
Dynamic regulation of interfacial adhesion in biomedical hydrogels
Hanjun
Sun
a,
Xinyu
Qu
a,
Qian
Wang
 *a,
Yuxin
Guo
b and
Xiaochen
Dong
*ab
*a,
Yuxin
Guo
b and
Xiaochen
Dong
*ab
aState Key Laboratory of Flexible Electronics (LoFE) & Institute of Advanced Materials (IAM), School of Flexible Electronics (Future Technologies), Nanjing Tech University, Nanjing 211816, China. E-mail: chelseawq@njtech.edu.cn; iamxcdong@njtech.edu.cn
bSchool of Chemistry & Materials Science, Jiangsu Normal University, Xuzhou 221116, China
First published on 1st December 2025
Abstract
Adhesive hydrogels represent a transformative technology in biomedicine due to their biocompatibility and multifunctionality. While extensive research has focused on improving their adhesion strength, the pursuit of long-term interfacial stability reveals a core conflict: strong adhesion often comes at the expense of easy removal. Dynamically regulating hydrogel adhesion is thus key to personalized medicine, allowing adaptation to complex clinical needs. Designing such systems demands a multifaceted approach that considers the physiological environment, medical requirements, stimulus-induced interfacial rearrangements, and mechanics-driven microstructure reconstruction. The dynamic regulation of hydrogel adhesion is more than a functional upgrade; it represents a paradigm shift for smart materials, from “static design” to “dynamic interaction”. This review first introduces the mechanisms of hydrogel adhesion. It then provides an in-depth analysis of strategies for dynamically regulating adhesion at the tissue–hydrogel interface and explores the latest progress and application potential in biomedicine.
 Qian Wang | Qian Wang received her PhD degree from the University of Chinese Academy of Sciences in 2015. Then, she continued her postdoctoral research at the Shanghai Institute of Microsystem and Information Technology, Chinese Academy of Sciences. In 2018, she joined Nanjing Tech University as an Associate Professor. Her current research interest focuses on the design and fabrication of flexible electronic devices. |
 Xiaochen Dong | Prof. Xiaochen Dong obtained his PhD degree from Zhejiang University in China in 2007. Then he joined the School of Materials Science and Engineering in Nanyang Technological University as a Postdoctor. In 2022, he was promoted to the vice president of Jiangsu Normal University. He has published more than 300 papers in journals including Angew. Chem., Adv. Mater., ACS Nano, etc. His current research mainly focuses on biophotonics and flexible electronic materials and devices. |
1. Introduction
Multifunctional biomedical hydrogels have enabled a profound transformation of traditional materials in biomedicine, owing to their excellent biocompatibility and low cost.1 Hydrogels consist of hydrophilic polymers that form a water-rich three-dimensional network, thereby providing a moist condition on the tissue interface. Their high flexibility and compliance allow them to conform to irregular human tissues, form stable bonds, and release drugs or bioactive molecules on demand for wound treatment.2 Attributed to their comparable elastic modulus to biological tissues, hydrogels can reduce mechanical stress and prevent damage to delicate tissues during adhesion.3 Furthermore, by incorporating conductive substances, biomedical hydrogels can achieve long-term, stable motion and wound monitoring, thereby contributing to patient rehabilitation.4,5 However, the human physiological environment is a dynamically complex system where multidimensional factors, from ubiquitous hydration barriers6 and biochemical variations7 to tissue modulus disparities,8 mechanical movements,9 and surface roughness,10 interact to pose significant challenges for biomedical hydrogel adhesives, creating critical clinical bottlenecks.The physiological environment imposes multidimensional physicochemical barriers to hydrogel adhesion, through its hyperosmotic-humid nature and biological complexity, with challenges arising from interconnected physical, chemical, and mechanical mechanisms. Firstly, tissue surfaces are universally covered by physical barriers of hydration layers (e.g., the aqueous matrix within the skin stratum corneum) and mucus layers (e.g., the viscoelastic glycoprotein network on the gastrointestinal tract). The hydration layer generates entropic repulsion to hinder molecular contact, while mucin lubrication significantly reduces interfacial adhesion efficiency.11 Internally, physiological water films such as synovial fluid in joints and mucosal secretions in the respiratory tract further physically isolate hydrogels from direct tissue contact, retard adhesive molecule diffusion and compromise bioadhesive groups through hydrolytic cleavage or protonation.12 Furthermore, the variations in the chemical microenvironment exacerbate additional challenges. Complex physiological factors of wet conditions, extreme pH gradients, high ionic strength and enzyme activity can all damage the molecular network of hydrogels and decrease their cohesive strength, thereby weakening the tissue adhesion.13,14 Additionally, this challenge is further compounded by the structural complexity and dynamic evolution of biological surfaces. For instance, factors like wrinkle dynamics, hair presence, and sweat secretion on the skin alter the interfacial microstructure and chemistry, thereby impeding intimate hydrogel contact and compromising adhesion performance.15
Furthermore, tissues exhibit significant mechanical disparities due to their compositional and functional diversity, with elastic moduli spanning 3 to 6 orders of magnitude, from the softness of skin (1–100 kPa) to the rigidity of bone (10–20 GPa).16–20 This modulus mismatch between hydrogels and tissue interfaces readily induces interfacial stress concentration and adhesion failure, often leading to clinical complications.21 In dynamic soft tissues subjected to high-frequency motions, mechanical mismatches exacerbate failure risks.22 For example, knee joint movements generate tensile strain (>50%), compressive stress (10–20 MPa), and shear force (0.1–10 kPa), causing rapid debonding of modulus-mismatched hydrogels and potential cartilage tears.23–25 In the cardiovascular system, the aorta's pulsatile pressure (120 mmHg, equivalent to 16 kPa cyclic loading) and myocardial strain (10–20%) can trigger interfacial tearing in low-modulus hydrogels under hemodynamic forces, while high-modulus hydrogels impede vascular vasodilation and induce thrombosis due to excessive rigidity.26 Therefore, addressing the mechanical mismatch necessitates tailoring the hydrogels' mechanical and adhesive properties to dynamically bridge the modulus gap across diverse tissues. Moreover, human tissues are constantly engaged in continuous dynamic movements (such as the cyclic mechanical forces from gastrointestinal peristalsis or the high-frequency motions in the oral and pharyngeal regions), generating periodic mechanical forces that further pose multidimensional challenges to the adhesion and durability of hydrogels.27,28 More importantly, once micro-cracks or localized adhesive failures are initiated, they tend to propagate irreversibly under cyclic loading, ultimately resulting in catastrophic failure. Current materials often experience disintegration or interfacial slippage due to insufficient fatigue cycles, highlighting the critical shortages in mechanical stability under dynamic loading.29 In conclusion, these adhesion challenges originate from a fundamental mismatch between the mechanical complexity of dynamic tissues, including motion frequency, stress types, the chemical microenvironment, and the static properties of conventional hydrogels, urgently necessitating the development of adaptable adhesive hydrogels.
Nevertheless, achieving strong adhesion in biomedical hydrogels presents a crucial challenge. Excessive adhesion not only complicates removal and risks tissue damage but also may further compromise the hydrogel's breathability and moisture balance, thereby undermining therapeutic efficacy. Conversely, inadequate adhesion restricts functional applications, resulting in short service life and entailing frequent replacement. Therefore, reconciling strong adhesion with easy and safe removal has become a central focus in the field. This on-demand dynamic adjustment can meet diverse applications, such as acute hemostasis, drug release, and biosensing, promoting the transformation of hydrogel materials from “static design” to “dynamic interaction”. In recent years, many reviews related to hydrogel adhesion have been published. Xiong et al.30 and Ma et al.31 summarized the adhesion mechanism of hydrogels. In terms of the adhesion interface structure design, Yang et al.32 discussed the topological cross-linking strategy of the hydrogel adhesion interface, and Bovone et al.33 summarized the chemical adhesion between hydrogels and tissues. Ouyang et al.34 discussed approaches to enhance the adhesion of biomedical hydrogels, and Wang et al.35 and Yuen et al.36 provided guidelines for enhancing the underwater adhesion properties of hydrogels. The development of principles and effective methods for the dynamic regulation of interfacial adhesion in biomedical hydrogels is imperative. This review first elucidates the adhesion mechanisms of biomedical hydrogels from chemical, topological, mechanical, and biological perspectives, establishing a theoretical foundation for dynamic regulation. It then focuses on strategies for controlling adhesion, aiming to provide innovative approaches for broadening hydrogel functionality through a systematic synthesis of existing knowledge (Fig. 1). Finally, the biomedical applications of these adhesive hydrogels are explored.
 | ||
| Fig. 1 A schematic illustration of hydrogel adhesion principles and dynamic regulation. | ||
2. Adhesion mechanisms of biomedical hydrogels
Generally, effective adhesion in flexible materials requires both strong adhesion and cohesion. Adhesion refers to the interfacial binding forces (e.g., covalent, non-covalent, and topological), whereas cohesion denotes the material's internal strength against fracture. Consequently, the macroscopic adhesion strength, the force needed for separation, is ultimately determined by the weaker of these two microscopic strengths.37 As shown in Fig. 2a, the loss of either adhesion or cohesion will lead to hydrogel adhesion failure. If cohesion is insufficient, the hydrogel undergoes internal fracture before detaching from the substrate. Conversely, weak interfacial bonding results in detachment at the interface. Furthermore, the effect of the complex physiological environment (physical, chemical, and mechanical factors) cannot be overlooked. Given the similar molecular interacting origins of adhesion and cohesion, this section summarizes the key adhesion mechanisms and the challenges posed by physiological conditions.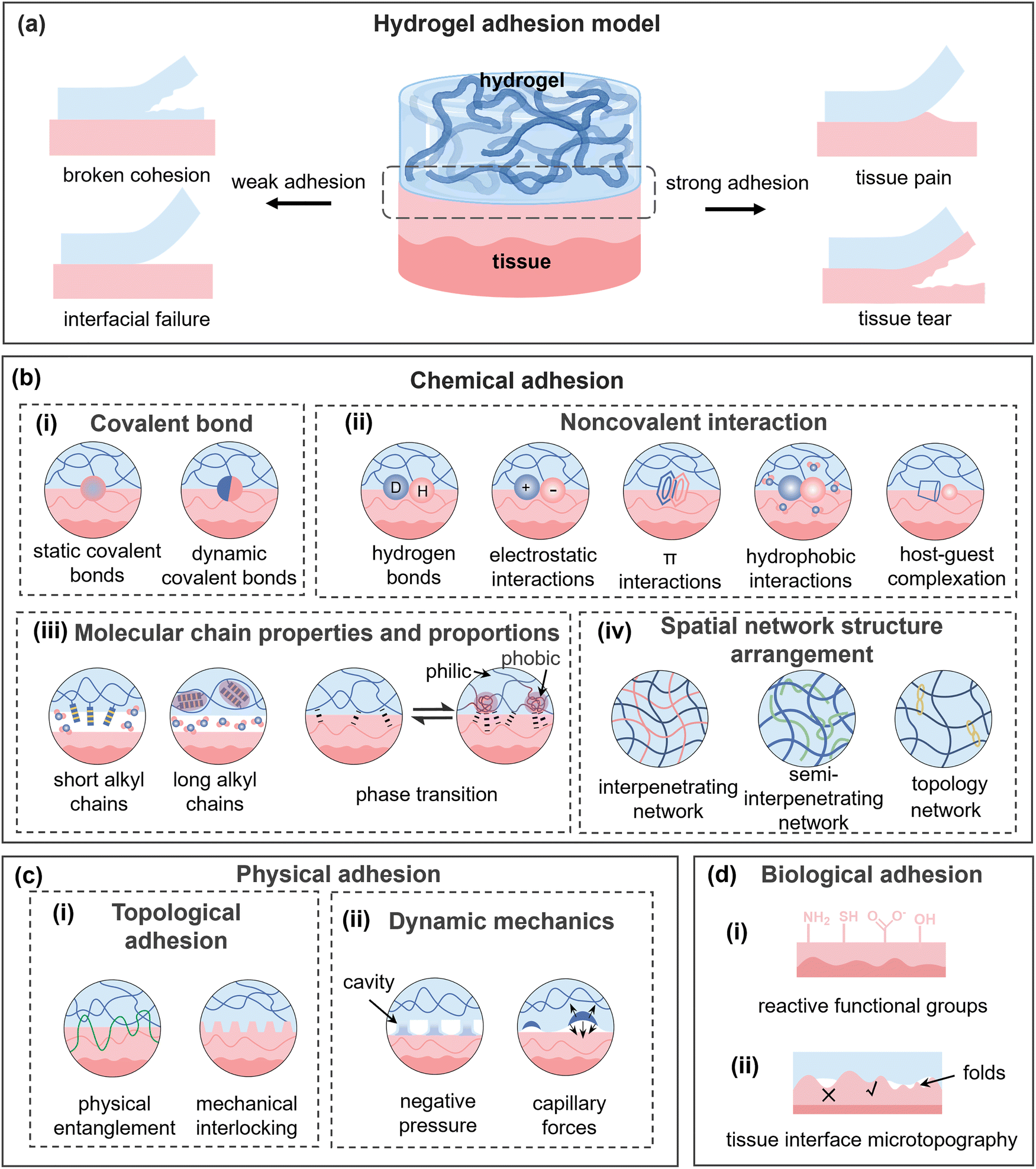 | ||
| Fig. 2 Illustration of hydrogel adhesion mechanisms and classification. (a) Models demonstrating failure behavior under strong and weak adhesion conditions. (b)–(d) Key mechanisms including chemical, physical, and biological adhesion. | ||
2.1. Chemical adhesion
Covalent adhesion offers strong bonding but is hampered by its need for specific conditions, slow kinetics, and the cytotoxicity of reagents like aldehydes or cyanides. Consequently, developing strategies for rapid, strong, and safe covalent adhesion on tissues represents a major clinical hurdle.
At the hydrogel–tissue interface, polar interactions (e.g., hydrogen bonds and electrostatic interactions.) rely on electric charges or dipoles, and they are susceptible to environmental interferences but flexibly regulated. Most hydrogels have numerous polar functional groups that can engage in hydrogen bonding with tissues, providing a stable adhesion interface.43 Unfortunately, water's high dielectric constant and ability to solvate polar functional groups can weaken the hydrogen bonds, preventing direct interactions between hydrogels and tissues.44 Thus, hydrogen bonds often need to cooperate with other non-covalent bonds to form stable adhesion. For organisms containing multiple ions, tough adhesion constructed from electrostatic interactions is routine yet critical. These interactions, relying on ambient pH value and ionic strength, are well-suited for adhering to charged biological tissues like mucous and cell membranes.28 Although electrostatic interactions are less affected by the aqueous environment and play a prominent role in underwater adhesion, ions in the physiological environment will inevitably interfere with the charge distribution on both the hydrogel and tissue surfaces, gradually decreasing the adhesion strength over time.45,46 van der Waals force usually acts in conjunction with electrostatic interaction at the adhesion interface. Unlike the direct charge-based nature of electrostatic forces, van der Waals forces are derived from the interaction of dipoles and are relatively weak. Their short operating distance makes them essential in scenarios where hydrogels and tissues are in close intimate contact or at a small scale.47 A notable example is the gecko feet, where van der Waals forces enabled by setae promotes the development of gecko-inspired adhesive hydrogels.48 Furthermore, irregularities on the surfaces increase the contact area and the number of van der Waals interactions, thereby promoting adhesion.49 Notably, water can greatly weaken the van der Waals force to one-hundredth, providing new insights for dynamically regulating hydrogel interfacial adhesion.50
In addition to the common polar non-covalent bonds mentioned above, other specific physicochemical interactions also advance in the interfacial adhesion. π-interactions can occur between aromatic or conjugated moieties in both the hydrogel and the substrate.29 For tissues with cations, excellent wet adhesion is often achieved through cation–π interactions with tissue amino groups.51 In addition, π-interactions usually coexist with other intermolecular forces and play an auxiliary role at the adhesion interface, such as disrupting the hydration layer.52,53 In an aqueous environment, the hydrophobic interactions can repel water molecules at the adhesion interface and disrupt the hydration layer, promoting localized aggregation of nonpolar molecules or hydrophobic chains, thus further enhancing the underwater adhesion performance.54–56 Host–guest complexation refers to the specific molecular recognition and bonding interaction between two or more molecules, a highly selective and dynamically responsive non-covalent interaction. In this interaction, the host molecules typically possess a large cavity (such as cyclodextrins), while the guest molecules (such as polyethylene glycol, adamantane, azobenzene, and ferrocene.) exhibit complementary shape or structure to the host molecules. Other non-covalent forces may also be present during the process of host–guest complexation.57,58 However, the application of host–guest complexation is often hindered by the absence of suitable host or guest molecules on the adhesive targets, consequently leading to their greater involvement in the internal crosslinking of hydrogels.59,60
On the hydrogel–tissue interfaces, multiple non-covalent interactions synergistically contribute to constructing a strong and multifunctional adhesion. Rational functional group optimization and microstructure design offer extensive prospects for noncovalent adhesion. However, noncovalent adhesion typically exhibits weaker adhesion forces compared to covalent adhesion and is more susceptible to external conditions. High temperatures, high humidity, and strong acidity/alkalinity in particular can disrupt noncovalent adhesion, limiting its reliability and stability.
Additionally, when hydrogels come into contact with substrates, the pore elasticity of the hydrogel directly affects the contact area, while greater crosslinking density is always accompanied by smaller and more rigid pore structures.69,76 Further research and quantification characterization are still in urgent demand to elucidate the relationship between the pore structure and the adhesion strength of hydrogels.
2.2. Physical adhesion
Physical entanglement is typically achieved through the diffusion of polymer chains, which demands two prerequisites: compatibility of the contact surfaces and good mobility of polymer chains. Typically, the polymer penetrates into the tissue matrix and forms an interpenetrating network to achieve chain entanglement.77 This adhesion method is substantially influenced by the molecular weight, concentration, and hydrophilic–hydrophobic properties of the polymer and is generally not applicable to pre-formed hydrogels.78 Mechanical interlocking refers to another type of mechanically interconnected adhesion where hydrogel precursors infiltrate into the rough substrate surface and stick to it through elaborate nested structures after polymerization.79 Especially, mechanical interlocking can connect two adherents through geometric shapes, such as a micro-needle array with barb structures, without requiring intermolecular interactions. However, it should be noted that due to substantial variations between substrates, the interfacial adhesion achieved through this anchoring approach will exhibit considerable differences.
Generally, both physical entanglement and mechanical interlocking can achieve strong adhesion, while they are largely irreversible. The separation process typically damages the molecular networks or microstructures, causing pain and tissue injury, as well as non-negligible residue upon detachment.
Since the hydrogels possess a porous structure and high water content, capillary forces are significant in the adhesion process, especially in wet adhesion.81 Capillary forces refer to the forces exerted by a liquid in a thin tubular or a narrow pore due to surface tension, being related to the elastic force or osmotic pressure at the adhesion interface. As shown in Fig. 2c(ii), capillary forces enable the hydrogel to adsorb liquids on the contact interface and enhance the contact with the substrate. On a tissue surface, capillary forces promote the hydrogels to adsorb the extracellular matrix, blood, or other biological fluids, facilitating the close contact between hydrogels and tissues.82 The capillary adhesion between hydrogels is thermodynamically driven and regulatable, requiring no surface modification.83 Although generally weak and influenced by interfacial liquids and the hydrogel's structure, the capillary force can be significantly enhanced through microstructure design. Additionally, capillary forces exhibit a certain annealing effect within hydrogels, which may alter the pore structure and thereby influence the physical properties and stability of the hydrogel.
2.3. Biological adhesion
The physiological tissue environment exhibits a complex interplay of liquid and solid components, where the fluid phase typically weakens adhesion through enhanced molecular mobility and solid components (proteins, minerals, etc.) serve as the core targets for adhesive interactions.84,85 As the primary constituents of tissue dry weight (proteins accounting for 20%), fibrous extracellular matrix (ECM) proteins, such as collagen and elastin, and cell membrane glycoproteins expose reactive functional groups, including primary amines (–NH2), thiols (–SH), carboxyl groups (–COOH), and hydroxyl groups (–OH) (Fig. 2d(i)), that engage in adhesion through nucleophilic addition, electrostatic interactions, or covalent coupling.33 Primary amines, owing to their high abundance (6% of amino acids) and strong nucleophilicity, serve as the most commonly targeted adhesion moieties, enabling efficient covalent crosslinking with electrophilic groups (e.g., aldehydes and acid anhydrides).86 Thiols, though less abundant (1%), facilitate rapid and efficient conjugation via thiol-ene click chemistry.87 It is worth noting that, unlike rigid substrates (e.g., glass and wood), biological tissues are dynamic and viscoelastic. This inherent feature, derived from various proteins in tissues (such as the sliding of collagen fibers, the deformation of elastin, and the water membrane of extracellular mechanisms), facilitates energy dissipation during adhesion, resulting in the tissue-dependent performance of hydrogel bioadhesives.88–90As shown in Fig. 2d(ii), tissue interface microtopography further presents physical adhesion opportunities. Tissue surface roughness (e.g., skin texture and mucosal folds) exerts dual effects: micron-scale features (e.g., skin texture and mucosal folds) create natural anchoring sites for mechanical interlocking, allowing hydrogels to form “topological entanglement” by filling interstitial spaces, thereby significantly enhancing adhesive strength. Excessive roughness, however, prevents complete filling of tissue interstices, resulting in insufficient contact area or stress concentration that compromises adhesion.91,92 Further, the interplay between biomolecule adsorption and tissue microstructure profoundly influences the adhesive performance and biocompatibility of hydrogels in a physiological environment. Polymers undergo featured biological reactions in these environments (e.g., acid/base hydrolysis and enzymatic catalysis), endowing the biomedical hydrogel with a series of specific adhesion requirements. For example, proteins like fibrinogen and immunoglobulins adsorb onto hydrogel surfaces to form a protein corona, where the as-formed corona may obstruct chemical bonding between hydrogel functional groups and tissue surfaces, alter interfacial adhesion energy through steric hindrance or charge screening effects, and trigger foreign body reactions.93 Notably, while in wet environments, its exposed amine and carboxyl groups also enable non-specific adsorption via electrostatic interactions or hydrogen bonding.94 Consequently, optimizing hydrogel bioadhesion requires suppressing contact defects from excessive roughness and non-specific protein adsorption while strategically leveraging moderate roughness to enhance interfacial bonding.
In summary, the tissue solid-phase characteristics (protein abundance and functional group distribution) and microstructural features (roughness and topological morphology) collectively define key adhesion opportunities. By designing covalent/noncovalent chemical strategies targeting reactive sites like amines and carboxyls and integrating physical interlocking via micron-scale topological adaptation, these synergistic mechanisms can overcome adhesion bottlenecks in wet environments, offering multi-dimensional solutions for biomedical interface integration.
Table 1 summarizes the advantages and disadvantages of common hydrogel–tissue adhesion mechanisms, with respect to their specificity, functionality, and applicable domains. For chemical adhesion, covalent and non-covalent interactions are decisive, with molecular and network design being auxiliary. While covalent bonds are strong, their irreversibility, need for specific conditions, and slow kinetics restrict their use at dynamic hydrogel–tissue interfaces, thus precluding applications requiring immediate adhesion. In contrast, non-covalent interactions, while weaker and more susceptible to the physiological environment, offer reversibility and stable adhesion under tissue deformation. Physical adhesion usually plays an auxiliary role as well. Among them, the topological adhesion tends to be invasive and irreversible, posing a potential risk of tissue damage during detachment, while achieving mechanical effects imposes stringent requirements on both hydrogel fabrication and tissue surface morphology. Physiological adhesion provides fundamental conditions for the stability of hydrogel–tissue adhesion interfaces and offers adaptive adhesion behavior in hydrogels, but this mechanism also compromises the universality and controllability, limiting its broad applicability.
| Adhesion mechanism | Types | Advantages | Disadvantages | |
|---|---|---|---|---|
| Chemical adhesion | Covalent bonds | High intensity | Potential biological toxicity | |
| Good stability | Usually irreversibility | |||
| High targetability | Slow formation speed | |||
| Specific reaction conditions | ||||
| Noncovalent interactions | Hydrogen bonds | Universality | Water-sensitivity | |
| Reversibility | pH-sensitivity | |||
| Electrostatic interactions | Higher intensity | pH-sensitivity | ||
| Reversibility | Ionic concentration-sensitivity | |||
| van der Waals force | Ubiquity | Extremely weak intensity | ||
| π-interactions | High targetability | Low applicability | ||
| Wet adhesion | ||||
| Hydrophobic interactions | Wet adhesion | Low applicability | ||
| Host–guest complexation | High targetability | Low applicability | ||
| Molecular chain | Base adhesion | Auxiliary adhesion | ||
| High adjustability | ||||
| Network structure | Base adhesion | Auxiliary adhesion | ||
| High adjustability | ||||
| Physical adhesion | Topological adhesion | High intensity | Unique surface morphology | |
| Slow formation speed | ||||
| Not relying on chemical action | Invasiveness | |||
| Usually irreversibility | ||||
| Dynamic mechanics | Negative pressure | Rapid adsorption | Unique structural requirements | |
| Reversibility | Make complex | |||
| Non-invasiveness | High sealing requirements | |||
| Capillary forces | Drain surface water | Weak intensity | ||
| Auxiliary adhesion | ||||
| Biological adhesion | Biologically adaptive | Low versatility | ||
| High biocompatibility | Uncontrollability | |||
| High targetability | ||||
3. Dynamic regulation of the interfacial adhesion of hydrogels
The robust adhesion of hydrogels has broadened their use in biomedicine, including biosensing, wound therapy, and tissue repair. However, a stable adhesion interface is often contradicted by easy peeling. This is particularly problematic for non-degradable hydrogels, as their removal via mechanical debridement poses a significant risk of secondary injury or infection. Conversely, biomedical hydrogels with insufficient adhesion fail to seal wounds effectively, which can lead to serious complications like bacterial infection. Thus, achieving precisely controlled adhesion and on-demand removal is crucial for safe biomedical applications. This section aims to thoroughly elucidate the principles, mechanisms, and applications of dynamic adhesion regulation in hydrogels.3.1. Stimulus-induced interfacial interaction rearrangement
Dynamic physical and reversible covalent bonds govern the cohesion of hydrogels and their interfacial interactions, thereby determining adhesion strength. By controlling the fracture and reformation of these dynamic bonds, the adhesion of hydrogels can be precisely regulated. As illustrated in Fig. 3a, various external stimuli (e.g., pH, temperature, light, electric fields, and biotrigger fluids) can trigger molecular-level changes in hydrogels, modulating their cohesion and adhesion to permit either robust bonding or facile detachment. The process is highly reversible, with adhesion properties recovering upon stimulus removal through the reorganization of dynamic bonds.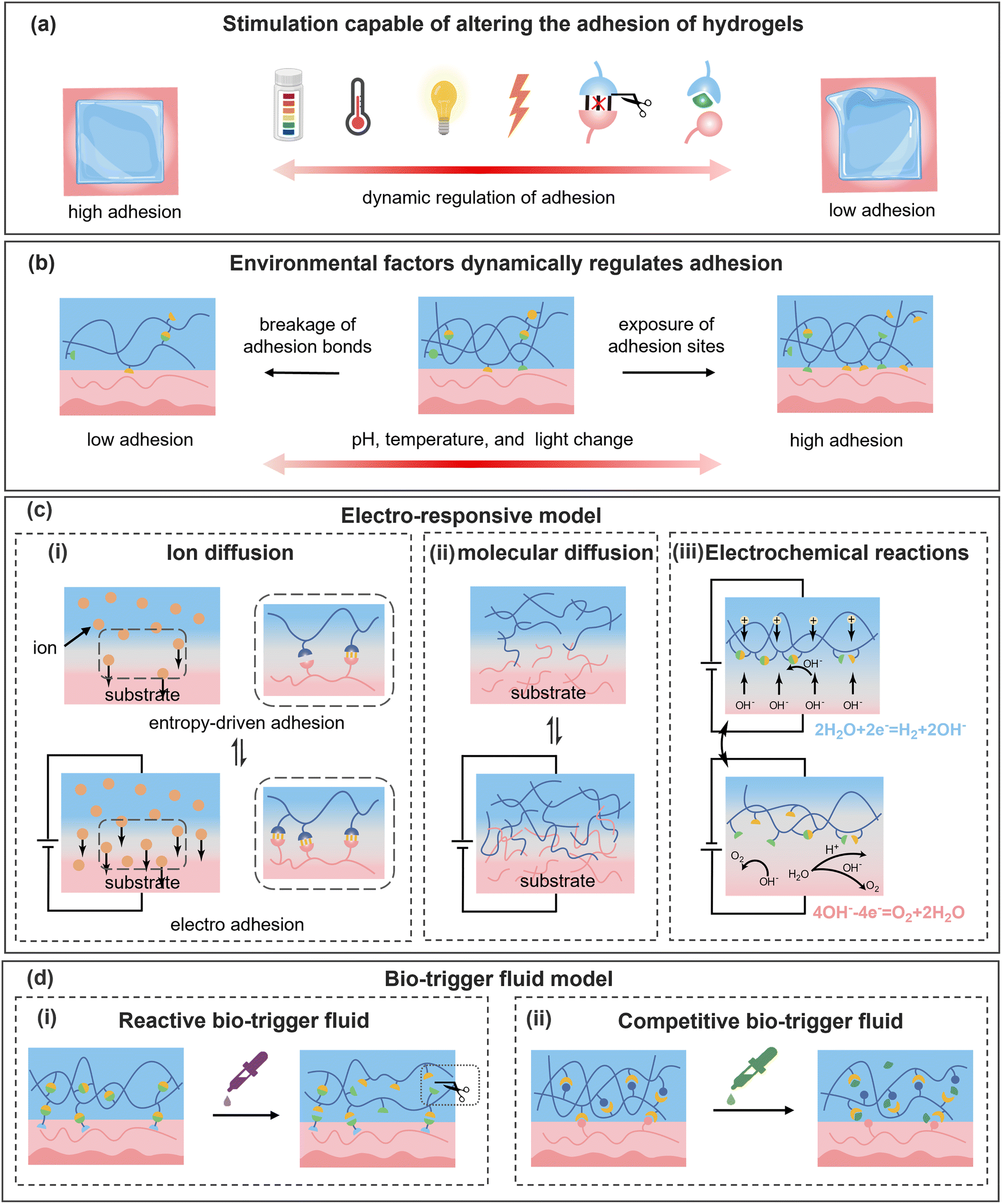 | ||
| Fig. 3 (a) and (b) Schematic illustration of dynamic hydrogel adhesion regulation in response to external stimuli and environmental factors. (c) Schematic of electric-field-responsive hydrogel adhesion mechanisms and representative models. (i) Ion diffusion occurs at the interface and changing the interfacial adhesive bonds. (ii) Molecular chain diffusion occurs at the adhesive interface. (iii) The electrochemical reactions under the electric field alter the pH value inside the hydrogel. (d) Schematic of hydrogel adhesion modulation by bio-fluids. (i) Reactive bio-trigger fluids break certain specific interfacial covalent bonds. (ii) Competitive bio-trigger fluids act on non-covalent bonds without changing the original functional groups. | ||
Carboxyl groups are commonly utilized as adhesive moieties in hydrogels, offering both hydrogen bonding and electrostatic interactions at the interface. The deprotonation of carboxyl groups increases negative charge density, thereby enhancing electrostatic attraction to positively charged substrates. However, this process concurrently weakens hydrogen bonds, which generally dominate the adhesion process, and the overall adhesion strength of the hydrogel is consequently decreased.95 As shown in Fig. 4a, Zhang et al.96 designed a nanofibrous cellulose/polyacrylic acid (PAA) hydrogel; the excessively hydrated hydrogen ions in acidic liquids disrupted the hydrogen bond network and facilitated the release and protonation of hydroxyl and carboxyl groups, presenting a notable enhancement in adhesion. The amino group is a common cationic pH-responsive group, which routinely exists in the form of –NH2 at a high pH value. Under acidic conditions, the majority of –NH2 groups are protonated to –NH3+, which will weaken or even eliminate the hydrogen bond between amino groups and enhance the hydrophilicity of the hydrogel. Wei et al.97 enhanced the deprotonation degree of tertiary amine groups on poly(2-(dimethylamino)ethyl methacrylate) in the hydrogel by increasing the alkalinity. This transformation induced a shift of the polymer network from hydrophilicity towards hydrophobicity, strengthening the underwater adhesion of the hydrogel. The catechol group's adhesion property is also correlated with the pH scale. By increasing the pH value at the adhesion interface, the oxidation of phenolic hydroxyl groups to quinones can be promoted, enabling a controlled detachment of the hydrogel.98 In addition to that common non-covalent bond, a Schiff base bond, a strong covalent bond, also possesses typical pH responsiveness. Mohanty et al.99 employed an acetic acid buffer solution (pH = 5) to disrupt the Schiff base bonds and intermolecular hydrogen bonds in a hydrogel, achieving a reduction of adhesion strength toward tissues. For the hydrogel–tissue interface linked by Schiff base bonds, it is also plausible to achieve on-demand detachment using a suitable pH buffer. Kang et al.100 concurrently introduced borate ester bonds and Schiff base bonds into a hydrogel, and the subsequent acid treatment significantly reduced the adhesion strength of the hydrogel by ∼92% (Fig. 4b).
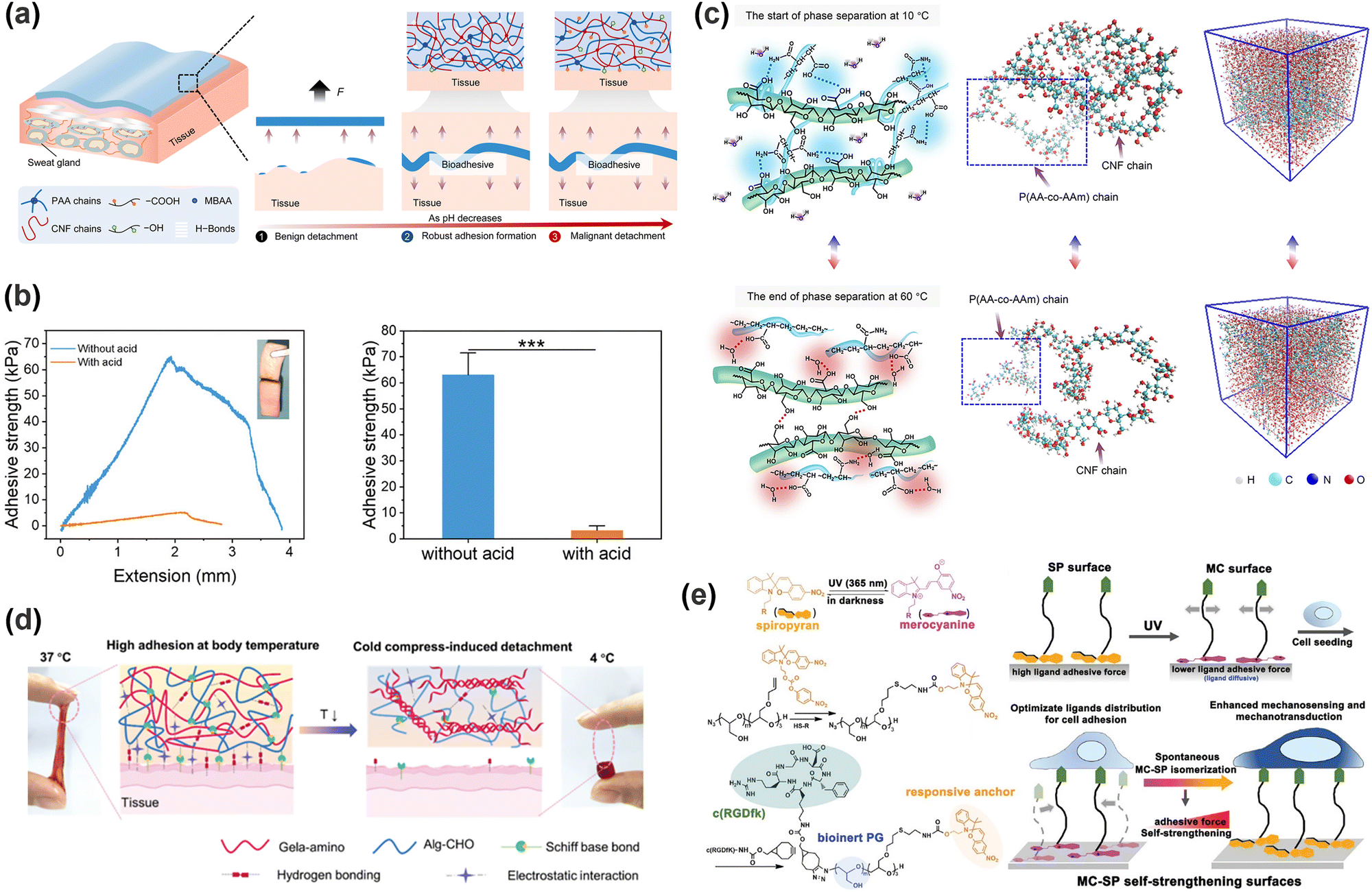 | ||
| Fig. 4 (a) Scheme of enhanced adhesion of hydrogels in an acidic environment. Reprinted with permission from ref. 96 Copyright 2023, Royal Society of Chemistry. (b) Scheme of the adhesion strength of hydrogels with or without acid treatment. Reprinted with permission from ref. 100 Copyright 2023, Wiley-VCH GmbH. (c) Scheme of the inter-chain hydrogen bond changes of CNF/P (AA-co-AM) hydrogels at 10 °C and 60 °C compared with MD snapshots. Reprinted with permission from ref. 102 Copyright 2023, American Chemical Society. (d) Schematic diagram of the helical structure of gelatin changing with temperature and the structure change of the hydrogel. Reprinted with permission from ref. 106 Copyright 2023, Wiley-VCH GmbH. (e) Photostimulation alters the isomerization of MC-SP and the interfacial adhesion strength of hydrogels. Reprinted with permission from ref. 116 Copyright 2020, John Wiley and Sons. | ||
Although pH-regulated adhesion operates via the straightforward mechanism of protonation/deprotonation, it is hampered by the slow diffusion of H+/OH− ions, resulting in sluggish response kinetics. Clinically, its application is challenging since exogenous pH adjustment often requires auxiliary solutions and is largely restricted to superficial tissues. More critically, excessive pH changes risk causing tissue damage, underscoring the need to maintain the interfacial pH within a safe range to prevent secondary harm. Furthermore, the dynamic and often unpredictable pH fluctuations at tissue surfaces, particularly in wounded areas, must be explicitly considered in hydrogel design. These inherent limitations collectively diminish the feasibility of pH regulation as a robust strategy for biomedical adhesives.
Hydrogen bonds, as one of the most common temperature-responsive non-covalent bonds, can dynamically regulate the degree of the crosslinking with temperature variation, thereby modulating the hydrogel adhesion strength.101 In Fig. 4c, the hydrogen bonds between cellulose nanofibers (CNF) and P(acrylic acid-co-acrylamide)(AA-co-AM) chains gradually decoupled with increasing temperature from 20 °C to 35 °C.102 The breaking of hydrogen bonds further led to the exposure of active functional groups at the interface, corresponding to the interfacial toughness increasing from 5 J m−2 to 1255 J m−2. Gelatin exhibits a representative temperature-responsive phase transition behavior, where the helical structure formed by hydrogen bonds gradually disintegrates with the temperature, leading to increased mobility of the molecular chains and enhanced collision probability of adhesion sites.103 Comparatively, the phase transition behavior of PNIPAM originates from the fracture of hydrogen bonds between polymer chains and water with temperature rise (LCST), exhibiting enhanced hydrophobic effect.104 Pang et al.105 designed a PNIPAM backbone to enable the hydrogel to exhibit distinct adhesion disparity at 25 °C and 40 °C. Schiff base bonds are another class of typical temperature-sensitive groups, as higher temperature increases the thermal motion within and between molecules and enhances the likelihood of carbon–nitrogen bond cleavage.74 Leveraging this principle, Geng et al.106 modified gelatin with ethylenediamine to increase its amino groups, which provided abundant active sites for the formation of Schiff base bonds with sodium alginate (Fig. 4d). In adhesive experiments with pig skin, as the temperature decreased from 37 °C to 4 °C, the adhesive strength diminished from 2800 Pa to 300 Pa. This approach, which utilizes low-temperature strengthening of cohesion to weaken adhesion, can significantly minimize residue, benefiting the treatment of fragile and sensitive wounds. Temperature can also cause phase transformation by affecting the crystallinity of the polymer chains. Tian et al.107 added crystallizable C18 chains to the polymer matrix and endowed temperature-controlled adhesion to the hydrogel. This strategy involves only reversible changes in chemical bonds without the need for additional chemical substances, exhibiting impressive reversibility, convenience, and flexibility.
Common approaches for temperature adjustment include cold/hot compresses, rinsing with cold/hot water, and stimulus-induced thermal conversion. Among these, stimulus-induced methods like photothermal, ultrasound, and magnetothermal effects provide greater tissue penetration depth and superior accuracy in programmability, whereas compresses and rinsing offer greater convenience.108,109 It is worth noting that thermo-sensitive hydrogels are highly affected by environmental temperature, which greatly restricts their applications under extreme conditions. In addition, significant performance variation may arise between the inner and outer sides of thermosensitive hydrogels when there is a distinct temperature difference with the human body, thereby affecting the stability of other functions.
Photochemical regulation involves chemical reactions under light irradiation (e.g., isomerization, cleavage,110,111 addition,112,113 reduction, exchange,114,115 and catalyze) to change the conformation, dipole moment, solubility, conductivity, or ion concentration of the hydrogel (Table 2). These changes subsequently lead to variations in cohesion or adhesive binding sites of the hydrogel, achieving management over its adhesion properties. Ultraviolet (UV) light and visible light are customary light sources for photochemical responses. As shown in Fig. 4e, UV irradiation induced the transformation of spiropyran to hydrophilic merocyanine form, compromised the hydrophobic interactions between the hydrogel and the substrate, and hindered the adhesive behavior of the hydrogel.116 Due to the photoisomerization properties of azobenzene, the host–guest interaction between azobenzene and β-cyclodextrin (β-CD) can be weakened under light irradiation.117–119 Inspired by this, Wu et al.120 designed a hyaluronic acid hydrogel modified with azobenzene and β-CD. After 5 min of red light irradiation, the adhesive strength of this hydrogel was reduced by 60%. The photocatalytic reaction can also change the hydrophilic/hydrophobic state of the hydrogel, achieving rapid switch in adhesion properties. Ryplida et al.121 utilized the photocatalytic effects of titanium oxide (TiO2) and silica-carbon dots (CDs) under UV or visible light irradiation, to modify the hydrogel into a hydrophilic state, facilitating the contact between resorcinol and the substrate. Interestingly, UV light is also capable of inducing redox reactions to modify the metal coordination in polymer crosslinking. Generally, UV light can easily reduce Fe3+ to Fe2+ and transform the stronger carboxylate–Fe3+ coordination interaction into a weaker carboxylate–Fe2+ coordination, motivating a reduction in hydrogel cohesion.42 Gao et al.122 coated Fe3+ and citric acid between two hydrogels containing PAA chains to achieve strong adhesion, while the two blocks can also be selectively separated by dissociation of strong coordination bonds under UV irradiation. Compared with UV light, near-infrared light (NIR) possesses deep penetration and minimal photodamage, being more suitable for biological applications. Jiang et al.123 used PAA-coated upconverting nanoparticles to convert NIR into UV light and manipulated the resulting UV irradiation to reduce Fe3+, weakening the interfacial adhesion strength of the hydrogel.
| Category | Example | Structural formula 1 | Structural formula 2 | Reaction conditions | Ref. |
|---|---|---|---|---|---|
| Isomerization | Spiropyran (merocyanine) |
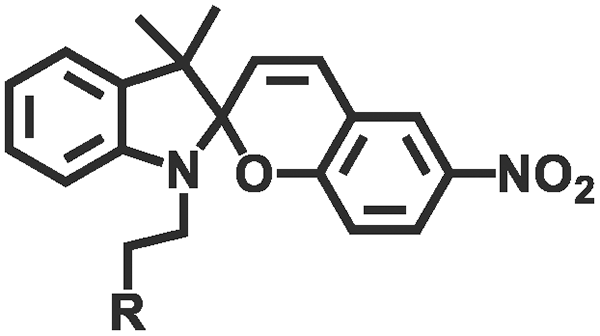
|
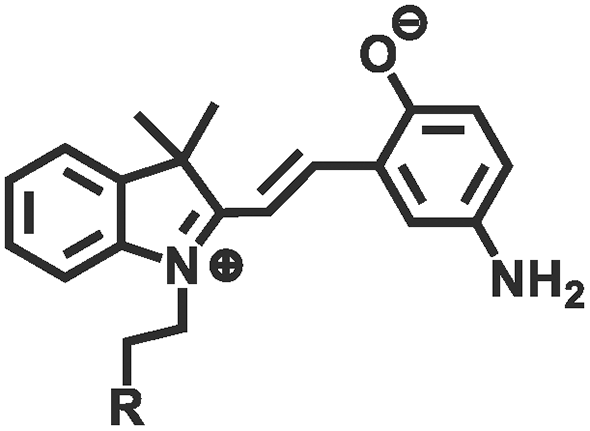
|
365 nm/in darkness | 126 |
| Azobenzene |
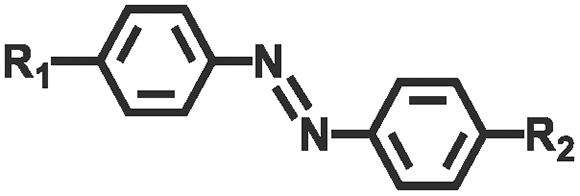
|
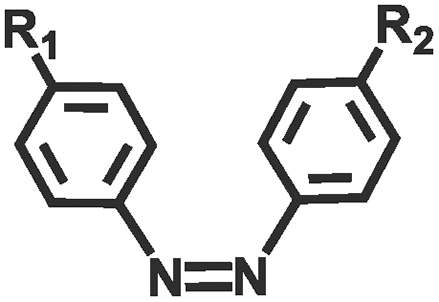
|
445 nm/445 nm | 120 | |
| Cleavage | Coumarin derivatives |
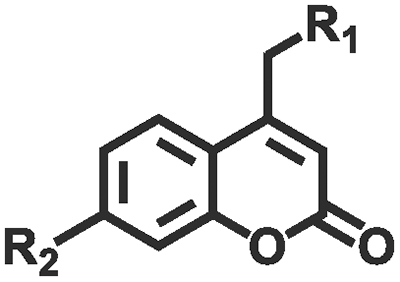
|
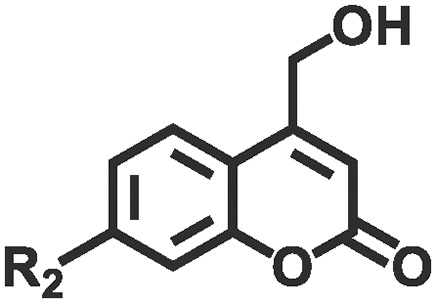
|
>365 nm | 110 |
| Derivatives of o-nitrobenzyl compounds |
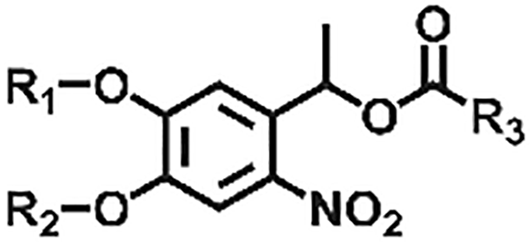
|
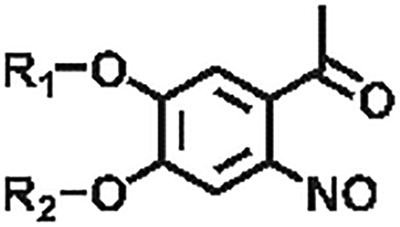
|
365 nm | 111 | |
| Addition | Styrene compounds |

|
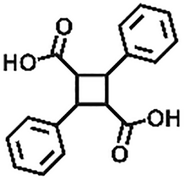
|
>300 nm/<280 nm | 112 |
| Thiol-ene “click” chemistry |

|

|
365 nm | 113 | |
| Metal coordination | Iron ion reduction | Fe3+ | Fe2+ | 365 nm | 121 |
| Ru–S coordination |
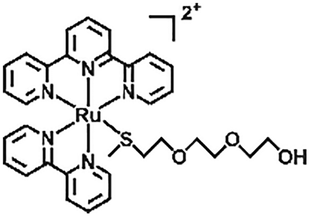
|
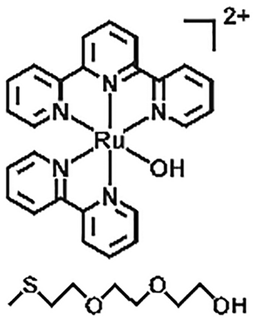
|
Light, H2O/heat | 120 | |
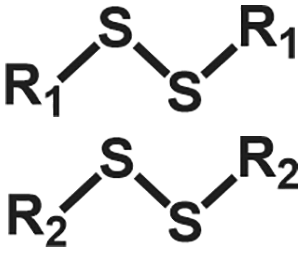
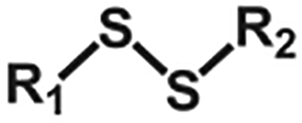
| Exchange | Disulfide |
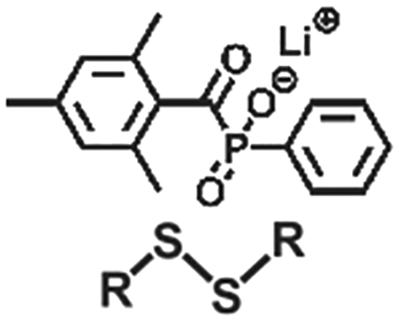
|
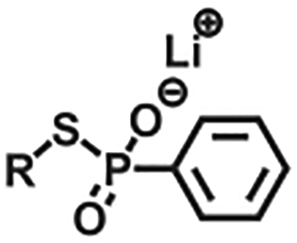
|
>365 nm | 114 |
|
|
365 nm | 115 | ||
| Catalyze | TiO2 | O2− and –OH | 365 nm | 121 | |
| Light conversion | NIR | UV | UCNPs | 123 |
Photothermal regulation involves the absorption of light energy by photosensitive groups, leading to a localized temperature increase. This process, once the internal temperature reaches a phase transition point, adapts the molecular interactions within the hydrogel and thereby modifies its adhesion properties.124 Compared to the conventional temperature-triggered approach, the photothermal conversion strategy offers remote controllability, higher targeting specificity, and a wider range of operating temperatures. Especially, hydrogels with photothermal response can be quickly restored to their initial state after the removal of stimulus, exhibiting notable reversibility. The customary photothermal-responsive functional groups or materials include coumarin derivatives, polydopamine, black phosphorus (BP), metal nanoparticles, metal–organic frameworks (MOFs), and carbon-based materials. Ding et al.125 added BP into the hydrogel and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C formed by the Knoevenagel condensation reaction was broken under NIR light irradiation. The increase in the fluidity of the hydrogel prompted a 68% reduction in shear adhesion strength.
C formed by the Knoevenagel condensation reaction was broken under NIR light irradiation. The increase in the fluidity of the hydrogel prompted a 68% reduction in shear adhesion strength.
The light-promoted adhesion regulation method has critical requirements on the photosensitivity and stability of the material, while shortcomings of complex operation and tissue damage after prolonged illumination should also be addressed. Moreover, the phototoxicity concerns of UV light and potential photodegradation from light-sensitive materials also cannot be ignored. Furthermore, the light stimulation is strictly influenced by the light source and biomedical equipment, which further limits the application scenarios. How to simplify the synthesis process of photosensitive materials and improve the prolonged photothermal conversion efficiency with delicate regulation of light stimulus (such as wavelength, intensity, and duration) remains a great challenge for the future of light-responsive adhesive hydrogels.
As shown in Fig. 3c(i), for ion diffusion in hydrogels, after precisely adjusting the magnitude and duration of the applied voltage, the diffusion rate and range of ions can be accurately regulated to meet specific requirements. In Fig. 5a, Liu et al.127 demonstrated that applying voltage to each end of a Li+-bearing hydrogel and a polyvinyl alcohol (PVA) hydrogel substantially enhanced the adhesive efficiency by 24-fold. The strong hydration of Li+ facilitated its co-diffusion with water molecules, increasing the adhesive sites and promoting interfacial adhesion. Notably, this electrostimulation-based adhesive method remained effective even at −20 °C. In addition, applying voltage can also drive the movement of charged molecular chains (Fig. 3c(ii)). When the hydrogel and substrate carried opposite charges, the directed diffusion of molecular chains enhanced the entanglement and electrostatic interaction at the adhered interface. Conversely, the reverse voltage effectively weakened this physical interaction and facilitated the rapid detachment. It has been observed that certain human tissues, such as the aorta, cornea, and lungs, exhibited anionic hydrogel properties. When a voltage is applied to each end of the specific tissue and a cationic hydrogel, the interfacial adhesion strength can be effectively enhanced or weakened on-demand.128 This flexible and reversible adhesion strategy provides new insights for tissue sealing during surgical procedures.
 | ||
| Fig. 5 (a) Schematic diagram of the interaction between hydrogels changed by Li+ migration under an electric field. Reprinted with permission from ref. 127 Copyright 2022, Wiley-VCH GmbH. (b) Schematic diagram of the changes in borate and ionic bonds in hydrogels under electric fields. Reprinted with permission from ref. 129 Copyright 2023, Wiley-VCH GmbH. (c) Illustration of adhesion reduction triggered by GSH solution via disulfide bond cleavage and cohesive strength weakening. Reprinted with permission from ref. 136 Copyright 2022, Wiley-VCH GmbH. (d) Schematic diagram of urea solution decreasing hydrogel adhesion. Reprinted with permission from ref. 147 Copyright 2023, American Chemical Society. (e) Schematic diagram of the reduction of hydrogel adhesion strength by DFO solution chelated with Fe3+. Reprinted with permission from ref. 151 Copyright 2023, Acta Materialia Inc. | ||
An extended electrical field can also provide energy for redox reactions, enabling electron transfer at the contact interface. In Fig. 3c(iii), when a voltage is applied, the water molecules are reduced to produce OH−, raising the pH value of the hydrogel. The resulting alkaline environment can promote the formation of borate ester bonds that exhibit high reactivity, reduce the exposure of phenolic groups, and ultimately decrease the adhesion properties. Carboxyl groups, in the presence of OH−, will deprotonate and complex with cations in the hydrogel, weakening the electrostatic interactions at the adhesion interface. Building on this, Yang et al.129 proposed an adhesion-regulating strategy based on electric-induced formation and dissociation of borate ester bonds and ionic bonds, which effectively enhanced the adhesion efficiency (20 s) and modulation range (112-fold) between the hydrogel and pig skin (Fig. 5b).
The electro-adhesion regulation effect is collectively determined by the distribution of current density, interfacial contact area, and the conductivity of the hydrogel, which may prevent achieving a wide range of stimulation. Deficient electric stimulation predicts inadequate regulation of hydrogel adhesion, while excessive voltage can not only damage the performance of the hydrogel, but also cause pain or burns to human tissues and even damage the nervous system. Furthermore, the employment of an external power source has certain inevitable operational inconveniences and complexity. By incorporating special materials or designing special structures to impel ultrasound-, light-, and magneto-induced electricity generation,130–132 and self-powering,133,134 the feasibility of hydrogels in the field of electro-adhesion can be substantially expanded.
| Category | Intermolecular interactions | Common biotrigger fluid | Reaction principle | Ref. |
|---|---|---|---|---|
| Reactivive | Disulfide bonds | GSH/DTT | Break disulfide bonds to form reduced thiol groups | 136 |
| Natural polymer | Protease | Specific recognition with the substrate | 138 | |
| Alginate lyase | 139 | |||
| Lysozyme | 141 | |||
| Competitive | Boronate ester bond | Glucose | Preferential binding of the boronic acid groups with cis-diols | 142 |
| Gorax | 144 | |||
| Hydrogen bond | Urea | Competes for polar functional groups | 146 | |
| Ionic bond | SO42− | Competes for amino groups | 148 | |
| Zn2+ | Competes for imidazole (breaks weak hydrogen bonds to form metal coordination bonds) | 149 | ||
| DFO | Competes for Fe3+ (forms stronger metal coordination bonds) | 151 | ||
| EDTA-4Na | Capture Cu2+ | 152 | ||
| Host–guest interaction | Adamantane amine | Compete for the CD (host molecule) | 58 | |
| Cu2+ | Compete for bpy (guest molecule) | 152 |
Hydrogels often contain natural polymers, such as proteins, whereas appropriate enzymes can selectively destroy the polymer network and enable controllable reduction of the adhesion strength on demand. Lee et al.138 used globular proteins as adhesion crosslinkers and enabled a firm adhesive energy of 750 J m−2. Meanwhile, a separated adhesion interface was facilely realized by using enzyme solutions without damaging the hydrogel network. Compared with GSH solution, enzyme solution is more degradable and can prevent secondary damage caused by hydrogel residues. R. Freedman et al.139 treated hydrogels containing alginate and chitosan with hydrogen peroxide, alginate lyase, and lysozyme, which significantly reduced the adhesive strength of the hydrogels. Chitosan can also be degraded by enzymes due to its amino groups.140 For example, Bao et al.141 used lysozyme and an acetic acid solution to dissolve chitosan molecular chains to remove the hydrogel adhesive. Actually, due to the enzyme induced degradation, hydrogels removed using enzymes cannot reversibly restore their original adhesion strength. Furthermore, in the case of tissue damage, abnormal expression of GSH-related metabolic enzymes and reactive oxygen species (ROS) can lead to an imbalance in the redox state of the wound, thereby impeding the wound healing.
Due to the preferential binding of the phenylboronic acid groups with adjacent cis-diols, the boronate ester linkage is a commonly utilized glucose-responsive dynamic crosslinking unit in hydrogels (Fig. 3d(ii)). An et al.142 introduced tannic acid into the hydrogel through dynamic boronate ester bonds, achieving a tough anchoring effect. When a glucose solution was sprayed onto the skin surface, glucose rapidly displaced catechol groups to form bonds with phenylboronic acid groups and promoted the detachment of tannic acid, reducing the adhesive strength of the hydrogel to 25%. Xue et al.143 demonstrated that the complexation between phenylboronic acid and polyols in hydrogels can be disrupted by glucose, decreasing the interfacial toughness of hydrogels from 400 J m−2 to 20 J m−2. Interestingly, it has also been evidenced that the competitive bio-trigger liquid of borax solution can disrupt hydrogen bonds among tannic acid, dopamine, and citric acid in hydrogels.144 It should be noted that in cases where boronic ester cross-linked adhesive hydrogels are utilized for wound healing or monitoring, the inherent glucose in the wound can lead to an unintended reduction in adhesive strength and engender potential detachment of the hydrogel.
Urea, a highly polar substance, exhibits excellent biocompatibility. Upon contact with an adhesive interface, urea molecules competitively interact with polar groups (e.g., carbonyl and hydroxyl), thereby disrupting the original hydrogen bonds.8,145 Chen et al.146 developed a hydrogel rich in amino cations and hydrogen bond donors, which achieved rapid sealing and closure of organs. Meanwhile, treatment with competitive removers, such as urea (disrupt hydrogen bonds) and NaCl (screen electrostatic effects), substantially reduced the interfacial adhesion strength by approximately 80%. Wang et al.147 observed a 7-fold decrease in the adhesion strength between a PAA/polyethylenimine (PEI) hydrogel and wet pig skin after treatment with 1 M urea solution (Fig. 5d). Actually, the biocompatibility of urea solution depends on its concentration, and individual differences and potential allergic reactions also need to be considered. Appropriate evaluations before use are recommended to ensure the demanded biocompatibility.
Specific ions can preferentially bind responsive groups in the hydrogel, altering the cohesion of the hydrogel. Cao et al.148 figured out that amino groups of chitosan can interact with multivalent anions (such as SO42−) to induce phase separation in the polymer network, thereby reducing the adhesion strength. The treatment of metal ion-containing solutions provides another innovative approach to alter the molecular interactions within the hydrogel by constructing stronger metal coordination. For example, Wang et al.149 synthesized a hydrogel from AA and 1-vinylimidazole with tough adhesion. After treatment with Zn2+ solution for 30 s, Zn2+ formed coordination with imidazole, reducing the number of imidazole functional groups at the adhesive interface and decreasing the adhesive strength of the hydrogel by 75%. DFO is a biocompatible iron chelating agent that is routinely used for the accumulation and deposition of iron in tissues. Accordingly, DFO is able to chelate Fe3+ from the catechol–Fe3+ network and disrupt the pristine metal-coordination bonds in hydrogels.150 Lv et al.151 proposed an underwater adhesive hydrogel derived from dopamine-modified hyaluronic acid and ε-poly-L-lysine, where the addition of Fe3+ not only enhanced the cohesion but also imparted the hydrogel with on-demand removal capability. However, upon treatment with DFO solution, the adhesive strength of the hydrogel declined to one-third (Fig. 5e).
By utilizing competitive guest molecules to replace the original guest molecules, the adhesion of the hydrogel can be selectively altered. Yang et al.58 treated a cyclodextrin (CD)-incorporated hydrogel with adamantane amine solution, and the host–guest interactions between CD and the tissue were massively disrupted, resulting in a decrease in the peeling force of the hydrogel from 93.7 N m−1 to 19.8 N m−1. Furthermore, utilizing more competitive liquids to capture guest molecules is also an effective means to reduce the adhesion strength of hydrogels. Nakamura et al.152 prepared a metal ion-responsive hydrogel containing 2,2′-bipyridyl (bpy) and β-CD, as well as a guest hydrogel (tBu gel) containing N-tert-butylacrylamide. Metal ions, such as copper ions, can complex with bpy to enable responsive adhesion, while an ion chelator, such as ethylenediamine-tetraacetic acid tetrasodium salt (EDTA-4Na), can capture Cu2+ and release bpy, leading to the competitive effect of bpy with the tBu gel towards β-CD and failing the adhesion between the two hydrogels. Unfortunately, due to the absence of host–guest molecules on the tissue surface, this method is not highly used at the hydrogel–tissue adhesion interface.
Table 3 briefly summarizes various reactive and competitive biological trigger fluids and their mechanisms for controlling tissue adhesion. For reactive fluids, consideration must be given to the inherent interfacial bonding chemistry and their specific applications. Comparatively, in the process of using competitive biological trigger fluids, changes occur only in non-covalent bonds, making it a safe regulatory strategy. However, critical requirements on the biocompatibility of the solution containing competitive groups are demanded. Additionally, the limited reaction rate arose from slow diffusion of body fluids in situ and potential non-specific adhesion should not be overlooked. In the future, it is imperative to enhance the composition and properties of biological trigger liquids to improve their biocompatibility, programmability, and reusability, advancing their applications in the field of biomedicine.
3.2. Mechanical effect-promoted microstructure reconstruction
In addition to the reversible intermolecular interactions induced by various stimuli, the objective mechanical transformations, such as sol–gel transitions and volume swelling or shrinking, originated from a more elaborate mechanical effect, revealing a more intuitional impact on the adhesion properties of hydrogels and enabling them to flourish in the field of “smart adhesion”. In this section, the mechanisms and applicable strategies of controllable adhesion of biomedical hydrogels are elucidated from the perspective of interfacial mechanical effects. | ||
| Fig. 6 (a)–(e) Schematic of hydrogel state transitions, adhesion mechanisms, and bio-interfacial interactions. | ||
The sol–gel transition of the hydrogel can be controlled by varying the pH scale,153 temperature,154,155 and light irradaiton.115,156 Su et al.157 synthesized a pH-responsive hydrogel using borate ester bonds and Schiff base bonds. Treatment with organic acid solutions such as citric acid disrupted the dynamic covalent bonds and hydrogen bonds in the hydrogel and promoted its transition to a sol state and even dissolution, achieving on-demand removal. By utilizing the temperature-induced sol–gel transition, the controllable adhesion can be combined with injectability, adapting to wounds of various shapes. In addition, specific solution flushing can directly prompt sol–gel transition and dissolve the hydrogel on demand. For instance, Shi et al.158 proposed a temperature-responsive hydrogel using F127, which exhibited a typical sol–gel–sol transition process within the temperature range of 25–47 °C, allowing easy removal with hot water (∼47 °C). In addition, specific solutions, such as DTT solution (cleaving disulfide bonds, Fig. 7a),135 amino acid solution (disrupting Schiff base bonds, Fig. 7b)159 and amantadine hydrochloride (AH) solution (interfering with host–guest interactions),160 all can effectively disrupt the crosslinked network by targeting interactions, enabling the dissolution and removal of the hydrogels. Nevertheless, the immersion approach may lead to skin saturation around the wound and increase the risk of infection, rendering the solution flushing method less suitable for unhealed wounds.
 | ||
| Fig. 7 (a) SEM morphology of hydrogels treated with DTT solution. Reprinted with permission from ref. 135 Copyright 2022, American Chemical Society. (b) Schematic diagram of removing a hydrogel using amino acid solution. Reprinted with permission from ref. 159 Copyright 2018, American Chemical Society. (c) Microscopic scheme of a hydrogel in full contact with a substrate under temperature stimulation. Reprinted with permission from ref. 162 Copyright 2022, The Author(s). (d) Schematic diagram of the change in the contact area of the bionic gecko toe hydrogel affected by temperature. (i)–(iv) Changes in the adhesion interface upon removal. Reprinted with permission from ref. 166 Copyright 2021, American Chemical Society. | ||
The stimuli-induced sol–gel transition in regulating the adhesion properties of hydrogels is essential to make their cohesion lower than the adhesion strength. The hydrogel is easy to remove by this approach, yet it will also cause residue formation. Concretely, when the hydrogel is transited to a sol state, small molecules forming the polymer network are more prone to diffuse into the contact interface, compromising the biocompatibility during the removal process.
The microphase transition behavior of hydrogels promises an effective strategy to adjust the interfacial contact area. During the processes of water absorption/desorption, the interactions between water molecules and polymer chains change, resulting in spontaneous contraction (dehydration) or expansion (water absorption) in hydrogels. The irregular contraction at the microscopic level reduces the interfacial adhesion area and decreases the collision probability of adhesive groups. Zhang et al.162 designed a hydrogel containing both catechol groups and polar carboxyl groups and established tough adhesion to various substrates (Fig. 7c), where NIPAM was incorporated to serve as a thermosensitive material. As the temperature rose beyond LCST, the hydrogel experienced distinct volume shrinkage, and simultaneously the hydrophobic PNIPAM extruded a large amount of water molecules to the hydrogel surface and impelled abundant carboxyl groups to migrate to the surface. The reduction of the contact area and the lubricating effect of carboxyl groups jointly weakened the adhesive properties of catechol groups. Liu et al.163 fabricated a dynamically cross-linked network using short-branched polyethyleneimine (b-PEI) and PVA, which was able to gradually fill the wrinkles on the skin as the temperature rose. At temperatures of 25 °C and 37 °C, the interfacial adhesion between the hydrogel and the skin differed by approximately 5-fold.
On a macroscopic scale, the dynamic regulation of the contact area can be realized by directing uneven deformations of the hydrogel, which is generally achieved from two perspectives: uneven stimulus intensity and uneven stimulus response. Variations in the degree of contraction at different positions will bring about uneven volume change, and the accumulated extra mechanical energy further results in the curling of the two ends. For instance, in a hydrogel synthesized from Feng’ group with asymmetric distribution of inertially settled PPy-PDA nanoparticles,164 an uneven deformation at both ends of the hydrogel was manifested after 5 min of NIR light irradiation, ultimately reducing the shear strength by 50%. However, the heterogeneous design may result in a significant performance difference at different parts of the hydrogel.165 To enhance the adhesive strength and controllability of hydrogels, researchers have turned their attention to the adhesive behavior of organisms. Inspired by the rapid transition between attachment and detachment states of geckos on various surfaces, Zhang et al.166 introduced a thermoresponsive hydrogel layer onto a dopamine (DA)-coated mushroom pillar hydrogel layer. This dual-layer design concurrently achieved strong underwater adhesion and thermal-induced self-peeling (Fig. 7d).
The influence of the contact area on the adhesive strength of hydrogels is significant, generally exhibiting a positive correlation. However, it is imperative to note that once the contact area surpasses a certain threshold, the enhancement in adhesive strength may approach saturation. In certain scenarios, adhesive failure may occur in regions beyond the interface, such as within the hydrogel itself. Under these circumstances, augmenting the contact area exerts a limited impact on the enhancement of adhesive strength. Furthermore, an increased contact area enhances other interactions, such as van der Waals forces, which can interfere with the dedicated regulation of adhesion properties. Hence, the application scenarios and the selection of specific adhesion bonds must be considered in the hydrogel design.
By combining flexible and adaptable biological structures that readily manipulate the pressure difference with the volume change of the hydrogel, the dynamic adhesion regulatory behavior of biological organisms can be simulated to the greatest extent (Fig. 6c). Lee and colleagues170 used a polyethylene glycol diacrylate (PEGDA) hydrogel to create a suction cup wall, where the dome-shaped protuberance structure inside the suction cup was made from the PNIPAM hydrogel. With increased temperature, the diameter of the protrusions decreased, the effective suction area on the top surface of the hydrogel reduced, and ultimately adhesion force at the contact interface declined (Fig. 8a). Additionally, Wang and colleagues171 designed a magnetically responsive adhesive hydrogel and manipulated the volume of the upper chamber by an external magnetic field, thereby facilely regulating the adhesive strength with higher precision. To obtain optimal adhesion regulation, delicate design on the suction cup structure is demanded. For instance, in the case of Clingfish-inspired nanostructure, the height, side length, and number of hexagonal microcolumns all impact.81 Unfortunately, biomimetics of suction cup structures cannot fully simulate the behavior of organisms, making the transition on the adhesion properties of hydrogels inflexible. To better mimic the behavior of octopus tentacles, an asymmetrically structured suction cup hydrogel was fabricated by 3D printing technology.169 The hydrogel allowed for gripping and releasing of heavy objects under the control of either an air pump or a water pump.
 | ||
| Fig. 8 (a) Schematic of temperature-dependent adhesion in a suction-cup hydrogel. Reprinted with permission from ref. 170 Copyright 2022, The Authors. (b) Polarized light micrograph of hydrogel crystallization. Reprinted with permission from ref. 48 Copyright 2023, Wiley-VCH GmbH. (c) Schematic of the effect of the ultrasonic device-substrate distance on interfacial pressure distribution. Reprinted with permission from ref. 183 Copyright 2022, The American Association for the Advancement of Science. (d) SEM images of hexagonal hydrogel micropillars in bulged and recovered states at the adhesion interface. Reprinted with permission from ref. 193 Copyright 2021, American Chemical Society. | ||
Auxiliary means can also be exploited to strengthen the negative pressure effect of hydrogels. After establishing a firm attachment between the hydrogel and the substrate, the forward hardening of the adhesion interface can reduce interface permeability and enhance interface stability and durability. Excessive cross-linking, crystallization172 and biomineralization173,174 are commonly employed strategies to enable interface hardening. Zhang et al.175 used the diffusion of amphiphilic ions in hydrogels and substrates to construct a biomineralization layer at the adhesion interface, resulting in a 50-fold enhancement in the interfacial shear adhesion. Inspired by snail mucus, Cho et al.176 proposed a poly(2-hydroxyethyl methacrylate) hydrogel that can undergo reversible transition between flexibility and rigidity through changes in water content, thereby altering the adhesion state. Unfortunately, the uncontrollability of water evaporation inevitably limited its application. Under external crystal seed stimulation, similarly, the crystallization arose in the hydrogel formed using the CH3COONa·3H2O salt and polyacrylamide soft networks, wherein the resulting crystalline structures exhibited thermomelting characteristics and imparted switchable adhesive properties to the hydrogel (Fig. 8b).48 By integrating the mechanisms of snail mucus and suction cup adhesion, Li et al.177 synthesized a bio-inspired slime comprised of tannic acid-grafted gelatin and AgNPs as the main constituents, with a PNIPAM-based sucker patch. Relatively, excessive NIR light irradiation softened the gelatinized mucus, while rinsing with cold PBS triggered suction cup water absorption and expansion, enabling controlled removal of the hydrogel.
In addition, the gas consumption at the adhesion interface can also create a negative pressure effect. For instance, by applying Fe electrodes to both ends of a hydrogel, the redox reaction at the interface could consume water and oxygen and generate a negative pressure.178 The adhesive energy of the hydrogel reached 200 J m−2 and 1400 J m−2 after 0.5 h and 3 h of electrochemical treatment, respectively. This electrochemical reaction-induced negative pressure can be combined with special structures, such as suction cup structures, to provide new insights for the development of programmable adhesion in hydrogels.
By adjusting the interfacial negative pressure effect, an intelligent hydrogel adhesion in wet environments with better reversibility and controllability can be achieved. However, the adhesion interface relying on the negative pressure effect imposes strict requirements for the material's sealing property, posing a huge challenge to the high throughput screening and synthesis of the hydrogels.
3.2.4.1. Skin barrier. The stratum corneum of skin, through its tightly packed cell arrangement and the lipid compounds, can impede the penetration of hydrogels into deeper layers of the skin. Sebum, a viscous liquid secreted by sebaceous glands, can form a protective film on the skin surface and resist hydrogel adhesion. In certain cases, such as plastic surgery or contact with specific materials, individuals may have to intentionally reduce the integrity of the skin barrier to enhance object adhesion. To reduce damage to the skin, pre-coating with a fixing solution can promote the diffusion and entanglement of molecular chains and facilitate the formation of a strong bonding layer, thereby enhancing the adhesion strength of the interface. The customary auxiliary anchoring solutions can be categorized into initiator solutions,179 nanoparticle suspensions,180,181 and polymer solutions.182 Ma et al.183 proposed a strategy to regulate the adhesion of hydrogels by using an ultrasound mediated anchoring agent. The mechanical effect generated by the rupture of microbubbles under ultrasonic oscillation propelled the anchoring agent into tissues, creating a bridging network at the interface. This ultrasonically induced cavitation strategy alleviated the barrier effect of the tissue and prevented anchoring agents from penetrating the tissue via passive diffusion (Fig. 6d(i)). Without chemical reactions, the employment of ultrasound increased the adhesion energy between the hydrogels and porcine skin by 10 to 100 times, whereas the adhesion strength and area between the hydrogels and skin could also be precisely regulated by changing the intensity of ultrasound (Fig. 8c). By integrating hydrogel injection, ultrasound cavitation, and ultraviolet curing devices into a detachable SkinPen, the translation of the ultrasound propulsive anchoring agent strategy into clinical applications has been greatly promoted.184 However, precise regulation of the adhesion strength between hydrogels and tissues is crucial to improve the feasibility of this strategy in clinical applications.
In nature, many organisms feature barb-like structures, such as the mantis' claws, the barbs of feline tongues, porcupine quills barbs, and bee stingers, which provide a powerful anchoring effect to enhance their adhesive capabilities through mechanical interlocking. Combining barb-like structures with an array of microneedles, which can disrupt tissue barriers in a minimally invasive manner, the hydrogel detachment can be facilely addressed.185 Moreover, controllable removal of hydrogels can simultaneously be achieved through mimicking the responsive expansion anchoring mechanism of the endoparasite Pomphorhynchus laevis (Fig. 6d(ii)). Yang et al.186 designed a dual-layered microneedle hydrogel array for skin graft fixation and drug delivery. The inner rigid layer (polystyrene, PS hydrogel) effectively penetrated the skin barrier and persuaded the hydrogel microneedles to directly come into contact with the tissue. The outer swellable layer (PS/PAA hydrogel) rapidly absorbed water from tissue and expanded radially, forming a mushroom-like structure to facilitate mechanical interlocking with the tissue. Upon removal of the rigid layer, the tips of the swollen microneedles rapidly recovered to the initial conical shape, relieving the mechanical interlocking of the hydrogel and facilitating its removal. However, this expansion screw-like adhesive mechanism may induce pain and damage to the tissue, and the destruction of the skin barrier also poses a potential risk of external pathogen invasion, limiting the potential clinical applications of this approach.
3.2.4.2. Hydration layer. In contact with the tissue, the interfacial hydration layer, acting as a barrier film, prevents the close contact of the adhesive functional groups with the substrate and decreases the probability of collision at adhesion sites. Methods for removing the hydration layer can be categorized into hydrophobic and hydrophilic approaches, corresponding to the exclusion and absorption of the interfacial water, respectively.187–189 The hydrophobic structure can maximize the chance of collision at the adhesive sites, enhancing the wet adhesion strength of the hydrogel.190 However, due to the inherent hydrophilicity of hydrogels, strategies involving absorption of the hydration layer are more feasible. The hydrogel designed by Qi et al.191 achieved disordered migration of polymer chains and enhanced adhesion property after absorbing the hydration layer. In practice, external forces can be applied to accelerate the removal of interface water, while, it is also necessary to avoid the compression of the surrounding tissue caused by the absorption of water.
By facilitating interfacial water drainage, the channel structure offers a novel strategy for mitigating the hydration layer's adverse effects on hydrogel adhesion (Fig. 6e(i)). Eklund et al.192 designed a PNIPAM hydrogel with microchannels, where the thermoresponsive hydrogel contracted to expel water through microchannels with the temperature rising. By directing the contraction and recovery of the hydrogel channels, the adhesion function can be switched agilely. In nature, organisms such as clingfish and tree frog with hexagonal suction structures can change the size of groove channels, disrupt the hydration layer, and establish good direct contact with wet surfaces (Fig. 6e(ii)). Inspired by this, Zhang et al.193 synthesized an anti-swelling hydrogel with a thermoresponsive hexagonal micro-column pattern. As shown in Fig. 8d, when the temperature reached 45 °C, the reversible interactions within the hydrogel weakened, and the surface hexagonal structure reverted to a smooth state. This smooth surface facilitated the diffusion of water towards the interface and the formation of a hydration layer, reducing the adhesive strength by 85%. Similarly, Meng et al.77 designed a hexagonal hydrogel whose adhesion strength was reduced through limited thermal expansion at the micro-column tops and decreased substrate contact area.
3.3. Endogenous factor-stimulated adaptive adhesion
Compared to exogenous regulation (which relies on external stimuli such as electric field, light, and heat), endogenous regulation leverages on inherent physiological signals like pH, enzymes, metabolites, water environment, tissue movement, etc. This approach can avoid potential tissue damage from external devices or energy input (such as ultraviolet rays and high temperatures) and greatly improve the convenience of operation. As mentioned in Section 2.3, wounds or specific tissues usually exhibit the characteristic factors of dysregulation, such as abnormal temperature, pH, and glucose level. Endogenous modulation can respond to local microenvironment changes in real time, and the inherent pathological specificity can enable targeted adhesion regulation, so as to achieve adaptive adjustment of adhesion strength with minimized impact on healthy tissues. Endogenous factors can be broadly categorized into two main types: chemical and physical factors. | ||
| Fig. 9 (a) Schematic of the physiological environment influencing the adhesion strength of hydrogels. (b) Schematic of the asymmetrical adhesion of the Janus hydrogel. (c) The hydrogel degrades on demand for controlled removal. (d) The design principles of controllable time-lapse adhesion evolution of hydrogels. | ||
Compared to other factors, the physical influences arising from mechanical movements of human tissues are often overlooked. Typically, cyclic stretching or compression of tissues (e.g., skin, muscle, joints, or internal organs) generates shear or peeling forces at the hydrogel–tissue interface, possibly leading to adhesive failure. High-frequency or large-amplitude motions (such as heartbeat or respiration) may further accelerate interfacial fatigue, thereby compromising the hydrogel's adhesive durability. Transforming this bodily-related detrimental effect into a mechanism to dynamically enhance hydrogel adhesion strength represents an important research frontier. He et al.200 addressed this issue by designing a double-layer hydrogel with asymmetric adhesion using N,N′-bis(acryloyl)cystamine (BAC) as a responsive dynamic crosslinker in the adhesive layer. When subjected to mechanical strain, the adhesive layer underwent responsive interaction reorganization and expelled the embedded water from the hydrogel, ultimately achieving self-hardening and adhesion enhancement (Fig. 10a). Inspiration can also be drawn from the biological “catch bond” residing between mammalian cells and the extracellular matrix. The catch bond exhibits an abnormal mechanical dependence: within a certain force range, the increase of external forces paradoxically enhances the bond stability (Fig. 9a(ii)). Under rapid force, the polymer network deforms to dissipate energy and delays interface failure, while the inducing local stress “drives” the formation of additional dynamic bonds in the stress-concentrated area, thereby enhancing adhesion. Crucially, in biomimetic designs, reversible bonds are key to replicate this behavior. Building on this principle, Yuan et al.201 designed a hydrogel with a “catch bond” mechanism for hydrogen bond-dominant adhesion, which facilitated long-term and tough adhesion in the physiological environment and enabled facile and clean detachment on-demand. It should be noted that such methods typically impose demanding requirements on the mechanical properties of hydrogels, exhibiting a combination of softness, toughness, stretchability and self-healing. Furthermore, it is also challenging to design hydrogels that can accurately reproduce the complex multi-axial stress environment encountered in vivo.
 | ||
| Fig. 10 (a) Schematic of a hydrogel inspired by the strain-hardening behavior of biological tissues to achieve self-hardening and improve adhesion strength. Reprinted with permission from ref. 200 Copyright 2022, The Author(s). (b) Schematic of an asymmetrically adhesive hydrogel to achieve adhesion between tissues and medical devices simultaneously under temperature triggering. Reprinted with permission from ref. 208 Copyright 2025, Wiley-VCH GmbH. (c) The hydrogel sprayed on the surface of the object at 37 °C to achieve in situ gelatinization. Reprinted with permission from ref. 210 Copyright 2025, the Author(s). (d) The hydrogel precisely positioned through weak adhesion to obtain strong adhesion on demand, resulting in high fault tolerance. Reprinted with permission from ref. 216 Copyright 2024, Wiley-VCH GmbH. | ||
It should be noted that although the endogenous regulation method offers greater convenience than that of exogenous control, the difficulty of precise control within the complex and variable physiological environments severely constrains its development. First, the intrinsic amplitude of physiological signals is small (e.g., a pH shift of ∼5–7.4) and the adhesion strength adjustment range is narrow, making it difficult to achieve strong adhesion under extreme conditions (e.g., >200 kPa for severe hemorrhage). Secondly, relying on molecular diffusion or enzyme-catalyzed reactions, the response time often ranges from minutes to hours, which fails to meet the critical requirements in emergency (hemostasis in seconds). Moreover, differences in the physiological microenvironment between patients (e.g., pH fluctuations in chronic wounds and varying enzyme expression levels) may also lead to failure of adhesion regulation. Currently, the predominant method is to combine endogenous signals with exogenous stimuli to achieve a broad-range and high-sensitivity dynamic regulation of hydrogel adhesion. In the future, machine learning is essential to be leveraged to analyze patient-specific physiological data and optimize hydrogel formulations to accommodate individual variability.
3.4. Application-oriented functional adhesion design
Due to the complexity of the human physiological environment and the extremely high requirements of biosafety for medical materials, in-depth research on the functionalization of biomedical hydrogel adhesion is flourishing. In this section, we mainly introduce the functional regulation strategies of asymmetric adhesion, degradability, and high fault tolerance.Integrating dynamical adhesion regulating into Janus hydrogels, the adhesion function of hydrogels can be further enhanced to meet diverse application needs.200 One typical approach is to design an adhesive layer with dynamic regulation. Wei et al.207 utilized the thermoplasticity of gelatin and constructed a non-adhesive shielding layer on top of a gelatin-based adhesive layer by casting a hot hydrogel precursor solution. The temperature sensitivity of gelatin prompted a dynamic enhancement of adhesion strength at physiological temperature. Yan et al.208 designed an adhesive bilayer Janus hydrogel, where a hexadecyl acrylate layer maintained rigidity at room temperature for easy handling and softened upon coming into contact with the human body, forming a stable adhesion interface between soft tissues and medical devices (Fig. 10b). Another effective approach is to design a side that can dynamically shield the adhesion group. Shen et al.209 incorporated PNIPAM micelles into a biomedical hydrogel, and the hydrogel quickly formed an adhesion interface upon coming into contact with the wound at the low-temperature sol state. Affected by body temperature, the PNIPAM micelles contracted and promoted the hydrogel to transform into a gel state, thereby reducing the exposure of adhesive groups on the outer surface and preventing contamination. Extending the fabrication strategies of Janus hydrogels, Shi et al.210 designed a hydrogel based on the triblock material (poly(D,L-lactide)/poly(ethylene glycol)). The precursor solution achieved full contact with the tissue surface by spraying and other methods and gelated in situ to form a hydrogel film at body temperature (Fig. 10c). The as-formed Janus hydrogel realized tough adhesion to the target site by increasing the contact area, as well as creating an outer smooth surface to prevent unnecessary adhesion to other tissues. Unfortunately, the complex structural design and environmental sensitivity of such hydrogels inevitably result in more fabrication difficulties and limit their development.
Actually, on-demand degradation of adhesion hydrogels can be triggered by both endogenous stimuli (e.g., pH, temperature, and enzyme activity) at the implant site and exogenous stimuli (e.g., light and magnetic field) applied in vitro. Ren et al.211 introduced disulfide bonds into an adhesion hydrogel designed for wound closure and enabled on-demand degradation. Importantly, the content of disulfide bonds can precisely regulate the degradation time, demonstrating extremely high flexibility in practical applications. Notably, various application scenarios demand distinct degradation characteristics of hydrogels; for instance, in an extended-release drug system, the degradation rate of the hydrogel should be synchronized with the release of the drug. In addition, the in vivo degradation process is greatly influenced by individual differences. Therefore, the degradation rate of hydrogels should be tailored to their specific functional requirements.
To mitigate the impact of repeated positioning or surgical errors on patients, the concept of high fault tolerance on strongly adhesive hydrogels is proposed, which arises the research interest in the relationship between adhesion time and adhesion strength at the interface. Generally, the relationship between the two is dependent on both the physicochemical properties of hydrogels and the adhesion environment not absolute.213 Precisely, the type of adhesive functional groups governs both the time required for interfacial bond formation and the time to achieve maximum adhesion strength. For example, adhesion formed through NHS esters typically takes only a few seconds, while polyurethane glue adhesion requires several hours; covalent adhesion takes a longer formation time than non-covalent adhesion and is therefore limited in applications that demand instantaneous adhesion.214 Interestingly, the time variance between covalent and noncovalent interactions during adhesion enables the hydrogel to promptly reposition itself in a short period and form a more stable adhesion interface once optimally positioned. This principle has inspired the consideration and design of highly fault-tolerant adhesive hydrogels on a time scale. Given this perspective, the hydrogel can be engineered with a weak-to-strong adhesion transition over time, enabling both initial accurate positioning and subsequent stable adhesion (Fig. 9d). For example, phenolic hydroxyl groups can be oxidized to quinones and subsequently react with tissue primary amines via Michael addition or Schiff base reactions, and Xue et al.215 employed the electro-oxidation method to achieve time-dependent adhesion in catechol-containing hydrogels, improving the fault tolerance of surgical hydrogel tapes. Chen et al.216 exploited the thermosensitivity of bovine serum protein (BSA) to design a hydrogel capable of achieving localization from weak adhesion to strong adhesion fixation. After heating for 10 min, the dissociation of BSA within the hydrogel increased the interfacial toughness by more than four times (Fig. 10d). Overall, while the adhesion properties of hydrogels are generally diminished over extended periods, the relationship between the adhesion strength and the adhesion time is complex. Therefore, a highly fault-tolerant design requires a comprehensive understanding of adhesion dynamics across multiple time scales.
Fig. 11 presents a comprehensive comparison of the strategies for dynamically modulating the biomedical hydrogel adhesion properties. Generally, the exogenous stimuli (e.g., light, temperature, and pH) offer superior spatiotemporal precision and reversibility, ideal for controlled manipulation; the endogenous factors (e.g., glucose, enzymes, and sweat) offer high biological relevance for targeted wound treatment or drug release. The choice of strategy involves a critical trade-off: while external methods excel in on-demand control, they may require specialized equipment; conversely, internally triggered methods are more autonomous but lack precision and speed. Furthermore, strategies based on mechanical effects exhibit high flexibility and rapid response, while they often require complex and sophisticated structural designs, posing fabrication challenges. Meanwhile, the application-oriented approaches demonstrate exceptional adaptability for specific scenarios, yet their highly customized nature results in significant usage limitations. Therefore, choosing an optimal strategy requires balancing critical factors like precision, speed, biocompatibility, and complexity against the specific needs of the biomedical application.
 | ||
| Fig. 11 A detailed analysis of the advantages, disadvantages and biomedical feasibility among various dynamic regulation strategies for biomedical hydrogel interfacial adhesion. | ||
4. Biomedical applications
With the flourishing of biomedical engineering and tissue engineering fields, scientists have engaged in the development of biocompatible adhesive materials for the repairment and regeneration of tissues. Beginning in the 1960s, many varieties of synthetic polymers and natural biomaterials were exploited to prepare hydrogels for tissue repair. With the continuous development in biomedical technology, the application of adhesive hydrogels in stem cell therapy and wound healing has gradually expanded. Meanwhile, the introduction of new technologies, such as nanotechnology and bioprinting, has also boosted adhesive hydrogels in the biomedical fields. In this chapter, we will succinctly introduce the advantages and applications of adhesive hydrogels in the biomedical fields of wearables, acute occlusion, wound healing, cell therapy, and soft robotics.4.1. Biomedical monitoring
The adhesive hydrogels play a key role in biomedical monitoring fields such as biosensing and bioimaging, and their unique interfacial bonding capabilities and functional designability provide innovative solutions for high-sensitivity detection and high-resolution imaging.217–219 Hydrogels are highly flexible and stretchable and can be tightly integrated with biological tissues (such as skin, muscles, and organs) to adapt to their deformations and movements, improving the stability and comfort of monitoring devices (e.g., sensors220–225 and bioimaging devices226) (Fig. 12a). The excellent adhesion properties enable the biomedical hydrogel to adhere firmly to the surface of human tissues, reduce the motion artifact, and improve the accuracy and convenience of signal acquisition.227 Meanwhile, stable adhesion with resistance to various harsh conditions such as low temperature,228,229 water environments,230–233 and abnormal pH scales234 is proposed to meet the needs of different application scenarios. Inevitably, the long-time exposure and cyclic deformations would compromise the adhesive strength, leading to a gradual deterioration in the timely responsiveness and accuracy of the human motion monitoring. Cyclically stable adhesion strength and controllable switching adhesion can increase the durability of wearable devices and reduce healthcare costs. Based on the temperature-sensitive properties of gelatin, Zhang et al.235 designed hydrogels with excellent adhesion stability over 200 adhesion/separation cycles. Xu et al.236 added the NIPAM monomer to the hydrogel sensor and achieved stable adhesion and easy removal concurrently under temperature stimulation. In addition to the monitoring of body surface signals, by introducing fluorescent markers or other detection signals into hydrogels, highly sensitive and high-resolution imaging of biomarkers can be achieved, which enables cell localization, protein expression analysis, and endogenous small molecule detection, to guide surgical procedures or interventional therapies.237,238 In future studies, hydrogels should be able to selectively adhere to specific biomarkers to improve detection sensitivity and imaging contrast. The regulatable adhesion strength also helps the implanted hydrogel biomonitoring device avoid non-specific adsorption and reduce the risk of secondary injury. | ||
| Fig. 12 (a)–(d) Schematic of biomedical applications of adhesive hydrogels, including wearable devices,220–226 acute closure,239,240 chronic wound treatment,254,257,268 and biomedical soft robotics.275 Reprinted with permission from ref. 220 Copyright 2024, Wiley-VCH GmbH. Reprinted with permission from ref. 221 Copyright 2025, American Chemical Society. Reprinted with permission from ref. 222 Copyright 2024, Wiley-VCH GmbH. Reprinted with permission from ref. 223 Copyright 2020, Royal Society of Chemistry. Reprinted with permission from ref. 224 Copyright 2022, The Authors, under exclusive license to Springer Nature America, Inc. Reprinted with permission from ref. 225 Copyright 2020, The Authors. Reprinted with permission from ref. 226 Copyright 2022, The American Association for the Advancement of Science. Reprinted with permission from ref. 239 Copyright 2024, Wiley-VCH GmbH. Reprinted with permission from ref. 240 Copyright 2024, Wiley-VCH GmbH. Reprinted with permission from ref. 254 Copyright 2025, American Chemical Society. Reprinted with permission from ref. 257 Copyright 2023, Wiley-VCH GmbH. Reprinted with permission from ref. 268 Copyright 2021, The Authors. Reprinted with permission from ref. 275 Copyright 2023, Wiley-VCH GmbH. | ||
4.2. Acute wound occlusion
Surgical bleeding and gastric perforation provide clinical applicable scenarios demanding rapid closure. Hydrogel-derived wound closure is a rapid, efficient, and less invasive method that can reduce the trauma and surgery time. Adhesive hydrogels can form a stable occlusion in a short period, stop bleeding, and inhibit spillage of blood and gastric contents, preventing infection and other complications (Fig. 12b).239,240 The premise of hydrogels for acute hemostasis is tough and fast wet adhesion properties, whereas hydrogels for rapid closure put excellent cohesion in priority to withstand higher pressures than conventional wounds (aorta, approximately 60–120 mmHg).241 The glycopeptide hydrogel designed by Teng et al.39 displayed a maximum tissue adhesion strength of 83.9 kPa and quickly stopped bleeding within 14 s. Compared with rapid hemostasis, the application of hydrogels in gastrointestinal closure faces more serious challenges, such as the gastric acid environment, mucus secretion, and frequent mechanical movements of the digestive tract.242 Xu et al.243 designed an adhesive hydrogel based on thiourea-catechol coupling, which can stabilize adhesion under acidic conditions for gastric ulcer treatment. However, hydrogels tailored with strong adhesion for rapid plugging also face great challenges in detaching after treatment, creating an urgent demand for responsive adhesion hydrogels for dynamic exfoliation.244,245 To address this challenge, Shi et al.246 developed a hydrogel that could rapidly seal wounds in situ and could be removed on demand by a thiol-thioester exchange reaction after wound healing. The hemostatic hydrogel designed by Chen et al.247 contained an environmentally sensitive metal coordination bond that can be easily removed by EDTA to avoid iatrogenic injury. Beyond accommodating conventional physiological environment, the hydrogel designed for acute occlusion should also prioritize rapid adhesion kinetics while minimizing the tissue damage and residue upon removal.4.3. Chronic wound treatment
With large amounts of water and a three-dimensional network structure, hydrogels can provide a humid environment for wound healing, as well as to absorb the exudate and sweat from the wound to reduce infection. With delicate incorporation of diverse bioactive components, hydrogels can also be featured with antibacterial248–251 and drug delivery252,253 properties to accelerate wound healing (Fig. 12c). In the chronic wounds, the hydrogel dressings can adhere to wound tissues for stable delivery of therapeutics over days.254 In addition, conductive hydrogels used for electrical stimulation therapy also demand robust adhesion to ensure adequate therapeutic efficacy.255 Incisional wounds usually heal more slowly than static wounds, where the flexibility and contraction of adhesion hydrogels make them widely used in the closure of cutting wounds.256,257 Yu et al.258 introduced cyclodextrin-modified chitosan into a PNIPAM-based hydrogel, which contracted more than 50% in 2 h (in a body temperature environment) through the assistance of hydrogen bonding and host–guest interaction. Treatment with urea and amantadine could reduce the adhesion strength of the hydrogel and facilitate its on-demand removal and replacement. In special circumstances, such as athletes, infants, or chronic patients, more strict adhesion requirements are enforced to ensure stable adhesion during high-frequent activities or prolonged wearing periods.259,260The three-dimensional network structure of hydrogels also provides sufficient space for cell growth and differentiation.261,262 By adjusting the pore structure, chemical composition, and physical features of hydrogels, the release rate of cells can be regulated to achieve continuous and progressive cell delivery.263,264 Especially, the hydrogels can solve the shortcoming of low tissue adhesion in traditional cell delivery platforms, so as to ensure stable and continuous release of carried cells at the target site.265 These unique cell adhesion hydrogels also limit cell movement and proliferation, prospecting an exceptional strategy for cancer treatment266 and antimicrobial therapy.267 The hydrogel designed by Cha and Kim268 was able to form thiol groups with the attached cancer cell membrane, enhancing the adhesive ability to cancer cells and limiting their movement. By manipulating the surface properties of hydrogels (e.g., chemical functional groups, hydrophilicity/hydrophobicity features, and network structures), cell adhesion can be further promoted.269 Sun et al.270 increased the number of cells attached to the hydrogel by increasing the polyamidoamine density on the hydrogel surface. Zhao et al.271 adjusted the pore size of the hydrogel and promoted cell attachment and proliferation. During the actual treatment process, specific adjustment of the viscosity of the hydrogel can further promote the cell growth and tissue repair.
4.4. Medical soft robots
Hydrogel soft robots, with excellent biocompatibility, sensitive response to the physiological environment, and low manufacturing costs, have attracted extensive attention in biomedicine and bioengineering.272,273 The dynamic, controllable adhesion regulation of hydrogels allows for stable gripping, locomotion or climbing on different surfaces of biomedical soft robots, expanding their use in minimally invasive surgery.274,275 For instance, the hydrogel soft robot designed by Xin et al.276 could manipulate the microstructure and movement direction under the synergistic stimulation of pH and magnetic fields, achieving targeted cancer cell killing in vivo. In the biomedicine field, hydrogel soft robots have encountered challenges due to insufficient adhesion, resulting in being unable to precisely affix to injured areas, or excessive adhesion, hindering mobility. In humoral washouts or tissue peristalsis (e.g., in the gastrointestinal tract), the hydrogel adhesion strength (typically < 100 kPa) is not sufficient to maintain long-term stability, resulting in a shift in the robot's position.277 The dynamically adjustable adhesion facilitates the biomedical hydrogel soft robots to move more flexibly to the lesion site and fix it firmly (Fig. 12d). Inspired by multifunctional pollen grains, Lee et al.278 designed a hydrogel soft robot capable of controllably adhering to tissues for targeted drug delivery. The hydrogel soft robot with a bilayer adhesion structure designed by Hu et al.275 could achieve in vivo targeted drug delivery under the action of a magnetic field. Lee et al.,170 inspired by octopus suction cups, designed a hydrogel soft robot that could seek in vivo and attach to target tissues for precise drug release.Currently, most biomedical hydrogel soft robots are limited to targeted drug delivery, and expanding applications such as non-invasive gastric perforation sealing, in-body signal monitoring, and biomedical imaging are still in their infancy, constrained by limitations including stimulus-response duration and single-mode actuation. Another primary constraint lies in the slow switching speed of the adhesion state, which demands a much higher performance than that in sensors and wound dressings. Actually, the response time of light/heat-triggered adhesion control (typically seconds to minutes) is inadequate for millisecond-scale surgical precision. With continuous innovation in materials, driving mechanisms, and control methods, hydrogel soft robots are poised to achieve extensive clinical applications.
With the development of science and technology, multifunctional biomedical hydrogels can be designed as an all-in-one biomedical platform with multiple functions such as motion monitoring, drug delivery, and emergency alarms. Despite the fact that tough adhesion of hydrogels can expand application scenarios, especially for in vivo applications, it is critical to adhere stably to the target tissue for a sustained time to achieve prolonged treatment or monitoring, without causing secondary trauma and infection. Hydrogels with dynamic controllable adhesion and on-demand removal features antecede a feasible research direction, rendering more feasible programmability in practice and holding more realistic practical value in reducing costs and material waste. Meanwhile, full consideration should be given to the loss of adhesion strength under high-frequency adhesion–detachment transitions in future research.
5. Conclusion and prospects
Hydrogels have exerted a significant influence across various aspects of biomedical fields due to their outstanding properties such as biocompatibility and adjustable adhesion, with certain hydrogels already being employed in clinical settings. In this review, we initially expounded upon the adhesion mechanisms of various hydrogels, offering insights into their adhesive properties. In particular, we systematically analyzed the influence of physiological factors on biomedical hydrogel adhesion behaviors. Subsequently, we summarized the adhesion regulation methods of various biomedical hydrogels and categorically organized their biomedical applications. The biological adhesion capability of hydrogels enabled them to firmly adhere to various tissues as required, presenting extensive possibilities within the biomedical domain. Importantly, the adjustable dynamic adhesion ability enabled hydrogels to be removed from tissues without tissue damage, offering novel avenues for post-treatment management.Although adhesive hydrogels have a wide range of biomedical application prospects, a few hydrogels can be applied in clinical settings on a large scale. The physiological environment of the human body is dynamic; how to maintain the adhesion reliability and function of hydrogels in the complex physiological environment over a long period remains an urgent problem. The controllable adhesion strength ensures a wide range of applicability of hydrogels for different fluid components (e.g., gastric acid and bile) and different physiological pressures (e.g., blood pressure and gastrointestinal motility). In Section 3, we provided a comprehensive summary of the dynamic regulation strategies and approaches for hydrogel adhesion properties, which offered new insights for the future development of biomedical hydrogels. However, strategies that enable rapid or gradual switching between adhesive and non-adhesive states remain scarce, and their complex preparation methods and limited functional expansibility greatly hinder their applications. Additionally, the current majority of dynamic adhesion regulation strategies still depend on external stimuli (e.g., temperature, light, and pH), with significant gaps in research on other modalities such as magnetic, enzyme, and mechanical motion responses. Furthermore, the conversion efficiency to stimuli should also be paid attention in practical applications. Moreover, the integration of multiple dynamic regulation strategies also faces challenges in the intelligent development of hydrogels. For instance, incorporating multi-responsive chemical components may compromise biocompatibility, while mutual interference between different response mechanisms can hinder synergistic functionality. The complex in vivo environment can further complicate the balance between response speed and actuation amplitude. Additionally, the efficient synthesis and large-scale production to meet hydrogel fabrication requirements remain primary challenges. Achieving scalability in production demands simplification of hydrogel fabrication processes, necessitating the introduction and design of more precise production methods. Consequently, the research on biomimetic hydrogels is still in its infancy, and it is crucial to advance in the direction of a safer, more standardized, smarter, more convenient, and larger scale.
Moreover, due to the large differences in the composition of hydrogels and between different individuals, the general quantified standard in the adhesion design of hydrogels is not applicable to individual needs and still to be analyzed on a case-by-case basis. Firstly, the standardization of adhesion testing methods (such as peel tests, shear tests, tensile adhesion tests, and burst pressure tests) is lacking, with undefined parameters like preload and adhesion time leading to inconsistent results. Secondly, a significant gap exists between the idealized laboratory conditions and the dynamic in vivo environments, which involves complex interfaces, physiological wetness, pH variations, and mechanical stresses that challenge adhesion stability. Moreover, biological processes such as protein adsorption and foreign body reactions can rapidly compromise adhesion, while current methods fail to monitor adhesion strength dynamically during regulation. Overcoming these standalization-related barriers is essential to bridge laboratory research and practical clinical applications. In addition, owing to the neglect of time-scale considerations in evaluating the adhesion properties of hydrogels, there is often a lack of clear and quantifiable standards for gelation time, effective adhesion time, and adhesion failure time, all of which will prevent their practical applications. Conclusively, a clear and quantitative characterization of the hydrogel adhesion properties and the relationship between the adhesion strength and the duration of action is essential, which is fundamental for guiding the selection of optimal hydrogels for specific application scenarios.
Biomedical adhesive hydrogels are increasingly favored across multiple frontier sectors, with their research and development holding significant importance in advancing the biomedical field. The dynamic regulation of biomedical hydrogel adhesion signifies a paradigm shift of smart materials from “static design” to “dynamic interaction”, demonstrating a transformative potential in clinics. It is hoped that with the advancement in principles and technology, more intelligent and practical adhesive hydrogels will be applied in clinical healthcare, providing greater impetus for the development of health management and biomedical research.
Conflicts of interest
The authors declare no competing interests.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Acknowledgements
This work was supported by the NNSF of China (62288102) and Key Project of Basic Research Program of Jiangsu Province (BK20243036).References
- S. Tang, K. Feng, R. Yang, Y. Cheng, M. Chen, H. Zhang, N. Shi, Z. Wei, H. Ren and Y. Ma, Adv. Healthcare Mater., 2025, 14, 2403734 CrossRef CAS PubMed.
- Y. Gong, P. Wang, R. Cao, J. Wu, H. Ji, M. Wang, C. Hu, P. Huang and X. Wang, ACS Nano, 2023, 17, 22355–22370 CrossRef CAS PubMed.
- W. Sheng, J. Zhou, Y. Jia, W. Li, R. Qiao, Z. Liu, W. Xu and T. Zhang, Small, 2025, 21, 2407957 CrossRef CAS PubMed.
- Y. Xu, X. Xu, Y. Zhao, Y. Tian, Y. Ma, X. Zhang, F. Li, W. Zhao, J. Ma, Q. Xu and Q. Sun, Acta Biomaterialia, 2025, 198, 63–84 CrossRef CAS PubMed.
- X. Hu, J. Chen, Z. Yan, D. Nie, F. Guan, C. Shi and N. Lin, ACS Appl. Mater. Interfaces, 2025, 17, 16453–16467 CrossRef CAS.
- H. An, M. Zhang, Z. Huang, Y. Xu, S. Ji, Z. Gu, P. Zhang and Y. Wen, Adv. Mater., 2024, 36, 2310164 CrossRef CAS.
- X. Yang, B. Li, F. Lou, Y. Y. Chau, W. Zhang, Z. Yang, Q. Zhang, J. Mo, H. Zheng, Y. Ding, Z. Xue, X. Jiao, J. C. Rodríguez-Cabello, W. Xu, K. W. Y. Chan, L. Bian, L. Si, Y. Zhang, Y. Cao and Z. Wang, Cell Rep. Phys. Sci., 2025, 6, 102772 CrossRef.
- Y. Zheng, A. Baidya and N. Annabi, Bioact. Mater., 2023, 29, 214–229 CAS.
- A. Roy, S. Zenker, S. Jain, R. Afshari, Y. Oz, Y. Zheng and N. Annabi, Adv. Mater., 2024, 36, 2404225 CrossRef CAS.
- J. Lee, B. S. Lee, S. Baik, D. W. Kim, N.-J. Park, J. W. Lee, S.-K. Bong, S. H. Lee, S.-N. Kim, J. H. Song, J. K. Kim, G.-R. Yi, K. H. Kim and C. Pang, Chem. Eng. J., 2022, 444, 136581 CrossRef CAS.
- Y. Liu, K. Li, J. Tian, A. Gao, L. Tian, H. Su, S. Miao, F. Tao, H. Ren, Q. Yang, J. Cao and P. Yang, Nat. Commun., 2023, 14, 5145 CrossRef CAS PubMed.
- J. Zhu, Y. Li, W. Xie, L. Yang, R. Li, Y. Wang, Q. Wan, X. Pei, J. Chen and J. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 53575–53592 CrossRef CAS PubMed.
- S. Bian, L. Hao, X. Qiu, J. Wu, H. Chang, G.-M. Kuang, S. Zhang, X. Hu, Y. Dai, Z. Zhou, F. Huang, C. Liu, X. Zou, W. Liu, W. W. Lu, H. Pan and X. Zhao, Adv. Funct. Mater., 2022, 32, 2207741 CrossRef CAS.
- X. Zhao, J. Luo, Y. Huang, L. Mu, J. Chen, Z. Liang, Z. Yin, D. Chu, Y. Han and B. Guo, ACS Nano, 2023, 17, 22015–22034 CrossRef.
- J. X. M. Chen, T. Chen, Y. Zhang, W. Fang, W. E. Li, T. Li, M. R. Popovic and H. E. Naguib, Adv. Funct. Mater., 2024, 34, 2403721 CrossRef CAS.
- C. F. Guimarães, L. Gasperini, A. P. Marques and R. L. Reis, Nat. Rev. Mater., 2020, 5, 351–370 CrossRef.
- G. Dang, Y. Wei, Q. Wan, J. Gu, K. Wang, M. Wan, C. Wang, J. Song, Z. Mu, F. R. Tay and L.-N. Niu, BMEMat, 2024, 2, e12069 CrossRef CAS.
- X. Jiang, Y. Sun, Y. Lyu, H. Kang, J. Zhang, K. Zhang and L. Bian, BMEMat, 2024, 2, 12078 CrossRef.
- D. E. Discher, P. Janmey and Y.-L. Wang, Science, 2005, 310, 1139–1143 CrossRef CAS PubMed.
- E. Song, Z. Xie, W. Bai, H. Luan, B. Ji, X. Ning, Y. Xia, J. M. Baek, Y. Lee, R. Avila, H.-Y. Chen, J.-H. Kim, S. Madhvapathy, K. Yao, D. Li, J. Zhou, M. Han, S. M. Won, X. Zhang, D. J. Myers, Y. Mei, X. Guo, S. Xu, J.-K. Chang, X. Yu, Y. Huang and J. A. Rogers, Nat. Biomed. Eng., 2021, 5, 759–771 CrossRef CAS PubMed.
- Y. Cao, C. Liu, W. Ye, T. Zhao and F. Fu, Adv. Healthcare Mater., 2025, 14, 2403079 CrossRef CAS.
- Y. Li, G. Li, Y. Chen, X. Zhao, Y. Wang, J. Liu and Z. Li, Adv. Funct. Mater., 2022, 32, 2207306 CrossRef CAS.
- P. Song, Q. Sun, X. Liang, J. Li, J. Shi, Y. Zhu and J. Du, ACS Nano, 2025, 19, 25466–25479 CrossRef CAS.
- Y. Li, W. Zhang, C. Zhao, W. Li, E. Dong, M. Xu, H. Huang, Y. Yang, L. Li, L. Zheng, M. Mao, S. Yao, L. Wang, J. Ma, X. Wang and W. Huang, Adv. Mater., 2024, 36, 2405405 CrossRef CAS.
- G. Yang, R. Lin, H. Li, Y. Chen, M. Liu, Z. Luo, K. Wang, J. Tu, Y. Xu, Z. Fan, Y. Zhou, Y. Pan, Z. Zhao and R. Liu, Sci. Adv., 2025, 11, eadt3811 CrossRef CAS PubMed.
- W. M. Abbott, J. Megerman, J. E. Hasson, G. L'Italien and D. F. Warnock, J. Vasc. Surg., 1987, 5, 376–382 CrossRef CAS.
- Y. Yang, X. Zhao, S. Wang, Y. Zhang, A. Yang, Y. Cheng and X. Chen, Nat. Commun., 2023, 14, 7771 CrossRef CAS PubMed.
- B. Ying, K. Nan, Q. Zhu, T. Khuu, H. Ro, S. Qin, S. Wang, K. Jiang, Y. Chen, G. Bao, J. Jenkins, A. Pettinari, J. Kuosmanen, K. Ishida, N. Fabian, A. Lopes, F. Codreanu, J. Morimoto, J. Li, A. Hayward, R. Langer and G. Traverso, Sci. Transl. Med., 2025, 17, eadq1975 CrossRef CAS PubMed.
- G. Pan, F. Li, S. He, W. Li, Q. Wu, J. He, R. Ruan, Z. Xiao, J. Zhang and H. Yang, Adv. Funct. Mater., 2022, 32, 2200908 CrossRef CAS.
- Y. Xiong, X. Zhang, X. Ma, W. Wang, F. Yan, X. Zhao, X. Chu, W. Xu and C. Sun, Polym. Chem., 2021, 12, 3721–3739 RSC.
- X. Ma, X. Zhou, J. Ding, B. Huang, P. Wang, Y. Zhao, Q. Mu, S. Zhang, C. Ren and W. Xu, J. Mater. Chem. A, 2022, 10, 11823–11853 RSC.
- J. Yang, R. Bai, B. Chen and Z. Suo, Adv. Funct. Mater., 2020, 30, 1901693 CrossRef CAS.
- G. Bovone, O. Y. Dudaryeva, B. Marco-Dufort and M. W. Tibbitt, ACS Biomater. Sci. Eng., 2021, 7, 4048–4076 CrossRef CAS.
- C. Ouyang, H. Yu, L. Wang, Z. Ni, X. Liu, D. Shen, J. Yang, K. Shi and H. Wang, Adv. Colloid Interface, 2023, 319, 102982 CrossRef CAS PubMed.
- S. Wang, J. Liu, L. Wang, H. Cai, Q. Wang, W. Wang, J. Shao and X. Dong, Adv. Mater. Technol., 2023, 8, 2201477 CrossRef CAS.
- H. Y. Yuen, H. P. Bei and X. Zhao, Chem. Eng. J., 2022, 431, 133372 CrossRef CAS.
- Y. Bu and A. Pandit, Bioact. Mater., 2022, 13, 105–118 CAS.
- C. Shi, Y. Zhang, G. Wu, Z. Zhu, H. Zheng, X. Sun, Y. Heng, S. Pan, H. Xiu, J. Zhang, Z. Yin, Z. Yu and B. Liang, Adv. Healthcare Mater., 2024, 13, 2302626 CrossRef CAS.
- L. Teng, Z. Shao, Q. Bai, X. Zhang, Y.-S. He, J. Lu, D. Zou, C. Feng and C.-M. Dong, Adv. Funct. Mater., 2021, 31, 2105628 CrossRef CAS.
- Y. Zheng, K. Shariati, M. Ghovvati, S. Vo, N. Origer, T. Imahori, N. Kaneko and N. Annabi, Biomaterials, 2023, 301, 122240 CrossRef CAS.
- J. He, Z. Zhang, Y. Yang, F. Ren, J. Li, S. Zhu, F. Ma, R. Wu, Y. Lv, G. He, B. Guo and D. Chu, Nano-Micro Lett., 2021, 13, 80 CrossRef CAS.
- S. Yang, J. Yu, Z. Zhang, H. Yang, Y. Wan, B. Yu, S. Ma, Y. Ma, F. Zhou and W. Liu, Chin. J. Chem., 2023, 41, 2679–2683 CrossRef CAS.
- S. Choi, J. R. Moon, N. Park, J. Im, Y. E. Kim, J.-H. Kim and J. Kim, Adv. Mater., 2023, 35, 2206207 CrossRef CAS.
- X. Liu, Q. Zhang, Z. Gao, R. Hou and G. Gao, ACS Appl. Mater. Interfaces, 2017, 9, 17645–17652 CrossRef CAS.
- G. Huang, Z. Tang, S. Peng, P. Zhang, T. Sun, W. Wei, L. Zeng, H. Guo, H. Guo and G. Meng, Macromolecules, 2022, 55, 156–165 CrossRef CAS.
- M. Xu, Y. Miao, J. Yu and L. Zhang, Adv. Mater. Interfaces, 2021, 8, 2101131 CrossRef CAS.
- Y. Zhang, B. Ren, S. Xie, Y. Cai, T. Wang, Z. Feng, J. Tang, Q. Chen, J. Xu, L. Xu and J. Zheng, ACS Appl. Polym. Mater., 2019, 1, 701–713 CrossRef CAS.
- Y. Fang, X. Xiong, L. Yang, W. Yang, H. Wang, Q. Wu, Q. Liu and J. Cui, Adv. Funct. Mater., 2023, 33, 2301505 CrossRef CAS.
- X. Liu, H. J. Zhang, S. Xi, Y. Zhang, P. Rao, X. You and S. Qu, Adv. Funct. Mater., 2025, 35, 2413464 CrossRef CAS.
- H. Yi, S. H. Lee, M. Seong, M. K. Kwak and H. E. Jeong, J. Mater. Chem. B, 2018, 6, 8064–8070 RSC.
- C.-Y. Zou, X.-X. Lei, J.-J. Hu, Y.-L. Jiang, Q.-J. Li, Y.-T. Song, Q.-Y. Zhang, J. Li-Ling and H.-Q. Xie, Bioact. Mater., 2022, 16, 388–402 CAS.
- W. Xu, Y. Nan, Y. Jin, X. Chen, M. Xie, C. Chen and C. Zhao, Chem. Mater., 2022, 34, 8740–8748 CrossRef CAS.
- Z. Ni, H. Yu, L. Wang, X. Liu, D. Shen, X. Chen, J. Liu, N. Wang, Y. Huang and Y. Sheng, Adv. Healthcare Mater., 2022, 11, 2101421 CrossRef CAS PubMed.
- Y. Yan, J. Huang, X. Qiu, X. Cui, S. Xu, X. Wu, P. Yao and C. Huang, J. Colloid Interface Sci., 2021, 582, 187–200 CrossRef CAS PubMed.
- L. Han, M. Wang, L. O. Prieto-López, X. Deng and J. Cui, Adv. Funct. Mater., 2020, 30, 1907064 CrossRef CAS.
- S. Wang, L. Wang, X. Qu, B. Lei, Y. Zhao, Q. Wang, W. Wang, J. Shao and X. Dong, ACS Appl. Mater. Interfaces, 2022, 14, 50256–50265 CrossRef CAS PubMed.
- J. Chen, Q. Peng, X. Peng, H. Zhang and H. Zeng, Chem. Rev., 2022, 122, 14594–14678 CrossRef CAS.
- M. Yang, J. Tian, K. Zhang, X. Fei, F. Yin, L. Xu, Y. Wang and Y. Li, Biomacromolecules, 2023, 24, 4843–4853 CrossRef CAS PubMed.
- X. Xu, Q. Feng, X. Ma, Y. Deng, K. Zhang, H. S. Ooi, B. Yang, Z.-Y. Zhang, B. Feng and L. Bian, Biomaterials, 2022, 289, 121802 CrossRef CAS PubMed.
- P. Ren, L. Yang, D. Wei, M. Liang, L. Xu, T. Zhang, W. Hu, Z. Zhang and Q. Zhang, Int. J. Biol. Macromol., 2023, 242, 124885 CrossRef CAS PubMed.
- Y. Hou, Y. Li, Y. Li, D. Li, T. Guo, X. Deng, H. Zhang, C. Xie and X. Lu, ACS Nano, 2023, 17, 2745–2760 CrossRef CAS PubMed.
- M. Matsuda, M. Ueno, Y. Endo, M. Inoue, M. Sasaki and T. Taguchi, Colloids Surf. B Biointerfaces, 2012, 91, 48–56 CrossRef CAS.
- Z. Wang, J. Zhao, W. Tang, T. He, S. Wang, X. He, Y. Chen, D. Yang and S. Peng, ACS Appl. Mater. Interfaces, 2021, 13, 3435–3444 CrossRef CAS.
- J. Wu, H. Yuk, T. L. Sarrafian, C. F. Guo, L. G. Griffiths, C. S. Nabzdyk and X. Zhao, Sci. Transl. Med., 2022, 14, eabh2857 CrossRef CAS PubMed.
- Y. Xie, Z. Li, Y. Zhang, Y. Lu, J. Zhang and L. Zong, Small, 2023, 19, 2303044 CrossRef CAS PubMed.
- Z. Wang, Y. Zhang, Y. Yin, J. Liu, P. Li, Y. Zhao, D. Bai, H. Zhao, X. Han and Q. Chen, Adv. Mater., 2022, 34, 2108300 CrossRef CAS.
- A. Eklund, H. Zhang, H. Zeng, A. Priimagi and O. Ikkala, Adv. Funct. Mater., 2020, 30, 2000754 CrossRef CAS.
- X. Shi and P. Wu, Small, 2021, 17, 2101220 CrossRef CAS.
- Y. Lai and Y. Hu, Mech. Mater., 2021, 159, 103877 CrossRef.
- Y. Li, L. Li, Z. Zhang, J. Cheng, Y. Fei and L. Lu, Chem. Eng. J., 2021, 420, 129736 CrossRef CAS.
- R. Song, X. Wang, M. Johnson, C. Milne, A. Lesniak-Podsiadlo, Y. Li, J. Lyu, Z. Li, C. Zhao, L. Yang, I. Lara-Sáez, S. A and W. Wang, Adv. Funct. Mater., 2024, 34, 2313322 CrossRef CAS.
- G. Wang, Y. Liu, B. Zu, D. Lei, Y. Guo, M. Wang and X. Dou, Chem. Eng. J., 2023, 455, 140493 CrossRef CAS.
- H. Jung, M. K. Kim, J. Y. Lee, S. W. Choi and J. Kim, Adv. Funct. Mater., 2020, 30, 2004407 CrossRef CAS.
- L. Zhou, C. Dai, L. Fan, Y. Jiang, C. Liu, Z. Zhou, P. Guan, Y. Tian, J. Xing, X. Li, Y. Luo, P. Yu, C. Ning and G. Tan, Adv. Funct. Mater., 2021, 31, 2007457 CrossRef CAS.
- B. Yi, T. Li, B. Yang, S. Chen, J. Zhao, P. Zhao, K. Zhang, Y. Wang, Z. Wang and L. Bian, Nat. Commun., 2024, 15, 239 CrossRef CAS.
- D. He and Y. Hu, Meccanica, 2021, 56, 1483–1504 CrossRef.
- J. Wu, Z. Pan, Z.-Y. Zhao, M.-H. Wang, L. Dong, H.-L. Gao, C.-Y. Liu, P. Zhou, L. Chen, C.-J. Shi, Z.-Y. Zhang, C. Yang, S.-H. Yu and D.-H. Zou, Adv. Mater., 2022, 34, 2200115 CrossRef CAS.
- Y. Gao, J. Chen, X. Han, Y. Pan, P. Wang, T. Wang and T. Lu, Adv. Funct. Mater., 2020, 30, 2003207 CrossRef CAS.
- Y. Yuan, H. Wu, X. Ren, J. Wang, R. Liu, B. Hu and N. Gu, Sci. China Mater., 2022, 65, 827–835 CrossRef CAS.
- S. Baik, D. W. Kim, Y. Park, T.-J. Lee, S. Ho Bhang and C. Pang, Nature, 2017, 546, 396–400 CrossRef CAS PubMed.
- F. Meng, Q. Liu, Z. Shi, D. Tan, B. Yang, X. Wang, K. Shi, M. Kappl, Y. Lei, S. Liu and L. Xue, Adv. Mater. Interfaces, 2021, 8, 2100528 CrossRef.
- R. Michel, L. Poirier, Q. van Poelvoorde, J. Legagneux, M. Manassero and L. Corté, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 738–743 CrossRef CAS.
- Z. Shao and Q. Liu, Extreme Mech. Lett., 2023, 61, 101996 CrossRef.
- G. Tian, D. Yang, C. Liang, Y. Liu, J. Chen, Q. Zhao, S. Tang, J. Huang, P. Xu, Z. Liu and D. Qi, Adv. Mater., 2023, 35, 2212302 CrossRef CAS.
- H. R. C. Screen, J. Mech. Behav. Biomed. Mater., 2008, 1, 51–58 CrossRef PubMed.
- X. Hu, X. Tan, I. Ullah, T. Jin, Z. Xu, J. Zhang, Z. Pan, Y. Yuan and J. Wang, ACS Nano, 2025, 19, 20128–20143 CrossRef.
- N. Sanz del Olmo, N. Molina, Y. Fan, F. Namata, D. J. Hutchinson and M. Malkoch, J. Am. Chem. Soc., 2024, 146, 17240–17249 CrossRef CAS PubMed.
- F. H. Silver, N. Kelkar and T. Deshmukh, Biomolecules, 2021, 11, 1018 CrossRef CAS.
- O. Chaudhuri, J. Cooper-White, P. A. Janmey, D. J. Mooney and V. B. Shenoy, Nature, 2020, 584, 535–546 CrossRef CAS.
- H. Trębacz and A. Barzycka, Biomolecules, 2023, 13, 574 CrossRef.
- K. Zheng, C. Zheng, L. Zhu, B. Yang, X. Jin, S. Wang, Z. Song, J. Liu, Y. Xiong, F. Tian, R. Cai and B. Hu, Nano-Micro Lett., 2025, 17, 281 CrossRef.
- Y. Fan, L. Zheng, M. Jin, X. Li, Z. A. Li and X. Wang, BMEMat, 2024, 2, e12089 CrossRef CAS.
- A. Fatehi Hassanabad, A. N. Zarzycki, K. Jeon, J. A. Dundas, V. Vasanthan, J. F. Deniset and P. W. M. Fedak, Biomolecules, 2021, 11, 1027 CrossRef CAS.
- Y.-J. Li, L. Zhang, P.-P. Yang, K. Zhang, X.-F. Gong, D.-Y. Hou, H. Cao, X.-C. Wu, R. Liu, K. S. Lam and L. Wang, Nano Lett., 2022, 22, 8076–8085 CrossRef CAS.
- M. Pan, M. Wu, T. Shui, L. Xiang, W. Yang, W. Wang, X. Liu, J. Wang, X.-Z. Chen and H. Zeng, J. Colloid Interface Sci., 2022, 622, 612–624 CrossRef CAS PubMed.
- L. Zhang, S. Wang, Z. Wang, Z. Huang, P. Sun, F. Dong, H. Liu, D. Wang and X. Xu, Mater. Horiz., 2023, 10, 2271–2280 RSC.
- X. Wei, D. Chen, X. Zhao, J. Luo, H. Wang and P. Jia, ACS Appl. Polym. Mater., 2021, 3, 837–846 CrossRef CAS.
- A. R. Narkar, B. Barker, M. Clisch, J. Jiang and B. P. Lee, Chem. Mater., 2016, 28, 5432–5439 CrossRef CAS.
- A. Mohanty, B. Chaw Pattnayak, L. Behera, A. Singh, S. K. Bhutia and S. Mohapatra, ACS Appl. Bio Mater., 2023, 6, 4314–4325 CrossRef CAS PubMed.
- X. Kang, P. Guan, C. Xiao, C. Liu, Y. Guan, Y. Lin, Y. Tian, K. Ren, Y. Huang, R. Fu, C. Ning, L. Fan, G. Tan and L. Zhou, Adv. Healthcare Mater., 2023, 12, 2203306 CrossRef CAS.
- L. Bai, Y. Jin, X. Shang, H. Jin, L. Shi, Y. Li and Y. Zhou, Nano Energy, 2022, 104, 107962 CrossRef CAS.
- L. Zhang, S. Wang, Z. Wang, Z. Liu, X. Xu, H. Liu, D. Wang and Z. Tian, ACS Nano, 2023, 17, 13948–13960 CrossRef CAS.
- Y. Liang, H. Xu, Z. Li, A. Zhangji and B. Guo, Nano-Micro Lett., 2022, 14, 185 CrossRef CAS.
- X. Zhang, H. Ding, Z. Li, Y. Bai and L. Zhang, Mater. Horiz., 2024, 11, 835–846 RSC.
- H. Pang, C. Ma, S. Li, H. Liu, C. Xia, J. Li, S. Zhang, W. Zhang, L. Cai and Z. Huang, Appl. Surf. Sci., 2021, 560, 149935 CrossRef CAS.
- H. Geng, X. Zheng, Y. Zhang, X. Cui, Z. Li, X. Zhang, J. Cui, F. Meng, L. Sun and S. Ni, Adv. Funct. Mater., 2023, 33, 2305154 CrossRef CAS.
- D. Tan, F. Meng, Y. Ni, W. Sun, Q. Liu, X. Wang, Z. Shi, Q. Zhao, Y. Lei, S. Luan and L. Xue, Chem. Eng. J., 2023, 471, 144625 CrossRef CAS.
- W.-S. Wu, X. Yan, S. Chen, Y. Du, J. Hu, Y. Song, Z. Zha, Y.-J. Xu, B. Cao, S.-H. Xuan, X. Liu, B. Chen, L. Dong, Y. Lu and S.-H. Yu, Adv. Mater., 2024, 36, 2309770 CrossRef CAS.
- Y. Shen, S. Li, X. Hou, J. Yu, Y. Zhu, C. Zhao, Z. Niu, X. Guan, B. Xiong, S. Wang, Y. Yang, X. Li, L. Sun, S. Wu, B. Huang, H. Xu and H. Yin, Adv. Sci., 2025, 12, 2500720 CrossRef CAS.
- W. Qiu, C. Gehre, J. P. Nepomuceno, Y. Bao, Z. Li, R. Müller and X.-H. Qin, Angew. Chem., Int. Ed., 2024, 63, e202404599 CrossRef CAS PubMed.
- Y. Xiang, P. Zhuge, X. Qi, X. Ge, J. Xiang, H. Xu, E. Cai, Y. Lan, X. Chen, Y. Li, Y. Shi, J. Shen and J. Liu, Bioact. Mater., 2024, 39, 562–581 CAS.
- W. Ji, M. Qin and C. Feng, Macromol. Chem. Phys., 2018, 219, 1700398 CrossRef.
- X. Gu, H. Niu, Q. Sun, S. Jiang, Y. Shi and Y. Cai, ACS Appl. Mater. Interfaces, 2025, 17, 3818–3828 CrossRef CAS.
- S. L. Banerjee, K. Bhattacharya, S. Samanta and N. K. Singha, ACS Appl. Mater. Interfaces, 2018, 10, 27391–27406 CrossRef CAS.
- N. Cui, K. Han, C. Zhou, M. Seong, T. Lu and H. E. Jeong, ACS Appl. Bio Mater., 2020, 3, 8338–8343 CrossRef CAS PubMed.
- L. Yu, Y. Hou, W. Xie, J. L. Cuellar-Camacho, Q. Wei and R. Haag, Adv. Mater., 2020, 32, 2006986 CrossRef PubMed.
- W. Zhao, H. Wang, H. Wang, Y. Han, Z. Zheng, X. Liu, B. Feng and H. Zhang, Nanoscale, 2021, 13, 6394–6399 RSC.
- F. He, L. Wang, S. Yang, W. Qin, Y. Feng, Y. Liu, Y. Zhou, G. Yu and J. Li, Carbohydr. Polym., 2021, 256, 117595 CrossRef CAS PubMed.
- A. S. Kuenstler, M. Lahikainen, H. Zhou, W. Xu, A. Priimagi and R. C. Hayward, ACS Macro Lett., 2020, 9, 1172–1177 CrossRef CAS PubMed.
- K. Wu, X. Wu, Y. Zhang, S. Chen, Z. Qiao, D. Wei, J. Sun and H. Fan, Biomacromolecules, 2022, 23, 1030–1040 CrossRef CAS.
- B. Ryplida, K. D. Lee, I. In and S. Y. Park, Adv. Funct. Mater., 2019, 29, 1903209 CrossRef.
- Y. Gao, K. Wu and Z. Suo, Adv. Mater., 2019, 31, 1806948 CrossRef PubMed.
- M. Jiang, X. Liu, Z. Chen, J. Li, S. Liu and S. Li, iScience, 2020, 23, 100832 CrossRef CAS.
- B. Lei, L. Cao, X. Qu, Y. Liu, J. Shao, Q. Wang, S. Li, W. Wang and X. Dong, Nano Research, 2023, 16, 5464–5472 CrossRef CAS.
- X. Ding, Y. Yu, L. Fan, W. Li, F. Bian, J. Wang and Y. Zhao, Adv. Healthcare Mater., 2024, 13, 2302588 CrossRef CAS.
- C. Li, A. Iscen, L. C. Palmer, G. C. Schatz and S. I. Stupp, J. Am. Chem. Soc., 2020, 142, 8447–8453 CrossRef CAS.
- Y. Liu, P. Wang, X. Su, L. Xu, Z. Tian, H. Wang, G. Ji and J. Huang, Adv. Mater., 2022, 34, 2108820 CrossRef CAS PubMed.
- L. K. Borden, A. Gargava and S. R. Raghavan, Nat. Commun., 2021, 12, 4419 CrossRef CAS PubMed.
- G. Yang, Y. Hu, W. Guo, W. Lei, W. Liu, G. Guo, C. Geng, Y. Liu and H. Wu, Adv. Mater., 2024, 36, 2308831 CrossRef CAS.
- F. Wang, J. Qiu, S. Guan, S. Chen, X. Nie, Z. Fu, F.-Z. Yao, W. Gong, K. Wang and X. Liu, Mater. Today, 2025, 84, 48–64 CrossRef CAS.
- C. Wu, Y. Pu, Y. Zhang, X. Liu, Z. Qiao, N. Xin, T. Zhou, S. Chen, M. Zeng, J. Tang, J. Pi, D. Wei, J. Sun, F. Luo and H. Fan, Adv. Healthcare Mater., 2022, 11, 2201255 CrossRef CAS PubMed.
- J. Liang, X. Huang, K. Qin, H. Wei, J. Yang, B. Liu and Z. Fan, Adv. Mater., 2025, 37, 2415417 CrossRef CAS PubMed.
- Y. Wang, P. Chen, Y. Ding, P. Zhu, Y. Liu, C. Wang and C. Gao, Adv. Funct. Mater., 2024, 34, 2409081 CrossRef CAS.
- L. Zhang, L. Chen, S. Wang, S. Wang, D. Wang, L. Yu, X. Xu, H. Liu and C. Chen, Nat. Commun., 2024, 15, 3859 CrossRef CAS.
- Y. Yang, H. Xu, M. Li, Z. Li, H. Zhang, B. Guo and J. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 41726–41741 CrossRef CAS.
- W. Li, L. Jiang, S. Wu, S. Yang, L. Ren, B. Cheng and J. Xia, Small, 2022, 18, 2107544 CrossRef CAS PubMed.
- W. Li, X. Liu, Z. Deng, Y. Chen, Q. Yu, W. Tang, T. L. Sun, Y. S. Zhang and K. Yue, Adv. Mater., 2019, 31, 1904732 CrossRef CAS.
- W. Lee, E. Heo, H. B. Koo, I. Cho and J.-B. Chang, Macromol. Rapid Commun., 2023, 44, 2200750 CrossRef CAS.
- B. R. Freedman, O. Uzun, N. M. M. Luna, A. Rock, C. Clifford, E. Stoler, G. Östlund-Sholars, C. Johnson and D. J. Mooney, Adv. Mater., 2021, 33, 2008553 CrossRef CAS.
- Z. Xu, H. Zhang, Y. Huang, H. Zhong, P. Qin, S. Cheng, Y. Wang and C. Yang, ACS Appl. Polym. Mater., 2024, 6, 1141–1151 CrossRef CAS.
- G. Bao, Q. Gao, M. Cau, N. Ali-Mohamad, M. Strong, S. Jiang, Z. Yang, A. Valiei, Z. Ma, M. Amabili, Z.-H. Gao, L. Mongeau, C. Kastrup and J. Li, Nat. Commun., 2022, 13, 5035 CrossRef CAS.
- H. An, M. Zhang, L. Zhou, Z. Huang, Y. Duan, C. Wang, Z. Gu, P. Zhang and Y. Wen, Adv. Funct. Mater., 2023, 33, 2211182 CrossRef CAS.
- Y. Xue, J. Zhang, X. Chen, J. Zhang, G. Chen, K. Zhang, J. Lin, C. Guo and J. Liu, Adv. Funct. Mater., 2021, 31, 2106446 CrossRef CAS.
- M. Fu, Y. Zhao, Y. Wang, Y. Li, M. Wu, Q. Liu, Z. Hou, Z. Lu, K. Wu and J. Guo, Small, 2023, 19, 2205489 CrossRef CAS PubMed.
- K. Chen, Q. Lin, L. Wang, Z. Zhuang, Y. Zhang, D. Huang and H. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 9748–9761 CrossRef CAS PubMed.
- Y. Chen, P. Ni, R. Xu, X. Wang, C. Fu, K. Wan, Y. Fang, H. Liu and Y. Weng, Adv. Healthcare Mater., 2023, 12, 2301913 CrossRef CAS.
- X. Wang, H. Huang, R. Xu, Y. Fang, Y. Weng, Z. Wang, X. Xiong and H. Liu, ACS Appl. Mater. Interfaces, 2023, 15, 45676–45688 CrossRef CAS.
- J. Cao, P. Wu, Q. Cheng, C. He, Y. Chen and J. Zhou, ACS Appl. Mater. Interfaces, 2021, 13, 24095–24105 CrossRef CAS PubMed.
- X. Wang, Y. Guo, J. Li, M. You, Y. Yu, J. Yang, G. Qin and Q. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 36166–36177 CrossRef CAS.
- Y. Liang, Z. Li, Y. Huang, R. Yu and B. Guo, ACS Nano, 2021, 15, 7078–7093 CrossRef CAS PubMed.
- Y. Lv, F. Cai, Y. He, L. Li, Y. Huang, J. Yang, Y. Zheng and X. Shi, Acta Biomater., 2023, 159, 95–110 CrossRef CAS.
- T. Nakamura, Y. Takashima, A. Hashidzume, H. Yamaguchi and A. Harada, Nat. Commun., 2014, 5, 4622 CrossRef.
- S.-H. Li, B.-B. Li, X.-L. Zhao, H. Wu, R.-L. Chai, G.-Y. Li, D. Zhu, G. He, H.-F. Zhang, K.-K. Xie, B. Cheng and Q. Zhao, Small, 2023, 19, 2301934 CrossRef CAS.
- S. Bi, C. He, R. Liu, X. Zhao, J. Liu, J. Gu, W. Liu and B. Yan, Ind. Eng. Chem. Res., 2023, 62, 7492–7503 CrossRef CAS.
- B. Guo, Y. Liang and R. Dong, Nat. Protoc., 2023, 18, 3322–3354 CrossRef CAS.
- P. Ran, H. Zheng, W. Cao, X. Jia, G. Zhang, Y. Liu and X. Li, ACS Appl. Mater. Interfaces, 2022, 14, 49375–49388 CrossRef CAS PubMed.
- K. Su, D. Deng, X. Wu, Y. Song, Y. Sun, X. Wang, Z. Zhang, J. Li, Z. Yan, X. Shang, C. Li, K. Zhang, S. Ang, K. Wu and P. Wu, Chem. Eng. J., 2024, 479, 147646 CrossRef CAS.
- X. Shi, Z. Chen, Y. He, Q. Lu, R. Chen, C. Zhao, D. Dong, Y. Sun and H. He, Carbohydr. Polym., 2022, 297, 120042 CrossRef CAS PubMed.
- W. Huang, Y. Wang, Z. Huang, X. Wang, L. Chen, Y. Zhang and L. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 41076–41088 CrossRef CAS PubMed.
- A. A. Gokaltun, L. Fan, L. Mazzaferro, D. Byrne, M. L. Yarmush, T. Dai, A. Asatekin and O. B. Usta, Bioact. Mater., 2023, 25, 415–429 CAS.
- Z. Zhou, Y. Jia, W. Lu, J. Lei and Z. Liu, J. Mech. Phys. Solids, 2024, 183, 105516 CrossRef CAS.
- Z. Zhang, C. Qin, H. Feng, Y. Xiang, B. Yu, X. Pei, Y. Ma and F. Zhou, Nat. Commun., 2022, 13, 6964 CrossRef CAS.
- Y. Liu, C. Wang, J. Xue, G. Huang, S. Zheng, K. Zhao, J. Huang, Y. Wang, Y. Zhang, T. Yin and Z. Li, Adv. Healthcare Mater., 2022, 11, 2200653 CrossRef CAS.
- L. Feng, W. Shi, Q. Chen, H. Cheng, J. Bao, C. Jiang, W. Zhao and C. Zhao, Adv. Healthcare Mater., 2021, 10, 2100784 CrossRef CAS PubMed.
- C. Cai, H. Zhu, Y. Chen, Y. Guo, Z. Yang, H. Li and H. Liu, ACS Nano, 2022, 16, 20044–20056 CrossRef CAS PubMed.
- Y. Zhang, S. Ma, B. Li, B. Yu, H. Lee, M. Cai, S. N. Gorb, F. Zhou and W. Liu, Chem. Mater., 2021, 33, 2785–2795 CrossRef CAS.
- W. Li, X. Hu, H. Liu, J. Tian, L. Li, B. Luo, C. Zhou and L. Lu, J. Mater. Chem. B, 2023, 11, 5010–5020 RSC.
- R. Huang, X. Zhang, W. Li, L. Shang, H. Wang and Y. Zhao, Adv. Sci., 2021, 8, 2100201 CrossRef CAS.
- Y. Wang, D. Liu, C. Wang, J. Wu, X. Xu, X. Yang, C. Sun, P. Jiang and X. Wang, Chem. Eng. J., 2023, 457, 141268 CrossRef CAS.
- Y.-W. Lee, S. Chun, D. Son, X. Hu, M. Schneider and M. Sitti, Adv. Mater., 2022, 34, 2109325 CrossRef CAS.
- S. Wang, H. Luo, C. Linghu and J. Song, Adv. Funct. Mater., 2021, 31, 2009217 CrossRef CAS.
- W.-T. Chen, L. Zeng, P. Li, Y. Liu, J.-L. Huang, H. Guo, P. Rao and W.-H. Li, J. Ind. Eng. Chem., 2023, 117, 103–108 CrossRef CAS.
- T. M. Lutz, C. Kimna, A. Casini and O. Lieleg, Mater. Today, 2022, 13, 100203 CrossRef CAS PubMed.
- W. Yang, X. Kang, X. Gao, Y. Zhuang, C. Fan, H. Shen, Y. Chen and J. Dai, Adv. Funct. Mater., 2023, 33, 2211340 CrossRef CAS.
- J. Zhang, Y. Wang, J. Zhang, I. M. Lei, G. Chen, Y. Xue, X. Liang, D. Wang, G. Wang, S. He and J. Liu, Small, 2022, 18, 2201796 CrossRef CAS.
- H. Cho, G. Wu, J. Christopher Jolly, N. Fortoul, Z. He, Y. Gao, A. Jagota and S. Yang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 13774–13779 CrossRef CAS.
- W. Li, Y. Yu, R. Huang, X. Wang, P. Lai, K. Chen, L. Shang and Y. Zhao, Adv. Sci., 2023, 10, 2301479 CrossRef CAS.
- Y. Miao, M. Xu and L. Zhang, Adv. Mater., 2021, 33, 2102308 CrossRef CAS PubMed.
- Y. Li, J. Liu, Q. Zhang, N. Hu, Z. Jiang, Q. Kan and G. Kang, ACS Adv. Mater. Interfaces, 2024, 16, 10822–10831 CrossRef CAS.
- Y. Zhang, Q. Chen, Z. Dai, Y. Dai, F. Xia and X. Zhang, J. Mater. Chem. B, 2021, 9, 585–593 RSC.
- Z. Pan, Q.-Q. Fu, M.-H. Wang, H.-L. Gao, L. Dong, P. Zhou, D.-D. Cheng, Y. Chen, D.-H. Zou, J.-C. He, X. Feng and S.-H. Yu, Nat. Commun., 2023, 14, 5378 CrossRef CAS PubMed.
- K. C. Wu, B. R. Freedman, P. S. Kwon, M. Torre, D. O. Kent, W. L. Bi and D. J. Mooney, Sci. Transl. Med., 2024, 16, eadj0616 CrossRef CAS PubMed.
- Z. Ma, C. Bourquard, Q. Gao, S. Jiang, T. De Iure-Grimmel, R. Huo, X. Li, Z. He, Z. Yang, G. Yang, Y. Wang, E. Lam, Z.-H. Gao, O. Supponen and J. Li, Science, 2022, 377, 751–755 CrossRef CAS PubMed.
- F. Zhou, L. Xin, S. Wang, K. Chen, D. Li, S. Wang, Y. Huang, C. Xu, M. Zhou, W. Zhong, H. Wang, T. Chen and J. Song, ACS Appl. Mater. Interfaces, 2023, 15, 27568–27585 CrossRef CAS.
- Z. Zeng, G. Jiang, T. Liu, G. Song, Y. Sun, X. Zhang, Y. Jing, M. Feng and Y. Shi, Bio-Des. Manuf., 2021, 4, 902–911 CrossRef CAS.
- S. Y. Yang, E. D. O'Cearbhaill, G. C. Sisk, K. M. Park, W. K. Cho, M. Villiger, B. E. Bouma, B. Pomahac and J. M. Karp, Nat. Commun., 2013, 4, 1702 CrossRef PubMed.
- X. Fan, Y. Fang, W. Zhou, L. Yan, Y. Xu, H. Zhu and H. Liu, Mater. Horiz., 2021, 8, 997–1007 RSC.
- Q. Peng, Q. Wu, J. Chen, T. Wang, M. Wu, D. Yang, X. Peng, J. Liu, H. Zhang and H. Zeng, ACS Appl. Mater. Interfaces, 2021, 13, 48239–48251 CrossRef CAS.
- Y. Zhou, C. Zhang, S. Gao, B. Zhang, J. Sun, J.-J. Kai, B. Wang and Z. Wang, Chem. Mater., 2021, 33, 8822–8830 CrossRef.
- S. Liu, Y. Xiang, Z. Liu, L. Li, R. Dang, H. Zhang, F. Wei, Y. Chen, X. Yang, M. Mao, Y. S. Zhang, J. Song and X. Zhang, Adv. Mater., 2024, 36, 2309774 CrossRef CAS PubMed.
- Y. Qi, C. Xu, Z. Zhang, Q. Zhang, Z. Xu, X. Zhao, Y. Zhao, C. Cui and W. Liu, Bioact. Mater., 2024, 35, 259–273 CAS.
- A. Eklund, O. Ikkala and H. Zhang, Adv. Funct. Mater., 2024, 34, 2214091 CrossRef CAS.
- B. Zhang, L. Jia, J. Jiang, S. Wu, T. Xiang and S. Zhou, ACS Appl. Mater. Interfaces, 2021, 13, 36574–36586 CrossRef CAS PubMed.
- S. Li, R. Li, X. Zhang, Y. Zhu, D. Wang, W. Cui, H. Chai, Y. Hou and S. Li, Adv. Funct. Mater., 2025, 2500329 CrossRef CAS.
- H. Zhou, M. Yang, W. He, Y. Gao, X. Zhu, J. Wu, L. Zhang and P. Wan, Matter, 2025, 102150 CrossRef CAS.
- J. Zhu, M. Zhang, R. Qiu, M. Li, L. Zhen, J. Li, J. Luo, J. Li, H. Wu and J. Yang, Acta Biomater., 2024, 188, 117–137 CrossRef CAS.
- Z. Zhou, J. Lei and Z. Liu, Polymer, 2022, 246, 124730 CrossRef CAS.
- J. Yuan, M. He, J. Yang, K. Li, K. Fan, H. Luo, B. Li and Y. Chen, Chem. Eng. J., 2025, 506, 160110 CrossRef CAS.
- M. Li, H. Lu, M. Pi, H. Zhou, Y. Wang, B. Yan, W. Cui and R. Ran, Adv. Sci., 2023, 10, 2304780 CrossRef CAS.
- Y. He, Q. Li, P. Chen, Q. Duan, J. Zhan, X. Cai, L. Wang, H. Hou and X. Qiu, Nat. Commun., 2022, 13, 7666 CrossRef CAS PubMed.
- Z. Yuan, X. Duan, X. Su, Z. Tian, A. Jiang, Z. Wan, H. Wang, P. Wei, B. Zhao, X. Liu and J. Huang, Bioact. Mater., 2023, 21, 566–575 CAS.
- Y. Duan, X. Zhao, M. Sun and H. Hao, Ind. Eng. Chem. Res., 2021, 60, 1071–1095 CrossRef CAS.
- Y. Liang, H. Xu, Q. Han, M. Xu, J. Zhang, J. Wang, X. Liu, Z. Yin and B. Guo, Nano Today, 2024, 54, 102105 CrossRef CAS.
- J. Lee, H. S. Seo, C. G. Park and M. Shin, BMEMat, 2024, 2, 12071 CrossRef.
- Y. Jia, J. Feng, Z. Feng, J. Liu, Y. Yang, X. Li, M. Lei, H. Guo, Z. Wei, Y. Lv and F. Xu, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2219024120 CrossRef CAS.
- S. He, W. Liang, Y. Tang, J. Zhang, R. Wang, L. Quan, Y. Ouyang, R. Huang, R. Dou and D. Wu, Nat. Commun., 2025, 16, 3198 CrossRef CAS PubMed.
- Y. Wei, Y. He, C. Wang, G. Chen and B. Zhao, Adv. Funct. Mater., 2023, 33, 2214366 CrossRef CAS.
- R. Yan, Z. Xu, H. Yi, C. Xu, H. Xu, X. Qi, X. Dong, C. Hu, Z. Li and X. Jiang, Adv. Funct. Mater., 2025, 35, 2422130 CrossRef CAS.
- J. Shen, S. Fu, X. Liu, S. Tian, Z. Yi and Y. Wang, Adv. Healthcare Mater., 2025, 14, 2500600 CrossRef CAS.
- K. Shi, T. Li, X. Hu, W. Chen, Y. Yu, Z. Bei, L. Yuan, Q. Tong, J. Liu, Q. Fan, Y. Qian and Z. Qian, Adv. Sci., 2025, 12, 2500731 CrossRef CAS.
- H. Ren, Z. Zhang, X. Cheng, Z. Zou, X. Chen and C. He, Sci. Adv., 2023, 9, eadh4327 CrossRef CAS.
- M. Chai, W. Zhong, S. Yan, T. Ye, R. Zheng, Z. Yang and X. Shi, BMEMat, 2024, 2, e12108 CrossRef CAS.
- X. Zhao, H. Wang, J. Luo, G. Ren, J. Wang, Y. Chen and P. Jia, ACS Appl. Polym. Mater., 2022, 4, 1784–1793 CrossRef CAS.
- H. Yuk, C. E. Varela, C. S. Nabzdyk, X. Mao, R. F. Padera, E. T. Roche and X. Zhao, Nature, 2019, 575, 169–174 CrossRef CAS PubMed.
- B. Xue, J. Gu, L. Li, W. Yu, S. Yin, M. Qin, Q. Jiang, W. Wang and Y. Cao, Nat. Commun., 2021, 12, 7156 CrossRef CAS.
- H. Chen, J. Yang, Z. Liu, Y. Li, Z. Tang, X. Shi and Q. Chen, Adv. Funct. Mater., 2025, 35, 2413171 CrossRef CAS.
- X. Qu, H. Sun, X. Kan, B. Lei, J. Shao, Q. Wang, W. Wang, Z. Ni and X. Dong, Nano Research, 2023, 16, 10348–10357 CrossRef CAS.
- W. Yuan, D. Gan, S. Wang, Q. Wang, W. Wang, C. Sun and X. Dong, Chem. Eng. J., 2022, 448, 137675 CrossRef CAS.
- X. Qu, J. Liu, S. Wang, J. Shao, Q. Wang, W. Wang, L. Gan, L. Zhong, X. Dong and Y. Zhao, Chem. Eng. J., 2023, 453, 139785 CrossRef CAS.
- K. Wu, J. Li, Y. Li, H. Wang, Y. Zhang, B. Guo, J. Yu and Y. Wang, Adv. Funct. Mater., 2024, 34, 2404451 CrossRef CAS.
- X. Cui, Y. Nie, S. A. Khan, X. Bo, N. Li, X. Yang, D. Wang, R. Cheng, Z. Yuan and H. Zhang, ACS Sens., 2025, 10, 537–544 CrossRef CAS.
- J. Shen, M. Cai, G. Li, C. F. Guo, X. Qiu and Y. Qian, Adv. Funct. Mater., 2025, 35, 2413597 CrossRef CAS.
- X. Pei, H. Zhang, Y. Zhou, L. Zhou and J. Fu, Mater. Horiz., 2020, 7, 1872–1882 RSC.
- Y. Jiang, A. A. Trotsyuk, S. Niu, D. Henn, K. Chen, C.-C. Shih, M. R. Larson, A. M. Mermin-Bunnell, S. Mittal, J.-C. Lai, A. Saberi, E. Beard, S. Jing, D. Zhong, S. R. Steele, K. Sun, T. Jain, E. Zhao, C. R. Neimeth, W. G. Viana, J. Tang, D. Sivaraj, J. Padmanabhan, M. Rodrigues, D. P. Perrault, A. Chattopadhyay, Z. N. Maan, M. C. Leeolou, C. A. Bonham, S. H. Kwon, H. C. Kussie, K. S. Fischer, G. Gurusankar, K. Liang, K. Zhang, R. Nag, M. P. Snyder, M. Januszyk, G. C. Gurtner and Z. Bao, Nature Biotechnol., 2023, 41, 652–662 CrossRef CAS PubMed.
- X. He, Z. Li, X. Huang, Q. Zhang, Y. Zeng, J. Li, C. K. Yiu, Y. Yang, J. Zhou, G. Xu, J. Wang, J. Li, Z. Xu, Z. Chen, Y. Liu, Y. Gao, B. Zhang, G. Zhao, Z. Gao, P. Wu, R. Shi, Y. Qiu, H. Zhang, L. Chow, D. Ye, Y. Huang and X. Yu, iScience, 2025, 3, e12124 CAS.
- C. Wang, X. Chen, L. Wang, M. Makihata, H.-C. Liu, T. Zhou and X. Zhao, Science, 2022, 377, 517–523 Search PubMed.
- D. Gan, Q. Wang, X. Zhang, X. Qu, H. Sun, Y. Cui, W. Wang, L. Qu and X. Dong, Adv. Funct. Mater., 2024, 34, 2411588 Search PubMed.
- X. Huang, Z. Zheng, H. Wang, W. Xu, M. Wu, M. Wang, C. Chen, L. Wan, R. Du, T. Zhu, Z. Huang, X. Wang, X. Wang, Q. Zhang and X. Jia, Adv. Funct. Mater., 2024, 34, 2312149 Search PubMed.
- J. Liu, X. Zhang, Y. Cui, Y. Liu, W. Wang, Y. Guo, Q. Wang and X. Dong, ACS Appl. Mater. Interfaces, 2024, 16, 5208–5216 CrossRef CAS.
- H. Zheng, M. Chen, Y. Sun and B. Zuo, Chem. Eng. J., 2022, 446, 136931 Search PubMed.
- G. Huang, H. Guo, Z. Tang, S. Peng, H. Liang, G. Meng and P. Zhang, Chem. Mater., 2023, 35, 5953–5962 CrossRef CAS.
- C. Wang, Y. Lu, H. Cui, J. Zhang and Y. Tan, Chem. Eng. J., 2024, 485, 149925 Search PubMed.
- Y. Zhao, D. Gan, L. Wang, S. Wang, W. Wang, Q. Wang, J. Shao and X. Dong, Adv. Mater. Technol., 2023, 8, 2201566 CrossRef.
- J. Wang, N. Zhang, Y. Tan, F. Fu, G. Liu, Y. Fang, X.-X. Zhang, M. Liu, Y. Cheng and J. Yu, ACS Appl. Mater. Interfaces, 2022, 14, 21945–21953 CrossRef CAS.
- Z. Zhang, J. Yang, H. Wang, C. Wang, Y. Gu, Y. Xu, S. Lee, T. Yokota, H. Haick, T. Someya and Y. Wang, Sci. Adv., 2024, 10, eadj5389 CrossRef CAS PubMed.
- X. Xu, Y. Lyu, D. Liu, X. Shi, Z. Ji, D. Liu, X. Jia and X. Wang, Small, 2025, 21, 2411808 CrossRef CAS PubMed.
- M. Gulfam, S.-H. Jo, T. T. Vu, I. Ali, A. Rizwan, S.-B. Joo, S.-H. Park and K. T. Lim, Carbohydr. Polym., 2023, 303, 120457 CrossRef CAS.
- X. Chen, J. Zhang, K. Wu, X. Wu, J. Tang, S. Cui, D. Cao, R. Liu, C. Peng, L. Yu and J. Ding, Small Methods, 2020, 4, 2000310 CrossRef CAS.
- K. Shen, Z. Lv, Y. Yang, H. Wang, J. Liu, Q. Chen, Z. Liu, M. Zhang, J. Liu and Y. Cheng, Adv. Mater., 2025, 37, 2414092 CrossRef CAS.
- Z. Wang, J. Xu, X. Wu, M. Han, R. Peng, R. Zhao, M. Qin, T. Li, J. Yin, L. Yu, Y. Li, H. Wu, Z. Lin, L. Wang, Y. Hu and Y. Wu, Adv. Funct. Mater., 2024, 34, 2408479 CrossRef CAS.
- X. Guo and G. S. Kassab, Am. J. Physiol. Heart Circ. Physiol., 2003, 285, H2614–H2622 CrossRef CAS.
- W. Han, B. Zhou, K. Yang, X. Xiong, S. Luan, Y. Wang, Z. Xu, P. Lei, Z. Luo, J. Gao, Y. Zhan, G. Chen, L. Liang, R. Wang, S. Li and H. Xu, Bioact. Mater., 2020, 5, 768–778 Search PubMed.
- X. Xu, X. Xia, K. Zhang, A. Rai, Z. Li, P. Zhao, K. Wei, L. Zou, B. Yang, W.-K. Wong, P. W.-Y. Chiu and L. Bian, Sci. Transl. Med., 2020, 12, eaba8014 CrossRef CAS.
- F. Song, Y. Kong, C. Shao, Y. Cheng, J. Lu, Y. Tao, J. Du and H. Wang, Acta Biomater., 2021, 136, 170–183 CrossRef CAS.
- A. Liu, Z. Huang, S. Cui, Y. Xiao, X. Guo, G. Pan, L. Song, J. Deng, T. Xu, Y. Fan and R. Wang, Mater. Horiz., 2024, 11, 5983–5996 RSC.
- J. Shi, D. Wang, H. Wang, X. Yang, S. Gu, Y. Wang, Z. Chen, Y. Chen, J. Gao, L. Yu and J. Ding, Acta Biomater., 2022, 145, 106–121 CrossRef CAS.
- J. Cheng, H. Wang, J. Gao, X. Liu, M. Li, D. Wu, J. Liu, X. Wang, Z. Wang and P. Tang, Adv. Healthcare Mater., 2023, 12, 2300312 CrossRef CAS.
- Y. Wang, C. He, C. Chen, W. Dong, X. Yang, Y. Wu, Q. Kong and B. Yan, ACS Appl. Mater. Interfaces, 2022, 14, 55342–55353 CrossRef CAS.
- M. Yan, S.-Y. Hu, Z.-G. Wang, R. Hong, X. Peng, M. Kuzmanović, M. Yang, R. Dai, Y. Wang, J. Gou, K. Li, J.-Z. Xu and Z.-M. Li, Biomacromolecules, 2024, 25, 2438–2448 CrossRef CAS PubMed.
- X. Qu, S. Wang, Y. Zhao, H. Huang, Q. Wang, J. Shao, W. Wang and X. Dong, Chem. Eng. J., 2021, 425, 131523 CrossRef CAS.
- C. Cao, N. Yang, Y. Zhao, D. Yang, Y. Hu, D. Yang, X. Song, W. Wang and X. Dong, Nano Today, 2021, 39, 101165 CrossRef CAS.
- Y. Chen, Y. Li, X. Yang, Z. Cao, H. Nie, Y. Bian and G. Yang, Acta Biomater., 2021, 125, 208–218 CrossRef CAS PubMed.
- T. Xiong, K. Yang, T. Zhao, H. Zhao, X. Gao, Z. You, C. Fan, X. Kang, W. Yang, Y. Zhuang, Y. Chen and J. Dai, Adv. Sci., 2023, 10, 2205997 CrossRef CAS.
- C. He, M. Yin, H. Zhou, J. Qin, S. Wu, H. Liu, X. Yu, J. Chen, H. Zhang, L. Zhang and Y. Wang, ACS Nano, 2025, 19, 1713–1731 CrossRef CAS PubMed.
- X. Qu, Q. Wang, D. Gan, H. Sun, Z. Ni and X. Dong, Nano Lett., 2025, 25, 4759–4766 CrossRef CAS PubMed.
- H. Liu, X. Hu, W. Li, M. Zhu, J. Tian, L. Li, B. Luo, C. Zhou and L. Lu, Chem. Eng. J., 2023, 452, 139368 CrossRef CAS.
- Y. Zhao, B. Yi, J. Hu, D. Zhang, G. Li, Y. Lu and Q. Zhou, Adv. Funct. Mater., 2023, 33, 2300710 CrossRef CAS.
- R. Yu, M. Li, Z. Li, G. Pan, Y. Liang and B. Guo, Adv. Healthcare Mater., 2022, 11, 2102749 CrossRef CAS.
- S. Li, L. Wang, W. Zheng, G. Yang and X. Jiang, Adv. Funct. Mater., 2020, 30, 2002370 CrossRef CAS.
- N. Zhao and W. Yuan, Compos. Part B Eng., 2022, 230, 109525 CrossRef CAS.
- M. A. Gionet-Gonzales, R. C. H. Gresham, K. H. Griffin, A. Casella, R. P. Wohlgemuth, D. H. Ramos-Rodriguez, J. Lowen, L. R. Smith and J. K. Leach, Acta Biomater., 2023, 155, 271–281 CrossRef CAS.
- H. Sun, M. Zhang, M. Liu, Y. Yu, X. Xu and J. Li, Biomacromolecules, 2020, 21, 4699–4708 CrossRef CAS PubMed.
- Q. Yu, H. Sun, Z. Yue, C. Yu, L. Jiang, X. Dong, M. Yao, M. Shi, L. Liang, Y. Wan, H. Zhang, F. Yao and J. Li, Adv. Healthcare Mater., 2023, 12, 2202309 CrossRef CAS.
- J. Chen, D. Wang, L.-H. Wang, W. Liu, A. Chiu, K. Shariati, Q. Liu, X. Wang, Z. Zhong, J. Webb, R. E. Schwartz, N. Bouklas and M. Ma, Adv. Mater., 2020, 32, 2001628 CrossRef CAS.
- M. M. Hasani-Sadrabadi, P. Sarrion, S. Pouraghaei, Y. Chau, S. Ansari, S. Li, T. Aghaloo and A. Moshaverinia, Sci. Transl. Med., 2020, 12, eaay6853 CrossRef CAS.
- M. Fan, L. Jia, M. Pang, X. Yang, Y. Yang, S. Kamel Elyzayati, Y. Liao, H. Wang, Y. Zhu and Q. Wang, Adv. Funct. Mater., 2021, 31, 2010587 CrossRef CAS.
- L. Schnaider, Z. Toprakcioglu, A. Ezra, X. Liu, D. Bychenko, A. Levin, E. Gazit and T. P. J. Knowles, Nano Lett., 2020, 20, 1590–1597 CrossRef CAS PubMed.
- J. Cha and P. Kim, ACS Appl. Mater. Interfaces, 2021, 13, 31371–31378 CrossRef CAS PubMed.
- H. Zhong, J. Huang, M. Luo, Y. Fang, X. Zeng, J. Wu and J. Du, Nano Res., 2023, 16, 599–612 CrossRef CAS.
- Q. Sun, K. Hong, L. Fan, X. Zhang, T. Wu, J. Du and Y. Zhu, Chem. Mater., 2023, 35, 9208–9224 CrossRef CAS.
- X. Zhao, S. Li, X. Du, W. Li, Q. Wang, D. He and J. Yuan, Bioact. Mater., 2022, 8, 196–209 CAS.
- G. Ge, Y.-Z. Zhang, W. Zhang, W. Yuan, J. K. El-Demellawi, P. Zhang, E. Di Fabrizio, X. Dong and H. N. Alshareef, ACS Nano, 2021, 15, 2698–2706 CrossRef CAS.
- G. Ge, Q. Wang, Y.-Z. Zhang, H. N. Alshareef and X. Dong, Adv. Funct. Mater., 2021, 31, 2107437 CrossRef.
- S. Yang, C. Qin, Z. Zhang, M. Zhang, B. Li, Y. Ma, F. Zhou and W. Liu, Chem. Bio. Eng., 2025, 2, 253–259 CrossRef CAS.
- X. Hu, Y. Zhou, M. Li, J. Wu, G. He and N. Jiao, Small, 2024, 20, 2306510 CrossRef CAS.
- C. Xin, D. Jin, Y. Hu, L. Yang, R. Li, L. Wang, Z. Ren, D. Wang, S. Ji, K. Hu, D. Pan, H. Wu, W. Zhu, Z. Shen, Y. Wang, J. Li, L. Zhang, D. Wu and J. Chu, ACS Nano, 2021, 15, 18048–18059 CrossRef CAS PubMed.
- Y. Zhong, X. T. Zheng, S. Zhao, X. Su and X. J. Loh, ACS Nano, 2022, 16, 19840–19872 CrossRef CAS PubMed.
- Y.-W. Lee, J.-K. Kim, U. Bozuyuk, N. O. Dogan, M. T. A. Khan, A. Shiva, A.-M. Wild and M. Sitti, Adv. Mater., 2023, 35, 2209812 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |