
Multi-level structural modulation enables fast lithium-ion transport in inorganic solid-state batteries
Tianpeng
Huang
abcd,
Yue
Zheng
acd,
Deye
Sun
acd,
Jun
Ma
 *acd,
Pengxian
Han
*acd and
Guanglei
Cui
*abcd
*acd,
Pengxian
Han
*acd and
Guanglei
Cui
*abcd
aQingdao Industrial Energy Storage Research Institute, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao 266101, P. R. China. E-mail: majun@qibebt.ac.cn; hanpx@qibebt.ac.cn; cuigl@qibebt.ac.cn
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, P. R. China
cShandong Energy Institute, Qingdao 266101, P. R. China
dQingdao New Energy Shandong Laboratory, Qingdao 266101, P. R. China
First published on 28th November 2025
Abstract
Solid-state lithium metal batteries (SSLMBs) are considered ideal candidates for the next-generation core technologies for development of clean energy storage and conversion systems owing to their inherent high energy density and exceptional safety. Nevertheless, the practical energy density, power characteristics, and cycling stability of SSLMBs are usually limited by sluggish charge transfer kinetics within and across solid-state components, including electrode, electrolyte, binder, and conductive additive materials. Therefore, understanding the intrinsic link between structure–charge transport–performance and improving charge transport kinetics in a heterogeneous solid system through structural modulation has become the key to comprehensively improving the electrochemical performance of SSLMBs. Herein, a unique perspective is proposed to optimize the short-range and long-range charge transport processes in SSLMBs through multi-level structural modulation at the electrode, solid electrolyte, and cell levels. We firstly summarize and evaluate the research progress in multi-level structural modulation. Then, the vital factors impacting structural regulation and regulation principles at the corresponding level are analyzed in depth. Furthermore, the extent of enhancement and limitations of various structural modulation approaches employed for charge transport are evaluated and compared. At the end, perspectives and suggestions were provided on principles for multi-level structural modulation toward fast charge transport kinetics in inorganic SSLMBs. This review will offer broadly applicable principles for the development of next-generation high-performance inorganic SSLMBs.
 Tianpeng Huang | Tianpeng Huang received his BS degree in New Energy Materials and Devices from Xiangtan University (XTU) in 2021. He is currently a PhD candidate majoring in Materials Science under the supervision of Prof. Guanglei Cui at the Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences (QIBEBT-CAS). Currently, his research interests lie in failure mechanisms in solid-state lithium batteries. |
 Jun Ma | Prof. Jun Ma received her PhD degree in Condensed Matter Physics from the Institute of Physics, Chinese Academy of Sciences (IOP-CAS) in 2014. Since 2014, she has been working at the Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences (QIBEBT-CAS). Her recent research interests include high-energy-density cathode materials, full solid-state batteries, energy storage mechanisms, and interface issues in batteries. |
 Pengxian Han | Prof. Pengxian Han received his Master's degree from Tianjin University in 2007. He then worked at BYD Co., Ltd. In July 2009, he joined the Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences (QIBEBT-CAS). His recent research interest is key materials and device design of high-energy-density solid-state lithium batteries. |
 Guanglei Cui | Prof. Guanglei Cui obtained his PhD degree from the Institute of Chemistry, Chinese Academy of Sciences (IC-CAS). He then did postdoctoral research at Max-Planck-Institute for Polymer Research and Max-Planck-Institute for Solid State Research. He is currently the leader of Solid Energy System Technology Center, the director of Energy Applied Technology Division of Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences (QIBEBT-CAS). His research topics include sustainable and highly efficient energy-storage materials, all-solid-state batteries, and novel energy devices. He has published more than 500 articles in international authoritative journals, which have been cited more than 40 |
1. Introduction
In order to accommodate the rapid improvement of energy-related technologies and devices, the energy density and safety of secondary batteries need to be urgently improved. Conventional lithium-ion batteries are based on oxide intercalation cathodes and carbon-based anodes, which cannot meet the aggressively challenging plan to reach an energy density of 500 Wh kg−1 by 2030.1 Solid-state lithium metal batteries (SSLMBs) exhibit great potential because lithium (Li) metal possesses the lowest electrochemical potential (−3.04 V versus a standard hydrogen electrode) and high specific theoretical capacity (3860 mAh g−1).2,3 Besides, the solid electrolytes (SEs) that replace liquid electrolytes are nonflammable with excellent high-temperature stability,4,5 thus avoiding the safety risk related to the liquid electrolytes fundamentally. Given this, SSLMBs are considered promising candidates for next-generation energy storage systems.However, the energy density, power characteristics, and cycling stability of most SSLMBs have been limited in practical applications due to the poor charge transport kinetics within and between battery components.6–12 SSLMBs typically feature a planar-type sandwich structure consisting of a high-capacity cathode, a solid electrolyte layer, and an appropriate lithium metal anode. The back-and-forth migration of Li+ between the cathode and the anode through SEs during charge and discharge is the premise of the regular operation of SSLMBs. This process involves short-range transport of charges within each component (composite cathodes, anodes, and SEs) and long-range transport between these components, as shown in Fig. 1. The fundamental mechanism of charge transport in SSLMBs is affected by the structure of individual components and the overall battery structure, which can be modulated by the multi-scale structure design and optimization.
 | ||
| Fig. 1 Schematic illustration of multi-level charge transport in inorganic SSLMBs. | ||
The majority of conventional cathode active materials (CAMs) exhibit sluggish Li+ diffusion coefficients (10−10–10−15 cm2 s−1) and insufficient electronic conductivities (10−4–10−10 S cm−1).13 Composite cathodes consist of active components and inactive substances, such as SEs, electronically conductive agents, and binders, commonly employed in SSLMBs to improve charge transport kinetics. SEs serve as the primary medium for Li+ conduction in SSLMBs and predominantly determine the power density and rate performance. The Li+ transport kinetics within SEs are determined by their inherent physicochemical properties, which are influenced by structures, such as crystal structures, pores, grain boundaries, and secondary phases.14–18 Composite solid electrolytes (CSEs) exhibit the highest application potential among the various extensively studied candidates for SEs.19–21 However, the complicated three-dimensional (3D) microstructures in composite cathodes or SEs formed by the random dispersion and disordered arrangement of different particles display intricate ionic and/or electronic percolation pathways. Furthermore, the electro-mechanical coupling effects during cycling may trigger structural changes in electrodes and SEs,22–24 such as deformation,25,26 crack formation,27–35 and dislocation introduction,36 affecting ionic transport. Therefore, it is essential to reorganize the microstructure of composite cathodes/SEs to create highly efficient and stable networks for carrier transfer.37–44
Besides the charge transport within electrodes and SEs, the charge transfer at the heterogeneous interface is also crucial for the performance of planar-type sandwich structure SSLMBs. The imperfect two-dimensional (2D) contact between rigid electrode layers and SE layers results in an insufficient charge transport channel at the interface.45–50 Realizing the fast and uniform Li+ transport throughout the 3D space at the battery structure level of SSLMBs is a crucial challenge.51–53 Besides, these interfaces involve numerous physical and chemical processes, including contact failure,31,54–57 (electro)chemical reaction,58–61 and space charge layer (SCL) formation,62–69 which increase the interfacial transport resistance and impede the Li+ transport at the interface, further leading to fast deterioration of rate performance and cyclability of SSLMBs. Recently, various structural optimization strategies have been suggested to tackle the interfacial issues, including buffer layer construction,45,70–75 electrolyte permeation,76–78 and multilayer SE construction.79–84 Nevertheless, some shortcomings are still found when the battery performance and the industrialization prospects of the structure preparation process are considered comprehensively.
To comprehensively enhance the electrochemical performance of SSLMBs, it is essential to reveal the impact of the structure on charge transport properties and facilitate efficient charge transport within the composite cathodes and SEs and across the electrode/electrolyte interface by appropriate structural modulation and designs. Previous reviews have summarized various failure mechanisms in solid-state batteries to explain the deterioration of electrochemical performance under real operating conditions and proposed corresponding solution strategies.6,7,61,85,86 However, there is currently a lack of in-depth discussion on the more fundamental charge transport processes. In addition, most modification strategies focus on a specific battery component or issue, which fails to integrate the various levels.18,20,38,87–91 This lack of understanding of the root scientific issues severely limits our ability to break through the bottleneck of high-energy secondary battery development in engineering technology. Herein, a unique perspective is proposed to optimize the short-range and long-range charge transport processes in SSLMBs through multi-scale structural modulation at the electrode, solid electrolyte, and cell levels. We first summarize and evaluate the research progress in the structural modulation of composite cathodes, SEs, and batteries. Then, we analyze the vital factors that impact structural regulation and regulation principles across various research scales. Furthermore, the extent of enhancement and limitations of various structural modulation approaches employed for charge transport are evaluated and compared. In the end, perspectives and suggestions were provided on structural modulation toward fast charge transport kinetics in inorganic SSLMBs.
2. Structural modulation of composite cathodes
The composite cathodes used in SSLMBs often exhibit intricate 3D microstructures as a result of the randomly dispersed and disordered arrangement of packed particles. Percolation theory states that the content and size of particles have an impact on the connectivity, tortuosity, dispersion, and contact area between particles, resulting in the alteration of charge transport channels.92–95 Hence, it is imperative to prioritize the examination of the interplay between these parameters and the microstructures, as well as the potential correlation among them. In order to achieve high-performance SSLMBs in terms of energy/power densities, rate capacity, and cyclability, it is crucial to manipulate the microstructure of cathodes through composition engineering, size engineering, and gradient design.2.1. Composition engineering
A composite cathode is generally composed of active materials and a combination of inactive components, including SEs, electronic conductive additives, and binders. The theoretical capacity of CAMs dictates the maximum achievable energy density of SSLMBs. Meanwhile, the incorporation of inactive materials plays a critical role in maintaining smooth operation of SSLMBs under practical operation. Charge transport in this multiphase composite system is governed by its heterogeneous microstructure and the effective transport properties peff can be determined through the volume averaging method proposed by Vadakkepatt et al.96 and are expressed as eqn (1):97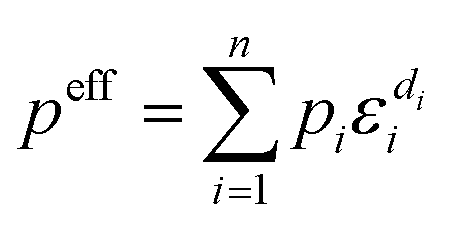 | (1) |
Vijayaraghavan et al.98 proposed a relation between the global structural properties, i.e., the porosity, the tortuosity, the electrolyte diffusion coefficient Del, and the total diffusion coefficient Deff, which has been widely used for conventional lithium-ion battery electrodes:
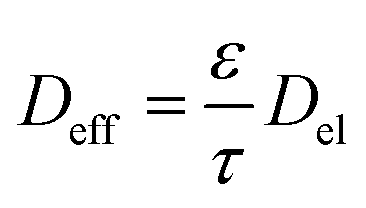 | (2) |
Based on eqn (1), the effective ionic conductivity 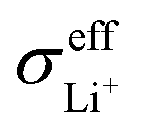 of the composite cathode can be expressed as eqn (3):
of the composite cathode can be expressed as eqn (3):
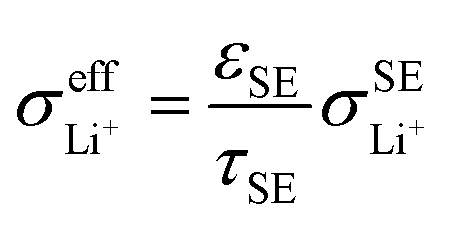 | (3) |
 represent the volume fraction, tortuosity factor, and ionic conductivity of SE, respectively. This equation emphasizes the role of the percolating electrolyte network in facilitating Li+ transport throughout the composite cathode, while the contribution of the CAM, which typically exhibits a much lower ionic conductivity than SEs, as shown in Table 1, is justifiably neglected.
represent the volume fraction, tortuosity factor, and ionic conductivity of SE, respectively. This equation emphasizes the role of the percolating electrolyte network in facilitating Li+ transport throughout the composite cathode, while the contribution of the CAM, which typically exhibits a much lower ionic conductivity than SEs, as shown in Table 1, is justifiably neglected.
| Materials | Ionic conductivity (S cm−1) | Electronic conductivity (S cm−1) | Ref. |
|---|---|---|---|
| Cathode material | |||
| LiFePO4 | 10−10–10−15 | 10−10–10−9 | 99 |
| LiCoO2 | 2.3 × 10−7 | 10−4–10−3 | 100 |
| LiNixCoyMnzO2 | 1.7–6.3 × 10−3 | 2.2 × 10−6–4.1 × 10−3 | 100 |
| LiMn2O4 | 5.25 × 10−7 | 10−6–10−5 | 101 and 102 |
| LiNi0.5Mn1.5O4 | 2.7 × 10−4 | 10−7–10−5 | 103 and 104 |
| Li1.2Ni0.13Co0.13Mn0.54O2 | 2.65 × 10−10 | 3.44 × 10−9 | 105 |
| Li2S | 10−5 | 10−13 | 106 and 107 |
| MoS6 | 2.5 × 10−7 | 6.49 × 10−9 | 108 |
| Li3TiCl6 | 1.04 × 10−3 | 3.32 × 10−7 | 109 |
| Li2Ru0.8S0.2O3.2 | 6.3 × 10−7 | 2.0 × 10−2 | 100 |
| Li1.75Ti2(Ge0.25P0.75S3.8Se0.2)3 | 2.0 × 10−4 | 2.25 × 10−1 | 111 |
| Li1.3Fe1.2Cl4 | 2.28 × 10−4 | 6.98 × 10−5 | 112 |
| Solid electrolyte | |||
| LiPON | 2.5 × 10−6 | 1.2 × 10−14 | 113 |
| β-Li3PS4 | 1.6 × 10−4 | 2.2 × 10−9 | 114 and 115 |
| Li2S–P2S5 | 1.7 × 10−2 | 1.3–2.2 × 10−9 | 115 and 116 |
| Li6PS5Cl | 2.4 × 10−3–6 × 10−5 | 3.4 × 10−8–6.4 × 10−9 | 117 and 118 |
| Li6PS5Br | 1.9 × 10−4–3.9 × 10−3 | 6.4 × 10−9–5.2 × 10−8 | 77 and 118 |
| Li6PS5I | 1.9–7.9 × 10−4 | 2.6 × 10−8 | 77 and 119 |
| Li10GeP2S12 | 1.2 × 10−2 | 5.7 × 10−9–1 × 10−8 | 120 and 121 |
| Li14SiP6 | 1.1 × 10−3 | 1.6 × 10−7 | 122 |
| Li1.3Al0.3Ti1.7(PO4)3 | 1.2 × 10−4 | 10−11 | 123 |
| Li1.5Al0.5Ge1.5(PO4)3 | 3.5 × 10−6 | 10−9 | 124 |
| Li7La3Zr2O12 (LLZO) | 10−6–10−4 | 5.5 × 10−8 | 115 and 125 |
| Al-doped LLZO | 3.4 × 10−4 | 2.7 × 10−9–1.7 × 10−7 | 126 |
| Ta-doped LLZO | 2.5 × 10−3 | 1 × 10−9–5 × 10−7 | 127 and 128 |
| Ga-doped LLZO | 2 × 10−3 | 1 × 10−9–7.1 × 10−8 | 129 and 130 |
| Li0.33La0.55TaO3 | 6.13 × 10−5 | 2.1 × 10−7 | 131 |
| Li3InCl6 | 8.49 × 10−4 | 7.3 × 10−9 | 132 |
| Li3YCl6 | 5.1 × 10−4 | 2.8 × 10−9 | 133 |
| Li3YBr6 | 1.7 × 10−3 | 1 × 10−9 | 133 |
| Li2Sc2/3Cl4 | 1.11 × 10−3 | 4.2 × 10−9 | 134 |
| Li2In1/3Sc1/3Cl4 | 1.99 × 10−3 | 4.7 × 10−10 | 134 |
Assuming that electrons travel primarily through the CAM or conductive agents, the effective electronic conductivity 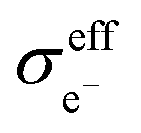 of the composite cathode can be expressed as eqn (4):
of the composite cathode can be expressed as eqn (4):
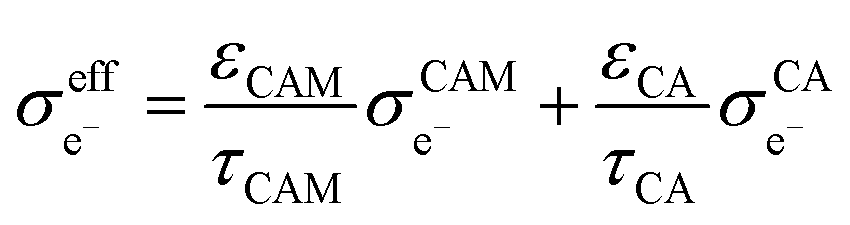 | (4) |
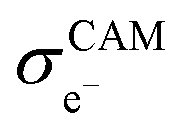 represent the volume fraction, tortuosity factor, and electronic conductivity of the CAM, respectively. εCA, τCA, and
represent the volume fraction, tortuosity factor, and electronic conductivity of the CAM, respectively. εCA, τCA, and 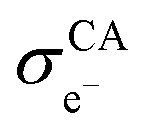 represent the volume fraction, tortuosity factor, and ionic conductivity of the conductive additive, respectively. εCAM and εCA are expressed as eqn (5) and (6), respectively:
represent the volume fraction, tortuosity factor, and ionic conductivity of the conductive additive, respectively. εCAM and εCA are expressed as eqn (5) and (6), respectively: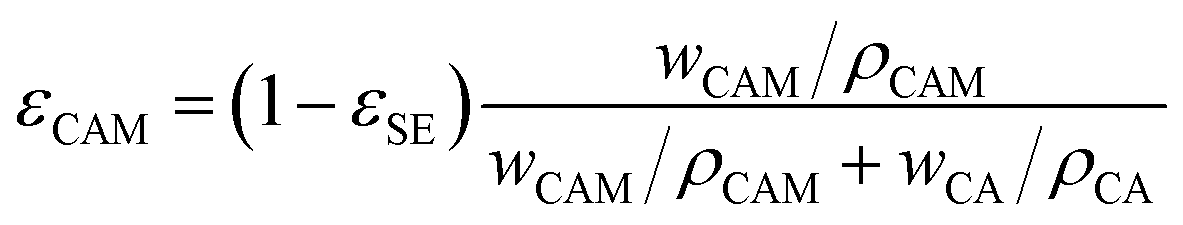 | (5) |
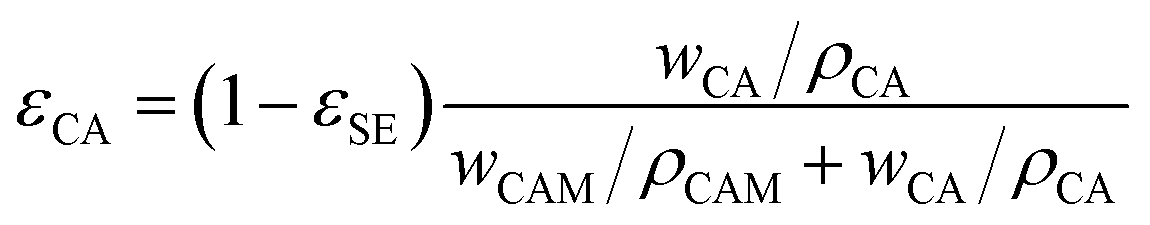 | (6) |
Based on eqn (3) and (4), the key factors governing the ionic and electronic conductivity of the composite cathode can be identified. The ionic conductivity of the SE is typically greater than 10−3 S cm−1, and its volume fraction in the composite cathode generally remains below 50%. In contrast, the tortuosity factor varies significantly with the cathode microstructure. Therefore, assuming the intrinsic ionic conductivity of the SE is content, any factor that modifies the tortuosity will strongly influence the effective ionic conductivity of the composite cathode. Similarly, the effective electronic conductivity of the composite cathode is primarily governed by the intrinsic conductivity of the conductive additives and the tortuosity of the electron transport pathways. Thus, the component composition and microstructure of the composite cathode play a decisive role in forming efficient ionic and electronic conduction networks. Key parameters can be optimized by adjusting the CAM/SE or CAM/conductive agent ratio, tailoring the morphology and size of constituent materials, incorporating selective conductive additives and binders, and developing all-electrochemically-active (AEA) cathode materials. These strategies directly enhance the overall electrode conductivity.
It is crucial to note that these conductivity parameters characterize the composite cathode in its initial, fully lithiated state. While the conductivity of the SE and conductive agents usually remains constant, the electronic and ionic conductivity of the CAM varies during (de)lithiation, leading to dynamic changes in the effective electronic conductivity throughout cycling. This creates a complex, state-dependent conductive network, leading to varied current distribution and polarization. Consequently, a cathode design optimized for its initial state may become highly inefficient at intermediate states of charge, potentially leading to localized over-discharge or underutilization of the active material, accelerated degradation, and capacity fade. Therefore, it is essential to model this evolution by machine learning to accurately predict cell performance, especially under high-rate conditions.
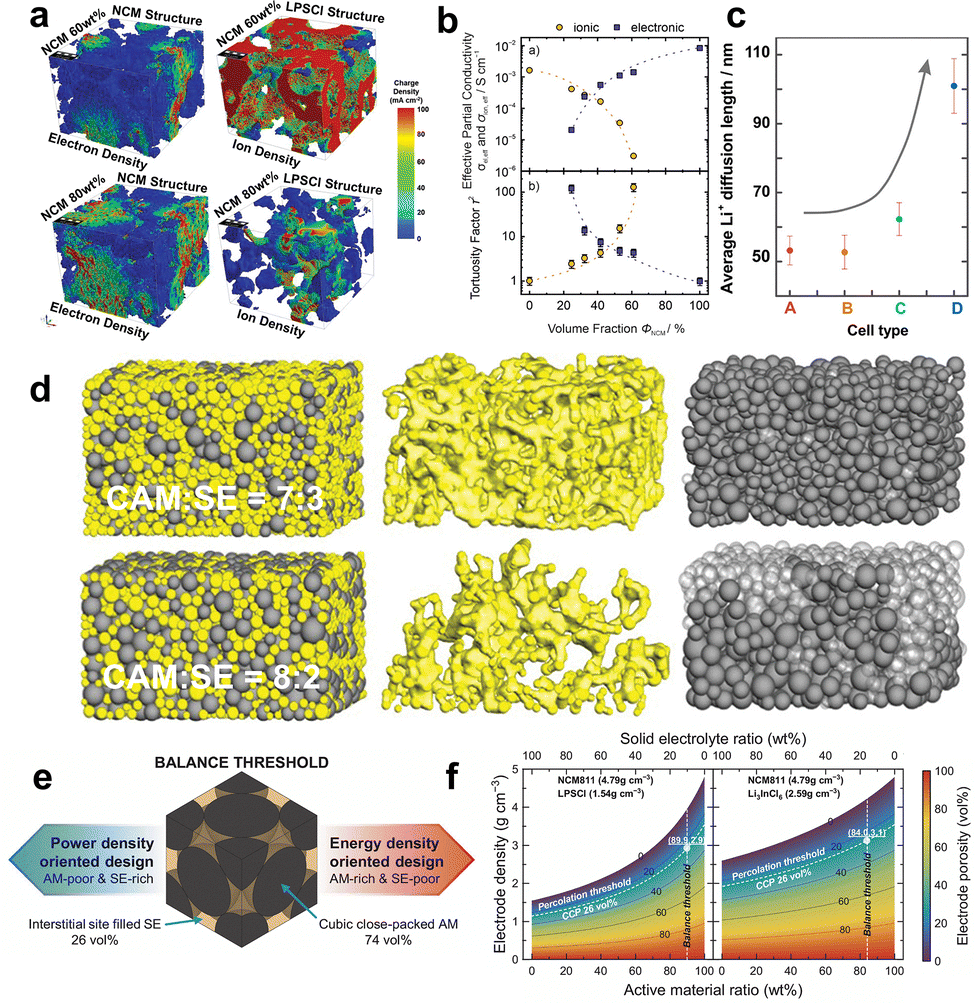 | ||
Fig. 2 (a) Simulation results of electron density and ion density in NCM 60 wt% and NCM 80 wt% composite cathodes. Reproduced with permission.135 Copyright 2020, Wiley-VCH. (b) Effective partial ionic and electronic conductivity and calculated tortuosity factors of composite cathodes with different volume fractions of the CAM. Reproduced with permission.92 Copyright 2021, IOP Publishing. (c) Average Li+ diffusion length calculated for cells with different CAM/SE fractions. (A) CAM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SE = 5:5, (B) CAM:SE = 6:4, (C) CAM:SE = 7:3, (D) CAM:SE = 8:2. Reproduced with permission.144 Copyright 2017, American Chemical Society. (d) Visualization of models for composite cathodes with different mass fractions of CAM. CAM is shown in gray and SE is shown in yellow. The composite microstructures, isolated SE percolation networks, and isolated CAM particles are shown in the left-hand, middle, and right-hand columns, respectively. The solid-gray and transparent-gray colors in the right-hand column represent the active and inactive CAM, respectively. Reproduced with permission.143 Copyright 2019, Wiley-VCH. (e) Schematic diagram showing the balance threshold between the CAM and the SE for desired characteristics (power density oriented vs. energy density oriented). (f) Electrode porosity related to its density as a function of the weight ratio of CAM and SE for various types of materials with different true densities. The white dotted lines represent the percolation threshold and balance threshold, respectively, according to the material true densities. Reproduced with permission.147 Copyright 2024, Springer Nature. :SE = 5:5, (B) CAM:SE = 6:4, (C) CAM:SE = 7:3, (D) CAM:SE = 8:2. Reproduced with permission.144 Copyright 2017, American Chemical Society. (d) Visualization of models for composite cathodes with different mass fractions of CAM. CAM is shown in gray and SE is shown in yellow. The composite microstructures, isolated SE percolation networks, and isolated CAM particles are shown in the left-hand, middle, and right-hand columns, respectively. The solid-gray and transparent-gray colors in the right-hand column represent the active and inactive CAM, respectively. Reproduced with permission.143 Copyright 2019, Wiley-VCH. (e) Schematic diagram showing the balance threshold between the CAM and the SE for desired characteristics (power density oriented vs. energy density oriented). (f) Electrode porosity related to its density as a function of the weight ratio of CAM and SE for various types of materials with different true densities. The white dotted lines represent the percolation threshold and balance threshold, respectively, according to the material true densities. Reproduced with permission.147 Copyright 2024, Springer Nature. | ||
Practically, a significant fraction (30–50 wt%) of SE is necessary for composite cathodes to ensure adequate ionic diffusion,138,139 which consequently leads to a decrease in energy density. Considering that the cathode loading of liquid cells is typically greater than 50 vol%,140 maximizing CAM loading over 50 vol% is a prerequisite to increasing the energy density of SSLMBs for commercial requirements. Sun et al.6 estimated that increasing the CAM content from 70% to 90% in composite cathodes will raise the specific energy of the LiNi0.8Co0.1Mn0.1O2 (NCM811)|Li6PS5Cl (LPSCl)|Li battery from 320 Wh kg−1 to about 426 Wh kg−1. Nevertheless, numerous investigations have shown that cathodes with high CAM loading experience a substantial decrease in capacity.141,142 Increasing the CAM content in the LiNixCoyMnzO2 (NCM)/75Li2S–25P2S5 (LPS) composite cathodes of the NCM|LPS|In battery from 60% to 80% resulted in a decrease in discharge capacity from 155 to 100 mAh g−1.143 This suggests that a significant amount of CAM was not activated in the high-loading cathodes, which can be attributed to the lack of sufficient carrier percolation pathways. As the amount of CAM increases, the SE percolating network becomes smaller, which hinders ionic percolation.93,143 The non-uniform distribution of SE particles leads to reduced connection, increased ionic tortuosity, and a more extended average ionic diffusion length.144,145 This is illustrated in Fig. 2c and results in poor rate capability. In addition, the proportion of CAM directly influences the extent of contact between the CAM and the SE. When CAM fractions exceed a certain threshold, additional CAM particles are isolated. The CAM particles not connected to the SE are not accessible to Li+, rendering them electrochemically inactive.145 Other CAM particles in contact with SE particles may undergo overcharging and become deactivated in subsequent electrochemical cycles.141 Put simply, a large CAM/SE proportion can lead to a reduced active interface between CAM particles and SE particles and Li+ (de)intercalation sites, drastically deteriorating CAM utilization. Ceder et al.143 observed a drop in CAM utilization from 98% to 52% when cathode loading increased from 70 wt% to 80 wt%. The researchers further visualize the corresponding ionic percolation network and active/inactive CAM particles, as depicted in Fig. 2d. Only CAM particles close to the SE separator can be lithiated at 80 wt% CAM loading, as represented by the solid-gray colors in the right-hand column of Fig. 2d. In order to maximize the CAM utilization of the cathode with high CAM loading, Wang et al.146 proposed utilizing low-density SE with small particle size at the same weight content to obtain a high SE volume ratio, construct sufficient ionic transport pathways, and realize adequate CAM utilization, hence achieving high specific energy. This reminds us that the factors affecting the microstructural characteristics of composite cathodes are intricate. It is essential to comprehend the correlation between various aspects in order to get optimum composite cathodes.
Although numerous studies have elucidated the relationship between the CAM/SE composition ratio and the ionic/electronic percolation networks, the optimal CAM/SE ratio for ideal electrode microstructure design remains unclear. To rationally determine the optimal ratio, Lee et al.147 proposed an idealized simplified model of core–shell composite particles with cubic close-packed spherical CAM particles and a ductile SE filling the voids, neglecting binders and conductive agents. Geometric analysis of this model revealed an optimal composition of 74 vol% CAM and 26 vol% SE (Fig. 2e). This value is defined as the balance threshold, indicating that the amounts of CAM and SE are well harmonized. From this threshold, the expected performance of SSLMBs in terms of energy density or power density can be determined: a CAM-rich design favors energy density, whereas an SE-rich design favors power density. Furthermore, considering the true densities of the materials (4.79 g cm−3 for NCM811, 1.54 g cm−3 for LPSCl, and 2.59 g cm−3 for Li3InCl6), a “parameter map” for different solid-state cathode designs was constructed, as shown in Fig. 2f. For the NCM811/LPSCl and NCM811/Li3InCl6 composite cathode systems, the balance thresholds correspond to 89.9 wt% CAM and 84.0 wt%, respectively. On the right side and the left side of the balance threshold, the electrode compositions can be determined toward specific energy and specific power orientations, respectively. It should be noted, however, that this design relies on the electrode density being above the percolation threshold, meaning that the core–shell structural units must maintain sufficient contact to establish long-range ionic percolation through interparticle interfaces. Even if these specific design parameters are based on idealized assumptions and do not account for actual particle morphology and size distribution, they still provide valuable guidance for rational electrode formulation.
Sangrós Giménez et al.150 investigated the impact of the carbon black (CB) conductor contents on the specific conductivity of the LiFeO4 composite cathode. In general lines, the percolation theory establishes a critical content (percolation threshold) of the conducting material that makes the cathode transform from an insulating system to a conducting one. This conclusion is also confirmed by Randau.95Fig. 3a shows that the electrical conductivity of a LiFeO4 cathode containing 0.5 wt% of CB derived from the percolation theory equals zero, which means the percolation probability is below the percolation threshold. In this situation, the clusters of electronically conductive agents within composite electrodes are isolated; thus, no substantial percolating network is established. After reaching the percolation threshold, the construction of effective electronic transport pathways begins, and electronic conductivity ceases to be zero. The conductivity continues to increase rapidly as the quantity of electronic percolation pathways increases. In fact, a rise in the content of CB only from 2 to 3.7 wt% is translated into an increase in electrical conductivity from 6.20 × 10−3 to 1.59 × 10−2 S cm−1, an 85.89% increase. However, once the amount of conductive agents is enough to form a complete electronic percolation network, the rise in conductivity becomes significantly slower. When the CB content was increased from 8 wt% to 10 wt%, the ionic conductivity only increased by 18.65%. This phenomenon is known as the percolation transition. In addition, the content of the conductive agents not only directly affects electronic percolation but also interferes with ionic percolation because of the change in tortuosity. An excessive amount of conductive agents increases the tortuosity of microstructures, impeding ionic diffusion and ultimately reducing the electrochemical activity of cathodes and the capacity of SSLMBs.
 | ||
| Fig. 3 (a) Specific electrical conductivity of the LiFeO4 cathode varied with the amount of CB: obtained experimentally, derived using the simulated data and the percolation theory, and determined via the simulated data and the implemented method. Reproduced with permission.150 Copyright 2020, Wiley-VCH. (b) Schematic illustration of interfaces in cathodes without conductive agents and with carbon-based agents or Ti2O3. Reproduced with permission.160 Copyright 2023, Elsevier Ltd. | ||
Conductive agents facilitate effective electron percolation. Unfortunately, recent research has shown that incorporating carbon-based conductive agents in composite electrodes diminishes the cycling stability of sulfide-based SSLMBs.151–153 Carbon conductors provide additional electron transport pathways and a lower interfacial kinetic barrier, facilitating Li+ with low chemical potential in the charged state to migrate deeper into the SE and extend the decomposition area.151,154,155 Typically, this electrochemical behavior is determined by the surface area and morphology of carbon conductors.95 Continuous interfacial growth and the accumulation of decomposition products lead to the local failure of Li+ diffusion pathways and a large interfacial charge transfer resistance inside cathodes. In a recent study, Strauss et al.156 evaluated the impact of usual conductive agents on the electrochemical performance of sulfide-based SSLMBs. They found that only the particulate conductive carbon additive Super C65 with a relatively limited specific area can increase the capacity and rate performance of SSLMBs, virtually without affecting the reversibility, while other conductive agents such as carbon nanofiber (CNF), Ketjen black (KB), and TiC degrade the cell cyclability. Ates et al.157 demonstrated that the cells employing vapor-grown carbon fibers (VGCF) exhibited a greater initial coulombic efficiency (79% vs. 62%) and capacity retention (90% vs. 40% after 50 cycles) than Super C65 in the LiNi0.6Co0.2Mn0.2O2 (NCM622)/Li3PS4 cathode systems. This is attributed to the larger aspect ratio of VGCF, which enables longer-distance electronic conduction and smaller specific surfaces, reducing the contact area and minimizing SE decomposition. To overcome the SE degradation caused by using carbon-based agents, Randau et al.95 and Lee et al.158 proposed suppressing decomposition reactions by introducing a coating on the surface of carbon fibers. Deng et al.159 introduced poly(3,4-ethylenedioxythiophene) to eliminate the detrimental effects of carbon conductors. Moreover, Goodenough et al.160 substituted carbon-based agents with Ti2O3, which has an ultra-narrow bandgap of approximately 0.1 eV and a high electronic conductivity (10–102 S cm−1) in cathodes. Hence, highly developed electronic transport pathways within composite cathodes are created, and charge transfer resistance is reduced, as shown in Fig. 3b. Besides, Ti2O3 exhibits an abundant tendency for the absorption of lattice oxygen released from the CAM, which helps to stabilize the CAM/SE interface and prevent SE oxidation. Compared to composite cathodes without conductive agents or with carbon-based agents, the as-modified cathode exhibits a greater initial specific capacity of 192 mAh g−1 at 0.1C and excellent cyclability (86.5% after 140 cycles) and rate capability.
Lee et al.162 employed acrylonitrile butadiene rubber (NBR) as a binder and exploited the nitrile groups to induce ion–dipole interactions with CAM and SE in the composite cathodes,163 improving their interfacial contact. Zhang et al.164 incorporated 1 wt% ethyl cellulose binder into composite cathodes, which not only strengthens the mutual contact among cathode components for constructing more diffusion channels for Li+ transfer but also effectively compensates for volume change and enhances structural stability during cycling. The assembled SSLMBs delivered a capacity retention of 89.7% after 100 cycles. In addition, polymethyl methacrylate, ethyl cellulose, styrene butadiene rubber, and nitrile rubber binders are also commonly used for preparing composite cathodes.165 Nevertheless, due to the inadequate ionic conductivity of most polymeric binders, an excessive amount of binder may lead to the encapsulation of individual CAM particles. This, in turn, hinders carrier transport at the three-phase interface, thus decreasing the utilization of CAMs.166,167 Therefore, it is imperative to develop advanced binders with both high ionic conductivity and strong adhesion to mitigate their adverse impact on percolation pathways.167,168
Jung et al.169 conducted optimization of the nonconductive (10−10–10−12 S cm−1) binder NBR and synthesized an ionic liquid polymer binder (NBR–Li(G3)TFSI, G3: triethylene glycol dimethyl ether, LiTFSI: lithium bis(trifluoromethanesulfony)imide). Owing to its high ionic conductivity (1.7 × 10−4 S cm−1), NBR–Li(G3)TFSI causes less degradation to the ionic conductivity of LPSCl when compounded (4.1 mS cm−1 to 3.3 mS cm−1), compared to NBR–LPSCl composites (1.7 mS cm−1), as shown in Fig. 4a. Also, the NCM622 cathode with the NBR–Li(G3)TFSI binder achieves an ionic conductivity of 2.0 × 10−4 S cm−1, higher than that of the NBR-based counterpart (1.2 × 10−4 S cm−1), indicating more efficient ionic transport pathways. Moreover, because of the excellent compatibility between Li(G3)TFSI and sulfide SE materials,170,171 the surface coverage value of CAMs increased from 27% to 42% when using the NBR–Li(G3)TFSI binder, facilitating better ionic contact (Fig. 4b). This enables more CAM participation in reactions, delivering higher capacity (160 mAh g−1vs. 76 mAh g−1). Hong et al.161 introduced a polytetrafluoroethylene (PTFE)-based ionomer (poly(tetrafluoroethylene-co-perfluoro(3-oxa-4-pentenesulfonic acid))lithium salt) as a binder for composite cathodes. Fig. 4a shows that the ionic conductivity of the LPSCl–ionomer composite is higher than that of the LPSCl–PTFE composite, indicating that the ionomer is a better conductor of Li+ (1.6 × 10−5 S cm−1) than PTFE. In addition, the ionomer binder can be dispersed uniformly throughout composite cathodes (Fig. 4c), facilitating interfacial Li+ transport by increasing the active surface area and creating additional Li+ diffusion channels. Its good adhesion properties ensure good interfacial contact between the components, effectively preventing the electronic and ionic percolation network distribution during cycling. However, the limited adhesion ability of PTFE leads to a temporal separation between AM and SE particles during cycling. Thus, the SSLMBs show rapid capacity degradation (24% capacity retention after 300 cycles). Conversely, the SSLMBs featuring the optimized cathode show 90% capacity retention after 300 cycles at 0.5C, exhibiting superior cycling stability for sulfide-based SSLMBs with composite cathodes employing binders reported to date.
 | ||
| Fig. 4 (a) Ionic conductivities of LPSCl–NBR, LPSCl–NBR–Li(G3)TFSI, LPSCl–PTFE, and LPSCl–PTFE–ionomer composites. The dashed line is the ionic conductivity of LPSCl. The structural formulae of different binders are also shown. Data sources: ref. 161 and 169. (b) Illustration of microstructures of NCM composite cathodes without and with Li(G3)TFSI. The blue arrows highlighted indicate Li+ ionic pathways enabled by Li(G3)TFSI. Reproduced with permission.169 Copyright 2019, Wiley-VCH. (c) Illustration of morphological changes experienced by composite cathodes prepared using PTFE and the ionomer, induced by cycling. Reproduced with permission.161 Copyright 2022, American Chemical Society. | ||
Nagao et al.110 developed an active material Li2Ru0.8S0.2O3.2 with a high theoretical specific capacity of 270 mAh g−1, composed of 80 mol% Li2RuO3 and 20 mol% Li2SO4. It exhibited mixed conductivity, showing an electronic conductivity of 2.0 × 10−2 S cm−1 and a relatively high ionic conductivity of 6.3 × 10−7 S cm−1. SSLMBs using this AEA cathode achieved an outstanding energy density of 220 Wh kg−1. Suo et al.13 reported two ideal candidates: layer-structured TiS2 and chevrel-phase Mo6S8. TiS2 and Mo6S8 exhibit a significant electronic conductivity of 10 S cm−1, which is comparable to commercial conductive carbon, and a high Li+ diffusion coefficient of 1.8–9.8 × 10−8 and 8 × 10−9–9 × 10−10 cm2 s−1, respectively, which is comparable to sulfide SE. Their stable host frameworks maintain relatively constant ionic/electronic conductivity across varying Li+ concentrations. The Mo6S8/S8 biphasic electrode shows exceptionally low tortuosity (1–4), significantly below conventional composite cathodes (10–100) and even surpassing commercial liquid battery electrodes (1.5–10).172 This AEA cathode delivers ultrahigh energy densities of 770 and 1900 Wh L−1 at the electrode level. Cui et al.111 reported a cathode material Li1.75Ti2(Ge0.25P0.75S3.8Se0.2)3 (LTG0.25PSSe0.2) that possesses both high ionic conductivity (0.2 mS cm−1) and electronic conductivity (225 mS cm−1). The high mixed conduction can be maintained during electrochemical cycling, with ionic conductivity and electronic conductivity being 0.22/242 mS cm−1 at the fully charged state and 0.66/412 mS cm−1 at the discharge state, as shown in Fig. 5a. Moreover, this homogeneous cathode exhibits zero-strain characteristics with merely 1.2% volume variation, completely eliminating mechanical and electrochemical incompatibility. The LTG0.25PSSe0.2|Li6PS5Cl|Li–Si cell demonstrated remarkable areal capacity (1 mAh cm−2), high energy density (390 Wh kg−1), and exceptional cycling stability (70% capacity retention after 20000 cycles). Sun et al.112 developed a halide cathode material Li1.3Fe1.2Cl4 showing a high ionic conductivity of 2.28 × 10−4 S cm−1, an electronic conductivity of 6.98 × 10−5 S cm−1 (Fig. 5b), and a theoretical capacity of 145 mAh g−1. It delivers a high electrode-level energy density of 529.3 Wh kg−1, which can be further enhanced to 725.6 Wh kg−1 when coupled with Ni-rich cathodes.
 | ||
| Fig. 5 (a) Ionic and electronic conductivity evolution of LTG0.25PSSe0.2 with Li+ extraction/insertion in the first cycle. Reproduced with permission.111 Copyright 2024, Springer Nature Limited. (b) Comparison of the ionic diffusion coefficients (red bars) and electronic conductivities (blue bars) of Li1.3Fe1.2Cl4 with those of different cathode materials and SE materials. Reproduced with permission.112 Copyright 2025, Springer Nature Limited. | ||
2.2. Size engineering
In addition to composition, the particle size is another crucial parameter of composite cathodes. The charge transport distance, porosity, tortuosity, and contact area of the composite cathode are closely related to the particle size. So, the particle size has a considerable impact on the microstructure of composite cathodes and the electrochemical performance of SSLMBs in terms of discharge capacity, cycling stability, and rate capability. Size engineering involves the separate control of CAM or SE particles and their interplay. | ||
| Fig. 6 (a) Diffusion coefficients of Li+ in the composite cathode with various particle sizes of NCM during discharge processes, (b) 3D digital-twin composite cathodes with various particle sizes of NCM and the calculated values of the Li percolation pathway. Reproduced with permission.173 Copyright 2023, Wiley-VCH GmbH. (c) Mean ionic and electronic partial conductivities of cathode composites with various particle sizes of NCM, (d) Rietveld plots of ex situ XRD data in the 2θ range of the (003) reflection of NCM622 for charged composite cathodes with various particle sizes of NCM. The gray shaded areas represent active and inactive fractions of CAMs. Reproduced with permission.179 Copyright 2018, American Chemical Society. (e) The schematic diagram of the microstructure of composite cathodes NCM/LSPSC. The NCM with different particle sizes (D50 = 3.0, 6.2, and 10.3 µm) is chosen. The blue balls represent CAM particles. The light purple regions represent SE. Reproduced with permission.180 Copyright 2022, Wiley-VCH GmbH. | ||
The microstructures of composite cathodes are significantly influenced by the particle size of SEs, which serves as the ionic transport medium. The particle size of SEs acquired through the standard ball milling process is typically in the microscale range, making it challenging to achieve uniform distribution within composite cathodes. The particle size of SEs is typically reduced to the nanoscale by a liquid-phase technique. The smaller SE particles make it easier to achieve homogenous dispersion within composite cathodes.181–184 Consequently, the interfacial contact area between CAM and SE particles is enlarged, leading to the formation of effective ionic conduction pathways, which positively influence the utilization of CAMs and the specific capacity of SSLMBs.181,182 The composite cathode with a homogeneous distribution of components also contributes to lowering the electrode polarization and improving the cycle performance.185
Although diminishing the particle size of components in composite cathodes can promote electronic and ionic percolation, size effects also induce other harmful factors, limiting the consistent enhancement of the electrochemical performance of SSLMBs. First, reducing the size of SE particles can decrease the width of the percolation channel and raise the impedance within the SE network. This is because there are more grain boundaries, which slow down the ionic conduction.136,183 This effect is especially prominent in oxide SEs, as it exhibits significantly higher grain boundary resistance in comparison to sulfide SEs.186 Second, the increased surface-to-volume ratio may lead to a greater interfacial area for competing decomposition reactions, which are severe in sulfide-based SSLMBs, resulting in high interfacial resistance182 and fast degradation of capacity.176 Third, the SE with a uniformly small size inside the composite cathode may evenly fill the empty spaces among CAM particles, hence impeding the contact between CAM particles. In this instance, the electronic conductivity of composite cathodes decreases, indicating an increase in the proportion of electrically isolated CAM particles.92 The electrically isolated CAM particles lack the ability to both donate and accept electrons, leading to an inadequate progression of the (de)intercalation process. This, in turn, negatively impacts the charge and discharge properties. In order to address this challenge, Yamada et al.136 applied a composite cathode that consisted of SEs with mixed and small-average particle sizes. This design promotes ionic percolation while also preventing complete electrical isolation of CAMs, ensuring sufficient electronic percolation. It emphasizes the significance of the particle size distribution of SEs in determining the microstructures of the composite cathodes.
 | ||
| Fig. 7 (a) Modeling results showing the effect of θCAM as a function of both λ and fCAM. θCAM represents the utilization of CAM. fCAM represents the mass fraction of CAM in composite cathodes. Reproduced with permission.143 Copyright 2019, Wiley-VCH. (b) Influence of the particle size of CAMs and SEs on the tortuosity. Arrows indicate the optimal particle diameter for achieving maximum tortuosity. Reproduced with permission.145 Copyright 2023, Wiley-VCH GmbH. | ||
2.3. Gradient design
High-mass-loading cathodes (>30 mg cm−2 or 4.0 mAh cm−2) are crucial for achieving high density in SSLMBs. However, electrode thickening leads to high tortuosity and sluggish longitudinal transport pathways, posing significant challenges in ionic transport kinetics. In SSLMBs, the ionic transport kinetics within each particle are governed by local electrochemical potentials, which show a strong dependence on the relative positioning of the individual particle within the electrode architecture.188,189 In the thick cathode, hindered Li+ transport between the SE separator and current collector results in accumulation of Li+ flux generated by the faradaic reaction of the CAM. This creates a longitudinal Li+ concentration gradient (Fig. 8a) and induces heterogeneous electrochemical reactions.190–193 Specifically, most of the Li+ first reacted in the area near the SE layer and experienced difficulty reaching the CAM close to the current collector. As shown in Fig. 8b, this leads to deeper delithiation of CAMs near the SE side than near the current collector. Such longitudinal delithiation heterogeneity causes localized potential buildup adjacent to the SE layer and reduced cathode utilization ultimately diminishing the capacity and rate capability of SSLMBs. | ||
| Fig. 8 (a) Schematic diagram of electrode degradation behavior in thick cathodes. (b) Zoomed-in neutron transmission change for the thick cathode layer during the charging process with the corresponding voltage profile. During the delithiation, the colors of the cathode layer gradually changed to blue, which resulted in a decrease in the neutron attenuation coefficient. (c) Schematic illustration and (d) volume rendering of the tortuosity-gradient hierarchical electrode and the 2D slice of the fast transport layer (FTL) and reaction equilibrium layer (REL). (e) The tortuosity and (f) active area of the FTL and REL. Reproduced with permission.199 Copyright 2022, Oxford University Press. (g) Schematic of the three-layer cathode with gradient design and the design detail of each layer corresponding to the Li+ flux over the thickness of the cathode. Reproduced with permission.192 Copyright 2025, Springer Nature. The SE electrochemical potential and the local ionic current in the SE within the (h) homogeneous cathode and (i) gradient cathode. The homogeneous cathode consists of 70 wt% CAM and 30 wt% SE. The gradient cathode is referred to as SE-60-70-80, whereby SE indicates the position of the separator, and the subsequent numbers specify the NCM content in wt% within the three layers. Reproduced with permission.193 Copyright 2025, American Chemical Society. | ||
In terms of the Li+ flux distribution, the ion transfer path requirement varies among the whole thick electrode. Therefore, the coupling of ionic transport and reaction kinetics should be considered as a criterion for designing the electrode structure. Inspired by gradient porosity designs in liquid electrolyte-based batteries,194–197 gradient microstructural engineering in solid-state electrodes is achieved by introducing deliberate spatial variations in cathode components or microstructures, which breaks the constraints of conventional homogenous electrodes to actively accommodate dynamic Li+ flux changes.81,192,193,198–202 For instance, Wang et al.199 fabricated a tortuosity-gradient hierarchical electrode composed of a fast transport layer (FTL) and a reaction equilibrium layer (REL) by strategically distributing CAM particles of various sizes along the electrode thickness, as shown in Fig. 8c. Synchrotron X-ray tomography reconstruction with volume rendering (Fig. 8d–f) reveals that the FTL, composed of small CAM particles (<8 µm), exhibits low tortuosity and shortened vertical ionic transport pathways, facilitating rapid Li+ transport from the SE side to the current collector side. In contrast, the REL, containing larger CAM particles (15–20 µm) with lower specific surface area, requires lower Li+ flux to achieve a specific delithiation level. This gradient architecture forms an efficient ionic permeation network, and the synergy between the FTL and the REL maintains a balance between ionic transport and delithiation, improving CAM utilization in thick electrodes and delivering a 15% increase in specific capacity. Zhu et al.192 designed a three-layer cathode with a catholyte content gradient, as shown in Fig. 8g. Layer A (adjacent to the SE) contains the highest SE fraction (33 wt%), establishing a high-speed ionic transport channel to facilitate initial Li+ ingress. Layer B (middle) has a standard SE content (23.5 wt%) to balance ionic conduction and energy density. Layer C (near the current collector) is formulated with a lower SE content (14 wt%) to minimize inactive volume and maximize CAM proportion. This tailored SE distribution addresses the mismatch in conventional composite cathodes between the uniform distribution of the catholyte and the non-uniform Li+ flux generated by the faradaic reaction of CAMs, enabling uniform reaction kinetics. At a high current density of 9.0 mA cm−2, a NCM811 gradient cathode with 30 mg cm−2 loading exhibited a 171% capacity improvement over its homogenous counterpart. A high-loading LCO gradient cathode (100 mg cm−2) achieved an areal capacity of 10.4 mAh cm−2, approaching practical requirements. Zeier et al.193 also developed a similar gradient cathode and provided additional insights into limiting processes through microstructure resolved simulations. As shown in Fig. 8h and i, the gradient catholyte design attenuated the drop of the electrochemical potential of Li+ in SE within the composite cathode and homogenized the local current density distribution, thereby mitigating localized high-current-induced degradation commonly observed in homogeneous thick cathodes.
In summary, gradient microstructure design enables efficient charge transport and balanced Li+ transport and consumption rates. A further optimization of the microstructure, for instance, by regulating particle size and content, along with enhanced ionic conductivity of the catholyte, may amplify the advantages of gradient cathode architectures.
3. Construction of conductive networks within SEs
3.1. Inorganic SEs
SEs with high ionic conductivity enable rapid and efficient Li+ transport between the electrodes, which is an essential requirement for the development of high-performance SSLMBs. While a few sulfide SEs exhibit ionic conductivities comparable to those of conventional liquid carbonate systems, the majority of inorganic SEs still fall significantly short and remain insufficient to meet the demands of commercial applications. The ionic conductivity σLi+ of inorganic SEs is typically described by the Nernst–Einstein relationship (eqn (7)):203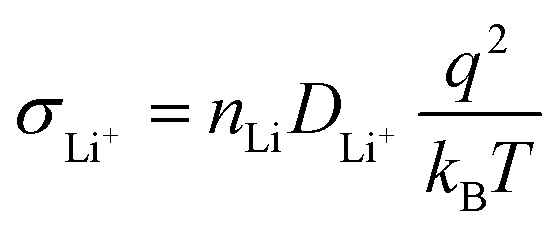 | (7) |
| DLi+ = D0e−Ea/kBT | (8) |
Based on mathematical analysis, it is evident that conductivity is primarily governed by active Li+ concentration and activation energy for Li+ transport in the inorganic crystal framework. These parameters are strongly affected by both microscopic features including Li+ site occupancy, Li+ vacancy concentration, Li+ content, atomic arrangement, and lattice defects, and mesoscopic/macroscopic structural elements, including boundaries, pores, cracks, and secondary phases, as shown in Fig. 9. Hence, a thorough understanding of how these multiscale structural factors influence Li+ transport is critical. Targeted structural engineering is required to construct tailored, high-efficiency pathways for Li+ migration, thereby enabling faster ion transport kinetics in inorganic SEs.
 | ||
| Fig. 9 Multi-scale factors affecting the Li+ conductivity of inorganic SEs. Li+ site occupancy: reproduced with permission.204 Copyright 2023, Springer Nature Limited. Li+ vacancy concentration: reproduced with permission.205 Copyright 2024, American Chemical Society. Li+ content: reproduced with permission.206 Copyright 2017, American Chemical Society. Phase structure: reproduced with permission.207 Copyright 2023, American Chemical Society. Atomic arrangement: reproduced with permission.208 Copyright 2023, Wiley-VCH GmbH. | ||
Based on the defect transport mechanism of ion diffusion, modulating the crystal structure can effectively enhance the ionic conductivity of SEs.209 Doping is the most conventional and effective method. First, doping can regulate the occupancy at different Li+ sites.204,210,211 Sun et al.211 increased the Li+ occupancy of LLZO at the 96h site by a Ca-W dual-substitution strategy, which can significantly lower the Li+ migration barrier and thus improve the ionic conductivity (5.74 × 10−4vs. 2.98 × 10−6 S cm−1). Lu et al.204 synthesized Li6.8Si0.8As0.2S5I by Si4+ doping to significantly enhance the occupancy of Li3 (48h) and Li4 (16e) new sites, whereas only Li1 (48h) and Li2 (24g) sites exist for the parent-phase Li6AsS5I, resulting in an increase in ionic conductivity from 3.92 × 10−6 S cm−1 to 1.04 × 10−2 S cm−1. Second, doping can increase the concentration of Li vacancies.205,212–215 Third, doping can increase the Li+ concentration in the SEs through charge compensation. Buannic et al.206 introduced the doping ion Sc3+ to partially populate the Zr site for finer control over the Li content in cubic Li7La3Zr2O12 according to the resulting stoichiometry (Li7−3x+yGaxLa3Zr2−yScyO12). Similarly, the ionic conductivity of Li6+xP1−xSixS5I (0 ≤ x ≤ 0.5) was increased from 3.13 × 10−6 S cm−1 to 7.34 × 10−3 S cm−1 by doping aliovalent Si4+ to increase the Li+ content and thereby facilitating Li+ diffusion.216 Fourth, doping can induce effective crystal structure changes, thereby increasing Li+ mobility. It has been reported that the Li+ sublattice is always ordered in tetragonal LLZO, while disordered in cubic-LLZO, that is, all Li symmetry sites are partially occupied.14 Therefore, substitution leads to an increase in the disordering of the Li+ sublattice to form a stable and highly conductive cubic garnet phase. It has been proved that single doping of Al3+, Ta5+, W6+, Ga3+, and Ge4+ is beneficial for stabilization of cubic-LLZO.206,217–221 Liang et al.222 demonstrated the feasibility of controllably regulating the crystal structure from trigonal (P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1) to orthorhombic (Pnma) in the Li3MCl6 (M represents a trivalent rare earth metal) system for increasing ionic conductivity. Fifth, doping can regulate the ordering of atomic arrangement, i.e., the crystallinity of the materials. Although the ionic conductivity of most inorganic SE systems depends on high crystallinity, some sulfide and halide systems show higher conductivity in an amorphous form.223–225 Balancing the amount and nature of amorphous and crystalline phases inside these materials is a crucial factor influencing the ionic conductivity. Zhu et al.226 dual-doped Li3BS3 SE with P5+ and Cl− and observed that with the increase of Cl− content (x = 0.2), the material transformed from the crystalline phase to the amorphous phase whose ionic conductivity is 3 orders of magnitude higher than that of the crystalline Li2.75−xP0.05BS3−xClx (x = 0.15).
m1) to orthorhombic (Pnma) in the Li3MCl6 (M represents a trivalent rare earth metal) system for increasing ionic conductivity. Fifth, doping can regulate the ordering of atomic arrangement, i.e., the crystallinity of the materials. Although the ionic conductivity of most inorganic SE systems depends on high crystallinity, some sulfide and halide systems show higher conductivity in an amorphous form.223–225 Balancing the amount and nature of amorphous and crystalline phases inside these materials is a crucial factor influencing the ionic conductivity. Zhu et al.226 dual-doped Li3BS3 SE with P5+ and Cl− and observed that with the increase of Cl− content (x = 0.2), the material transformed from the crystalline phase to the amorphous phase whose ionic conductivity is 3 orders of magnitude higher than that of the crystalline Li2.75−xP0.05BS3−xClx (x = 0.15).
In recent years, use of high-entropy materials has gained significant research attention as a promising approach for enhancing the ionic conductivity of SEs.15,16,208,227–235 The incorporation of multiple cationic species increases structural entropy and reduces Gibbs free energy, maintaining long-range order while introducing pronounced local lattice distortions due to variations in ionic radii and bonding states, resulting in short-range disorder. Unlike conventional ordered crystals, the disordered lattice in high-entropy electrolytes forms intricate Li+ migration pathways. Chemical disorder further disrupts lattice homogeneity, broadening the energy distribution of local sites and facilitating energy overlap between adjacent positions, which enhances ion hopping and ultimately leads to superior ionic conductivity.
Strategies ranging from conventional doping to high-entropy design can effectively tune crystal structures to enhance ionic conductivity. However, the quest for superior SEs now confronts the challenge of combinatorial exploration in the chemical space. Machine learning emerges as a pivotal tool to navigate this complexity.236–241 By training on computational and experimental datasets, machine learning models, such as crystal graph convolutional neural networks and genetic algorithms, enable the targeted screening of high-performance SEs. Beyond mere prediction, machine learning deciphers elemental roles and ion migration pathways, revealing hidden composition–structure–property relationships and identifying non-intuitive descriptors that govern ionic transport. This approach represents a paradigm shift in materials screening, transforming the design of advanced SEs from a process reliant on trial-and-error and isolated theoretical analysis to a rational design workflow guided by data-driven insight.
In addition, introducing defects such as point defects242 or stacking faults243 into the crystal structure also reduces the Li+ migration barriers and creates additional transport sites, therefore increasing the ionic conductivity of SEs. While the introduction of defects and structure disorder being beneficial for many poor ionic conductors, the introduction of higher dimensional defects is detrimental for fast ion diffusion dynamics in materials with crystallograpically well-defined diffusion or transport pathways, and thus a proper control of the defect chemistry and the defect concentration represents a key factor to manipulate fast ionic dynamics in SEs.244 In addition to crystal structure engineering, recent studies have revealed that the nature of chemical bonding also significantly influences ionic transport. Sun et al.245 demonstrated for the first time that the covalent character of metal–halogen bonds plays a critical role in driving the superionic transition in halide SEs, revising the conventional view that structural phase transitions are solely responsible. This enhanced covalence triggers contraction of [GdBr6]3− octahedra and collective motion of Br− ions, facilitating efficient ionic transport pathways and enabling Li3GdBr6 to achieve a room-temperature conductivity of 5.2 mS cm−1, which ranks among the highest values reported for bromide-based SEs.
The presence of pores reacts macroscopically to the incomplete densification of SEs. The ionic conductivity linearly increases with increasing SE density, which is attributed to the larger contact area between the SE particles.246 The combined effects of grain and grain boundary resistance determine the conductive property of inorganic SEs. The magnitude of grain boundary resistance determines the overall ionic conductivity of the SEs compared to grain resistance. The abundance of grain boundaries in oxide SEs results in a reduction in total ionic conductivity.247 Canepa et al.248 suggested that volume changes are introduced in grain boundary regions to accommodate the distinct coordination environments of atoms during the formation of grain boundaries from the bulk. These volume changes are called grain boundary excess free volumes per area. Unlike oxide SEs, sulfide and chloride SEs have a significant excess volume, indicating the presence of ample free space for interstitial atoms, especially small atoms such as Li+, making their grain boundaries have a minimal impact on their ionic conductivity. The occurrence of grain boundaries, pores, and cracks in oxide SEs is attributed to the inadequate development of grains during the sintering process. These microstructural defects can hinder Li+ migration, resulting in a decrease in ionic conductivity. Furthermore, the evaporation of lithium during the sintering process generates secondary phases that typically exist at grain boundaries and impede the Li+ movement.
The internal structure of SEs mainly depends on the sintering process. Recent studies have shown that the reduced grain boundary number, modified and optimized grain boundaries, and improved densification can be achieved by changing the sintering conditions, including pressure, temperature, atmosphere, and sintering technique, resulting in enhanced ionic conductivity. The sulfide SEs exhibit low Young's modulus (18–25 GPa) and good compressibility, which implies that their densification behavior is closely related to pressure.249 Microstructural defects, such as pores, can be reduced by high pressure250–252 or hot pressing.253,254 When increasing the fabrication pressure from 50 to 250 MPa, the relative density of LPSCl increases from 68.3 to 75.2%, and the ionic conductivity shows a similar trend, increasing from 0.99 to 2.06 mS cm−1.252 By optimizing the molding pressure (270 MPa) and temperature (200 °C), Sakamoto et al.253 achieved a near theoretical density (98% relative density) of Li2S–P2S5 glassy SEs, which increased the ionic conductivity (1.1 mS cm−1) by a factor of five compared to conventional room temperature molding conditions. However, this conventional pressurization method is unsuitable for ceramic SEs with high Young's modulus because of their limited elastic deformation under particular pressure. Even if the pressure of cold pressing is increased to 800 GPa, the relative density of the LLZO SE is still only 73%.251 Field-assisted sintering techniques255 and oscillating pressure sintering256 are characterized by controllable microstructure, improving the densification of ceramic SEs. Besides, selecting a suitable sintering atmosphere257 is conductive to the optimization of grain boundaries and the reduction of pores at the grain boundaries, which results in a high density of 96% and a high ionic conductivity of 7.4 × 10−4 S cm−1 for Li6.75La3Zr1.75Ta0.25O12.258 Moreover, introducing low melting point sintering additives such as Li3PO4, B2O3, LiBF4, Li3BO3, and LiOH also benefits the densification and ionic conductivity of ceramic pellets.259–261 There are three mechanisms. First, sintering additives promote the growth of the grains and increase the contact area between grains to reduce the grain boundaries and pores. Second, sintering additives solidly dissolve at the grain boundaries to improve densification by weakening the grain boundaries and filling the pores. Third, sintering additives promote grain rearrangement and mass transfer, ensuring small grain size and uniform distribution. However, sintering additives may result in the formation of undesirable secondary phases.
In summary, the ionic conductivity of inorganic SEs is governed by a synergistic interplay between ionic transport kinetics and multi-scale structural characteristics. At the most fundamental level, the crystal structure defines the prerequisite migration pathways for Li+ transport. According to eqn (7) and (8), ionic conductivity depends exponentially on activation energy Ea, such that a minimal reduction in the latter can enhance the former by orders of magnitude. Therefore, a rationally designed crystal framework facilitates low activation energy and thus enables efficient Li+ diffusion. However, when considering the practical application of SEs in ASSLMBs, meso- and macroscopic structural features such as grain boundaries and defects significantly influence ionic transport. These imperfections often account for the discrepancy between theoretical predictions and experimental measurements of ionic conductivity. Therefore, achieving ultra-fast ionic transport represents a system level challenge that requires a dual strategy: the rational design of crystalline materials with superior migration pathways through methods such as doping, coupled with advanced processing techniques that optimize grain boundaries and enhance densification. Only through such an integrated approach can the high intrinsic ionic conductivity be fully realized in macroscopic samples.
Moreover, in volume-constrained SSLMBs, electrode active materials undergo significant volume changes during lithium (de)intercalation processes, inducing dynamic stress/strain changes within the batteries.262–265 This leads to structural changes in SEs, including crack evolution, elastic/plastic strain, and lattice softening, further affecting ionic conductivity. Gu et al.266 demonstrated significant stress concentration at grain boundaries of LPSCl SE in the NCM|LPSCl|Li cell during charging, triggering crack formation. Such structural degradation impedes both intra-grain and inter-grain ionic transport. Yildiz et al.267 revealed that elastic strain promotes Li+ disordering in β-Li3PS4 SE, enhancing ionic conductivity similarly to chemical doping. Zeier et al.36 investigated how plastic strain affects the ionic conductivity of Li6PS5Br SE. Plastic strain introduces dislocations that distort the local lattice structure. On the one hand, vacancies and interstitials can rapidly diffuse by using dislocation cores as diffusion channels. On the other hand, the open space at the dislocation core offers fast diffusion channels for defects. Consequently, the dislocation density increases with increasing pressure, and thus, the ionic conductivity of Li6PS5Br increases. In addition, internal strain has also been shown to induce lattice softening,268 which may result in a decreased activation barrier for ionic transport.269 Ding et al.270 investigated lattice softening in LPSCl and found that the low-energy phonon modes (<10 meV) associated with Li facilitate Li+ migration. Moreover, since the thermodynamic stabilization windows of SEs are often smaller than the actual operating range of the batteries, the decomposition of the SEs during the electrochemical process also affects the actual electronic conductivity.271
3.2. Composite solid electrolytes
Composite solid electrolytes (CSEs) combining inorganic SEs and organic polymer SEs have attracted much attention because they combine excellent ionic conductivity with mechanical properties. On the one hand, the CSEs with high ionic conductivity ensure rapid Li+ migration within the SE layer. On the other hand, their rigid-flexible coupling mechanical properties can overcome the impeded Li+ transport at the interface caused by the rigid contact and buffer the volume change during charge/discharge cycling, which means that the CSEs exhibit excellent interface compatibility with electrodes. Therefore, ASSLMBs using CSEs can realize excellent cyclability and rate capability. Moreover, under the premise of the same material system, the ionic conduction characteristics of the CSEs can be significantly improved through clever structural design.41,42,272Indeed, two-phase CSEs can essentially be viewed as quasi-three-phase systems.273–276 The ionic conductivity of such materials is governed by the dynamic transport of Li+ through the inorganic filler, polymer matrix, and interfacial regions. It has been demonstrated that the fast Li+ transport channels at the continuous ceramic/polymer interface can even be directly connected to the electrodes on both sides of it, allowing for rapid transport of Li+ between the two electrodes along the interfacial phase,277 thus realizing higher ionic conductivity of SEs (1.25 × 10−2 S cm−1).278 The inorganic particle and the interface layer covered on the particle can be taken as a unit: a composite grain. The effective ionic conductivity σ3 of this unit can be described based on average t-matrix approximation in quantum scattering theory279 (eqn (9)):
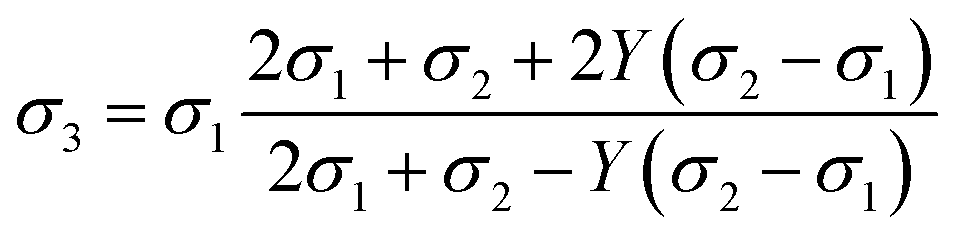 | (9) |
| Y = 1/(1 + t/R)3 | (10) |
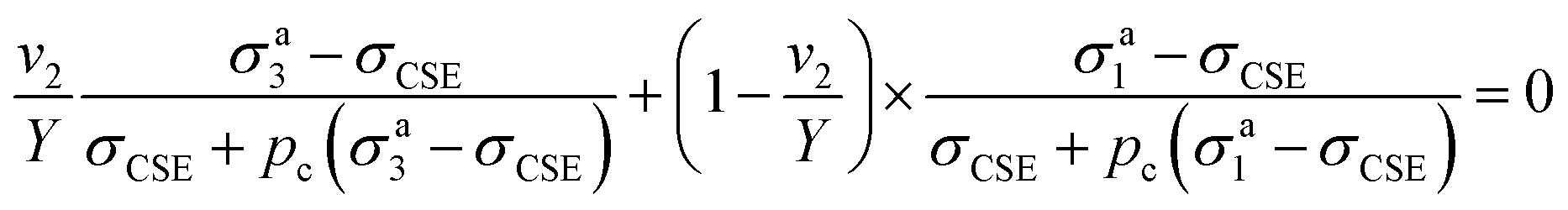 | (11) |
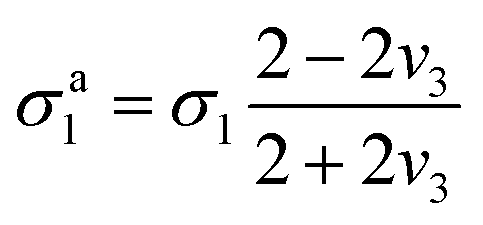 | (12) |
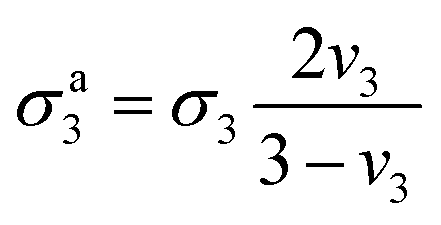 | (13) |
The transition from a single-phase percolation structure of single SEs to a three-phase percolation structure of CSEs not only overcomes the low ionic transport efficiency caused by the physicochemical properties of the polymer SEs but also constructs new ionic transport pathways, thus facilitating the Li+ transport within the SE layer and significantly increasing the ionic conductivity of the SEs. For example, Wang et al.274 formed a 3D CSE by in situ polymerizing poly(ethylene glycol) methyl ether acrylate (PEGMEA) on a 3D self-supported porous skeleton (p-LPSCl). Fig. 10a–c reveals that 6Li resonance in LPSCl and poly(PEGMEA) is at 1.38 and −1.23 ppm, with measured 6Li concentrations of 136.7 and 12.4, respectively. In addition, a new resonance of pristine CSE appears at 0.15 ppm, which is related to the LPSCl/poly(PEGMEA) interfacial phase with the 6Li concentration of 9.24. After polarization, the 6Li concentration in the inorganic phase, organic phase, and interfacial phase increased by 256.7%, 27.3%, and 180.2%, respectively, as shown in Fig. 10c. This demonstrated that Li+ mostly migrates along the continuous LPSCl phase and that the interfacial phase facilitates Li+ migration better than the organic phase. The CSEs show a high ionic conductivity of 4.6 × 10−4 S cm−1 due to the highly conductive continuous inorganic phase and interfacial phase, which is 128 times greater than that of the corresponding polymer. Moreover, Cui et al.283 experimentally quantified for the first time that the ionic conductivity of the interfacial phase reaches as high as 2.5 mS cm−1, which is 33 times that of the bulk composite SEs. Further optimization of the interface through LPSCl modification of the LLZTO framework increased the interfacial conductivity to 12 mS cm−1.
 | ||
| Fig. 10 (a) Schematic representation of the Li+ conduction pathway in the 3D p-LPSCl/poly(PEGMEA) CSE. (b) 6Li solid-state nuclear magnetic resonance (SSNMR) spectra of the pristine p-LPSCl/poly(PEGMEA) composite, LPSCl, and poly(PEGMEA). (c) Quantitative fitting of the 3D composite 6Li SSNMR spectra before and after polarization. Insets show the magnified parts of the spectra. Reproduced with permission.274 Copyright 2021, Wiley-VCH GmbH. | ||
In addition, the structural design of CSEs by adjusting the content, size, morphology, and distribution of components enables the rearrangement of Li+ transport channels in the same material system, as shown in Fig. 11, which significantly improves ionic conductivity. Inorganic SSEs are usually introduced into the polymer matrix as zero-dimensional (0D) nanoparticle fillers to facilitate Li+ transportation by reducing the crystallinity of the polymer matrix,284 building surface groups, and regulating the interaction of Li+ with the polymer chains.285 It is worth noting that although nanoscale ceramic fillers can significantly improve ionic conductivity, an increase in ceramic content does not always correlate positively with ionic conductivity. When the filler content is too large, it can lead to particle aggregation, preventing the formation of an effective Li+ transport path.286 In addition, the different microstructures of the ceramic particles also contribute to the differences in the ionic conductivity of the CSEs. The highly conductive dense ceramic particles act as conductors in CSEs and can increase the ionic conductivity of CSEs. In contrast, porous ceramic particles form an overall insulating solid structure due to the blocked grain–grain contacts, resulting in a systematic decrease in conductivity.287
 | ||
| Fig. 11 Schematic illustration of CSEs composed of inorganic fillers (green) of distinct morphologies and distributions and the polymer matrix (orange). Solid lines and dashed lines represent rapid and slow Li+ transport, respectively. | ||
Compared with the Li+ transport behavior across a lot of particle–particle junctions in 0D nanoparticle-filled CSEs, the ionic transport performance of CSEs filled by one-dimensional (1D) nanowires is more favorable because 1D fillers with a high aspect ratio have longer Li+ transport channels and effectively reduce the interfacial resistance between inorganic particles.288,289 Liu et al.278 evaluated the ionic conductivity improvement of polyacrylonitrile (PAN)-based CSE by various structural Li0.33La0.557TiO3 (LLTO) ceramic fillers. The ionic conductivity of the filler-free polymer SE is 3.62 × 10−7 S cm−1, which is improved to 1.02 × 10−6 S cm−1 by adding LLTO nanoparticles and is further increased to 5.4 × 10−6 S cm−1 by the incorporation of randomly dispersed LLTO nanowires. Furthermore, the CSE that contains well-aligned nanowires, which replace the disordered LLTO nanowires, shows another order of magnitude enhancement in conductivity, reaching 6.05 × 10−5 S cm−1. This is due to the absence of surface crossing junctions in the aligned nanowires, which provide fast ionic conduction pathways.
Moreover, 3D nanostructured inorganic frameworks filled with polymers have been developed, which provide continuous three-dimensional Li+ percolation channels. These 3D framework-based CSEs achieve greater ionic conductivity enhancement than CSEs based on 0D nanoparticles or 1D nanofibers, as shown in Fig. 12. Besides high ionic conductivity, the mechanical strength and long-term stability of CSEs can also be improved. The advanced 3D inorganic frameworks used in CSEs are divided into three main categories: interconnected fiber networks,290–292 continuous frameworks,274–276,283,293–295 and vertically aligned frameworks.277,296–299 Yan et al.291 reported a CSE composed of Li6.28La3Al0.24Zr2O12 nanofibers and poly(ethylene glycol) diacrylate. Acrylate functional groups were covalently bonded on the surface of nanofibers, which enabled the chemical grafting of functional monomers directly from the nanofiber surfaces, thus leading to the interconnection of separate 1D inorganic nanofibers to form 3D Li+ conductive pathways. The highest ionic conductivity of the CSE with a 3D interconnected fiber network was 4.9 × 10−4 S cm−1, an order of magnitude higher than that of the pristine polymer SE (2 × 10−5 S cm−1). In addition, it is also possible to transform nanowires or nanofibers into 3D nanomats or textiles through artificial processing, which have better regularity and symmetry than the 3D networks formed by random interconnections of nanowires.300,301 Gong et al.301 constructed a flexible Li7La2.75Ga0.25Zr1.75Nb0.25O12 ceramic textile, retaining the structural characteristics of the original textile template, which consisted of a network of continuous interlocked fibers and interlaced yarns and was further combined with the PEO polymer matrix to form a CSE. The measured ionic conductivity of the CSE was 2.7 × 10−5 S cm−1, which is an order of magnitude higher than that of the PEO–LiX polymer SE system with 10−6 S cm−1.302 In addition to the 3D interconnected nanowires, there is another more advanced structure, a continuous porous inorganic framework, in which the significant impact of the Li+ transportation pathway stemmed from the continuous inorganic framework and newly generated ceramic phase/polymer phase interfacial phase.275,276,293,294 Yan et al.275 reported a rigid-flexible coupling CSE combining a self-supported porous Li1.3Al0.3Ti1.7(PO4)3 (LATP) framework and poly(ethylene glycol) methyl ether acrylate SE, which exhibits a high ionic conductivity of 2.0 × 10−4 S cm−1, 56 times higher than that of the pristine polymer SE. In such a configuration, the conductivity is also 23 times higher than that of the polymer/0D LATP composite with 8.6 × 10−6 S cm−1, suggesting fast and efficient transport of the porous framework. Although the first two 3D frameworks constructed effective Li+ percolation networks, the random pathways are still long and tortuous. Comparatively, the composite of the 3D inorganic framework with vertically aligned pathways and the polymer SE can realize short and fast Li+ transport pathways, further improving the ionic migration efficiency.277,296–298 Wang et al.296 prepared a porous Li1.5Al0.5Ge1.5(PO4)3 ceramic skeleton with vertically aligned structures by ice templating and further introduced the PEO-based polymer SE into the vertical pores to form a CSE. The highly oriented and low curvature vertical structure directly linked the electrodes to form channels with fast ionic conduction, and the ionic conductivity of the CSE is 1.6 × 10−4 S cm−1, which is an order of magnitude higher than that of the PEO-based polymer SE (1.92 × 10−5 S cm−1) and nearly 6.9 times that of the CSE with LAGP randomly dispersed inside.
 | ||
| Fig. 12 Promotional factors for the conductivity of CSE obtained by mixing the polymer matrix and inorganic fillers with different structures (0D, 1D, and 3D). The promotion factor is defined as the ratio of the ionic conductivity of CSE to that of the corresponding polymer SE. The polymer matrix includes P(PEGMEA), PAN, PVDF, and PEO. Inorganic fillers include sulfide SEs, LLZO, LLTO, and LATP. Data sources: ref. 274, 278, 289, 294, and 303–315. | ||
4. Battery structure optimization and design
Generally, SSLMBs have sandwich structures stacked individually with prepared electrode and electrolyte layers in a precisely controlled environment. Although preparing this structure is simple, there are several crucial issues. First, the SSE layer needs to be made thin enough, which is extremely challenging; otherwise, it will result in the quite high length of the ion conduction pathway between electrodes, leading to high cell resistance. Next, the limited integration between planar electrodes and SEs leads to a non-complete interfacial contact with insufficient carrier transport pathways. Particularly, these limited interfacial Li+ transport pathways may be destroyed or hindered by interfacial contact separation, interfacial reaction, and space-charge layers (SCLs). Last, the long and tortuous carrier transport channels limit the carrier diffusion efficiency, and the limited 2D interfacial contact leads to slow electrochemical kinetics in conventional cathodes with disordered stacked particles. Given this, the general planar-type sandwich structure of SSLMBs fails to match the volumetric performance characteristics of commercial high-energy cells. In recent years, a series of approaches, including SE layer thinning, buffer layer construction, electrolyte permeation, multilayer SE construction, and 3D integrated battery structure design, have been employed to enhance the Li+ transport dynamics of SSLMBs effectively.4.1. SE layer thinning
The inorganic SE layer in the general planar-type sandwich structure of SSLMBs needs to be relatively thick to maintain the mechanical integrity of the SE layer, the thickness of which is typically over 500 µm and in some cases can even reach 1000 µm. The thickness is 30–60 times that of conventional polyolefin separators (≈15 µm) used in commercial lithium-ion batteries. However, such a thick SE layer hinders the efficiency of Li+ long-range transport and thus limits the rate performance. Besides, the heavy SE layer would occupy a large volume and mass portion of the full cell, significantly sacrificing the energy density.316 For example, with the same configuration of electrodes, the volumetric energy density of SSLMBs can be increased from 120 Wh kg−1 to 280 Wh kg−1 when the thickness of SE is reduced from 100 µm to 20 µm.317 Therefore, it is indispensable to move on to preparing the ultrathin SE layer to reduce the distance and time of Li+ diffusion and to assemble SSLMBs with high energy density and rate capability.113,318–322Table 2 summarizes the current methods for preparing thin inorganic SE layers and the corresponding properties. Zhao et al.323 reported an ultrathin freestanding Li9.88GeP1.96Sb0.04S11.88Cl0.12 film (8 µm) possessing an ionic conductivity of 1.9 × 10−3 S cm−1, an ultrahigh ionic conductance of 1860 mS, and an ultralow areal resistance of 0.68 Ω cm2. Jiang et al.324 prepared a 25 µm-thick Li0.34La0.56TiO3 freestanding film by tape-casting, having the potential for fabrication of NCM|Li batteries with 400 Wh kg−1 energy density. The ionic conductivity of this film increases from 9.6 × 10−6 S cm−1 to 2.0 × 10−5 S cm−1, owing to the reduced grain boundary resistance, as compared to a thick SE (>200 µm) formed by cold-pressing. Besides, the area-specific resistance of the LLTO SE film decreased from 2153 Ω cm2 to 207 Ω cm2 as thickness decreased from 160 µm to 41 µm.
| Method | Solid electrolyte | Thickness (µm) | Ionic conductivity (S cm−1) | Energy density | Ref. |
|---|---|---|---|---|---|
| Note: NCM111, LiNi0.3Co0.3Mn0.3O2; Gr, graphite; NCM711, LiNi0.7Co0.15Mn0.15O2; LZO, Li2O–ZrO2; NCM9055, LiNi0.9Co0.05Mn0.05O2; LTO, Li4Ti5O12; LZCF, Li2ZrCl5F; NCM83, LiNi0.83Co0.11Mn0.06O. | |||||
| Slurry casting | Li3PS4 | 2 × 10−4 | 155 Wh kg−1 (NCM111@Gr) | 333 | |
| Slurry casting | Li6PS5Cl | 45 | 5.1 × 10−4 | 314 Wh kg−1 (NCM711|Li) | 327 |
| Slurry casting | Li6PS5Cl | 30 | 1.31 × 10−3 | >900 Wh L−1 (LZO@NCM9055|Ag–C) | 334 |
| Slurry casting | Li7La3Zr2O12 | 5 | 5.55 × 10−3 | 329 | |
| Tape casting | Li0.34La0.56TiO3 | 25 | 2 × 10−5 | 324 | |
| Tape casting | Li6.4La3Zr1.4Ta0.6O12 | 40 | 4.3 × 10−4 | 320 | |
| Tape casting | Li9.88GeP1.96Sb0.04S11.88Cl0.12 | 8 | 1.9 × 10−3 | 323 | |
| Solution infusion | Li6PS5Cl | 60 | 6.3 × 10−3 | 394 Wh kg−1 (S|LiIn) | 330 |
| Solution infusion | Li6PS5Cl0.5Br0.5 | 40 | 2 × 10−3 | 330 | |
| Solution infusion | Li3InCl6 | 25 | 5 × 10−4 | 316 | |
| Cold-pressing | Li3PS4 | 70 | 2 × 10−4 | 42 Wh kg−1 (LCO|LTO) | 335 |
| Cold-pressing | Li6PS5Cl | 35 | 2 × 10−4 | 284.4 Wh kg−1 (Co3S4|Li) | 332 |
| Hot-pressing | Li5.4PS4.4Cl1.6 | 30 | 8.4 × 10−3 | 331 | |
| Hot-pressing | Li5.7PS4.7Cl1.3 | 40 | 1.1 × 10−3 | 354.4 Wh kg−1 (NCM811@LZCF|Li3N–Li) | 328 |
| Hot-pressing | Li6PS5Cl | 25 | 2.1 × 10−3 | 390 Wh kg−1 (NCM83|µSi) | 325 |
| Radio-frequency magnetron sputtering | Li2.9La0.68ZrO8 | 0.56 | 4 × 10−7 | 336 | |
| Pulsed laser deposition | Li7La3Zr2O12 | 1 | 7.36 × 10−7 | 337 | |
| Spin coating | Li7La3Zr2O12 | 0.31 | 1.67 × 10−6 | 338 | |
| Hydrothermal approach | Li1.2Al0.2Ti1.8(PO4)3 | 88 | 1.65 × 10−4 | 339 | |
In the process of electrolyte layer thinning, we should also pay attention to the mechanical properties of the SE film, as this parameter is crucial for the actual assembly and operation of SSLMBs under low or no working pressure conditions. Hu et al.325 developed a fusion bonding technique to prepare an ultrathin LPSCl film (≤25 µm) by exploiting the viscosity of thermoplastic polyamide under low thermos-pressure (≤5 MPa). This film exhibited excellent toughness, low modulus, and high ionic conductivity (2.1 × 10−3 S cm−1). Notably, it also has unique stress-dissipation, which facilitates the cycling stability of NCM83|LPSCl|Li–In SSLMBs (>80% capacity retention after 707 cycles). In addition, the cell delivers high energy densities of 390 Wh kg−1 and 1020 Wh L−1. Moreover, introducing a supporting scaffold can also reduce the thickness of the SE layer while enhancing its mechanical properties.324,326–330 Some studies have achieved the cathode-supported SE film by casting the homogeneous slurry containing SE powder and the polymeric binder directly on the cathode surface. In this case, not only the thickness of the SE film can be further reduced to 5–10 µm,329 but also the interfacial resistance can be significantly decreased as a result of the high integration of cathode and SE, resulting in better electrochemical performance of SSLMBs.326 Zhu et al.330 prepared a 60 µm ultrathin LSPCl SE film using a chemically compatible cellulose membrane as the self-limiting skeleton. The thin cellulose promises high mechanical strength, good flexibility, and excellent adhesion to SE particles to construct a continuous and complete 3D ionic conducting network. The self-limited LPSCl film exhibits an ionic conductivity of 6.3 × 10−3 S cm−1, which is slightly lower than that of the cold-pressed LPSCl pellet (8.6 × 10−3 S cm−1). However, owing to the sharp decrease in the thickness from 500 µm for the SE pellet to 60 µm for the SE film, a significant reduction in the bulk ionic transport resistance from 45.8 to 14.6 Ω is achieved. It is worth noting that such scaffolds require specialized fabrication methods, such as electrospinning, which have limitations in producing large-scale films. Kim et al.327 introduced a 45 µm LPSCl SE film with an ionic conductivity of 5.1 × 10−4 S cm−1 based on a perforated polyethylene separator as a mechanical support. Besides, the frame-based SE film shows a superior tensile strength of 44.1 MPa compared to those of freestanding SE films (not applicable), which ensures mechanical robustness for commercial-level cell assembly and is suitable for large-scale commercial production, such as roll-to-roll processes. Moreover, the mono-cell-level energy density of the SSLMBs with the LiNi0.7Co0.15Mn0.15O2 cathode, frame-based SE film, and 200 µm Li metal achieved 314 Wh kg−1 and 404 Wh L−1, surpassing those of commercial liquid electrolyte-based batteries (260–270 Wh kg−1). However, the ionic conductivity of this scaffold-based SE film is typically slightly lower than that of the corresponding SE pellets due to the non-intimate contact between SE particles.
Moreover, solvents and/or binders are inevitably present in the conventional film-making process, which can reduce the ionic conductivity of SE films and affect the electrochemical stability of the interface due to the ionic–electronic mixed conductive phase formed by reaction between binders and electrodes.331 Thus, developing a solvent- and binder-free film-making strategy is urgently needed. Liu et al.332 coated a 5 nm homogenous polydopamine layer on the LPSCl SE particles, resulting in a modified adhesive particle surface. A free-standing LPSCl thin film with a thickness of 35 µm and an ionic conductivity of 2 × 10−4 S cm−1 could be achieved by cold-pressing. In this situation, the oxygen atom in o-benzoquinone is suitably located for coordination with Li+, and the catechol units could also chelate with Li+. Based on the charge transfer interaction between o-benzoquinone and catechol units in polydopamine, Li+ could be transferred through the π systems, thus achieving smooth ionic conduction in the LPSCl SE film. When assembled with a Co3S4 cathode of 6.37 mg cm−2 loading and Li metal anode, the full-cell level energy density could reach 284.4 Wh kg−1.
4.2. Battery structure optimization
Regarding the general surface roughness characteristics of the planar electrode layer and the planar SE layer, point-to-point contact is usually found in the planar-type sandwich structure of SSLMBs, thus showing a limited interfacial contact area and inefficient Li+ transport pathways across the interface. However, due to the complex solid–solid interfacial properties, these limited interfacial Li+ transport pathways are further hindered by interfacial problems, including interfacial contact separation, interfacial reactions, and space-charge layers, which severely limit the Li+ transport kinetics at the interface, leading to high interfacial resistance and consequent rapid degradation of SSLMBs in terms of capacity, rate capability, and cyclability.58,340 Therefore, rational optimization of the cell structure is required to enhance the electrode/electrolyte integration in a planar-type sandwich structure, including buffer layer construction, electrolyte permeation, and multilayer SE layer construction. It is worth noting that the realization of fast interfacial ionic transport kinetics is closely related to improving the preparation method.Han et al.45 introduced an Li2.3C0.7B0.3O3 solder with a low melting point between the LCO cathode and LLZO SE, which enhances the interfacial contact due to the intrinsic mobility of the molten state Li2.3C0.7B0.3O3 during sintering. The solder could ensure fast Li+ diffusion at the interface by forming the high-ionic conductive Li2.3−xC0.7+xB0.3−xO3 interfacial phase with Li2CO3. Benefiting from the optimized interfacial structure, the LCO|LLZO|Li cell delivered a large initial capacity of 94 mAh g−1 with stable cycling for 100 cycles. To improve wettability at the initial SE/Li anode interface, a buffer layer with lithophilic properties is typically introduced.345–347 Han et al.345 introduced an ultrathin Al2O3 coating at the Li7La2.75Ca0.25Zr1.75Nb0.25O12/Li interface using an atomic layer deposition technique, which significantly reduced the interfacial impedance from 1710 Ω to 1 Ω. However, introducing a buffer layer may not be effective in resolving interfacial dynamic contact failure due to localized stress concentration caused by large volume changes when Li+ intercalation/deintercalation occurs within electrodes. The low toughness of the buffer layer cannot address interfacial contact separation due to shrinkage of the electrode particles,55 and the rigidity of the buffer layer may not be able to accommodate the large volume expansion of the electrode material or to perform effective depressurization, leading to cracking of the buffer layer.56 In this case, modification of the material itself is a better option, such as developing and designing low- or zero-strain cathode materials348,349 that exhibit small or even zero volume changes upon intercalation/deintercalation of Li+ or using doping to build strong chemical bonds to enhance the interfacial adhesion.350
The inorganic SE typically exhibits a lower oxidative stability limit than the high operating voltage (>3.5 V) of SSLMBs59 and is inherently unstable to lithium metal,351 which leads to unavoidable side reactions between the solid electrolyte and the electrode.352–357 In addition, side reactions between the electrolyte and electrode materials may occur due to elemental interdiffusion.358–360 The low ionic conductivity of the interface decomposition mainly contributes to the larger interface resistance by severely blocking the effective Li+ transport channels at the interface. The interfacial (electro)chemical stability can be significantly improved by designing stabilized Li+ conductive buffer layers. On the one hand, the buffer layer can prohibit direct contact between electrodes and SEs and broaden the voltage stabilization window to avoid side reactions when the operating voltage exceeds the electrochemical stabilization window of the SEs. On the other hand, the buffer layer with high ionic conductivity ensures fast Li+ transport at the interface, and the low electronic conductivity at the interface can further inhibit the side reactions.
Cao et al.361 applied amorphous Li0.35La0.5Sr0.05TiO3 as a buffer layer at the NCM/LPSCl interface. Raman spectroscopy and first-principles thermodynamic calculations show that LPSCl decomposes during charging, forming a passivation layer with poor ionic conductivity at the interface, which hinders Li+ transport across the interface. However, the Li0.35La0.5Sr0.05TiO3 layer effectively inhibits the side reactions and enhances the interfacial thermodynamic stability, and the high Li+ conductivity improves the interfacial Li+ transport kinetics. The LiNi1/3Co1/3Mn1/3O2 (NCM111)|LPSCl|Li–In battery delivered a capacity retention of 91.5% after 850 cycles. In addition, fast ionic conductors such as Li4Ti5O12, LiNbO3, Li2SiO3, LiTaO3, LiZr2(PO4)3, and Li3PO4 exhibit a wide electrochemical window with less than 2 V reduction potential and about 4 V oxidation potential, and thus these materials could be used as conductive buffer layers to maintain stability during electrochemical cycling and stabilize the cathode/SE interface.351,358,362,363 Moreover, Kim et al.364 utilized energy band diagrams to design a titanium compound self-induced interlayer (TSI) by controlling the energy level of the LATP/Li interface. Because of the lower Fermi level of LATP relative to that of Li metal, facile electron flow occurs across the interface, which provokes the titanium reduction of LATP, generating the interfacial deterioration. However, the energy band of TSI is located at a relatively lower position than the Fermi level of Li metal, which means electrons can be transferred from the Li anode to TSI and subsequently pinned at TSI. In addition, because the lowest value of the conduction band of TSI is located at a relatively lower position than that of LATP, electrons cannot easily climb the barrier to reach LATP. Therefore, the buffer layer precludes the undesirable electron transport from the Li metal anode to the LATP surface and enhances their interfacial stability. The Li symmetric cell with a TSI successfully maintains its constant overpotential over 1000 cycles.
When electrodes with significant differences in Li+ chemical potentials are in contact with SEs in SSLMBs, Li+ diffuses from the SE to the cathode with a high chemical potential to reach equilibrium at the interface.365 The redistribution of Li+ gradually reduces the Li+ concentration at the SE interface and creates a Li-deficient SCL.64,67,68 Due to the lack of sufficient charge carriers for efficient ion transport, the interfacial resistance and polarization increase and the overall electrochemical performance of the SSLMBs diminishes.65,66
Introducing an inert buffer layer with good electronic insulation and ionic conductivity at the oxide cathode/sulfide SE interface to prevent their direct contact effectively suppresses SCL formation.366–371 Ohta et al.367 interposed a LiNbO3 (LNO) layer between the LCO cathode and Li3.25Ge0.25P0.75S4 (LGPS) SE as a buffer layer, which generates new LCO/LNO and LNO/LGPS interfaces. The similar lithium chemical potential between LCO and LNO minimizes Li+ transfer across their interface, while the electronically insulating nature of both LNO and LGPS prevents electron flow. This combination fundamentally suppresses the formation of a SCL at the LCO/LGPS interface, leading to a significant reduction in interface resistance from 910 Ω to 20 Ω. Theoretical calculations demonstrated that the insertion of a buffer layer ensures stable structure and electronic states of the interface and eliminates the lithium adsorption sites, therefore suppressing the space charge separation.368 In addition, introducing dielectric materials is also a promising approach to mitigate SCL formation at the interface, which could create a reverse electric field capable of offsetting SCL formation.67,75,372–374 It is worth noting that the dielectric properties of the material will also affect the SCL formation and Li+ transport behavior through the SCL. The selected materials should exhibit a large dielectric constant and paraelectric properties, which means the dipole moment direction can be changed in real-time based on the electric field under actual battery conditions.373 Besides, since dielectric materials usually have poor ionic conductivity, they cannot be uniformly coated as in the case of the fast ionic conductors, but rather island coating can be performed. It is noteworthy that ferroelectric materials also demonstrate significant potential in modulating the SCL.375
In order to overcome the hindrance of limited interfacial transport resulting from interface degradation, constructing an appropriate buffer layer is a relatively adaptive and effective measure, as shown in Fig. 13. The specific interfacial challenges vary across different types of inorganic SEs, depending on their intrinsic properties.376–378 For oxide SEs, which exhibit high oxidation stability and mechanical strength, the primary interfacial concerns lie in poor physical contact and side reaction at the anodic interface. In contrast, sulfide and halide SEs exhibit favorable deformability, enabling good interfacial contact through cold pressing. However, their chemical potential mismatch with conventional oxide cathode materials leads to inevitable formation of SCLs. In addition, most sulfide SEs suffer from poor electrochemical compatibility with electrodes, triggering severe side reactions. Halide SEs, while possessing an exceptionally wide voltage window and good compatibility with cathodes, demonstrate poor electrochemical stability against lithium metal. For different interfacial behaviors, such as contact loss, side reaction, and SCL, materials with good wettability, electrochemical stability, and dielectric properties can be selected as buffer layers, respectively. At the same time, it is worth our attention that introducing a buffer layer implies the insertion of new interfaces, which needs to be considered in terms of new interfacial issues and the properties of the buffer layer itself.
 | ||
| Fig. 13 Schematic illustration of solving the blocked Li+ transport at the electrode/SE interface in SSLMBs via buffer layer construction. LLSTO, LCBO, and GO represent Li0.35La0.5Sr0.05TiO3, Li2.3–xC0.7+xB0.3−xO3, and the graphene oxide buffer layer, respectively. Side reaction in the cathode/SE interface: reproduced with permission from ref. 361. Copyright 2020, American Chemical Society. Contact loss in the cathode/SE interface: reproduced with permission from ref. 45. Copyright 2018, Elsevier Inc. The space-charge layer in the cathode/SE interface: reproduced with permission from ref. 75. Copyright 2023, Wiley-VCH GmbH. Contact loss in the Li anode/SE interface: reproduced with permission from ref. 345. Copyright 2016, Springer Nature Limited. Side reaction in the Li anode/SE interface: reproduced with permission from ref. 379. Copyright 2020, Wiley-VCH. | ||
The first is the SE precursor penetration method, which uses a suitable solvent to dissolve the SE to obtain a liquefied electrolyte, followed by the removal of the solvent to realize the solidification of the battery.76,77,380 This method not only reduces interfacial resistance but also decreases the proportion of catholyte, enabling higher energy density. Kim et al.76 integrated the electrode and the SE by dissolving the LSPCl SE in ethanol and subsequently injecting the solution into a porous electrode, as shown in Fig. 14a. Favorable ionic percolation was achieved due to the conformal contact interface between the electrode and the SE. The LSPCl-infiltrated LCO and graphite electrodes show high reversible capacities (141 mAh g−1 and 364 mAh g−1) at 0.14 mA g−1, which are not only superior to those of conventional dry-mixed and slurry-mixed solid-state electrodes but also comparable to those of liquid electrolyte cells. This method is also commonly used in polymer SE-based SSLMBs.381 In addition, it is compatible with conventional Li+ battery electrodes, which can be directly converted into all-solid-state configurations without complex processing. Moreover, this method can be adapted to various roll-to-roll and large-area printing techniques, such as spray coating and gravure printing, showing promise for cost-effective production. Nevertheless, several limitations remain. First, the use of solvents may adversely affect the ionic conductivity of SEs. In addition, repeated infiltration and slow drying cycles in a moisture-free environment limit the scalability. Second, infiltration of solution-based SEs tends to be incomplete and inefficient. To improve SE penetration, elevated temperatures are often applied to enhance the molecular mobility of the SE. This, however, requires the SEs to exhibit heat-recoverable properties, which entails maintaining high ionic conductivity after thermal treatment, such as Li3InCl6, LPSCl, and LiI–Li4SnS4.380,382,383
 | ||
| Fig. 14 Schematic illustration of electrode/electrolyte conformal contact interfaces established by electrolyte permeation. (a) SE precursor penetration. Reproduced with permission.76 Copyright 2017, American Chemical Society. (b) Melt penetration. Reproduced with permission.78 Copyright 2021, Springer Nature Limited. (c) In situ polymerization. Reproduced with permission.391 Copyright 2021, Wiley-VCH GmbH. | ||
The second is the melt penetration method, melting at a high temperature for liquefied electrolytes and subsequently cooling at a low temperature to achieve solidification. This strategy enables both high volumetric energy density and low interfacial resistance. Commonly used SEs include metal halides and lithium hydrides. Xiao et al.78 first deposited a layer of the Li1.9OHCl0.9 SE powder with low melting points (300 °C) on the slurry-cast electrode, then the SE was thermally melted during heating and infiltrated into the electrode through capillary forces, and finally, a dense electrode with a melt-infiltrated SE was formed by cooling and solidification, as shown in Fig. 14b. The infiltrated-structure NCM|graphite cell delivered a large initial capacity of 155 mAh g−1 at 25 mA g−1 and a capacity retention of 75% after 100 cycles, which is comparable to that in liquid electrolytes. This approach mimics the low-cost fabrication of commercial Li+ cells with liquid electrolytes. As such, nearly the same commercial equipment could be used for electrode and cell manufacturing, which reduces the barrier for industry adoption. However, the general applicability of this approach remains limited, as it is suitable only for a few SEs that combine low melting points with high ionic conductivity such as lithium metal halides,133,384 lithium hydrides,385–387 and lithium closo-borates.388 Furthermore, the high-temperature melting process raises concerns regarding the thermal stability of key cell components, including electrode materials, binders, and current collectors, which constrains its broader applicability.
In addition, in situ polymerization is another effective strategy to realize the highly integrated structural features of electrodes and SEs in SSLMBs with planar-type sandwich structures. Liquid precursors with high mobility/wettability can adequately fill voids at the electrode/SE interface and infiltrate porous cathodes, and then precursors are in situ polymerized from a liquid-state to a solid-state in the filled area under mild thermal, light or electric treatments, which not only guarantees continuous contacts among solid components for fast Li+ transport, but also maximally maintains the solid-state of batteries.389 Nie et al.390in situ constructed a uniform and thin (10 µm) interfacial layer at the interface of the Li6.4La3Zr1.4Ta0.6O12 SE and the LiFe0.4Mn0.6PO4 cathode based on the thermal polymerization reaction of poly(ethylene glycol) diacrylate, resulting in no micro-cracks, and voids were observed at the interface. The conformal interfacial layer achieved a stable and tight interfacial contact between the electrode and the SE with an interfacial resistance of only 202.6 Ω, which is very close to the liquid battery. At the same time, its high ionic conductivity with 1.4 × 10−4 S cm−1 ensured the fast Li+ transport at the interface. Zheng et al.391 formed a conductive polythiophene layer at electrode/SE interfaces by in situ polymerization of 2,2′-bithiophene in a polyethylene oxide SE. Although an excellent initial interfacial contact can be achieved between polymer SEs and electrodes, interfacial voids and micro-cracks cannot be eliminated entirely. In contrast, the in situ formed polythiophenes with different sizes can fill the non-contact space remaining after the pressing of batteries, as shown in Fig. 14c, thus realizing high interfacial compatibility, which leads to a significant decrease in the interfacial resistance from 13040 Ω to 1765 Ω. In addition, the interface formed by in situ polymerization can enhance the electrochemical stability of electrode/electrolyte interfaces by changing the interfacial electric potential distribution, thus realizing the long lifespan of SSLMBs. In situ polymerization streamlines the fabrication process by eliminating the separate steps of electrolyte synthesis and cell assembly required in conventional methods, thereby reducing manufacturing complexity and potentially lowering costs. Unlike electrolyte precursor infiltration and melt infiltration techniques, in situ polymerization imposes no specific requirements on electrolyte properties, offering superior generality and feasibility. Furthermore, substantial progress in related research and existing reviews provide solid guidance for its implementation. However, incomplete monomer conversion during the polymerization process remains a challenge. Future efforts should focus on developing residue-free initiators or initiator-free in situ polymerization systems to avoid introducing additional transport limitations.392,393
Different inorganic SE chemistries indeed significantly differ in their thermodynamic stability against oxidation and reduction. Given this, employing heterogeneous electrolytes on the cathode and anode sides to construct an asymmetric, multilayer SE architecture has emerged as a promising strategy, as shown in Fig. 15a. This design expands the electrochemical stability window and enhances interfacial chemical and electrochemical stability. This configuration is frequently implemented in halide-based SSLMBs.205,394–396 Halides are commonly positioned at the cathode due to their high oxidative stability,134,397 whereas sulfides are favored at the anode because of their superior reductive stability.398 For instance, Hu et al.205 used a Li3ScCl6–LPSCl bi-layer SE in an NCM9055|Li–In cell, achieving a capacity retention of 82.7% after 1000 cycles. The observed cycling stability stems not only from enhanced interfacial electrochemical compatibility but also from the excellent ductility of both sulfide and halide electrolytes.397 This mechanical compliance ensures intimate interfacial contact with electrode layers and minimizes interfacial resistance. Nevertheless, some studies have uncovered chemical incompatibilities between these heterogeneous electrolytes.399–401 When sulfides are used as the bulk electrolyte and halides are incorporated into the composite cathode, indium sulfide species may form at their interface.401 The Li3InCl6@NCM622|LPSCl|Li–In cells exhibit severe capacity fading, reaching 50% after only 20 cycles at 1C.399 This adverse effect can be alleviated by introducing an additional halide electrolyte layer to spatially separate the halide-based cathode from the sulfide electrolyte.
 | ||
| Fig. 15 Schematic illustration of multilayer solid electrolyte structures constructed using (a) heterogeneous inorganic SEs, (b) heterogeneous polymer SEs, (c) polymer SEs modified with distinct plasticizers, (d) CSEs with a gradient distribution of inorganic fillers, and (e) CSEs with heterogeneous inorganic fillers. | ||
In addition, multilayer polymer/CSE structures have been widely investigated by researchers due to the flexible and tunable physicochemical properties of polymers. Specifically, heterogenous SE structures in the depth direction are achieved by adjusting the type of polymer or plasticizer and the type or content of inorganic fillers. A multilayer SE structure facilitates overcoming the shortcomings of the individual polymer components and works either independently or synergistically. When working independently, each polymer layer is individually targeted to enhance cathode or lithium metal anode electrochemical compatibility. Meanwhile, in a synergistic mode, the multilayer structural design can provide reinforced performance in terms of ionic conductivity and mechanical properties.
First, multilayer electrolyte structural design is achieved by varying the composition of the polymer SE in the depth direction based on differences in the physicochemical properties of polymers. As shown in Fig. 15b, a high-voltage stable polymer SE and a low-voltage stable polymer SE are placed at the cathode side and anode side, respectively, which broaden the electrochemical window under real operational conditions. In other words, this multilayer design solves the problem inherent to a single polymer system, whose energy gap between the empty and occupied electronic states is not large enough to maintain stability against lithium metal plating on the anode side and Li+ extraction on the cathode side. Zhou et al.402 designed a double-layer polymer SE with a poly(N-methyl-malonic amide) in contact with the cathode and a poly(ethylene oxide) polymer in contact with the lithium metal anode, improving the electrochemical stability of both cathode/SE and SE/anode interfaces. The double-layer SE has an upper oxidation voltage limit of 4.75 V, which matches with the high-voltage LCO cathode. The assembled SSLMB shows a capacity retention of 91.2% after 100 cycles. It has been reported that polymer backbones without unstable groups and introducing electron-withdrawing groups are promising choices to achieve high-voltage stability, such as acetonitrile, poly(acrylonitrile), polyvinyl alcohol-b-cyanide ethyl ether, and poly(ethyl a-cyanoacrylate),403–405 which can be placed at the cathode side to construct multilayer polymer SEs. Additionally, the composition of polymers involves the in situ generation of an ideal CEI and SEI via reactions between the SE and the cathode/anode, whose microstructure and composition are crucial for improving interfacial stability.406,407 Wang et al.407 reported a multilayer polymer SE in which an organic–inorganic CSE was used as the intermediate layer to provide sufficient mechanical strength, while different polymer SE layers are placed on two sides. Specifically, the poly(ethylene oxide)-based SE is placed in contact with the cathode to induce the generation of a dense and uniform LiF-rich CEI film and the poly(propylene carbonate)-based SE reacts with lithium anode to promote the formation of the SEI film consisting of inorganic lithium species to improve the interfacial stability. The multilayer SE-based SSLMBs exhibit a remarkable capacity retention of 94.5% over 150 cycles. Therefore, using distinct plasticizers to specifically modify polymer SE is feasible to construct ideal multilayer SEs,408,409 as shown in Fig. 15c. Lv et al.409 selected tetramethylene sulfone (TMS) and methyl (2,2,2-trifluoromethyl) carbonate (FEMC) as plasticizers in the polymer matrix on the cathode and anode sides, respectively, to construct different interfacial modification layers, thereby enhancing the high-voltage compatibility of the cathode/SE interface and reducing the reactivity of the SE/Li interface. The designed multilayer SE exhibits a high ionic conductivity of 8.8 × 10−4 S cm−1, a wide electrochemical window of 4.8 V, excellent Li dendrite resistance, and superior interfacial stability. The SSLMB with the LFP cathode can maintain a stable capacity of over 125 mAh g−1 after 500 cycles at 1C. Unlike the previously mentioned designs, each layer of this multilayer polymer SE has essentially the same composition except for the plasticizers, effectively promoting fast Li+ migration by eliminating the compositional mismatch between different polymer matrices and additional interface resistance due to the macroscopic phase separation. Moreover, desirable multilayer CSE structures can also be achieved by changing the concentration and structure of inorganic fillers in the polymer matrix,410–413 as shown in Fig. 15d and e, respectively. Deng et al.412 designed a multilayer SE with a gradient distribution of inorganic fillers. The Li6.4La3Zr1.4Ta0.6O12 (LLZTO) inorganic filler-rich CSE is located on the cathode side, compatible with high-voltage cathodes, while the poly(ethylene glycol)diacrylate polymer-rich CSE is located on the anode side, enabling the excellent interfacial contact with the anode and facilitating uniform Li deposition. When paired with LFP and the lithium metal anode, it delivered a capacity of 161.0 mAh g−1 at 0.1C and retained 82.4% capacity after 200 cycles. Huo et al.411 designed a hierarchical sandwich-type CSE. The polymer-rich outer layers (20 vol% LLZTO particles) are flexible to ensure good interfacial contact with electrodes and show a high Li+ transference number of 0.47, which can immobilize anions and guide a uniform distribution of electrical field, whereas the inorganic particle-rich middle layer (80 vol% LLZTO particles) enhances the mechanical strength and inhibits lithium dendrites effectively. These designs with gradient inorganic fillers offer continuous ionic transport pathways. For constructing multilayer SEs by adjusting the type of inorganic fillers, Kim et al.413 selected Li6.28Al0.24La3Zr2O12 and LiTa2PO8 as active fillers in a poly(vinylidene fluoride-co-hexafluoropropylene) matrix for the cathode-side and anode-side, respectively. In particular, Li6.28Al0.24La3Zr2O12 exhibited stable interfacial properties with the cathode due to its excellent electrochemical oxidative stability and mechanical strength. Besides, LiTa2PO8 displayed improved interfacial stability with the lithium metal anode resulting from its high ionic conductivity and effective suppression ability against lithium dendrite growth. The multilayer CSE demonstrated a high ionic conductivity of 8.7 × 10−4 S cm−1, an electrochemical window of 5.1 V, and a high critical current density of >1.0 mA cm−2. The assembled SSLMBs delivered a capacity retention of 91% at 0.2C after 140 cycles.
Based on the differences in mechanical properties, ionic conductivity, and thermodynamic stability against oxidation and reduction of different electrolyte chemistries, selecting suitable material systems for combining and designing multilayer electrolyte structures can satisfy the distinctive demands of cathodes and anodes simultaneously, thus realizing the high performance of SSLMBs. However, some of the multilayer electrolytes face issues such as discontinuous Li+ migration pathways and low compatibility of extra electrolyte/electrolyte interfaces. Most importantly, such multilayer electrolyte structures seem to disagree with the overall goal of creating thinner separator layers for practical use of SSLMBs. It is also in stark contrast to simple processing on a large scale. Therefore, the design and construction of thin multilayer electrolyte films with sufficient ionic conductivity and low internal interfacial resistance still face significant challenges.
4.3. Three-dimensional integrated structure design
In conventional composite cathodes with disorderly stacked particles, the diffusion of ions and electrons in long and tortuous pathways limits the charge transport efficiency. Moreover, the limited 2D contact between electrode and electrolyte makes the electrochemical active area insufficient, accompanied by inhomogeneous carrier distribution and spatial reaction heterogeneity.49 To solve these issues, the construction of SSLMBs with a 3D electrode/electrolyte integrated structure was proposed. This structural design not only has a large interfacial contact area, which provides sufficient reactive active sites to accelerate electrochemical kinetics at the interface and thus improves the rate capability of cells, but also has low tortuous 3D interconnected ionic and electronic transport channels, which are conducive to the realization of high loading cathodes and avoid the low utilization of the CAM in the conventional thick powder composite cathode due to the insufficient electronic and ionic conductivities. Besides, this 3D structural design also mitigates large volume variations of cathode materials, which eliminates the capacity attenuation caused by mechanical degradation. The realization of 3D integrated structures includes two aspects: to begin with, constructing 3D structured cathodes by using special techniques, including interconnected,414 porous,415–417 tunneled,52,53 and vertically aligned structures.51,418–421 Next, using direct infiltration, ion implantation, and in situ polymerization methods to achieve 3D electrode/electrolyte integration.Jin et al.414 prepared a 3D interconnected LiNi0.5Co0.2Mn0.3O2 framework through an electrospinning technique, followed by in situ polymerization of the polymer precursor solution infiltration into the electrodes to achieve a 3D integrated structure, as depicted in Fig. 16a. This design facilitates the construction of 3D continuous ionic and electronic transport channels and enlarged and intimate electrode/electrolyte interfaces, thus ensuring fast electrochemical kinetics, with the Li+ diffusion coefficient increasing from 2.49 × 10−12 cm2 s−1 (powder composite structure) to 7.9 × 10−12 cm2 s−1. The assembled SSLMBs demonstrated a high areal capacity of 1.19 mAh cm−2 at a high cathode loading of 9.28 mg cm−2. Nie et al.415 employed a carbon nanotube coated conductive melamine formaldehyde (CMF) sponge as an active material host to construct porous channels in the electrodes and then penetrated gel polymer SEs into the electrodes by in situ thermal induced polymerization. The 3D reconstructions show that LiFe0.4Mn0.6PO4 (LMFP) active particles are mainly loaded on the CMF skeleton and gel polymer SEs are filled into the entire pores of the sponge (Fig. 16b). COMSOL Multiphysics simulation (Fig. 16c) of the wettability of the electrolyte precursor in different structures of electrodes suggests that this porous structure improves the infiltration speed and penetrability of the precursor, thus constructing fast ionic and electronic transport channels. It has a significant impact on improving the electrochemical reaction kinetics, especially for high-loading electrodes. In contrast, as for the case of conventional cathodes mentioned in Section 4.2.2, the electrolyte precursor is merely in contact with the surface electrode particles and does not infiltrate into the interior of the electrode, as shown in Fig. 16c. This can be attributed to the existence of tortuous channels between the disorderly stacked particles, which reduces the porosity and contact area with the electrolyte precursors, lowering the wettability. The highly efficient integration of the porous electrode with the electrolyte and the homogenous distribution of the electrolyte enable a uniform distribution of Li+ flux and effectively homogenize the local current density, thereby alleviating the concentration polarization inside the electrode and achieving a more uniform and sufficient reaction, ultimately leading to an increase in the utilization of the active materials. In contrast, the traditional electrodes suffer from high tortuosity, resulting in sluggish Li+ transport behavior. It will inevitably cause significant concentration polarization and the local depletion of Li+ within the electrodes, especially for the region far away from the SE. Under the same areal mass loading of 12 mg cm−2, the porous LiFe0.4Mn0.6PO4 cathode shows higher discharge capacity (156.5 vs. 123.6 mAh g−1) and better rate capability. Besides, the porous electrode with a high area active mass loading of 25.0 mg cm−2 can deliver a high discharge capacity of 128.7 mAh g−1. Xia et al.53 deposited a LiPON SE onto an amorphous MnO2−x nanosheet array film, achieving the directional transition from MnO2−x to LixMnO2 and the integration of the 3D tunnel structured electrode with the electrolyte, as shown in Fig. 16d. This 3D integrated structure enables the formation of a large cathode/electrolyte interface and a short Li+ diffusion length, and thus LixMnO2|LiPON|Li SSLMBs exhibits a large specific capacity of 185 mAh g−1 and excellent cycle performance (81.3% capacity retention after 1000 cycles). A similar nanosheet array electrode structure is also capable of 3D fully infiltration by electrolyte precursor solution penetration and subsequent in situ polymerization.52 Nie et al.419 produced the electrodes with vertically aligned channels by an ice-templating method, and then a solid polymer electrolyte was penetrated into aligned porous channels of the electrodes by in situ UV-curing polymerization, thus enabling the 3D electrode/electrolyte integration. The vertically aligned porous structure of cathodes is covered by the polymer SE, which maximizes the contact area between the electrode and the SE, thus ensuring rapid Li+ diffusion at the interface. The resistance of the aligned electrode/SE interface is 41.6 Ω cm2, which is much smaller than that of the traditional electrode/SE interface (720.1 Ω cm2). Besides, the electrode with the vertically aligned porous channels shortened the Li+ transport distance and promoted Li+ diffusion. As shown in Fig. 16e, the electrochemical reaction in the aligned electrode reaches rapid equilibrium in 10 s and then maintains a low and stable concentration gradient. In contrast, there is a large concentration difference after 500 s in the traditional electrode, which has a serious impact on electrochemical reaction kinetics in the whole electrode. Similar vertically aligned cathodes with electrolytes by in situ polymerization to achieve 3D integrated structure have also been reported by Sun et al.418 and Huang et al.51 In addition, the cathodes with vertically aligned structures can also be attained by magnetically assisted templating,422,423 wood templating,424 solvent evaporation,425 and phase inversion.426
 | ||
| Fig. 16 (a) Schematic illustration of the synthesis process of a 3D integrated cross-linked NCM cathode and a SE. Reproduced with permission.414 Copyright 2022, Elsevier B.V. (b) The 3D reconstruction of the integration of a porous electrode and an in situ polymerized SE. (c) Simulation of the wettability of the liquid electrolyte precursors in traditional LMFP and porous LMFP electrodes. Reproduced with permission from ref. 415. Copyright 2023, Wiley-VCH GmbH. (d) Schematic illustration of the synthesis process of integrated 3D LixMnO2 nanosheet arrays and LiPON SE. Reproduced with permission from ref. 53. Copyright 2020, Wiley-VCH GmbH. (e) Simulation of Li+ transport kinetics in the traditional-LFP and aligned-LFP samples. Reproduced with permission from ref. 419. Copyright Elsevier B.V. | ||
3D integrated structures effectively promote Li+ transport kinetics and enable high cathode loading, which is a significant strategy to realize SSLMBs with fast charging and high energy density. However, the difficult preparation methods of 3D structured cathodes and uncontrollable electrolyte filling behavior limit the realization of large-scale applications.
5. Summary and outlook
As the demand for secondary lithium-ion batteries increases in both the electric vehicle and stationary storage sector, so does the demand for higher energy density, power density, and lifespan of SSLMBs. However, it remains a great challenge for SSLMBs to ensure effective carrier transport through the whole battery. Improving carrier transport properties by structural modulation is an ideal solution to tackle this issue. Nevertheless, the intricacy of various systems and different transport behaviors at multiple levels brings about more difficulties, which need to be thoroughly explored and well-solved. This review comprehensively summarizes the research progress in the structural modification of cathodes, SEs, and batteries for improving ionic transport, as shown in Fig. 17. Besides, the crucial factors that need attention for structural modulation are analyzed and the modulation results and limitations of various modulation strategies are evaluated. The main contents of this review are shown as follows: | ||
| Fig. 17 Schematic illustration of fast ionic transport in practical batteries through multi-level structural modulation. | ||
(1) Microstructural modulation of composite cathodes involves composition engineering and size engineering, which affect carrier percolation pathways by altering the connectivity, tortuosity, dispersion, and contact area between particles. The addition of SEs is a prerequisite for the construction of an effective ionic transport network. Insufficient ionic transport channels occur when the SE content is too low, which hinders the participation of some CAMs in the electrochemical process, resulting in a decrease in the capacity of SSLMBs. For the composite cathode without electronically conductive agents, the trends of electronic and ionic conductivity with the CAM/SE proportion are opposite. Conductive agents facilitate the fast migration of electrons, guaranteeing efficient electrochemical conversion of CAMs, especially under high current density. It is worth noting that an effective electronic transport network can only be formed since the percolation threshold is exceeded. However, if the content of the conductive agent is excessively high, it hinders ionic transport and decreases the capacity of SSLMBs. In addition, the utilization of carbon-based conductive agents accelerates the decomposition of sulfide SEs, thereby diminishing the cycling stability of SSLMBs. Applying a tiny quantity of binder can improve the adhesion between particles and thus ensure the efficiency of carrier transport during battery operation. Nevertheless, an excessive amount of binder can impede the carrier transfer at the three-phase interface and diminish the CAM utilization. But the inherent insulating properties of typical binders contribute to the elevated barrier of ionic migration. Moreover, the application of these inactive materials decreases the proportion of CAMs in cathodes, hence restricting the energy density of SSLMBs. Therefore, developing all-electrochemically-active cathodes is of great significance as they feature a self-supported integrated electronic–ionic conductive network.
(2) A basic idea of constructing rapid carrier transport channels by size engineering involves decreasing the particle size of the components in composite cathodes. Applying small-sized CAMs can shorten the percolation pathways for Li+ transport, promote electronic percolation, and increase the active sites, leading to higher utilization of CAMs. Reducing the size of SE particles enhances their uniform dispersion within composite cathodes and increases the contact area with CAM particles, thus facilitating ionic transport and lowering electrode polarization. Nevertheless, there are multiple inconsistencies. First, reducing the size of SE particles can decrease inherent ionic conductivity, hence directly impacting the efficiency of transport along the percolation channel in composite cathodes. Second, the enhanced interfacial contact area can potentially trigger more detrimental interfacial side reactions. Third, the uniform distribution of small-sized SE particles within composite cathodes can lead to the isolation of certain CAM particles and thus decrease CAM utilization. Size engineering also requires consideration of the interplay between the size effects of various components. A higher particle size ratio of CAM to SE promotes the construction of uniform ionic transport networks and enhances the utilization of CAMs. Higher loading of cathodes necessitates larger particle size ratios to achieve high energy density. Nevertheless, the power density of SSLMBs may be restricted by the presence of too-large CAM particles or very small SE particles.
(3) The insufficient ionic conductivity of inorganic SEs is typically determined by their crystal structure, and the primary strategy to manipulate their microstructure is doping. The modulation principle includes regulation of the occupancy at different Li+ sites, increasing the Li+ concentration through charge compensation, inducing favorable local crystal structural changes, increasing the disordering of the Li+ sublattice, and achieving controllable transitions from crystalline to amorphous phases. Besides, the incorporation of point defects or stacking faults into the crystal structure can also diminish the Li+ migration barriers and facilitate the Li+ transfer inside SEs. It is crucial to recognize that these strategies for modulating microstructures depend on the defect transport mechanism of ionic diffusion, which is beneficial for many poor ionic conductors, but is detrimental for fast ion dynamics in materials with crystallographically well-defined diffusion. On the other hand, the ionic conductivity of inorganic SE is also constrained by micro-defects such as pores, grain boundaries, cracks, and secondary phases, which are related to the synthesis process. Hence, it is essential to optimize the preparation process by controlling the sintering temperature and atmosphere, employing advanced sintering techniques and regulating the pressing pressure. The ionic transport kinetics are improved through the reduction, modification and optimization of grain boundaries, increased relative density or densification of SE pellets, and suppression of secondary phase formation. Moreover, since SSLMBs are limited by the confined volume, the stresses and strains generated during the cycling process may cause changes in the structure of SEs, including volume deformation, defect introduction, and lattice distortion, and further affect the ionic transport properties.
(4) The ionic conductivity of CSEs is closely related to the morphology and content of inorganic SE fillers. By manipulating these two parameters, the structural design of CSEs can achieve a significant improvement in ionic conductivity in the same material system. In the case of 0D nanoparticles or 1D nanowires dispersed in the polymer matrix to form CSEs, the ionic conductivity of SEs initially increases and then decreases as the content of inorganic fillers increases. This is because an excessive content of inorganic fillers will be agglomerated, thus hindering ionic transport. When inorganic SEs are incorporated as 3D frameworks, they provide continuous pathways for ionic transport, resulting in a substantial increase in ionic conductivity. However, the process of preparing these frameworks is arduous and relatively demanding, which limits their large-scale production.
(5) To optimize the typical SSLMBs with a planar-type sandwich structure, the initial step is to reduce the thickness of SE layers, which can be achieved by slurry cast, tape cast, solution infusion, cold/hot pressing, and other methods. The prepared SE films are typically classified as freestanding, cathode-supported, and skeleton-supported. Freestanding SE films have enhanced ionic conductivity because of reduced ionic transport distances and minimized grain boundaries, although they possess inadequate mechanical properties. Cathode-supported SE films exhibit a significant level of fusion between cathodes and SEs, resulting in a substantial reduction in interfacial resistance. Skeleton-supported SE films have enhanced mechanical properties, but their ionic conductivity is slightly lower than that of cold-pressed SE pellets. And such skeletons usually require unique preparation techniques. More importantly, all of these conventional film manufacturing processes mentioned above inevitably involve the utilization of solvents and/or binders, which may adversely affect ionic conductivity and interfacial chemical stability. Solvent- and binder-free film fabrication approaches offer tremendous benefits. In addition, the two-dimensional contact between the electrode layer and the SE layer in SSLMBs with sandwich-structures creates limited Li+ transport pathways, which may be further hampered by interfacial issues such as contact failure, interfacial reactions, and SCL formation. Constructing a buffer layer at the electrode/electrolyte interface or building a multilayer SE structure can effectively solve these interfacial problems and ensure rapid Li+ transport at the interface during cycling. However, the introduction of the buffer layer implies the formation of new interfaces, which may lead to more intricate interfacial situations, and thus puts high requirements on the intrinsic characteristics of buffer layers. The multilayer SE structure may lead to discontinuous Li+ migration channels and is not compatible with the objective of thinning SEs. Alternatively, electrolyte permeation approaches have been proposed to create 3D electrode/electrolyte conformal interfaces, thereby increasing the effective permeation pathways for interfacial Li+ transport. In contrast to SE precursor infiltration and melt penetration, which suffer from non-scalability and material limitations, respectively, in situ polymerization shows potential for large-scale production.
(6) 3D integrated structure design provides a novel research idea for improving ionic transport at the battery level. It consists of two processes, firstly, the construction of 3D structured cathodes utilizing unique technologies, and then 3D electrode/electrolyte integration through electrolyte permeation previously discussed. This design constructs continuous conduction pathways for Li+ through entire cathodes and generates sufficient reactive sites, which effectively promotes ionic transport kinetics and is expected to realize higher electrochemical properties of SSLMBs in terms of fast charging and high energy density. However, the preparation process is complex and difficult to achieve on a large scale.
Based on the great progress achieved so far, we put forward several possible directions for structural modulation to achieve fast charge transport kinetics in SSLMBs.
(1) From single-factor analysis to multi-scale, predictive design principles: future research on SSLMBs must transition from isolated factor studies to integrated multi-scale design frameworks. A critical quantitative target is establishing the synergistic effects between at least two key variables (e.g., particle size, content, and distribution) to derive generalized design rules. Methodologically, this requires developing cross-scale multi-physics models that couple electrochemical kinetics with mechanical behavior, validated against in situ/operando characterization data. Specific unresolved challenges include determining the critical size threshold for CAM particles under operational stresses and optimizing the particle size ratio for maximum ionic/electronic percolation to fulfill the requirement of fast charging. The field would benefit from the intelligent-assisted structural design of electrodes and batteries.
(2) Quantitative indicators and implementation approaches for high-loading cathodes: achieving the targets of exceeding 500 Wh kg−1 and 1200 Wh L−1 in SSLMBs critically depends on the successful development of composite cathodes with high areal capacity (>4.0 mAh cm−2). However, current research generally lacks quantitative design principles linking the microstructure to macroscopic performance. The key to future breakthroughs lies in systematically revealing and establishing the quantitative structure–property relationships between critical cathode parameters (e.g., thickness, porosity, and CAM volume fraction) and transport properties (ionic/electronic conductivity) as well as performance outputs (areal capacity and reaction homogeneity). This requires integrating multi-physics simulations, advanced three-dimensional microstructure characterization techniques (such as X-ray tomography), and machine learning to create a closed-loop ‘design-fabrication-performance’ optimization system. Such an approach will provide a clear theoretical foundation and actionable process for the precise manufacturing of high-performance, high-loading cathodes.
(3) Methodological innovations for solid-state electrolyte development: future research must transcend the singular pursuit of ionic conductivity and pivot towards the systematic establishment of quantitative structure–property relationships. Specifically, for CSEs, it is essential to achieve an ionic conductivity >10−3 S cm−1 while synergistically optimizing their electrochemical stability window (>4.3 V vs. Li+/Li) and mechanical strength to meet the demands of ultra-thin processing applications. To achieve these targets, research methodologies must be revolutionized. First, there should be a strong emphasis on rational design based on atomic-scale simulations and high-throughput computations to precisely guide structural modifications of inorganic/polymer electrolytes and interfacial component optimization. Second, it is necessary to establish process control standards suitable for mass production of CSEs. For example, appropriate solvents and binders should be selected for the wet process to maintain the high ionic conductivity of CSEs. And precise slurry rheology indicators and drying kinetics curves should also be defined to ensure the stable preparation of large-area, defect-free ultra-thin electrolyte films.
(4) Quantifying interface compatibility: a paradigm shift from qualitative observations to quantitative metrics is imperative in future studies. Beyond the mere identification of interfacial reaction products, it is paramount to establish standardized quantitative descriptors. These should include not only the interfacial resistance and its growth rate during cycling but also their correlation with critical performance parameters, such as rate capability and cycle life. Establishing such quantitative relationships is crucial for a rigorous assessment of the compatibility between electrode and solid electrolyte materials.
Author contributions
Tianpeng Huang: investigation, data curation, formal analysis, writing – original draft and writing – reviewing and editing; Yue Zheng: resources; Deye Sun: resources; Jun Ma: conceptualization, funding acquisition and writing – reviewing and editing; Pengxian Han: funding acquisition and writing – reviewing and editing; Guanglei Cui: conceptualization, funding acquisition and writing – reviewing and editing.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
This work was financially supported by the National Key R&D Program of China (No. 2023YFC2812700), National Natural Science Foundation of China (No. 22379155 and 52037006), the Youth Innovation Promotion Association of CAS (No. 2021210), Key R&D Program of Shandong Province, China (No. 2023CXGC010302), the Emerging Industry Cultivation Plan of Qingdao Future Industry Cultivation Project (No. 21-1-4-xxgg-7-gx), the Qingdao New Energy Shandong Laboratory (No. QIBEBT/SEI/QNESLS202304), the Taishan Scholars Program (No. ts201511063, No. tsqn202306308), and the Key Scientific and Technological Innovation Project of Shandong (No. 2024CXGC010305).Notes and references
- L. Zhao, A. E. Lakraychi, Z. Chen, Y. Liang and Y. Yao, ACS Energy Lett., 2021, 6, 3287–3306 CrossRef CAS.
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X.-Q. Yang and J.-G. Zhang, Nat. Energy, 2019, 4, 180–186 CrossRef CAS.
- X. Zhang, Y. Yang and Z. Zhou, Chem. Soc. Rev., 2020, 49, 3040–3071 RSC.
- T. Inoue and K. Mukai, ACS Appl. Mater. Interfaces, 2017, 9, 1507–1515 CrossRef CAS.
- L. Wang, Z. Chen, Y. Liu, Y. Li, H. Zhang and X. He, eTransportation, 2023, 16, 100239 CrossRef.
- C. Wang, J. Liang, Y. Zhao, M. Zheng, X. Li and X. Sun, Energy Environ. Sci., 2021, 14, 2577–2619 RSC.
- L. Xu, S. Tang, Y. Cheng, K. Wang, J. Liang, C. Liu, Y.-C. Cao, F. Wei and L. Mai, Joule, 2018, 2, 1991–2015 CrossRef CAS.
- A. Banerjee, X. Wang, C. Fang, E. A. Wu and Y. S. Meng, Chem. Rev., 2020, 120, 6878–6933 CrossRef CAS.
- J. Ma, B. Chen, L. Wang and G. Cui, J. Power Sources, 2018, 392, 94–115 CrossRef CAS.
- X. Miao, S. Guan, C. Ma, L. Li and C. W. Nan, Adv. Mater., 2023, 35, e2206402 CrossRef PubMed.
- J. Liu, H. Yuan, H. Liu, C. Z. Zhao, Y. Lu, X. B. Cheng, J. Q. Huang and Q. Zhang, Adv. Energy Mater., 2021, 12, 2100748 CrossRef.
- J. Janek and W. G. Zeier, Nat. Energy, 2023, 8, 230–240 CrossRef.
- M. Li, T. Liu, Z. Shi, W. Xue, Y. S. Hu, H. Li, X. Huang, J. Li, L. Suo and L. Chen, Adv. Mater., 2021, 33, 2008723 CrossRef CAS.
- Y. Wang, Y. Wu, Z. Wang, L. Chen, H. Li and F. Wu, J. Mater. Chem. A, 2022, 10, 4517–4532 RSC.
- Y. Zeng, B. Ouyang, J. Liu, Y.-W. Byeon, Z. Cai, L. J. Miara, Y. Wang and G. Ceder, Science, 2022, 378, 1320–1324 CrossRef CAS.
- Y. Li, S. Song, H. Kim, K. Nomoto, H. Kim, X. Sun, S. Hori, K. Suzuki, N. Matsui, M. Hirayama, T. Mizoguchi, T. Saito, T. Kamiyama and R. Kanno, Science, 2023, 381, 50–53 CrossRef CAS.
- M. K. Tufail, P. Zhai, M. Jia, N. Zhao and X. Guo, Energy Mater. Adv., 2023, 4, 0015 CrossRef CAS.
- T. Song, Y. Lin, D. Wang, Q. Chen, C. Ling and S. Shi, Adv. Energy Mater., 2024, 14, 2302440 CrossRef CAS.
- Y. Zheng, Y. Yao, J. Ou, M. Li, D. Luo, H. Dou, Z. Li, K. Amine, A. Yu and Z. Chen, Chem. Soc. Rev., 2020, 49, 8790–8839 RSC.
- L.-Z. Fan, H. He and C.-W. Nan, Nat. Rev. Mater., 2021, 6, 1003–1019 CrossRef CAS.
- H. Liang, L. Wang, A. Wang, Y. Song, Y. Wu, Y. Yang and X. He, Nano-Micro Lett., 2023, 15, 42 CrossRef CAS.
- S. Ohno, R. Koerver, G. Dewald, C. Rosenbach, P. Titscher, D. Steckermeier, A. Kwade, J. Janek and W. G. Zeier, Chem. Mater., 2019, 31, 2930–2940 CrossRef CAS.
- H.-K. Tian, A. Chakraborty, A. A. Talin, P. Eisenlohr and Y. Qi, J. Electrochem. Soc., 2020, 167, 090541 CrossRef CAS.
- Z. Xu, M. M. Rahman, L. Mu, Y. Liu and F. Lin, J. Mater. Chem. A, 2018, 6, 21859–21884 RSC.
- F. Sun, C. Wang, M. Osenberg, K. Dong, S. Zhang, C. Yang, Y. Wang, A. Hilger, J. Zhang, S. Dong, H. Markötter, I. Manke and G. Cui, Adv. Energy Mater., 2022, 12, 2103714 CrossRef CAS.
- J. Adjah, K. I. Orisekeh, R. A. Ahmed, M. Vandadi, B. Agyei-Tuffour, D. Dodoo-Arhin, E. Nyankson, J. Asare, N. Rahbar and W. O. Soboyejo, J. Power Sources, 2024, 594, 234022 CrossRef CAS.
- J. Zhao, Y. Tang, Q. Dai, C. Du, Y. Zhang, D. Xue, T. Chen, J. Chen, B. Wang, J. Yao, N. Zhao, Y. Li, S. Xia, X. Guo, S. J. Harris, L. Zhang, S. Zhang, T. Zhu and J. Huang, Energy Environ. Mater., 2021, 5, 524–532 CrossRef.
- Z. Ning, D. S. Jolly, G. Li, R. De Meyere, S. D. Pu, Y. Chen, J. Kasemchainan, J. Ihli, C. Gong, B. Liu, D. L. R. Melvin, A. Bonnin, O. Magdysyuk, P. Adamson, G. O. Hartley, C. W. Monroe, T. J. Marrow and P. G. Bruce, Nat. Mater., 2021, 20, 1121–1129 CrossRef CAS PubMed.
- Z. Ning, G. Li, D. L. R. Melvin, Y. Chen, J. Bu, D. Spencer-Jolly, J. Liu, B. Hu, X. Gao, J. Perera, C. Gong, S. D. Pu, S. Zhang, B. Liu, G. O. Hartley, A. J. Bodey, R. I. Todd, P. S. Grant, D. E. J. Armstrong, T. J. Marrow, C. W. Monroe and P. G. Bruce, Nature, 2023, 618, 287–293 CrossRef CAS PubMed.
- W. Wang, J. Wang, C. Lin and H. Ruan, Adv. Funct. Mater., 2023, 33, 2303484 CrossRef CAS.
- X. Sun, L. Wang, J. Ma, X. Yu, S. Zhang, X. Zhou and G. Cui, ACS Appl. Mater. Interfaces, 2022, 14, 17674–17681 CrossRef CAS.
- S. Lee, L. Su, A. Mesnier, Z. Cui and A. Manthiram, Joule, 2023, 7, 2430–2444 CrossRef CAS.
- L. Romano Brandt, J.-J. Marie, T. Moxham, D. P. Förstermann, E. Salvati, C. Besnard, C. Papadaki, Z. Wang, P. G. Bruce and A. M. Korsunsky, Energy Environ. Sci., 2020, 13, 3556–3566 RSC.
- Y. Bi, J. Tao, Y. Wu, L. Li, Y. Xu, E. Hu, B. Wu, J. Hu, C. Wang, J.-G. Zhang, Y. Qi and J. Xiao, Science, 2020, 370, 1313–1317 CrossRef CAS PubMed.
- Y. Zhou, H. Zhang, Y. Wang, T. Wan, P. Guan, X. Zhou, X. Wang, Y. Chen, H. Shi, A. Dou, M. Su, R. Guo, Y. Liu, L. Dai and D. Chu, ACS Nano, 2023, 17, 20621–20633 CrossRef CAS.
- V. Faka, M. T. Agne, M. A. Lange, D. Daisenberger, B. Wankmiller, S. Schwarzmuller, H. Huppertz, O. Maus, B. Helm, T. Boger, J. Hartel, J. M. Gerdes, J. J. Molaison, G. Kieslich, M. R. Hansen and W. G. Zeier, J. Am. Chem. Soc., 2024, 146, 1710–1721 CrossRef CAS PubMed.
- P. Minnmann, F. Strauss, A. Bielefeld, R. Ruess, P. Adelhelm, S. Burkhardt, S. L. Dreyer, E. Trevisanello, H. Ehrenberg, T. Brezesinski, F. H. Richter and J. Janek, Adv. Energy Mater., 2022, 12, 202201425 Search PubMed.
- X. Yang, K. Doyle-Davis, X. Gao and X. Sun, eTransportation, 2022, 11, 100152 CrossRef.
- W. Jiang, X. Zhu, Y. Liu, S. Zhao, R. Huang, M. Ling, L. Wang and C. Liang, eTransportation, 2023, 17, 100246 CrossRef.
- Z. Zeng, J. Cheng, Y. Li, H. Zhang, D. Li, H. Liu, F. Ji, Q. Sun and L. Ci, Mater. Today Phys., 2023, 32, 101009 CrossRef CAS.
- X. Yu, J. Ma, C. Mou and G. Cui, Acta Phys.-Chim. Sin., 2022, 38, 1912061 Search PubMed.
- Z. Zhang, H. Chen, Z. Hu, S. Zhou, L. Zhang and J. Luo, Front. Energy, 2022, 16, 706–733 CrossRef.
- F. Zheng, C. Li, Z. Li, X. Cao, H. Luo, J. Liang, X. Zhao and J. Kong, Small, 2023, 19, e2206355 CrossRef.
- Z. Chen, Q. Ai, A. E. Lakraychi, C. Wu, L. Zhao, L. Guo, V. G. Hadjiev, H. Guo, Z. Fan, J. Lou, Y. Liang and Y. Yao, Adv. Energy Mater., 2024, 14, 2403050 CrossRef CAS.
- F. Han, J. Yue, C. Chen, N. Zhao, X. Fan, Z. Ma, T. Gao, F. Wang, X. Guo and C. Wang, Joule, 2018, 2, 497–508 CrossRef CAS.
- H. Guo, F. Shen, W. Guo, D. Zeng, Y. Yin and X. Han, Mater. Lett., 2021, 301, 130302 CrossRef CAS.
- J. Zhang, C. Wang, M. Zheng, M. Ye, H. Zhai, J. Li, G. Tan, X. Tang and X. Sun, Nano Energy, 2022, 102, 107672 CrossRef CAS.
- K. Chen, S. Shinjo, A. Sakuda, K. Yamamoto, T. Uchiyama, K. Kuratani, T. Takeuchi, Y. Orikasa, A. Hayashi, M. Tatsumisago, Y. Kimura, T. Nakamura, K. Amezawa and Y. Uchimoto, J. Phys. Chem. C, 2019, 123, 3292–3298 CrossRef CAS.
- N. Hu, Y. H. Zhang, Y. Yang, H. Wu, Y. Liu, C. Hao, Y. Zheng, D. Sun, W. Li, J. Li, Z. Hu, T. S. Chan, C. W. Kao, Q. Kong, X. Wang, S. C. Haw, J. Ma and G. Cui, Adv. Energy Mater., 2024, 14, 2303797 CrossRef CAS.
- Y. Kimura, M. Fakkao, T. Nakamura, T. Okumura, N. Ishiguro, O. Sekizawa, K. Nitta, T. Uruga, M. Tada, Y. Uchimoto and K. Amezawa, ACS Appl. Energy Mater., 2020, 3, 7782–7793 CrossRef CAS.
- C. Huang, C. L. A. Leung, P. Leung and P. S. Grant, Adv. Energy Mater., 2020, 11, 2002387 CrossRef.
- W. Liu, C. Yi, L. Li, S. Liu, Q. Gui, D. Ba, Y. Li, D. Peng and J. Liu, Angew. Chem., Int. Ed., 2021, 60, 12931–12940 CrossRef CAS.
- Q. Xia, Q. Zhang, S. Sun, F. Hussain, C. Zhang, X. Zhu, F. Meng, K. Liu, H. Geng, J. Xu, F. Zan, P. Wang, L. Gu and H. Xia, Adv. Mater., 2021, 33, e2003524 CrossRef.
- W. J. Li, M. Hirayama, K. Suzuki and R. Kanno, Mater. Trans., 2016, 57, 549–552 CrossRef CAS.
- R. Koerver, I. Aygün, T. Leichtweiß, C. Dietrich, W. Zhang, J. O. Binder, P. Hartmann, W. G. Zeier and J. Janek, Chem. Mater., 2017, 29, 5574–5582 CrossRef CAS.
- D. Wang, Q. Sun, J. Luo, J. Liang, Y. Sun, R. Li, K. Adair, L. Zhang, R. Yang, S. Lu, H. Huang and X. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 4954–4961 CrossRef CAS.
- E. J. Cheng, Y. Kushida, T. Abe and K. Kanamura, ACS Appl. Mater. Interfaces, 2022, 14, 40881–40889 CrossRef CAS.
- Y. Xiao, Y. Wang, S.-H. Bo, J. C. Kim, L. J. Miara and G. Ceder, Nat. Rev. Mater., 2020, 5, 105–126 CrossRef CAS.
- Y. Zhu, X. He and Y. Mo, ACS Appl. Mater. Interfaces, 2015, 7, 23685–23693 CrossRef CAS.
- T. K. Schwietert, V. A. Arszelewska, C. Wang, C. Yu, A. Vasileiadis, N. J. J. de Klerk, J. Hageman, T. Hupfer, I. Kerkamm, Y. Xu, E. van der Maas, E. M. Kelder, S. Ganapathy and M. Wagemaker, Nat. Mater., 2020, 19, 428–435 CrossRef CAS.
- T. Krauskopf, F. H. Richter, W. G. Zeier and J. Janek, Chem. Rev., 2020, 120, 7745–7794 CrossRef CAS PubMed.
- K. Yamamoto, Y. Iriyama, T. Asaka, T. Hirayama, H. Fujita, C. A. Fisher, K. Nonaka, Y. Sugita and Z. Ogumi, Angew. Chem., Int. Ed., 2010, 49, 4414–4417 CrossRef CAS.
- N. J. J. de Klerk and M. Wagemaker, ACS Appl. Energy Mater., 2018, 1, 5609–5618 CAS.
- Y. Nomura, K. Yamamoto, T. Hirayama, S. Ouchi, E. Igaki and K. Saitoh, Angew. Chem., Int. Ed., 2019, 58, 5292–5296 CrossRef CAS.
- J. Zhang, C. Zheng, L. Li, Y. Xia, H. Huang, Y. Gan, C. Liang, X. He, X. Tao and W. Zhang, Adv. Energy Mater., 2019, 10, 1903311 CrossRef.
- Z. Cheng, M. Liu, S. Ganapathy, C. Li, Z. Li, X. Zhang, P. He, H. Zhou and M. Wagemaker, Joule, 2020, 4, 1311–1323 CrossRef CAS.
- L. Wang, R. Xie, B. Chen, X. Yu, J. Ma, C. Li, Z. Hu, X. Sun, C. Xu, S. Dong, T. S. Chan, J. Luo, G. Cui and L. Chen, Nat. Commun., 2020, 11, 5889 CrossRef CAS PubMed.
- L. Katzenmeier, L. Carstensen, S. J. Schaper, P. Muller-Buschbaum and A. S. Bandarenka, Adv. Mater., 2021, 33, e2100585 CrossRef PubMed.
- Z. Gu, J. Ma, F. Zhu, T. Liu, K. Wang, C. W. Nan, Z. Li and C. Ma, Nat. Commun., 2023, 14, 1632 CrossRef CAS.
- S. P. Culver, R. Koerver, W. G. Zeier and J. Janek, Adv. Energy Mater., 2019, 9, 1900626 CrossRef.
- H. J. Guo, Y. Sun, Y. Zhao, G. X. Liu, Y. X. Song, J. Wan, K. C. Jiang, Y. G. Guo, X. Sun and R. Wen, Angew. Chem., Int. Ed., 2022, 61, e202211626 CrossRef CAS.
- J. Liang, Y. Zhu, X. Li, J. Luo, S. Deng, Y. Zhao, Y. Sun, D. Wu, Y. Hu, W. Li, T. K. Sham, R. Li, M. Gu and X. Sun, Nat. Commun., 2023, 14, 146 CrossRef CAS.
- L. Wang, A. Mukherjee, C.-Y. Kuo, S. Chakrabarty, R. Yemini, A. A. Dameron, J. W. DuMont, S. H. Akella, A. Saha, S. Taragin, H. Aviv, D. Naveh, D. Sharon, T.-S. Chan, H.-J. Lin, J.-F. Lee, C.-T. Chen, B. Liu, X. Gao, S. Basu, Z. Hu, D. Aurbach, P. G. Bruce and M. Noked, Nat. Nanotechnol., 2023, 19, 208–218 CrossRef PubMed.
- U. Nisar, N. Muralidharan, R. Essehli, R. Amin and I. Belharouak, Energy Storage Mater., 2021, 38, 309–328 CrossRef.
- W. Li, S. Zhang, W. Zheng, J. Ma, L. Li, Y. Zheng, D. Sun, Z. Wen, Z. Liu, Y. Wang, G. Zhang and G. Cui, Adv. Funct. Mater., 2023, 33, 2300791 CrossRef CAS.
- D. H. Kim, D. Y. Oh, K. H. Park, Y. E. Choi, Y. J. Nam, H. A. Lee, S. M. Lee and Y. S. Jung, Nano Lett., 2017, 17, 3013–3020 CrossRef CAS PubMed.
- S. Yubuchi, M. Uematsu, M. Deguchi, A. Hayashi and M. Tatsumisago, ACS Appl. Energy Mater., 2018, 1, 3622–3629 CrossRef CAS.
- Y. Xiao, K. Turcheniuk, A. Narla, A. Y. Song, X. Ren, A. Magasinski, A. Jain, S. Huang, H. Lee and G. Yushin, Nat. Mater., 2021, 20, 984–990 CrossRef CAS PubMed.
- G. T. Hitz, D. W. McOwen, L. Zhang, Z. Ma, Z. Fu, Y. Wen, Y. Gong, J. Dai, T. R. Hamann, L. Hu and E. D. Wachsman, Mater. Today, 2019, 22, 50–57 CrossRef CAS.
- J. Zhu, X. Li, C. Wu, J. Gao, H. Xu, Y. Li, X. Guo, H. Li and W. Zhou, Angew. Chem., Int. Ed., 2021, 60, 3781–3790 CrossRef CAS PubMed.
- J. Wu, Z. Ju, X. Zhang, A. C. Marschilok, K. J. Takeuchi, H. Wang, E. S. Takeuchi and G. Yu, Adv. Mater., 2022, 34, e2202780 CrossRef.
- Y. Hu, X. Xie, W. Li, Q. Huang, H. Huang, S.-M. Hao, L.-Z. Fan and W. Zhou, ACS Sustainable Chem. Eng., 2023, 11, 1253–1277 CrossRef CAS.
- B. Hu, S. Zhang, Z. Ning, D. Spencer-Jolly, D. L. R. Melvin, X. Gao, J. Perera, S. D. Pu, G. J. Rees, L. Wang, L. Yang, H. Gao, S. Marathe, G. Burca, T. J. Marrow and P. G. Bruce, Joule, 2024, 8, 2623–2638 CrossRef CAS.
- Z. Zhang, J. Gou, K. Cui, X. Zhang, Y. Yao, S. Wang and H. Wang, Nano-Micro Lett., 2024, 16, 181 CrossRef CAS.
- E. P. Alsaç, D. L. Nelson, S. G. Yoon, K. A. Cavallaro, C. Wang, S. E. Sandoval, U. D. Eze, W. J. Jeong and M. T. McDowell, Chem. Rev., 2025, 125, 2009–2119 CrossRef.
- S. E. Sandoval, C. G. Haslam, B. S. Vishnugopi, D. W. Liao, J. S. Yoon, S. H. Park, Y. Wang, D. Mitlin, K. B. Hatzell, D. J. Siegel, P. P. Mukherjee, N. P. Dasgupta, J. Sakamoto and M. T. McDowell, Nat. Mater., 2025, 24, 673–681 CrossRef CAS PubMed.
- S. Dong, L. Sheng, L. Wang, J. Liang, H. Zhang, Z. Chen, H. Xu and X. He, Adv. Funct. Mater., 2023, 33, 2304371 CrossRef CAS.
- G. Yoon, S. Kim and J.-S. Kim, Adv. Sci., 2023, 10, 2302263 CrossRef CAS PubMed.
- J. Lu, C. Xu, W. Dose, S. Dey, X. Wang, Y. Wu, D. Li and L. Ci, Chem. Soc. Rev., 2024, 53, 4707–4740 RSC.
- Z. Song, Y. Ma, X. Cheng, Z. Zhu, Y. Zhong, J. He, T. Wang, D. Xu, Q. Zhang, K. I. Ozoemena, T. Brezesinski, Y. Ma, S. Passerini and Y. Wu, Mater. Today, 2025, 88, 1005–1027 CrossRef CAS.
- F. Yu, Y. Wang, J. Liu, Y. Zhang, Z. Yang, H. Shen, J. Li, G. Zeng, K. Zhang, D. Cui, J. Xia, M. Yang, H. Liu, C. Guo, W. Bao, K. Sun and J. Li, Small, 2025, 21, 2505434 CrossRef CAS PubMed.
- P. Minnmann, L. Quillman, S. Burkhardt, F. H. Richter and J. Janek, J. Electrochem. Soc., 2021, 168, 040537 CrossRef CAS.
- H. Erabhoina and M. Thelakkat, Sci. Rep., 2022, 12, 5454 CrossRef CAS PubMed.
- A. Bielefeld, D. A. Weber and J. Janek, ACS Appl. Mater. Interfaces, 2020, 12, 12821–12833 CrossRef CAS.
- S. Randau, F. Walther, A. Neumann, Y. Schneider, R. S. Negi, B. Mogwitz, J. Sann, K. Becker-Steinberger, T. Danner, S. Hein, A. Latz, F. H. Richter and J. Janek, Chem. Mater., 2021, 33, 1380–1393 CrossRef CAS.
- A. Vadakkepatt, B. Trembacki, S. R. Mathur and J. Y. Murthy, J. Electrochem. Soc., 2016, 163, A119–A130 CrossRef CAS.
- B. Seo and C. Kim, Int. J. Precis. Eng. Manuf.-Green Technol., 2024, 12, 227–243 CrossRef.
- B. Vijayaraghavan, D. R. Ely, Y.-M. Chiang, R. García-García and R. E. García, J. Electrochem. Soc., 2012, 159, A548–A552 CrossRef CAS.
- D.-K. Kim, H.-M. Park, S.-J. Jung, Y. U. Jeong, J.-H. Lee and J.-J. Kim, J. Power Sources, 2006, 159, 237–240 CrossRef CAS.
- S. Wang, M. Yan, Y. Li, C. Vinado and J. Yang, J. Power Sources, 2018, 393, 75–82 CrossRef CAS.
- J. Xie, T. Tanaka, N. Imanishi, T. Matsumura, A. Hirano, Y. Takeda and O. Yamamoto, J. Power Sources, 2008, 180, 576–581 CrossRef CAS.
- C. Y. Ouyang, S. Q. Shi and M. S. Lei, J. Alloys Compd., 2009, 474, 370–374 CrossRef CAS.
- M. Kunduraci and G. G. Amatucci, J. Electrochem. Soc., 2006, 153, A1345 CrossRef CAS.
- J. W. Kim, D. H. Kim, D. Y. Oh, H. Lee, J. H. Kim, J. H. Lee and Y. S. Jung, J. Power Sources, 2015, 274, 1254–1262 CrossRef CAS.
- W. Du, Q. Shao, Y. Wei, C. Yan, P. Gao, Y. Lin, Y. Jiang, Y. Liu, X. Yu, M. Gao, W. Sun and H. Pan, ACS Energy Lett., 2022, 7, 3006–3014 CrossRef CAS.
- Z. Lin, Z. Liu, N. J. Dudney and C. Liang, ACS Nano, 2013, 7, 2829–2833 CrossRef CAS PubMed.
- F. Liu, L. Wang, Z. Zhang, P. Shi, Y. Feng, Y. Yao, S. Ye, H. Wang, X. Wu and Y. Yu, Adv. Funct. Mater., 2020, 30, 202001607 Search PubMed.
- M. Yang, Y. Yao, M. Chang, F. Tian, W. Xie, X. Zhao, Y. Yu and X. Yao, Adv. Energy Mater., 2023, 13, 2300962 CrossRef CAS.
- K. Wang, Z. Gu, Z. Xi, L. Hu and C. Ma, Nat. Commun., 2023, 14, 1396 CrossRef CAS.
- K. Nagao, Y. Nagata, A. Sakuda, A. Hayashi, M. Deguchi, C. Hotehama, H. Tsukasaki, S. Mori, Y. Orikasa, K. Yamamoto, Y. Uchimoto and M. Tatsumisago, Sci. Adv., 2020, 6, eaax7236 CrossRef CAS PubMed.
- L. Cui, S. Zhang, J. Ju, T. Liu, Y. Zheng, J. Xu, Y. Wang, J. Li, J. Zhao, J. Ma, J. Wang, G. Xu, T.-S. Chan, Y.-C. Huang, S.-C. Haw, J.-M. Chen, Z. Hu and G. Cui, Nat. Energy, 2024, 9, 1084–1094 CAS.
- J. Fu, C. Wang, S. Wang, J. W. Reid, J. Liang, J. Luo, J. T. Kim, Y. Zhao, X. Yang, F. Zhao, W. Li, B. Fu, X. Lin, Y. Hu, H. Su, X. Hao, Y. Gao, S. Zhang, Z. Wang, J. Liu, H. Abdolvand, T. K. Sham, Y. Mo and X. Sun, Nature, 2025, 643, 111–118 CrossRef CAS.
- D. Cheng, T. Wynn, B. Lu, M. Marple, B. Han, R. Shimizu, B. Sreenarayanan, J. Bickel, P. Hosemann, Y. Yang, H. Nguyen, W. Li, G. Zhu, M. Zhang and Y. S. Meng, Nat. Nanotechnol., 2023, 18, 1448–1455 CrossRef CAS PubMed.
- Z. Liu, W. Fu, E. A. Payzant, X. Yu, Z. Wu, N. J. Dudney, J. Kiggans, K. Hong, A. J. Rondinone and C. Liang, J. Am. Chem. Soc., 2013, 135, 975–978 CrossRef CAS PubMed.
- F. Han, A. S. Westover, J. Yue, X. Fan, F. Wang, M. Chi, D. N. Leonard, N. J. Dudney, H. Wang and C. Wang, Nat. Energy, 2019, 4, 187–196 CrossRef CAS.
- Y. Seino, T. Ota, K. Takada, A. Hayashi and M. Tatsumisago, Energy Environ. Sci., 2014, 7, 627–631 RSC.
- H. J. Deiseroth, J. Maier, K. Weichert, V. Nickel, S. T. Kong and C. Reiner, Z. Anorg. Allg. Chem., 2011, 637, 1287–1294 CrossRef CAS.
- L. Zhou, K.-H. Park, X. Sun, F. Lalère, T. Adermann, P. Hartmann and L. F. Nazar, ACS Energy Lett., 2018, 4, 265–270 CrossRef.
- Z.-Y. He, Z.-Q. Zhang, M. Yu, C. Yu, H.-T. Ren, J.-Z. Zhang, L.-F. Peng, L. Zhang, S.-J. Cheng and J. Xie, Rare Met., 2021, 41, 798–805 CrossRef.
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto and A. Mitsui, Nat. Mater., 2011, 10, 682–686 CrossRef CAS PubMed.
- S. Wenzel, S. Randau, T. Leichtweiß, D. A. Weber, J. Sann, W. G. Zeier and J. Janek, Chem. Mater., 2016, 28, 2400–2407 CrossRef CAS.
- S. Strangmüller, H. Eickhoff, D. Müller, W. Klein, G. Raudaschl-Sieber, H. Kirchhain, C. Sedlmeier, V. Baran, A. Senyshyn, V. L. Deringer, L. van Wüllen, H. A. Gasteiger and T. F. Fässler, J. Am. Chem. Soc., 2019, 141, 14200–14209 CrossRef.
- Z.-B. Xiao, M.-Y. Ma, X.-M. Wu, Z.-Q. He and S. Chen, Trans. Nonferrous. Metal. Soc., 2006, 16, 281–285 CrossRef CAS.
- J. K. Feng, L. Lu and M. O. Lai, J. Alloys Compd., 2010, 501, 255–258 CrossRef CAS.
- J. Li, H. Luo, K. Liu, J. Zhang, H. Zhai, X. Su, J. Wu, X. Tang and G. Tan, ACS Appl. Mater. Interfaces, 2023, 15, 7165–7174 CrossRef CAS PubMed.
- Y.-T. Chen, A. Jena, W. K. Pang, V. K. Peterson, H.-S. Sheu, H. Chang and R.-S. Liu, J. Phys. Chem. C, 2017, 121, 15565–15573 CrossRef CAS.
- K. Ishiguro, H. Nemori, S. Sunahiro, Y. Nakata, R. Sudo, M. Matsui, Y. Takeda, O. Yamamoto and N. Imanishi, J. Electrochem. Soc., 2014, 161, A668–A674 CrossRef CAS.
- W. Ping, C. Wang, Z. Lin, E. Hitz, C. Yang, H. Wang and L. Hu, Adv. Energy Mater., 2020, 10, 2000702 CrossRef CAS.
- S. Qin, X. Zhu, Y. Jiang, M. E. Ling, Z. Hu and J. Zhu, Appl. Phys. Lett., 2018, 112, 113901 CrossRef.
- M. Philipp, B. Gadermaier, P. Posch, I. Hanzu, S. Ganschow, M. Meven, D. Rettenwander, G. J. Redhammer and H. M. R. Wilkening, Adv. Mater. Interfaces, 2020, 7, 202000450 Search PubMed.
- Y. Huang, Y. Jiang, Y. Zhou, X. Liu, X. Zeng and X. Zhu, Ionics, 2020, 27, 145–155 CrossRef.
- Z. Song, T. Wang, H. Yang, W. H. Kan, Y. Chen, Q. Yu, L. Wang, Y. Zhang, Y. Dai, H. Chen, W. Yin, T. Honda, M. Avdeev, H. Xu, J. Ma, Y. Huang and W. Luo, Nat. Commun., 2024, 15, 1481 CrossRef CAS PubMed.
- T. Asano, A. Sakai, S. Ouchi, M. Sakaida, A. Miyazaki and S. Hasegawa, Adv. Mater., 2018, 30, e1803075 CrossRef.
- L. Zhou, T.-T. Zuo, C. Y. Kwok, S. Y. Kim, A. Assoud, Q. Zhang, J. Janek and L. F. Nazar, Nat. Energy, 2022, 7, 83–93 CrossRef CAS.
- J. Park, K. T. Kim, D. Y. Oh, D. Jin, D. Kim, Y. S. Jung and Y. M. Lee, Adv. Energy Mater., 2020, 10, 2001563 CrossRef CAS.
- Y. Yamada, K. Watanabe, H. S. Kim, K. Suzuki, S. Hori, R. Kanno and M. Hirayama, Batteries Supercaps, 2023, 6, e202300261 CrossRef CAS.
- S. Deng, Y. Wang, T. Sun, W. Li, M. Ge, J. Wang, P. Cloetens, P. Pianetta, D. Mitlin and Y. Liu, Angew. Chem., Int. Ed., 2025, 64, e202511534 CrossRef CAS.
- Y. Kato, S. Hori, T. Saito, K. Suzuki, M. Hirayama, A. Mitsui, M. Yonemura, H. Iba and R. Kanno, Nat. Energy, 2016, 1, 16030 CrossRef CAS.
- Y. Kato, S. Shiotani, K. Morita, K. Suzuki, M. Hirayama and R. Kanno, J. Phys. Chem. Lett., 2018, 9, 607–613 CrossRef CAS.
- M. Ebner, F. Geldmacher, F. Marone, M. Stampanoni and V. Wood, Adv. Energy Mater., 2013, 3, 845–850 CrossRef CAS.
- M. Otoyama, Y. Ito, A. Hayashi and M. Tatsumisago, J. Power Sources, 2016, 302, 419–425 CrossRef CAS.
- A. Tron, R. Hamid, N. Zhang and A. Beutl, Nanomaterials, 2023, 13, 327 CrossRef CAS.
- T. Shi, Q. Tu, Y. Tian, Y. Xiao, L. J. Miara, O. Kononova and G. Ceder, Adv. Energy Mater., 2019, 10, 201902881 Search PubMed.
- W. Zhang, D. A. Weber, H. Weigand, T. Arlt, I. Manke, D. Schroder, R. Koerver, T. Leichtweiss, P. Hartmann, W. G. Zeier and J. Janek, ACS Appl. Mater. Interfaces, 2017, 9, 17835–17845 CrossRef CAS PubMed.
- M. Clausnitzer, R. Mücke, F. Al-Jaljouli, S. Hein, M. Finsterbusch, T. Danner, D. Fattakhova-Rohlfing, O. Guillon and A. Latz, Batteries Supercaps, 2023, 6, e202300167 CrossRef CAS.
- D. Wang, L. J. Jhang, R. Kou, M. Liao, S. Zheng, H. Jiang, P. Shi, G. X. Li, K. Meng and D. Wang, Nat. Commun., 2023, 14, 1895 CrossRef CAS.
- W. Lee, J. Kee, T. Yu, H.-J. Kim, M. K. Kim, S. Jang, J. Kim, Y.-J. Han, S. Choi, S. Choi, T.-H. Kim, S.-H. Park, W. Jin, G. Song, D.-H. Seo, S.-K. Jung and J. Kim, Nat. Commun., 2024, 15, 5860 CrossRef CAS.
- W. J. Li, M. Hirayama, K. Suzuki and R. Kanno, Solid State Ionics, 2016, 285, 136–142 CrossRef CAS.
- F. Nobili, F. Croce, B. Scrosati and R. Marassi, Chem. Mater., 2001, 13, 1642–1646 CrossRef CAS.
- C. Sangrós Giménez, L. Helmers, C. Schilde, A. Diener and A. Kwade, Chem. Eng. Technol., 2020, 43, 819–829 CrossRef.
- W. Zhang, T. Leichtweiss, S. P. Culver, R. Koerver, D. Das, D. A. Weber, W. G. Zeier and J. Janek, ACS Appl. Mater. Interfaces, 2017, 9, 35888–35896 CrossRef CAS.
- F. Walther, S. Randau, Y. Schneider, J. Sann, M. Rohnke, F. H. Richter, W. G. Zeier and J. Janek, Chem. Mater., 2020, 32, 6123–6136 CrossRef CAS.
- D. H. S. Tan, Y.-T. Chen, H. Yang, W. Bao, B. Sreenarayanan, J.-M. Doux, W. Li, B. Lu, S.-Y. Ham, B. Sayahpour, J. Scharf, H. E. Han, H. J. Han, H. Jeong, J. B. Lee, Z. Chen and Y. S. Meng, Science, 2021, 373, 1494–1499 CrossRef CAS.
- F. Han, T. Gao, Y. Zhu, K. J. Gaskell and C. Wang, Adv. Mater., 2015, 27, 3473–3483 CrossRef CAS PubMed.
- K. Yoon, J. J. Kim, W. M. Seong, M. H. Lee and K. Kang, Sci. Rep., 2018, 8, 8066 CrossRef PubMed.
- F. Strauss, D. Stepien, J. Maibach, L. Pfaffmann, S. Indris, P. Hartmann and T. Brezesinski, RSC Adv., 2020, 10, 1114–1119 RSC.
- T. Ates, M. Keller, J. Kulisch, T. Adermann and S. Passerini, Energy Storage Mater., 2019, 17, 204–210 CrossRef.
- N. Lee, J. Lee, T. Lee, J. Oh, I. Hwang, G. Seo, H. Kim and J. W. Choi, ACS Appl. Mater. Interfaces, 2023, 15, 34931–34940 CrossRef CAS.
- S. Deng, Y. Sun, X. Li, Z. Ren, J. Liang, K. Doyle-Davis, J. Liang, W. Li, M. Norouzi Banis, Q. Sun, R. Li, Y. Hu, H. Huang, L. Zhang, S. Lu, J. Luo and X. Sun, ACS Energy Lett., 2020, 5, 1243–1251 CrossRef CAS.
- R. Fang, Y. Liu, Y. Li, A. Manthiram and J. B. Goodenough, Mater. Today, 2023, 64, 52–60 CrossRef CAS.
- S.-B. Hong, Y.-J. Lee, U.-H. Kim, C. Bak, Y. M. Lee, W. Cho, H. J. Hah, Y.-K. Sun and D.-W. Kim, ACS Energy Lett., 2022, 7, 1092–1100 CrossRef CAS.
- Y.-J. Lee, S.-B. Hong and D.-W. Kim, J. Ind. Eng. Chem., 2023, 122, 341–348 CrossRef CAS.
- K. Lee, S. Kim, J. Park, S. H. Park, A. Coskun, D. S. Jung, W. Cho and J. W. Choi, J. Electrochem. Soc., 2017, 164, A2075–A2081 CrossRef CAS.
- J. Zhang, H. Zhong, C. Zheng, Y. Xia, C. Liang, H. Huang, Y. Gan, X. Tao and W. Zhang, J. Power Sources, 2018, 391, 73–79 CrossRef CAS.
- K. Wang, Q. Ye, J. Zhang, H. Huang, Y. Gan, X. He and W. Zhang, Front. Mater., 2021, 8, 727617 CrossRef.
- N. C. Rosero-Navarro, T. Kinoshita, A. Miura, M. Higuchi and K. Tadanaga, Ionics, 2017, 23, 1619–1624 CrossRef CAS.
- Y. J. Nam, D. Y. Oh, S. H. Jung and Y. S. Jung, J. Power Sources, 2018, 375, 93–101 CrossRef CAS.
- D. Y. Oh, D. H. Kim, S. H. Jung, J.-G. Han, N.-S. Choi and Y. S. Jung, J. Mater. Chem. A, 2017, 5, 20771–20779 RSC.
- D. Y. Oh, Y. J. Nam, K. H. Park, S. H. Jung, K. T. Kim, A. R. Ha and Y. S. Jung, Adv. Energy Mater., 2019, 9, 1802927 CrossRef.
- K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 13121–13129 CrossRef CAS PubMed.
- K. Ueno, J. Murai, K. Ikeda, S. Tsuzuki, M. Tsuchiya, R. Tatara, T. Mandai, Y. Umebayashi, K. Dokko and M. Watanabe, J. Phys. Chem. C, 2015, 120, 15792–15802 CrossRef.
- M. Li, H. Pan, T. Liu, X. Xiong, X. Yu, Y.-S. Hu, H. Li, X. Huang, L. Suo and L. Chen, ACS Energy Lett., 2022, 7, 766–772 CrossRef CAS.
- S. B. Hong, Y. J. Lee, H. J. Lee, H. T. Sim, H. Lee, Y. M. Lee and D. W. Kim, Small, 2023, 20, 2304747 CrossRef.
- M. Nagao, A. Hayashi and M. Tatsumisago, Electrochim. Acta, 2011, 56, 6055–6059 CrossRef CAS.
- M. Nagao, A. Hayashi, M. Tatsumisago, T. Ichinose, T. Ozaki, Y. Togawa and S. Mori, J. Power Sources, 2015, 274, 471–476 CrossRef CAS.
- G. F. Dewald, Z. Liaqat, M. A. Lange, W. Tremel and W. G. Zeier, Angew. Chem., Int. Ed., 2021, 60, 17952–17956 CrossRef CAS PubMed.
- Y.-W. Song, K. Heo, D. Hwang, M.-Y. Kim, H. Lee, B.-S. Kang, Y. Hong, H.-S. Kim, J. Kim and J. Lim, Ionics, 2022, 28, 5421–5431 CrossRef CAS.
- X. Yao, D. Liu, C. Wang, P. Long, G. Peng, Y. S. Hu, H. Li, L. Chen and X. Xu, Nano Lett., 2016, 16, 7148–7154 CrossRef CAS PubMed.
- F. Strauss, T. Bartsch, L. de Biasi, A. Y. Kim, J. Janek, P. Hartmann and T. Brezesinski, ACS Energy Lett., 2018, 3, 992–996 CrossRef CAS.
- W. Jiang, X. Zhu, R. Huang, S. Zhao, X. Fan, M. Ling, C. Liang and L. Wang, Adv. Energy Mater., 2022, 12, 2103473 CrossRef CAS.
- N. C. Rosero-Navarro, A. Miura and K. Tadanaga, J. Power Sources, 2018, 396, 33–40 CrossRef CAS.
- M. Calpa, N. C. Rosero-Navarro, A. Miura and K. Tadanaga, Electrochim. Acta, 2019, 296, 473–480 CrossRef CAS.
- M. Cronau, M. Duchardt, M. Szabo and B. Roling, Batteries Supercaps, 2022, 5, e202200041 CrossRef CAS.
- A. Sakuda, T. Takeuchi and H. Kobayashi, Solid State Ionics, 2016, 285, 112–117 CrossRef CAS.
- E. Hayakawa, H. Nakamura, S. Ohsaki and S. Watano, Adv. Powder Technol., 2022, 33, 103705 CrossRef.
- S. Yu and D. J. Siegel, Chem. Mater., 2017, 29, 9639–9647 CrossRef CAS.
- A. K. Sharma, B. S. Vishnugopi, A. Ayyaswamy, A. Nath, D. Chatterjee, D. Tewari, M. S. Ng, W. S. Tang, V. Premnath, J. A. Jeevarajan and P. P. Mukherjee, ACS Appl. Mater. Interfaces, 2025, 17, 49520–49532 CrossRef CAS PubMed.
- G. Tan, F. Wu, J. Lu, R. Chen, L. Li and K. Amine, Nanoscale, 2014, 6, 10611 RSC.
- X. Lu, X. Zhang, C. Tan, T. M. M. Heenan, M. Lagnoni, K. O’Regan, S. Daemi, A. Bertei, H. G. Jones, G. Hinds, J. Park, E. Kendrick, D. J. L. Brett and P. R. Shearing, Energy Environ. Sci., 2021, 14, 5929–5946 RSC.
- R. Bradbury, G. F. Dewald, M. A. Kraft, T. Arlt, N. Kardjilov, J. Janek, I. Manke, W. G. Zeier and S. Ohno, Adv. Energy Mater., 2023, 13, 2203426 CrossRef CAS.
- A. M. Stavola, X. Sun, D. P. Guida, A. M. Bruck, D. Cao, J. S. Okasinski, A. C. Chuang, H. Zhu and J. W. Gallaway, ACS Energy Lett., 2023, 8, 1273–1280 CrossRef CAS.
- T. Ji, Y. Zhang, J. Torres, A. S. Mijailovic, Y. Tang, X. Zhao, J.-C. Bilheux, J. Wang, B. W. Sheldon, O. Oyedeji and H. Zhu, Nat. Commun., 2025, 16, 7667 CrossRef CAS PubMed.
- E. Schlauymann, J. Drews, L. Ketter, M. A. Lange, T. Danner, A. Latz and W. G. Zeier, ACS Energy Lett., 2025, 10, 1664–1670 CrossRef.
- A. Shodiev, M. Chouchane, M. Gaberscek, O. Arcelus, J. Xu, H. Oularbi, J. Yu, J. Li, M. Morcrette and A. A. Franco, Energy Storage Mater., 2022, 47, 462–471 CrossRef.
- M. Chouchane, W. Yao, A. Cronk, M. Zhang and Y. S. Meng, ACS Energy Lett., 2024, 9, 1480–1486 CrossRef CAS.
- X. Niu, Y. Lu, P. Chen, C. Cao, X. Liao, L. He and Y. Ni, Adv. Energy Mater., 2025, 15, 2502245 CrossRef CAS.
- Y. Lin, H. Huang, Y. Xia, X. Zhang, H. Pan, M. Yan and Y. Jiang, Adv. Energy Mater., 2025, 15, e03269 CrossRef CAS.
- M. Rosen, M. Finsterbusch, O. Guillon and D. Fattakhova-Rohlfing, J. Mater. Chem. A, 2022, 10, 2320–2326 RSC.
- Q.-S. Liu, H.-W. An, X.-F. Wang, F.-P. Kong, Y.-C. Sun, Y.-X. Gong, S.-F. Lou, Y.-F. Shi, N. Sun, B. Deng, J. Wang and J.-J. Wang, Natl. Sci. Rev., 2023, 10, nwac272 CrossRef CAS.
- M. Clausnitzer, T. Danner, B. Prifling, M. Neumann, V. Schmidt and A. Latz, Batteries Supercaps, 2024, 7, e202300522 CrossRef CAS.
- V. Kiyek, C. Schwab, W. S. Scheld, C. Roitzheim, A. Lindner, W. Menesklou, M. Finsterbusch, D. Fattakhova-Rohlfing and O. Guillon, Adv. Sci., 2024, 11, 2404682 CrossRef CAS.
- G. Cheng, J. Yu, Y. Wang, Z. Ju, Y. Zhu, W. Tian, J. Chen, H. Wang and G. Yu, Angew. Chem., Int. Ed., 2025, 64, e202425357 CrossRef CAS PubMed.
- J. C. Bachman, S. Muy, A. Grimaud, H. H. Chang, N. Pour, S. F. Lux, O. Paschos, F. Maglia, S. Lupart, P. Lamp, L. Giordano and Y. Shao-Horn, Chem. Rev., 2016, 116, 140–162 CrossRef CAS.
- P. Lu, Y. Xia, G. Sun, D. Wu, S. Wu, W. Yan, X. Zhu, J. Lu, Q. Niu, S. Shi, Z. Sha, L. Chen, H. Li and F. Wu, Nat. Commun., 2023, 14, 4077 CrossRef CAS.
- R. Li, P. Lu, X. Liang, L. Liu, M. Avdeev, Z. Deng, S. Li, K. Xu, J. Feng, R. Si, F. Wu, Z. Zhang and Y.-S. Hu, ACS Energy Lett., 2024, 9, 1043–1052 CrossRef CAS.
- L. Buannic, B. Orayech, J.-M. López Del Amo, J. Carrasco, N. A. Katcho, F. Aguesse, W. Manalastas, W. Zhang, J. Kilner and A. Llordés, Chem. Mater., 2017, 29, 1769–1778 CrossRef CAS.
- J. Wang, F. Chen, L. Hu and C. Ma, Nano Lett., 2023, 23, 6081–6087 CrossRef CAS.
- X. Li, Y. Xu, C. Zhao, D. Wu, L. Wang, M. Zheng, X. Han, S. Zhang, J. Yue, B. Xiao, W. Xiao, L. Wang, T. Mei, M. Gu, J. Liang and X. Sun, Angew. Chem., Int. Ed., 2023, 62, e202306433 CrossRef CAS.
- K. Jun, Y. Chen, G. Wei, X. Yang and G. Ceder, Nat. Rev. Mater., 2024, 9, 887–905 CrossRef CAS.
- A. Morscher, B. B. Duff, G. Han, L. M. Daniels, Y. Dang, M. Zanella, M. Sonni, A. Malik, M. S. Dyer, R. Chen, F. Blanc, J. B. Claridge and M. J. Rosseinsky, J. Am. Chem. Soc., 2022, 144, 22178–22192 CrossRef CAS PubMed.
- F. Sun, Y. Ynag, S. Zhao, Y. Wang, M. Tang, Q. Huang, Y. Ren, H. Su, B. Wang, N. Zhao, X. Guo and H. Yu, ACS Energy Lett., 2022, 7, 2835–2844 CrossRef CAS.
- P. Adeli, J. D. Bazak, A. Huq, G. R. Goward and L. F. Nazar, Chem. Mater., 2021, 33, 146–157 CrossRef CAS.
- Z. Wang, Y. Jiang, J. Wu, Y. Jiang, W. Ma, Y. Shi, X. Liu, B. Zhao, Y. Xu and J. Zhang, Nano Energy, 2021, 84, 105906 CrossRef CAS.
- J. Liu, J. Zhang, R. You, D. Chen, Y. Li, Y. Lu and Q. Yang, Mater. Res. Express, 2022, 9, 055505 CrossRef CAS.
- J. Yang, S. Chen, Q. Yuan, G. Tan, Q. Liu, S. Yang, Y. Deng, Y. Zhao, W. Liu, Y. Yu, Y. Cui, J. Wang, S.-H. Bo and C. Xu, J. Am. Chem. Soc., 2025, 147, 36557–36569 CrossRef CAS PubMed.
- J. Zhang, L. Li, C. Zheng, Y. Xia, Y. Gan, H. Huang, C. Liang, X. He, X. Tao and W. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 41538–41545 CrossRef.
- H. Buschmann, S. Berendts, B. Mogwitz and J. Janek, J. Power Sources, 2012, 206, 236–244 Search PubMed.
- M. A. Howard, O. Clemens, E. Kendrick, K. S. Knight, D. C. Apperley, P. A. Anderson and P. R. Slater, Dalton Trans., 2012, 41, 12048–12053 RSC.
- E. Rangasamy, J. Wolfenstine and J. Sakamoto, Solid State Ionics, 2012, 206, 28–32 Search PubMed.
- Y. Li, Z. Wang, Y. Cao, F. Du, C. Chen, Z. Cui and X. Guo, Electrochim. Acta, 2015, 180, 37–42 CrossRef CAS.
- R. H. Brugge, J. A. Kilner and A. Aguadero, Solid State Ionics, 2019, 337, 154–160 CrossRef CAS.
- J. Liang, E. van der Maas, J. Luo, X. Li, N. Chen, K. R. Adair, W. Li, J. Li, Y. Hu, J. Liu, L. Zhang, S. Zhao, S. Lu, J. Wang, H. Huang, W. Zhao, S. Paranell, R. I. Smith, S. Canapathy, M. Wagemaker and X. Sun, Adv. Energy Mater., 2022, 12, 2103921 Search PubMed.
- H. Tsukasaki, S. Mori, H. Morimoto, A. Hayashi and M. Tatsumisago, Sci. Rep., 2017, 7, 4142 CrossRef PubMed.
- H. Tsukasaki, S. Mori, S. Shiotani and H. Yamamura, Solid State Ionics, 2018, 317, 122–126 CrossRef CAS.
- F. Zhao, S. Zhang, S. Wang, J. W. Reid, W. Xia, J. Liu, G. King, J. A. Kaduk, J. Liang, J. Luo, Y. Gao, F. Yang, Y. Zhao, W. Li, S. H. Alahakoon, J. Guo, Y. Huang, T.-K. Sham, Y. Mo and X. Sun, Science, 2025, 390, 199–204 CrossRef CAS.
- X. Zhu, P. Lu, D. Wu, Q. Gao, T. Ma, M. Yang, L. Chen, H. Li and F. Wu, Nano Lett., 2023, 23, 10290–10296 CrossRef CAS PubMed.
- S.-K. Jung, H. Gwon, H. Kim, G. Yoon, D. Shin, J. Hong, C. Jung and J.-S. Kim, Nat. Commun., 2022, 13, 7638 CrossRef CAS PubMed.
- S. Li, J. Lin, M. Schaller, S. Indris, X. Zhang, T. Brezesinski, C.-W. Nan, S. Wang and F. Strauss, Angew. Chem., Int. Ed., 2023, 62, e202314155 CrossRef CAS.
- F. Strauss, J. Lin, A. Kondrakov and T. Brezesinski, Matter, 2023, 6, 1051–1070 CrossRef.
- Q. Zhao, Z. Cao, X. Wang, H. Chen, Y. Shi, Z. Cheng, Y. Guo, B. Lin, Y. Gong, Z. Du and S. Yang, J. Am. Chem. Soc., 2023, 145, 21242–21245 CrossRef CAS.
- D. Li, D. Yu, G. Zhang, A. Du, Z. Ye, Y. Jia, W. Hou, T. Xu, F. Li, S. Chi, Y. Zhu and C. Yang, Angew. Chem., Int. Ed., 2025, 64, e202419735 CrossRef CAS PubMed.
- Z. Song, T. Wang, H. Yang, W. H. Kan, Y. Chen, L. Wang, Y. Zhang, Y. Dai, H. Chen, W. Yin, T. Honda, M. Dvdeev, H. Xu, J. Ma, Y. Huang and W. Luo, Nat. Commun., 2024, 5, 1481 CrossRef.
- Y. Feng, Z. Zhu, L. Yang, Y. Mu, Z. Yan, H. Cui, Y. Ye, D. Zuo, Q. Hu, Y. Zhu, L. Zeng and J. Wan, Adv. Mater., 2025, 37, e09838 CrossRef CAS.
- Y. Ye, Z. Gu, J. Geng, K. Niu, P. Yu, Y. Zhou, J. Lin, H. J. Woo, Y. Zhu and J. Wan, Nano Lett., 2025, 25, 3747–3755 CrossRef CAS.
- F. Zhao, S. Zhang and X. Sun, Adv. Mater., 2025, 37, 2501544 CrossRef CAS.
- Q. Hu, K. Chen, J. Li, T. Zhao, F. Liang and D. Xue, Next Energy, 2024, 5, 100159 CrossRef.
- Z. Zheng, J. Zhou and Y. Zhu, Chem. Soc. Rev., 2024, 53, 3134 RSC.
- T. Cai, A. Chen, S. Liang, J. Mu, L. Wang, W. He, K. Tao, J. Li and F. Huang, Adv. Mater., 2025, e08717 CrossRef CAS.
- A. C. C. Dutra, B. A. Goldmann, M. S. Islam and J. A. Dawson, Nat. Rev. Mater., 2025, 10, 566–583 CrossRef CAS.
- Q. Wang, F. Yang, Y. Wang, D. Zhang, R. Sato, L. Zhang, E. J. Cheng, Y. Yan, Y. Chen, K. Kisu, S.-I. Orimo and H. Li, Angew. Chem., Int. Ed., 2025, 64, e202506573 CrossRef CAS.
- S. Wang, J. Liu, X. Song, H. Xu, Y. Gu, J. Fan, B. Sun and L. Yu, Nano-Micro Lett., 2025, 17, 287 CrossRef.
- E. Kim and Y. Kim, Dalton Trans., 2022, 51, 18159 RSC.
- E. Sebti, H. A. Evans, H. Chen, P. M. Richardson, K. M. White, R. Giovine, K. P. Koirala, Y. Xu, E. Gonzalez-Correa, C. Wang, C. M. Brown, A. K. Cheetham, P. Canepa and R. J. Clément, J. Am. Chem. Soc., 2022, 144, 5795–5811 CrossRef CAS PubMed.
- L. Schweiger, K. Hogrefe, B. Gadermaier, J. L. M. Rupp and H. M. R. Wilkening, J. Am. Chem. Soc., 2022, 144, 9597–9609 CrossRef CAS.
- J. Fu, H. Su, J. Luo, X. Li, J. Liang, C. Wang, J. T. Kim, Y. Hu, F. Zhao, S. Zhang, H. Duan, X. Hao, W. Li, J. Peng, J. Liu, S. Wang, T.-K. Sham and X. Sun, Angew. Chem., Int. Ed., 2025, 64, e202508835 CrossRef CAS PubMed.
- M. S. Diallo, T. Shi, Y. Zhang, X. Peng, I. Shozib, Y. Wang, L. J. Miara, M. C. Scott, Q. H. Tu and G. Ceder, Nat. Commun., 2024, 15, 858 CrossRef CAS.
- J. A. Dawson, P. Canepa, M. J. Clarke, T. Famprikis, D. Ghosh and M. S. Islam, Chem. Mater., 2019, 31, 5296–5304 CrossRef CAS.
- W. Xie, Z. Deng, Z. Liu, T. Famprikis, K. T. Butler and P. Canepa, Adv. Energy Mater., 2024, 14, 2304230 CrossRef CAS.
- M. Cronau, M. Szabo, C. König, T. B. Wassermann and B. Roling, ACS Energy Lett., 2021, 6, 3072–3077 CrossRef CAS.
- P. Perrenot, A. Fauchier-Magnan, M. Mirolo, L. Lecarme, P. H. Jouneau, A. Boulineau, P. Bayle-Guillemaud and C. Villevieille, Adv. Funct. Mater., 2023, 34, 2310739 CrossRef.
- F. Zhang, Y. Guo, L. Zhang, P. Jia, X. Liu, P. Qiu, H. Zhang and J. Huang, eTransportation, 2023, 15, 100220 CrossRef.
- J.-M. Doux, Y. Yang, D. H. S. Tan, H. Nguyen, E. A. Wu, X. Wang, A. Banerjee and Y. S. Meng, J. Mater. Chem. A, 2020, 8, 5049–5055 RSC.
- R. Garcia-Mendez, J. G. Smith, J. C. Neuefeind, D. J. Siegel and J. Sakamoto, Adv. Energy Mater., 2020, 10, 2000335 CrossRef CAS.
- Y. Wang, B. Hoang, J. Hoerauf, C. Lee, C.-F. Lin, G. W. Rubloff, S. B. Lee and A. C. Kozen, J. Electrochem. Soc., 2021, 168, 010533 CrossRef CAS.
- Y. Zhang, D. Luo, W. Luo, S. Du, Y. Deng and J. Deng, Electrochim. Acta, 2020, 359, 136965 CrossRef CAS.
- H.-Y. Li, B. Huang, Z. Huang and C.-A. Wang, Ceram. Int., 2019, 45, 18115–18118 CrossRef.
- F. Aguesse, J. M. López del Amo, V. Roddatis, A. Aguadero and J. A. Kilner, Adv. Mater. Interfaces, 2014, 1, 1300143 CrossRef.
- Y. Li, Z. Wang, C. Li, Y. Cao and X. Guo, J. Power Sources, 2014, 248, 642–646 CrossRef CAS.
- A. Curcio, A. G. Sabato, M. Nuñez Eroles, J. C. Gonzalez-Rosillo, A. Morata, A. Tarancón and F. Ciucci, ACS Appl. Energy Mater., 2022, 5, 14466–14475 CrossRef CAS.
- D. A. I. Lijing, W. Jing, S. H. I. Zhongxiang, Y. U. Lina and S. H. I. Jun, J. Funct. Mater., 2022, 53, 1117–1122 Search PubMed.
- X. Zhang, T.-S. Oh and J. W. Fergus, J. Electrochem. Soc., 2019, 166, A3753–A3759 CrossRef CAS.
- W. Huang, Y. Ye, H. Chen, R. A. Vilá, A. Xiang, H. Wang, F. Liu, Z. Yu, J. Xu, Z. Zhang, R. Xu, Y. Wu, L.-Y. Chou, H. Wang, J. Xu, D. T. Boyle, Y. Li and Y. Cui, Nat. Commun., 2022, 13, 7091 CrossRef.
- J. Gu, Z. Liang, J. Shi and Y. Yang, Adv. Energy Mater., 2022, 13, 202203153 Search PubMed.
- S. Y. Han, C. Lee, J. A. Lewis, D. Yeh, Y. Liu, H.-W. Lee and M. T. McDowell, Joule, 2021, 5, 2450–2465 CrossRef CAS.
- S. Kalnaus, N. J. Dudney, A. S. Westover, E. Herbert and S. Hackney, Science, 2023, 381, eabg5998 CrossRef CAS.
- J. Gu, X. Chen, R. Ma, Z. He, Z. Liang, H. Zhong, Y. Su, J. Shi and Y. Yang, Energy Storage Mater., 2023, 63, 103052 CrossRef.
- P. Žguns and B. Yildiz, PRX Energy, 2022, 1, 023003 CrossRef.
- R. Hanus, M. T. Agne, A. J. E. Rettie, Z. Chen, G. Tan, D. Y. Chung, M. G. Kanatzidis, Y. Pei, P. W. Voorhees and G. J. Snyder, Adv. Mater., 2019, 31, 1900108 CrossRef PubMed.
- P. Till, M. T. Agne, M. A. Kraft, M. Courty, T. Famprikis, M. Ghidiu, T. Krauskopf, C. Masquelier and W. G. Zeier, Chem. Mater., 2022, 34, 2410–2421 CrossRef CAS.
- J. Ding, PhD thesis, Duke University, 2022.
- B. Shao, Y. Huang and F. Han, Adv. Energy Mater., 2023, 13, 2204098 CrossRef CAS.
- T. Yang, C. Wang, W. Zhang, Y. Xia, H. Huang, Y. Gan, X. He, X. Xia, X. Tao and J. Zhang, J. Energy Chem., 2023, 84, 189–209 CrossRef CAS.
- K. Liu, X. Li, J. Cai, Z. Yang, Z. Chen, B. Key, Z. Zhang, T. L. Dzwiniel and C. Liao, ACS Energy Lett., 2021, 6, 1315–1323 CrossRef CAS.
- Y. Wang, J. Ju, S. Dong, Y. Yan, F. Jiang, L. Cui, Q. Wang, X. Han and G. Cui, Adv. Funct. Mater., 2021, 31, 2101523 CrossRef CAS.
- Y. Yan, J. Ju, S. Dong, Y. Wang, L. Huang, L. Cui, F. Jiang, Q. Wang, Y. Zhang and G. Cui, Adv. Sci., 2021, 8, 2003887 CrossRef CAS.
- A. G. Nguyen, R. Verma, G. C. Song, J. Kim and C. J. Park, Adv. Sci., 2023, 10, 2207744 CrossRef CAS.
- X. Zhang, J. Xie, F. Shi, D. Lin, Y. Liu, W. Liu, A. Pei, Y. Gong, H. Wang, K. Liu, Y. Xiang and Y. Cui, Nano Lett., 2018, 18, 3829–3838 CrossRef CAS PubMed.
- W. Liu, S. W. Lee, D. Lin, F. Shi, S. Wang, A. D. Sendek and Y. Cui, Nat. Energy, 2017, 2, 17035 CrossRef CAS.
- C.-W. Nan, Prog. Mater. Sci., 1993, 37, 1–116 CrossRef CAS.
- C. G. Granqvist and O. Hunderi, Phys. Rev. B: Condens. Matter Mater. Phys., 1978, 18, 1554–1561 CrossRef CAS.
- M. Nakamura, Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 29, 3691–3693 CrossRef.
- J. C. M. Garnett and J. Larmor, Proc. R. Soc. London, 1904, 73, 443–445 Search PubMed.
- L. Cui, S. Zhang, J. Ju, S. Liu, H. Wang, J. Xu, W. Zhang, P. Mu, Y. Zhang, L. Liu, P. Xu, P. Han, Z. Lv and G. Cui, Angew. Chem., Int. Ed., 2025, e202517153 CAS.
- C. Shen, Y. Huang, J. Yang, M. Chen and Z. Liu, Energy Storage Mater., 2021, 39, 271–277 CrossRef.
- M. Marcinek, J. Syzdek, M. Marczewski, M. Piszcz, L. Niedzicki, M. Kalita, A. Plewa-Marczewska, A. Bitner, P. Wieczorek, T. Trzeciak, M. Kasprzyk, P. Łężak, Z. Zukowska, A. Zalewska and W. Wieczorek, Solid State Ionics, 2015, 276, 107–126 CrossRef CAS.
- W. Wang, E. Yi, A. J. Fici, R. M. Laine and J. Kieffer, J. Phys. Chem. C, 2017, 121, 2563–2573 CrossRef CAS.
- J. A. Isaac, D. Devaux and R. Bouchet, Nat. Mater., 2022, 21, 1412–1418 CrossRef CAS PubMed.
- W. Liu, N. Liu, J. Sun, P. C. Hsu, Y. Li, H. W. Lee and Y. Cui, Nano Lett., 2015, 15, 2740–2745 CrossRef CAS PubMed.
- R. Fan, C. Liu, K. He, S. Ho-Sum Cheng, D. Chen, C. Liao, R. K. Y. Li, J. Tang and Z. Lu, ACS Appl. Mater. Interfaces, 2020, 12, 7222–7231 CrossRef CAS.
- K. Fu, Y. Gong, J. Dai, A. Gong, X. Han, Y. Yao, C. Wang, Y. Wang, Y. Chen, C. Yan, Y. Li, E. D. Wachsman and L. Hu, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7094–7099 CrossRef CAS.
- C. Yan, P. Zhu, H. Jia, Z. Du, J. Zhu, R. Orenstein, H. Cheng, N. Wu, M. Dirican and X. Zhang, Energy Storage Mater., 2020, 26, 448–456 CrossRef.
- S. Yu, Q. Xu, X. Lu, Z. Liu, A. Windmüller, C.-L. Tsai, A. Buchheit, H. Tempel, H. Kungl, H.-D. Wiemhöfer and R.-A. Eichel, ACS Appl. Mater. Interfaces, 2021, 13, 61067–61077 CrossRef CAS.
- J. Bae, Y. Li, J. Zhang, X. Zhou, F. Zhao, Y. Shi, J. B. Goodenough and G. Yu, Angew. Chem., Int. Ed., 2018, 57, 2096–2100 CrossRef CAS.
- C. Liu, J. Wang, W. Kou, Z. Yang, P. Zhai, Y. Liu, W. Wu and J. Wang, Chem. Eng. J., 2021, 404, 126517 CrossRef CAS.
- P. Wang, J. Tan, Z. Liu, C. Wang, C. Bao, X. Xia, B. Li and Q. Liu, Adv. Funct. Mater., 2025, e12441 CrossRef.
- X. Wang, H. Zhai, B. Qie, Q. Cheng, A. Li, J. Borovilas, B. Xu, C. Shi, T. Jin, X. Liao, Y. Li, X. He, S. Du, Y. Fu, M. Dontigny, K. Zaghib and Y. Yang, Nano Energy, 2019, 60, 205–212 CrossRef.
- Z. Niu, Y. Wang, Y. Wang, Y. Wu, Z. Zhao, G. Li, J.-K. Kim, X. Liu, Z. Shen, M. Chen, S. Passerini and Z. Chen, Energy Storage Mater., 2025, 81, 104492 CrossRef.
- H. Zhang, X. An, Y. Yang, Y. Long, S. Nie, L. Liu, G. Yang, Z. Tian, H. Cao, Z. Cheng, H. Liu and Y. Ni, EcoMat, 2022, 5, e12317 CrossRef.
- Z. Niu, Y. Wang, Y. Wang, Y. Wu, Z. Zhao, G. Li, J.-K. Kim, X. Liu, Z. Shen, M. Chen, S. Passerini and Z. Chen, Energy Storage Mater., 2025, 81, 104492 CrossRef.
- C. Wang, X.-W. Zhang and A. J. Applebyb, J. Electrochem. Soc., 2005, 152, A205 CrossRef CAS.
- Y. Gong, K. Fu, S. Xu, J. Dai, T. R. Hamann, L. Zhang, G. T. Hitz, Z. Fu, Z. Ma, D. W. McOwen, X. Han, L. Hu and E. D. Wachsman, Mater. Today, 2018, 21, 594–601 CrossRef CAS.
- F. Croce, G. B. Appetecchi, L. Persi and B. Scrosati, Nature, 1998, 394, 456–458 CrossRef CAS.
- Y. Zhao, C. Wu, G. Peng, X. Chen, X. Yao, Y. Bai, F. Wu, S. Chen and X. Xu, J. Power Sources, 2016, 301, 47–53 CrossRef CAS.
- T. Yang, J. Zheng, Q. Cheng, Y. Y. Hu and C. K. Chan, ACS Appl. Mater. Interfaces, 2017, 9, 21773–21780 CrossRef CAS PubMed.
- H. Zhai, P. Xu, M. Ning, Q. Cheng, J. Mandal and Y. Yang, Nano Lett., 2017, 17, 3182–3187 CrossRef CAS.
- D. Li, L. Chen, T. Wang and L. Z. Fan, ACS Appl. Mater. Interfaces, 2018, 10, 7069–7078 CrossRef CAS PubMed.
- P. Zhu, C. Yan, M. Dirican, J. Zhu, J. Zang, R. K. Selvan, C.-C. Chung, H. Jia, Y. Li, Y. Kiyak, N. Wu and X. Zhang, J. Mater. Chem. A, 2018, 6, 4279–4285 RSC.
- B. Li, Q. Su, L. Yu, D. Wang, S. Ding, M. Zhang, G. Du and B. Xu, ACS Appl. Mater. Interfaces, 2019, 11, 42206–42213 CrossRef CAS.
- P. Sivaraj, K. P. Abhilash, B. Nalini, P. Perumal, K. Somasundaram and P. C. Selvin, Macromol. Res., 2020, 28, 739–750 CrossRef CAS.
- S. Zhang, Z. Li, Y. Guo, L. Cai, P. Manikandan, K. Zhao, Y. Li and V. G. Pol, Chem. Eng. J., 2020, 400, 25996 Search PubMed.
- Y. Jin, X. Zong, X. Zhang, C. Liu, D. Li, Z. Jia, G. Li, X. Zhou, J. Wei and Y. Xiong, J. Power Sources, 2021, 501, 230027 CrossRef CAS.
- Y. Li and H. Wang, Ind. Eng. Chem. Res., 2021, 60, 1494–1500 CrossRef CAS.
- R. Wang, Q. Dong, C. Wang, M. Hong, J. Gao, H. Xie, M. Guo, W. Ping, X. Wang, S. He, J. Luo and L. Hu, Adv. Mater., 2021, 33, e2100726 CrossRef PubMed.
- M. Zhang, P. Pan, Z. Cheng, J. Mao, L. Jiang, C. Ni, S. Park, K. Deng, Y. Hu and K. K. Fu, Nano Lett., 2021, 21, 7070–7078 CrossRef CAS PubMed.
- X. Liang, X. Shi, L. Lan, Y. Qing, B. Zhang, Z. Fang and Y. Wang, Gels, 2024, 10, 199 CrossRef CAS PubMed.
- S. Wang, Y. Liao, S. Li, C. Cui, J. Liang, G. Du, Z. Tong, L. Yuan, T. Zhai and H. Li, Adv. Energy Mater., 2023, 14, 2303641 CrossRef.
- S. Randau, D. A. Weber, O. Kötz, R. Koerver, P. Braun, A. Weber, E. Ivers-Tiffée, T. Adermann, J. Kulisch, W. G. Zeier, F. H. Richter and J. Janek, Nat. Energy, 2020, 5, 259–270 CrossRef CAS.
- L. Chen, X. Huang, R. Ma, W. Xiang, J. Ma, Y. Wu, D. Yang, C. Wang, W. Ping and H. Xiang, Energy Storage Mater., 2024, 65, 103140 CrossRef.
- Y. Li, Y. Wu, Z. Wang, J. Xu, T. Ma, L. Chen, H. Li and F. Wu, Mater. Today, 2022, 55, 92–109 CrossRef CAS.
- W. Xiang, R. Ma, X. Liu, X. Kong, S. Shen, L. Wang, Z. Jin, Z. Zhan, C. Chen and C. Wang, Nano Energy, 2023, 116, 108816 CrossRef CAS.
- X. Yang, K. R. Adair, X. Gao and X. Sun, Energy Environ. Sci., 2021, 14, 643–671 RSC.
- M. Zhang, Z. Huang, J. Cheng, O. Yamamoto, N. Imanishi, B. Chi, J. Pu and J. Li, J. Alloys Compd., 2014, 590, 147–152 CrossRef CAS.
- X. Zhao, P. Xiang, J. Wu, Z. Liu, L. Shen, G. Liu, Z. Tian, L. Chen and X. Yao, Nano Lett., 2023, 23, 227–234 CrossRef CAS.
- Z. Jiang, S. Wang, X. Chen, W. Yang, X. Yao, X. Hu, Q. Han and H. Wang, Adv. Mater., 2019, 32, 1906221 CrossRef.
- L. Hu, Y. Ren, C. Wang, J. Li, Z. Wang, F. Sun, J. Ju, J. Ma, P. Han, S. Dong and G. Cui, Adv. Mater., 2024, 36, 2401909 CrossRef CAS PubMed.
- X. Chen, W. He, L.-X. Ding, S. Wang and H. Wang, Energy Environ. Sci., 2019, 12, 938–944 RSC.
- D. Kim, H. Lee, Y. Roh, J. Lee, J. Song, C. B. Dzakpasu, S. H. Kang, J. Choi, D. H. Kim, H. J. Hah, K. Y. Cho, Y. G. Lee and Y. M. Lee, Adv. Energy Mater., 2023, 14, 2302596 CrossRef.
- D. Li, H. Liu, C. Wang, C. Yan, Q. Zhang, C. W. Nan and L. Z. Fan, Adv. Funct. Mater., 2024, 34, 2315555 CrossRef CAS.
- X. Yan, Z. Li, H. Ying, F. Nie, L. Xue, Z. Wen and W.-Q. Han, Ionics, 2017, 24, 1545–1551 CrossRef.
- G. L. Zhu, C. Z. Zhao, H. J. Peng, H. Yuan, J. K. Hu, H. X. Nan, Y. Lu, X. Y. Liu, J. Q. Huang, C. He, J. Zhang and Q. Zhang, Adv. Funct. Mater., 2021, 31, 2101985 CrossRef CAS.
- Z. Zhang, L. Wu, D. Zhou, W. Weng and X. Yao, Nano Lett., 2021, 21, 5233–5239 CrossRef CAS.
- G. Liu, J. Shi, M. Zhu, W. Weng, L. Shen, J. Yang and X. Yao, Energy Storage Mater., 2021, 38, 249–254 CrossRef.
- A. Sakuda, K. Kuratani, M. Yamamoto, M. Takahashi, T. Takeuchi and H. Kobayashi, J. Electrochem. Soc., 2017, 164, A2474–A2478 CrossRef CAS.
- Y.-G. Lee, S. Fujiki, C. Jung, N. Suzuki, N. Yashiro, R. Omoda, D.-S. Ko, T. Shiratsuchi, T. Sugimoto, S. Ryu, J. H. Ku, T. Watanabe, Y. Park, Y. Aihara, D. Im and I. T. Han, Nat. Energy, 2020, 5, 299–308 CrossRef CAS.
- Y. J. Nam, S.-J. Cho, D. Y. Oh, J.-M. Lim, S. Y. Kim, J. H. Song, Y.-G. Lee, S.-Y. Lee and Y. S. Jung, Nano Lett., 2015, 15, 3317–3323 CrossRef CAS.
- D. J. Kalita, S. H. Lee, K. S. Lee, D. H. Ko and Y. S. Yoon, Solid State Ionics, 2012, 229, 14–19 CrossRef CAS.
- J. Tan and A. Tiwari, ECS Solid State Lett., 2012, 1, Q57–Q60 CrossRef CAS.
- R.-J. Chen, M. Huang, W.-Z. Huang, Y. Shen, Y.-H. Lin and C.-W. Nan, J. Mater. Chem. A, 2014, 2, 13277 RSC.
- C. Peng, Y. Kamiike, Y. Liang, K. Kuroda and M. Okido, ACS Sustainable Chem. Eng., 2019, 7, 10751–10762 CrossRef CAS.
- Y. Zhao, K. Zheng and X. Sun, Joule, 2018, 2, 2583–2604 CrossRef CAS.
- H. Guo, F. Shen, W. Guo, D. Zeng, Y. Yin and X. Han, Mater. Lett., 2021, 301, 130302 CrossRef CAS.
- A. Sharafi, S. Yu, M. Naguib, M. Lee, C. Ma, H. M. Meyer, J. Nanda, M. Chi, D. J. Siegel and J. Sakamoto, J. Mater. Chem. A, 2017, 5, 13475–13487 RSC.
- M. B. Dixit, A. Verma, W. Zaman, X. Zhong, P. Kenesei, J. S. Park, J. Almer, P. P. Mukherjee and K. B. Hatzell, ACS Appl. Energy Mater., 2020, 3, 9534–9542 CrossRef CAS.
- J. A. Lewis, F. J. Q. Cortes, Y. Liu, J. C. Miers, A. Verma, B. S. Vishnugopi, J. Tippens, D. Prakash, T. S. Marchese, S. Y. Han, C. Lee, P. P. Shetty, H. W. Lee, P. Shevchenko, F. De Carlo, C. Saldana, P. P. Mukherjee and M. T. McDowell, Nat. Mater., 2021, 20, 503–510 CrossRef CAS PubMed.
- X. Han, Y. Gong, K. K. Fu, X. He, G. T. Hitz, J. Dai, A. Pearse, B. Liu, H. Wang, G. Rubloff, Y. Mo, V. Thangadurai, E. D. Wachsman and L. Hu, Nat. Mater., 2017, 16, 572–579 CrossRef CAS PubMed.
- C. Cui, Q. Ye, C. Zeng, S. Wang, X. Xu, T. Zhai and H. Li, Energy Storage Mater., 2022, 45, 814–820 CrossRef.
- S. Guo, T. T. Wu, Y. G. Sun, S. D. Zhang, B. Li, H. S. Zhang, M. Y. Qi, X. H. Liu, A. M. Cao and L. J. Wan, Adv. Funct. Mater., 2022, 32, 2201498 CrossRef CAS.
- F. Strauss, L. de Biasi, A. Y. Kim, J. Hertle, S. Schweidler, J. Janek, P. Hartmann and T. Brezesinski, ACS Mater. Lett., 2019, 2, 84–88 CrossRef.
- X. Zhao, Y. Tian, Z. Lun, Z. Cai, T. Chen, B. Ouyang and G. Ceder, Joule, 2022, 6, 1654–1671 CrossRef CAS.
- L. Zhou, M. K. Tufail, N. Ahmad, T. Song, R. Chen and W. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 28270–28280 CrossRef CAS.
- Y. Zhu, X. He and Y. Mo, J. Mater. Chem. A, 2016, 4, 3253–3266 RSC.
- S. Wenzel, D. A. Weber, T. Leichtweiss, M. R. Busche, J. Sann and J. Janek, Solid State Ionics, 2016, 286, 24–33 CrossRef CAS.
- H. Chung and B. Kang, Chem. Mater., 2017, 29, 8611–8619 CrossRef CAS.
- A. Kato, H. Kowada, M. Deguchi, C. Hotehama, A. Hayashi and M. Tatsumisago, Solid State Ionics, 2018, 322, 1–4 CrossRef CAS.
- F. Walther, R. Koerver, T. Fuchs, S. Ohno, J. Sann, M. Rohnke, W. G. Zeier and J. Janek, Chem. Mater., 2019, 31, 3745–3755 CrossRef CAS.
- X. Li, H. Guan, Z. Ma, M. Liang, D. Song, H. Zhang, X. Shi, C. Li, L. Jiao and L. Zhang, J. Energy Chem., 2020, 48, 195–202 CrossRef.
- X. Wu, M. Mirolo, C. A. F. Vaz, P. Novak and M. El Kazzi, ACS Appl. Mater. Interfaces, 2021, 13, 42670–42681 CrossRef CAS PubMed.
- A. Sakuda, A. Hayashi and M. Tatsumisago, Chem. Mater., 2009, 22, 949–956 CrossRef.
- L. Miara, A. Windmuller, C. L. Tsai, W. D. Richards, Q. Ma, S. Uhlenbruck, O. Guillon and G. Ceder, ACS Appl. Mater. Interfaces, 2016, 8, 26842–26850 CrossRef CAS.
- K. Park, B.-C. Yu, J.-W. Jung, Y. Li, W. Zhou, H. Gao, S. Son and J. B. Goodenough, Chem. Mater., 2016, 28, 8051–8059 CrossRef CAS.
- D. Cao, Y. Zhang, A. M. Nolan, X. Sun, C. Liu, J. Sheng, Y. Mo, Y. Wang and H. Zhu, Nano Lett., 2020, 20, 1483–1490 CrossRef CAS.
- L. Wang, X. Sun, J. Ma, B. Chen, C. Li, J. Li, L. Chang, X. Yu, T. S. Chan, Z. Hu, M. Noked and G. Cui, Adv. Energy Mater., 2021, 11, 2100881 CrossRef CAS.
- X. Zheng, Z. Xue, H. Hao, Y. Cho, Y. Li, C. Kim, P. Czaja, S. S. Lee, S. Bone, E. Spielman-Sun, S. Jiang, X. W. Gu, J. N. Weker, G. Yang and J. Nanda, Sci. Adv., 2025, 11, eady7189 CrossRef CAS.
- H. Kim, C. Im, S. Ryu, Y. J. Gong, J. Cho, S. Pyo, H. Yun, J. Lee, J. Yoo and Y. S. Kim, Adv. Funct. Mater., 2021, 32, 2107555 CrossRef.
- K. Takada, Langmuir, 2013, 29, 7538–7541 CrossRef CAS.
- N. Ohta, K. Takada, L. Zhang, R. Ma, M. Osada and T. Sasaki, Adv. Mater., 2006, 18, 2226–2229 CrossRef CAS.
- N. Ohta, K. Takada, I. Sakaguchi, L. Zhang, R. Ma, K. Fukuda, M. Osada and T. Sasaki, Electrochem. Commun., 2007, 9, 1486–1490 CrossRef CAS.
- J. Haruyama, K. Sodeyama, L. Han, K. Takada and Y. Tateyama, Chem. Mater., 2014, 26, 4248–4255 CrossRef CAS.
- F. S. Gittleson and F. El Gabaly, Nano Lett., 2017, 17, 6974–6982 CrossRef CAS.
- J. Y. Liang, X. D. Zhang, X. X. Zeng, M. Yan, Y. X. Yin, S. Xin, W. P. Wang, X. W. Wu, J. L. Shi, L. J. Wan and Y. G. Guo, Angew. Chem., Int. Ed., 2020, 59, 6585–6589 CrossRef CAS PubMed.
- J. Yi, P. He, H. Liu, H. Ni, Z. Bai and L.-Z. Fan, J. Energy Chem., 2021, 52, 202–209 CrossRef CAS.
- C. Yada, A. Ohmori, K. Ide, H. Yamasaki, T. Kato, T. Saito, F. Sagane and Y. Iriyama, Adv. Energy Mater., 2014, 4, 1301416 CrossRef.
- B. K. Park, H. Kim, K. S. Kim, H. S. Kim, S. H. Han, J. S. Yu, H. J. Hah, J. Moon, W. Cho and K. J. Kim, Adv. Energy Mater., 2022, 12, 2201208 CrossRef CAS.
- N. Li, K. Xie and H. Huang, ACS Energy Lett., 2023, 8, 4357–4370 CrossRef CAS.
- Q. Wang, Z. A. Grady, C. R. Bowen and J. I. Roscow, J. Materiomics, 2025, 11, 100912 CrossRef.
- B. He, F. Zhang, Y. Xin, C. Xu, X. Hu, X. Wu, Y. Yang and H. Tian, Nat. Rev. Chem., 2023, 7, 826–842 Search PubMed.
- K. Chen, Y. Zhu, J. Li, J. Zhang, H. Luo, S. Yin, L. Zhou, D. Guo, X. A. Chen and S. Wang, Angew. Chem., Int. Ed., 2025, e202510602 CAS.
- T. Yu, Y. Liu, H. Li, Y. Sun, S. Guo and H. Zhou, Chem. Rev., 2025, 125, 3595–3662 CrossRef CAS PubMed.
- M. Wang, Z. Peng, W. Luo, Q. Zhang, Z. Li, Y. Zhu, H. Lin, L. Cai, X. Yao, C. Ouyang and D. Wang, Adv. Sci., 2020, 7, 2000237 CrossRef CAS.
- M.-J. Kim, J.-W. Park, B. G. Kim, Y.-J. Lee, Y.-C. Ha, S.-M. Lee and K.-J. Baeg, Sci. Rep., 2020, 10, 11923 CrossRef CAS PubMed.
- Z. Bi, S. Mu, N. Zhao, W. Sun, W. Huang and X. Guo, Energy Storage Mater., 2021, 35, 512–519 CrossRef.
- K. H. Park, D. Y. Oh, Y. E. Choi, Y. J. Nam, L. Han, J.-Y. Kim, H. Xin, F. Lin, S. M. Oh and Y. S. Jung, Adv. Mater., 2016, 28, 1874–1883 CrossRef CAS.
- Y. E. Choi, K. H. Park, D. H. Kim, D. Y. Oh, H. R. Kwak, Y.-G. Lee and Y. S. Jung, ChemSusChem, 2017, 10, 2605–2611 CrossRef CAS.
- X. Li, J. Liang, N. Chen, J. Luo, K. R. Adair, C. Wang, M. N. Banis, T.-K. Sham, L. Zhang, S. Zhao, S. Lu, H. Huang, R. Li and X. Sun, Angew. Chem., Int. Ed., 2019, 58, 16427–16432 CrossRef CAS.
- H. Maekawa, M. Matsuo, H. Takamura, M. Ando, Y. Noda, T. Karahashi and S.-I. Orimo, J. Am. Chem. Soc., 2009, 131, 894–895 CrossRef CAS.
- M. B. Ley, D. B. Ravnsbæk, Y. Filinchuk, Y.-S. Lee, R. Janot, Y. W. Cho, J. Skibsted and T. R. Jensen, Chem. Mater., 2012, 24, 1654–1663 CrossRef CAS.
- F. Lu, Y. Pang, M. Zhu, F. Dong, J. Yang, F. Fang, D. Sun, S. Zheng and C. Wang, Adv. Funct. Mater., 2019, 29, 1809219 CrossRef.
- L. Duchêne, A. Remhof, H. Hagemann and C. Battaglia, Energy Stroage Mater., 2020, 25, 782–794 Search PubMed.
- Z. Li, J. Fu and X. Guo, Natl. Sci. Open, 2022, 2, 20220036 CrossRef.
- L. Nie, S. Chen, M. Zhang, T. Gao, Y. Zhang, R. Wei, Y. Zhang and W. Liu, Nano Res., 2023, 17, 2687–2692 CrossRef.
- J. Zheng, C. Sun, Z. Wang, S. Liu, B. An, Z. Sun and F. Li, Angew. Chem., Int. Ed., 2021, 60, 18448–18453 CrossRef CAS.
- S. Zhang, B. Xie, X. Zhuang, S. Wang, L. Qiao, S. Dong, J. Ma, Q. Zhou, H. Zhang, J. Zhang, J. Ju, G. Xu, Z. Cui and G. Cui, Adv. Funct. Mater., 2023, 34, 2314063 CrossRef.
- S. Zou, Y. Yang, J. Wang, X. Zhou, X. Wan, M. Zhu and J. Liu, Energy Environ. Sci., 2024, 17, 4426–4460 RSC.
- X. Shi, Z. Zeng, M. Sun, B. Huang, H. Zhang, W. Luo, Y. Huang, Y. Du and C. Yan, Nano Lett., 2021, 21, 9325–9331 CrossRef CAS.
- C. Wang, J. Liang, J. Luo, J. Liu, X. Li, F. Zhao, R. Li, H. Huang, S. Zhao, L. Zhang, J. Wang and X. Sun, Sci. Adv., 2021, 7, eabh1896 CrossRef CAS.
- X. Li, J. Liang, J. Luo, M. Norouzi Banis, C. Wang, W. Li, S. Deng, C. Yu, F. Zhao, Y. Hu, T.-K. Sham, L. Zhang, S. Zhao, S. Lu, H. Huang, R. Li, K. R. Adair and X. Sun, Energy Environ. Sci., 2019, 12, 2665–2671 RSC.
- L. M. Riegger, R. Schlem, J. Sann, W. G. Zeier and J. Janek, Angew. Chem., Int. Ed., 2021, 60, 6718–6723 CrossRef CAS PubMed.
- Y. Kato, S. Hori and R. Kanno, Adv. Energy Mater., 2020, 10, 2002153 CrossRef CAS.
- T. Koç, F. Marchini, G. Rousse, R. Dugas and J.-M. Tarascon, ACS Appl. Energy Mater., 2021, 4, 13575–13585 CrossRef.
- T. Koç, M. Hallot, E. Quemin, B. Hennequart, R. Dugas, A. M. Abakumov, C. Lethien and J.-M. Tarascon, ACS Energy Lett., 2022, 7, 2979–2987 CrossRef.
- C. Rosenbach, F. Walther, J. Ruhl, M. Hartmann, T. A. Hendriks, S. Ohno, J. Janek and W. G. Zeier, Adv. Energy Mater., 2022, 13, 2203673 CrossRef.
- W. Zhou, Z. Wang, Y. Pu, Y. Li, S. Xin, X. Li, J. Chen and J. B. Goodenough, Adv. Mater., 2019, 31, e1805574 CrossRef.
- Z. Zhang, L. Hu, H. Wu, W. Weng, M. Koh, P. C. Redfern, L. A. Curtiss and K. Amine, Energy Environ. Sci., 2013, 6, 1806–1810 RSC.
- P. Hu, J. Chai, Y. Duan, Z. Liu, G. Cui and L. Chen, J. Mater. Chem. A, 2016, 4, 10070–10083 RSC.
- Z.-Y. Wang, C.-Z. Zhao, S. Sun, Y.-K. Liu, Z.-X. Wang, S. Li, R. Zhang, H. Yuan and J.-Q. Huang, Matter, 2023, 6, 1096–1124 CrossRef CAS.
- L. Li, J. Wang, L. Zhang, H. Duan, Y. Deng and G. Chen, Energy Storage Mater., 2022, 45, 1062–1073 CrossRef.
- Z. Wang, J. Tan, J. Cui, K. Xie, Y. Bai, Z. Jia, X. Gao, Y. Wu and W. Tang, J. Mater. Chem. A, 2024, 12, 4231–4239 RSC.
- J. Pan, Y. Zhang, J. Wang, Z. Bai, R. Cao, N. Wang, S. Dou and F. Huang, Adv. Mater., 2022, 34, e2107183 CrossRef.
- Q. Lv, Y. Jing, B. Wang, B. Wu, S. Wang, C. Li, L. Wang, L. Xiao, D. Wang, H. Liu and S. Dou, Energy Storage Mater., 2024, 65, 103122 CrossRef.
- H. Duan, M. Fan, W. P. Chen, J. Y. Li, P. F. Wang, W. P. Wang, J. L. Shi, Y. X. Yin, L. J. Wan and Y. G. Guo, Adv. Mater., 2019, 31, e1807789 CrossRef.
- H. Huo, Y. Chen, J. Luo, X. Yang, X. Guo and X. Sun, Adv. Energy Mater., 2019, 9, 1804004 CrossRef.
- C. Deng, N. Chen, C. Hou, H. Liu, Z. Zhou and R. Chen, Small, 2021, 17, e2006578 CrossRef.
- J.-H. Kim, J. Sun, J.-S. Jang, D.-H. Park, S.-Y. Ahn, W.-C. Kim, K. Min and K.-W. Park, Energy Storage Mater., 2024, 64, 103080 CrossRef.
- Y. Jin, X. Zong, X. Zhang, Z. Jia, S. Tan and Y. Xiong, J. Power Sources, 2022, 530, 231297 CrossRef CAS.
- L. Nie, R. Gao, M. Zhang, Y. Zhu, X. Wu, Z. Lao and G. Zhou, Adv. Energy Mater., 2024, 14, 2302476 CrossRef CAS.
- J.-K. Hu, Y.-C. Gao, S.-J. Yang, X.-L. Wang, X. Chen, Y.-L. Liao, S. Li, J. Liu, H. Yuan and J.-Q. Huang, Adv. Funct. Mater., 2024, 34, 2311633 CrossRef CAS.
- F. W. Yang, Y. J. Shen, Z. P. Zhang, W. H. Ruan, M. Z. Rong and M. Q. Zhang, Adv. Mater., 2025, 37, e09057 CrossRef CAS PubMed.
- X. Yang, Q. Sun, C. Zhao, X. Gao, K. R. Adair, Y. Liu, J. Luo, X. Lin, J. Liang, H. Huang, L. Zhang, R. Yang, S. Lu, R. Li and X. Sun, Nano Energy, 2019, 61, 567–575 CrossRef CAS.
- L. Nie, S. Chen, C. Zhang, L. Dong, Y. He, T. Gao, J. Yu and W. Liu, Cell Rep. Phys. Sci., 2022, 3, 100851 CrossRef CAS.
- A. Song, W. Zhang, L. Ma, Y. Lai, Y. Zhao, J. Zhu, M. Huang, L. Wang, L. Dong, N. Li, C. Shen and K. Xie, ACS Energy Lett., 2024, 9, 5027–5036 CrossRef CAS.
- Z. Cao, X. Yao, S. Park, K. Deng, C. Zhang, L. Chen and K. Fu, Sci. Adv., 2025, 11, eadr4292 CrossRef CAS.
- J. Billaud, F. Bouville, T. Magrini, C. Villevieille and A. R. Studart, Nat. Energy, 2016, 1, 16097 CrossRef CAS.
- J. S. Sander, R. M. Erb, L. Li, A. Gurijala and Y. M. Chiang, Nat. Energy, 2016, 1, 16099 CrossRef CAS.
- L. L. Lu, Y. Y. Lu, Z. J. Xiao, T. W. Zhang, F. Zhou, T. Ma, Y. Ni, H. B. Yao, S. H. Yu and Y. Cui, Adv. Mater., 2018, 30, e1706745 CrossRef PubMed.
- Y. Zhu, Z. Ju, X. Zhang, D. M. Lutz, L. M. Housel, Y. Zhou, K. J. Takeuchi, E. S. Takeuchi, A. C. Marschilok and G. Yu, Adv. Mater., 2020, 32, e1907941 CrossRef PubMed.
- Z. Lv, M. Yue, M. Ling, H. Zhang, J. Yan, Q. Zheng and X. Li, Adv. Energy Mater., 2021, 11, 2003725 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |