ferroelectric, piezoelectric, and optical order parameters.
The advancement of this field is characterized by a three-tiered progression: H.-Y. Ye et al. pioneered the establishment of two-dimensional chiral superlattices (R3HQ)4KCe(NO3)8, utilizing organic interfacial engineering to induce the polar point group P1, achieving a ferroelectric polarization of 5.5 µC cm−2 coupled with photoluminescence, thereby revealing multiferroic mechanisms mediated by symmetry breaking.308 Subsequently, by overcoming dimensional constraints, they achieved a piezoelectric coefficient enhancement to d33 = 106 pC N−1 (a 300% improvement over pure phases) in three-dimensional europium-based perovskites (3HQ)2RbEuBr5 through Eu3+/Tb3+ lattice-level hybridization, empirically demonstrating the amplification effects of high-dimensional covalent networks on electromechanical–optical quantum efficiency.309 Addressing the bottleneck of thermally induced decoupling, Y. Zhang et al. employed 4FHQ+ fluorinated cations to reconstruct hydrogen-bonding networks, leveraging strong C–F⋯H–N dipolar hybridization to elevate the phase transition temperature to 423 K while maintaining 2.1% ferroelastic strain switching capability, thereby establishing the compatibility principle between kinetic stability and ferroelasticity.310 Furthermore, they pioneered chiral quantum dimensions, realizing spin–orbit–lattice triple-degree-of-freedom hybridization in R/S-3-hydroxylquinuclidinium perovskites, coupling ferroelastic domain wall switching with intense circularly polarized luminescence (|glum| = 0.003), constituting the first empirical demonstration of asymmetric quantum energy transfer.311 (HQ = quinuclidium, FHQ = 4-fluoro-quinuclidium)
This paradigm shift, progressing from interfacial modification (inducing symmetry breaking) to lattice engineering (level hybridization/stability breakthroughs) and further to chiral quantum control (triple-degree coupling), drives the dimensional extension of inorganic networks through the precision design of organic cations, ultimately achieving cross-dimensional manipulation of multiferroic order parameters, thereby providing an atomic-scale manipulation platform for the design of perovskite functional building blocks.
3.6. Comparative analysis of RE coordination-based photofunctional materials
Having systematically examined each class of RE coordination-based photofunctional materials, from discrete molecular complexes to extended frameworks and hybrid systems, it is instructive to provide a unified perspective that highlights their relative strengths, limitations, and functional niches. Such comparative analysis is essential for guiding material selection toward specific applications and for identifying future research opportunities. Table 1 summarizes the key structural, photophysical, and functional characteristics of the major material categories discussed in this section (Table 2).| Material category | Structural features | Key photophysical advantages | Synthetic control and processability | Stability | Functional versatility | Representative applications |
|---|---|---|---|---|---|---|
| Mononuclear complexes | Discrete, well-defined coordination sphere; single RE center | High color purity; long luminescence lifetime; precise structure–property correlations | Excellent synthetic tunability; solution-processable | Moderate (subject to ligand exchange and photobleaching) | High for sensing; limited for multifunctionality | Mechanistic studies; bioimaging probes; quantum sensors |
| Polynuclear clusters | Atomically precise; high RE density; rigid ligand shells | Enhanced luminescence via collective effects; upconversion; high quantum yields (up to 99%) | Challenging synthesis; requires precise stoichiometry | High (rigid shell protects the core) | High (multimodal imaging, drug delivery, thermometry) | Bioimaging; upconversion; nanothermometry |
| Supramolecular assemblies | Hierarchical structures via non-covalent interactions; dynamic | Chiral amplification; circularly polarized luminescence (CPL); adaptive responses | Moderate (relies on self-assembly pathways) | Moderate (environment-dependent) | High (chiral, dynamic, responsive) | CPL; chiral sensing; anti-counterfeiting |
| 1D coordination polymers | Extended chain structures; anisotropic topology | Directional energy transfer; polarized emission | Moderate (crystalline growth control) | Moderate to high (depends on chain rigidity) | Moderate (anisotropy, chirality) | Polarized light sources; magneto-optical devices |
| MOFs | 3D porous crystalline networks; tunable pore environments | High surface area; synergistic metal-node/linker emission; guest encapsulation | High (modular synthesis; post-synthetic modification) | High (often robust; water-stable designs available) | Very high (sensing, catalysis, scintillation) | Ratiometric sensing; white LEDs; scintillators; gas sensing |
| COFs | 2D/3D porous covalent networks; high chemical stability | High stability; modular functionalization; RE post-synthetic grafting | Moderate (imine/hydrazone linkage control) | High (robust covalent bonds) | High (sensing; photocatalysis) | Ratiometric thermometry; chemical sensing |
| Organic–inorganic hybrids | Heterogeneous interfaces; RE-NPs with organic ligands | Synergistic properties; extended excitation windows; device integration | Moderate (interface control critical) | Moderate to high (depends on coating) | Very high (device integration, theranostics) | LEDs; bioimaging; solar cells; electroluminescent devices |
| Material category | Design strategy | Encryption dimensions | Work (Ref.) |
|---|---|---|---|
| Stimuli-responsive molecules/assemblies | Light/heat/pH-triggered changes in ligand structure or energy transfer pathways | Dynamic color/intensity switching, ratiometric response, logic gates | 220, 241, 274, 312 and 318–320 |
| Temporal dimension modulation materials | Controlling triplet exciton migration kinetics or energy transfer to RE ions | Programmable fluorescence lifetime (µs to hundreds of ms) | 113, 321 and 322 |
| Spatial/chiral structure materials | Chiral induction, host–guest assembly, multidimensional structure construction | Circularly polarized luminescence (CPL), 3D shape/structural color response | 323 and 324 |
| Physically unclonable function tags | Leveraging intrinsic microscopic disorder from random assembly processes | Uncopyable microscopic patterns, dual-key (intensity/lifetime) authentication | 273, 325 and 326 |
| Multifunctional integrated systems | Integrating multiple luminescent centers/response mechanisms into a single platform | Up/down-conversion multimodal emission, multi-stimuli synergistic response, self-healing | 208, 327 and 328 |
Several overarching themes emerge from this comparison. First, a clear trade-off between structural precision and functional complexity exists across material classes. Discrete mononuclear complexes offer unparalleled atomic-level control, making them ideal for mechanistic studies and fundamental structure–property elucidation, but their functional complexity is limited by the constraints of a single metal center. Conversely, extended frameworks like MOFs enable sophisticated multifunctionality (e.g., combined sensing, catalysis, and guest encapsulation) at the cost of increased structural heterogeneity and more complex structure–property relationships. Second, the dimensionality of the material architecture critically influences photophysical properties. The transition from 0D clusters to 1D chains to 3D frameworks progressively enables anisotropic energy transfer, directional emission, and collective exciton dynamics that are inaccessible in isolated molecules. This dimensional control is a powerful design lever for engineering specific optical functions. Third, the stability–processability–functionality triangle represents a key design consideration. While crystalline frameworks (MOFs and COFs) often exhibit superior chemical and thermal stability, their poor solution processability limits certain applications. Conversely, molecular complexes and supramolecular assemblies, while offering excellent solution processability, may face stability challenges in demanding environments. Hybrid materials strategically combine the advantages of different components, offering a pathway to balance these competing requirements. Finally, the emerging frontiers in the field are increasingly characterized by the integration of multiple functionalities within single material systems, such as combining luminescence with magnetism (magneto-optical materials), chirality (CPL), or electrical responsivity (electroluminescence), and by the development of intelligent, adaptive materials capable of responding to complex environmental cues. The future of RE coordination-based photofunctional materials lies in harnessing the unique structural and electronic properties of RE ions while leveraging the full spectrum of synthetic and assembly strategies to create materials that are not only highly efficient but also smart, multifunctional, and application-ready.
4. Emerging applications
Building upon the systematic elaboration of controlled synthesis and performance modulation strategies for RE coordination-based photofunctional materials, their remarkable properties ultimately need to be translated into the ability to solve practical problems, thereby demonstrating their scientific and technological value. With the continuous enrichment of material systems and the precise optimization of performance, a series of groundbreaking emerging applications are flourishing. These applications not only fully exploit the unique photophysical characteristics of RE ions (such as narrow-band emission, long lifetime, up/down-conversion capabilities, and stimulus responsiveness) but also, through ingenious material design and system integration, extend the performance advantages from the molecular level to macroscopic functional devices and systems.This section first highlights the commercially mature applications of RE-based luminescent materials, providing a benchmark for understanding technology readiness levels. It then showcases representative frontiers under active laboratory investigation, including optical anti-counterfeiting and information encryption, biomedicine, radiation detection (scintillators), optical wireless communication, and machine learning-aided design. By bridging molecular-level control with macroscopic functionality, these materials address real-world challenges and open new avenues for next-generation photonic and electronic devices. Key challenges and future perspectives are also discussed to guide continued innovation in the field.
4.1. Optical anti-counterfeiting applications for multilevel information encryption
RE coordination-based photofunctional materials, with their unique luminescent properties, such as high color purity, long lifetimes, up/down-conversion capabilities, and stimulus responsiveness, serve as an ideal platform for constructing high-security optical anti-counterfeiting and information encryption systems. Their evolution has progressed from early one-dimensional security relying on static emission color to multilevel systems that comprehensively utilize the temporal dimension (lifetime), dynamic responsiveness, spatial dimension (structural or chiral photoluminescence), and physically unclonable functions (PUFs). These advancements significantly enhance information density and security levels.312–316 In contrast to the static emission color and fixed intensity of commercial inorganic phosphors used in conventional anti-counterfeiting inks, the systems discussed below exploit dynamic, multi-responsive, and time-dependent optical behaviors, offering fundamentally higher security levels at the cost of increased material and readout complexity. | ||
| Fig. 51 (a) Schematic of DAE chromophores embedded in ZJU-88 and their reversible photoisomerization under UV/vis light. (b) Normalized emission spectra of ZJU-88⊃OF-DAE and absorption spectra of OF-DAE and CF-DAE. (c) Emission spectra of ZJU-88⊃OF-DAE under 300 nm UV irradiation, with the inset showing intensity changes at 613 nm. (d) Emission spectra of ZJU-88⊃CF-DAE under visible light (>450 nm), with the inset showing intensity variation at 613 nm. (e) Photographs of ZJU-88⊃OF-DAE in daylight (i) and under a 365 nm UV lamp and (ii) after 60 s UV irradiation in daylight (iii) and under a 365 nm UV lamp (iv). Reprinted with permission from ref. 241. Copyright 2019, Wiley-VCH. (f) Schematic illustration of the chemical structures of the photo-responsive samples. (g) Digital photos and (h) UV-vis absorption spectra showing the photo-responsive behavior of the representative sample P5. (i) Digital images of a series of Tb3+-containing polymers (from left to right): with SP feed fixed at 0.5 mg and Tpy feed gradually increasing from 0.5, 1, 2, 4, to 8 mg. Reprinted with permission from ref. 312. Copyright 2024, Wiley-VCH. | ||
Supramolecular self-assembly or composite construction can not only overcome concentration quenching and enhance luminescence efficiency but also confer novel properties. Y. Tang et al. constructed Eu3+-based nanoparticles (Eu-NPs) via J-aggregation-driven self-assembly of a tailored β-diketone complex (Eu (THA)3) [HTHA: 4,4,4-trifluoro-1-(9-amylcarbazole-3-yl)-1,3-butanedione]. Spatial confinement within the nanostructure suppresses non-radiative decay, boosting both the photoluminescence quantum yield and lifetime. Crucially, this assembly integrates three orthogonal stimulus responses: pH-triggered reversible switching between the red emission of Eu3+ and the green luminescence of the AIE ligand; linear thermal quenching for dual-mode (intensity/lifetime) thermometry; and UV/vis-light-modulated FRET between dithienylethene (DTE) and Eu3+ for reversible emission on/off switching (Fig. 52a–c).274 T. Chen et al. finely manipulated room-temperature phosphorescence (RTP) via dynamic RE coordination. Embedding terpyridine phenylboronic acid (TPYBOH) phosphors into a polyvinyl alcohol (PVA) matrix yielded ultralong organic RTP with a lifetime up to 0.629 s. Introducing Eu3+ or Tb3+ enables precise modulation of the phosphorescence intensity and lifetime through competitive photosensitized energy transfer (PSET), allowing on-demand manipulation of RTP performance (Fig. 52d and e).317
 | ||
| Fig. 52 (a) Schematic of DAE chromophores embedded in ZJU-88 and their reversible photoisomerization under UV/vis light. (b) Normalized emission spectra of ZJU-88⊃OF-DAE and absorption spectra of OF-DAE and CF-DAE. (c) Emission spectra of ZJU-88⊃OF-DAE under 300 nm UV irradiation, with the inset showing intensity changes at 613 nm. Reprinted with permission from ref. 274. Copyright 2025, Wiley-VCH. (d) The introduction of Ln3+ (Eu3+ and Tb3+) into PVA-terpyridine phenylboronic acids (TPYBOH@PVA) for finely manipulating RTP properties via the ligand-to-metal photosensitized energy transfer process, with the resulting Ln3+-manipulated materials showing muticolor fluorescence and mutual blue phosphorescence. (e) The proof-of-concept demonstrations of the advanced multi-level encryption and anti-counterfeiting based on Ln3+-manipulated RTP materials, revealing a greatly enhanced security level. Reprinted with permission from ref. 317, CC BY 4.0. | ||
In summary, through synergistic ligand and assembly engineering, RE-based photofunctional materials have evolved into versatile platforms capable of multidimensional, stimulus-responsive, and time-encoded information encryption. The following table summarizes typical material systems and their respective dimensions in information encryption.113,208,220,241,274,312,318–328
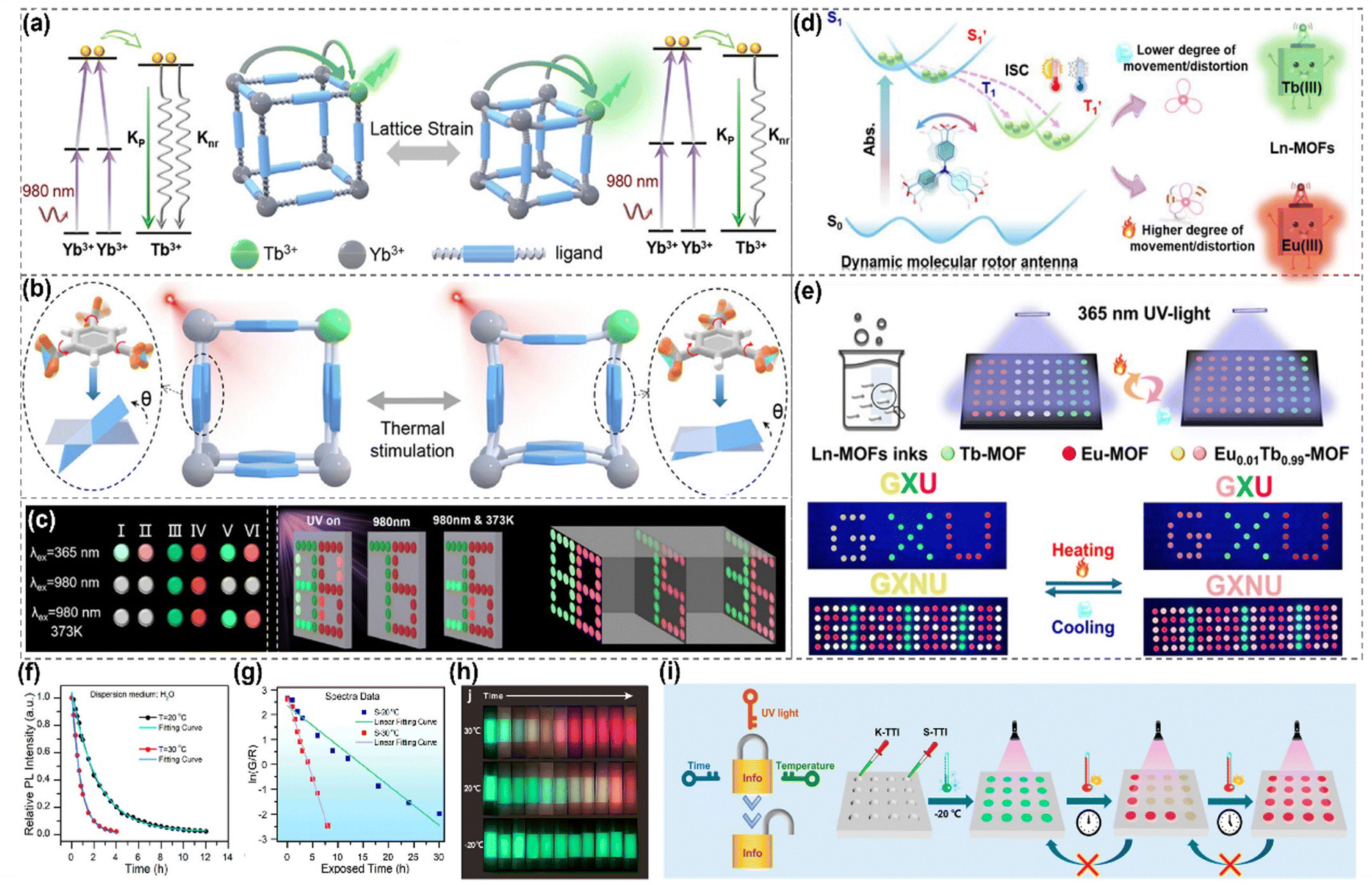 | ||
| Fig. 53 (a) Schematic sketch of responsive UC Ln-MOFs via lattice strain regulation. (b) Design of dynamic responsive UC MOFs via thermally-induced lattice strain regulation. (c) Images of “88” made with the different types of MOFs for information encryption. Redrawn from data in ref. 319, Copyright 2025, Wiley-VCH. (d) Schematic of temperature changes regulating the degree of motion of the TPA-COOH ligands, resulting in an adaptive antenna effect that selectively sensitizes different lanthanide ions. (e) Luminescence changes in the “GXU” and “GXNU” patterns under a 365-nm UV light and after heating. Redrawn from data in ref. 220, Copyright 2025, Wiley-VCH. (f) Relative PL intensity changes over time at T = 20 and 30 °C and fitting curves. (g) The ln(G/R) values versus exposure time and linear fitting curves. (h) Photographs of S-TTIs over time at different storage temperatures under 365 nm UV light. (i) Schematic diagram of the fabrication of a multilocking encryption system based on S-TTIs and K-TTIs. Reprinted with permission from ref. 318. Copyright 2025, Wiley-VCH. | ||
Achieving richer and more intuitive luminescence color switching under a single stimulus is beneficial for constructing higher-performance anti-counterfeiting and information encryption materials. Zhu et al. introduced a dynamic molecular rotor, 4,4′,4″-triphenylamine tricarboxylic acid (TPA-COOH), as a smart responsive unit to construct stable Ln-MOFs. The propeller-like TPA-COOH ligand responds to temperature changes by modulating the rotational freedom and torsional angle. This molecular-scale conformational change directly modifies its matching degree with the excited-state energy levels of different RE ions (such as Tb3+ and Eu3+) (Fig. 53d). Consequently, temperature stimulation drives the switching of the energy transfer pathway between Tb3+ and Eu3+, leading to a reversible change in the macroscopic luminescence color between green and red (Fig. 53e).220 Accurately studying the distinct response dynamics of luminescence performance under different external stimuli can lead to a locking encryption system with multiple logics. Y. Tang et al. utilized the irreversible fluorescence quenching of CsPbBr3@SiO2 nanoparticles in water. By tuning parameters like temperature, nanoparticle concentration and salt addition, the quenching kinetics were precisely modulated (Fig. 53f and g). Combining these with long-lived red-emitting Eu3+ complexes created colorimetric fluorescent time–temperature indicators (TTIs) showing a dynamic green-to-red color change over time/temperature (Fig. 53h). Furthermore, combining TTI units with different kinetics enabled the construction of a multi-logic encryption system with “time-lock” and “temperature-lock” features (Fig. 53i).318 Encrypted information becomes readable only within a specific time-temperature window (when the colors of different TTI units match a predefined combination) and is subsequently self-erased upon reaction completion, achieving “one-time” high-security decryption.
 | ||
| Fig. 54 (a) Schematic of the conceptual design, where black and red spheres denote Lu3+ and Eu3+ (or Tb3+), respectively. (b) Comparison of emission lifetime tuning ranges: previous studies (0.01–1 ms) versus this work (1–152 ms). (c) Chemical structures of the Eu3+/Tb3+-incorporated Lu3+ molecular crystals used for proof-of-concept. (d) Emission photographs of Eu0.010Lu1.990-dpph, Tb0.013Lu1.987-dpph, Eu0.005Lu1.995-dcph, and Tb0.011Lu1.989-dcph. (e) Energy-level diagrams of Eu0.010Lu1.990-dpph and Tb0.013Lu1.987-dpph. Reprinted with permission from ref. 321. Copyright 2025, Wiley-VCH. | ||
 | ||
| Fig. 55 (a) Schematic of the assembly of negatively charged Ag-NPs mediated by positively charged [EuL3]3+. (b) Preparation of physically unclonable function (PUF) labels. (c) Readout of label codes using fluorescence intensity, Raman signal, and fluorescence lifetime. Reprinted with permission from ref. 326, CC BY 4.0. (d) [YbL3]3+-mediated self-assembly of CsPbBr3 nanocrystals into ordered 3D cubic structures (Yb-CsPbBr3 SNCs). (e) Confocal microscope images of Yb-CsPbBr3 SNCs. (f) and (g) Raw intensity, binary, and quaternary encoding maps for the fluorescence intensity of each pixel in 10 × 10 (f) and 20 × 20 (g) arrays. (h)–(j) Evaluation of 15 PUFs (10 × 10 and 20 × 20): (h) randomness, (i) uniqueness, and (j) complementary error function analysis. (k) and (l) Histogram of reproducibility frequency distribution after pairing 15 encoded labels of (k) binary and (l) quaternary. Each pixel's fluorescence intensity level was used for pairing; x- and y-axes correspond to the first and second measurements, respectively; color bars indicate the similarity index. Reprinted with permission from ref. 325. Copyright 2025, Wiley-VCH. | ||
 | ||
| Fig. 56 (a) Operating principle of the stimuli-responsive CDs–Eu–HL nanocomposite for multidimensional optical recording and encryption. (b) Schematic of information coding, reading, encryption, and decryption. Reprinted with permission from ref. 327. Copyright 2017, Wiley-VCH. (c) Illustration of upconversion/downshifting circularly polarized luminescence beyond 1200 nm in a single nanoparticle for optical anti-counterfeiting and information encryption. Reprinted with permission from ref. 323. Copyright 2025, Wiley-VCH. (d) Synthesis of a novel 2D Yb-PMA MOF achieving upconversion luminescence under 980 nm excitation by introducing Ho3+, Tb3+, or Eu3+ as a luminescent center and using Yb3+ as a sensitizer; such designed RE-based 2D MOFs show promising potential for information-encryption applications. Reprinted with permission from ref. 208. Copyright 2025, Wiley-VCH. (e)–(f) Dual-mode hydrogel for multistage information encryption: (e) structural diagram showing the self-assembly and photopolymerization of the pDGI/p(AAm-DMA-6APA) hydrogel, followed by Ln3+ coordination to yield varied fluorescence emissions. (f) Schematic of a dual-mode hydrogel for multistage information encryption. Reprinted with permission from ref. 324. Copyright 2024, Wiley-VCH. | ||
CPL combines luminescence with chirality, adding a signal dimension and decryption difficulty. P. Duan et al. achieved full-color gamut, multimodal CPL by co-assembling multilayer core–shell RE nanoparticles (exhibiting broad UV to NIR-II up/down-conversion emission) with chiral liquid crystal polymer (CLP) films. Notably, composite films prepared by the drop-casting method exhibited view-side-dependent CPL signals, originating from the photonic bandgap effect of the CLP film and the asymmetric distribution of nanoparticles. This property is exploitable for advanced image anti- counterfeiting and binary information encryption systems (Fig. 56c).323 L. Sun et al. successfully achieved upconversion red emission under 980 nm excitation in 2D RE-MOFs (Yb-PMA) by introducing Ho3+ as the activator and Yb3+ as the sensitizer. Further co-doping with Tb3+ or Eu3+ created multimode-emitting MOFs with both upconversion and downshifting luminescence (Fig. 56d).208 Recent studies have identified two types of intelligent responsive MOF/CP-based luminescent materials. ZJNU-1003-RE enables dynamic spectral modulation under dual-mode excitation (UV and X-ray) and facilitates photonic barcode anti-counterfeiting by coupling the regulation of Y3+/Tb3+/Eu3+ ratios with thermal stimulation.329 Additionally, Eu/Tb@CP2 employs an in situ encapsulation strategy for rare earth ions, achieving a 37-fold enhancement in quantum yield, and constructs a time-resolved multilevel encryption system that integrates the photochromic properties of the TPT ligand.330 Furthermore, this encapsulation methodology is also applicable to the rare earth supramolecular systems.
Furthermore, this encapsulation approach is also applicable to the RE supramolecular domain: T. Chen et al. reported a dual-mode hydrogel featuring independently tunable structural color and fluorescence. Shear flow-induced self-assembly created a rigid lamellar structure of poly (dodecylglyceryl itaconate) (pDGI) as a confined domain, within which a polymer network was formed, generating structural colors. Simultaneously, a fluorescent monomer (6APA) capable of coordinating with RE ions (Eu3+/Tb3+) was introduced to endow the system with fluorescence. The structural color is tunable by crosslinking density and water content, while the fluorescence color is modulated by the RE ion ratio. Information from the two modes can be displayed separately in different channels or synergistically overlaid to read the final message, enabling a programmable multi-level information encryption output (Fig. 56e and f).324
Notably, the incorporation of inert ions (e.g., Y3+, and Sc3+) critically modulates luminescence properties in information encryption materials, which substantially enhances decryption reliability and security performance. The core mechanism lies in their stable d0 configuration and compatible ionic radii, which modulate the host lattice field and passivate quenching centers, thereby enhancing the radiative transition probability of activator ions. For instance, J. Xie et al. demonstrated that Y3+ and Sc3+ doping can enhance the upconversion luminescence of Na3ScF6:Yb/Er and NaYF4:Yb/Er samples.331 Additionally, L. Sun et al. reported that Y3+ doping not only improves the crystallinity of Eu(1−x)Yx-BTC MOFs (BTC: 1,3,5-benzenetricarboxylic acid), but also optimizes the luminescence performance of RE MOFs by reducing cross-relaxation and minimizing energy dissipation.332 These enhancements enable better application of such materials in photonic barcodes and fingerprint detection.
In summary, optical anti-counterfeiting technology based on RE photofunctional molecular materials is rapidly advancing towards greater dynamism, multidimensionality, integration, and intelligence. Based on the progress demonstrated above, future research efforts should focus on: (1) developing more efficient and stable dynamic response units, particularly upconversion-responsive systems excitable by NIR light for deep tissue penetration, and smart materials with excellent cycling stability and fast response times, (2) exploring the theoretical and practical limits of lifetime encoding and striving for the orthogonal and independent control of lifetime alongside color, intensity, and polarization to maximize encoding capacity, (3) designing intelligent encryption materials with logic operation and environmental interaction capabilities, enabling materials to make judgmental responses to sequences, intensities, or combinations of multiple stimuli, thereby achieving higher-level cryptographic logic and dynamic “one-time-pad” style anti-counterfeiting, (4) advancing materials towards flexible, wearable, and printable devices, while addressing challenges in the consistency of material properties (e.g., PUF randomness and luminescence performance) and long-term stability during scale-up from lab to production, to facilitate their practical application in commodity, document, and pharmaceutical security. Altogether, these advances will propel optical information encryption technology to new heights, providing a more robust and intelligent material foundation for protecting high-value information and safeguarding economic and public security.
4.2. Biological applications
Compared with clinically approved Gd-based MRI agents and Eu/Tb chelates used in time-resolved immunoassays, the RE coordination-based platforms described here remain predominantly at the stage of proof-of-concept in animal models. Their key advantages lie in integrated functionalities, such as multimodal imaging, stimuli-responsive drug release, and NIR-II deep-tissue detection, that are difficult to achieve with commercial agents. The evolution of RE coordination-based photofunctional materials in biomedicine vividly demonstrates the integrated logic of “molecular design → properties →applications”. From the earliest regulation of light absorption and energy-transfer efficiency, this field has progressively addressed central biomedical challenges, including sensitivity, penetration depth, targeting specificity, and therapeutic controllability. Its development trajectory can be summarized as a continuous progression from performance control at the molecular level, through functional realization via supramolecular and framework engineering, to system integration into intelligent theranostic platforms.This section systematically narrates this progression. We first introduce probes that achieve advanced sensing and labeling through precise molecular design. We then describe multifunctional delivery and imaging platforms constructed via supramolecular and framework engineering. Finally, we focus on intelligent theranostic systems integrated through innovative mechanisms. Altogether, these examples demonstrate how sustained advances in fundamental photophysics and coordination chemistry are translating into increasingly sophisticated biomedical functions and, ultimately, enabling the principles of precision medicine.333–338
Following these pioneering molecular-design principles and proof-of-concept biological demonstrations,346–348 the “electronic-symmetry-tuned emission” strategy proposed by F. Zhang et al. exemplifies this. This work systematically modifies the peripheral substituents of Er3+-phthalocyanine complexes to finely tune the ligand's electronic symmetry, thereby reshaping its excitation spectrum while maintaining efficient triplet energy transfer and bright 1530 nm emission (Fig. 57a and b). This design cleverly shifts the detection signal from the emission wavelength to the excitation wavelength, enabling probes with a single emission band to achieve dynamic, quantitative sensing of pH and Cu2+via excitation-ratiometric methods (Fig. 57c–e),349 opening a new avenue for high-precision biosensing in the deeper tissue-penetrating NIR-IIb window (>1500 nm). Combining the optical properties of RE complexes with bioorthogonal chemistry enables specific labeling and imaging of biomolecules. J.-L. Zhang et al. reported hydrophilic NIR RE complexes functionalized with click chemistry groups (azide/alkyne). (Fig. 58a). These probes exhibited a “turn-on” fluorescence property, showing significant enhancement upon conjugation to biomolecules, and were successfully used for specific NIR fluorescence imaging of nucleic acids, proteins, and glycans within cells (Fig. 58b). Their outstanding contribution lies in being the first to achieve dual-modal NIR fluorescence and time-of-flight secondary ion mass spectrometry (ToF-SIMS) imaging with the same RE molecular probe (Fig. 58c),55 providing a powerful tool for multidimensional cellular research from morphological observation to chemical composition analysis.
 | ||
| Fig. 57 (a) Generalized photophysical mechanism of molecular Er3+ complexes. ISC: intersystem crossing. Analyte-induced tuning of ligand energy levels alters the excitation profile while maintaining emission at 1530 nm. (b) Chemical structures of the symmetric AAAA-type complex EP0 and asymmetric AAAB-type complexes EP1–EP4. (c) Schematic illustration of pH and Cu2+ detection using Er3+ complexes. (d) pH-dependent normalized emission spectra of EP2 (5 µM) under 690 and 705 nm excitation. (e) Cu2+-dependent normalized emission spectra of EP5 (5 µM) under 690 and 700 nm excitation. Reprinted with permission from ref. 349. Copyright 2025, American Chemical Society. | ||
 | ||
| Fig. 58 (a) Synthetic routes to the Yb-1/Nd-1/Er-1 complexes and to the water-soluble, clickable derivatives Yb-2, -3, and -4. (b) Schematic illustration of Yb3+ complexes for labeling metabolically tagged biomolecules via bioorthogonal chemistry. (c) Workflow for correlated near-infrared fluorescence (NIRF) and time-of-flight secondary ion mass spectrometry (ToF-SIMS) imaging of the same cell. Reprinted with permission from ref. 55. Copyright 2022, Wiley-VCH. | ||
High-contrast, multiparameter imaging in the NIR-II window, particularly the 1500–1900 nm band, is the holy grail for deep-tissue in vivo studies. The Er3+–bacteriochlorin complex reported by F. Zhang et al. achieved bright 1530 nm emission under 760 nm excitation via efficient ligand → Er3+ triplet energy transfer (Fig. 59a–e).351 Building on this, the “Lanbow” platform represents a qualitative leap. Through systematic modification of Er3+-phthalocyanine complexes, a family of molecules with tunable absorption peaks but uniform 1530 nm emission were obtained (Fig. 59f–i).352 This strategy of “excitation encoding, single-emission detection”, combined with deep learning, for the first time enabled real-time multispectral imaging up to nine excitations and surgical navigation in deep living tissues, providing a transformative tool for the dynamic analysis of complex biological processes.
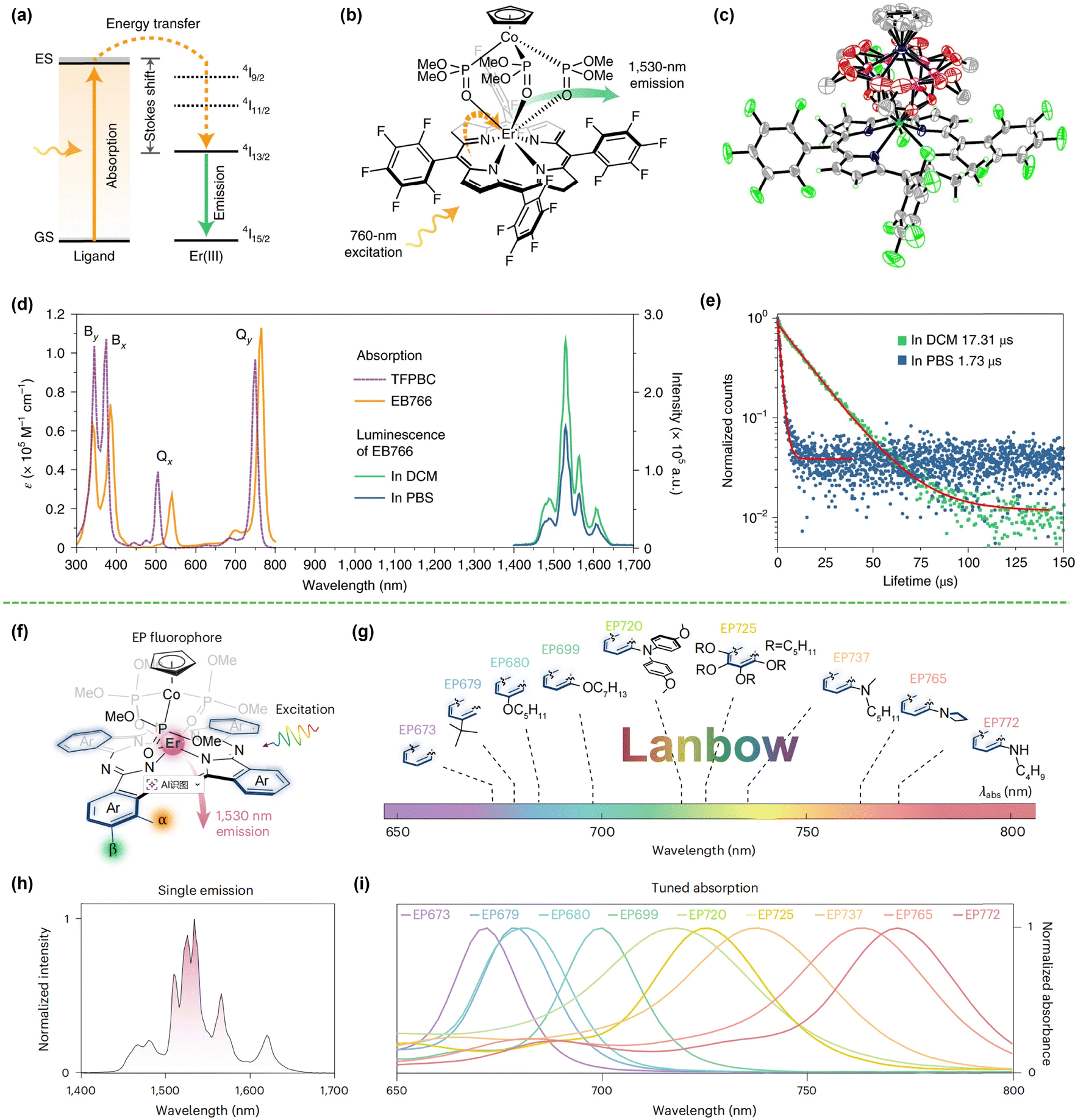 | ||
| Fig. 59 (a) Er3+ complex sensitization: ligand absorption, energy transfer and Er-centred emission. GS, ground state; ES, excited state. (b) and (c) Chemical (b) and X-ray (c) structures of EB766 (30% ellipsoids). (d) Absorption (left) and emission (right, 760 nm ex., 1400–1700 nm) spectra of TFPBC/EB766 in DCM and PBS (pH 7.4). ε, molar extinction coefficient. (e) 1530 nm decay curves: lifetimes 17.3 ± 3.5 µs (DCM) and 1.73 ± 0.06 µs (PBS). Red lines, fits. Reprinted with permission from ref. 351. Copyright 2021, Springer Nature. (f) EP fluorophore structures. (g) α/β substitution patterns on four aromatic rings – the “RE rainbow” (Lanbow). (h) and (i) Emission (h) and absorption (i) profiles of EP fluorophores. Reprinted with permission from ref. 352. Copyright 2025, Springer Nature. | ||
Constructing polynuclear RE supramolecular systems combining high stability and excellent performance is a significant direction. J. -L. Zhang et al. reported a series of Gd3+ complexes for the MRI and phototherapy multimodalities using a single coordination complex, opening an avenue for multimodal imaging.353–357 Q.-F. Sun et al. demonstrated the construction of a series of anionic, water-stable RE organic polyhedra via a ligand deprotonation self-assembly strategy. Among them, the Gd8L12 cube, leveraging its immense molecular weight and rigid structure, exhibited an extraordinarily high longitudinal relaxivity (r1) of 400.53 mM−1s−1 at 0.47 T, far exceeding clinical small-molecule contrast agents, and showed longer retention time at tumor sites, highlighting the tremendous enhancement of magnetic resonance imaging performance by supramolecular structural engineering. Developing single molecular scaffolds compatible with multiple functional modalities (e.g., optical imaging, nuclear imaging, and therapy) is a crucial path towards clinical translation (Fig. 60a).358 E. Boros et al. constructed a paradigmatic trifunctional single-molecule RE theranostic platform. Its core lies in an optimized pyridyl-alkynyl-aryl antenna ligand (Fig. 60b), a design that achieves: (1) high luminescence quantum yield when complexed with Eu3+, activatable via Cerenkov Radiation Energy Transfer (CRET) for intraoperative navigation; (2) excellent pharmacokinetics (rapid renal clearance and high in vivo stability) for its Y3+ complex (86Y-labeled) used in PET diagnosis; (3) efficient chelation of therapeutic radionuclides like 177Lu3+ and 161Tb3+ (Fig. 60c).359 This work seamlessly integrates diagnosis, navigation, and therapy into a single molecular entity, representing a systematic philosophy in theranostic agent design.
 | ||
| Fig. 60 (a) Schematic of RE8L12 assembly-based fluorescence and magnetic resonance (MR) imaging. Reprinted with permission from ref. 358. Copyright 2020, American Chemical Society. (b) Chemical structure of the RE–L ligand. (c) Schematic illustration of the applications of different RE complexes in luminescence imaging, radiotherapy, and PET/CT imaging. Reprinted with permission from ref. 359, CC BY 4.0. | ||
Balancing imaging brightness and therapeutic efficiency is a core challenge in phototheranostic design.360 J.-L. Zhang et al. provided an elegant and powerful solution, metal ion modulation (Fig. 61a).361 By synthesizing a homologous series of carbazole-porphyrin complexes with different RE ions (Gd3+, Yb3+, and Er3+), the researchers demonstrated that the energy gap between the ligand triplet state and the RE3+ excited state directly dictates the energy dissipation pathway: a large gap (Er) favors luminescence imaging, a small gap (Gd) favors singlet oxygen generation for photodynamic therapy, and a moderate gap (Yb) can balance photoacoustic imaging, photothermal, and photodynamic therapy. Utilizing clinical radionuclides as an intrinsic excitation source can completely overcome light penetration depth limitations, enabling deep integration of radiotherapy and phototherapy. E. Boros et al. pioneered the CRET activation of targeted molecular RE probes, successfully applied for luminescence image-guided surgery of tumors in mice (Fig. 61b).362 G. Liu et al. proposed a more efficient novel mechanism for “radio-photodynamic” therapy. They constructed a hybrid system of RE nanoparticles and an aggregation-induced emission photosensitizer. The decay energy of radionuclide 18F is captured by the RE nanoparticles and then directly sensitizes the photosensitizer's triplet state via near-100% efficient triplet energy transfer (TET), greatly circumventing the energy loss associated with intersystem crossing in traditional photodynamic therapy (Fig. 61c),363 significantly improving singlet oxygen yield and tumor suppression efficacy.
 | ||
| Fig. 61 (a) Schematic of the energy dissipation pathways in LnL complexes and the proposed YbL@MSN-based phototheranostic platform for photoacoustic imaging (PAI)-guided synergistic therapy. Reprinted with permission from ref. 361. Copyright 2021, American Chemical Society. (b) Schematic of targeted molecular Eu3+ probes enabling luminescence-guided surgery and postoperative one-photon luminescence microscopy of solid tumors. Reprinted with permission from ref. 360. Copyright 2023, American Chemical Society. (c) Mechanism and workflow of the LnNP–TQ NP hybrid system: direct sensitization of the photosensitizer triplet state via a TET-enhanced pathway harnesses radiative decay energy for efficient ROS generation and radio-photodynamic tumor therapy. Reprinted with permission from ref. 363. Copyright 2025, American Chemical Society. | ||
In summary, RE coordination-based photofunctional materials are triggering a series of paradigm shifts in biomedicine through ingenious molecular design and multi-scale structural engineering. Future development in this field will place greater emphasis on the deep integration and innovation in the following directions: (1) based on a profound understanding of energy transfer mechanisms (e.g., TET, CRET), design next-generation ligands to pursue higher NIR-II luminescence efficiency and more precise logic-gated responsiveness. (2) Develop “smart” RE theranostic agents capable of sensing multiple biological signals and acting according to complex programmed logic, enabling truly personalized adaptive therapy. (3) Systematically evaluate the long-term biological effects of materials, develop green and scalable preparation processes, and promote deep integration between materials science and clinical medicine. (4) Investigate non-traditional mechanisms such as ultrasound excitation and bioluminescence resonance energy transfer, and explore the application of RE molecules in emerging fields like neuroscience and microbiomics. Following this clear path from molecular design to system integration, RE coordination-based photofunctional materials are steadily advancing towards the construction of a more powerful, intelligent, and diverse next-generation biomedical technology system, laying a solid foundation for the future of precision medicine.
4.3. Scintillators
Commercial X-ray scintillators such as CsI:Tl and Bi4Ge3O12 are fabricated by high-temperature melt-growth techniques, offering high light yield and radiation hardness but limited tunability and high production cost. The molecular and framework RE scintillators highlighted here, while not yet commercially deployed, promise solution processability, spectral tunability, and room-temperature synthesis, addressing niche applications such as flexible imaging and multicolor radiography. Scintillators are materials that convert high-energy radiation (e.g., X-rays and γ-rays) into lower-energy photons, holding significant importance in medical diagnostics, industrial inspection, and advanced anti-counterfeiting.367,368 Traditional commercial inorganic scintillators, such as CsI: Tl, Bi4Ge3O12, PbWO4, and YAlO3: Ce, typically rely on expensive bulk crystals grown under stringent conditions via methods like the Czochralski process. Currently, most researched high-performance scintillators are largely confined to ceramics and perovskite materials, which face challenges including complex fabrication, environmental toxicity, self-absorption, and stability issues. RE ions possess relatively high atomic numbers and rich energy level structures, enabling them to function simultaneously as radiation absorption centers and luminescent centers.369 They exhibit excellent scintillation performance in various host materials, such as RE complexes, RE-MOFs,200,212,370,371 RE-clusters,372 and organic–inorganic hybrid RE materials.373 These materials offer several advantages such as non-reabsorbing emission, tunable excited-state lifetimes, spectral tenability, low toxicity, and high thermal stability. Upon X-ray irradiation, RE coordination-based photofunctional materials interact primarily through the photoelectric effect and Compton scattering, generating numerous secondary electrons. These secondary electrons subsequently produce abundant charge carriers, which excite both RE ions and their corresponding organic ligands. For RE complexes, organic ligands with high absorption cross-sections effectively harvest excitation energy, generating singlet and triplet excitons. These excitons then sensitize RE ion luminescence via non-radiative resonance energy transfer. The energy harvesting efficiency largely depends on the molecular triplet energy levels within the RE complex; optimizing these levels can significantly enhance radioluminescence efficiency.A groundbreaking demonstration of this principle was reported in ultrabright molecular scintillators designed via RE-assisted near-unity triplet exciton recycling. By precisely tailoring the triplet energy of organic ligands to match the emitting level of the RE center (e.g., Eu3+), dark triplet excitons generated from secondary X-rays can be captured and transferred to the RE with >99% efficiency (Fig. 62a–d).135 This strategy led to an enhancement in radioluminescence by more than three orders of magnitude, achieving a light yield comparable to commercial CsI:Tl crystals. Moreover, this molecular design enables emission color tuning from ultraviolet to near-infrared and lifetime control from nanoseconds to hundreds of microseconds, facilitating high-resolution radiographic imaging and X-ray-mediated photodynamic therapy. Recent advancements in specific RE complexes further underscore their practical potential. For instance, Eu3+-based hybrid ternary complexes, such as Eu(TTA)3Phen, leverage synergistic interactions between dual organic ligands and the RE ion to achieve exceptional scintillation performance (Fig. 62e and f).374 These materials exhibit a high photoluminescence quantum yield (∼84.5%), an outstanding light yield (∼24900 photons per MeV), and an ultra-low detection limit of 19.97 nGy s−1, which is substantially below the typical dose for medical diagnostics. Furthermore, they demonstrate excellent thermal stability and remarkable robustness under prolonged X-ray irradiation. When integrated into flexible polymer matrices like PMMA, they form highly transparent composite films that enable high-spatial-resolution radiographic imaging (<10 µm), showcasing their versatility for next-generation flexible and sensitive X-ray imaging systems. Molecular RE complexes have demonstrated their potential as scintillators through efficient triplet exciton harvesting and the antenna effect. However, their discrete molecular structures limit energy migration and utilization in three-dimensional space. To overcome this limitation and synergize with the excellent luminescent properties of RE ions, RE-based metal–organic frameworks (MOFs) with long-range ordered crystal structures have attracted widespread attention. MOFs not only inherit the efficient energy transfer pathways found in complexes, but their unique periodic crystal structures provide channels for exciton and charge carrier delocalization.375–378 This paradigm shift is convincingly evidenced by Zheng et al., which demonstrates that in materials like Tb-MOF-76, the crystalline nature facilitates the formation of high-density molecular triplet excitons and offers a delocalized electronic feature, enabling RE emitters to directly trap X-ray-generated charge carriers (Fig. 63a–c).370 This transition from “molecular-unit luminescence” to “bulk-lattice luminescence” positions RE-MOFs as highly promising scintillators for applications requiring high sensitivity and spatial resolution.
 | ||
| Fig. 62 (a) Schematic of triplet exciton-mediated energy transfer from a molecular antenna to RE centres. (b) Triplet exciton recycling in organolanthanide molecules. (c) Relative alignment of ligand triplet energy levels and the emissive 5D0 level of Eu3+. (d) X-ray-excited RL intensity versus UV-excited PL intensity for various organoeuropium molecules. Reprinted with permission from ref. 135. Copyright 2024, Springer Nature. (e) Proposed mechanism of the X-ray-induced scintillation process in the hybrid RE ternary complex. (f) Structural formulas of the Eu3+-based hybrid ternary complexes, Eu(TTA)3Phen, Eu(DBM)3Phen, and Eu(AcAc)3Phen. Reprinted with permission from ref. 374. Copyright 2025, Wiley-VCH. | ||
 | ||
| Fig. 63 (a) Scintillation process in Ln-MOFs. (b) Calculated energies of RE 4f orbitals (red) relative to host bands (blue). Solid and dotted lines denote occupied and empty 4f orbitals, respectively. VBM, valence band maximum; CBM, conduction band minimum. (c) Partial charge densities (green) of the VBM (left) and CBM (right) in Tb-MOF-76 microcrystals. Reprinted with permission from ref. 370, CC BY 4.0. (d) Rational design and corresponding X-ray-excited luminescence (XEL) processes in Ln-Cu4I4 MOFs. (e) Perspective view of the 3D crystal structure of Tb-Cu4I4 MOFs. Reprinted with permission from ref. 379. Copyright 2023, Wiley-VCH. (f) Scintillation mechanism under X-ray irradiation, showing energy transfer via ligand 1 and ligand 2 to Eu3+, along with LLCT/LMCT transitions and 5D0 → 7FJ (J = 0–4) emission. (g) Coordination environments of Eu3+ with ligand 1 (Oba) and ligand 2 (Phen or Bphen). Reprinted with permission from ref. 212. Copyright 2025, Wiley-VCH. (h) Proposed energy-puzzle strategy for fabricating efficient MOF-based scintillators by doping REs with matched energy levels into Pb-MOFs. Reprinted with permission from ref. 380. Copyright 2024, American Chemical Society. | ||
Furthermore, the readily tunable organic and inorganic building blocks of MOFs offer great flexibility for optical property modulation. To further enhance the radioluminescence performance of RE-MOFs, researchers have leveraged this tunability to pursue optimizations primarily from two complementary aspects: metal node/cluster engineering and sophisticated ligand sensitization. First, regarding the enhancement of X-ray absorption and the construction of efficient energy transfer channels: the core X-ray absorption process in RE MOFs can still be limited by the relatively low effective atomic number (Z) of the organic components. Therefore, strengthening X-ray absorption and ensuring efficient energy transfer from the absorber to the luminescent chromophore are crucial. Q. Zhao et al. exemplified an innovative “cluster-based antenna” sensitization strategy by constructing Ln–Cu4I4 heterometallic MOFs. In this design, the heavy-atom [Cu4I4] cluster serves as an efficient X-ray absorber, generating triplet excitons via halide-to-ligand charge transfer that matches the energy level of Tb3+ (3X/MLCT state), thereby sensitizing the RE ion. This approach achieved a high light yield of ∼29400 photons MeV−1, demonstrating the feasibility of synergistically optimizing absorption and energy transfer by incorporating heavy atom cluster units (Fig. 63d and e).379
Second, concerning advanced ligand engineering and energy transfer networks: to maximize energy transfer efficiency and suppress non-radiative losses, more intricate ligand designs have been employed. M.-J. Lin et al. presented a highly effective “dual-ligand antenna” strategy. By synergistically coordinating Eu3+ with two types of ligands, 4,4′-oxybis (benzoic acid) and a phenanthroline derivative (Bphen), to form MOFs like Eu–O–Bphen, a hierarchical energy transfer network (Ligand 1 → Ligand 2 → Eu3+) was constructed. This design significantly minimizes energy loss. Concurrently, the steric hindrance introduced by Bphen suppresses detrimental π–π stacking among ligands, elevating the photoluminescence quantum yield to 86.3% and resulting in a record-high light yield of ∼60300 photons MeV−1. This highlights the breakthrough performance achievable by optimizing ligand synergy and intermolecular interactions (Fig. 63f and g).212 Third, the modular architecture of RE-MOFs offers a broadly applicable platform for performance tuning, which enhances the versatility of materials and facilitates novel emission properties. S. Wang et al. proposed a universal “energy-puzzle” strategy. By doping minute amounts of RE ions (e.g., Tb3+ and Eu3+), whose energy levels “match” the triplet state of the organic linker, into non- or weakly-emissive lead-based MOFs (e.g., SCU-200), the inert MOFs were successfully converted into efficient scintillators, improving the detection limit by over two orders of magnitude (Fig. 63h).380 More importantly, by selecting NIR-emitting REs (e.g., Yb3+) matched with low-energy linkers, X-ray-induced NIR luminescence was achieved within the MOF framework for the first time, significantly expanding the material scope and functional boundaries of MOF scintillators.
Beyond the infinite frameworks of MOFs, researchers are also exploring finite, atomically precise structures. S.-Q. Zang et al. reported a groundbreaking fluoride-bridged terbium cluster, Tb16, which represents a novel class of metal cluster scintillators (Fig. 64a–f).372 This cluster features a precise 16-nuclear Tb core stabilized by organic ligands and interconnected by µ3- and µ4-fluoride bridges. The design ingeniously combines triplet exciton recycling (enabled by the heavy Tb atoms) with fluoride-bridge-induced carrier traps. This dual mechanism endows Tb16 with a high light yield of ∼41400 photons MeV−1 and, most remarkably, temperature-inert radioluminescence with intensity variation less than 8.63% over a wide range of 300 to 540 K. The deep traps (1.45 eV) capture charge carriers at low temperatures and thermally release them at high temperatures, compensating for thermal quenching loss.
 | ||
| Fig. 64 (a) Crystal structure and coordination mode of Tb16 and inter-cluster distances. (b) X-ray excited luminescence (XEL) intensity spectra of Tb16 and BGO wafer with the same thickness and sectional area. (c) XEL intensity spectra of Tb16 at different X-ray dose rates ranging from 0.688 to 278 µGy s−1. (d) X-ray response sensitivities of Tb16 over the range of 0.688–278µGy s−1. (e) Variation of XEL intensity with temperature for Tb16. (f) Temperature-dependent integrated curves of luminescence intensity for Tb16. Reprinted with permission from ref. 372, CC BY 4.0. | ||
Parallel to the pursuit of structural order and precision, the exploration of amorphous RE materials opens another promising avenue, emphasizing processability and new functionalities.371,381 D.-B. Kuang et al. introduced the first organic–inorganic hybrid RE halide glasses, Bzmim3LnCl6 (Ln = Tb, Eu), fabricated via a low-temperature melt-quenching method. Unlike crystalline MOFs or clusters, these materials possess a disordered glassy network, which offers distinct advantages: high optical transparency (>85% in visible light), ease of forming large-area flexible films, and low-temperature, energy-efficient synthesis.382 By simply adjusting the Tb3+/Eu3+ ratio, the radioluminescence color can be tuned from green to red, enabling multicolor visualization of X-ray dose distribution. Despite their amorphous nature, these glasses achieve a high spatial resolution exceeding 25 lp mm−1 in X-ray imaging, rivaling their crystalline counterparts. This work significantly broadens the material form factor of RE scintillators, highlighting the potential of hybrid glasses for low-cost, scalable, and color-tunable radiation detection applications.
In summary, the landscape of RE-based scintillators is expanding rapidly beyond traditional crystalline systems. From the long-range order of MOFs to the atomic precision of metal clusters like Tb16, and further to the functional disorder of hybrid glasses like Bzmim3LnCl6, each material form provides unique levers for property modulation, whether it be exceptional thermal stability, ultra-high resolution, or facile color tunability and processability. These advances collectively underscore that the strategic design of RE materials, whether by engineering crystalline frameworks, precise molecular clusters, or tunable amorphous networks, is key to developing next-generation scintillators tailored for challenging environments, advanced imaging modalities, and cost-sensitive applications.
4.4. Optical wireless communications
Optical wireless communication (OWC) technology has emerged as a key candidate to address the surging demand for wireless data traffic, owing to its advantages such as high speed, enhanced security, and freedom from spectrum licensing. In up-conversion materials, particularly their fluorescence lifetime and modulation bandwidth directly determine the achievable data rate. Conventional materials, such as phosphor ceramics and perovskites, often face challenges including demanding synthesis conditions or the presence of toxic elements. RE-MOFs have attracted considerable attention due to their tunable structures and excellent luminescence properties.191,383 However, their typically long fluorescence lifetimes in the millisecond range severely limit their application in high-speed OWC.384Recently, a breakthrough study successfully transformed RE-MOFs into high-performance color converters through a sophisticated energy-transfer strategy. In this work, the Tb-BTC MOF served as the energy donor, while encapsulated deep-red-emitting organic chromophores (A1/A2) acted as energy acceptors. Leveraging the confinement effect of the MOF pores and well-matched energy levels between donors and acceptors, an energy transfer efficiency as high as 99% was achieved. This dramatically shortened the fluorescence lifetime of the system from 1.3 ms for the pristine MOF to 4.6 ns for the composite material. This fundamental change increased its −3 dB modulation bandwidth from below 0.1 MHz to 65.7 MHz. In a practical OWC link test based on DC-biased optical orthogonal frequency-division multiplexing (DCO-OFDM) modulation, the composite achieved a net data rate of 1.076 Gbit s−1 (Fig. 65).385 This demonstration marks the first instance of a RE MOF-based color converter surpassing Gbit s−1 data transmission, outperforming the majority of reported organic and inorganic luminescent materials and establishing a new paradigm for high-speed, MOF-enabled OWC systems. While still at the laboratory stage, such systems illustrate how molecular-level energy transfer engineering can overcome intrinsic limitations of RE materials, opening a pathway toward high-bandwidth devices that complement commercial EDFAs and inorganic phosphor-based visible light communication systems.
 | ||
| Fig. 65 Illustration of the benefits of combining RE-based MOFs with an efficient energy transfer strategy for the development of high-performance optical wireless communication applications. Reprinted with permission from ref. 385, CC BY 4.0. | ||
4.5. Emerging application scenarios
Beyond these applications, RE coordination-based photo-functional materials demonstrate significant utility across diverse fields including LEDs, photocatalysis, solar cells, etc. Of significant current interest are emerging multifunctional materials that integrate luminescence with other properties. Rare earth-based systems are now extensively explored for applications such as luminescent thermometers utilizing molecular nanomagnets,386 luminescent ferroelectric materials,387 and lumino-magnets (photo-magnets), which demonstrate the vast potential of these materials beyond pure emission.388 In the field of LEDs, RE coordination materials continue to push technological boundaries through innovative design.389 G. Qian et al. achieved single-component white-light emission (CIE coordinates (0.33,0.34)) via controllable self-assembly of DPA ligands with lanthanide ions,390 while C.-X. Shan et al. constructed a high-stability UV-LED luminescent layer (color purity >95%) through covalent grafting of PMA carriers onto diamond surfaces.391 Notably, W. Liu et al. pioneered a dynamic coordination hydrogel platform enabling photo-mechano-electrical trifunctional synergy.392 These strategies establish new paradigms for lanthanide luminescent materials from molecular energy-level modulation, interfacial covalent anchoring, and dynamic network cooperation perspectives.Transitioning to photocatalysis, RE elements' unique 4f electronic configurations prove crucial in overcoming energy efficiency limitations: their strong reducing active species (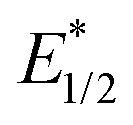 reaching −3.45 V) activate inert chemical bonds, while precise ligand-field engineering significantly broadens spectral response.393 Research progression reveals a clear hierarchical evolution—from molecular-scale Ce(III) guanidinate complexes developed by E. J. Schelter et al. enabling efficient electron transfer,394 to Nd-MOF/TiO2 core–shell structures constructed by A. Zaleska-Medynska enhancing degradation efficiency (87.5% in 60 min) via upconversion,395 culminating in the Ce-MOF-Ru(II)-bpy system developed by T. K. Maji's group achieving highly selective CO2 reduction (99.5% selectivity) through pore confinement and electron transfer synergy.396 Particularly noteworthy are the high-entropy alloy photocatalysts (H2 evolution rate: 13.4 mmol·g−1·h−1) fabricated via a “weaving strategy” by Y. Tang et al.,397 signifying the field's advancement into multi-metallic atomic-level cooperation.
reaching −3.45 V) activate inert chemical bonds, while precise ligand-field engineering significantly broadens spectral response.393 Research progression reveals a clear hierarchical evolution—from molecular-scale Ce(III) guanidinate complexes developed by E. J. Schelter et al. enabling efficient electron transfer,394 to Nd-MOF/TiO2 core–shell structures constructed by A. Zaleska-Medynska enhancing degradation efficiency (87.5% in 60 min) via upconversion,395 culminating in the Ce-MOF-Ru(II)-bpy system developed by T. K. Maji's group achieving highly selective CO2 reduction (99.5% selectivity) through pore confinement and electron transfer synergy.396 Particularly noteworthy are the high-entropy alloy photocatalysts (H2 evolution rate: 13.4 mmol·g−1·h−1) fabricated via a “weaving strategy” by Y. Tang et al.,397 signifying the field's advancement into multi-metallic atomic-level cooperation.
In solar cells, the technological evolution exhibits equally distinct trajectories: an early work conducted by J.-Y. Liu et al. enhanced efficiency by 72% using Tb/Dy complex co-sensitization.398 Building on this, Y. Tang et al. innovatively introduced europium–porphyrin complexes to construct 2D/3D heterostructures, not only achieving a breakthrough efficiency of 18.2% but also demonstrating 3000-hour environmental stability through thermodynamic self-healing mechanisms.399 Subsequently, Zhang et al. developed polynuclear rare-earth cluster-based materials as interfacial additives, further pushing the efficiency to 21.31%.400 These achievements systematically demonstrate RE coordination-based photofunctional materials' diversified application pathways—from sensitizers and grain-boundary passivators to interfacial functional agents—providing sustained momentum for clean energy technology advancement.
5. Machine learning-aided design
The discovery and optimization of advanced luminescent materials increasingly demand time- and cost-efficient research paradigms capable of navigating vast compositional and structural design spaces. While molecular simulations may be feasible for each material and application, analyzing high-capacity, high-dimensional complex “big data” remains arduous without the application of data science technologies. In recent years, data-driven modeling has emerged as a powerful tool in materials science,401 enabling not only advanced analysis but also the design and synthesis of functional metamaterials.402,403Machine learning has become increasingly important for discovering and optimizing RE luminescent materials, particularly in designing systems with enhanced luminous efficiency. One notable application is in the optimization of upconversion phosphors. R. Lv et al. employed machine learning approaches, such as support vector machines (SVM), to optimize the enhancement of upconversion luminescence in multimetallic-sensitized phosphors.404 The study applied these methods to assess the luminescent stability of RE phosphors and to evaluate the significance of various doping elements. Results demonstrated that RE complexes synthesized via hydrothermal methods exhibit robust stability against environmental influences, with Yb and Er identified as the most influential dopants for luminescence enhancement.
Beyond phosphor optimization, machine learning also aids in the development of advanced optical materials and imaging systems. L. Sun et al. developed a novel dumbbell-shaped RE-doped nanocrystal capable of modulating energy migration pathways through controlled energy transfer processes and Ce3+-mediated energy transfer.405 This design enables excitation wavelength-dependent dual-emission modes spanning both the visible and NIR-II spectral regions. Significantly, by integrating these multimodal nanocrystals with deep learning algorithms, the team combined visible fluorescence with deep-penetrating NIR-II emission to develop a novel fluorescence imaging technique. This approach successfully converts blurred visible images from tissue imaging into high-resolution outputs with narrow emission bands and high signal-to-noise ratios, establishing a new framework for intelligent optical imaging.
In parallel, machine learning has accelerated the design of micro- and nano-optical devices. Y. Liu et al. proposed a novel framework that leverages machine learning models to facilitate the design of micro- and nano-optical devices. By employing machine learning algorithms to extract the statistical relationships between structural parameters and optical responses from training datasets, and integrating them with iterative optimization methods, the approach enables large-scale, systematic optimization and design of metasurface-based micro- and nano-optical devices. Fig. 66406 illustrates (i) defined design objectives; (ii) a generative model that produces (iii) a candidate metasurface pool; and (d) a predictive model that evaluates optical performance. This closed-loop system integrates forward prediction and inverse design models within an iterative optimizer, allowing autonomous device design once objectives are specified.
 | ||
| Fig. 66 (a) Based on the specified design goals, (b) a retrieval model generates (c) a pool of candidate metasurface structures that satisfy the target phase requirements. (d) A prediction model then evaluates the optical response of each candidate to assess device performance. Reprinted with permission from ref. 406. Copyright 2022, Wiley-VCH. | ||
Machine learning further addresses limitations in traditional luminescence theory and high-throughput discovery. According to classical theory, the emission of Eu2+ 5d → 4f transitions depends heavily on the host matrix structure, yet trial-and-error search is inefficient. R.-J. et al. overcame this by establishing a quantitative model correlating Eu2+ emission with six host matrix parameters, covering composition, local coordination, and electronic structure—achieving a prediction error below 7 nm. As shown in Fig. 67,407 this model guided high-throughput screening and identified five promising near-infrared hosts, leading to the discovery of Eu2+-doped materials with record-long emission wavelengths. Overall, machine learning effectively overcomes bottlenecks in traditional luminescence prediction, such as high computational cost and long development cycles. By coupling predictive emission models with high-throughput screening and inverse design, ML not only extends traditional luminescence theory but also enables targeted, rational discovery of RE-based photonic materials. These methodologies are poised to dramatically accelerate material innovation, reduce experimental trial-and-error, and optimize synthesis pathways, ultimately reshaping how next-generation luminescent materials and devices are conceived and realized.
 | ||
| Fig. 67 Discovery of NIR-emitting Eu2+ phosphors via high-throughput screening. (a) Root-mean-square error between emission peaks predicted by the HCEP model and experimental values. (b) Workflow of the HCEP-assisted high-throughput screening process. (c) Crystal structures and space groups of the five selected NIR Eu2+ phosphor candidates. Reprinted with permission from ref. 407 Copyright 2022, Elsevier. | ||
6. Summary, challenges, and future directions
This review systematically delineates the landscape of RE coordination-based photofunctional materials, spanning from molecular engineering strategies to emerging applications. The field is currently undergoing a profound paradigm shift that the focus is moving away from the traditional pursuit of isolated photophysical parameters, such as emission color and efficiency, but toward exploring how to bridge the gaps between energy forms and material states to achieve precise, efficient, and intelligent conversion of energy and information. Consequently, the functional role of these materials is evolving from passive “emitters” to active “intelligent energy-information hubs”. However, this ambitious transition faces three intertwined fundamental scientific challenges: the insulating barriers in energy channels, the biocompatibility gap in material cycling, and the dimensional limits of information processing. Overcoming these challenges requires not only addressing specific technical bottlenecks but also propelling material systems toward new frontiers characterized by functional decoupling and synergy, integration of biological logic, and information intelligence (Fig. 68). | ||
| Fig. 68 Strategic roadmap for next-generation bio-integrated materials: from fundamental challenges to multidisciplinary solutions. | ||
First, the insulating barrier in energy channels represents a fundamental physical limit restricting both efficiency and practical applications. This manifests as a bottleneck in photophysical mechanisms: the competition and synergy among multiple energy transfer pathways, such as Dexter electron exchange, Förster resonance energy transfer (FRET), and charge-transfer states, remain insufficiently understood, impeding progress toward theoretical limits in energy transfer efficiency. Moreover, excited states of RE ions, especially near-infrared emitters like Er3+ and Yb3+, are highly susceptible to quenching by high-energy molecular vibrations (e.g., C–H and O–H) and surface defects, making it challenging to fully suppress nonradiative decay channels. This barrier is particularly acute when integrating RE materials into devices, as their intrinsic electrical insulation prevents direct electrical excitation.
AI-driven molecular simulations and high-throughput virtual screening can systematically explore the vast design space of ligands and interface structures, predicting optimal energy-level alignment and energy transfer pathways to guide the design of efficient “energy conversion cloaks” and “optoelectronic bridges”. Recent breakthroughs in “organic–inorganic hybrid” interface engineering exemplify a path forward: by designing molecularly precise “energy conversion cloaks” on insulating RE nanocrystals, a level-matched “optoelectronic bridge” can be established at the interface, enabling exciton energy (rather than charge current) to be efficiently and non-destructively transferred from the organic layer to the RE luminescent centers, resulting in dramatic efficiency enhancements. This insight highlights that future breakthrough hinges on “functional decoupling and synergy”, delegating energy harvesting, transfer, and emission to the most competent units within heterogeneous systems and achieving ultrahigh-efficiency coupling through atomic- and molecular-scale interface engineering. Combined with AI-powered inverse design, optimal heterogeneous structural schemes can be generated automatically for target optoelectronic performance, accelerating the discovery of novel, high-efficiency materials. Nonetheless, realizing this vision on a broad scale entails overcoming engineering challenges related to scalable synthesis and device integration, including the controllability and cost of complex architectures, macroscopic processability, and precise energy-level alignment and charge management at heterogeneous interfaces.
Second, the biocompatibility gap in material cycling necessitates a fundamental reconsideration of material synthesis, application, and lifecycle from a chemical stability perspective. Most high-performance RE complexes suffer from inadequate stability, prone to ligand dissociation and photodegradation under aqueous, oxidative, or photonic conditions. Introducing dynamic elements for stimuli-responsiveness often compromises long-term stability, especially in biomedical contexts. Insights from RE biogeochemical cycling reveal that organisms regulate directional crystallization of RE ions into biogenic minerals, inspiring “environmentally intelligent” formation routes. Synthetic biology tools could engineer enzymes or artificial cells to synthesize, assemble, and repair RE materials under physiological conditions. Biomimetic synthesis approaches, such as predicting artificial molecules or peptide sequences with enhanced stability, provide blueprints for biocompatible materials. These not only support “living” photonic devices for in vivo applications but also promote sustainable goals, including mild extraction and recycling of RE elements and exploration of light RE or non-RE substitutes. Computational methods can optimize synthesis pathways and assess degradation behavior, fostering green design across the lifecycle. Furthermore, the development of multifunctional integrated devices based on RE coordination-based photofunctional materials holds significant promise as an important future direction. Four integration strategies show great potential: (1) sensing-display integration, where RE complexes serve dual roles as responsive sensors and emissive layers for direct visualized detection; (2) energy conversion-lighting integration, which combines upconversion/downconversion luminescence with photovoltaic effects to enable self-powered illumination; (3) information storage-encryption-transmission integration, capitalizing on multiple emission channels and long-lived excited states for high-security optical processing and (4) the integration of luminescence with magnetism, enabling applications such as luminescent thermometers, ferroelectric devices, and photo-magnets that exploit coupled optical-magnetic properties. In the realm of flexible electronics, RE coordination polymers offer the unique capability to facilitate mechanical sensing, optical signaling, and energy conversion, thereby advancing the field of wearable smart devices. Additionally, chiral RE complexes enable the integration of circularly polarized emission with magnetic/electric responsiveness, opening new avenues for quantum information processing and advanced optoelectronic applications.
Finally, the dimensional limit of information processing challenges the upgrade of these materials from static “optical barcodes” to dynamic “intelligent processors”. Although multiplexed fluorescence encoding based on unique RE energy levels has been exploited for bioimaging, the information dimensions (color, intensity, lifetime, etc.) remain underutilized, and logical decoding of complex signals is lacking. Cutting-edge visions aim to build “RE molecular photoelectrochemical logic gates” by coupling distinct molecular recognition units with RE luminescent centers. Utilizing mechanisms based on energy transfer, electron transfer, or switching coordination fields, these systems perform Boolean logic operations optically, directly reporting complex states such as “AND” or “OR”. Realizing this demands deepening fundamental mechanistic understanding, resolving full excitation dynamics under multiple stimuli with ultrafast spectroscopy, and leveraging artificial intelligence (especially machine learning) to analyze complex multi-dimensional luminescence response data, establishing the mapping models between signals and logical states and thereby enabling the inverse design of molecular systems that achieve desired logic functions. Furthermore, the exceptionally narrow linewidths and long spin coherence times of RE ions render them ideal quantum interfaces between photons and qubits, laying a foundation for disruptive information technologies, including quantum sensing and quantum memory. AI algorithms show great potential in optimizing the control of quantum systems, processing quantum information, and correlating material structure with quantum properties.
In summary, the future of RE coordination-based photofunctional materials unfolds along the main axis of “overcoming insulating barriers → integrating biological logic → embedding information intelligence”. Their evolutionary logic elevates from “making brighter materials” to “designing smarter energy and information conversion paradigms”. Artificial intelligence, as a powerful auxiliary and enabling technology, will permeate the entire chain from material design and performance optimization to constructing intelligent responses and system integration, accelerating the realization of this paradigm. Addressing the specific challenges outlined above and seizing the frontier opportunities urgently require deep interdisciplinary integration: chemists must devise sophisticated interfaces, molecular recognition, and logic units; physicists must elucidate ultrafast energy/charge transfer and quantum coherence mechanisms; biologists must decipher and exploit biological mineralization codes; computational and data scientists need to develop and apply advanced AI models and algorithms, collaborating with experimental scientists to navigate the vast materials design space and build intelligent bridges from molecular blueprints to functional devices. Only through such profound collaboration can the full potential of REs, as the “soul of light” and “carriers of information”, be unlocked, enabling them to assume indispensable roles in next-generation displays, bio-integrated photonics, quantum information science, adaptive camouflage, and beyond.
Author contributions
F.-J. Song, X. Li, P. Su and Y. Tang conceived the contents and prepared the manuscript. Y. Tang, J.-L. Zhang and C.-H Yan contributed to the supervision and polish of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Data availability
This article is a review and does not report any new primary data. All data discussed and analysed herein are derived from previously published research articles, which are cited throughout the text. Readers are directed to the cited references for access to the underlying data.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Projects 22221001, 22131007, 22475091, 22577006, 22131003, and T2550067), the 111 Project (B20027), the Science and Technology Program of Gansu Province (24ZD13GA015, 23ZDGA012, and 26JDRA008), the Fundamental Research Funds for the Central Universities (lzujbky-2025-jdzx13) and the fellowship of China Postdoctoral Science Foundation (2025M780946).Notes and references
- J. Thompson, R. I. R. Blyth, G. Gigli and R. Cingolani, Adv. Funct. Mater., 2004, 14, 979–984 CrossRef CAS.
- S. I. Weissman, J. Chem. Phys., 1942, 10, 214–217 CrossRef CAS.
- G. A. Crosby, R. E. Whan and J. J. Freeman, J. Phys. Chem., 1962, 66, 2493–2499 CrossRef CAS.
- T. Cheisson and E. J. Schelter, Science, 2019, 363, 489–493 CrossRef CAS PubMed.
- Virender, A. Chauhan, A. Kumar, G. Singh, A. A. Solovev, J. Xiong, X. Liu and B. Mohan, J. Rare Earths, 2024, 42, 16–27 CrossRef CAS.
- P. Su, F. Song, J. Cao, C.-H. Yan and Y. Tang, Acc. Chem. Res., 2025, 58, 218–230 CrossRef CAS PubMed.
- Z. Zeng, Y. Xu, Z. Zhang, Z. Gao, M. Luo, Z. Yin, C. Zhang, J. Xu, B. Huang, F. Luo, Y. Du and C. Yan, Chem. Soc. Rev., 2020, 49, 1109–1143 RSC.
- C.-H. Yan and X. Huang, J. Rare Earths, 2025, 43, 2029–2052 CrossRef.
- H. Jia, N. Li, H. He, X. Zhang, Y. Teng and W. Qin, Adv. Opt. Mater., 2024, 12, 2302583 CrossRef CAS.
- J.-C. G. Bünzli, Trends Chem., 2019, 1, 751–762 CrossRef.
- Y. Yang, P. Wang, L. Lu, Y. Fan, C. Sun, L. Fan, C. Xu, A. M. El-Toni, M. Alhoshan and F. Zhang, Anal. Chem., 2018, 90, 7946–7952 CrossRef CAS PubMed.
- J. He, C. S. Bonnet, S. V. Eliseeva, S. Lacerda, T. Chauvin, P. Retailleau, F. Szeremeta, B. Badet, S. Petoud, É. Tóth and P. Durand, J. Am. Chem. Soc., 2016, 138, 2913–2916 CrossRef CAS PubMed.
- S. Sajid, S. Alzahmi, N. Tabet, M. Y. Al-Haik, M. Abdel-Hafiez, Y. Haik and I. M. Obaidat, J. Mater. Chem. C, 2024, 12, 18575–18590 RSC.
- C. Chen, S. Zheng and H. Song, Chem. Soc. Rev., 2021, 50, 7250–7329 RSC.
- L. Hu, Y. Yang, Y. Gao, Y. Wei, J. Zhu and W. Wu, Chem. Eng. J., 2024, 488, 150965 CrossRef CAS.
- Y. Xu, X. Zhan, J. Du, Z. Wu and D. Zhang, Chem. Eng. J., 2024, 489, 151303 CrossRef CAS.
- L. Wang, S. Chen, Y. Jiang, Y. Gong, W. Yang, Y. Lin, Y. Liu, M. Xiao and Z. Su, Chin. Chem. Lett., 2025, 112149 CrossRef.
- K. Pei, J. Wu, M. Zhao, X. Feng, Y. Li, Y. Ma, H. Li and T. Zhai, Adv. Opt. Mater., 2022, 10, 2102143 CrossRef CAS.
- Y. Yang, X. Hu, Z. Yang and W. Huang, Adv. Funct. Mater., 2025, 35, 2412970 CrossRef CAS.
- S. Li, L. Zhou and H. Zhang, Light: Sci. Appl., 2022, 11, 177 CrossRef CAS PubMed.
- G.-Q. Jin, C. V. Chau, J. F. Arambula, S. Gao, J. L. Sessler and J.-L. Zhang, Chem. Soc. Rev., 2022, 51, 6177–6209 RSC.
- D. E. Barry, D. F. Caffrey and T. Gunnlaugsson, Chem. Soc. Rev., 2016, 45, 3244–3274 RSC.
- F. Saraci, V. Quezada-Novoa, P. R. Donnarumma and A. J. Howarth, Chem. Soc. Rev., 2020, 49, 7949–7977 RSC.
- S. A. Younis, N. Bhardwaj, S. K. Bhardwaj, K.-H. Kim and A. Deep, Coord. Chem. Rev., 2021, 429, 213620 CrossRef CAS.
- R. Xue, Y.-S. Liu, W. Yang and G.-Y. Yang, Coord. Chem. Rev., 2024, 501, 215577 CrossRef CAS.
- J.-Q. Liu, Z.-D. Luo, Y. Pan, A. Kumar Singh, M. Trivedi and A. Kumar, Coord. Chem. Rev., 2020, 406, 213145 CrossRef CAS.
- Y. Zhang, P. Wei, Z. Li, Y. Sun, Y. Liu and S. Huang, Coord. Chem. Rev., 2024, 514, 215905 CrossRef CAS.
- M. Zhang, X. Zhai, M. Sun, T. Ma, Y. Huang, B. Huang, Y. Du and C. Yan, Chem. Soc. Rev., 2020, 49, 9220–9248 RSC.
- Y. Sun, M. Kong, J. Ke, Y. Gu, F. Li and W. Feng, Coord. Chem. Rev., 2025, 523, 216222 CrossRef CAS.
- B. Zheng, J. Fan, B. Chen, X. Qin, J. Wang, F. Wang, R. Deng and X. Liu, Chem. Rev., 2022, 122, 5519–5603 CrossRef CAS PubMed.
- Z. Wei, Y. Liu, B. Li, J. Li, S. Lu, X. Xing, K. Liu, F. Wang and H. Zhang, Light: Sci. Appl., 2022, 11, 175 CrossRef CAS PubMed.
- J. M. Robinson, Phys. Rep., 1979, 51, 1–62 CrossRef CAS.
- R. Janicki, A. Mondry and P. Starynowicz, Coord. Chem. Rev., 2017, 340, 98–133 CrossRef CAS.
- A. G. Bispo-Jr, N. A. Oliveira, I. M. S. Diogenis and F. A. Sigoli, Coord. Chem. Rev., 2025, 523, 216279 CrossRef CAS.
- D. Zhao, Q. Li and H. Li, Acc. Mater. Res., 2025, 6, 1133–1146 CrossRef CAS.
- J. Sun, H. Fu, H. Jing, X. Hu, D. Chen, F. Li, Y. Liu, X. Qin and W. Huang, Adv. Mater., 2025, 37, 2417397 CrossRef CAS PubMed.
- J.-C. G. Bünzli, S. Comby, A.-S. Chauvin and C. D. B. Vandevyver, J. Rare Earths, 2007, 25, 257–274 CrossRef.
- K. Binnemans, Chem. Rev., 2009, 109, 4283–4374 CrossRef CAS PubMed.
- A. K. R. Junker, L. R. Hill, A. L. Thompson, S. Faulkner and T. J. Sørensen, Dalton Trans., 2018, 47, 4794–4803 RSC.
- M. Latva, H. Takalo, V.-M. Mukkala, C. Matachescu, J. C. Rodríguez-Ubis and J. Kankare, J. Lumin., 1997, 75, 149–169 CrossRef CAS.
- E. G. Moore, A. P. S. Samuel and K. N. Raymond, Acc. Chem. Res., 2009, 42, 542–552 CrossRef CAS PubMed.
- C.-L. Liu, R.-L. Zhang, C.-S. Lin, L.-P. Zhou, L.-X. Cai, J.-T. Kong, S.-Q. Yang, K.-L. Han and Q.-F. Sun, J. Am. Chem. Soc., 2017, 139, 12474–12479 CrossRef CAS PubMed.
- L. Y. Mironov, E. B. Sveshnikova and V. L. Ermolaev, Opt. Spectrosc., 2014, 116, 933–940 CrossRef CAS.
- C. Alexander, Z. Guo, P. B. Glover, S. Faulkner and Z. Pikramenou, Chem. Rev., 2025, 125, 2269–2370 CrossRef CAS PubMed.
- A. G. Martynov, Y. Horii, K. Katoh, Y. Bian, J. Jiang, M. Yamashita and Y. G. Gorbunova, Chem. Soc. Rev., 2022, 51, 9262–9339 RSC.
- X.-Z. Li, C.-B. Tian and Q.-F. Sun, Chem. Rev., 2022, 122, 6374–6458 CrossRef CAS PubMed.
- Y. Li, F. Guo, S. Wei, D. Li, J. Zhang, K. Xu and D. Li, J. Rare Earths, 2024, 42, 278–285 CrossRef CAS.
- X. Zhang, X. Jin and Y. Li, Chin. Chem. Lett., 2022, 33, 2117–2120 CrossRef CAS.
- A. Nagar, A. Sengupta, M. A. Sk and P. K. Mohapatra, Inorg. Chem., 2023, 62, 19631–19647 CrossRef CAS PubMed.
- M. Mirzakhani, S. Naseri, C. Egger, A. Rosspeintner, H. Nozary and C. Piguet, Small, 2023, 19, 2303721 CrossRef CAS PubMed.
- T.-A. Phan, F. Gendron, S. Kiraev, O. Galangau, Y. Fréroux, H. Al Sabea, C. Mittelheisser, M. Dallon, A. Bouchet, F. Riobé, R. Métivier, M. Sliwa, B. Le Guennic, O. Maury, A. Banyasz, B. Marekha, L. Norel and S. Rigaut, Chem. Sci., 2025, 16, 22504–22516 RSC.
- W. Lv, J. Han, F. Wu, S. Wang, M. Lin, Q. Ling and Z. Lin, Chem. Eng. J., 2026, 527, 171577 CrossRef CAS.
- J. Shi, Y. Li, S. Yin, Z. Song, Y. Zhou, T. Gao, P. Yan and H. Li, J. Rare Earths, 2026, 44, 15–24 CrossRef CAS.
- J.-Y. Hu, Y. Ning, Y.-S. Meng, J. Zhang, Z.-Y. Wu, S. Gao and J.-L. Zhang, Chem. Sci., 2017, 8, 2702–2709 RSC.
- G.-Q. Jin, D.-e Sun, X. Xia, Z.-F. Jiang, B. Cheng, Y. Ning, F. Wang, Y. Zhao, X. Chen and J.-L. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202208707 CrossRef CAS PubMed.
- N. Sapkota, E. Mäkilä, A. Lehtonen and A. Peuronen, Cryst. Growth Des., 2025, 25, 3119–3127 CrossRef CAS.
- D. Jin, C. Uhlmann, E. K. Schneider, T. Seifert, D. Graf, S. Lebedkin, P. Weis, M. M. Kappes and P. W. Roesky, Inorg. Chem., 2024, 63, 24762–24770 CrossRef CAS PubMed.
- L. C. Adi, A. Santria and N. Ishikawa, Dalton Trans., 2024, 53, 628–639 RSC.
- T. Liu, X. Zhang, H. Zhang, H. Zhao, Z. Zhang and Y. Tian, J. Rare Earths, 2021, 39, 1187–1193 CrossRef CAS.
- L. Godec, R. C. Knighton, N. Hamon, W. Thor, K.-L. Wong, R. Tripier and L. J. Charbonnière, J. Am. Chem. Soc., 2025, 147, 31187–31197 CrossRef CAS PubMed.
- N. Hamon, L. Godec, E. Jourdain, F. Lucio-Martínez, C. Platas-Iglesias, M. Beyler, L. J. Charbonnière and R. Tripier, Inorg. Chem., 2023, 62, 18940–18954 CrossRef CAS PubMed.
- L. Collobert, L. Bridou, Z. Garda, O. Maury, É. Tóth, C. Platas-Iglesias, R. Tripier and M. Beyler, Inorg. Chem., 2025, 64, 9330–9341 CrossRef CAS PubMed.
- N. Hamon, A. Roux, M. Beyler, J.-C. Mulatier, C. Andraud, C. Nguyen, M. Maynadier, N. Bettache, A. Duperray, A. Grichine, S. Brasselet, M. Gary-Bobo, O. Maury and R. Tripier, J. Am. Chem. Soc., 2020, 142, 10184–10197 CrossRef CAS PubMed.
- C. Alexander, H. Li, P. Harvey, G. E. Pavlovskaya, N. J. Rogers and D. Parker, Chem. Biomed. Imaging, 2025 DOI:10.1021/cbmi.5c00144.
- Q. Miao, R. Dekkers, K. B. S. S. Gupta, M. Overhand, R. Dasgupta and M. Ubbink, Inorg. Chem., 2023, 62, 3776–3787 CrossRef CAS PubMed.
- Z. Garda, V. Nagy, A. Rodríguez-Rodríguez, R. Pujales-Paradela, V. Patinec, G. Angelovski, É. Tóth, F. K. Kálmán, D. Esteban-Gómez, R. Tripier, C. Platas-Iglesias and G. Tircsó, Inorg. Chem., 2020, 59, 8184–8195 CrossRef CAS PubMed.
- M. T. Kaczmarek, M. Zabiszak, M. Nowak and R. Jastrzab, Coord. Chem. Rev., 2018, 370, 42–54 CrossRef CAS.
- A. Kamboj, B. Phogat, A. Yadav and K. Poonia, Inorg. Chem. Commun., 2025, 179, 114714 CrossRef CAS.
- J. Corredoira-Vázquez, C. González-Barreira, P. Oreiro-Martínez, A. M. García-Deibe, J. Sanmartín-Matalobos and M. Fondo, J. Rare Earths, 2024, 42, 1–15 CrossRef.
- S. Chi, Y. Xu, B. Xie and T. Gao, J. Mol. Struct., 2022, 1260, 132715 CrossRef CAS.
- K. Andiappan, A. Sanmugam, E. Deivanayagam, K. Karuppasamy, H.-S. Kim and D. Vikraman, Inorg. Chem. Commun., 2023, 150, 110510 CrossRef CAS.
- S. Gholizadeh Dogaheh, J. Soleimannejad and E. C. Sanudo, J. Mol. Struct., 2020, 1219, 129060 CrossRef CAS.
- C. Kruck, T. Neumann, A. Schäfer, E. Kreidt, P. Weis, C. Holzer, M. M. Kappes and M. Seitz, Inorg. Chem., 2025, 64, 22048–22058 CrossRef CAS PubMed.
- D. Zhao, C. Yue, Q. Li, L. Guo, J. Yang and H. Li, ACS Appl. Polym. Mater., 2023, 5, 9375–9384 CrossRef CAS.
- D. Zhao, J. Yang, X. Tian, J. Wei, Q. Li and Y. Wang, Chem. Eng. J., 2022, 434, 134806 CrossRef CAS.
- L. Lu, X. Shao, X. Lin, L. Ding, B. Song and J. Sun, Chin. Chem. Lett., 2023, 34, 107203 CrossRef CAS.
- C. Wei, B. Sun, Z. Zhao, Z. Cai, J. Liu, Y. Tan, H. Wei, Z. Liu, Z. Bian and C. Huang, Inorg. Chem., 2020, 59, 8800–8808 CrossRef CAS PubMed.
- M. L. Brown, T. E. Karpiuk and D. B. Leznoff, CrystEngComm, 2023, 25, 4658–4668 RSC.
- T. Liu, A. S. Ivanov, I. Popovs, S. Jansone-Popova and D.-E. Jiang, RSC Adv., 2023, 13, 764–769 RSC.
- M. Pietraszkiewicz, J. Karpiuk and O. Pietraszkiewicz, J. Alloys Compd., 2000, 300–301, 141–146 CrossRef CAS.
- Z. K. Huffman, J. M. Sperling, C. J. Windorff, B. N. Long, L. Cordova, H. Ramanantoanina, C. Celis-Barros and T. E. Albrecht-Schönzart, Chem. Commun., 2022, 58, 11791–11794 RSC.
- S. Pailloux, C. E. Shirima, A. D. Ray, E. N. Duesler, K. A. Smith, R. T. Paine, J. R. Klaehn, M. E. McIlwain and B. P. Hay, Dalton Trans., 2009, 7486–7493, 10.1039/B905947D.
- Y. Kitagawa, M. Kumagai, P. P. Ferreira da Rosa, K. Fushimi and Y. Hasegawa, Chem. – Eur. J., 2021, 27, 264–269 CrossRef CAS PubMed.
- Y. Hirai, T. Nakanishi, Y. Kitagawa, K. Fushimi, T. Seki, H. Ito and Y. Hasegawa, Angew. Chem., Int. Ed., 2017, 56, 7171–7175 CrossRef CAS PubMed.
- O. G. Willis, A. Pucci, E. Cavalli, F. Zinna and L. Di Bari, J. Mater. Chem. C, 2023, 11, 5290–5296 RSC.
- A. Sickinger, B. Baguenard, A. Bensalah-Ledoux, Y. Guyot, L. Guy, F. Pointillart, O. Cador, M. Grasser, B. Le Guennic, F. Riobé, O. Maury and S. Guy, J. Mater. Chem. C, 2024, 12, 4253–4260 RSC.
- X.-L. Li, A. Wang, Y. Li, C. Gao, M. Cui, H.-P. Xiao and L. Zhou, Inorg. Chem., 2023, 62, 4351–4360 CrossRef CAS PubMed.
- N. F. M. Mukthar, N. D. Schley and G. Ung, J. Am. Chem. Soc., 2022, 144, 6148–6153 CrossRef CAS PubMed.
- K. Xu, X. Xie and L.-M. Zheng, Coord. Chem. Rev., 2022, 456, 214367 CrossRef CAS.
- J.-M. Herrera, S. J. A. Pope, A. J. H. M. Meijer, T. L. Easun, H. Adams, W. Z. Alsindi, X.-Z. Sun, M. W. George, S. Faulkner and M. D. Ward, J. Am. Chem. Soc., 2007, 129, 11491–11504 CrossRef CAS PubMed.
- J. Y. Liew, J. J. Brown, E. G. Moore and M. Schwalbe, Chem. – Eur. J., 2016, 22, 16178–16186 CrossRef CAS PubMed.
- S. I. Klink, H. Keizer, H. W. Hofstraat and F. C. J. M. van Veggel, Synth. Met., 2002, 127, 213–216 CrossRef CAS.
- J. J. Zakrzewski, B. Sieklucka and S. Chorazy, Inorg. Chem., 2020, 59, 1393–1404 CrossRef CAS PubMed.
- D. Rajah, M. C. Pfrunder, B. S. K. Chong, A. R. Ireland, I. M. Etchells and E. G. Moore, Dalton Trans., 2021, 50, 7400–7408 RSC.
- D. Mara, Z. Cai, S. Bonabello, S. Penna, R. Van Deun, P. Deplano, L. Marchiò, L. Pilia and F. Artizzu, Cryst. Growth Des., 2024, 24, 3798–3808 CrossRef CAS.
- P. F. Muldoon, G. Collet, S. V. Eliseeva, T.-Y. Luo, S. Petoud and N. L. Rosi, J. Am. Chem. Soc., 2020, 142, 8776–8781 CrossRef PubMed.
- B. Golesorkhi, S. Naseri, L. Guénée, I. Taarit, F. Alves, H. Nozary and C. Piguet, J. Am. Chem. Soc., 2021, 143, 15326–15334 CrossRef CAS PubMed.
- O. G. Willis, F. Petri, D. F. De Rosa, A. Mandoli, R. Pal, F. Zinna and L. Di Bari, J. Am. Chem. Soc., 2023, 145, 25170–25176 CrossRef CAS PubMed.
- O. G. Willis, F. Petri, G. Pescitelli, A. Pucci, E. Cavalli, A. Mandoli, F. Zinna and L. Di Bari, Angew. Chem., Int. Ed., 2022, 61, e202208326 CrossRef CAS PubMed.
- H. Shimoji, T. Fujii, A. Sumida, Y. Kitagawa, Y. Hasegawa, H. Imoto and K. Naka, J. Mater. Chem. C, 2023, 11, 15608–15615 RSC.
- N. A. Avagyan, P. S. Lemport, T. A. Polikovskiy, A. V. Tsorieva, M. T. Metlin, I. V. Taydakov, R. V. Zonov, K. A. Lyssenko, M. F. Vokuev, I. A. Rodin, V. A. Roznyatovsky, Y. A. Ustynyuk and V. G. Nenajdenko, Rare Met., 2025, 44, 4279–4293 CrossRef CAS.
- J. A. Adewuyi and G. Ung, J. Am. Chem. Soc., 2024, 146, 7097–7104 CrossRef CAS PubMed.
- J.-Y. He, Y. Wang, X. Chen, W.-P. Chen, G. Zhou and Y.-Z. Zheng, Adv. Mater., 2024, 36, 2406882 CrossRef CAS PubMed.
- O. Kotova, S. Comby, C. Lincheneau and T. Gunnlaugsson, Chem. Sci., 2017, 8, 3419–3426 RSC.
- H.-J. Chen, L.-Q. Chen, L.-R. Lin, L.-S. Long and L.-S. Zheng, Inorg. Chem., 2021, 60, 6986–6990 CrossRef CAS PubMed.
- X. Yu, A. A. Ryadun, D. I. Pavlov, T. Y. Guselnikova, A. S. Potapov and V. P. Fedin, Angew. Chem., Int. Ed., 2023, 62, e202306680 CrossRef CAS PubMed.
- J. Xue, Y. Wang, G. Yang and Y. Wang, J. Rare Earths, 2024, 42, 446–454 CrossRef CAS.
- A. de Bettencourt-Dias, S. Viswanathan and A. Rollett, J. Am. Chem. Soc., 2007, 129, 15436–15437 CrossRef CAS PubMed.
- A. de Bettencourt-Dias, P. S. Barber, S. Viswanathan, D. T. de Lill, A. Rollett, G. Ling and S. Altun, Inorg. Chem., 2010, 49, 8848–8861 CrossRef CAS PubMed.
- A. de Bettencourt-Dias, P. S. Barber and S. Bauer, J. Am. Chem. Soc., 2012, 134, 6987–6994 CrossRef CAS PubMed.
- A. de Bettencourt-Dias and J. S. K. Rossini, Inorg. Chem., 2016, 55, 9954–9963 CrossRef CAS PubMed.
- H. Jiang, Y.-Y. He, H.-C. Liang, M.-J. Shang, H.-H. Lu, C.-H. Liu, Y.-S. Meng, T. Liu and Y.-Y. Zhu, Chin. J. Struct. Chem., 2024, 43, 100354 CAS.
- J. I. Deneff, L. E. S. Rohwer, K. S. Butler, B. Kaehr, D. J. Vogel, T. S. Luk, R. A. Reyes, A. A. Cruz-Cabrera, J. E. Martin and D. F. Sava Gallis, Nat. Commun., 2023, 14, 981 CrossRef CAS PubMed.
- M. Tsurui, R. Takizawa, Y. Kitagawa, M. Wang, M. Kobayashi, T. Taketsugu and Y. Hasegawa, Angew. Chem., Int. Ed., 2024, 63, e202405584 CrossRef CAS PubMed.
- X.-D. Huang, X.-F. Ma, T. Shang, Y.-Q. Zhang and L.-M. Zheng, Inorg. Chem., 2023, 62, 1864–1874 CrossRef CAS PubMed.
- Y. Pei, Y. Fan, K. Sun, D. Hu, Y. Liu, J. Yin, L. Chen, M. Xu, W. Yan, X. Liu and F. Li, Angew. Chem., Int. Ed., 2025, 64, e202423791 CrossRef CAS PubMed.
- A. M. Kaczmarek, H. S. Jena, C. Krishnaraj, H. Rijckaert, S. K. P. Veerapandian, A. Meijerink and P. Van Der Voort, Angew. Chem., Int. Ed., 2021, 60, 3727–3736 CrossRef CAS PubMed.
- X.-Q. Guo, L.-P. Zhou, S.-J. Hu, L.-X. Cai, P.-M. Cheng and Q.-F. Sun, J. Am. Chem. Soc., 2021, 143, 6202–6210 CrossRef CAS PubMed.
- P.-R. Su, T. Wang, P.-P. Zhou, X.-X. Yang, X.-X. Feng, M.-N. Zhang, L.-J. Liang, Y. Tang and C.-H. Yan, Natl. Sci. Rev., 2022, 9, nwab016 CrossRef CAS PubMed.
- S. P. K. Panguluri, E. Jourdain, P. Chakraborty, S. Klyatskaya, M. M. Kappes, A. M. Nonat, L. J. Charbonnière and M. Ruben, J. Am. Chem. Soc., 2024, 146, 13083–13092 CrossRef CAS PubMed.
- T. Jia, P.-M. Cheng, M.-X. Zhang, W.-D. Liu, C.-Y. Li, H.-F. Su, L.-S. Long, L.-S. Zheng and X.-J. Kong, J. Am. Chem. Soc., 2024, 146, 28618–28623 CrossRef CAS PubMed.
- M. I. Schönherr, A. Biewald, A. Mähringer, T. J. Koller, P. Mayer, M. Döblinger, A. Hartschuh and D. D. Medina, Adv. Funct. Mater., 2025, 2507464 CrossRef.
- S.-R. Zhang, W.-T. Zhang, X. Li, G.-J. Xu, W. Xie, Y.-H. Xu, N. Xu and Z.-M. Su, Inorg. Chem., 2025, 64, 2990–2999 CrossRef CAS PubMed.
- Y. Zhong, Z. Wu, Y. Zhang, B. Dong and X. Bai, InfoMat, 2023, 5, e12392 CrossRef CAS.
- M. W. Mara, D. S. Tatum, A.-M. March, G. Doumy, E. G. Moore and K. N. Raymond, J. Am. Chem. Soc., 2019, 141, 11071–11081 CrossRef CAS PubMed.
- K. P. Ruoff, M. K. Gish, E. Song, I. Douair, P. Pandey, M. Steger, J. C. Johnson, P. J. Carroll, M. Gau, C. H. Chang, R. E. Larsen, A. J. Ferguson and E. J. Schelter, J. Am. Chem. Soc., 2023, 145, 16374–16382 CrossRef CAS PubMed.
- C. H. Simms, V. R. M. Nielsen, T. J. Sørensen, S. Faulkner and M. J. Langton, Phys. Chem. Chem. Phys., 2024, 26, 18683–18691 RSC.
- R. K. Wilharm, M. R. Gau, D. Trauner and E. J. Schelter, J. Am. Chem. Soc., 2025, 147, 27143–27147 CrossRef CAS PubMed.
- D. M. Driscoll, F. D. White, S. Pramanik, J. D. Einkauf, B. Ravel, D. Bykov, S. Roy, R. T. Mayes, L. H. Delmau, S. K. Cary, T. Dyke, A. Miller, M. Silveira, S. M. VanCleve, S. M. Davern, S. Jansone-Popova, I. Popovs and A. S. Ivanov, Nature, 2024, 629, 819–823 CrossRef CAS PubMed.
- B. Brennecke, Q. Wang, Q. Zhang, H.-Y. Hu and M. Nazaré, Angew. Chem., Int. Ed., 2020, 59, 8512–8516 CrossRef CAS PubMed.
- J. D. Routledge, X. Zhang, M. Connolly, M. Tropiano, O. A. Blackburn, A. M. Kenwright, P. D. Beer, S. Aldridge and S. Faulkner, Angew. Chem., Int. Ed., 2017, 56, 7783–7786 CrossRef CAS PubMed.
- D. F. Caffrey, T. Gorai, B. Rawson, M. Martínez-Calvo, J. A. Kitchen, N. S. Murray, O. Kotova, S. Comby, R. D. Peacock, P. Stachelek, R. Pal and T. Gunnlaugsson, Adv. Sci., 2024, 11, 2307448 CrossRef CAS PubMed.
- B.-A. N. Willis, D. Schnable, N. D. Schley and G. Ung, J. Am. Chem. Soc., 2022, 144, 22421–22425 CrossRef CAS PubMed.
- J. Liu, J. Yin, H. Yuan, Y. Zhao, S. Luo and F. Li, J. Rare Earths, 2022, 40, 1382–1388 CrossRef CAS.
- J. Xu, R. Luo, Z. Luo, J. Xu, Z. Mu, H. Bian, S. Y. Chan, B. Y. H. Tan, D. Chi, Z. An, G. Xing, X. Qin, C. Gong, Y. Wu and X. Liu, Nat. Photonics, 2025, 19, 71–78 CrossRef CAS.
- X. Liao, Y. Chen, T. Xie, R.-J. Sun, L.-Z. Yang, C.-F. Liu, R. Wang, S. Klyatskaya, M. Ruben, W. Zhang and Y.-S. Fu, Nat. Commun., 2025, 16, 6263 CrossRef CAS PubMed.
- A. J. Shin, C. Zhao, Y. Shen, C. E. Dickerson, B. Li, H. Roshandel, D. Bím, T. L. Atallah, P. H. Oyala, Y. He, L. K. Alson, T. A. Kerr, A. N. Alexandrova, P. L. Diaconescu, W. C. Campbell and J. R. Caram, Science, 2024, 385, 651–656 CrossRef CAS PubMed.
- D. Serrano, S. K. Kuppusamy, B. Heinrich, O. Fuhr, D. Hunger, M. Ruben and P. Goldner, Nature, 2022, 603, 241–246 CrossRef CAS PubMed.
- H. B. Wineinger, J. M. Sperling and T. E. Albrecht, Coord. Chem. Rev., 2025, 545, 217004 CrossRef CAS.
- G. Zhan, L. Wang, Z. Zhao, P. Fang, Z. Bian and Z. Liu, Angew. Chem., Int. Ed., 2020, 59, 19011–19015 CrossRef CAS PubMed.
- J. Li, L. Wang, Z. Zhao, B. Sun, G. Zhan, H. Liu, Z. Bian and Z. Liu, Nat. Commun., 2020, 11, 5218 CrossRef CAS PubMed.
- R. M. Diaz-Rodriguez, D. A. Gálico, D. Chartrand, E. A. Suturina and M. Murugesu, J. Am. Chem. Soc., 2022, 144, 912–921 CrossRef CAS PubMed.
- R. M. Diaz-Rodriguez, D. A. Gálico, D. Chartrand and M. Murugesu, J. Am. Chem. Soc., 2024, 146, 34118–34129 CrossRef CAS PubMed.
- L. Aboshyan-Sorgho, C. Besnard, P. Pattison, K. R. Kittilstved, A. Aebischer, J.-C. G. Bünzli, A. Hauser and C. Piguet, Angew. Chem., Int. Ed., 2011, 50, 4108–4112 CrossRef CAS PubMed.
- A. Nonat, C. F. Chan, T. Liu, C. Platas-Iglesias, Z. Liu, W.-T. Wong, W.-K. Wong, K.-L. Wong and L. J. Charbonnière, Nat. Commun., 2016, 7, 11978 CrossRef CAS PubMed.
- J. Wang, Y. Jiang, J.-Y. Liu, H.-B. Xu, Y.-X. Zhang, X. Peng, M. Kurmoo, S. W. Ng and M.-H. Zeng, Angew. Chem., Int. Ed., 2021, 60, 22368–22375 CrossRef CAS PubMed.
- T. Lathion, A. Fürstenberg, C. Besnard, A. Hauser, A. Bousseksou and C. Piguet, Inorg. Chem., 2020, 59, 1091–1103 CrossRef CAS PubMed.
- N. Deorukhkar, C. Egger, L. Guénée, C. Besnard and C. Piguet, J. Am. Chem. Soc., 2024, 146, 308–318 CrossRef CAS PubMed.
- G. Bao, K.-L. Wong, D. Jin and P. A. Tanner, Light: Sci. Appl., 2018, 7, 96 CrossRef PubMed.
- Y. Yamaguchi, T. Nakai, S. Omagari, M. Wang, Y. Hasegawa and Y. Kitagawa, Commun. Chem., 2025, 8, 269 CrossRef CAS PubMed.
- A. G. Cosby, S. H. Ahn and E. Boros, Angew. Chem., Int. Ed., 2018, 57, 15496–15499 CrossRef CAS PubMed.
- J. Wang, J. J. Zakrzewski, M. Heczko, M. Zychowicz, K. Nakagawa, K. Nakabayashi, B. Sieklucka, S. Chorazy and S.-I. Ohkoshi, J. Am. Chem. Soc., 2020, 142, 3970–3979 CrossRef CAS PubMed.
- J. Wang, J. J. Zakrzewski, M. Zychowicz, V. Vieru, L. F. Chibotaru, K. Nakabayashi, S. Chorazy and S.-I. Ohkoshi, Chem. Sci., 2021, 12, 730–741 RSC.
- R. C. Knighton, L. K. Soro, L. Francés-Soriano, A. Rodríguez-Rodríguez, G. Pilet, M. Lenertz, C. Platas-Iglesias, N. Hildebrandt and L. J. Charbonnière, Angew. Chem., Int. Ed., 2022, 61, e202113114 CrossRef CAS PubMed.
- D. A. Gálico, C. M. Santos Calado and M. Murugesu, Chem. Sci., 2023, 14, 5827–5841 RSC.
- F.-F. Liu, D. Wang, G.-H. Chen, Y. Qiao, F. Luo, J. Zhang and L. Zhang, Sci. China: Chem., 2023, 66, 1731–1736 CrossRef CAS.
- M.-H. Du, L.-Q. Chen, L.-P. Jiang, W.-D. Liu, L.-S. Long, L. Zheng and X.-J. Kong, J. Am. Chem. Soc., 2022, 144, 5653–5660 CrossRef CAS PubMed.
- F.-F. Wang, Y. Zhang, T. Zhao, P.-F. Feng, Z. Lu, S.-Q. Zang and T. C. W. Mak, Angew. Chem., Int. Ed., 2023, 62, e202305693 CrossRef CAS PubMed.
- D. A. Gálico and M. Murugesu, Angew. Chem., Int. Ed., 2022, 61, e202204839 CrossRef PubMed.
- S.-Q. Wang, Y. Wang, X. Yang, Y. Liu, H. Li, Z. Yang, W.-Y. Sun and J. L. Sessler, Angew. Chem., Int. Ed., 2024, 63, e202317775 CrossRef CAS PubMed.
- D. A. Gálico, A. A. Kitos, J. S. Ovens, F. A. Sigoli and M. Murugesu, Angew. Chem., Int. Ed., 2021, 60, 6130–6136 CrossRef PubMed.
- H.-L. Wang, Z. Ni, Z.-Y. Ruan, Z.-H. Zhu, P.-Y. Liao, G. Feng, J.-H. Jia and M.-L. Tong, Nano Res., 2023, 16, 11495–11502 CrossRef CAS.
- D. A. Gálico and M. Murugesu, Adv. Opt. Mater., 2024, 12, 2401064 CrossRef.
- W.-D. Liu, H. Xu, C.-Y. Li, L.-S. Long, L.-S. Zheng and X.-J. Kong, Inorg. Chem. Front., 2025, 12, 253–260 RSC.
- D. A. Gálico and M. Murugesu, J. Mater. Chem. C, 2024, 12, 15413–15417 RSC.
- C.-T. Yeung, K.-H. Yim, H.-Y. Wong, R. Pal, W.-S. Lo, S.-C. Yan, M. Yee-Man Wong, D. Yufit, D. E. Smiles, L. J. McCormick, S. J. Teat, D. K. Shuh, W.-T. Wong and G.-L. Law, Nat. Commun., 2017, 8, 1128 CrossRef PubMed.
- Y. B. Tan, Y. Okayasu, S. Katao, Y. Nishikawa, F. Asanoma, M. Yamada, J. Yuasa and T. Kawai, J. Am. Chem. Soc., 2020, 142, 17653–17661 CrossRef CAS PubMed.
- T. Zhao, Y.-F. Zhang, G.-H. Wang, X.-X. Wang, P.-F. Feng and S.-Q. Zang, Angew. Chem., Int. Ed., 2025, 64, e202421426 CrossRef CAS PubMed.
- X.-F. Duan, L.-P. Zhou, H.-R. Li, S.-J. Hu, W. Zheng, X. Xu, R. Zhang, X. Chen, X.-Q. Guo and Q.-F. Sun, J. Am. Chem. Soc., 2023, 145, 23121–23130 CrossRef CAS PubMed.
- X.-F. Duan, H. Ji, L.-P. Zhou, S.-J. Hu, J. Fu, P. Duan, X.-Q. Guo and Q.-F. Sun, CCS Chem., 2025, 7, 1972–1980 CrossRef CAS.
- M. Rancan, J. Tessarolo, A. Carlotto, S. Carlotto, M. Rando, L. Barchi, E. Bolognesi, R. Seraglia, G. Bottaro, M. Casarin, G. H. Clever and L. Armelao, Cell Rep. Phys. Sci., 2022, 3, 100692 CrossRef CAS.
- X.-Z. Li, L.-P. Zhou, S.-J. Hu, L.-X. Cai, X.-Q. Guo, Z. Wang and Q.-F. Sun, Chem. Commun., 2020, 56, 4416–4419 RSC.
- X. Tang, D. Chu, W. Gong, Y. Cui and Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 9099–9105 CrossRef CAS PubMed.
- S.-J. Hu, X.-Q. Guo, L.-P. Zhou, D.-N. Yan, P.-M. Cheng, L.-X. Cai, X.-Z. Li and Q.-F. Sun, J. Am. Chem. Soc., 2022, 144, 4244–4253 CrossRef CAS PubMed.
- M. Boone, F. Artizzu, J. Goura, D. Mara, R. Van Deun and M. D'Hooghe, Coord. Chem. Rev., 2024, 501, 215525 CrossRef CAS.
- J. Zhao, J. Yuan, Z. Fang, S. Huang, Z. Chen, F. Qiu, C. Lu, J. Zhu and X. Zhuang, Coord. Chem. Rev., 2022, 471, 214735 CrossRef CAS.
- B. B. Rath, M. Gupta and J. J. Vittal, Chem. Mater., 2022, 34, 178–185 CrossRef CAS.
- X. Sun, Y. Zheng, X. Liu, X. He, Z. Zeng, X. Wang, P. Cen, D. Tian, D. Peng and X. Liu, Sci. China: Chem., 2025, 68, 4912–4921 CrossRef CAS.
- R. Maouche, S. Belaid, B. Benmerad, S. Bouacida, C. Daiguebonne, Y. Suffren, S. Freslon, K. Bernot and O. Guillou, Inorg. Chem., 2021, 60, 3707–3718 CrossRef CAS PubMed.
- H. Yao, G. Calvez, C. Daiguebonne, Y. Suffren, K. Bernot, T. Roisnel and O. Guillou, Inorg. Chem., 2022, 61, 4895–4908 CrossRef CAS PubMed.
- J. Huang, H.-m Ding, Y. Xu, D. Zeng, H. Zhu, D.-M. Zang, S.-S. Bao, Y.-q Ma and L.-M. Zheng, Nat. Commun., 2017, 8, 2131 CrossRef PubMed.
- Z.-M. Zhai, T. Hou, Y. Xu, Q. Teng, S.-S. Bao and L.-M. Zheng, Chem. – Eur. J., 2025, 31, e202403699 CrossRef CAS PubMed.
- M. Atzori, K. Dhbaibi, H. Douib, M. Grasser, V. Dorcet, I. Breslavetz, K. Paillot, O. Cador, G. L. J. A. Rikken, B. Le Guennic, J. Crassous, F. Pointillart and C. Train, J. Am. Chem. Soc., 2021, 143, 2671–2675 CrossRef CAS PubMed.
- K. Dhbaibi, M. Grasser, H. Douib, V. Dorcet, O. Cador, N. Vanthuyne, F. Riobé, O. Maury, S. Guy, A. Bensalah-Ledoux, B. Baguenard, G. L. J. A. Rikken, C. Train, B. Le Guennic, M. Atzori, F. Pointillart and J. Crassous, Angew. Chem., Int. Ed., 2023, 62, e202215558 CrossRef CAS PubMed.
- M.-X. Zhang, M.-Y. Ye, L.-S. Long and L.-S. Zheng, Adv. Opt. Mater., 2025, 13, 2402191 CrossRef CAS.
- M.-X. Zhang, T. Jia, P.-M. Cheng, W.-D. Liu, L.-S. Long and L.-S. Zheng, Angew. Chem., Int. Ed., 2025, 64, e202501349 CrossRef CAS PubMed.
- J. Heine and K. Müller-Buschbaum, Chem. Soc. Rev., 2013, 42, 9232–9242 RSC.
- R.-J. Wei, X. Luo, G.-H. Ning and D. Li, Acc. Chem. Res., 2025, 58, 746–761 CrossRef CAS PubMed.
- J. Dong, X. Han, Y. Liu, H. Li and Y. Cui, Angew. Chem., Int. Ed., 2020, 59, 13722–13733 CrossRef CAS PubMed.
- B. Li, H.-M. Wen, Y. Cui, W. Zhou, G. Qian and B. Chen, Adv. Mater., 2016, 28, 8819–8860 CrossRef CAS PubMed.
- J. Rocha, L. D. Carlos, F. A. A. Paz and D. Ananias, Chem. Soc. Rev., 2011, 40, 926–940 RSC.
- B. Mohanty, S. Kumari, P. Yadav, P. Kanoo and A. Chakraborty, Coord. Chem. Rev., 2024, 519, 216102 CrossRef CAS.
- M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC.
- Z.-J. Lin, J. Lü, M. Hong and R. Cao, Chem. Soc. Rev., 2014, 43, 5867–5895 RSC.
- F. Haj Sadeghi, H. Heydarinasab, V. Haddadi-Asl, H. Eivaz Mohammadloo and B. Ramezanzadeh, Coord. Chem. Rev., 2026, 549, 217221 CrossRef CAS.
- A. D. G. Firmino, F. Figueira, J. P. C. Tomé, F. A. A. Paz and J. Rocha, Coord. Chem. Rev., 2018, 355, 133–149 CrossRef CAS.
- L. Feng, C. Dong, M. Li, L. Li, X. Jiang, R. Gao, R. Wang, L. Zhang, Z. Ning, D. Gao and J. Bi, J. Hazard. Mater., 2020, 388, 121816 CrossRef CAS PubMed.
- L. Li, Y. Zhu, X. Zhou, C. D. S. Brites, D. Ananias, Z. Lin, F. A. A. Paz, J. Rocha, W. Huang and L. D. Carlos, Adv. Funct. Mater., 2016, 26, 8677–8684 CrossRef CAS.
- Y. Cui, H. Xu, Y. Yue, Z. Guo, J. Yu, Z. Chen, J. Gao, Y. Yang, G. Qian and B. Chen, J. Am. Chem. Soc., 2012, 134, 3979–3982 CrossRef CAS PubMed.
- H. Li, Y. Li, L. Zhang, E. Hu, D. Zhao, H. Guo and G. Qian, Adv. Mater., 2024, 36, 2405535 CrossRef CAS PubMed.
- A. E. Psalti, S. V. Eliseeva, A. Hatzidimitriou, S. Oikonomidis, S. Petoud and T. Lazarides, J. Am. Chem. Soc., 2026, 148, 4020–4031 CrossRef CAS PubMed.
- X. Feng, S. Mou, Y. Pang, R. Long, C. Liu, Z. Bai, X. Liu and S. Liu, Chem. Eng. J., 2025, 509, 161474 CrossRef CAS.
- J. I. Deneff, K. S. Butler, L. E. S. Rohwer, C. J. Pearce, N. R. Valdez, M. A. Rodriguez, T. S. Luk and D. F. Sava Gallis, Angew. Chem., Int. Ed., 2021, 60, 1203–1211 CrossRef CAS PubMed.
- J. Yang, Q. Yue, G.-D. Li, J.-J. Cao, G.-H. Li and J.-S. Chen, Inorg. Chem., 2006, 45, 2857–2865 CrossRef CAS PubMed.
- P. Mahata, K. V. Ramya and S. Natarajan, Chem. – Eur. J., 2008, 14, 5839–5850 CrossRef CAS PubMed.
- X. Chen, X. Zhang and Y. Zhao, Chem. Soc. Rev., 2025, 54, 152–177 RSC.
- Y. Xie, G. Sun, G. A. Mandl, S. L. Maurizio, J. Chen, J. A. Capobianco and L. Sun, Angew. Chem., Int. Ed., 2023, 62, e202216269 CrossRef CAS PubMed.
- J. Chen, Y. Xie, W. Yang, R. Sun, F. Xing, G. A. Mandl, J. A. Capobianco and L. Sun, Angew. Chem., Int. Ed., 2025, 64, e202509093 CrossRef CAS PubMed.
- X. Yu, Z. Zhou, N. Zhestkij, Y. Kenzhebayeva, S. A. Povarov, J. Chen, D. I. Pavlov, N. A. Burzak, A. R. Knyazeva, S. S. Lazarev, V. A. Dyachuk, A. S. Potapov, L. Sun, V. A. Milichko and V. P. Fedin, J. Am. Chem. Soc., 2026, 148, 14197–14204 CrossRef CAS PubMed.
- N. Song, K. Lv, X. Zhang, T. Zhu, Z. Zhao, Z. Liu, Y. Gong, C. Cheng and C. Yang, Angew. Chem., Int. Ed., 2026, 65, e19226 CrossRef CAS PubMed.
- Y. Zhang, X. Wang, K. Xu, F. Zhai, J. Shu, Y. Tao, J. Wang, L. Jiang, L. Yang, Y. Wang, W. Liu, J. Su, Z. Chai and S. Wang, J. Am. Chem. Soc., 2023, 145, 13161–13168 CrossRef CAS PubMed.
- H. Chen, X. Yang, Y. Ye, H. Hong, Q. Wei, Y. Zhu and M.-J. Lin, Angew. Chem., Int. Ed., 2025, 64, e202506118 CrossRef CAS PubMed.
- Y. Xin, J. Wang, M. Zychowicz, J. J. Zakrzewski, K. Nakabayashi, B. Sieklucka, S. Chorazy and S.-I. Ohkoshi, J. Am. Chem. Soc., 2019, 141, 18211–18220 CrossRef CAS PubMed.
- J. Wang, J. J. Zakrzewski, M. Zychowicz, Y. Xin, H. Tokoro, S. Chorazy and S.-I. Ohkoshi, Angew. Chem., Int. Ed., 2023, 62, e202306372 CrossRef CAS PubMed.
- J.-H. Fu, S.-Y. Wang, Y.-S. Chen, S. Prusty and Y.-T. Chan, J. Am. Chem. Soc., 2019, 141, 16217–16221 CrossRef CAS PubMed.
- L.-J. Han, W. Yan, S.-G. Chen, Z.-Z. Shi and H.-G. Zheng, Inorg. Chem., 2017, 56, 2936–2940 CrossRef CAS PubMed.
- L. Su, Q. Niu, J. Liu, X. Liu, Y. Wang, Z. Ge, Y. Ye and Z. Li, Chem. Eng. J., 2025, 520, 166124 CrossRef CAS.
- D. Zhao, D. Yue, L. Zhang, K. Jiang and G. Qian, Inorg. Chem., 2018, 57, 12596–12602 CrossRef CAS PubMed.
- X. Liu, W. Liu, Y. Kou, X. Yang, Z. Ju and W. Liu, Inorg. Chem. Front., 2022, 9, 4065–4074 RSC.
- H.-L. Wang, Y.-L. Li, H.-H. Zou, F.-P. Liang and Z.-H. Zhu, Adv. Mater., 2025, 37, 2502742 CrossRef CAS PubMed.
- L.-L. Ma, G.-P. Yang, G.-P. Li, P.-F. Zhang, J. Jin, Y. Wang, J.-M. Wang and Y.-Y. Wang, Inorg. Chem. Front., 2021, 8, 329–338 RSC.
- C. Liang, S. Tan, L. Shao, X. Xue, J. Liu, N. Liu, W. Zhang and Q. Shi, Inorg. Chem., 2023, 62, 9508–9517 CrossRef CAS PubMed.
- X. Zhai, X. Mu, G. Tan, L. Liang, Y. Kou, P. Su, C.-H. Yan and Y. Tang, Sci. China: Chem., 2025, 68, 924–934 CrossRef CAS.
- A.-G. Liu, Y. Chen, Z.-T. Chen, X.-H. Liang, H.-X. Xia and B. Li, Adv. Mater., 2025, 37, e10964 CrossRef CAS PubMed.
- J. Wang, D. Li, Y. Ye, Y. Qiu, J. Liu, L. Huang, B. Liang and B. Chen, Adv. Mater., 2021, 33, 2008020 CrossRef CAS PubMed.
- C. Sun, X. Liu, H. Zhuo, X. He, Z. Ge, Y. Zhang, Z. Li and Q. Xiong, J. Hazard. Mater., 2025, 484, 136793 CrossRef CAS PubMed.
- X.-Y. Xu, X. Lian, J.-N. Hao, C. Zhang and B. Yan, Adv. Mater., 2017, 29, 1702298 CrossRef PubMed.
- H.-Q. Zheng, Y. Cui and G. Qian, Acc. Mater. Res., 2023, 4, 982–994 CrossRef CAS.
- R. J. Drout, L. Robison and O. K. Farha, Coord. Chem. Rev., 2019, 381, 151–160 CrossRef CAS.
- Y. Cui, T. Song, J. Yu, Y. Yang, Z. Wang and G. Qian, Adv. Funct. Mater., 2015, 25, 4796–4802 CrossRef CAS.
- Y. Cui, R. Song, J. Yu, M. Liu, Z. Wang, C. Wu, Y. Yang, Z. Wang, B. Chen and G. Qian, Adv. Mater., 2015, 27, 1420–1425 CrossRef CAS PubMed.
- T. Liang, Z. Guo, Y. He, Y. Wang, C. Li, Z. Li and Z. Liu, Adv. Sci., 2022, 9, 2104561 CrossRef CAS PubMed.
- J. An, C. M. Shade, D. A. Chengelis-Czegan, S. Petoud and N. L. Rosi, J. Am. Chem. Soc., 2011, 133, 1220–1223 CrossRef CAS PubMed.
- H.-L. Wang, Q.-X. Yu, Z.-H. Zhu, X.-L. Lu, Y.-L. Li, F.-P. Liang and H.-H. Zou, Inorg. Chem., 2023, 62, 5863–5871 CrossRef CAS PubMed.
- R. Zhai, Y. Xiao, Z. Gu and J. Zhang, Nano Res., 2022, 15, 1102–1108 CrossRef CAS.
- H. N. Abdelhamid and A. J. Chamkha, Anal. Chem., 2025, 97, 23721–23742 CrossRef CAS PubMed.
- C. Zhang, Z.-S. Li, X.-Y. Dong, Y.-Y. Niu and S.-Q. Zang, Adv. Mater., 2022, 34, 2109496 CrossRef CAS PubMed.
- X. Tong, J. Wang, M. Wei, J. Qiang, Z. Gao, F. Hu and Y. S. Zhao, Aggregate, 2025, 6, e70188 CrossRef CAS.
- W. Liu, X. Huang, C. Chen, C. Xu, J. Ma, L. Yang, W. Wang, W. Dou and W. Liu, Chem. – Eur. J., 2019, 25, 1090–1097 CrossRef CAS PubMed.
- R. Gao, Q. Zou, Q.-Q. Su, X.-F. Ma, Y.-H. Qin, R. Liao, S.-S. Bao and L.-M. Zheng, Chin. Chem. Lett., 2025, 36, 110404 CrossRef CAS.
- Z. Li, G. Wang, Y. Ye, B. Li, H. Li and B. Chen, Angew. Chem., Int. Ed., 2019, 58, 18025–18031 CrossRef CAS PubMed.
- M.-Y. Guo, P. Li, S.-L. Yang, R. Bu, X.-Q. Piao and E.-Q. Gao, ACS Appl. Mater. Interfaces, 2020, 12, 43958–43966 CrossRef CAS PubMed.
- K. Wang, Y.-L. Zhu, T.-F. Zheng, X. Xie, J.-L. Chen, Y.-Q. Wu, S.-J. Liu and H.-R. Wen, Anal. Chem., 2023, 95, 4992–4999 CrossRef CAS PubMed.
- T. Xia, W. Cao, Y. Cui, Y. Yang and G. Qian, Opto-Electron. Adv., 2021, 4, 200063 CrossRef CAS.
- A. M. Kaczmarek, Y.-Y. Liu, M. K. Kaczmarek, H. Liu, F. Artizzu, L. D. Carlos and P. Van Der Voort, Angew. Chem., Int. Ed., 2020, 59, 1932–1940 CrossRef CAS PubMed.
- Y. Yu, Q. Jin, Y. Ren, Y. Wang, D. Zhu and J. Wang, Chem. Eng. J., 2023, 465, 142819 CrossRef CAS.
- X.-L. Mao, Y.-J. Cai, Q.-X. Luo, X. Liu, Q.-Q. Jiang, C.-R. Zhang, L. Zhang, R.-P. Liang and J.-D. Qiu, Anal. Chem., 2024, 96, 5037–5045 CrossRef CAS PubMed.
- X. Shen and B. Yan, Talanta, 2023, 265, 124869 CrossRef CAS PubMed.
- Z. Zhang and B. Yan, Sens. Actuators, B, 2025, 426, 137107 CrossRef CAS.
- C. Wang, Z. Xiong, X. Feng, S. Xu, Y. Zhang, Y. Zhang, L. Zeng, J. Tan and Y. Ren, Sens. Actuators, B, 2024, 418, 136314 CrossRef CAS.
- Y. Yu and G. Li, J. Hazard. Mater., 2022, 422, 126927 CrossRef CAS PubMed.
- Q. Ren, P. Yang, J. Liu, Y. Chen, S. Ouyang, Y. Zeng, P. Zhao and J. Tao, Anal. Chim. Acta, 2021, 1188, 339191 CrossRef CAS PubMed.
- K. Li, X. Li, D. Wang, Z. Li and C. Li, Microchem. J., 2021, 170, 106764 CrossRef CAS.
- Q. Jin, Y. Hou, D. Zhu, Y. Yu and Y. Ren, Langmuir, 2024, 40, 13596–13602 CrossRef CAS PubMed.
- C. Wang, R. Zhang and H. Möhwald, Langmuir, 2010, 26, 11987–11990 CrossRef CAS PubMed.
- F. Chen, J. Wang, W. Xu, Z. Ren, G. Peng, T. Huang and F. Zhao, J. Alloys Compd., 2025, 1010, 177421 CrossRef CAS.
- Y. Kitagawa, M. Kumagai, K. Fushimi and Y. Hasegawa, Chem. Phys. Lett., 2020, 749, 137437 CrossRef CAS.
- P. Su, L. Liang, T. Wang, P. Zhou, J. Cao, W.-S. Liu and Y. Tang, Chem. Eng. J., 2021, 413, 127408 CrossRef CAS.
- A. Ruiz-Arias, F. Fueyo-González, C. Izquierdo-García, A. Navarro, M. Gutiérrez-Rodríguez, R. Herranz, C. Burgio, A. Reinoso, J. M. Cuerva, A. Orte and J. A. González-Vera, Angew. Chem., Int. Ed., 2024, 63, e202314595 CrossRef CAS PubMed.
- R. Nishiyabu, N. Hashimoto, T. Cho, K. Watanabe, T. Yasunaga, A. Endo, K. Kaneko, T. Niidome, M. Murata, C. Adachi, Y. Katayama, M. Hashizume and N. Kimizuka, J. Am. Chem. Soc., 2009, 131, 2151–2158 CrossRef CAS PubMed.
- Y. Yang, Y. Liu, D. Tu, M. Chen, Y. Zhang, H. Gao and X. Chen, Angew. Chem., Int. Ed., 2022, 61, e202116983 CrossRef CAS PubMed.
- Z. Luo, X. Liang, T. He, X. Qin, X. Li, Y. Li, L. Li, X. J. Loh, C. Gong and X. Liu, J. Am. Chem. Soc., 2022, 144, 16366–16377 CrossRef CAS PubMed.
- S. Fernández-Fariña, O. Kotova, S. R. Donohoe and T. Gunnlaugsson, Chem. Soc. Rev., 2025, 54, 11226–11265 RSC.
- O. Kotova, C. O’Reilly, S. T. Barwich, L. E. Mackenzie, A. D. Lynes, A. J. Savyasachi, M. Ruether, R. Pal, M. E. Möbius and T. Gunnlaugsson, Chem, 2022, 8, 1395–1414 CAS.
- J. Liu, M.-a Morikawa, H. Lei, K. Ishiba and N. Kimizuka, Langmuir, 2016, 32, 10597–10603 CrossRef CAS PubMed.
- J. A. Kitchen, D. E. Barry, L. Mercs, M. Albrecht, R. D. Peacock and T. Gunnlaugsson, Angew. Chem., Int. Ed., 2012, 51, 704–708 CrossRef CAS PubMed.
- C. Kim, K. Y. Kim, J. H. Lee, J. Ahn, K. Sakurai, S. S. Lee and J. H. Jung, ACS Appl. Mater. Interfaces, 2017, 9, 3799–3807 CrossRef CAS PubMed.
- S. Lim, Y. Cho, J. H. Kang, M. Hwang, Y. Park, S. K. Kwak, S. H. Jung and J. H. Jung, J. Am. Chem. Soc., 2024, 146, 18484–18497 CrossRef CAS PubMed.
- A. J. Savyasachi, O. Kotova, E. T. Luis, A. D. Lynes, S. Mills, S. A. Bright, G. J. McManus, M. E. Möbius, D. C. Williams, R. Pal, J. J. Boland and T. Gunnlaugsson, Chem, 2025, 11, 102321 CAS.
- Y.-L. Li, H.-L. Wang, Z.-H. Zhu, Y.-F. Wang, F.-P. Liang and H.-H. Zou, Nat. Commun., 2024, 15, 2896 CrossRef CAS PubMed.
- W.-L. Zhou, Y.-G. Wu, W. Qin, R.-X. Chen, S. Wang, J. Liu, X. Xu, Y. Chen and Y. Liu, Sci. China: Chem., 2026, 69, 1300–1308 CrossRef CAS.
- J. R. Kumpfer and S. J. Rowan, J. Am. Chem. Soc., 2011, 133, 12866–12874 CrossRef CAS PubMed.
- Z. Li, G. Wang, Y. Wang and H. Li, Angew. Chem., Int. Ed., 2018, 57, 2194–2198 CrossRef CAS PubMed.
- L. Liang, X. Yang, X. Yan, Y. Kou, Y. Zhang, P. Su and Y. Tang, Adv. Mater., 2026, 38, e14252 CrossRef CAS PubMed.
- R. Wang, Y. Zhang, W. Lu, B. Wu, S. Wei, S. Wu, W. Wang and T. Chen, Angew. Chem., Int. Ed., 2023, 62, e202300417 CrossRef CAS PubMed.
- F.-J. Song, K. Wang, X.-Y. Yan, K. Cui, N. Song, X. Zhu, C.-H. Yan, P. Su and Y. Tang, Angew. Chem., Int. Ed., 2026, e9901177, DOI:10.1002/anie.9901177.
- G. Pu, J. Song, Z. Cheng, Y. Tang, C. Kang and J. Wang, Laser Photonics Rev., 2025, 19, 2500156 CrossRef.
- S. Wen, J. Zhou, P. J. Schuck, Y. D. Suh, T. W. Schmidt and D. Jin, Nat. Photonics, 2019, 13, 828–838 CrossRef CAS.
- G. Bao, R. Deng, D. Jin and X. Liu, Nat. Rev. Mater., 2025, 10, 28–43 CrossRef CAS.
- X. Wang, R. R. Valiev, T. Y. Ohulchanskyy, H. Ågren, C. Yang and G. Chen, Chem. Soc. Rev., 2017, 46, 4150–4167 RSC.
- J. Ming, J. Zhou, X. Liu and F. Zhang, Adv. Mater., 2025, e08521, DOI:10.1002/adma.202508521.
- X. Cheng, J. Zhou, J. Yue, Y. Wei, C. Gao, X. Xie and L. Huang, Chem. Rev., 2022, 122, 15998–16050 CrossRef CAS PubMed.
- B. Li, M. Zhao, J. Lin, P. Huang and X. Chen, Chem. Soc. Rev., 2022, 51, 7692–7714 RSC.
- W. Zou, C. Visser, J. A. Maduro, M. S. Pshenichnikov and J. C. Hummelen, Nat. Photonics, 2012, 6, 560–564 CrossRef CAS.
- W. Shao, G. Chen, A. Kuzmin, H. L. Kutscher, A. Pliss, T. Y. Ohulchanskyy and P. N. Prasad, J. Am. Chem. Soc., 2016, 138, 16192–16195 CrossRef CAS PubMed.
- J. Lee, B. Yoo, H. Lee, G. D. Cha, H.-S. Lee, Y. Cho, S. Y. Kim, H. Seo, W. Lee, D. Son, M. Kang, H. M. Kim, Y. I. Park, T. Hyeon and D.-H. Kim, Adv. Mater., 2017, 29, 1603169 CrossRef PubMed.
- J. Zhou, S. Wen, J. Liao, C. Clarke, S. A. Tawfik, W. Ren, C. Mi, F. Wang and D. Jin, Nat. Photonics, 2018, 12, 154–158 CrossRef CAS.
- J. Liu, P. Geiregat, L. Pilia, R. Van Deun and F. Artizzu, Adv. Opt. Mater., 2021, 9, 2001678 CrossRef CAS.
- F. Zhao, J. Hu, D. Guan, J. Liu, X. Zhang, H. Ling, Y. Zhang and Q. Liu, Adv. Mater., 2023, 35, 2304907 CrossRef CAS PubMed.
- L. van Turnhout, D. G. Congrave, Z. Yu, R. Arul, S. A. Dowland, E. Sebastian, Z. Jiang, H. Bronstein and A. Rao, J. Am. Chem. Soc., 2024, 146, 22612–22621 CrossRef CAS PubMed.
- P. Zhang, J. Ke, D. Tu, J. Li, Y. Pei, L. Wang, X. Shang, T. Guan, S. Lu, Z. Chen and X. Chen, Angew. Chem., Int. Ed., 2022, 61, e202112125 CrossRef CAS PubMed.
- S. Han, Z. Yi, J. Zhang, Q. Gu, L. Liang, X. Qin, J. Xu, Y. Wu, H. Xu, A. Rao and X. Liu, Nat. Commun., 2021, 12, 3704 CrossRef CAS PubMed.
- Z. Ju and R. Deng, Angew. Chem., Int. Ed., 2025, 64, e202422575 CrossRef CAS PubMed.
- L. van Turnhout, A. Tew, S. A. Dowland, E. Sebastian, Z. Jiang, R. Arul, Z. Yu and A. Rao, J. Am. Chem. Soc., 2025, 147, 37788–37797 CrossRef CAS PubMed.
- M. Zhao, B. Li, Y. Wu, H. He, X. Zhu, H. Zhang, C. Dou, L. Feng, Y. Fan and F. Zhang, Adv. Mater., 2020, 32, 2001172 CrossRef CAS PubMed.
- C. Sun, M. Zhao, X. Zhu, P. Pei and F. Zhang, CCS Chem., 2022, 4, 476–486 CrossRef CAS.
- H. Xu, S. Han, R. Deng, Q. Su, Y. Wei, Y. Tang, X. Qin and X. Liu, Nat. Photonics, 2021, 15, 732–737 CrossRef CAS.
- H. Suo, P. Zhao, X. Zhang, Y. Guo, D. Guo, J. Chang, J. Chen, P. Li, Z. Wang, H. Wei, W. Zheng and F. Wang, Nat. Commun., 2025, 16, 3249 CrossRef CAS PubMed.
- J. Tao, D. Zheng, Y. Tang, H. He, Y. Zhang, Y. Yang, L. Dai, H. Zha, Y. Sang and Z. Nie, Angew. Chem., Int. Ed., 2025, 64, e202419640 CrossRef CAS PubMed.
- M. Zhao, R. Wang, B. Li, Y. Fan, Y. Wu, X. Zhu and F. Zhang, Angew. Chem., Int. Ed., 2019, 58, 2050–2054 CrossRef CAS PubMed.
- M. Zhao, B. Li, P. Wang, L. Lu, Z. Zhang, L. Liu, S. Wang, D. Li, R. Wang and F. Zhang, Adv. Mater., 2018, 30, 1804982 CrossRef PubMed.
- Y.-L. Lin, S. Zheng, C.-C. Chang, L.-R. Lee and J.-T. Chen, Nat. Commun., 2024, 15, 916 CrossRef CAS PubMed.
- Y. Fu, K. Okuro, J. Ding and T. Aida, Angew. Chem., Int. Ed., 2025, 64, e202416541 CrossRef CAS PubMed.
- B. Huang, H. Dong, K.-L. Wong, L.-D. Sun and C.-H. Yan, J. Phys. Chem. C, 2016, 120, 18858–18870 CrossRef CAS.
- Y. Shang, T. Chen, T. Ma, S. Hao, W. Lv, D. Jia and C. Yang, J. Rare Earths, 2022, 40, 687–695 CrossRef CAS.
- Z. Yu, Y. Deng, J. Ye, L. van Turnhout, T. Liu, A. Tew, R. Arul, S. Dowland, Y. Sun, X. Li, L. Dai, Y. Lu, C. Ducati, J. J. Baumberg, R. H. Friend, R. L. Z. Hoye and A. Rao, Nature, 2025, 647, 625–631 CrossRef CAS PubMed.
- J. Tan, P. Zhang, X. Song, C. Han, F. Wang, J. Zhang, C. Duan, Z. Zhang, S. Han, H. Xu and X. Liu, Nature, 2025, 647, 632–638 CrossRef CAS PubMed.
- C. Shi, L. Ye, Z.-X. Gong, J.-J. Ma, Q.-W. Wang, J.-Y. Jiang, M.-M. Hua, C.-F. Wang, H. Yu, Y. Zhang and H.-Y. Ye, J. Am. Chem. Soc., 2020, 142, 545–551 CrossRef CAS PubMed.
- C. Shi, J.-J. Ma, J.-Y. Jiang, M.-M. Hua, Q. Xu, H. Yu, Y. Zhang and H.-Y. Ye, J. Am. Chem. Soc., 2020, 142, 9634–9641 CAS.
- Q. Xu, L. Ye, R.-M. Liao, Z. An, C.-F. Wang, L.-P. Miao, C. Shi, H.-Y. Ye and Y. Zhang, Chem. – Eur. J., 2022, 28, e202103913 CrossRef CAS PubMed.
- C.-F. Wang, C. Shi, A. Zheng, Y. Wu, L. Ye, N. Wang, H.-Y. Ye, M.-G. Ju, P. Duan, J. Wang and Y. Zhang, Mater. Horiz., 2022, 9, 2450–2459 RSC.
- D. Zhao, L. Guo, Q. Li, C. Yue, B. Han, K. Liu and H. Li, Adv. Mater., 2024, 36, 2405164 CrossRef CAS PubMed.
- S. Zhou, Y. Wang, P. Hu, W. Zhong, H. Jia, J. Qiu and J. Fu, Laser Photonics Rev., 2023, 17, 2200531 CrossRef CAS.
- Y. Shao, H. Liu, M. Chen, Z. Song and Q. Liu, Laser Photonics Rev., 2025, 19, 2401467 CrossRef CAS.
- L. E. MacKenzie and R. Pal, Nat. Rev. Chem., 2021, 5, 109–124 CrossRef CAS PubMed.
- R. Arppe and T. J. Sørensen, Nat. Rev. Chem., 2017, 1, 0031 CrossRef CAS.
- L. Li, J. Zhou, J. Han, D. Liu, M. Qi, J. Xu, G. Yin and T. Chen, Nat. Commun., 2024, 15, 3846 CrossRef CAS PubMed.
- G. Zhao, Y. Kou, N. Song, X. Wei, X. Zhai, P. Feng, F. Wang, C.-H. Yan and Y. Tang, ACS Nano, 2023, 17, 7624–7635 CrossRef CAS PubMed.
- Y. Xu, R. Lin, S. Chen, Z. Wang, W. Zhang, C. Zhu, K. Wang, Y. S. Zhao and Z. Gao, Adv. Funct. Mater., 2025, e13640 Search PubMed.
- W. Feng, X. Liao, S. Lu, D. Han, Q. Guo, Y. Zhao, W. Tian and H. Yan, Chem. Sci., 2025, 16, 22517–22526 RSC.
- T. Nakai, K. Shima, M. Wang, S. Shoji, T. Nakanishi, K. Fushimi, Y. Hasegawa and Y. Kitagawa, Angew. Chem., Int. Ed., 2025, 64, e202513236 CrossRef CAS PubMed.
- X. Chen, J. Wan, W. Li, Y. Pan, Z. Xia, M. Lu, Z. Yuan and X. Xie, Angew. Chem., Int. Ed., 2025, e24478 Search PubMed.
- B. Yang, X. Yang, Y. Shi, X. Jin, T. Li, M. Liu and P. Duan, Angew. Chem., Int. Ed., 2025, 64, e202417223 CrossRef CAS PubMed.
- Y. Sun, X. Le, H. Shang, Y. Shen, Y. Wu, Q. Liu, P. Théato and T. Chen, Adv. Mater., 2024, 36, 2401589 CrossRef CAS PubMed.
- Y. Kou, Z. Yang, G. Tan, L. Liang, H. Cheng, P. Su and Y. Tang, Adv. Funct. Mater., 2025, e18377 Search PubMed.
- Y. Kou, Y. Guo, L. Liang, X. Li, Y. Wang, P. Su, C.-H. Yan and Y. Tang, Aggregate, 2025, 6, e701 CrossRef CAS.
- X. Li, Y. Xie, B. Song, H.-L. Zhang, H. Chen, H. Cai, W. Liu and Y. Tang, Angew. Chem., Int. Ed., 2017, 56, 2689–2693 CrossRef CAS PubMed.
- G. Yin, G. Huo, M. Qi, D. Liu, L. Li, J. Zhou, X. Le, Y. Wang and T. Chen, Adv. Funct. Mater., 2024, 34, 2310043 CrossRef CAS.
- H. Li, Q. Dong, J. Guo, E. Hu, Y. Li, R. Shan, L. Zhang, D. Zhao, H. Guo and G. Qian, Adv. Mater., 2026, 38, e21289 CrossRef CAS PubMed.
- Y.-F. Yuan, Y. Lin, M.-T. He, J.-J. Zhai, Y.-Q. Xu, J.-X. Liu, Z.-Y. Cao, P.-F. Zhang and M.-H. Li, Inorg. Chem., 2026, 65, 3560–3569 CrossRef CAS PubMed.
- M.-M. Liu, G.-C. Zheng, Y. Wei, D. Tian, Q.-B. Zheng, L. Huang and J. Xie, Rare Met., 2023, 42, 1018–1027 CrossRef CAS.
- J. Chen, M. Li, R. Sun, Y. Xie, J. R. Reimers and L. Sun, Adv. Funct. Mater., 2024, 34, 2315276 CrossRef CAS.
- E. L. Schmidt, Z. Ou, E. Ximendes, H. Cui, C. H. C. Keck, D. Jaque and G. Hong, Nat. Rev. Methods Primers, 2024, 4, 23 CrossRef CAS.
- T. Zhang, X. Qu, J. Shao and X. Dong, Chem. Soc. Rev., 2025, 54, 8406–8433 RSC.
- B. J. Tickner, G. J. Stasiuk, S. B. Duckett and G. Angelovski, Chem. Soc. Rev., 2020, 49, 6169–6185 RSC.
- X. Qiu, J. Xu, M. Cardoso Dos Santos and N. Hildebrandt, Acc. Chem. Res., 2022, 55, 551–564 CrossRef CAS PubMed.
- Y. Jiao, S. Yi, F. Liu, D. Zhen and Q. Peng, J. Lumin., 2025, 288, 121534 CrossRef CAS.
- J. Zhou, X. Hu, C. Liu, Y. Liu, N. Tian, F. Wu, W. Li, J. Lei and Z. Dai, Coord. Chem. Rev., 2025, 534, 216574 CrossRef CAS.
- X.-S. Ke, B.-Y. Yang, X. Cheng, S. L.-F. Chan and J.-L. Zhang, Chem. – Eur. J., 2014, 20, 4324–4333 CrossRef CAS PubMed.
- Y. Ning, X.-S. Ke, J.-Y. Hu, Y.-W. Liu, F. Ma, H.-L. Sun and J.-L. Zhang, Inorg. Chem., 2017, 56, 1897–1905 CrossRef CAS PubMed.
- J.-Y. Hu, Z.-Y. Wu, K. Chai, Z.-S. Yang, Y.-S. Meng, Y. Ning, J. Zhang and J.-L. Zhang, Inorg. Chem. Front., 2017, 4, 1539–1545 RSC.
- Y. Ning, G.-Q. Jin and J.-L. Zhang, Acc. Chem. Res., 2019, 52, 2620–2633 CrossRef CAS PubMed.
- Y. Ning, J. Tang, Y.-W. Liu, J. Jing, Y. Sun and J.-L. Zhang, Chem. Sci., 2018, 9, 3742–3753 RSC.
- Y. Ning, S. Chen, H. Chen, J.-X. Wang, S. He, Y.-W. Liu, Z. Cheng and J.-L. Zhang, Inorg. Chem. Front., 2019, 6, 1962–1967 RSC.
- Y. Ning, S. Cheng, J.-X. Wang, Y.-W. Liu, W. Feng, F. Li and J.-L. Zhang, Chem. Sci., 2019, 10, 4227–4235 RSC.
- G.-Q. Jin, Y. Ning, J.-X. Geng, Z.-F. Jiang, Y. Wang and J.-L. Zhang, Inorg. Chem. Front., 2020, 7, 289–299 RSC.
- X.-X. Peng, X.-F. Zhu and J.-L. Zhang, J. Inorg. Biochem., 2020, 209, 111118 CrossRef CAS PubMed.
- X.-F. Zhu, G.-Q. Jin and J.-L. Zhang, Sci. Sin.: Chim., 2020, 50, 1575–1584 Search PubMed.
- B. Wu, L. Zhang, K. Yan, M. Mei, W. Wu, Z. He, S. Wang and F. Zhang, J. Am. Chem. Soc., 2025, 147, 44185–44190 CrossRef CAS PubMed.
- A. Kovalenko, S. V. Eliseeva, G. Collet, S. El Abdellaoui, S. Natkunarajah, S. Lerondel, L. Guénée, C. Besnard and S. Petoud, J. Am. Chem. Soc., 2024, 146, 12913–12918 CrossRef CAS PubMed.
- T. Wang, S. Wang, Z. Liu, Z. He, P. Yu, M. Zhao, H. Zhang, L. Lu, Z. Wang, Z. Wang, W. Zhang, Y. Fan, C. Sun, D. Zhao, W. Liu, J.-C. G. Bünzli and F. Zhang, Nat. Mater., 2021, 20, 1571–1578 CrossRef CAS PubMed.
- L. Zhang, R. Cheng, Z. He, M. Mei, B. Wu, W. Tan, B. Yan, S. Wang and F. Zhang, Nat. Photonics, 2025, 19, 1209–1218 CrossRef CAS.
- X.-S. Ke, Y. Ning, J. Tang, J.-Y. Hu, H.-Y. Yin, G.-X. Wang, Z.-S. Yang, J. Jie, K. Liu, Z.-S. Meng, Z. Zhang, H. Su, C. Shu and J.-L. Zhang, Chem. – Eur. J., 2016, 22, 9676–9686 CrossRef CAS PubMed.
- Z.-S. Yang, Y. Ning, H.-Y. Yin and J.-L. Zhang, Inorg. Chem. Front., 2018, 5, 2291–2299 RSC.
- Y. Yao, H.-Y. Yin, Y. Ning, J. Wang, Y.-S. Meng, X. Huang, W. Zhang, L. Kang and J.-L. Zhang, Inorg. Chem., 2019, 58, 1806–1814 CrossRef CAS PubMed.
- Z.-S. Yang, X.-F. Zhu and J.-L. Zhang, ACS Appl. Mater. Interfaces, 2025, 17, 35221–35229 CrossRef CAS PubMed.
- X.-S. Ke, J. Tang, Z.-S. Yang and J.-L. Zhang, J. Porphyrins phthalocyanines, 2014, 18, 950–959 CrossRef CAS.
- Z. Wang, L. He, B. Liu, L.-P. Zhou, L.-X. Cai, S.-J. Hu, X.-Z. Li, Z. Li, T. Chen, X. Li and Q.-F. Sun, J. Am. Chem. Soc., 2020, 142, 16409–16419 CrossRef CAS PubMed.
- M. A. Joaqui-Joaqui, G. G. Sands, D. Śmiłowicz, M. J. Gork, E. Aluicio-Sarduy, T. E. Barnhart, J. W. Engle and E. Boros, J. Am. Chem. Soc., 2025, 147, 45303–45314 CrossRef CAS PubMed.
- Z.-S. Yang, Y. Yao, A. C. Sedgwick, C. Li, Y. Xia, Y. Wang, L. Kang, H. Su, B.-W. Wang, S. Gao, J. L. Sessler and J.-L. Zhang, Chem. Sci., 2020, 11, 8204–8213 RSC.
- M. Zhu, H. Zhang, G. Ran, D. N. Mangel, Y. Yao, R. Zhang, J. Tan, W. Zhang, J. Song, J. L. Sessler and J.-L. Zhang, J. Am. Chem. Soc., 2021, 143, 7541–7552 CrossRef CAS PubMed.
- R. Lengacher, K. E. Martin, D. Śmiłowicz, H. Esseln, P. Lotlikar, A. Grichine, O. Maury and E. Boros, J. Am. Chem. Soc., 2023, 145, 24358–24366 CrossRef CAS PubMed.
- Y. An, D. Xu, P. He, Z. Wang, Y. Li, J. Ming, R. Liu, J. Li, Z. Lu and G. Liu, J. Am. Chem. Soc., 2025, 147, 11964–11974 CrossRef CAS PubMed.
- J.-X. Zhang, W.-L. Chan, C. Xie, Y. Zhou, H.-F. Chau, P. Maity, G. T. Harrison, A. Amassian, O. F. Mohammed, P. A. Tanner, W.-K. Wong and K.-L. Wong, Light: Sci. Appl., 2019, 8, 46 CrossRef PubMed.
- P.-L. Lam, Y. Wu, L. Yao, H.-Y. Kai, H.-F. Chau, Q. Zhang, W. Zhou, J.-C. G. Bünzli and K.-L. Wong, Angew. Chem., Int. Ed., 2025, 64, e202519204 CrossRef CAS PubMed.
- G. Rizzo and B. Marelli, J. Am. Chem. Soc., 2025, 147, 24857–24869 CrossRef CAS PubMed.
- S.-Y. Yang, L. Zhang, F.-C. Kong, Y. Chen, W.-J. Li, F. Wang, C. Liu, X. He, X. Xiao, J. Wang, J. Sun, P. C. Y. Chow, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem, 2025, 11, 102534 CAS.
- P. Singh, G. Dosovitskiy and Y. Bekenstein, ACS Nano, 2024, 18, 14029–14049 CrossRef CAS PubMed.
- L. Yang, Z. Li, L. He, J. Sun, J. Wang, Y. Wang, M. Li, Z. Zhu, X. Dai, S.-X. Hu, F. Zhai, Q. Yang, Y. Tao, Z. Chai, S. Wang and Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202306465 CrossRef CAS PubMed.
- X. Zhang, H. Qiu, W. Luo, K. Huang, Y. Chen, J. Zhang, B. Wang, D. Peng, Y. Wang and K. Zheng, Adv. Sci., 2023, 10, 2207004 CrossRef CAS PubMed.
- C. Zhao, Y. Wang, S. Bao, Y. Zang, X. Liu and W. Huang, Adv. Mater., 2025, 37, 2500925 CrossRef CAS PubMed.
- Y.-S. Hu, R.-Y. Fang, Y.-H. Liu, M.-H. Fu, S.-H. Wang, J.-W. Yuan, Q. Yang, Q.-C. Peng, Z.-Y. Wang, X.-Q. Li, K. Li and S.-Q. Zang, Aggregate, 2025, 6, e70140 CrossRef CAS.
- L. Li, J. Chen, G. He and H. Guo, Laser Photonics Rev., 2025, e02368 Search PubMed.
- Y. Zhang, T. Du, H. Duan, B. Chen, J. Zhang, Q. Xiao, G. Chen, D. Guo, H. Liao, S. Zhou and K. Zheng, Angew. Chem., Int. Ed., 2025, 64, e202423155 CrossRef CAS PubMed.
- J. Lu, D. Zhou, X. Hao, H. Lu, Z. Zhang, Y. Jia, S. Mei, Y. Huang, S. Tang, Z. Peng, L. Chen, M. Yuan, L. Chen, S. Wang, M. Feng, Y. Wang and S. Wang, Adv. Sci., 2025, e21054 Search PubMed.
- C. Wang, H. Li, J. Xue, Y. Chen, L. Wang, X. Xu, S. Wang and R. Li, Chem. Eng. J., 2025, 526, 171125 CrossRef CAS.
- H. Li, Y. Cai, Q. Dong, L. Zhang, E. Hu, H. Guo and G. Qian, Adv. Opt. Mater., 2025, 13, e01708 CrossRef CAS.
- J.-X. Wang, I. Nadinov, S. Thomas, O. Shekhah, X. Zhu, T. He, T. He, X. Yuan, S. Wang, H. Jiang, O. M. Bakr, H. N. Alshareef, M. Eddaoudi and O. F. Mohammed, Chem, 2025, 11, 102646 CAS.
- X. Liu, R. Li, X. Xu, Y. Jiang, W. Zhu, Y. Yao, F. Li, X. Tao, S. Liu, W. Huang and Q. Zhao, Adv. Mater., 2023, 35, 2206741 CrossRef CAS PubMed.
- J. Lu, Y. Fan, B. Zhou, Z. Ye, S. Mei, D. Zhou, L. Chen, L. Chen, H. Liu, H. Zhu, X. Dai, Y. Wang, J. Diwu and S. Wang, ACS Mater. Lett., 2024, 6, 3951–3957 CrossRef CAS.
- P. Ran, L. Yang, T. Jiang, X. Xu, J. Hui, Y. Su, C. Kuang, X. Liu and Y. Yang, Adv. Mater., 2022, 34, 2205458 CrossRef CAS PubMed.
- T.-C. Wang, Z.-L. He, J.-B. Luo, Q.-P. Peng, J.-H. Wei, K.-L. Chen, J.-H. Chen, X.-X. Guo and D.-B. Kuang, Angew. Chem., Int. Ed., 2025, 64, e202504658 CrossRef CAS PubMed.
- J.-X. Wang, L. Gutiérrez-Arzaluz, X. Wang, M. Almalki, J. Yin, J. Czaban-Jóźwiak, O. Shekhah, Y. Zhang, O. M. Bakr, M. Eddaoudi and O. F. Mohammed, Matter, 2022, 5, 253–265 CrossRef CAS.
- S. Wu, Y. Lin, J. Liu, W. Shi, G. Yang and P. Cheng, Adv. Funct. Mater., 2018, 28, 1707169 CrossRef.
- X. Zhu, Y. Wang, T. He, S. Thomas, H. Jiang, O. Shekhah, J.-X. Wang, T. K. Ng, H. N. Alshareef, O. M. Bakr, B. S. Ooi, M. Eddaoudi and O. F. Mohammed, J. Am. Chem. Soc., 2025, 147, 6805–6812 CrossRef CAS PubMed.
- D. Errulat, R. Marin, D. A. Gálico, K. L. M. Harriman, A. Pialat, B. Gabidullin, F. Iikawa, O. D. D. Couto, Jr., J. O. Moilanen, E. Hemmer, F. A. Sigoli and M. Murugesu, ACS Cent. Sci., 2019, 5, 1187–1198 CrossRef CAS PubMed.
- N. Wang, Z.-J. Xu, H.-F. Ni, W. Luo, H.-K. Li, M.-L. Ren, C. Shi, H.-Y. Ye, X.-B. Fu, Y. Zhang and L.-P. Miao, Angew. Chem., Int. Ed., 2024, 63, e202409796 CrossRef CAS PubMed.
- J. J. Zakrzewski, R. Jankowski, M. Romanowska, J. Wang, D. Pinkowicz, B. Sieklucka, S.-I. Ohkoshi and S. Chorazy, Angew. Chem., Int. Ed., 2025, 64, e202424651 CrossRef CAS PubMed.
- Y. Tang, H. Wu, W. Cao, Y. Cui and G. Qian, Adv. Opt. Mater., 2021, 9, 2001817 CrossRef CAS.
- R. Shi, L. Yu, Y. Tian, X. Wang, Z. Sun, B. Qi and F. Luo, Mater. Chem. Phys., 2022, 280, 125806 CrossRef CAS.
- D. Zhang, Q. Zhao, J. Zang, Y.-J. Lu, L. Dong and C.-X. Shan, Carbon, 2018, 127, 170–176 CrossRef CAS.
- K. Yao, Y. Shen, J. Li, S. Song and W. Liu, Small, 2026, 22, e13586 CrossRef CAS PubMed.
- Y. Qiao and E. J. Schelter, Acc. Chem. Res., 2018, 51, 2926–2936 CrossRef CAS PubMed.
- Y. Qiao, T. Cheisson, B. C. Manor, P. J. Carroll and E. J. Schelter, Chem. Commun., 2019, 55, 4067–4070 RSC.
- P. Parnicka, W. Lisowski, T. Klimczuk, J. Łuczak, A. Żak and A. Zaleska-Medynska, Appl. Catal., B, 2021, 291, 120056 CrossRef CAS.
- S. Karmakar, S. Barman, F. A. Rahimi, S. Biswas, S. Nath and T. K. Maji, Energy Environ. Sci., 2023, 16, 2187–2198 RSC.
- S. Xie, C. Wang, Y. Xi, Y. Hu, J. Wei, H. Lin, L. Xu, P. Su and Y. Tang, Angew. Chem., Int. Ed., 2026, 65, e22878 CrossRef CAS PubMed.
- X. Wang, Y.-L. Yang, P. Wang, L. Li, R.-Q. Fan, W.-W. Cao, B. Yang, H. Wang and J.-Y. Liu, Dalton Trans., 2012, 41, 10619–10625 RSC.
- X. Feng, R. Chen, Z.-A. Nan, X. Lv, R. Meng, J. Cao and Y. Tang, Adv. Sci., 2019, 6, 1802040 CrossRef PubMed.
- J. Zhang, J. Li, Y. Yang, C. Yang, Y. Dong, K. Lin, D. Xia and R. Fan, ACS Appl. Energy Mater., 2022, 5, 13318–13326 CrossRef CAS.
- K. T. Butler, D. W. Davies, H. Cartwright, O. Isayev and A. Walsh, Nature, 2018, 559, 547–555 CrossRef CAS PubMed.
- W. Ma, Z. Liu, Z. A. Kudyshev, A. Boltasseva, W. Cai and Y. Liu, Nat. Photonics, 2021, 15, 77–90 CrossRef CAS.
- Y. Xu, X. Zhang, Y. Fu and Y. Liu, Photonics Res., 2021, 9, B135–B152 CrossRef.
- F. Yang, Y. Wang, X. Jiang, B. Lin and R. Lv, ACS Comb. Sci., 2020, 22, 285–296 CrossRef CAS PubMed.
- Y. Song, M. Lu, Y. Xie, G. Sun, J. Chen, H. Zhang, X. Liu, F. Zhang and L. Sun, Adv. Funct. Mater., 2022, 32, 2206802 CrossRef CAS.
- W. Ma, Y. Xu, B. Xiong, L. Deng, R.-W. Peng, M. Wang and Y. Liu, Adv. Mater., 2022, 34, 2110022 CrossRef CAS PubMed.
- S. Li, M. Amachraa, C. Chen, L. Wang, Z. Wang, S. P. Ong and R.-J. Xie, Matter, 2022, 5, 1924–1936 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |