DOI:
10.1039/D5CS00020C
(Review Article)
Chem. Soc. Rev., 2026,
55, 504-555
Advances of rare earth-based catalysts for recycling CO2 and plastic waste
Received
2nd August 2025
First published on 3rd December 2025
Abstract
The crisis caused by the excessive use of fossil fuels—emissions of billions of tonnes of CO2 and the accumulation of plastic waste—is imminent. Conventional disposal technologies, such as physical storage, face risks of leakage, capacity limitations, and secondary pollution (such as microplastics). In contrast, chemical recycling, especially thermal catalytic technology, is considered a key alternative solution due to its high resource recovery potential. However, its large-scale implementation remains hindered by the absence of efficient and durable catalysts. Rare earth-based catalysts, with their unique 4f/5d electronic structure and tunable coordination environments, demonstrate significant advantages in activating inert C–C/C–H bonds, promoting CO2 adsorption and conversion, inhibiting coking and deactivation, and making them highly competitive for CO2 hydrogenation and plastic catalytic conversion. Despite rapid progress, challenges related to cost, long-term stability, and mechanistic understanding persist, impeding their industrial application. This review systematically summarises the controlled synthesis and in situ characterisation methods of rare earth-based catalysts and thoroughly explores their applications, performance regulation mechanisms, and challenges in CO2 hydrogenation and plastic recycling, aiming to provide insights for designing efficient, stable, and industrially scalable rare earth catalytic systems.

Liangliang Zhang
| Liangliang Zhang received his BSc and PhD from Jilin University in 2017 and 2022, respectively. He is currently working under the direction of Prof. Xiao Wang at the Changchun Institute of Applied Chemistry, Chinese Academy of Sciences (CAS). His current research interests focus on rare earth-based catalytic nanomaterials for heterogeneous catalytic reactions. |

Shuyan Song
| Shuyan Song received his BSc degree in Chemistry in 2003 and MSc in inorganic chemistry in 2006 both from Northeast Normal University. He joined the group of Prof. Hongjie Zhang at the Changchun Institute of Applied Chemistry, Chinese Academy of Sciences (CAS), where he received his PhD in inorganic chemistry in 2009. He is working as a professor under the direction of Prof. Zhang at the Changchun Institute of Applied Chemistry, CAS. His research focus is primarily on the development of porous functional materials for heterogeneous catalysis, proton conduction, chemical sensing and detection. |

Hongjie Zhang
| Hongjie Zhang received his BSc degree from Peking University in 1978. He then worked as a research assistant in the Changchun Institute of Applied Chemistry, where he received his MSc degree in Inorganic Chemistry in 1985. Then, he worked as an assistant professor at the same institute from 1985 to 1989. He then studied at Universite de Bordeaux I, Laboratoire de Chimie du Solide du CNRS (France), where he received his PhD degree in Solid State Chemistry and Materials Sciences in 1993. He joined the Changchun Institute of Applied Chemistry, CAS, as a professor in 1994. His current research interests include lanthanide organic–inorganic hybrid materials, electroluminescent devices, functional nanomaterials, and the structure and properties of rare earth magnesium alloys. |

Xiao Wang
| Xiao Wang received his BSc degree in Chemistry in 2008 from Jilin University. Then, he joined the group of Prof. Hongjie Zhang at the Changchun Institute of Applied Chemistry, Chinese Academy of Sciences (CAS), and received his PhD degree in Inorganic Chemistry in 2013. His research focus is primarily on the fabrication of functional inorganic materials for heterogeneous catalytic reactions and energy-related applications. |
1. Introduction
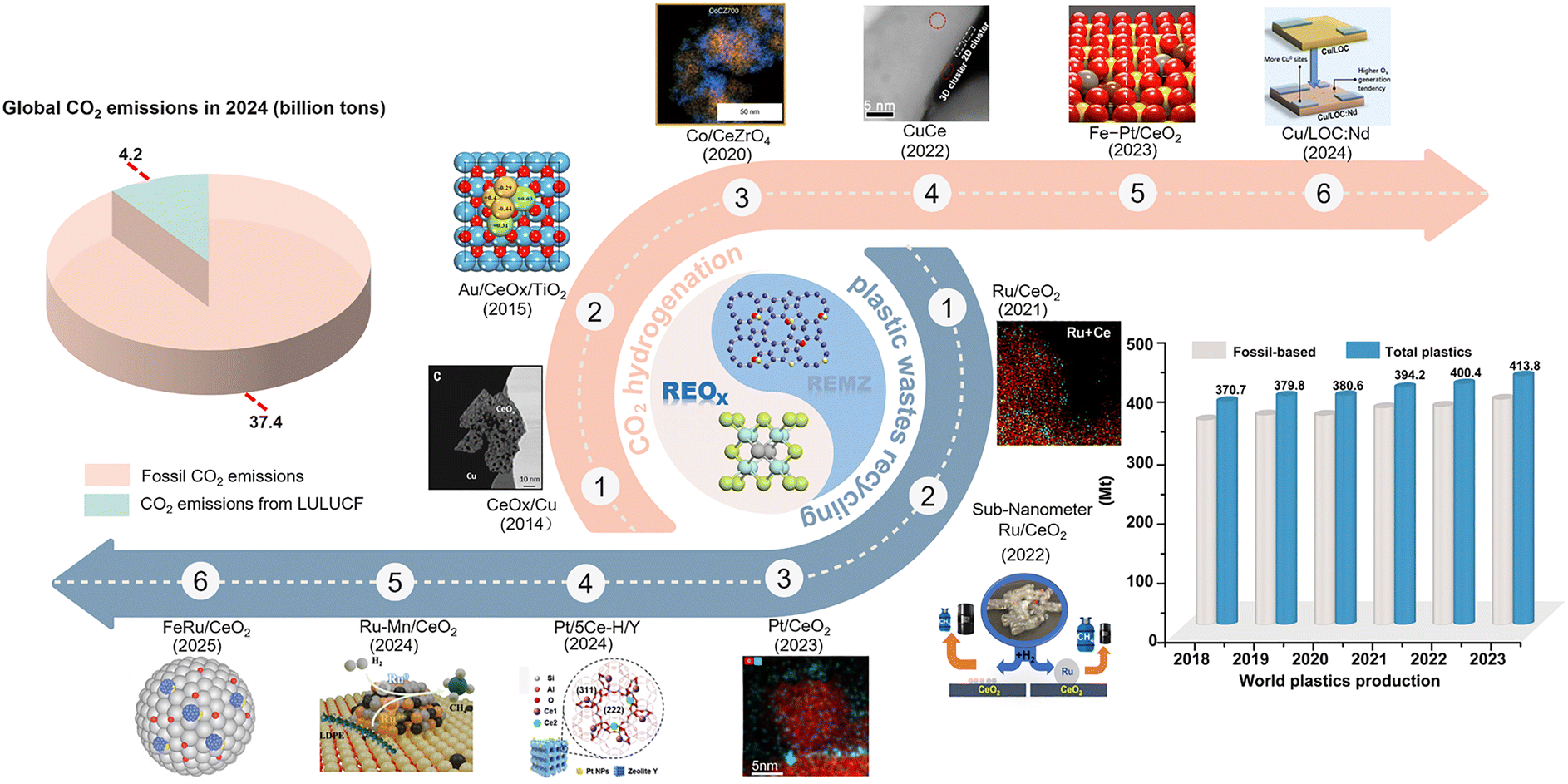
Against the backdrop of rapid development in human society, the excessive extraction and use of fossil fuels have triggered a dual carbon waste crisis: CO2 emissions and plastic waste, both of which urgently require immediate global action. On the one hand, CO2 emissions are expected to reach 41.6 billion tons in 2024 (Global Carbon Budget 2024) with fossil fuels contributing 90% (37.4 billion tons) (Fig. 1),1 directly intensifying the greenhouse effect to trigger extreme weather, ice-sheet collapse, sea-level rise, and so on. On the other hand, according to Plastics Europe's analysis, total global plastics production reached 413.8 million metric tonnes in 2023 (Fig. 1), an increase of 11.7% relative to 2018.2 With the rapid growth in plastics production, a large amount of solid waste will also be generated each year, and it is expected that by 2050, about 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 million metric tonnes of plastic waste will be accumulated globally.3–5 Worse still, this discarded plastic waste not only seriously pollutes water and soil, but also poses a lasting threat to humans. To prevent irreversible tipping points in Earth's ecosystems, large-scale deployment of carbon recycling technologies is urgently needed to address the systemic environmental crisis triggered by these two types of waste carbon.6,7
000 million metric tonnes of plastic waste will be accumulated globally.3–5 Worse still, this discarded plastic waste not only seriously pollutes water and soil, but also poses a lasting threat to humans. To prevent irreversible tipping points in Earth's ecosystems, large-scale deployment of carbon recycling technologies is urgently needed to address the systemic environmental crisis triggered by these two types of waste carbon.6,7
 |
| | Fig. 1 Current status of waste carbon emissions and development of rare earth-based catalysts for waste carbon recycling. Reprinted with permission from ref. 27. Copyright (2014) American Association for the Advancement of Science. Reprinted with permission from ref. 28. Copyright (2015) American Chemical Society. Reprinted with permission from ref. 29. Copyright (2020) Springer Nature. Reprinted with permission from ref. 30. Copyright (2022) Springer Nature. Reprinted with permission from ref. 31. Copyright (2023) American Chemical Society. Reprinted with permission from ref. 32. Copyright (2024) American Association for the Advancement of Science. Reprinted with permission from ref. 33. Copyright (2021) Elsevier. Reprinted with permission from ref. 34. Copyright (2022) American Chemical Society. Reprinted with permission from ref. 35. Copyright (2023) Springer Nature. Reprinted with permission from ref. 25. Copyright (2024) Wiley-VCH. Reprinted with permission from ref. 36. Copyright (2024) American Chemical Society. Reprinted with permission from ref. 37. Copyright (2025) Wiley-VCH. (Abbreviations: LULUCF – land-use, land-use change, and forestry; REOx – rare earth oxides; REMZ – Rare earth-modified zeolites). | |
Currently, among engineered end-of-life management options, physical storage still dominates for both CO2 and plastic waste.8,9 Taking CO2 as an example, CCS (Carbon Capture and Storage) projects utilizing physical storage have reached historic highs in terms of the number of facilities and CO2 capture capacity. Currently, there are 47 operational CCS projects globally, with an annual CO2 capture capacity of 64 million metric tonnes.10 In the realm of plastic waste, global production reached 425 million tonnes in 2020. The distribution of end-of-life management was as follows: approximately 39% underwent physical storage via landfilling, 24% were treated through formal incineration (with some energy recovery), 22% were recycled, and the remaining 15% were mismanaged.11 However, this approach faces systemic risks: CCS is hindered by long-term liability concerns, leakage risks, and limited cost-effectiveness of storage capacity, while deep-sea sequestration poses potential risks of CO2 leakage, leading to localized marine acidification and ecological imbalance.12 Simultaneously, plastics in landfills degrade into microplastics (MPs) under complex physical, chemical, and biological conditions, which are subsequently released via leachate and airborne pathways. These MPs act as carriers for pollutants including heavy metals, pathogens, and other hazardous substances, posing severe threats to environmental integrity and human health. In contrast, chemical recycling, which converts carbon waste into new chemicals or base monomers through chemical reactions, is more attractive.6,7,13,14 It not only effectively reduces carbon-based waste but also reduces dependence on fossil fuels. More importantly, CO2 and waste plastics (especially polyolefins) exhibit potential complementarity and synergistic effects in chemical recycling pathways, offering the possibility of coupled conversion between the two.15–17 On the one hand, the pyrolysis or catalytic pyrolysis of waste plastics can produce hydrogen-rich light hydrocarbons or hydrogen gas; on the other hand, the hydrogenation reaction of CO2 requires a significant amount of hydrogen gas. Using waste plastics as a hydrogen source or a reducing agent for CO2 conversion, or introducing CO2 as a mild oxidant and carbon source into the plastic conversion process, hold promise for achieving “waste-to-waste” management and efficient resource utilization, producing higher-value fuels or chemicals. If this synergistic recycling strategy can be realized, its economic and environmental benefits will far exceed those of treating either waste carbon source individually. Many methods have been developed for chemical recycling, such as thermal catalysis, electrocatalysis, and photocatalysis. Among these, thermal catalysis technology has advantages in terms of energy density, mass transfer efficiency, and catalytic activity, making it more promising for industrial-scale production and attracting widespread attention. Undoubtedly, catalysts play a vital role in the thermal catalysis processes. Therefore, the design and fabrication of high-performance heterogeneous catalysts to improve selectivity and efficiency is a top-priority task, though it remains a great challenge.
Rare earth materials, owing to their unique 4f/5d electronic structures and tunable coordination environments, exhibit excellent performance in catalysis, including reactions such as carbon waste recycling, organic synthesis, petroleum cracking, automobile exhaust purification, fuel cells, and biomass transformation.18–20 We take ceria as an example, as an important rare earth oxide, it shows particularly prominent performance in the CO2 hydrogenation reaction. This superior activity primarily stems from the reversible Ce4+/Ce3+ redox cycle, which dynamically generates oxygen vacancies and endows the surface with an electron-rich character, thereby greatly facilitating CO2 adsorption and activation.20–24 In the field of resource conversion for plastic waste (such as polyolefins), the core value of rare earth materials similarly originates from their unique electronic and structural properties. These plastics consist primarily of stable C–C and C–H bonds, and their catalytic cracking heavily relies on acid sites. Within this process, rare earth ions (e.g., La3+ and Ce4+) can effectively modulate the strength and density of the catalyst's acid sites,25,26 thereby promoting the cleavage of C–C/C–H bonds. Furthermore, a key challenge in this conversion process lies in the fact that unsaturated intermediates tend to undergo polymerization or condensation, leading to catalyst deactivation via coking. Addressing this challenge, rare earth oxides—particularly CeO2—play a vital role through their abundant and tunable oxygen vacancies, which promote the hydrogenation of unsaturated intermediates and effectively suppress coke formation, thereby significantly enhancing the stability and durability of catalysts in complex reaction environments. Given these properties and considering recent research hotspots and prospects for large-scale application, this review focuses on the use of rare-earth molecular sieves and rare-earth-based oxide catalysts in waste carbon recycling.
Despite the promising performance of rare earth-based catalysts (Fig. 1),25,27–37 there are numerous challenges in applying them to the chemical recycling of waste carbon. A comprehensive understanding of reaction mechanisms is crucial for designing efficient and selective catalytic systems. Additionally, scalability and economic viability must be addressed to facilitate the transition from laboratory to industrial applications. This paper reviews the controlled synthesis methods for rare earth-based catalysts and their in situ characterization techniques in waste carbon recycling and discusses their applications in the hydrogenation of CO2 to produce CO, methane, methanol, ethanol, and olefins and aromatics, as well as in plastic recycling. Through an in-depth analysis of the current state of research on rare earth-based catalysts for the chemical recycling of waste carbon, we explore the influence of the catalyst structure and composition on their performance and mechanism and summarise the opportunities and future research directions in this area of sustainable chemistry. We highlight the transformative potential of rare earth-based catalysts in addressing waste carbon emissions and aim to stimulate innovation and collaboration in finding sustainable solutions.
2. Preparation of rare earth-based catalysts
Rare earth-based catalysts have garnered significant attention in the field of carbon waste recovery. Among them, rare earth oxides and rare earth-modified zeolites represent the two most widely explored categories. Despite both containing rare earth elements, the former directly uses rare earth oxide materials as the active component or support matrix, whereas the latter incorporates rare earth ions or oxide clusters into zeolitic frameworks as additives or structural stabilizers. These intrinsic distinctions in functionality and material architecture lead to markedly different synthetic methods. The following section provides a comparative overview of the key synthesis technologies for these two types of catalysts.
2.1. Preparation of rare earth oxides
Compared to some common divalent transition metal ions, rare earth ions, due to their higher oxygen affinity and stronger Lewis acidity, readily react with common precipitating agents in solution, triggering uncontrollable nucleation processes and rapidly forming insoluble hydroxides or carbonates. This rapid and uneven precipitation process severely disrupts the molecular-level uniformity of the precursor mixture, making it difficult to control the morphology, crystal phase, and chemical uniformity of the final rare earth oxides. To overcome this challenge, researchers have developed various advanced strategies by leveraging the significant advances in synthetic chemistry in recent years. These methods mainly include solvothermal, sol–gel, co-precipitation, solution combustion synthesis, microemulsion, template-assisted, and mechanosynthesis methods, among others. They provide possibilities for improving the microstructure and physicochemical properties of catalysts by controlling the reaction kinetics, spatial confinement, or the introduction of specific structure-directing agents. Despite these advances, achieving truly precise control at the atomic/molecular level remains a major challenge and a core direction for future research. This chapter will focus on several representative synthetic strategies that are widely used in the preparation of rare earth oxides. Specifically, these strategies include solvothermal, co-precipitation, template-assisted, MOF-pyrolysis, mechanosynthesis, sol–gel, and microemulsion methods (Table 1).
Table 1 Methods for preparing rare earth oxides
| Methods |
Core reaction conditions |
Advantages |
Disadvantages |
| Solvothermal |
High temperature and pressure |
Controlled size, shape, crystallinity, and composition |
Longer-time reaction |
| Hard to control crystal growth |
| Co-precipitation |
Aqueous solution with pH control |
Simple and easy to operate |
Difficult to control particle size and morphology |
| Low cost and mild conditions |
| Versatile and widely used |
| Template-assisted |
Precursor coating on templates or penetrating the template pores |
Precise control of size, morphology, and structure |
Difficulty in template design and preparation |
| Creates CeO2 with abundant defects |
Multi-step and time-consuming |
| MOF pyrolysis |
High temperature calcination of MOF |
Uniform metal dispersion |
Requires high-temperature treatment |
| High surface area |
Time-consuming and high cost |
| Mechanosynthesis |
High-energy ball milling or grinding |
Low cost and environmentally friendly |
Limited control over crystal structure |
| Simple and fast process |
Possible contamination from milling media |
| Sol–gel |
Hydrolysis and condensation in mixed solvents |
High specific surface area |
Process can be time-consuming (gelation + drying) |
| Produces highly homogeneous and porous nanomaterials |
| Microemulsion |
Surfactant-stabilized microemulsions |
Precise control of size and morphology |
Requires large amounts of surfactants |
| Produces monodisperse nanoparticles |
Complex solution composition |
2.1.1. Solvothermal method.
The solvothermal method is a technique under high-temperature and high-pressure conditions in a sealed environment, using water or organic solvents as the reaction medium.38–42 In this method, rare earth precursors (such as cerium nitrate) are dissolved in a solvent and heated to or above the solvent's boiling point under autogenous pressure, driving the processes of nucleation and crystal growth. By adjusting parameters such as temperature, precipitant concentration, pH value, reaction time, and precursor type, the composition, morphology, size, and crystallinity of rare earth-based oxides can be controlled.43 Mai et al. succeeded in producing CeO2 nanocrystals with distinguishable morphologies, including nanopolyhedron, nanorod, and nanocube structures, by accurately tuning the hydrothermal reaction temperature and the concentration of NaOH.44 Ke et al. employed a modified hydrothermal approach, introducing precursors of other lanthanide elements (Ln, such as La, Nd, Pr, and Lu) into the Ce precursor solution during the hydrothermal synthesis process, thereby achieving the synthesis of Ln-doped CeO2 nanowires (Fig. 2a–d).45 It should be emphasised that this hydrothermal synthesis strategy is not only applicable to rare-earth oxide catalysts, but can also be extended to the controlled preparation of rare-earth oxide-derived compounds. Zhang et al. used La(NO3)3 6H2O as a precursor to directly synthesise La(OH)3 intermediates by the hydrothermal method and successfully obtained La2O2CO3 materials after calcination at 500 °C.46 Zhou et al. further extended this method and, through the same hydrothermal–calcination route, efficiently synthesized not only La2O2CO3 but also Sm2O2CO3 materials.42
 |
| | Fig. 2 (a) and (b) TEM images of CeO2:Nd and CeO2:Lu nanocrystals. (c) and (d) HAADF-STEM EDS elemental mapping of CeO2:Nd and CeO2:Lu nanocrystals. Reprinted with permission from ref. 45. Copyright (2013) American Chemical Society. (e) HR-TEM image and elemental mappings of the used 1.5Ce/HEOs catalyst. Reprinted with permission from ref. 51. Copyright (2024) Elsevier. | |
2.1.2. Co-precipitation method.
The co-precipitation has gradually become one of the most commonly employed approaches for the synthesis of rare-earth oxides, thanks to its advantages of readily available raw materials, simple operation, low cost, fast synthesis, and excellent scalability.47–50 The process consists of three key steps: (1) dissolving soluble salts containing rare earth and target metal cations in a predetermined molar ratio; (2) adding alkaline precipitating agents (such as NH3·H2O, NaOH, urea, or K2CO3) to form the target compound precipitate; and (3) subjecting the precipitate to thermal treatment. Its core competitive advantages are as follows: (1) process simplicity—no need for complex kinetic control or expensive organic reagents; and (2) suitable for constructing multi-component systems. For example, Xue et al. synthesized Co–CeO2 catalysts by coprecipitating a mixture of Co/Ce precursors with K2CO3 precipitant.50 The precipitate was then filtered, washed, and calcined in a programmed manner to obtain Co–CeO2 catalysts with a uniformly dispersed structure. This process did not require the use of organic additives or precision control equipment, fully demonstrating the advantages of its simplicity. Xu et al.'s research confirmed the applicability of coprecipitation in multi-metal systems: using equimolar ratios of Fe, Co, Ni, Cr, and Mn salts as multimetal precursors, they introduced cerium as a dopant and successfully synthesized a Ce/(FeCoNiCrMn)3O4 catalyst through co-precipitation and high-temperature calcination (Fig. 2e).51 However, this method has become the preferred choice for large-scale industrial production due to its ease of operation and cost advantages; its inherent uncontrollable reaction kinetics result in insufficient control of microstructural uniformity, making it difficult to meet the design requirements for high-precision catalytic materials.
2.1.3. Template-assisted method.
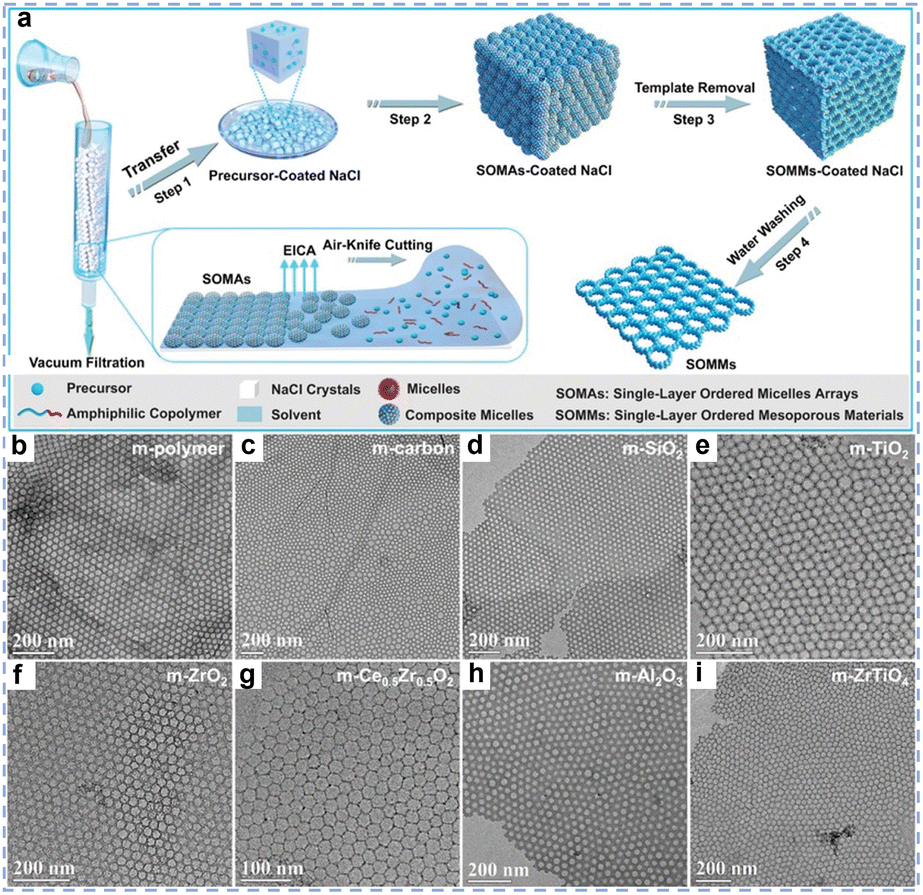
Template-assisted synthesis of rare earth oxides is a method that utilizes structurally specific “templates” to precisely control the pore structure and morphology of the resulting material.52,53 Its core principle relies on the spatial confinement or surface-directing effects of the template to guide the ordered deposition and assembly of rare earth precursors on the surface or within the interior of the template, forming a composite material that replicates template's structure. The target porous or nanostructured catalytic material is ultimately yielded by removing the template. Based on the nature of the templating agents, template methods are primarily categorized into hard template methods and soft template methods. Hard template methods utilize rigid materials (such as silica, polymer colloids, or carbon spheres) as templates.39,54,55 By coating rare earth precursors or allowing them to penetrate the template pores to form a core@shell or pore-fille structure, which is then calcined or etched to remove the template, hollow or porous rare earth catalytic materials are obtained. Zhang et al. prepared uniformly dispersed MnO2/CeO2–MnO2 composite microspheres, wherein carbon spheres served as sacrificial templates.56 The method requires that the hard templates remain inert during synthesis, do not react with the product, and are easily removed. The soft template method primarily utilizes surfactant-formed supramolecular aggregates (e.g., cetyltrimethylammonium bromide CTAB, PS-b-PEO, Pluronic series P123 or F127, etc.) as templates to construct mesoscopic structures through self-assembly between amphiphilic molecules and rare earth-based precursors. Subsequent removal of template molecules via thermal treatment or solvent extraction yields mesoporous rare earth-based catalytic materials.39,57 For example, Liu et al. used the surface of inorganic salts (such as NaCl or KCl) as the assembly interface, amphiphilic block copolymers (such as PEO-b-PS) as soft templates, and metal salts as precursors to synthesize monolayer-ordered mesoporous materials,58 including mesoporous carbon, metal oxides (such as TiO2, CeO2, Al2O3, and ZrO2), and functionally mesoporous complex mesoporous materials with compositions like Ce0.5Zr0.5O2 (Fig. 3a–i). The key to this method lies in precisely controlling the interactions (such as electrostatic forces, hydrogen bonding, or coordination bonds) between rare earth precursors and amphiphilic molecules. Compared with the hard template method, the soft template method requires no pre-preparation of templates, and the process is more simplified. However, it requires high selectivity of precursors to ensure sufficient intermolecular interactions for cooperative self-assembly.
 |
| | Fig. 3 Single-layer ordered mesoporous materials (SOMMs). (a) Schematic illustration of the fabrication process of SOMMs. (b)–(i) TEM images of single-layer ordered mesoporous materials of different compositions: (b) m-polymer, (c) m-carbon, (d) m-SiO2, (e) m-TiO2, (f) m-ZrO2, (g) m-Ce0.5Zr0.5O2, (h) m-Al2O3, (i) m-ZrTiO4 synthesized by using PS10000-b-P4VP2000 as an SDA. Reprinted with permission from ref. 58. Copyright (2020) Wiley-VCH. | |
2.1.4. MOF pyrolysis method.
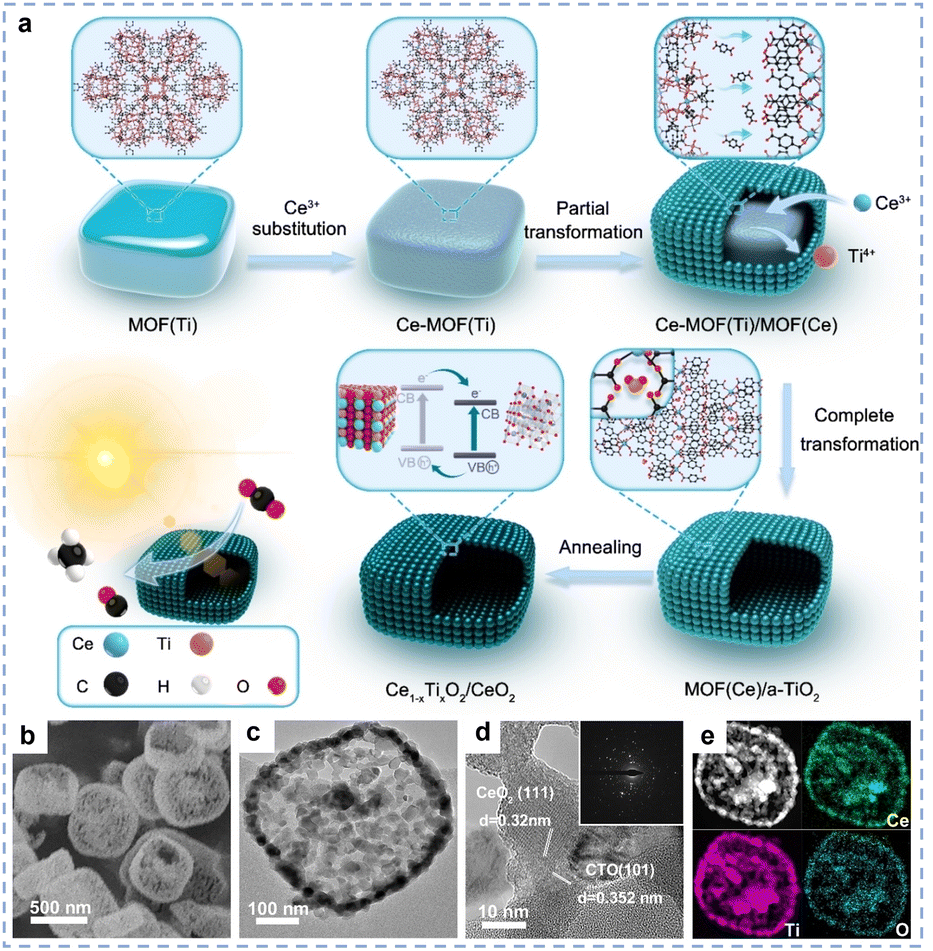
The MOF pyrolysis method is an advanced strategy that involves precisely designing metal–organic framework (MOF) precursors and converting them into high-performance rare earth oxide materials through controlled pyrolysis.38,59,60 This method starts with MOFs containing rare earth nodes (such as RE-MOFs), which are pyrolyzed under controlled atmospheres to selectively convert atomically dispersed rare earth species into highly dispersed rare earth oxide nanoparticles.61 By precisely tuning the pyrolysis conditions, metal oxide materials with pore structures can be obtained while partially maintaining the high specific surface area of the MOF materials, which can significantly improve the atomic utilisation of the catalysts. For example, Wei et al. first synthesized titanium-based MOF (MOF(Ti)),62 and then used Ce3+ as an etchant to convert it into a composite material composed of cerium-based MOF (MOF(Ce)) and amorphous TiO2 (Fig. 4a). This composite material was used as a sacrificial template, and after calcination, a mesoporous Ce1−xTixO2/CeO2-0.8 catalyst was obtained. Thanks to the abundance of Ce1−xTixO2 solid solution/CeO2 heterojunction and mesoporous structure (Fig. 4b–e), the catalyst exhibits excellent catalytic performance.
 |
| | Fig. 4 (a) Schematic illustration of preparing Ce1−xTixO2/CeO2 photocatalysts. (b) FESEM image of CTO/CeO2-0.8. (c) TEM image of CTO/CeO2-0.8. (d) HRTEM image with the corresponding SAED pattern (inset) of CTO/CeO2-0.8. (e) HAADF-STEM image with elemental mapping images for Ce, Ti and O of CTO/CeO2-0.8. Reprinted with permission from ref. 62. Copyright (2023) Wiley-VCH. | |
2.1.5. Mechanosynthesis method.
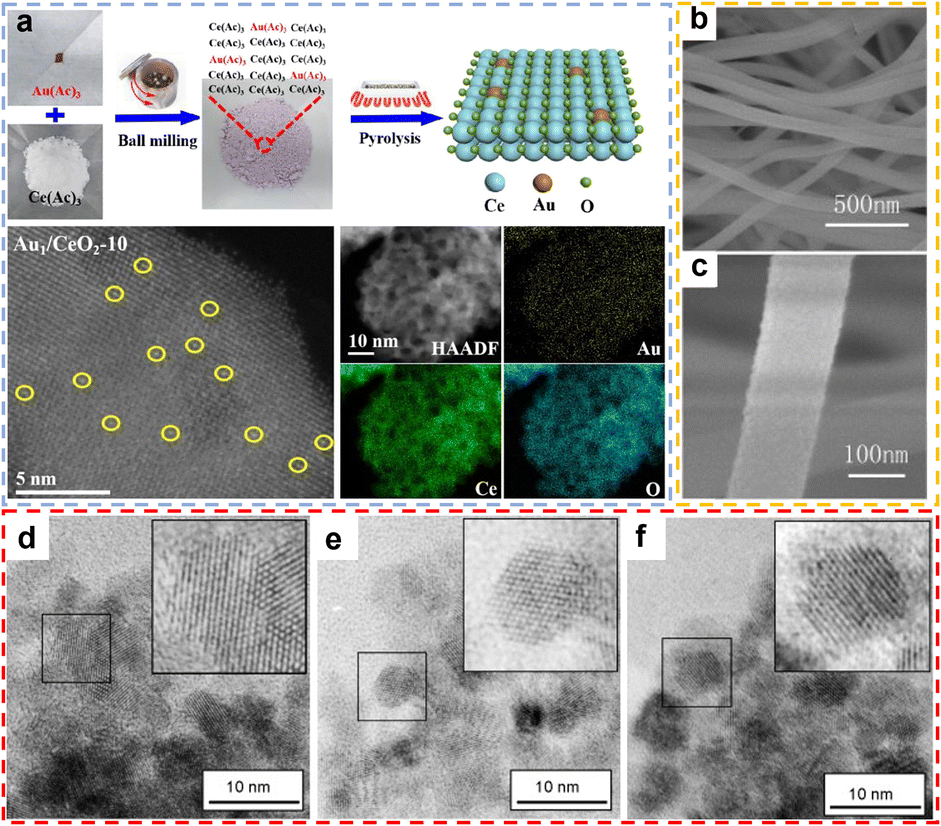
Mechanosynthesis is a method of driving chemical reactions through mechanical energy, widely used in the preparation of rare earth oxide materials.63 Its core lies in utilizing mechanical means such as ball milling and grinding to provide energy through collisions, friction, and shear forces, enabling physical and chemical transformations of materials.63–65 The main advantages of mechanical synthesis include solvent-free conditions, fast reaction rates, simple operation, low cost, and environmental friendliness, making it a crucial technology for the synthesis of rare earth oxide materials.63 For example, Liu et al. synthesized CuOx–ZrO2–CeO2 composite oxide materials via a simple grinding method.66 Specifically, Cu, Ce, and Zr salts were mixed with KOH and ground in a mortar for 30 minutes, followed by washing, filtration, drying, and calcination to obtain the final product. The resulting composite exhibited highly dispersed Cu species and a high concentration of oxygen vacancies. Although manual grinding methods offer simplicity of equipment and ease of operation, they present inherent limitations: (i) poor reproducibility due to operator dependence on physical inputs; and (ii) safety hazards due to open system exposure to reactants/products. Recent advances in laboratory-grade automated ball milling technology overcome these drawbacks by precisely controlling the energy input through an adjustable milling frequency, enabling (i) high reproducibility of the experiments, (ii) inherent safety due to the physical containment of hazardous materials, and (iii) high catalyst generation efficiency. For instance, Gan et al. synthesised atomically dispersed gold-based catalysts,67 Au1/CeO2, by homogeneously mixing gold and cerium precursors using ball milling and subsequently calcining them (Fig. 5a). This method not only effectively ensures the homogeneous distribution of the metal precursors but also has good scalability for the synthesis of kilogram-scale single-atom catalysts. Compared with the grinding method, the ball milling method avoids prolonged contact with highly corrosive substances such as NaOH and significantly improves the repeatability of experiments by precisely setting ball milling parameters.
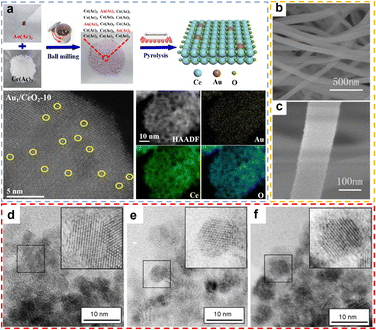 |
| | Fig. 5 (a) Schematic illustration of the preparation process for Au1/CeO2 and AC HAADF-STEM images of Au1/CeO2 catalysts with different preparation scales, and elemental mapping images of Au1/CeO2-10. Reprinted with permission from ref. 67. Copyright (2020) Elsevier. (b) and (c) SEM images of CeO2 nanofibers. Reprinted with permission from ref. 72. Copyright (2012) Elsevier. (d)–(f) Nanocrystals of CeO2 (d), CePrOx (e), CeTbOx (f). Reprinted with permission from ref. 74. Copyright (2008) Elsevier. | |
2.1.6. Sol–gel method.
The sol–gel method is a significant technique for preparing rare earth oxides.49,68–70 This approach involves the hydrolysis and condensation of rare earth-based precursors (such as metal alkoxides or inorganic salts) in solution to gradually construct a three-dimensional network gel structure, which is subsequently transformed into rare earth oxide materials through drying and calcination processes.47 Compared to solvothermal and co-precipitation methods, this approach demonstrates significant advantages in product characteristics and morphology control: the resulting rare earth oxides not only generally exhibit larger specific surface areas and superior compositional homogeneity, but also provide greater flexibility for producing specialized morphologies (e.g., thin films and fibers). For example, Tsiotsias et al. employed cerium nitrate and praseodymium (Pr) nitrate as precursors with citric acid as a chelating agent, and successfully synthesized mesoporous Pr-doped CeO2 materials via the sol–gel method.71 The obtained material demonstrates a high specific surface area, with Pr elements exhibiting atomic-level uniform dispersion within the CeO2 lattice. Tang et al. used Ce(NO3)3·6H2O as the cerium source and PVP as the spinning aid to prepare nanofiber precursors using a sol–gel method combined with electrospinning technology. After calcination at 400 °C,72 fluorite-type CeO2 nanofibers with a diameter ranging from 80 to 120 nm and a porous structure were obtained (Fig. 5b and c). Nevertheless, the development of sol–gel technology faces constraints from its time-consuming synthesis protocols and the high cost of precursors (e.g., certain metal alkoxide).
2.1.7. Microemulsions method.
Microemulsions are homogeneous and thermodynamically stable dispersion systems composed of water, oil, and surfactants.73 They are classified into two types: O/W (oil-in-water) and W/O (water-in-oil). In the synthesis of rare earth oxide nanoparticles, the W/O system is commonly used. Rare earth ion precursors react, nucleate, and grow controllably within the droplets, and the resulting nanoparticles are obtained through thermal treatment.49 The rare earth oxides prepared by this method exhibit excellent particle size uniformity and surface properties. For example, Małecka et al. employed a W/O microemulsion system,74 where the organic phase consisted of cyclohexane/1-pentanol, and the aqueous phase was an aqueous solution containing rare earth salts and ammonia. Using this system, they synthesized small CeO2 particles (4–5 nm) with a fluorite structure (Fig. 5d), which exhibit the two most common shapes: an octahedron defined by eight crystal faces, and a truncated octahedron defined by eight (111) and six (100) crystal faces. Additionally, by introducing other rare earth salt precursors into the system, CeLnOx (Ln = Pr, Tb, Lu) mixed oxide materials of similar size were simultaneously prepared (Fig. 5e and f). Zhang et al. conducted an analysis of how annealing temperature governs the crystallinity and morphology of CeO2 nanocrystals synthesized through a microemulsion approach. At 350 °C, the sample predominantly exhibited aggregated spherical particles around 65 nm in size. When the temperature was elevated to 600 °C, well-defined CeO2 nanocrystals with diameters of 6–8 nm were obtained. Furthermore, an increase in temperature induced a phase transformation from triclinic to cubic symmetry.75 Furthermore, Hadi et al. demonstrated that the synthesis pathway markedly influences the structural characteristics of CeO2 nanocrystals. The microemulsion method yielded significantly smaller particles (5.2 nm) than the mechanochemical process (6.9 nm), confirming the dependence of crystallite size on preparation strategy.76
2.2. Preparation of rare earth-modified zeolites
Rare earth-modified zeolites possess characteristics such as large specific surface area, stable zeolite structure, and regular pore structure, making them widely applied in processes such as catalytic cracking, hydrocracking, alkylation, and hydrodeoxygenation.25,77,78 Their preparation primarily involves strategies such as ion exchange, impregnation, and hydrothermal synthesis to introduce rare earth elements (La, Ce, Pr, etc.) into the zeolite framework or channels.79,80 Among these, the ion exchange method is the most widely used preparation technique due to its high operational efficiency and significant modification effects. Compared to the impregnation method, the ion exchange method enables high dispersion of rare earth ions in zeolite channels and on surfaces, and there is a strong electrostatic interaction between rare earth ions and the zeolite framework, resulting in a strong binding force and good stability.81 This section will systematically elaborate on the principles and parameters affecting this method. Thermodynamic studies indicate that the ion exchange process first dehydrates hydrated rare earth ions ([Re(H2O)n3+]), followed by the entry of Re3+ into the β-cage structure of the zeolite unit cell. Since the ion exchange process is an endothermic reaction, an increase in system temperature facilitates ion exchange. It is important to note that achieving deep modification often requires multi-step exchange operations.82 The structural stability of rare earth-modified zeolites prepared via ion exchange is closely related to the type of rare earth elements, modification temperature, pH value, and rare earth content. Hu et al. systematically investigated the effects of rare earth element type (Sm, La, and Gd),83 modification temperature (30 °C, 50 °C, 70 °C, and 90 °C), pH value (3, 4, 5, and 6), and rare earth loading (1%, 2%, 3%, and 4%) on zeolite structural stability. The results demonstrated that the Sm/HZSM-5 zeolite prepared with a 2 wt% Sm loading at an ion-exchange temperature of 70 °C, pH 3, and an exchange duration of 2 h exhibited the optimal structural stability.
3.
In situ characterization techniques
Although ex situ characterization methods have been extensively employed to investigate the static properties of rare earth-based catalysts, significant limitations remain when relying solely on pre- or post-reaction ex situ analyses for probing their structure–activity relationships—particularly in CO2 hydrogenation reactions. This constraint originates from a critical reality: most rare earth-based catalysts undergo substantial dynamic structural evolution under actual reaction conditions. For instance, in CeO2-supported metal catalysts, dynamic transformations may occur in metal site size (e.g., transitions from atomically dispersed states to clusters/nanoparticles), oxygen vacancy concentration, and electronic structure (e.g., interfacial charge transfer at metal–support interfaces). Crucially, the genuine active sites of certain rare earth-based catalysts are generated only during the catalytic operation. Such real-time structural dynamics provide crucial insights that ex situ techniques fail to capture, thus preventing effective monitoring of structural evolution, active-site behaviours, and reaction pathways in operating rare earth-based catalysts, which hinders the rational design of highly efficient and stable systems. To fundamentally understand the dynamic structural evolution and catalytic mechanisms of rare earth-based catalysts in realistic environments, this review systematically summarizes key in situ characterization techniques—in situ transmission electron microscopy (TEM), in situ infrared (IR) spectroscopy, in situ X-ray absorption spectroscopy (XAS), and in situ X-ray photoelectron spectroscopy (XPS)—providing a robust theoretical foundation for designing high-performance rare earth catalyst systems.
3.1.
In situ XPS
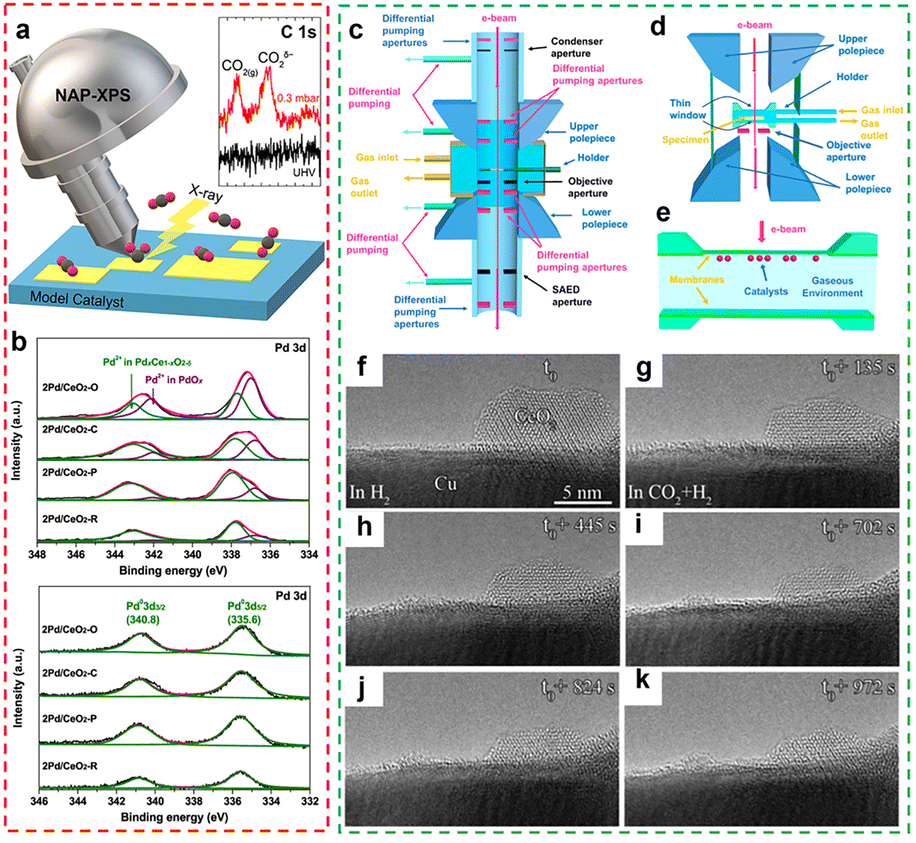
XPS is a highly sensitive surface analysis technique (with a detection depth of approximately <10 nm) that can characterize the elemental composition, chemical state, and electronic interactions of material surfaces.84,85 For rare earth-based catalysts, XPS is particularly important, as the incorporation of rare earth elements significantly modifies interatomic binding energy or generates new binding-energy features within the system.86 By analysing the shifts or emergence of binding energy peaks in XPS spectra, the valence states of rare earth elements can be determined, and their electronic interactions with neighbouring atoms (such as supports or active components), including charge transfer and bonding characteristics, can be inferred. In situ XPS further advances this capability by enabling X-ray excitation of core-level or valence electrons under reaction-relevant conditions (e.g., specific temperature, pressure, and atmosphere), while detecting the emitted photoelectrons in real time.87 This allows researchers to monitor the dynamic evolution of the surface chemical states of rare earth-based catalysts (Fig. 6a). Compared to traditional XPS, it significantly reduces surface reconstruction or contamination caused by environmental changes (such as transferring from the reaction chamber to the analysis chamber), thereby enabling more accurate and realistic capture of the instantaneous changes on the catalyst surface during the reaction process. For example, Jiang et al. employed in situ XPS to investigate the changes in the chemical state of Pd species after hydrogen reduction. As shown in Fig. 6b, all calcined samples before reduction showed two types of Pd 3d5/2 peaks at 337.8 eV and 336.8 eV,88 attributed to Pd2+ in PdxCe1−xO2−δ solid solution and PdO particles, respectively. For the 2Pd/CeO2-R and 2Pd/CeO2-P samples, the PdxCe1−xO2−δ solid solution dominates the ceria surface, and the (–Pd2+–O2−–Ce4+–) linkage suggests that the Pd ions present an atomically dispersed state in the ceria matrix, whereas the remaining two samples show PdO particles distributed on the surface of the support. In situ XPS characterization after reduction revealed that the PdO and PdxCe1−xO2−δ phases on the catalyst surface were converted into metallic Pd. Meanwhile, a noticeable decline in the surface Pd content and the Pd/Ce atomic ratio was observed, suggesting partial aggregation of Pd species during the reduction treatment. To date, various quasi in situ and in situ XPS techniques have been developed, providing powerful characterization tools for revealing and real-time tracking of the evolution of elemental valence states (particularly those of rare earth elements) and key electron-transfer behaviours in rare earth-based catalysts under near-real reaction conditions.
 |
| | Fig. 6 (a) A diagram of in situ NAP-XPS. Reprinted with permission from ref. 87. Copyright (2022) American Chemical Society. (b) Pd 3d XPS curves of the calcined Pd/CeO2 catalysts and in situ reduced Pd/CeO2 catalysts. Reprinted with permission from ref. 88. Copyright (2020) American Chemical Society. (c) Architecture of differentially pumping environmental TEM (E-TEM). (d) Windowed gas cell holder compatible with TEM. (e) A cross-sectional drawing of the holder tip. Reprinted with permission from ref. 90. Copyright (2023) American Chemical Society. (f)–(k) E-TEM images for a 5% CeOx/Cu catalyst after switching from (f) reducing to (g)–(k) CO2 hydrogenation conditions (5 mTorr CO2, 5 mTorr H2, 250 °C). All of the images were collected in the same spot of the sample. Reprinted with permission from ref. 93. Copyright (2023) American Chemical Society. | |
3.2.
In situ TEM
In recent years, the development of in situ TEM technology, integrated with an environmental control system, has enabled researchers to directly observe the structural dynamics of rare earth-based catalytic materials in real time under reaction-relevant conditions, with high spatial resolution (up to angstrom level) and elemental analysis capability (Fig. 6c–e).89–92In situ TEM can reveal the dynamic relationship between environmental conditions and the structure of rare earth-based catalysts, providing deeper insights into their structural reconstruction and activity changes during catalytic reactions. For example, Zhang et al. used in situ transmission electron microscopy to investigate the effect of temperature on Pd@CeO2 catalysts under a 150 torr O2 atmosphere.91 When the temperature was below 500 °C, the catalyst's nanostructure remained stable, with typical 2–3 nm particle clusters (mostly ceria nanocrystals). However, when the temperature reaches 500 °C, the catalyst's structure begins to transform, with atoms on smaller and more isolated particles becoming mobile and starting to leave the clusters. At 650 °C, the temporal evolution of the CeO2 nanostructure is clearly observable, ultimately forming a truncated octahedron surrounded by eight (111) planes (the lowest surface energy planes) and six (100) planes. Moncada et al. utilized in situ TEM technology to systematically investigate the effects of gas conditions on the structure of the 5% CeOx/CuO catalyst.93 Experimental observations show that the catalyst exhibits significant morphological changes under CO2 hydrogenation conditions compared to a pure H2 environment or a pristine CeOx/CuO system, confirming the decisive influence of the chemical environment on the surface structure of the catalyst. In this process, amorphous layers of Ce2O3 were uniformly dispersed on the copper substrate. The researchers found that these morphological changes were not caused by electron beam irradiation. The flattening and redispersion process of the ceria overlayer can be observed when trace amounts of CO2 are introduced in an H2-rich environment (Fig. 6f–k). These experimental results confirm that the observed morphological changes originate from a near real catalytic reaction process. This finding provides evidence for understanding the surface remodelling mechanism of oxide/metal catalysts in CO2 hydrogenation reactions.
3.3.
In situ IR spectroscopy
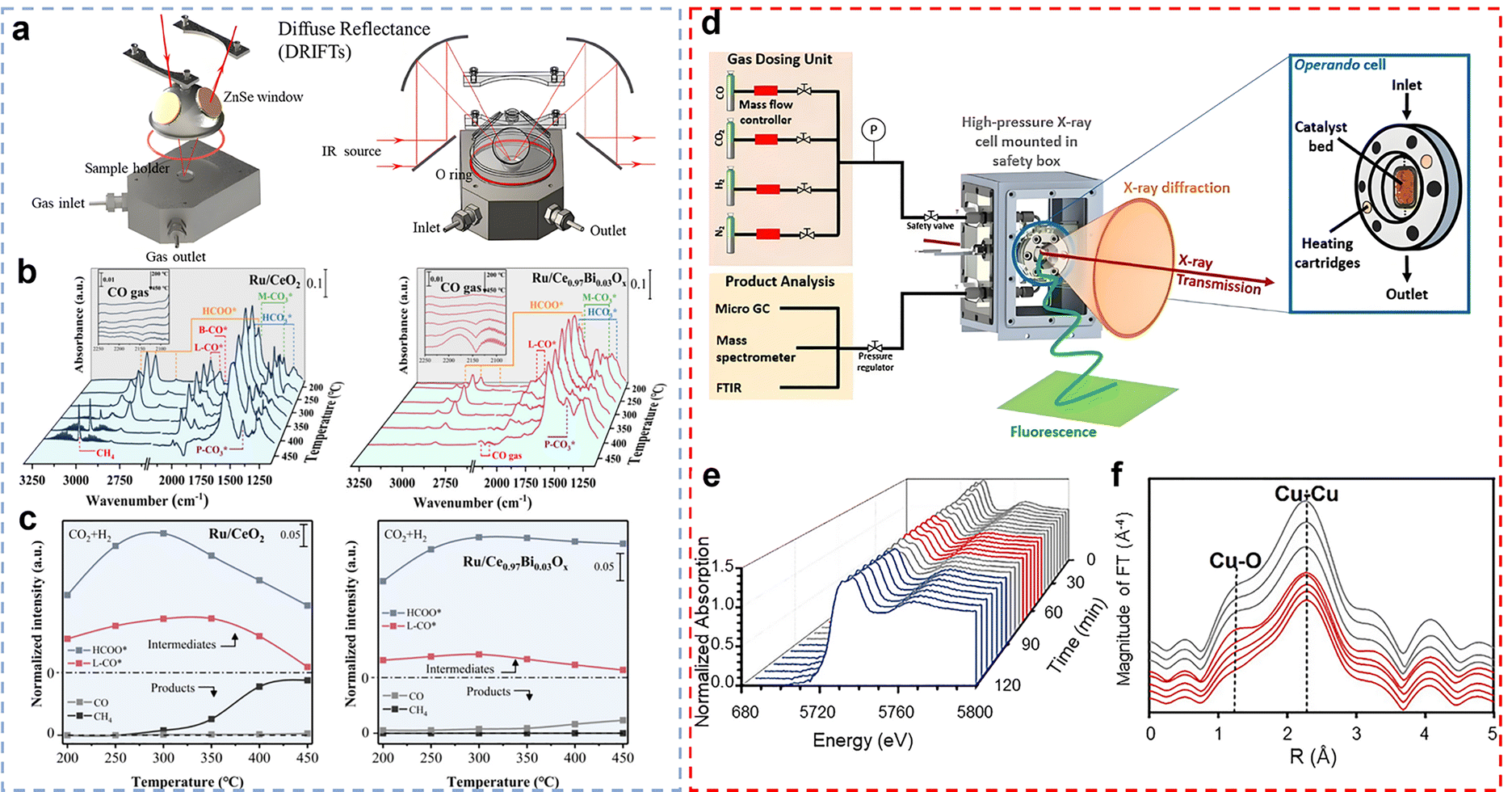
In situ IR spectroscopy is an advanced dynamic characterization technique, particularly suitable for real-time monitoring of the surface chemical structure of catalytic materials and the evolution of reactants under actual CO2 hydrogenation reaction conditions.94 This technique monitors variations in the characteristic vibrational absorption bands of molecular or surface species (such as stretching and bending vibrations of chemical bonds) under changes in temperature, pressure, and reaction atmosphere, enabling sensitive capture of dynamic information on reactant structural evolution, formation of key intermediates, and chemical bond breaking/formation.95 Based on measurement modes, in situ IR spectroscopy can be categorized into transmission, diffuse reflectance (DRIFTS) (Fig. 7a), attenuated total reflection (ATR), and reflection–absorption types.94,96 Among these, DRIFTS has become an indispensable technical tool for studying the mechanism of CO2 hydrogenation reactions due to its high-efficiency detection capability for powder catalysts and surface-adsorbed species. Compared to traditional infrared spectroscopy, in situ IR spectroscopy, by integrating an in situ reaction cell that precisely controls temperature and pressure, can directly observe the adsorption and activation of reactants (such as CO2), the formation and transformation of key intermediates (such as HCOO* and H3CO*), and the evolution of final products (such as CO, CH4, and CH3OH) during the CO2 hydrogenation process. This unique ability to track reaction pathways in real-time and dynamically at the molecular level makes in situ IR spectroscopy (particularly DRIFTS) a powerful tool for revealing the microscopic mechanisms of CO2 hydrogenation catalytic reactions, the chemical behaviour of active sites on catalyst surfaces, and structure–activity relationships, providing crucial experimental evidence for optimizing catalyst design and reaction processes. Xie et al. monitored the intermediate product changes in the CO2 hydrogenation reaction catalysed by Ru/CeO2 and Ru/Ce0.97Bi0.03Ox by the in situ DRIFTS technique(Fig. 7b and c).97 For Ru/CeO2, a gradual increase in the intensity of HCOO* was detected as the temperature was increased from 200 °C to 300 °C. Subsequently, HCOO* species were consumed by H2 and converted to CH4* at 350 °C, while L-CO* decreased, resulting in a relatively high intensity of CH4* and a weakening of the CO gas signal. The band intensities of M-CO3*, bridged-CO*, and polydentate carbonate did not change much at 350 °C, suggesting that their contribution was small, and thus the route for the generation of CH4 involves the carbonyl and formate route. For Ru/Ce0.97Bi0.03Ox, significant CO gas bands were detected but no CH4* was observed, consistent with its CO2 hydrogenation performance. Further analysis showed that L-CO* and HCOO* on Ru/CeO2 were converted to CH4 above 300 °C, while Ru/Ce0.97Bi0.03Ox had difficulty in hydrogenating HCOO* and L-CO* to CH4, probably due to the weak interaction of Ru with CO*.
 |
| | Fig. 7 (a) Illustration of infrared accessories and light pathway of diffuse reflectance infrared Fourier transform spectroscopy (DRIFTs). Reprinted with permission from ref. 94. Copyright (2019) Springer Nature. (b) In situ DRIFTS collected at different temperatures on the Ru/CeO2 and Ru/Ce0.97Bi0.03Ox catalysts. (c) Normalized intensities for key adsorption species on Ru/CeO2 and Ru/Ce0.97Bi0.03Ox catalysts. Reprinted with permission from ref. 97. Copyright (2024) American Chemical Society. (d) Scheme of the operando high-pressure X-ray cell used for methanol and Fischer–Tropsch syntheses at the synchrotron radiation source. The spectroscopic cell was designed in a way that it is applicable for X-ray absorption spectroscopy (XAS). Reprinted with permission from ref. 99. Copyright (2021) Wiley-VCH. (e) XANES spectra at the Ce L3-edge during the complete thermal treatment. Red, blue, and gray spectra correspond to isothermal reduction in CO, isothermal CO oxidation, and heating/cooling steps, respectively. (f) FT-EXAFS measured at the Cu K-edge during the reduction treatment from 400 °C to 250 °C. Reprinted with permission from ref. 105. Copyright (2022) Wiley-VCH. | |
In addition, in situ infrared spectroscopy, particularly CO-DRIFTS, serves as an effective tool for probing the dispersion state and electronic properties of metal particles in supported catalysts. By analysing the positions and intensities of CO adsorption bands, the adsorption configurations of CO molecules on metal surfaces can be identified, thereby providing indirect insight into the dispersion and electronic environment of the metal species. Single-site Rh species and Rh aggregates were identified by the CO-DRIFTS technique by Lee et al.98 According to CO-DRIFTS measurements, CO interacts with Rh centers via linear, bridge, and gem-dicarbonyl adsorption modes. The absorption bands at 2084–2088 cm−1 and 2012–2016 cm−1 correspond to gem-dicarbonyl species located on isolated Rh atoms, which constitute the main adsorption configuration in Rh/CeO2–Al2O3 catalysts. Notably, the Rh/15-CeO2–Al2O3 catalysts, on the other hand, showed Rh bridge peaks (ca. 1910 cm−1) and linear peaks (ca. 2046 cm−1) indicating the formation of Rh nanoparticles. With its unique advantages of high sensitivity, non-destructiveness and real-time monitoring, in situ IR spectroscopy has been widely used in the fields of catalytic chemistry, energy materials and environmental science. This technology not only provides an important basis for the in-depth understanding of the structure-property relationship of materials, but also provides a key technical support for the optimal design of reaction processes.
3.4.
In situ XAS
Given the rich energy level structure, multiple electron shells, and strong shielding of inner-shell electrons by outer-shell electrons characteristic of rare earth elements, X-ray absorption spectroscopy (XAS) plays a crucial role in the investigation of rare earth-based catalytic materials. This technique analyses variations in the absorption coefficient near and above the absorption edge, providing insights into the oxidation state, electronic structure, and local atomic environment of the absorbing element. In situ XAS offers unique advantages in catalyst characterization—not only providing key information on the geometric configuration and electronic structure of active sites but also enabling in situ monitoring of changes in active sites and structure of catalytic materials during reactions (Fig. 7d).99In situ XAS spectrum primarily consists of two regions: X-ray absorption near edge structure (XANES) enables real-time tracking of the valence state evolution of active components during the catalytic process and their electronic interactions with the support, thereby revealing the dynamic evolution of elemental valence and electronic structures; extended X-ray absorption fine structure (EXAFS), on the other hand, quantitatively fits spectral data to obtain structural parameters such as the types of coordinating atoms, coordination numbers, and bond lengths, dynamically monitoring the structural evolution of catalytic sites under real reaction conditions (e.g., temperature, pressure, and gas composition).100–103 Most importantly, in situ XAS can accurately capture changes in the electronic structure of rare earth catalysts during the reaction process (especially the evolution of the valence state of key rare earth elements), providing direct evidence to reveal the structure–property relationship between microstructure and macroscopic catalytic performance. For example, Yan et al.'s research clearly demonstrates this advantage: they used in situ XAS technology to track the dynamic evolution of the valence state (Ce3+/Ce4+) of the key rare earth component Ce under pressurized CO2 hydrogenation conditions in real time. Through linear combination fitting (LCF) analysis,104 the molar fraction changes of Ce3+ in the catalyst were quantitatively revealed: the air-calcined CuO/CeO2 precursor contained 0% Ce3+ (initially dominated by Ce4+), while after hydrogenation at 250 °C and 10 bar CO2, the Ce3+ content in Cu/CeO2 significantly increased to 15%; in contrast, the air-calcined CuO/CeW0.25Ox precursor already contained 49% Ce3+, and under the same reaction conditions, its Ce3+ content further increased to 59%. These in situ observations enabled the monitoring of the valence state of the rare earth Ce under actual reaction conditions. Moreover, in situ XAS can further monitor the formation and evolution of oxygen vacancies (VO) in rare earth-based catalysts:105 as shown in Fig. 7e, the increase in Ce3+ concentration in the Ce L3 edge XANES spectrum under CO atmosphere directly indicates the formation of VO; simultaneously, in situ EXAFS at the Cu K edge (Fig. 7f) captures an increase in Cu–O coordination number during cooling, revealing that part of Cu is reoxidized by oxygen from the CeO2 lattice—this is the key structural evidence for the existence of VO. It is worth emphasizing that XAS can detect sub-surface oxygen vacancies and bulk redox behaviour, which are difficult to access using surface-sensitive techniques such as XPS or Raman spectroscopy, thereby providing more comprehensive and accurate structural dynamics information of rare earth-based catalysts. These studies collectively demonstrate that in situ XAS (combining XANES and EXAFS) is an indispensable tool for elucidating the valence states, coordination structures, and oxygen vacancy dynamics of rare earth catalysts under real reaction conditions.
4. Application of rare earth-based catalysts to CO2 hydrogenation
The reaction of renewable green H2 with CO2 to produce high-value chemicals or fuels is an effective strategy for carbon reduction and CO2 utilization. The selectivity of hydrogenation products is influenced by a variety of factors, including reaction temperature, pressure, and characteristics like active metal type, support, and additives. In this section, we will systematically summarize the applications of rare earth-based catalysts in CO2 hydrogenation reactions, including the RWGS reaction and CO2 hydrogenation to methane, methanol, ethanol, and olefins and aromatics. By combining the latest research advances, this chapter will reveal the structure–activity relationships of rare earth-based catalysts in these reactions and offer theoretical guidance for catalyst design and optimization.
4.1. RWGS reaction
The RWGS reaction is a pivotal catalytic process that converts CO2 and H2 into CO and H2O, following the chemical equation CO2 + H2 ↔ CO + H2O (ΔH298K = +41.2 kJ mol−1). As an endothermic reaction,106 RWGS reaction favors higher CO2 conversion and CO selectivity at elevated temperatures (typically in the range of 400–700 °C),107,108 though such conditions also introduce challenges like active metal sintering and coke depositions. The core value of the RWGS reaction lies in transforming CO2 into CO,108 which—when combined with H2 to form syngas—serves as a versatile industrial feedstock for Fischer–Tropsch synthesis (FTS), methanol production, and other high-value chemical processes,109 thereby bridging carbon recycling into the circular economy. Current research focuses on developing efficient and stable catalysts, optimizing reaction conditions to suppress side reactions (e.g., CO2 methanation), and exploring low-cost, high-performance non-noble metal catalytic systems. Among many catalysts, rare earth-based catalysts are a common class of catalytic materials for the RWGS reaction due to their excellent redox properties.110
4.1.1. Cerium-based catalysts for the RWGS reaction.
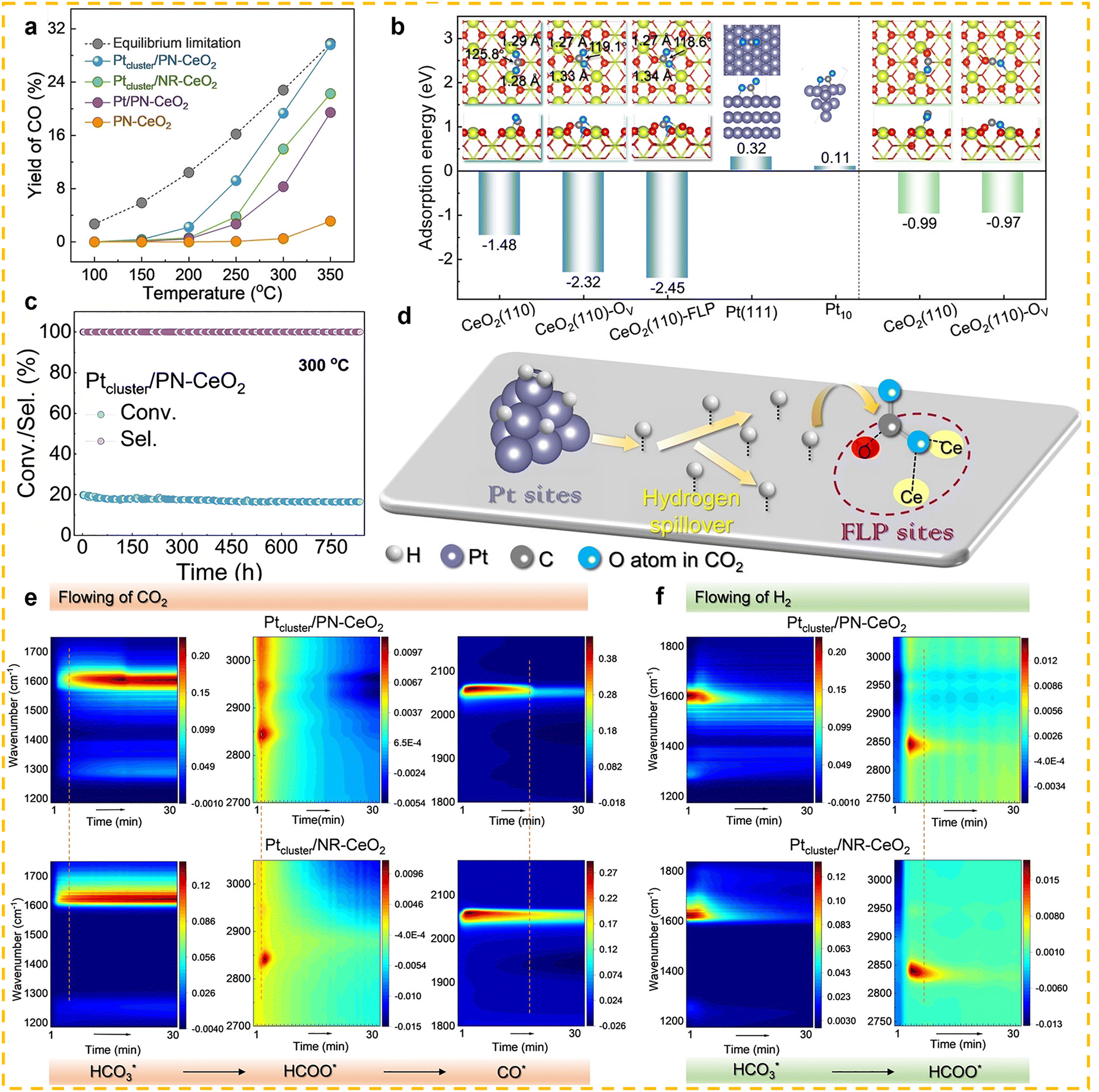
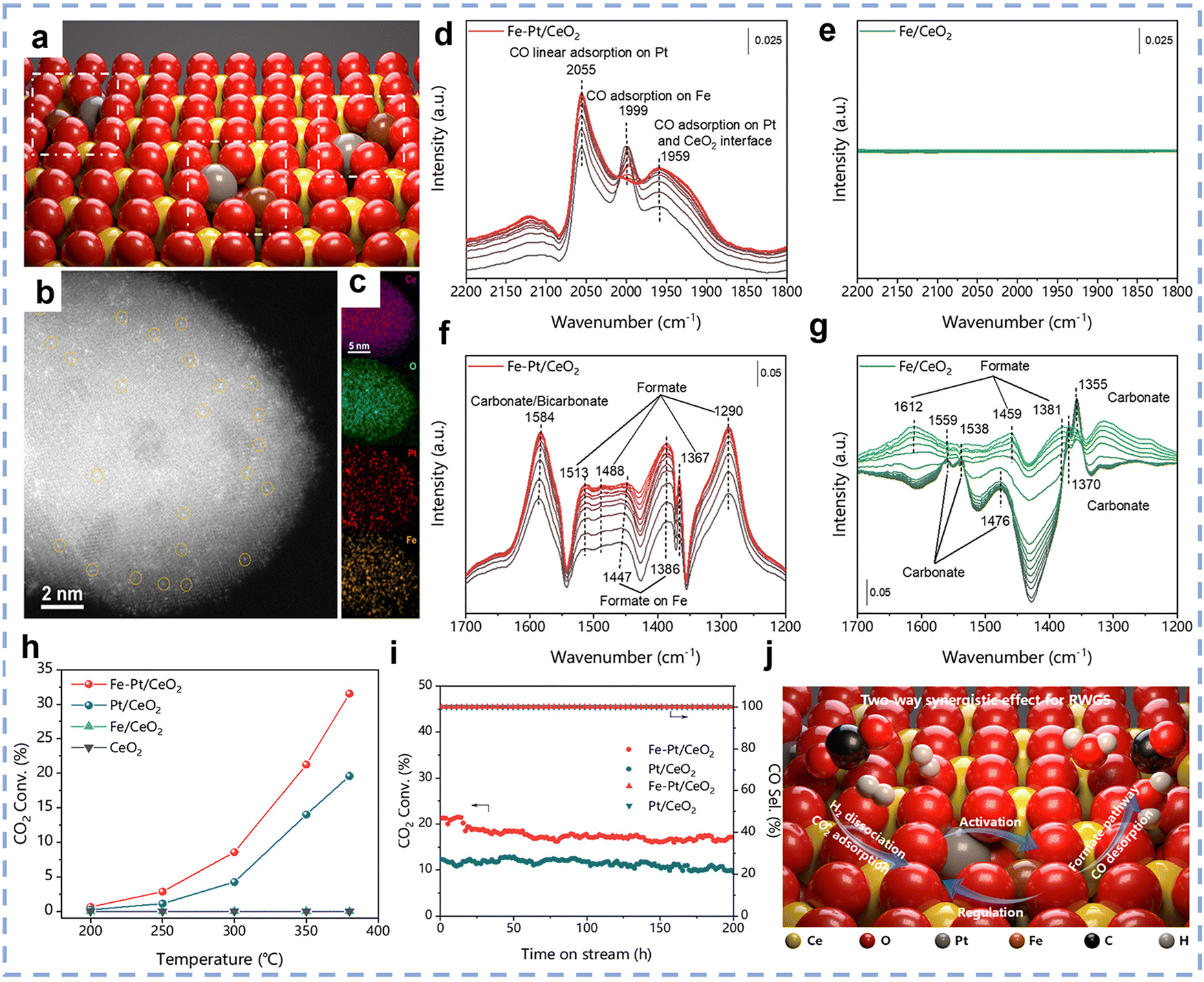
Ce-based catalysts are one of the most commonly used catalysts in the RWGS reaction. Among them, CeO2 exhibits outstanding activity in the RWGS reaction owing to its exceptional redox flexibility. This behavior contrasts sharply with conventional oxide supports such as Al2O3 and SiO2, which generally show limited redox capability in heterogeneous catalysis. Cao et al. systematically explored the dynamic mechanism of oxygen vacancy in the CO2 hydrogenation reaction using CeO2 nanorods with tunable surface oxygen vacancy concentration as a model catalyst.111 By adjusting the concentration of oxygen vacancies on the surface of CeO2, it was found that the catalytic CO2 hydrogenation to generate CO showed a volcano-shaped activity curve, indicating that a moderate concentration of oxygen vacancies can effectively sustain H2 activation and promote CO2 dissociation, thus improving the reaction efficiency. Compared to pure CeO2 catalysts, the catalytic performance in the RWGS reaction is significantly enhanced when metal is loaded onto CeO2 as a support. Zhao et al. synthesized three Pt/CeO2 catalysts with different dispersions of Pt species.112 The results showed that the performance of the metal-loaded catalysts was significantly improved compared to pure CeO2. Additionally, the authors systematically investigated the effect of the dispersion state of Pt species on CO selectivity in the RWGS reaction. The study demonstrated that atomically dispersed Pt species could achieve over 98% CO selectivity within the temperature range of 200–450 °C and remain stable during the reaction, significantly outperforming Pt nanoparticle catalysts. Through CO-TPD and in situ FTIR experiments, it was found that atomically dispersed Pt species exhibited weaker CO adsorption, which suppressed excessive hydrogenation and prevented CH4 formation. In contrast, larger Pt clusters or particles exhibited stronger CO adsorption, leading to a decrease in CO selectivity. This finding reveals the critical role of atomically dispersed Pt species in enhancing CO selectivity in the RWGS reaction and provides new insights for designing Pt-based catalysts with excellent CO selectivity. Li et al. developed an interface-independent, efficient low-temperature RWGS reaction catalyst by constructing platinum (Pt) clusters and frustrated Lewis pair (FLP) dual active sites on the surface of porous CeO2 nanorods (PN–CeO2).113 The study found that Pt clusters, with their high electron density, efficiently dissociate H2 and facilitate the overflow of hydrogen species to the support. Meanwhile, the FLP sites, with their unique spatial/electronic structure, strongly adsorb and activate CO2, while weakening the adsorption of CO to promote its desorption, thus suppressing methane formation (Fig. 8b and d). Under 350 °C conditions, the catalyst exhibited a CO2 conversion rate of 30.1%, CO selectivity of 98.5%, and a CO yield of 29.6% (close to the thermodynamic equilibrium value of 29.8%) (Fig. 8a). The turnover frequency (TOF) reached as high as 8720 h−1, with no loss of activity over 840 h of continuous operation (Fig. 8c). The in situ DRIFTS experiments not only confirmed that CO2 hydrogenation on CeO2 follows the formate pathway, i.e., “HCO3* → HCOO* → CO*”, but also revealed the enhancement mechanism of the FLP sites at the molecular level (Fig. 8e). Specifically, compared with conventional catalysts, the FLP sites on Ptcluster/PN–CeO2 significantly accelerated the conversion rate of CO2 to formate species. Simultaneously, in a hydrogenation environment, the intermediates on its surface were more rapidly consumed by the spilled-over H* species (Fig. 8f). Collectively, these findings demonstrate the superior CO2 hydrogenation capability of Ptcluster/PN–CeO2. Compared to single-metal site rare earth-based catalysts, dual-metal site rare earth-based catalytic materials significantly enhance catalytic activity due to higher metal loading and versatile active sites. A dual-metal site typically consists of one metal as the active site (active metal) and another as the auxiliary metal. In this case, the auxiliary metal influences the surface electronic structure of the active metal through electronic effects or directly bonds with reactants, thereby affecting the reaction intermediates. This aids in CO2 activation and the formation/desorption of intermediate products, resulting in faster reaction kinetics and improved catalytic efficiency. Wang et al. investigated the design of a hetero-dual-site catalyst, Fe–Pt/CeO2, to enhance the catalytic performance of the RWGS reaction.31 The Fe atoms are controllably anchored on the CeO2 surface in proximity to Pt atoms (Fig. 9a–c), resulting in the Fe–Pt/CeO2 catalyst showing a significant CO2 conversion rate (21.3%) at 350 °C, which is 1.5 times higher than Pt/CeO2, with CO selectivity approaching 100% (Fig. 9h). In situ DRIFTS further elucidated the reaction mechanism: three strong characteristic peaks at 1999, 1447, and 1386 cm−1 were observed in the Fe–Pt/CeO2 sample, which can be attributed to the adsorption of CO and formate species on Fe sites, indicating the direct involvement of Fe atoms in the reaction (Fig. 9d and f). In contrast, in situ DRIFTS analysis of the single-site Fe/CeO2 catalyst (Fig. 9e and g) revealed no obvious CO-related peaks, suggesting that the catalytic activity of Fe atoms in Fe–Pt/CeO2 is not intrinsic, but mainly originates from their synergistic interaction with Pt atoms. Further studies demonstrated that Fe atoms not only promote CO desorption by regulating the charge density of Pt atoms but can also be activated by the excess active hydrogen species generated on Pt, thereby giving rise to the so-called “two-way synergistic effect” (Fig. 9j). In addition, long-term durability tests confirmed the excellent stability of Fe–Pt/CeO2, which retained more than 80% of its initial activity even after 200 h of continuous operation at 350 °C (Fig. 9i). Compared to precious metal catalysts, non-precious metal catalysts have attracted increasing attention from researchers due to their low cost, abundant raw materials, easy availability, and lower industrial application costs. These advantages make non-precious metal catalysts highly promising for a wide range of applications in fields such as energy, environmental protection, and the chemical industry. Additionally, non-precious metal catalysts generally offer greater sustainability by reducing reliance on rare and expensive metals. Therefore, the development of efficient and stable non-precious metal catalysts has become an important direction in current catalytic research. Liu et al. developed Cu/CeO2 catalysts and investigates the unique catalytic mechanism of the partially sintered structure in the high-temperature RWGS reaction.30 Under extreme conditions (600 °C and high space velocity of 400000 mL gcat−1 h−1), the ceria nanorod support undergoes partial sintering. However, due to the strong interaction between Cu and Ce, Cu stabilizes on the catalyst surface in the form of two-dimensional layered clusters and three-dimensional hemispherical clusters, forming abundant active metal sites. This structure imparts the catalyst with exceptionally high activity, exhibiting superior performance compared to most non-precious and precious metal catalysts in high-temperature reducing atmospheres, while maintaining outstanding stability for over 240 h. The study suggests that the Cu–Ce interface stability and the dynamic cycling of oxygen vacancies are the main reasons for the catalyst's high efficiency and long-term stability.
 |
| | Fig. 8 (a) CO yields at various temperatures with a WHSV of 12000 mL gcat−1 h−1. (b) The adsorption behaviors of CO2 and CO on the CeO2(110) surface with various active sites. The red and yellow balls represent the O and Ce atoms of CeO2, respectively. The purple gray and blue balls represent the C and O atoms of CO2, respectively. (c) Catalytic stability of Ptcluster/PNCeO2 with a WHSV of 12000 mL gcat−1 h−1 at 300 °C. (d) The proposed dual-active sites of Pt and FLP for the RWGS reaction. In situ DRIFTS spectra of Ptcluster/PN–CeO2 and Ptcluster/NR–CeO2 under flowing (e) CO2 and (f) H2. Both Ptcluster/PN–CeO2 and Ptcluster/NR–CeO2 catalysts were pretreated by a flow of 50% H2/Ar for 1h. After removal of the free H2 by a flow of Ar in the catalytic environments, a flow of 50% CO2/Ar was introduced and the DRIFTS signals were collected at 350 °C every 20 s for 30 min (e). Subsequently, the 50% CO2/Ar gas was switched to a flow of 50% H2/Ar, which was kept for another 30 min to collect the DRIFTS signal every 20 s (f). Reprinted with permission from ref. 113. Copyright (2023) Wiley-VCH. | |
 |
| | Fig. 9 (a) Schematic illustration of the Fe–Pt/CeO2. The yellow, red, gray, and brown balls represent Ce, O, Pt, and Fe atoms, respectively. (b) HAADF-STEM image of Fe–Pt/CeO2 with the Pt SAs highlighted by the orange circles. (c) EDX-mapping images of Fe–Pt/CeO2. (d)−(g) In situ DRIFT spectra of the RWGS reaction over Fe–Pt/CeO2 (d) and (f), and Fe/CeO2 (e) and (g). (h) Temperature-programmed RWGS reaction over Fe–Pt/CeO2, Pt/CeO2, Fe/CeO2, and pure CeO2. Reaction conditions: 30 mg of the catalyst; CO2/H2/Ar = 24/72/4 (v/v/v); total feed gas flow rate is 100 mL min−1; ordinary pressure. (i) Long-term stability test at 350 °C. (j) Schematic illustration of the two-way synergistic effect over Fe–Pt/CeO2 catalyst. Reprinted with permission from ref. 31. Copyright (2023) American Chemical Society. | |
In the RWGS reaction, CeO2 is not only used as a catalyst support but also commonly serves as a promoter. They significantly enhance the catalyst's activity by adjusting the surface properties of the catalyst, optimizing interactions with substrate molecules, or forming defect sites on the surface. Xu et al. integrated the CeO2 promoter onto Cu2O nanocubes through a controlled surface deposition method to prepare the CuOx–5CeO2 catalyst, which was applied in the RWGS reaction.114 The study showed that the catalyst exhibited excellent catalytic performance under 380 °C reaction conditions, with a CO2 conversion rate of 31%, a CO selectivity of 98%, and the ability to maintain stable activity over 50 h.
Beyond its excellent thermal catalytic properties, CeO2 has also been recognized as a versatile platform for photothermal RWGS catalysis. When combined with plasmonic metals such as Au or with photoactive transition metals like Fe, cerium-based systems exhibit remarkable potential. Lu et al. developed a photothermal catalyst (Au/CeO2) composed of Au nanoparticles supported on CeO2 nanorods,115 which achieved a CO2 conversion rate of 40% under photothermal conditions at 400 °C (compared to 3.2% in the thermal process) with near 100% CO selectivity, and the CO production rate under photothermal conditions reached 12.5 times that of the thermal process. This shows that photothermal catalysis has significantly improved performance compared with traditional thermal catalysis. Based on the success of precious metal catalysts, the researchers further explored the potential of non-precious metal systems. Zhao et al. studied the performance of FeO–CeO2 nanocomposite catalysts in photothermal CO2 hydrogenation reactions.116 The study showed that the FeCe-300 catalyst achieved a CO2 conversion rate of 43.63%, CO selectivity of 99.87%, and a CO yield of 19.61 mmol h−1 gcat−1 under Xe lamp irradiation, with no performance degradation observed during 50 h of continuous operation. The FeCe-300 catalyst was composed of FeO and CeO2 nanoparticles, and the FeO effectively promoted CO formation. As the reduction temperature increased, the formation of metallic Fe0 in the catalyst promoted the Sabatier reaction, which in turn reduced the selectivity for CO.
4.1.2. Non-cerium-based catalysts for the RWGS reaction.
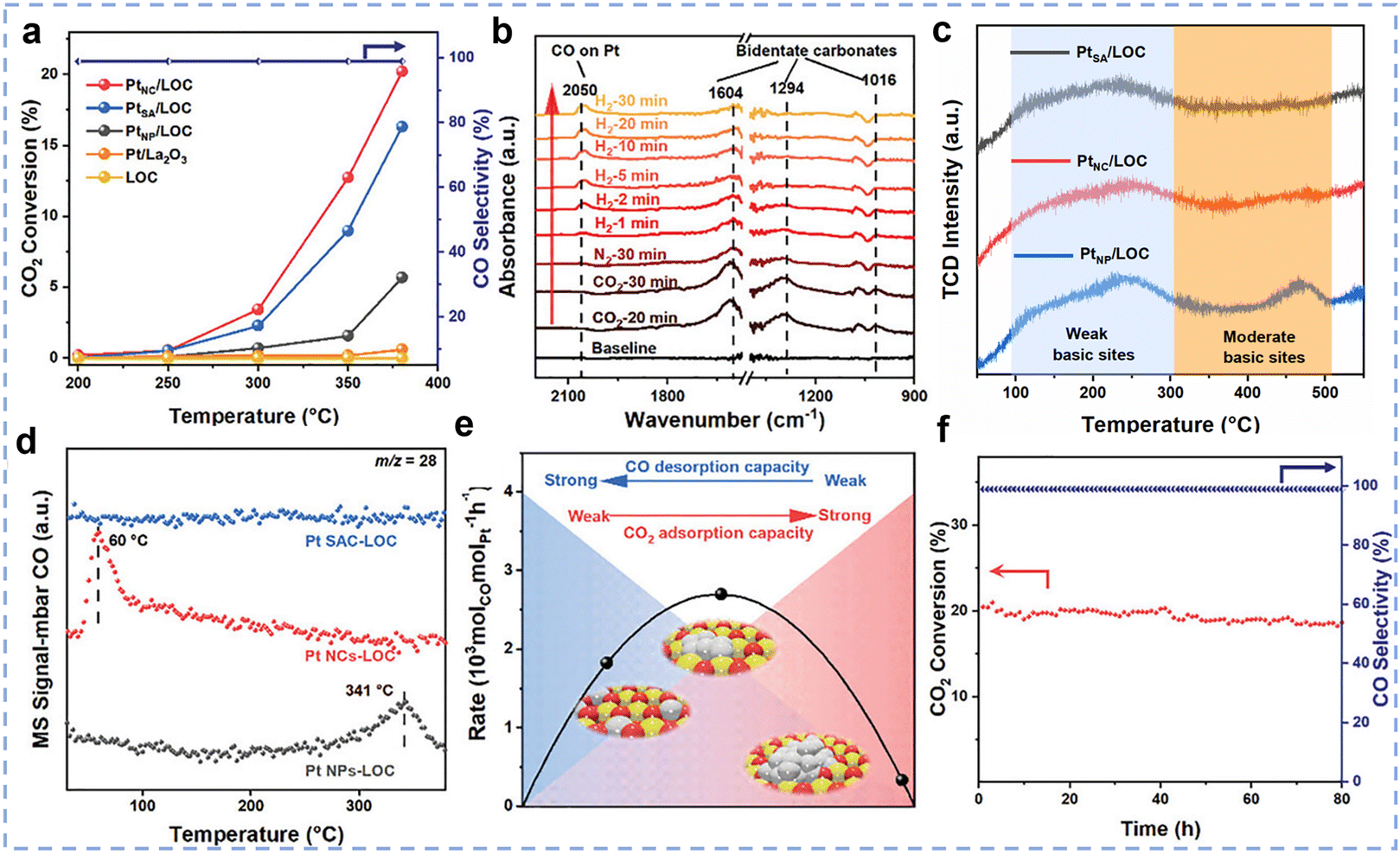
While cerium-based catalysts are widely used due to their excellent redox properties and high stability, other rare earth metals, such as lanthanum (La) and yttrium (Y), exhibit outstanding catalytic activity in the RWGS reaction due to their unique surface properties. Zhang et al. prepared a highly efficient catalyst for the RWGS reaction by loading ultra-small Pt clusters (average size 1.38 nm) onto a La2O2CO3 (LOC) support,46 named PtNC/LOC. At 300 °C, the PtNC/LOC catalyst demonstrated an exceptionally high CO generation rate (2678 molCO molPt−1 h−1) and nearly 100% CO selectivity (Fig. 10a), with catalytic activity significantly superior to that of LOC supported Pt single atom and nanoparticle catalysts (PtSA/LOC and PtNP/LOC). Further studies demonstrated that the hydrogenation of CO2 on PtNC LOC catalysts proceeds via a typical carbonate pathway (Fig. 10b). The particle size of Pt plays a decisive role in this process: compared with PtSA/LOC and PtNP/LOC, the PtNC/LOC catalyst ensures moderate CO2 adsorption while simultaneously promoting efficient CO desorption, thereby delivering superior catalytic activity (Fig. 10c–e). In addition, the catalyst showed good stability by maintaining the activity above 87.7% of the initial value after 80 h of continuous operation at 380 °C, which was attributed to the strong interaction between the LOC supports and the Pt clusters (Fig. 10f). Li et al. developed an efficient and stable Ru–Sn/La2O2CO3 catalytic system for the RWGS reaction,117 achieving over 99% CO selectivity at 400 °C. Characterization results indicate that the electron transfer from Sn to Ru not only suppressed H2 dissociation but also enhanced the ability of Ru to adsorb oxygen species, thereby promoting the adsorption and activation of CO2 and increasing CO yield. The research showed that an optimal Ru/Sn atomic ratio of 5:0.43 achieves the best balance between CO2 activation and H2 dissociation rates, resulting in the highest CO yield. Ultimately, a Ru–Sn/La2O2CO3 catalyst with an ultra-low Ru loading (0.01 wt%) achieved a CO production rate of 3.6 × 106 mmolCO gRu−1 h−1, which is 103 times higher than the best data reported in the literature. Li et al. developed an inverse-structured Y2O3/Cu catalyst with a Y2O3/CuOx/Cu multiphase interface by sintering copper species,118 which exhibited excellent performance under harsh reaction conditions (600 °C, a gas hourly space velocity of 400000 mL gcat−1 h−1). The catalyst achieved an initial CO2 conversion of 52.3% and a reaction rate of 5.3 × 10−8 mol gcat−1 s−1, outperforming conventional Cu/Y2O3 catalysts (31.6%, 1.4 × 10−8 mol gcat−1 s−1), respectively, and surpassing commercial Cu-Zn-Al catalysts (2.6 × 10−8 mol gcat−1 s−1). Additionally, the inverse catalyst exhibited superior cycling stability, with only 1.4% activity decay after 7 cycles compared to 6.2% degradation in conventional catalysts, highlighting the effectiveness of the inverse structural design in balancing high activity and thermal stability for RWGS reaction applications. Among non-cerium-based rare earth elements, while La and Y have already been applied in catalyst systems for the RWGS reaction, Sm2O3 also demonstrates significant potential as a highly efficient catalyst support. Zhao et al. constructed a model Ni/Sm2O3 catalyst by depositing monodisperse Ni nanoparticles onto a Sm2O3 support for CO2 hydrogenation.119 By modulating the composition of the reduction gas mixture, they successfully achieved in situ control over the encapsulation state of the strong metal–support interaction (SMSI). When the catalyst was reduced in H2, a complete Sm2O3 overlayer formed, fully encapsulating the Ni nanoparticles. This structural configuration resulted in high CO selectivity within the 225–300 °C temperature range: CO2 conversion increased with temperature, reaching 4.7% at 300 °C, while CO selectivity remained close to 100% across the entire temperature range.
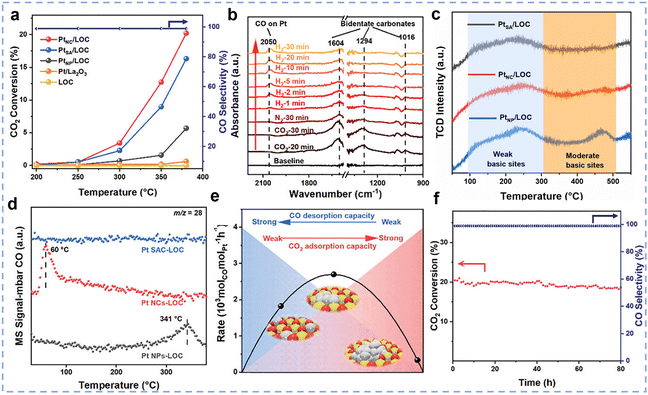 |
| | Fig. 10 (a) The profile of CO2 conversion and CO selectivity versus temperature with various catalysts. (b) In situ DRIFT spectra and evolution of surface intermediate species of PtNC/LOC. (c) CO2-TPD and (d) CO-TPD-MS spectra of PtSA/LOC, PtNC/LOC, and PtNP/LOC. (e) Schematic diagram of the relationship between the CO formation rate (300 °C) with CO desorption and CO2 adsorption. (f) The long-term stability of PtNC/LOC. Reprinted with permission from ref. 46. Copyright (2023) Wiley-VCH. | |
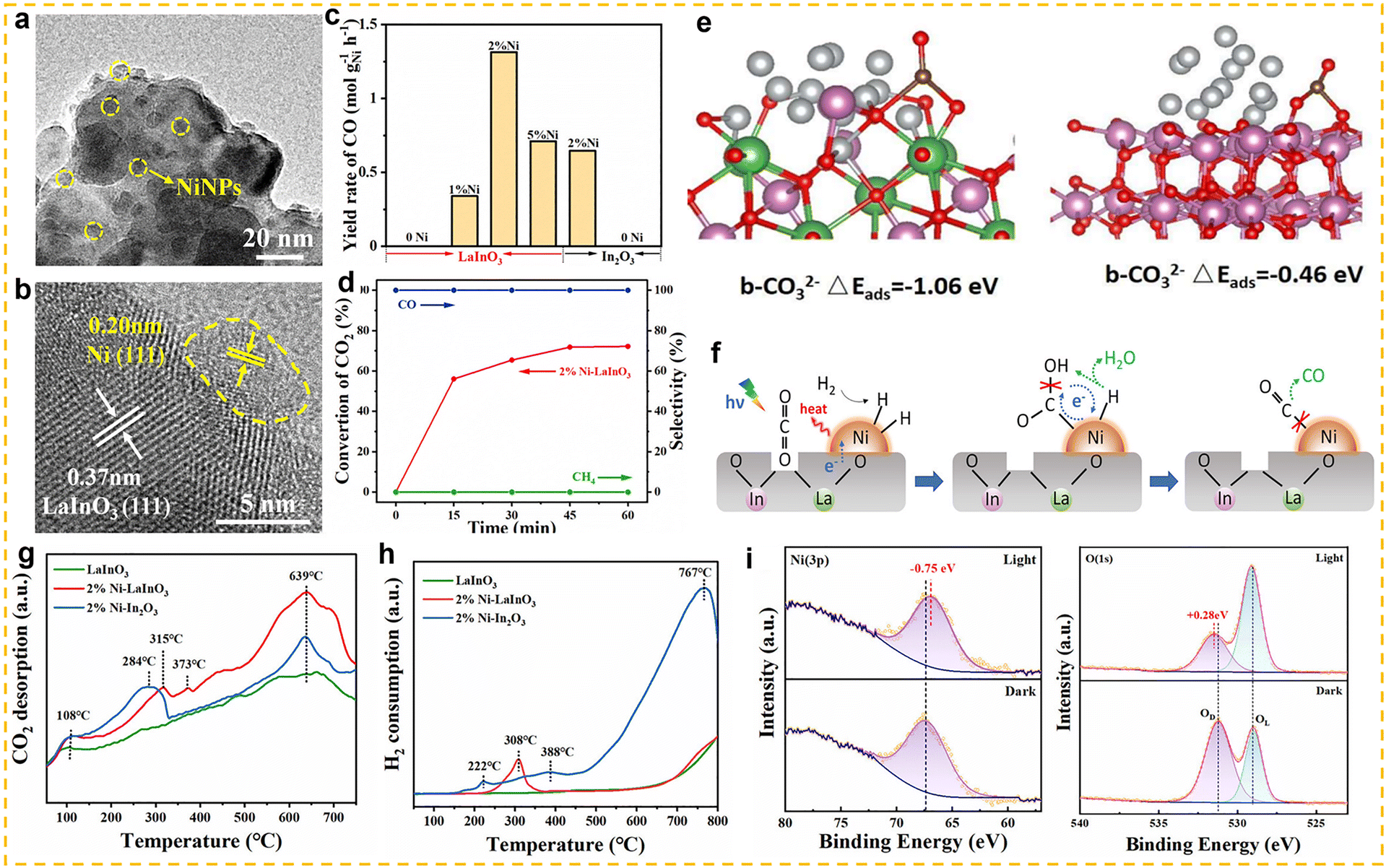
Non-cerium-based rare earth elements can also be used as promoters for the RWGS reaction to significantly enhance the catalytic performance by improving the electronic structure, acid-base properties, and stability of the catalyst. Ranjbar et al. investigated the effects of different promoters (Ce, La, Mg and K) on the Ni/Al2O3 catalyst in the RWGS reaction.120 By incorporating elements such as Ce, La, Mg, and K, they studied the role of these promoters in enhancing the catalyst's performance. The results showed that La and K significantly improved the CO2 conversion rate and CO selectivity. Further investigation revealed that the introduction of promoters improved the dispersion of Ni and enhanced the catalyst's ability to adsorb CO2, thereby improving catalytic activity. Wang et al. successfully prepared an efficient RWGS catalyst by highly dispersing metallic nickel onto a gadolinium-doped ceria support (Ni–GDC).121 The results showed that with increasing Gd doping ratio, the concentration of oxygen vacancies in the catalyst first increased and then decreased, reaching a maximum when the Gd/Ce molar ratio was 1. Further analysis revealed a significant positive correlation between the oxygen vacancy concentration and CO2 conversion. The resulting Ni–GDC catalyst exhibited excellent catalytic activity and long-term stability in the RWGS reaction, achieving a high CO production rate of 19.4 mmol g−1 min−1 at 700 °C and maintaining stable performance during 20 h of continuous operation with almost no loss of activity. In addition, researchers also extended the application of non-cerium-based catalysts from conventional thermal catalysis to photothermal systems. Yu et al. designed a Ni nanoparticle-loaded LaInO3 photothermal catalyst (Ni/LaInO3) (Fig. 11a and b).122 Under hydrogen-rich conditions (H2:CO2 = 4:1), this catalyst achieved highly efficient RWGS performance, with a CO production rate of 1314 mmol gNi−1 h−1, approximately twice that of Ni–In2O3, while maintaining nearly 100% CO selectivity and more than 72% CO2 conversion (Fig. 11c and d). CO2-TPD and H2-TPR analyses revealed that Ni/LaInO3 exhibited superior CO2 adsorption and H2 activation capabilities compared with Ni–In2O3 (Fig. 11g and h). Combining theoretical calculations with in situ irradiation XPS (ISI-XPS) analysis, the researchers further elucidated the mechanism behind this performance enhancement: abundant oxygen vacancies in Ni/LaInO3 effectively enhance CO2 adsorption and activation (Fig. 11e), while photoinduced charge transfer (PCT) promotes efficient H2 activation and drives subsequent CO2 hydrogenation (Fig. 11f and i).
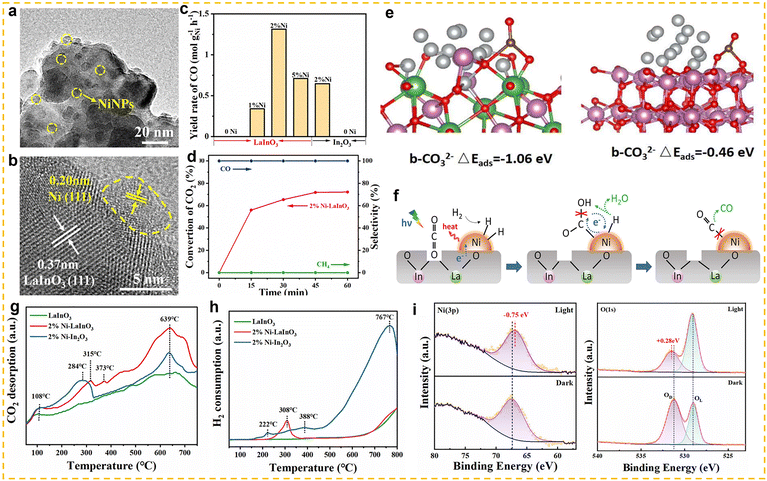 |
| | Fig. 11 (a) TEM image of 2% Ni–LaInO3. (b) HRTEM image of 2% Ni–LaInO3. (c) Catalytic performance of LaInO3 or In2O3 before and after loading Ni NPs. (d) Photothermal CO2 conversion test and selectivity of 2% Ni–LaInO3. (e) The adsorption energy of b-CO32− over 2% Ni–LaInO3 and 2% Ni–In2O3. (f) Schematic diagram for light-driven RWGS reaction over 2% Ni–LaInO3. (g) CO2-TPD spectra of LaInO3, 2% Ni–LaInO3 and 2% Ni–In2O3. (h) H2-TPR spectra of LaInO3, 2% Ni–LaInO3 and 2% Ni–In2O3. (i) ISI-XPS spectra of 2% Ni–LaInO3 with or without light irradiation. Reprinted with permission from ref. 122. Copyright (2023) Wiley-VCH. | |
4.2. CO2 hydrogenation to methane
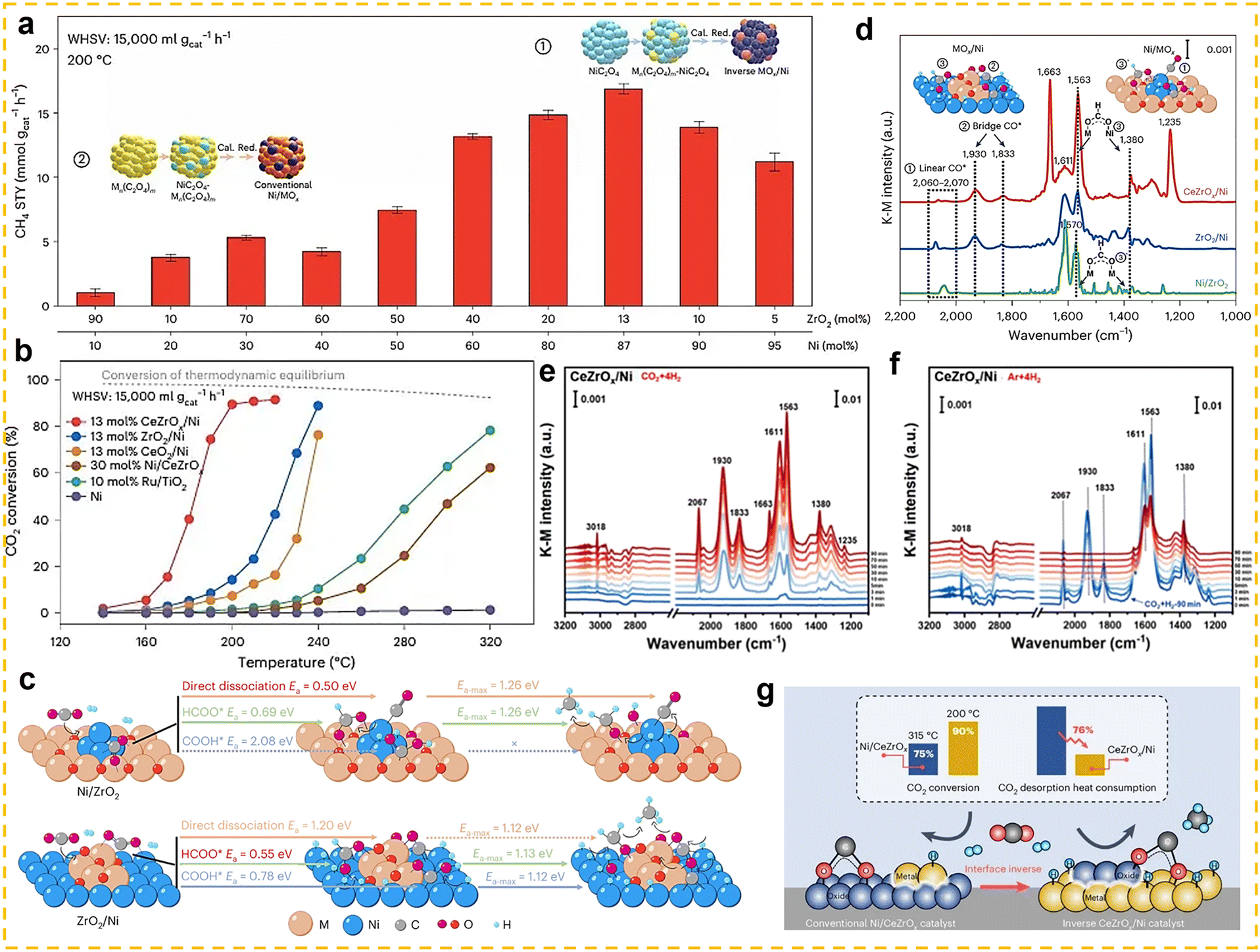
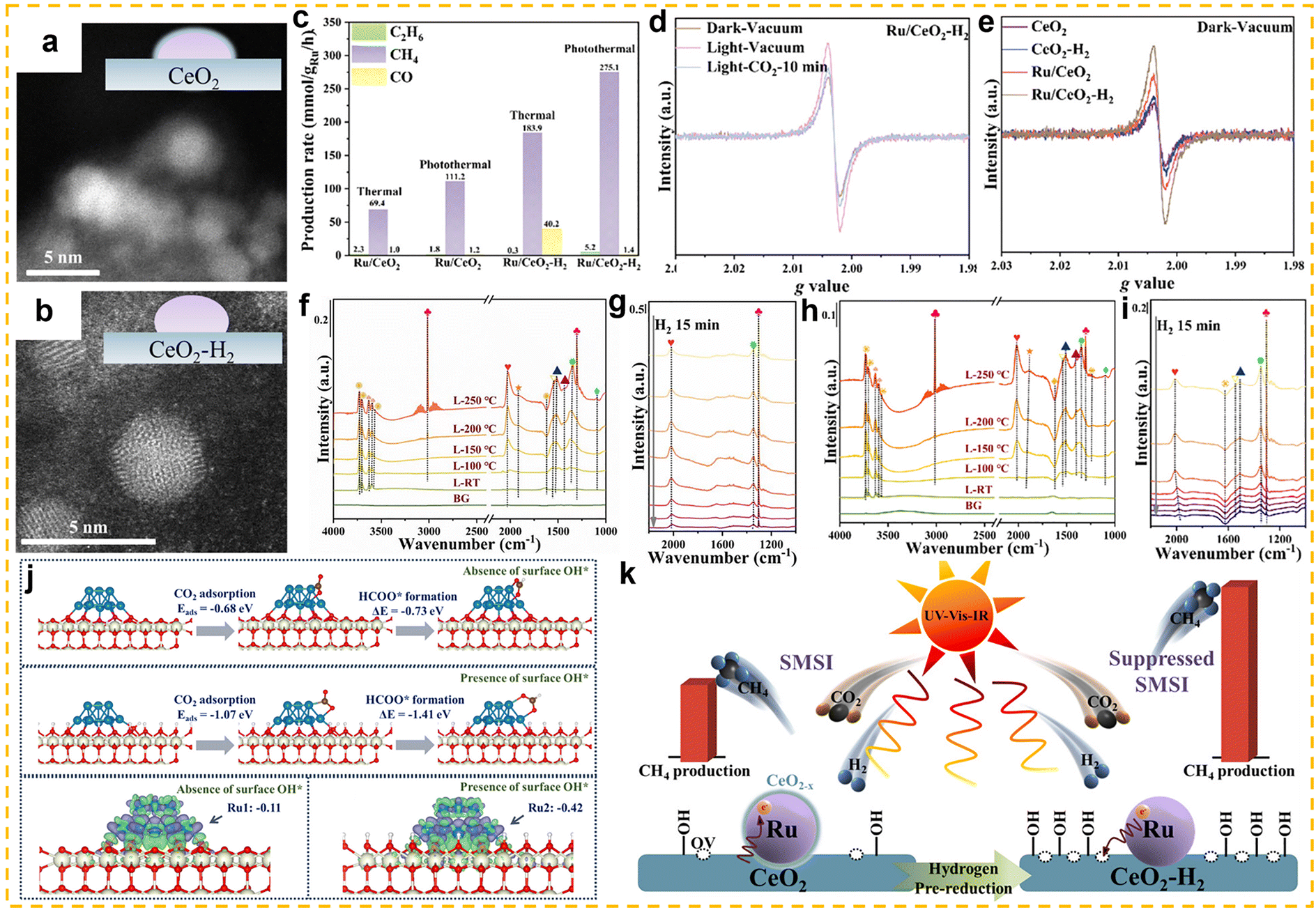
The CO2 hydrogenation to methane, also known as the Sabatier reaction,123,124 proceeds according to the equation: CO2 + 4H2 ↔ CH4 + 2H2O (ΔH298K = −165.03 kJ mol−1).125 From the perspective of reaction mechanisms, CO2 methanation proceeds through two main routes: the formate route (associative route) and the CO route (dissociative route).125,126 In the formate route, CO2 first adsorbs onto the catalyst surface and then undergoes a stepwise hydrogenation to form a bidentate formate (HCOO*) intermediate, and is finally hydrogenated to form CH4. In the CO route, CO2 is converted into a CO intermediate via the RWGS reaction, and the CO intermediate is then hydrogenated to form methane. The competition between these two routes is primarily influenced by the properties of the catalyst. Thermodynamically, this reaction is highly exothermic, and low-temperature conditions (25–400 °C) are more favourable for the forward reaction.125 However, the high bond energy of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in CO2 molecules (806 kJ mol−1) results in extremely strong chemical stability,126 leading to a substantial kinetic barrier for activation. Therefore, the development of highly active low-temperature CO2 methanation catalysts is of critical importance. Typical CO2 methanation catalyst structures consist of active metals dispersed on oxide supports. Common active metals include precious metals (such as Rh, Pd, and Ru) and non-precious metals (such as Ni, Co, and Fe).119,127–134 Among these, nickel-based catalysts have attracted widespread attention due to their high methane yield and lower cost compared to precious metals. Additionally, various support materials have been studied for this catalytic reaction, ranging from traditional zeolites to more complex structures (such as core-shell structures). Among these, rare earth-based oxides (such as CeO2,135 La2O2CO3,136 CeZrOx,137 La2O3,138,139 and Sm2O3140) have been extensively studied as supports or promoters and have demonstrated high catalytic activity.
O bond in CO2 molecules (806 kJ mol−1) results in extremely strong chemical stability,126 leading to a substantial kinetic barrier for activation. Therefore, the development of highly active low-temperature CO2 methanation catalysts is of critical importance. Typical CO2 methanation catalyst structures consist of active metals dispersed on oxide supports. Common active metals include precious metals (such as Rh, Pd, and Ru) and non-precious metals (such as Ni, Co, and Fe).119,127–134 Among these, nickel-based catalysts have attracted widespread attention due to their high methane yield and lower cost compared to precious metals. Additionally, various support materials have been studied for this catalytic reaction, ranging from traditional zeolites to more complex structures (such as core-shell structures). Among these, rare earth-based oxides (such as CeO2,135 La2O2CO3,136 CeZrOx,137 La2O3,138,139 and Sm2O3140) have been extensively studied as supports or promoters and have demonstrated high catalytic activity.
4.2.1. Cerium-based catalysts for the CO2 hydrogenation to methane reaction.
Compared with inert supports (such as Al2O3 or SiO2), the reducible support CeO2 exhibits higher catalytic activity in many cases. Martin et al. investigated the catalytic properties of different supports (SiO2, Al2O3, CeO2, ZSM-5 and MCM-41) loaded with Pd, Rh and Ni metal particles.127 The results demonstrate that Rh/CeO2 exhibits superior activity for CO2 hydrogenation to methane at relatively low temperatures. In addition to the type of supports, the active metal size and support morphology also significantly affect the catalytic activity and product selectivity. Liao et al. systematically investigated the effects of Rh particle size and CeO2 morphology on the reaction of CO2 hydrogenation for methane preparation. The study shows that changes in the CeO2 morphology significantly affect the density of interface oxygen vacancies, which in turn determine the catalytic activity.141 Meanwhile, variations in the Rh particle size mainly influence the product selectivity. As the Rh particle size decreases, the metallic character of the catalyst weakens, leading to reduced methane selectivity. Alloying is one of the effective strategies to modulate the catalytic performance of reactive metals. Through alloying effects (e.g. electron transfer and lattice strain), the electronic structure and geometrical coordination environments of the metal components are synergistically modulated to optimise the adsorption barriers and conversion pathways of the reaction intermediates, and the catalytic activity is significantly enhanced. Zhang et al. developed an argon-annealing redispersion method that reduced the particle size of NiRu alloys in NiRu/CeO2 from 30–40 nm to 3–5 nm (denoted as NiRu/CeO2–Ar), leading to significantly enhanced catalytic performance compared to the pristine sample.135 Overall, the CO2 conversion and CH4 selectivity on NiRu/CeO2–Ar is markedly higher than those of the NiRu/CeO2 catalyst and other reported Ni-based catalysts, and the reaction rate reached 2.28 molCO2 g−1 h−1. Additionally, the activation energy was 67.09 kJ mol−1, significantly lower than 82.14 kJ mol−1 of NiRu/CeO2, demonstrating reduced energy requirements. In addition to these catalysts, inverse catalysts promote the asymmetric adsorption of intermediates, exhibiting high activity in the hydrogenation of CO2, and have been widely applied in CO2 methanation reactions in recent years. Song et al. synthesized nickel-supported oxide catalysts and oxide-supported nickel catalysts using a stepwise co-precipitation method.137 They found that the oxide/nickel inverse catalysts (such as ZrO2/Ni and CeO2/Ni) exhibited significantly higher activity in CO2 methanation, particularly the 13 mol% ZrO2/Ni catalyst, which achieved a methane space-time yield of 16.9 mmolCH4 gcat−1 h−1, much higher than the conventional catalyst's 5.3 mmolCH4 gcat−1 h−1 (Fig. 12a). Based on the good CO2 methanation activity of CeO2 and ZrO2 particles, the study adopted a mixed oxide strategy to design the interface structure. The developed CeZrOx/Ni inverse structure catalyst demonstrated excellent performance in low-temperature CO2 methanation, achieving 90% CO2 conversion and >99% CH4 selectivity at 200 °C and atmospheric pressure, significantly outperforming Ni/CeZrOx (Fig. 12b). In situ DRIFTS results revealed that CO* species were mainly linearly adsorbed on conventional Ni/MOx interfaces, occupying the active sites of the catalyst. In contrast, on inverse MOx/Ni interfaces, CO* predominantly exhibited bridged adsorption, while only a small fraction of linearly adsorbed CO* originated from the direct dissociation of CO2 on the metal. This adsorption mode effectively suppressed the formation of Ni(CO)4 and the loss of Ni (Fig. 12d). When CO2 was cut off from the feed, rapid consumption of CO* and formate species along with the formation of CH4 was observed on the inverse MOx/Ni catalyst by in situ DRIFTS (Fig. 12e and f). Combined with theoretical calculations, these results indicate that CO2 can be hydrogenated to CH4 on inverse MOx/Ni catalysts via two pathways: the formate route and the CO route (via carboxylate intermediates). In contrast, on conventional Ni/MOx interfaces, carboxylate intermediates are difficult to form, so CH4 can only be produced through formate intermediates and directly dissociated linear CO*. Since the hydrogenation of both formate species and linear CO* is a strong endothermic process and their reverse reactions are more favorable, CO2 hydrogenation to CH4 is less efficient at low temperatures on conventional Ni/MOx catalysts (Fig. 12c and g). Therefore, constructing inverse MOx/Ni structures appears to be a feasible strategy for enhancing the low-temperature CO2 methanation activity of Ni-based catalysts.
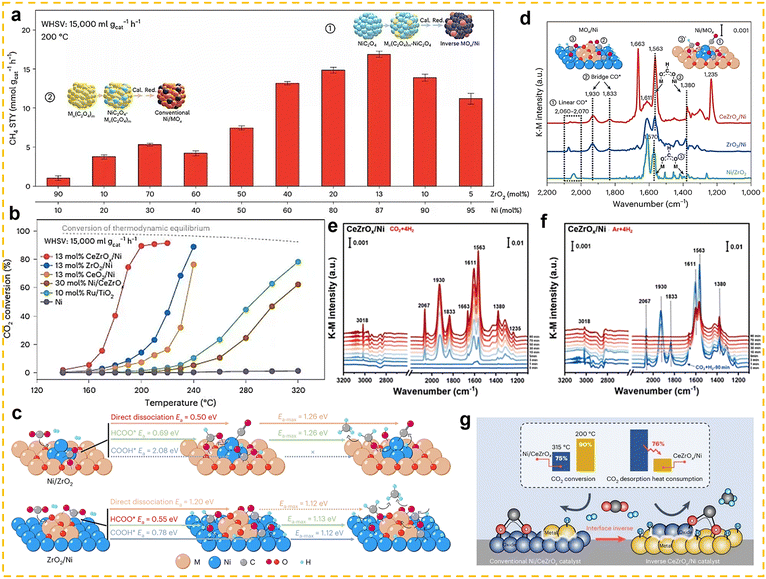 |
| | Fig. 12 (a) STY of CH4 at 200 °C as a function of the percentage of ZrO2 in the Ni–ZrO2 catalysts with CO2 conversion <15%, the error bars show the deviation of STY based on three repeated experiments (Cal., calcination; Red., reduction in catalyst preparation processes). (b) Temperature-dependent activities of the 13mol% CeZrOx/Ni, 13mol% ZrO2/Ni, 13mol% CeO2/Ni, 30mol% Ni/CeZrOx (15 wt% Ni/ CeZrOx) and Ni catalysts. (c) A schematic diagram of the methanation process at the interface of Ni/ZrO2 and ZrO2/Ni. (d) In situ DRIFTS spectra of the CO2/H2 reaction on Ni/ZrO2, ZrO2/Ni and CeZrOx/Ni catalysts, and the catalysts are exposed to 80% H2/20% CO2 (10 mL min−1) atmosphere at 140°C for 90min, using Kubelka–Mumk (K–M) transform to process the spectral data. (e) In situ DRIFTS spectra of the CO2/H2 reaction on CeZrOx/Ni catalyst. The catalysts were exposed to an 80% H2/20% CO2 (10 mL min−1) atmosphere at 140 °C for 90 min. (f) In situ DRIFTS spectra of the H2 atmosphere on CeZrOx/Ni catalyst (pretreated 90 mins in 80% H2/20% CO2 atmosphere at 140 °C and the inlet was switched to 80% H2/20% Ar and maintained at the same temperature for 90 min. (g) Schematic diagram showing that CeZrOx/Ni catalysts exhibit significant activity advantages over conventional Ni/CeZrOx catalysts. Reprinted with permission from ref. 137. Copyright (2024) Springer Nature. | |
In addition, the application of Ce as a promoter in the CO2 methanation reaction was investigated by Daroughegi et al.142 They synthesized Ni based Al2O3 catalysts using an ultrasound-assisted co-precipitation method and modified them with promoters such as Zr, Ce, La, and Mo. The study systematically examined the effects of these promoters on the catalyst's structure, performance, and low-temperature activity. The results showed that the introduction of promoters significantly enhanced the interaction between NiO and Al2O3, promoted the dispersion of Ni species, and reduced the crystallite size. Catalytic performance testing revealed that the Ce promoter had the most significant effect on enhancing low-temperature activity. Specifically, the 25Ni–5CeO2–Al2O3 catalyst achieved a CO2 conversion rate of 76.4% and CH4 selectivity of 99.1% at 350 °C.
In addition to conventional thermal catalysis, cerium-based catalysts have also exhibited excellent performance in photothermal CO2 methanation. In recent years, metal catalysts supported on CeO2 (M/CeO2) have become a research hotspot in the field of photothermal catalytic CO2 methanation reactions. Various M/CeO2 catalyst systems, such as Co/CeO2,143,144 CuNi/CeO2,145 Ru/Mg–CeO2,146 Ni/CeO2,147–149 and Ru/CeO2,150 have all demonstrated excellent catalytic performance. Zhou et al. revealed the performance differences and mechanisms of Ru/CeO2 catalysts in photothermal CO2 methanation by modulating the strong metal–support interaction (SMSI).150 Ru/CeO2 with SMSI effect and Ru/CeO2–H2 with suppressed the SMSI were prepared using an impregnation method (Fig. 13a and b). Experiments show that the methane production rate of the latter under 200 °C and light irradiation (275.1 mmol gRu−1 h−1) is 2.5 times that of the former (111.2 mmol gRu−1 h−1) (Fig. 13c) in situ EPR measurements showed that the oxygen vacancy (g = 2.003) signal of Ru/CeO2–H2 was significantly stronger than that of Ru/CeO2 (Fig. 13e). Under vacuum irradiation, localized surface plasmon resonance (LSPR) induced the migration of hot electrons from Ru nanoparticles to CeO2–H2, reducing Ce4+ to Ce3+ and generating abundant surface oxygen vacancies (OVs). Upon subsequent CO2 introduction, the OV signal intensity decreased markedly, indicating that OVs further promoted CO2 adsorption and activation through photoinduced electron trapping (Fig. 13d). In situ DRIFTS further confirmed that both catalysts exhibited characteristic peaks of intermediates under photothermal and thermal conditions, but the intermediate signals on Ru/CeO2 were consistently weaker than those on Ru/CeO2–H2, indicating the superior activity of the latter in CO2 methanation (Fig. 13f–i). Both catalysts followed the formate and CO pathways. DFT calculations revealed that on Ru/CeO2 enriched with surface –OH groups (Ru/CeO2–H2), the formation of HCOO* was more favorable (ΔE = −1.41 eV) than on Ru/CeO2 lacking –OH groups (Ru/CeO2) (ΔE = −0.73 eV). Moreover, Bader charge analysis indicated that the charge density difference of Ru sites in Ru/CeO2 with –OH was −0.42, higher than that in Ru/CeO2 without –OH (−0.11), facilitating H2 dissociation (Fig. 13j). Collectively, these results confirm that suppressing SMSI optimizes interfacial electron transfer, enhances the generation of oxygen vacancies, and promotes favorable intermediate pathways, thereby boosting the photothermal CO2 methanation activity of Ru/CeO2 catalysts (Fig. 13k). This study further demonstrates that rational modulation of the metal–support interaction provides an effective strategy for the design of highly efficient photothermal catalysts. Moreover, Yue et al. developed a CuNi alloy catalyst supported on CeO2 (CuNi/CeO2).145 This catalyst exhibits different product selectivities under dark reaction and visible light catalytic conditions, with visible light excitation significantly promoting the further hydrogenation of CO2 to CH4. Extensive experimental results demonstrate that, under visible light excitation, hot electron transfer from plasmonic Cu to Ni not only modifies the conventional thermal reduction pathway but also enriches surface hydroxyl groups and oxygen vacancies, thereby providing effective sites for CO2 capture.
 |
| | Fig. 13 (a) HAADF-STEM image of Ru/CeO2. (b) HAADF-STEM image Ru/CeO2–H2. (c) Average production rates of CH4, CO, and C2H6 in Ru/CeO2 and Ru/CeO2–H2 at different conditions (thermal: 200 °C, dark; photothermal: 200 °C, light). (d) and (e) OV signals of catalysts during in situ EPR spectroscopy in the dark and light irradiation, in vacuum and CO2 atmosphere conditions, respectively. In situ DRIFTS results on Ru/CeO2 and Ru/CeO2–H2 by introducing a continuous flow of reaction gas 72% H2/18% CO2/10% Ar (f) and (h) and switching to gas 10% H2/Ar (g) and (i) (light: L; background: BG; room temperature: RT). (j) CO2 adsorption processes and formation pathways of HCOO* in the absence and the presence of surface OH* on the Ru/CeO2 catalyst. Charge density difference plots and Bader charge analyses in the absence and the presence of surface OH* on the Ru/CeO2 catalyst. (k) Electron migration pathways and performance of Ru/CeO2 and Ru/CeO2–H2 under the photothermal methanation reaction. Reprinted with permission from ref. 150. Copyright (2024) American Chemical Society. | |
4.2.2. Non-cerium-based catalysts for the CO2 hydrogenation to methane reaction.
As a support material for the catalyst, lanthanum oxide provides basic sites that facilitate the adsorption and activation of CO2, thereby enhancing the activity of the CO2 methanation reaction. Tang et al. successfully prepared mesoporous 50% Ni–La2O3 catalysts via the colloidal solution combustion method and applied them to the CO2 methanation reaction.139 The catalysts prepared by this method exhibited small nickel particle sizes (approximately 4.5 nm) and a rich metal-support interface, along with a good mesoporous structure. The study showed that this catalyst demonstrated higher activity in the CO2 methanation reaction compared to the 50% Ni–La2O3 catalyst prepared by conventional methods. Specifically, at 300 °C, the CO2 conversion rate of the mesoporous 50% Ni–La2O3 catalyst reached 51%, significantly higher than the 9% achieved by the 50% Ni–La2O3 catalyst. In recent years, another La-based compound, La2O2CO3, has attracted significant attention in CO2 methanation reactions. Dai et al. developed a Ni/ La2O2CO3 catalyst via a surface carbonate modification strategy.136 Catalytic performance tests revealed that the Ni/La2O2CO3 catalyst achieved a CO2 conversion rate of 91% at 350 °C, significantly higher than the 62% achieved by Ni/La2O3. Meanwhile, the CH4 selectivity exceeded 99.9%, surpassing the maximum of 97.7% for Ni/La2O3. In the stability test conducted at 300 °C for 120 h, the CO2 conversion rate of Ni/La2O2CO3 decreased slowly from 85% to 74%, whereas the conversion rate of Ni/La2O3 sharply dropped from 32% to 10% within 60 h. In situ DRIFT analysis further revealed that the surface of Ni /La2O2CO3 generated both monodentate and bidentate formate species, which rapidly converted to CH4 within 1 minute in the gas switching test. In contrast, Ni/La2O3 only generated inert bidentate formate species, which exhibited lower catalytic activity due to hindered further conversion. Among non-cerium-based rare earth catalysts, in addition to La, rare earth elements such as samarium (Sm), Pr, and Y have also been widely applied in the CO2 methanation reaction. Ilsemann et al. investigated the application of Sm in this reaction.140 They successfully prepared Ni–Sm2O3 catalysts with nickel loadings ranging from 4–89 wt% by modifying the epoxide addition method and systematically explored the relationship between their structures and the performance of the CO2 methanation reaction. Catalytic performance tests showed that the catalyst loaded with 39 wt% Ni exhibited the best performance in the CO2 methanation reaction. Li et al. prepared a series of xCA–Ni/Y2O3 catalysts with excellent activity and stability using a citric-acid-assisted method.151 These catalysts exhibited outstanding performance in the CO2 methanation reaction. In particular, the 12CA–Ni/Y2O3 catalyst achieved a high CO2 conversion of 92% and a CH4 selectivity of 100% at 350 °C, with catalytic activity far superior to that of the Ni/Y2O3 catalyst without citric acid. Further studies revealed that the citric-acid-assisted xCA–Ni/Y2O3 catalysts not only promoted the reduction and dispersion of NiO species but also significantly increased the number of surface basic sites. The abundant basic sites facilitated CO2 activation at low temperatures, while the smaller Ni particles provided more metallic Ni active sites for H2 dissociation. The synergistic effect between these two types of active sites effectively enhanced the overall catalytic activity for CO2 methanation. Alcalde-Santiago et al. synthesized Ni catalysts supported on PrOx by an impregnation method.152 The catalyst exhibited good catalytic activity in the CO2 methanation reaction, showing higher activity than the Ni/LaOx catalyst prepared by the same method but slightly lower than that of Ni/CeO2. This difference was mainly attributed to the relatively low efficiency of active site formation and the slightly higher stability of chemisorbed CO2, which to some extent limited the overall catalytic activity.
Concurrently, non-cerium rare earth elements such as Pr, La, neodymium (Nd), Sm, Europium (Eu), and gadolinium (Gd) are frequently employed as promoters in CO2 methanation reactions. Garbarino et al. prepared a series of Ni/La–γ-Al2O3 catalysts with varying La contents using an impregnation method and a silica-free γ-Al2O3 support.153 The results showed that this series of catalysts exhibited high catalytic activity in the CO2 methanation reaction. Among them, the Ni/La–γ-Al2O3 catalyst with 14% La loading displayed the best performance at 350 °C, achieving nearly 100% methane selectivity and a yield of about 90%. Further studies revealed that the introduction of lanthanum as a promoter effectively enhanced the surface basicity of the catalyst, thereby facilitating CO2 adsorption and significantly improving the overall catalytic activity. Gac et al. investigated the effect of Nd as a promoter on nickel catalysts in the CO2 methanation reaction.154 Two series of catalysts were prepared using nanoceria and γ-Al2O3 as supports via an impregnation method, with a nickel content of 20 wt% and Nd addition ranging from 1 to 10 wt%. The results indicated that ceria-supported nickel catalysts exhibited high activity in the reaction, with minimal changes in activity as the Nd content increased. The CO2 conversion rate of the alumina-supported catalyst was significantly enhanced with Nd promotion, particularly at low Nd content (5 wt%), where a noticeable improvement in activity was observed. Mechanistic studies reveal that for the ceria-supported system, the Nd promoter induces complex changes in redox properties and acid-base characteristics, collectively modulating the activation method of CO2 and the transformation process of surface intermediates, whereas for the alumina-supported system, the activity enhancement is associated with unique changes in its basicity. Namvar et al. investigated the performance of a series of xNi–5M–Al2O3 (x = 15, 25, 35 wt%, M = Tb, Dy, Nd) catalysts promoted by non-cerium rare earth elements in the CO2 methanation reaction.155 The results showed that the catalyst modified with terbium (Tb) exhibited the highest catalytic activity among all samples. In particular, the 25 wt% Ni–5 wt% Tb–Al2O3 catalyst achieved a CO2 conversion of 66.93% and a CH4 selectivity of 100% at 400 °C, demonstrating excellent catalytic performance. Further studies revealed that the introduction of Tb not only significantly improved the dispersion of nickel on the support surface but also facilitated CO2 adsorption and activation through the generation of oxygen vacancies, thereby endowing the catalyst with superior reaction activity.
Analogous to cerium-based catalysts, non-cerium-based catalysts have likewise demonstrated outstanding activity and stability in photothermal CO2 methanation reactions. Wang et al. prepared a series of Pt/LaCoO3 catalysts that achieved highly efficient photo-thermal CO2 hydrogenation to CH4.156 At a light intensity of 1.2 W cm−2 at 250 °C, the 0.6Pt/LaCoO3 catalyst exhibited outstanding catalytic performance, with a CH4 production rate as high as 119.8 mmol gcat−1 h−1 and a CH4 selectivity of 87%. DRIFTS results revealed that the reaction proceeded via the formate pathway, while light irradiation accelerated the transformation of the intermediate species (*HCOO) without altering the reaction route, thereby significantly enhancing the CH4 formation rate.
4.3. CO2 hydrogenation to methanol
The hydrogenation of CO2 to methanol (CO2 + 3H2 ↔ CH3OH + H2O, ΔH298K = −49.5 kJ mol−1) is an exothermic reaction with significant environmental and energy value,157 yet it faces inherent thermodynamic and kinetic contradictions. The CO bond dissociation energy of CO2 molecules is extremely high, resulting in a high activation energy barrier for the reaction.158,159 Therefore, the reaction must be carried out at high temperatures to accelerate the reaction kinetics. However, as this reaction is an exothermic process involving a reduction in the number of gas molecules, thermodynamics requires low temperatures to maintain a high equilibrium constant, while high pressure (>3 MPa) is needed to compensate for entropy loss. While low temperature and high pressure synergistically optimize thermodynamic equilibrium, low temperature inhibits CO2 activation kinetics, leading to a dilemma in temperature control. Moreover, a major competing reaction is the endothermic RWGS reaction, where the byproduct CO not only reduces methanol selectivity but may also induce subsequent catalyst poisoning. Thus, catalyst design must balance efficient CO2 activation and selective regulation.6,160 However, the CZA catalyst is prone to sintering and deactivation at high temperatures and moisture exposure, which limits its long-term stability.160 This limitation necessitates the exploration of novel support and promoter systems to enhance catalytic stability and performance. Consequently, various oxide materials have been extensively studied, including ZnO,161 Al2O3,162 TiO2,163–165 ZrO2,166 SiO2,167,168 In2O3,169 and rare earth oxides.88,104,166,170 Among these, rare earth-based oxides have emerged as a research hotspot due to their unique catalytic properties, tunable redox properties, dynamic oxygen vacancy formation, and strong metal–support interactions that facilitate CO2 adsorption and H2 activation (Table 2).
Table 2 Summary of rare earth-based catalysts for the hydrogenation of CO2 to methanol
| Catalysts |
H2/CO2 ratio |
Space velocitya |
T/°C |
P (MPa) |
CO2 conv. (%) |
MeOH sel. (%) |
Ref. |
|
(W) = WHSV = mass flow rate/catalyst mass, mL gcat−1 h−1, (G) = GHSV = volume flow rate/bed volume, h−1.
|
| Er0.2CuZnO |
3 |
1800 |
190 |
5 |
3 |
90 |
171
|
| Ca-doped PdZn/CeO2 |
3 |
2400 |
220 |
3 |
7.7 |
100 |
172
|
| Cu/AlCeO-7 |
3 |
6000 |
280 |
4 |
22.5 |
34 |
173
|
| CZC/10TNTs |
3 |
7500 |
260 |
3 |
23.3 |
59.8 |
174
|
| 1.0PdZn/CeO2 |
3 |
2400 |
220 |
2 |
14 |
95 |
175
|
| Pd/CeO2-NPH |
3 |
60000 |
260 |
5 |
8.7 |
76.8 |
176
|
| Pd/CeO2-NR |
3 |
60000 |
260 |
5 |
7.4 |
74.9 |
176
|
| Pd/CeO2-NC |
3 |
60000 |
260 |
5 |
6.6 |
50.1 |
176
|
| Pd/CeO2-NPG |
3 |
60000 |
260 |
5 |
7.4 |
69.9 |
176
|
| In2O3/CeO2 |
4 |
17681 |
300 |
5 |
32.9 |
94.34 |
177
|
| NiGa/Zr5Ce5 |
3.3 |
3600 |
260 |
4 |
4 |
48 |
178
|
| Cu/CeO2 |
3 |
2400 |
280 |
3 |
8 |
77 |
179
|
| CuCeZr |
3 |
20000 |
240 |
3 |
6.5 |
64.8 |
180
|
| CuZr + Ce |
3 |
20000 |
240 |
3 |
4.4 |
63.6 |
180
|
| CuCe + Zr |
3 |
20000 |
240 |
3 |
1.7 |
72.3 |
180
|
| CeZr + Cu |
3 |
20000 |
240 |
3 |
0.9 |
60.9 |
180
|
| CeO2–Pd/ZrO2 |
3 |
12000 |
330 |
3 |
4.7 |
6.8 |
181
|
| 75Cu–25Ga/Ce0.9Zr0.1O2 |
3 |
15000 |
270 |
3 |
4 |
98 |
182
|
| Ni5Ga3/CeO2 |
3 |
8000 |
270 |
1 |
6.15 |
87.6 |
183
|
| Au/ZrCe0.05 |
3 |
48000 |
320 |
4 |
11.2 |
9 |
184
|
| 2 wt% Pd/CeO2 |
3 |
3600 |
240 |
3 |
49.6 |
69.5 |
185
|
| Cu/AlCeO |
3 |
14400 |
200 |
3 |
2.9 |
85 |
186
|
| Pd–Cu/Ti0.8Ce0.2O2 |
3 |
3600 |
250 |
4.1 |
12.4 |
27.1 |
187
|
| Pd–Cu/Ti0.2Ce0.8O2 |
3 |
3600 |
250 |
4.1 |
8.5 |
31.4 |
187
|
| Pd–Cu/CeO2–C |
3 |
3600 |
250 |
4.1 |
6.7 |
29.4 |
187
|
| CuZn/CeO2-3 |
3 |
15000 |
260 |
3 |
14.5 |
68.0 |
188
|
| CuGa/CZ |
3 |
4500 |
240 |
4.2 |
7.2 |
50 |
189
|
| Cu/CeW0.25Ox |
3 |
15000 |
250 |
3.5 |
13 |
87 |
104
|
| CuCeZr |
3 |
20000 |
280 |
3 |
10.5 |
45.6 |
180
|
| Cu/Ce0.2Zr0.8O2 |
3 |
12000 |
280 |
3 |
5.7 |
94.4 |
190
|
| Cu1La0.2/SBA-15 |
3 |
12000 |
240 |
3 |
6 |
80 |
191
|
| InCe-h |
3 |
6000 |
250 |
0.1 |
∼ |
16 |
192
|
| CZCe-10 |
3 |
3000 |
250 |
3 |
12.4 |
81 |
193
|
| CuO/CeO2-H |
3 |
10000 |
260 |
3 |
15.6 |
92.4 |
194
|
| CuO/CeO2-R |
3 |
10000 |
260 |
3 |
14.7 |
22.5 |
194
|
| CuO/CeO2-C |
3 |
10000 |
260 |
3 |
14.9 |
21.6 |
194
|
| In–Co/Ce |
2.7 |
2400 |
280 |
3 |
9.4 |
74 |
195
|
| In–Co/Ce |
2.7 |
12000 |
280 |
3 |
6.5 |
78.5 |
195
|
| In–Co/Ce |
2.7 |
12000 |
260 |
3 |
3.6 |
83.7 |
195
|
| 50CuLaZr |
3 |
10000 |
240 |
3 |
11 |
61.9 |
196
|
| 80CuLaZr |
3 |
10000 |
240 |
3 |
16.4 |
54.6 |
196
|
| Cu/CeO2 nanorod |
3 |
3000 |
300 |
2 |
6.7 |
19.8 |
197
|
| CuZnY-2 |
3 |
9000 |
200 |
3 |
2.7 |
88.8 |
198
|
| Cu/CeO2 |
3 |
10000 |
280 |
3 |
10.1 |
∼ |
166
|
| Cu0.3Ce0.3Zr0.7 |
3 |
30000 |
240 |
3 |
4.1 |
55.3 |
199
|
| La0.9Sr0.1CuO |
2.9 |
10000 |
300 |
3 |
8.59 |
49 |
200
|
| 0.5Pd–10Cu/CeO2 |
3 |
3000 |
210 |
3 |
3.9 |
67.3 |
201
|
| 1Pd–10Cu/CeO2 |
3 |
3000 |
210 |
3 |
11.2 |
36.2 |
201
|
| 2Pd–10Cu/CeO2 |
3 |
3000 |
210 |
3 |
4.3 |
52.4 |
201
|
| PdIn/CeO2-300 |
3 |
30000 |
300 |
3 |
5 |
78.9 |
202
|
| CuCe/S1 |
3 |
8000 |
240 |
3 |
∼ |
58 |
203
|
| Cu/CeO2-CP |
3 |
2400 |
240 |
3 |
3.8 |
80.5 |
204
|
| Cu/CeO2-SCP |
3 |
2400 |
240 |
3 |
5.9 |
84.6 |
204
|
| Cu/CeO2-SG |
3 |
2400 |
240 |
3 |
6.4 |
89.1 |
204
|
| CZC-3 |
3 |
12000 |
280 |
3 |
15.6 |
64.5 |
205
|
| CuCeIn5 |
3 |
8000 |
275 |
3 |
9.7 |
46.4 |
206
|
| CuCeIn10 |
3 |
8000 |
275 |
3 |
10.0 |
54.6 |
206
|
| CuCe |
3 |
8000 |
250 |
3 |
9.4 |
35.4 |
206
|
| CuZnCe-P |
3 |
6000 |
230 |
4 |
5.6 |
72.78 |
207
|
| CuZnCe-C |
3 |
6000 |
230 |
4 |
4.6 |
74.96 |
207
|
| 2%Pd/CeO2-R |
3 |
6000 |
240 |
3 |
4.6 |
25.9 |
88
|
| 2%Pd/CeO2-R |
3 |
2000 |
240 |
5 |
5.9 |
47.7 |
88
|
| 15%Cu/CeO2-small NR |
3 |
24000 |
280 |
3 |
5.8 |
92.0 |
208
|
| Cu0.6Zn0.3La0.3 |
3 |
30000 |
240 |
3 |
1.0 |
64.2 |
209
|
| Cu0.6Zn0.2La0.4 |
3 |
30000 |
240 |
3 |
2.5 |
78.3 |
209
|
| Cu0.6Zn0.15La0.45 |
3 |
30000 |
240 |
3 |
1.6 |
80.7 |
209
|
| Ce–CuZn |
3 |
20000 |
260 |
2.8 |
8 |
71.1 |
210
|
| Cu/LOC:Nd |
3 |
12000 |
260 |
3 |
2.17 |
|
32
|
| In–Cu/CeO2 |
3 |
7200 |
200 |
3 |
7.6 |
95 |
211
|
| In–Cu/CeO2 |
3 |
7200 |
300 |
3 |
15.1 |
52.1 |
211
|
| LOC/Cu-1 |
3 |
∼ |
200 |
8 |
∼ |
85.5 |
212
|
| CuNi2/CeO2-NT |
3 |
6000 |
260 |
3 |
17.8 |
78.8 |
213
|
| Cu/CeO2/In2O3@mSiO2 |
3 |
12000 |
250 |
3 |
7.8 |
88.9 |
214
|
| 1Cu2Ni/CeO2-NR |
3 |
6000 |
240 |
3 |
16.78 |
72.91 |
215
|
| 1Cu2Ni/CeO2-NR |
3 |
6000 |
260 |
3 |
18.35 |
73.33 |
215
|
| La0.8Zr0.2Cu0.7Zn0.3Ox |
3 |
3600 |
250 |
5 |
12.6 |
52.5 |
216
|
| Co/La4Ga2O9 |
3 |
3000 |
270 |
3.5 |
4.6 |
15.3 |
217
|
| Co/La4Ga2O9 |
3 |
3000 |
280 |
3 |
9.6 |
13.7 |
217
|
| 0.1% Rh/CeO2 |
3 |
∼ |
250 |
3 |
3.2 |
22.2 |
218
|
| 2Rh0.1Fe0.5Na/CeO2 |
3 |
6000 |
250 |
3 |
9.6 |
32.8 |
219
|
| 2Rh0.5Fe0.5Na/CeO2 |
3 |
6000 |
250 |
3 |
9.8 |
27.3 |
219
|
| 2Rh0.5La/CeO2 |
3 |
6000 |
250 |
3 |
6.9 |
39.6 |
219
|
| 2Rh0.5Fe/CeO2 |
3 |
6000 |
250 |
3 |
8.4 |
30.8 |
219
|
| 2Rh0.5Na/CeO2 |
3 |
6000 |
250 |
3 |
7.9 |
30.3 |
219
|
| Rh/CeO2-r |
3 |
6000 |
250 |
3 |
11.2 |
31.4 |
220
|
| Rh/CeO2-c |
3 |
6000 |
250 |
3 |
5.3 |
27.2 |
220
|
| Rh/CeO2-p |
3 |
6000 |
250 |
3 |
12.0 |
2.2 |
220
|
| Nano-Pd/CeO2 |
3 |
3000 |
240 |
3 |
5.3 |
27.0 |
221
|
4.3.1. Cerium-based catalysts for the CO2 hydrogenation to methanol reaction.
The crystal facet of CeO2-based catalysts plays an important role in the methanol synthesis reaction, as demonstrated by synthesizing Pd supported on CeO2 with distinct morphologies (rod-like, cubic, polyhedral, and octahedral).88 Experimental results revealed that the rod-like CeO2-supported 2Pd/CeO2-R catalyst exhibited optimal performance, achieving a CO2 conversion rate of 1.3 mmol g−1 h−1 and a methanol space-time yield (STY) of 22.8 mg g−1 h−1, significantly surpassing catalysts with other morphologies. Further research has found that the density and amount of oxygen vacancies, which vary with the morphology of CeO2, serve as crucial factors determining the catalytic activity. The rod-like CeO2 exposed (110) crystal facets, which exhibited the highest density and the highest amount of oxygen vacancies, thereby enhancing CO2 adsorption and activation. Effect of interactions between different types of supports and metals on the reaction of CO2 hydrogenation to methanol was studied by Wang et al. It was shown that Cu/CeO2 exhibited significant advantages in the range of 200–300 °C: its methanol selectivity was 42.4% higher than that of Cu/ZrO2, while the CO2 conversion (10.0%) was similar to that of Cu/ZrO2 (12.4%).166 Structural characterization indicates that, compared to Cu/ZrO2 catalysts, Cu/CeO2 possesses more abundant metal–oxide interfacial sites and a larger number of oxygen vacancies. These interfacial regions enhance CO2 adsorption and activation, while the vacancies facilitate charge localization and redistribution during the reaction, thereby strengthening CO2 adsorption capacity and stabilizing the key carbonate intermediates in the methanol synthesis process, ultimately resulting in higher methanol selectivity. Compared with single metal active centres, bimetallic reaction systems exhibit higher catalytic activity in methanol synthesis. Choi et al. employed Cu/CeO2 and Pd–Cu/CeO2 catalysts for the hydrogenation of CO2 to methanol.201 The results showed that the Pd–Cu/CeO2 catalyst exhibited a higher methanol yield compared to the Cu/CeO2 catalyst. The enhanced activity of the Pd–Cu/CeO2 catalyst primarily originated from the increased reactivity of the active Cu sites, which benefited from the role of Pd: Pd not only increased the dispersion and surface concentration of Cu but also facilitated the reduction of Cu active sites through its electron-donating effect, thereby enhancing the overall catalytic performance. Furthermore, multi-metal catalyst systems possessing synergistic effects, rich interfaces, and metal-support interactions also demonstrate high catalytic activity. Ye et al. constructed a Ce–CuZn ternary catalyst by atomic-level substitution based on the crystal engineering strategy of MOFs (Fig. 14a and e),210 The catalyst was prepared by first synthesizing a Ce–MOF, followed by sequential introduction of Cu/Zn to form Cu+-rich Cu/Zn–OV–Ce active sites. The catalyst exhibited 71.1% methanol selectivity, a space–time yield of 400.3 g kgcat−1 h−1 and 170 h of stable operation (8.0% CO2 conversion) at 260 °C and 2.8 MPa, with performance comparable to that of commercial CuZnAl catalysts, while the CO selectivity (26.7%) was lower (Fig. 14f). In situ CO-DRIFTS and Quasi in situ XPS analyses revealed the coexistence of Cu0 and Cu+ species on the catalyst surface, with the Cu+ fraction reaching as high as 56.5%, indicating that the abundant Cu+ species play a crucial role in the formation of Cu/Zn–OV–Ce active sites (Fig. 14g and i). In situ DRIFTS further confirmed that formate is the dominant reaction intermediate, and the reaction primarily follows the formate pathway (Fig. 14h). DFT calculations showed that, compared with OV–CeO2−x, Cu+–CeO2−x significantly lowers the activation barrier for CO2 hydrogenation to HCOO*; moreover, the incorporation of Zn greatly facilitates H2 dissociation and HCOO* formation, thereby enhancing CO2 hydrogenation via the formate pathway (Fig. 14j). Consequently, the Ce–CuZn catalyst exhibits outstanding methanol synthesis activity. This study proposes an atomic-level regulation approach to progressively construct efficient active sites, thereby developing high-performance multimetallic catalysts for methanol synthesis. Similar to the RWGS reaction and CO2 methanation reactions, Ce as a promoter also effectively facilitates the CO2 hydrogenation to methanol. Xu et al. developed an efficient CuCe/S1 catalyst by co-loading copper species and CeO2 onto pure silica zeolite S1, which demonstrated excellent performance in the CO2 hydrogenation to methanol reaction.203 Under conditions of 240 °C, 3 MPa, and 8000 mL gcat−1 h−1, the methanol space-time yield (STYMeOH) reached 87.23 g kgCu−1 h−1, which is 6.9 times higher than Cu/S1 (12.60 g kgCu−1 h−1), with a methanol selectivity of 58%. Compared with Cu/S1, the introduction of Ce species effectively suppressed the growth of CuO, stabilized Cu+ sites, and significantly enhanced CO2 adsorption capacity, thereby endowing CuCe/S1 with superior catalytic activity during the reaction.
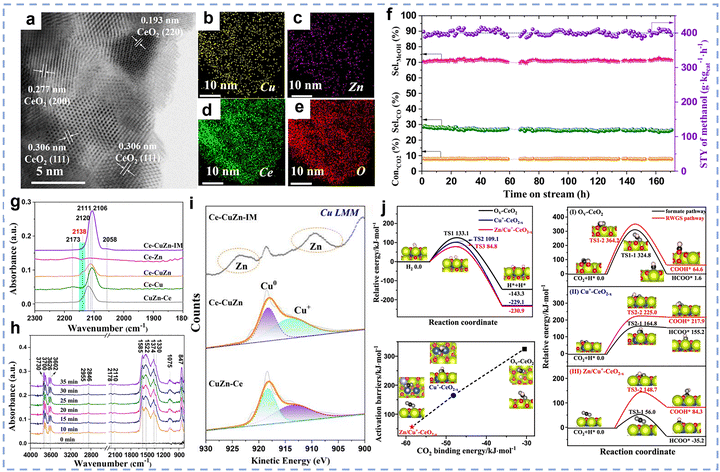 |
| | Fig. 14 (a)–(e) The HRTEM images of reduced Ce–CuZn sample with corresponding elemental mapping. (f) Catalytic stability of Ce–CuZn catalyst. (g) CO-DRIFTS results of CuZnCe after purging by He for 20min at 30 °C. (h) The result of in situ DRIFTS for CO2 hydrogenation at 260 °C. (i) Cu LMM XAES spectra of the reduced CuZnCe catalysts obtained by quasi in situ XPS. (j) The potential energy profiles and the corresponding structures involved in the dissociation of molecular adsorption H2 and CO2 activation. The relationship of CO2 binding energy with the activation barrier of CO2 hydrogenation to HCOO*. Green: Ce, purple: Zn, blue: Cu, black: C, white: H; red represents the surface O and gray represents O in adsorbed molecules. TS transition state. Reprinted with permission from ref. 210. Copyright (2024) Springer Nature. | |
Due to the strong exothermic nature of the CO2 hydrogenation to methanol reaction, high temperature conditions can promote CO2 activation but lead to the reaction equilibrium shifting in the reverse direction. To address this contradiction, photothermal catalysis provides an innovative strategy to achieve efficient CO2 activation and reaction equilibrium regulation simultaneously under mild conditions, thus significantly reducing the energy consumption of conventional thermal catalysis.223 Li et al. synthesized the 0.5Cu@CeO2 catalyst using a coordination complex precipitation method.170 In this catalyst, atomically dispersed Cu2+ ions replace Ce3+, forming new Frustrated-Lewis-pairs. Under conditions of 140 °C, 2 MPa (CO2/H2 = 1:3), and illumination, the catalyst achieved a methanol yield of 1896.02 µmol g−1 h−1, which is 4.6 times higher than that under non-illuminated conditions. In contrast, the 0.5Cu/CeO2 catalyst prepared by the impregnation method achieved a yield of only 1339.46 µmol g−1 h−1 under the same light conditions, which is 2.2 times that under non-light conditions. Both catalysts exhibited significantly enhanced performance under light exposure (5.6-fold vs. 2.2-fold), strongly demonstrating the critical role of light in reducing the reaction activation energy barrier. At the same time, the significantly higher light response factor and absolute yield (1896.02 vs. 1339.46 µmol g−1 h−1) of 0.5Cu@CeO2 further highlights the advantages of its unique atomic-level dispersion structure. Xie et al. explored the structural modulation mechanism of cerium-modified Cu/ZnO/Al2O3 catalysts in low-temperature photothermal catalysis of CO2 to methanol.222 The CuZn0.4Ce0.1 catalyst achieved a methanol yield of 822 g kgCu−1 h−1 (94% selectivity) at 225 °C and 20 bar under light illumination. The effect of cerium on the catalytic reaction activity of the Cu/ZnO-based catalyst was found by modulating the cerium loading: at low loading (≤10 at%), CeO2 preferentially formed a ZnO/CeO2 interface with ZnO, which enhanced the CO2 chemisorption and facilitated the formate intermediate route; when the loading was increased to 20 wt%, strong Cu–CeO2 interactions led to the formation of Cu nanoclusters and overstabilised surface species, reduced methanol-generating activity.
4.3.2. Non-cerium-based catalysts for the CO2 hydrogenation to methanol reaction.
Recent studies have shown that non-cerium-based catalysts exhibit unique advantages in CO2 hydrogenation to methanol reaction. For example, La2O2CO3 (LOC), a highly regarded alkaline oxide material, is considered a promising support or promoter due to its moderate surface alkalinity and excellent thermal stability.212 He et al. prepared La2O2CO3/Cu nanorod composite catalysts (LOC/Cu-x, where x represents the mass ratio of La to Cu) via the coprecipitation-calcination method,212 and investigated their performance in CO2 hydrogenation to methanol. The experiments showed that copper content significantly regulated product selectivity under conditions of 200 °C, 8 MPa: at low Cu content (x = 5, 3), methane was the main product, while at high copper content (x = 1), the methanol selectivity reached 85.5% (200 °C) with a reaction rate of 13.3 mmol gCu−1 h−1, and the activity remained stable without degradation after three cycles. However, La2O2CO3, as an irreducible, stable, and basic metal oxide, has a limited ability to generate surface oxygen vacancies (OVs), which restricts its efficiency in activating CO2 molecules at low temperatures. Zhang et al. disrupted the dyadicity of the La2O2CO3 lattice by doping rare earth elements, thereby significantly enhancing the ability to form OVs and ultimately achieving efficient conversion of CO2 to methanol.32 Among these rare earth-doped catalysts, Nd uniformly doped La2O2CO3 catalyst showed the best performance (Fig. 15a). Under the conditions of 260 °C, 3 MPa, and a space velocity of 36000 mL gcat−1 h−1, the methanol space-time yield (STY) of Cu/LOC:Nd reached 9.9 molMA h−1 molCu−1, significantly higher than that of undoped Cu/LOC (7.2 molMA h−1 molCu−1) and commercial CuZnAl (3.3 molMA h−1 molCu−1) (Fig. 15b). Mechanistic studies indicated that the reaction follows a dual-route: (1) The formate route (CO2 → HCO3* → HCOO* → H3CO* → CH3OH), with HCOO* (1600, 1330 cm−1) detected at 140 °C and H3CO* (2930 cm−1) appearing at 180 °C in DRIFTS (Fig. 15c); (2) the RWGS + CO hydrogenation route, confirmed by CO/H2 switching experiments (Fig. 15d). Upon switching from CO to H2, distinct peaks of H3CO* at 2932 and 2933 cm−1 were observed, indicating the occurrence of the RWGS reaction followed by CO hydrogenation. The CO hydrogenation activity of Cu/LOC:Nd (5.35 molMA h−1 molCu−1) was higher than that of Cu/LOC (3.24 molMA h−1 molCu−1) (Fig. 15e). Structural characterization revealed that Nd doping disrupted the lattice symmetry of pure LOC, reducing the oxygen vacancy formation energy from 0.62 eV to 0.04 eV (Fig. 15f), thereby significantly promoting oxygen vacancy generation and increasing the Cu0 content. The enhanced CO2 adsorption capacity, together with the efficient promotion of H2 dissociation by Cu0, synergistically accelerated the methanol formation rate (Fig. 15g and h). This strategy provides a new paradigm for defect regulation and metal state optimization in the design of irreducible oxide-supported catalysts. Similar to La2O2CO3, PrOx, as an important non-cerium rare earth oxide, has attracted increasing attention in recent years for CO2 hydrogenation to methanol. Unlike La2O2CO3, PrOx possesses a reversible Pr3+/Pr4+ redox property, which, combined with oxygen vacancies and metal–support interfacial charge transfer, synergistically promotes CO2 adsorption and the activation of key intermediates. Zhang et al. developed a ZnO-modified, defect-rich Pr2O3 nanosheet–supported copper-based catalyst for efficient CO2 hydrogenation to methanol.224 The catalyst exhibited excellent catalytic performance under reaction conditions of 260 °C and 3.0 MPa, achieving a CO2 conversion of 12.7% and a methanol selectivity of 75%. Structural characterization revealed that the catalyst possessed highly dispersed metallic Cu0 active sites, abundant defective Pr3+–Vo–Pr3+ structures, and favorable interfacial Cu–O–Pr sites. Further investigations demonstrated that CO2 hydrogenation over this catalyst proceeded via both the formate route and the RWGS + CO-Hydro route. The synergistic interaction between surface defect structures and Cu–Pr2O3 interfacial sites significantly enhanced the adsorption and activation of CO2 and CO intermediates, thereby accelerating the CO2 hydrogenation process toward methanol formation.
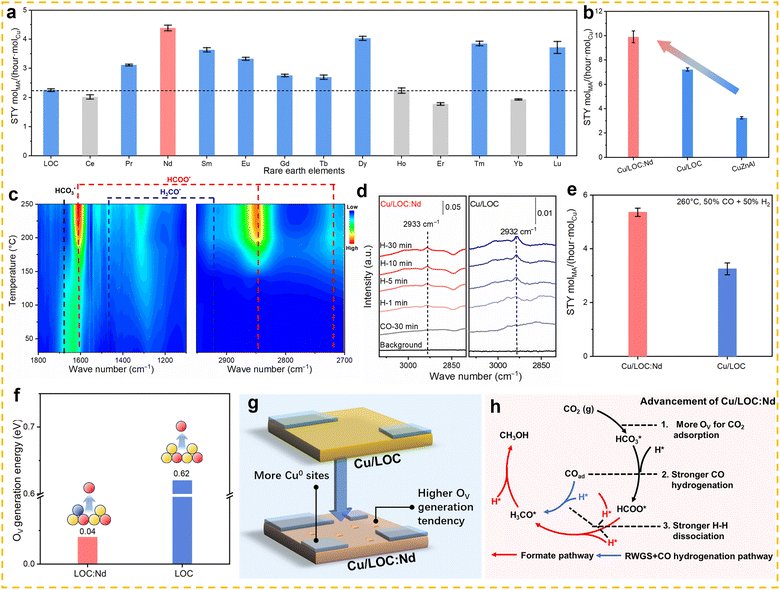 |
| | Fig. 15 (a) Normalized STY by Cu weight for Cu/LOC and rare earth atom–doped Cu/LOC:X catalysts. (b) Normalized STY by Cu weight for Cu-based catalysts on different supports. (c) Operando DRIFTS and corresponding variation of the Cu/LOC:Nd catalyst with the temperature ranging from 30 to 260 °C. (d) DRIFTS spectra of switching the feed gas from CO to H2 for Cu/LOC and Cu/LOC:Nd catalysts. (e) Catalytic performance of CO hydrogenation over Cu/LOC and Cu/LOC:Nd catalysts. (f) Ov generation energy of LOC:Nd and LOC. The yellow, red, blue, and black balls represent La, O, Nd, and C atoms. (g) Nd doping effect and (h) the proposed reaction loop for the Cu/LOC:Nd catalyst. Reprinted with permission from ref. 32. Copyright (2024) American Association for the Advancement of Science. | |
Non-cerium rare earth promoters (such as La and Sm) exhibit remarkable promotional effects in the CO2 hydrogenation to methanol process. Their introduction can enhance the dispersion of active metal species and strengthen the adsorption and activation of CO2, thereby effectively improving the catalytic activity. Chen et al. constructed a highly dispersed Cu-LaOx interface catalyst (Cu1La0.2/SBA-15) by embedding LaOx into the pore walls of SBA-15 and loading copper nanoparticles.191 This catalyst demonstrated excellent performance in CO2 hydrogenation to methanol. Under conditions of 240 °C and 3 MPa, the methanol selectivity reached 81.2%, and the space-time yield was 190.8 mg gcat−1 h−1 (nearly 10 times higher than Cu1La0/SBA-15 without La), with stable activity over 100 h. Structural characterization indicates that La species not only significantly enhance CO2 adsorption capacity but also improve Cu dispersion, forming uniformly dispersed active sites. The Cu–LaOx interfaces formed play a crucial role in methanol synthesis rates. Kinetic analysis revealed that the apparent activation energy for methanol production on Cu1La0.2/SBA-15 was 62.5 kJ mol−1 (compared to 96.6 kJ mol−1 for pure Cu), with a TOF of 20.4 h−1. Yamamura et al. prepared a series of 14 wt% Cu/a-ZrO2 (a-: amorphous) catalysts modified with different amounts of the Sm additive by a co-impregnation method and applied them to CO2 hydrogenation to methanol.225 The results showed that as the Sm content increased, the CO2 conversion first increased and then gradually decreased. Among these catalysts, the Cu/a-ZrO2 catalyst doped with 5–6 mol% Sm exhibited the best performance, achieving a methanol production rate of 3.7 mmol gcat−1 h−1, which was approximately 20% higher than that of Cu/a-ZrO2. Further investigation revealed that an appropriate amount of Sm doping enhanced the dispersion of copper nanoparticles, increased the number of Cu–ZrO2 interfacial active sites, and improved CO2 adsorption and activation capabilities, thereby significantly boosting the catalytic performance. However, excessive Sm loading led to the sintering of Cu nanoparticles and a reduction in active sites, resulting in a decline in overall catalytic activity.
4.4. CO2 hydrogenation to ethanol
Ethanol is an important basic chemical with multiple applications: it serves not only as a clean fuel additive, but also as a fundamental raw material for chemical products such as acetaldehyde and acetic acid, while additionally functioning as a highly effective disinfectant and industrial solvent.226,227 Notably, the technological route of producing ethanol through CO2 hydrogenation offers a promising industrial solution for converting greenhouse gases into high-value fuels and chemicals. This reaction process typically involves three key steps: First, CO2 is adsorbed and activated on the catalyst surface to form C1 intermediates (e.g., *HCOO, * CHxO, *CO, and *CHx); Second, the C1 intermediates undergo high-energy-barrier C–C coupling to generate C2 intermediates (e.g., *CH3CO and *CH3COO), a process requiring specific catalytic active sites to facilitate bond formation; finally, the C2 intermediates are hydrogenated to ethanol.228 However, the activation of CO2 requires a large energy input due to its extremely high thermodynamic stability, and the complex reaction network involving multiple intermediates makes selective control challenging. How to synergistically optimise the two energy-intensive steps of CO2 activation and C–C coupling, while effectively suppressing competing side reactions such as CO2 methanation, methanol synthesis, and RWGS reaction, is a key issue for this conversion process.229,230 To this end, catalyst design requires the construction of a multifunctional active centre system to achieve the following three key functions: (1) efficient CO2 activation; (2) lower the energy barrier of the C–C coupling step; (3) selective inhibition of side-reaction routes, which can comprehensively enhance the selectivity of the ethanol synthesis and the overall process efficiency. In recent years, researchers have developed various catalysts for the hydrogenation of CO2 to produce ethanol, including Cu-based,231–233 Co-based,217,234 Fe-based,235,236 precious metal-based catalysts,237,238 and multi-component composite catalytic systems.219,239 Among these, rare earth components are frequently used as supports or promoters due to their variable oxidation states and strong coordination abilities, enabling the regulation of catalyst structure and enhancing the activity and selectivity of the CO2 hydrogenation to ethanol reaction. Currently, these rare earth-containing catalysts are generally classified into two major categories: cerium-based catalysts and non-cerium-based catalysts (Table 3).
Table 3 Summary of the rare earth-based catalysts for the hydrogenation of CO2 to ethanol
| Catalysts |
H2/CO2 ratio |
Space velocitya |
T/°C |
P (MPa) |
CO2 conv. (%) |
EtOH sel. (%) |
Ref. |
|
(W) = WHSV = mass flow rate/catalyst mass, mL gcat−1 h−1, (G) = GHSV = volume flow rate/bed volume, h−1.
|
| Pd2Ce@Si |
3 |
3000 |
240 |
3 |
6.9 |
97.8 |
241
|
| La-CoGaO |
3 |
3000 |
240 |
3 |
9.8 |
65.8 |
242
|
| Co/La4Ga2O9 |
3 |
3000 |
270 |
3.5 |
4.6 |
34.7 |
217
|
| Co/La4Ga2O9 |
3 |
3000 |
280 |
3 |
9.6 |
23.3 |
217
|
| Rh1/CeTiOx |
3 |
∼ |
250 |
3 |
6.3 |
99.1 |
240
|
| 0.1% Rh/CeO2 |
3 |
∼ |
250 |
3 |
3.2 |
74.3 |
218
|
| 5La-CuFeOx |
3 |
3000 |
320 |
3 |
39.8 |
33.8 |
235
|
| 5La-CuFeOx |
3 |
6000 |
320 |
3 |
40.32 |
28.53 |
235
|
| 5La-CuFeOx |
3 |
12000 |
320 |
3 |
35.01 |
26.14 |
235
|
| 2Rh0.5Fe0.5Na/CeO2 |
3 |
4000 |
250 |
3 |
13.0 |
29.8 |
219
|
| 2Rh0.5La/CeO2 |
3 |
6000 |
250 |
3 |
6.9 |
10.3 |
219
|
| 2Rh0.5Fe/CeO2 |
3 |
6000 |
250 |
3 |
8.4 |
16.5 |
219
|
| 2Rh0.5Na/CeO2 |
3 |
6000 |
250 |
3 |
7.9 |
23.0 |
219
|
| 2Rh0.1Fe0.5Na/CeO2 |
3 |
6000 |
250 |
3 |
9.6 |
15.3 |
219
|
| 2Rh0.5Fe0.5Na/CeO2 |
3 |
6000 |
250 |
3 |
9.8 |
21.5 |
219
|
| Rh/CeO2–SiO2 |
3 |
∼ |
240 |
5 |
8.3 |
6.1 |
243
|
| Rh/CeO2-r |
3 |
6000 |
250 |
3 |
11.2 |
20.9 |
220
|
| Rh/CeO2-c |
3 |
6000 |
250 |
3 |
5.3 |
7.6 |
220
|
| Rh/CeO2-p |
3 |
6000 |
250 |
3 |
12.0 |
0.8 |
220
|
| Pd2/CeO2 |
3 |
3000 |
240 |
3 |
9.2 |
99.2 |
221
|
4.4.1. Cerium-based catalysts for CO2 hydrogenation to ethanol reaction.
Lou et al. reported a CeO2-supported Pd dimer catalyst (Pd2/CeO2) that efficiently catalyses CO2 hydrogenation to ethanol under conditions of 240 °C and 3 MPa.221 The catalyst achieved an ethanol selectivity of 99.2%, a space-time yield of 45.6 g gPd−1 h−1, a CO2 conversion rate of 9.2%, and a turnover frequency (TOF) based on Pd atoms of 211.7 h−1. To further improve the performance of cerium-based catalysts, Zheng et al. tuned the CeO2 supports to obtain Rh1/CeTiOx catalysts that exhibited record-breaking catalytic performance in the hydrogenation of CO2 to ethanol (Fig. 16f):240 an ethanol selectivity of 99.1%, a turnover frequency (TOF) of 493.1 h−1, which was significantly higher than those of Rh1/TiO2 and Rh1/CeO2, respectively, and it maintained stability over 5 cycles (Fig. 16a–c). In situ DRIFTS results indicate that at low temperatures (50–150 °C), the Rh1/CeTiOx catalyst mainly exhibits intermediates generated from CO2 adsorption and subsequent hydrogen transfer, including CO3*, HCO3*, and HCOO*. When the temperature increases above 200 °C, these intermediates gradually disappear, while new species such as CO*, C2H5O*, and CH3* emerge (Fig. 16d). Combined with theoretical calculations, it is revealed that CO2 is first adsorbed and activated at the Lewis-acid-base pairs, forming the key intermediate HCOO*, which is subsequently hydrogenated stepwise into HCOOH*, H2COOH*, CH2O*, CH2OH*, CH3O, and CH3OH*. Among these intermediates, CH2OH* possesses the longest and weakest C–O bond, making it the critical precursor for generating CHx* (Fig. 16e). Notably, CO2* can also be hydrogenated into COOH*, which further produces CO*. The resulting CO* can then couple with CHx* to form CHxCO*, which undergoes a series of hydrogenation steps to ultimately yield ethanol (Fig. 16g and h). This work provides a new paradigm for atomic-level active site design and support regulation in the directional conversion of CO2. In addition, researchers have also attempted to design multi-component synergistic catalytic systems to improve the performance of CO2 hydrogenation to ethanol. Ji et al. successfully developed the 2Rh0.5Fe0.5Na/CeO2 catalyst by systematically screening different transition metals and alkali metal promoters and optimizing the doping levels of Rh, Fe, and Na.219 Under reaction conditions of 250 °C and 3 MPa, the optimized catalyst demonstrated excellent performance: 29.8% ethanol selectivity, 13.0% CO2 conversion rate, and an ethanol space-time yield (STY) of 116.7 mmol gRh−1 h−1. Characterization results revealed that the modification of Fe and Na not only significantly promoted the dispersion and stabilization of Rh species, but also enhanced the adsorption and dissociation of CO2, thereby improving the catalytic activity. In situ DRIFT analysis further confirmed that the catalyst could stabilize adsorbed species, such as m-CO32− and CO3H−, and promote the coupling of HCOO* and CO* with CHx* species. Compared to other catalysts, the optimized catalyst significantly outperformed existing Rh-based catalysts, demonstrating superior performance in the CO2 hydrogenation to ethanol process. Graciani et al. studied the surface chemical mechanisms of the Pt/CeOx/TiO2(110) catalyst in CO2 hydrogenation to alcohols using AP-XPS and DFT calculations.238 The catalyst demonstrates a unique ability to activate CO2, with the Pt–CeOx–TiO2 interface achieving strong CO2 adsorption through the formation of Pt–C and C–O–Ce bonds. Under CO2/H2 conditions, the active sites exhibit a mixed state of Ti4+/Ti3+, Ce3+, and Pt0/Pt+, resulting in a 21% selectivity for ethanol (relative to methanol), with catalytic activity 2.6–3.8 times that of the Cu/ZnO system. After the introduction of H2O, the surface coverage of carbon-containing intermediates (CH3O, HCOO, CO3, and CHx) significantly increases, and ethanol selectivity rises to 38%. DFT calculations confirm that the synergistic interaction between Ce3+ and Pt promotes methanol synthesis through the CHxO intermediate route, while the addition of H2O facilitates the formation of CHx species, further promoting C–C coupling and nearly doubling the EtOH/MeOH ratio.
 |
| | Fig. 16 (a) Comparison of the activity and selectivity (based on the number of moles of carbon) on the various catalysts. Reaction conditions: catalyst (30 mg), H2O (20 mL), 250 °C, initial pressure (3.0 MPa, H2/CO2 = 3:1), 5 h, 400 rpm. (b) Comparison of the space-time yield of ethanol obtained in this work with other Rh-based catalysts reported in literature. (c) Recycled testing of the synthesized Rh1/CeTiOx catalyst for a reaction of 5 hours at five runs. Reaction conditions: catalyst (30 mg), H2O (20 mL), 250 °C, initial pressure (3.0 MPa, H2/CO2 = 3:1), 5 h, 400 rpm. (d) In situ DRIFT spectra of CO2 hydrogenation with Rh1/CeTiOx catalyst bubbled in aqueous phase at different temperatures. (e) The calculated length of the C–O bond in CHxO* and CHxOH* over the Rh1/CeTiOx model. (f) EDX mapping of Rh (green), Ti (yellow), Ce (red), and O (blue) in Rh1/CeTiOx. (g) Free-energy diagram of the CO2 hydrogenation reaction pathways over the Rh1/CeTiOx catalyst. (h) The illustrated catalytic cycle of ethanol formation from CO2 hydrogenation on the Rh1/CeTiOx catalyst. The inset figure shows the structure of the Rh1/CeTiOx catalyst. Reprinted with permission from ref. 240. Copyright (2022) Wiley-VCH. | |
4.4.2. Non-cerium-based catalysts for CO2 hydrogenation to ethanol reaction.
In the catalytic system for CO2 hydrogenation to ethanol, there has been little research on rare earth element catalysts other than cerium, with existing work mainly focusing on La-based catalysts. An et al. prepared the Co/La4Ga2O9 catalyst by reducing the LaCo0.5Ga0.5O3 precursor.217 Under conditions of 270 °C, 3.5 MPa, and 3000 mL g−1 h−1, the catalyst achieved 34.7% ethanol selectivity and 4.6% CO2 conversion, with a total alcohol selectivity of 50%. Characterization showed that the La4Ga2O9 support facilitated the RWGS reaction, converting CO2 to CO. The CO then migrated to Co0–Co2+ on Co NPs, where Co0–Co2+ active sites (formed through electron transfer from Co to the supports) catalyzed the hydrogenation of CO to produce ethanol. The ethanol selectivity was significantly better than that of Co-based systems reported in the literature. As pressure increased, the selectivity for alcohols significantly improved, while the selectivity for hydrocarbons decreased. However, temperatures above 280 °C promoted methane formation. CO-TPD confirmed that the CO desorption temperature from the La4Ga2O9 surface (400 °C) was higher than the reaction temperature (270 °C), which facilitated CO migration to the Co sites for synthesis. In the 100 h stability test, the catalysts maintained a stable CO2 conversion rate, but the ethanol/methanol ratio decreased due to an increase in the Co0/Co2+ ratio. In another study, He et al. prepared La-doped CuFeOx catalysts using a mechanochemical method, which demonstrated excellent performance in CO2 hydrogenation to ethanol.235 The optimized 5La-CuFeOx catalyst achieved a CO2 conversion rate of 39.8% and an ethanol selectivity of 33.8%, with an ethanol yield of 13.5% under conditions of 320 °C, 3 MPa, and 3000 mL gcat−1 h−1—significantly outperforming the undoped CuFeOx. Structural characterization revealed that La doping not only enhanced the interaction between Cu and Fe species (as evidenced by EDX mapping), thereby exposing more Cu0–Fe5C2 interfaces, but also increased the oxygen vacancy content from 31.0% to 42.0% as quantified by XPS, which in turn markedly promoted CO2 adsorption and activation. Key reaction mechanism studies showed that the introduction of La promotes electron transfer and optimizes the adsorption behavior of CO intermediates, leading to an optimal match between the formation rates of CHx and C(H)O intermediates. The subsequent C–C coupling at the abundant Cu0–Fe5C2 interfaces enables the highly efficient synthesis of ethanol. Moreover, this catalyst maintained 36.66% CO2 conversion and 27.65% ethanol selectivity in a 120 h stability test. Moreover, Zheng et al. employed LaCo1−xGaxO3 perovskite as a precursor and prepared a novel Co/La2O3–La4Ga2O9 catalyst via reduction, which was applied to the direct hydrogenation of CO2 to ethanol.242 When the Co/Ga atomic ratio was 7:3, the obtained catalyst exhibited the best performance, achieving a CO2 conversion of 9.8%, a total alcohol selectivity of 74.7%, and an ethanol fraction as high as 88.1% among the alcohol products. The study revealed that the synergistic interaction between surface Co0 and Coδ+ species is the key factor enabling the catalyst to achieve high activity and selectivity.
4.5. CO2 hydrogenation to olefins and aromatics
The production of olefins and aromatics using CO2 hydrogenation provides considerable economic benefits and helps to promote industrial carbon neutrality and renewable carbon cycling, making it an important way to achieve sustainable development. The products obtained in this process, such as ethylene, propylene, butene and aromatics,244,245 can be used as high-value feedstocks for petrochemicals,246 with market values much higher than those of C1 products such as CO and CH4.247,248 Recently, the hydrogenation of CO2 under heterogeneous catalysis to produce olefins and aromatics has been extensively studied. This process involves two primary routes: (1) the methanol route, where CO2 is reduced to methanol intermediates (via formate/CO) and subsequently, on acidic sites (e.g., zeolites), methanol is transformed through methanol-to-olefins (MTO) or methanol-to-aromatics (MTA) to form olefins and aromatics; (2) the CO2-FTS route, where CO2 is first converted to CO via RWGS reaction, followed by chain propagation through Fischer-Tropsch synthesis (FTS).248,249 However, both routes face critical challenges. The major challenge of the methanol route lies in the mismatch of optimal reaction conditions between the two steps. Methanol synthesis typically requires low temperatures and high pressures, whereas the subsequent MTO/MTA reactions demand higher temperatures, moderately strong acidity, and low water activity. The significant mismatch between the optimal conditions of the two steps often leads to the accumulation or depletion of methanol intermediates, thus reducing the selectivity and yield of olefins and aromatics.248 In the CO2-FTS route, the product distribution of FTS follows the Anderson–Schulz–Flory (ASF) distribution law, resulting in limited selectivity for the target hydrocarbons. In addition, the route is usually accompanied by higher CO and CH4 selectivity,249–251 increasing the difficulty and cost of product separation. Together, these unfavourable factors constrain its industrial application.244 In recent years, to address these challenges, researchers have extensively investigated the use of rare earth-based catalysts for CO2 hydrogenation to produce olefins and aromatic hydrocarbons.
4.5.1. Cerium-based catalysts for CO2 hydrogenation to olefins and aromatics reaction.
In the CO2 hydrogenation to olefins and aromatics reaction, the morphology of the CeO2 support is crucial for catalytic performance. Torrente-Murciano et al. synthesized Fe/CeO2-particle, Fe/CeO2-rod, and Fe/CeO2-cube catalysts,252 which exhibited distinct patterns of hydrocarbon selectivity variation: the selectivity of the former two (particles and rods) generally increased with rising reaction temperature; conversely, the selectivity of Fe/CeO2-NC remained largely unaffected by temperature changes. Notably, Fe/CeO2-NR demonstrated the highest hydrocarbon selectivity among the three catalysts investigated. In recent years, tandem catalysts have garnered significant attention from researchers due to their higher selectivity and catalytic activity. Classic tandem catalysts typically contain two different active centers with specific functions, which are arranged adjacent to each other. Sedighi et al. developed a CuCe/SAPO-34 hybrid catalyst system,253 achieving the direct conversion of CO2 by coupling the CO2-to-methanol and methanol-to-olefins (MTO) processes. Under the optimum reaction conditions, olefin selectivity and CO2 conversion were 61.83% and 13.15%, respectively. The NiCu/CeO2-SAPO-34 bifunctional catalyst developed by Ghasemi et al. further enhances catalytic activity and selectivity,254 and the catalyst achieved 76.6% selectivity for light olefins (C2H4 22.7%, C3H6 35.5%, and C4H8 18.4%), a CO2 conversion rate of 15.3%. Additionally, the catalyst maintained stable activity during 90 h of continuous operation, demonstrating its potential for industrial applications. Wang et al. prepared a Zn0.5Ce0.8Zr1.8O4/H-RUB-13 catalyst, which achieved a CO2 conversion rate of 30.1%, C2–C4 olefins selectivity of 72.7% (C3 = 40.3%, C4 = 25.0%), and CO selectivity of 26.5% under conditions of 350 °C, 3.5 MPa, and H2/CO2 = 6 (Fig. 17b and c).255 The C2–C4 olefins yield was 2.3–3.3 times higher than the values in the literature at that time. In situ DRIFTS results revealed that, during the reaction, the Zn0.5Ce0.2Zr1.8O4 solid solution surface sequentially generated carbonate species (1440–1540 cm−1 and 1336 cm−1), formate species (1598 and 1373 cm−1), and methoxy species (1072 cm−1), with their intensities gradually increasing over time (Fig. 17d). This indicates that CO2 was activated at oxygen vacancies and converted to methanol via the formate–methoxy pathway. Meanwhile, the CO peaks associated with the RWGS route (2076, 2110, 2180 cm−1) were relatively weak, suggesting that CO formation was suppressed (Fig. 17e). Consistently, the results from 13C CP/MAS NMR spectroscopy and GC-MS further corroborated this conclusion: at 200 °C, a strong peak at 167 ppm corresponded to formate species, while a weaker peak at 55 ppm corresponded to methoxy species (Fig. 17f). Moreover, significant amounts of 13C-labeled methanol and DME were detected in the products, whereas the 13CO signal was very weak (Fig. 17g and h). These results consistently demonstrate that this catalytic system predominantly produces methanol through the formate–methoxy pathway, while the RWGS reaction is markedly suppressed. DFT calculations showed that the free energy barrier of this route (161 kJ mol−1) is significantly lower than that of the RWGS reaction route (216 kJ mol−1), highlighting its superiority in the reaction process (Fig. 17i and j). Subsequently, the methanol generated was further converted into light olefins over H-RUB-13. Moreover, by tuning the amount, strength, and distribution of acid sites in H-RUB-13, the alkene-based cycle in MTO was significantly enhanced, thereby promoting the formation of more propylene and butylene. This study paves the way for rational design of high-efficiency catalysts to regulate product distribution in CO2-to-light-olefins conversion (Fig. 17a). The above conversion processes all follow the methanol route, in which CO2 is first converted into methanol on metal oxides, while zeolites are primarily responsible for subsequently converting methanol into olefins and aromatics. In contrast to this conversion pathway, research on CeO2-based catalysts that directly synthesize olefins and aromatics via the RWGS-FTS route has also attracted widespread attention. Xie et al. developed a CeO2–Pt@mSiO2–Co tandem catalyst with a well-defined interface spatial arrangement (Fig. 18a–g).256 The Pt/CO2 interface catalyzed the RWGS reaction to produce CO, while the Co/mSiO2 interface catalyzed the Fischer–Tropsch (F–T) reaction to generate C2–C4 hydrocarbons. Under the conditions of 250 °C and H2/CO2 = 3, the catalyst achieved a selectivity of 40% for C2–C4 hydrocarbons and 60% for methane (Fig. 18h). When the H2/CO2 ratio was optimized to 0.3, the selectivity for C2–C4 hydrocarbons increased to 59%, while the selectivity for methane decreased to 41% (Fig. 18i). Comparative experiments showed that single-interface catalysts (CeO2–Pt@mSiO2, CeO2@mSiO2–Co) and physical mixtures were unable to efficiently generate C2–C4 hydrocarbons (Fig. 18h). Moreover, the CeO2–Pt@mSiO2–Co tandem catalyst maintained stable catalytic activity and product selectivity during continuous operation for up to 40 hours, showing no noticeable deactivation or fluctuation (Fig. 18j). This work confirms the critical role of interface spatial arrangement in multi-step tandem reactions.
 |
| | Fig. 17 (a) Schematic illustration of the catalytic reaction of Zn0.5Ce0.2Zr1.8O4/HRUB-13 converting CO2 into light olefins. (b) and (c) CO2 conversion and product distribution at different H2/CO2 ratios over the Zn0.5Ce0.2Zr1.8O4/HRUB-13(200) composite catalyst. (d) and (e) Time-Dependent DRIFT Spectra for CO2 Hydrogenation on Zn0.5Ce0.2Zr1.8O4 Solid Solution in the range of 1000–1800 cm−1 (d) and in the range of 2000–2200 cm−1 (e). (f) 13C CP/MAS NMR spectrum of the organic species formed on the Zn0.5Ce0.2Zr1.8O4 solid solution in 13CO2 hydrogenation at 200 °C for 30 min (the peaks at about 167 and 55 ppm are attributed to formate and methoxyl species, respectively). (g) GC-MS diagram of CO2 and CO in effluents (insert is the MS spectra of 13C-labeled CO2). (h) GC-MS diagram of methanol and DME in effluents (insert is the MS spectra of 13C-labeled methanol and DME). The integration peak area in GC-MS, representing the relative content, of 13C-labeled carbon monoxide and (methanol + DME) is 0.05 and 0.47, respectively. (i) Free energy, enthalpy, and entropy profiles for CO2 hydrogenation to methanol at 300 °C. (j) Free energy, enthalpy, and entropy profiles for RWGS reaction at 300 °C. Reprinted with permission from ref. 255. Copyright (2020) Elsevier. | |
 |
| | Fig. 18 (a) Schematic illustration of the catalytic reaction of CeO2–Pt@mSiO2–Co for converting CO2 into light olefins. (b)–(g) Elemental mapping of CeO2–Pt@mSiO2–Co with energy dispersive X-ray spectroscopy (EDS) (b). Corresponding EDS elemental mapping for (c) Ce, (d) Pt, (e) O, (f) Si, and (g) Co, respectively. Scale bar: 20 nm. (h) Catalytic performance of single-interface catalysts CeO2–Pt@mSiO2, CeO2@mSiO2–Co, physical mixture catalyst and tandem catalyst CeO2–Pt@mSiO2–Co. (i) CO2 conversion and hydrocarbons distribution at different H2/CO2 ratios over the tandem catalyst at 250 °C. (j) Stability test of CeO2–Pt@mSiO2–Co catalyst. Reprinted with permission from ref. 256. Copyright (2017) American Chemical Society. | |
4.5.2. Non-cerium-based catalysts for CO2 hydrogenation to olefins and aromatics reaction.
Recent studies demonstrate that composite catalytic systems incorporating non-Ce-based materials with zeolites (e.g., HZSM-5 and SAPO-34) exhibit superior performance in CO2 hydrogenation to olefins and aromatics, where their synergistic effects further enhance selectivity. Li et al. developed a 6.7 wt% ZnO-Y2O3/SAPO-34 bifunctional catalyst prepared by a coprecipitation method,257 which achieved a CO2 conversion rate of 27.6% and a light olefin selectivity of 83.9% in hydrocarbon products at a reaction temperature of 390 °C, with methane selectivity only at 1.8%. Comparative experiments revealed that the performance of this catalyst was significantly better than that of the physically mixed ZnO + Y2O3 (with a CO2 conversion rate of 17.4%) and the 6.7 wt% ZnO/Y2O3 system prepared by impregnation. The study found that ZnO and Y2O3 play different roles in the mixed oxide, with Y2O3 promoting the adsorption and activation of CO2, while ZnO is related to the adsorption and activation of H2, and the synergistic effect of both enhances catalytic activity.
Non-cerium rare earth metals (such as La) are also commonly used as catalyst promoters to enhance catalytic activity and increase the proportion of olefins and aromatic hydrocarbons in the product. Metal promoters are typically introduced through impregnation, co-impregnation, or co-precipitation methods. Ferraz et al. investigated the effect of catalyst basicity on CO2 hydrogenation to methanol and subsequent MTG reactions for converting to gasoline-range hydrocarbons by preparing In2O3 catalysts modified with different promoters (Ga, Nb, La, and Mg).258 The results showed that the La/In2O3/HZSM-5 catalyst with moderate basicity also performs well in generating C5+ hydrocarbons, especially under high-temperature conditions, where its C5+ selectivity is similar to that of Ga/In2O3/HZSM-5. Zhang et al. synthesized La-modified ZnZrOx oxides using a one-pot sol–gel method and coupled them with H-SAPO-34 or H-SAPO-18 zeolites to form bifunctional catalysts.259 The study found that the introduction of an appropriate amount of La promotes the formation of more surface oxygen vacancies in ZnZrOx, enhancing CO2 adsorption and activation, and facilitating methanol production. In the CO2 hydrogenation reaction, ZnZrOx(0.3La) displayed the highest CO2 conversion rate and methanol selectivity, reaching 6.9% and 98.3%, respectively. When ZnZrOx(0.3La) was combined with H-SAPO-34 or H-SAPO-18 molecular sieves, the catalyst's C2=–C4= hydrocarbon selectivity was 83.2% and 77.5% in hydrocarbons, respectively. These results suggest that regulating the concentration of surface oxygen vacancies in metal oxides is an effective strategy for designing efficient bifunctional catalysts, thus advancing research on CO2 hydrogenation to produce light olefins. Additionally, the study demonstrated that the ZnZrOx(0.3La)/H-SAPO-34 composite catalyst exhibited excellent catalytic stability, maintaining a high olefin selectivity of 76.2% after 200 h of reaction, proving its effectiveness and stability in long-term reactions.
5. Application of rare earth-based catalysts to plastic waste recycling
With the advancement of plastics recycling technology, rare earth-based catalysts have emerged as a key technology driving the chemical recycling process for a variety of plastics. At present, oxygen-containing polymers—including polylactic acid, polycarbonates, and polyurethanes—can be efficiently broken down into their monomeric units via nucleophilic attack on the carbonyl (CO) bonds, and these monomers can then be repolymerized to regenerate high-quality, virgin-equivalent plastics. In contrast, due to the stability of the C–C bond, polyolefins can only be used for the production of commercially valuable liquid fuels and hydrogen-enriched gases. In this section, we will systematically summarise the applications of rare earth-based catalysts in the chemical recycling of different types of plastics, including polyethylene (PE), polypropylene (PP), polyethylene terephthalate (PET), polylactic acid (PLA), polyamides (PA), polycarbonate (PC), polyurethane (PU) and mixed plastic. We will also illustrate the recent research advances of these catalysts in these applications and focus on the functional mechanism of the rare earth components in enhancing reactivity, selectivity, and stability.
5.1. PE recycling
PE, as the most widely produced plastic globally, has garnered significant attention for its chemical recycling technologies due to the challenges posed by its inert C–C/C–H bonds.260 The chemical structure of PE consists of repeating –[CH2–CH2]– units, with its C–C and C–H bonds exhibiting high bond energy, resulting in strong chemical inertness and resistance to degradation.261 Depending on synthesis conditions and molecular structure, PE is primarily categorized into low-density polyethylene (LDPE), linear low-density polyethylene (LLDPE), and high-density polyethylene (HDPE).261,262 LDPE, characterized by long-chain branching, offers excellent flexibility and is commonly used in films and packaging.261,263 LLDPE, with predominantly short-chain branching, combines strength and toughness,261 while HDPE, with its linear structure and high crystallinity, is suitable for manufacturing containers and pipes.264 Currently, the chemical recycling technologies for PE include pyrolysis, catalytic pyrolysis, hydrogenolysis, hydrocracking, and oxidation.265,266 This section will focus on providing an overview of three general techniques: catalytic pyrolysis, hydrogenolysis, and hydrocracking.
5.1.1. Catalytic pyrolysis.
Conventional pyrolysis requires high temperatures (500–800 °C) to cleave C–C bonds,266,281 producing mixed hydrocarbon fuels but suffering from high energy consumption and poor product selectivity. In contrast, catalytic pyrolysis utilizing acid active sites can not only reduce reaction temperatures to 280–500 °C but also tailor product distribution.266 Currently, the reported catalytic cracking catalysts primarily include various acidic zeolites (such as ZSM-5, HZSM-5, HY, and USY),282 clays, and fluid catalytic cracking (FCC) systems.266 The catalytic pyrolysis process primarily follows the carbocation mechanism,283 and its product distribution is not only influenced by reaction temperature and time but also closely related to the catalyst's specific surface area, pore structure, and acid strength. In this process, rare earth elements (such as Ce and La) are often used as additives to participate in the catalytic pyrolysis reaction. Luo et al. developed a CuCe/ZSM-5 catalyst for efficient catalytic pyrolysis of PE into high-quality oil products.284 The bimetallic Cu/Ce modification enabled uniform dispersion of metal species at the nanoscale while creating optimal steric hindrance effects. Compared to unmodified and single-metal-modified ZSM-5, the Cu–Ce dual modification significantly enhanced short-chain olefin selectivity, increasing aliphatic product yield from 58.5% to 76.1%. Mechanistic studies revealed that the synergistic effect between Cu0 and CeOx promoted C–C bond scission activity to facilitate short-chain olefin formation, while the well-tuned steric hindrance effectively suppressed polycyclic aromatic hydrocarbon (PAH) generation. Building on this, in the present review, the term catalytic pyrolysis is used in a broad sense to encompass not only the conventional oxygen-free thermal catalytic decomposition of polymers but also pyrolysis–reforming systems, where the pyrolysis products are further converted into value-added gaseous products (such as syngas or hydrogen) under specific atmospheres (e.g., steam-rich or mildly oxidative). Such processes combine the advantages of catalytic cracking and reforming, allowing flexible production of liquid hydrocarbons, carbon materials, or hydrogen-rich gases depending on the reaction conditions and catalyst design. Huang et al. explored the influence of different oxide and zeolite supports (Al2O3, MgO, CeO2, Y2O3, TiO2, SiO2, and ZSM-5) on the performance of Ni-based catalysts in the steam reforming of LDPE-derived pyrolysis vapors for syngas production.285 The study revealed that the catalytic performance was highly dependent on the physicochemical properties of the supports, which significantly influenced both activity and carbon deposition formation. Among the examined catalysts, Ni/Y2O3 exhibited excellent syngas yield and reactivity, below Ni/CeO2, while Ni/ZSM-5, characterized by moderate metal–support interactions, showed the lowest activity but superior resistance to carbon deposition. Moreover, Suarez et al. systematically investigated the catalytic performance of Ni-based catalysts supported on various oxides for the oxidative steam reforming (OSR) of HDPE-derived pyrolysis vapors and further examined the influence of introducing rare-earth oxide promoters on catalytic activity and stability.286 In the unpromoted system, the Ni/ZrO2 catalyst exhibited the highest reforming performance at 700 °C and a space velocity of 3.12 gcat min gHDPE−1, achieving a conversion rate of 92.2% and an H2 yield of 12.8%, both markedly higher than those of the commercial Ni/Al2O3 and Ni/SiO2 catalysts. This superior performance was attributed to the outstanding redox capability of the ZrO2 support and its oxygen-vacancy-mediated activation of reactive oxygen species, which effectively promoted hydrocarbon conversion and the reforming reaction. When La2O3 and CeO2 were introduced as promoters, the resulting composite catalysts (Ni/La2O3–Al2O3, Ni/CeO2–Al2O3, and Ni/CeO2–ZrO2) achieved similar conversion levels (∼92%) but exhibited slightly lower hydrogen production. Notably, in the promoted systems, the rate of carbon deposition was significantly reduced, suggesting that the incorporation of rare-earth oxides improved the catalyst's resistance to coking and enhanced its overall stability.
5.1.2. Hydrogenolysis.
The hydrogenolysis process utilizes catalysts such as Ru, Ni, Pt, and Rh-based catalysts to cleave the C–C bonds in polyethylene in a hydrogen atmosphere.278,287–291 Unlike hydrocracking reactions, the activation of C–H bonds and the cleavage of C–C bonds in polyolefins primarily occur at metal active sites. The reactivity and product selectivity of this process can be controlled by regulating the size, dispersion, valence state of metal sites and properties of the supports. Various oxide supports have been explored, with common types including CeO2, TiO2,292 SiO2,293 SrTiO3294 and Al2O3,295 among others.283 Among these, rare earth oxides with unique electronic structures are commonly used as supports, exhibiting excellent catalytic activity and selectivity due to their distinctive properties, thereby providing new solutions for the chemical recycling of polyethylene (Table 4). Significant progress has been made in the study of Ru/CeO2 catalysts for polyethylene hydrogenolysis reactions. Research teams have gradually refined the design strategies of the catalytic system by systematically regulating the structure of active sites and the properties of the support. Nakaji et al. developed an efficient and reusable heterogeneous catalyst, Ru/CeO2, which showed excellent performance in the hydrogenolysis reaction of LDPE.33 The catalyst was able to catalyse the reaction efficiently under mild reaction conditions (473 K, 2 MPa H2) to produce C5–C21 liquid fuel and C22–C45 wax in yields of 77% and 15%, respectively, for a total yield of 92%. The catalytic activity was significantly better than that of other metal/CeO2 catalysts and Pt/zeolite catalysts, while the selectivity was also better than that of other Ru/support catalysts. More importantly, the catalyst performs well in real-world applications, converting commercial plastic bags and waste polyethylene into valuable chemicals at high yields of 91% and 88%, respectively. Based on the confirmation of the superiority of the Ru/CeO2 system, researchers have begun to deeply explore the influence patterns of the active metal site structure. Ji et al. investigated the effect of Ru size on the hydrogenolysis of LDPE and found that CeO2-supported Ru nanoclusters exhibited the highest conversion efficiency and optimal selectivity for liquid alkanes, significantly outperforming Ru single-atom and nanoparticle catalysts.271 Further spectroscopic characterization revealed the intrinsic correlation between ruthenium size and catalytic performance: as the Ru size increases, the metal-support interaction (MSI) weakens, which is unfavorable for the activation of C–H and C–C bonds, while the hydrogen spillover effect is significantly enhanced, weakening the hydrogenation of alkane species. The balance achieved between these two countervailing effects enables the Ru nanocluster catalyst to deliver optimal catalytic performance. Buhori et al. systematically investigated the performance of Ru/CeO2 catalysts in polyethylene hydrogenolysis reaction by modulating CeO2 morphology (cubic and rod-like) and Ru impregnation methods (wet impregnation and hydrothermal method).273 The results showed that the Ru/CeO2-C–HT catalyst prepared by the hydrothermal method exhibited higher liquid yield in a shorter period of time, while the Ru/CeO2-R-HT catalyst was effective in inhibiting CH4 generation. The number of oxygen vacancies in CeO2 and the size of the Ru particles are the key factors influencing the catalytic performance, with the more oxygen vacancies and the small the Ru particles, thus promoting liquid product selectivity and reducing gaseous by-products. In addition, the preparation of Ru alloy catalysts by combining a single Ru active metal with other metals can further enhance the inhibition of terminal cracking, which further reduces the selectivity of methane. Hu et al. synthesised Ru9Pt91/C, Ru9Pt91/SiO2 and Ru9Pt91/CeO2 catalysts and investigated their polyethylene hydrogenolysis performance at 300 °C and 0.5 MPa H2.289 These catalysts exhibited similar methane selectivity (<2.3%), suggesting that the inhibition of terminal cracking by the dilute Ru9Pt91 alloy is universal across different carriers. Since CeO2 displayed a significant promoting effect, Ru9Pt91/CeO2 was evaluated at a lower temperature (250 °C). The results showed that Ru9Pt91/CeO2 could completely convert polyethylene within 3 h at 2.0 MPa, and at 0.5 MPa, complete conversion could be achieved within 8 h with methane selectivity consistently below 3.7%. In contrast to Hu et al.'s study, Zhao et al. successfully developed an efficient polyethylene hydrogenolysis system for methane production by codoping Mn into RuO2 and CeO2.36 They found that incorporating heteroatoms into the RuO2 lattice significantly increases the content of oxidized Ru species in a reducing atmosphere. The catalyst was able to achieve more than 99% polyethylene conversion in only 8 h at 250 °C, with a methane product selectivity close to 99% (Fig. 19a). The characteristic vibrations of LDPE (2930 cm−1 and 2850 cm−1) were investigated by DRIFT spectroscopy, and their intensity varied in correlation with the rate of polymer cracking, with a significant decrease in the intensity of vibrations on the RuMn/CeO2 catalyst and a smaller change on the Ru/CeO2 catalyst, suggesting that Mn facilitated the PE conversion (Fig. 19b). The H2-TPD results revealed a distinct difference: RuMn/CeO2 exhibited a prominent desorption at 222 °C, while Ru/CeO2 showed a much stronger peak at peak 396 °C, indicating that the introduction of Mn significantly lowered the H2 desorption temperature, thereby enhancing the H-spillover effect and promoting the hydrogenation process (Fig. 19c). Meanwhile, the C3H8-TPD results demonstrated that the desorption peak of C3H8 on RuMn/CeO2 shifted to higher temperatures, reflecting a stronger alkane adsorption capacity compared with Ru/CeO2 (Fig. 19d). Furthermore, DFT calculations showed that Ru NPs served as the primary sites for H2 adsorption and dissociation (−1.33 eV), whereas the Ru–RuOx interface exhibited weaker H2 adsorption (−0.31 eV) but provided the most stable adsorption site for C3H8 (−1.43 eV) (Fig. 19e). These findings imply that the coeffect effect between Ru0 and Ruδ+ species effectively strengthens the interaction between LDPE molecules and the catalyst surface, thereby endowing RuMn/CeO2 with superior hydrogenolysis activity (Fig. 19f). In order to investigate the guiding principle of methane generation, Wang et al. performed LDPE hydrogenolysis by using doped zirconium catalysts (Ru-XZr, X = Ti, Nb, Ce, W, V, Mo, and Fe).296 For the hydrogenolysis reaction, the authors found that there was an inverted pyramid correlation between the inhibition of methane generation and the reducibility of the doped oxides and that doped oxides with moderately reducible catalysts with moderate reducibility (e.g., W, V, and Mo) stored and supplied the extra hydrogen to Ru through an inverse hydrogen spillover effect and most effectively inhibited methane production under hydrogen-poor conditions. In recent years, remarkable progress has been made in the field of plastics recycling technology. In addition to the traditional thermal catalysis technology, a series of new catalytic technologies have emerged, including microwave-assisted catalysis and photothermal catalysis. These innovative methods provide diversified solutions for the efficient depolymerisation of plastics through different mechanisms. In terms of microwave-assisted catalysis, Wang's team pioneered the development of an atmospheric pressure microwave catalytic system based on the CsRu/CeO2 catalyst.297 The system was able to achieve efficient deconstruction of LDPE under a mild condition of 300 °C, with a gas product yield of 57.6% and a residue yield of 28.9%. The product analysis showed that the system had a high selectivity of 55% for low-carbon olefins (C2=–C4=), with ethylene accounting for 34% of the total, along with benzene tetramer (BTX) selectivity of 18%. This result confirms that the synergistic effect of the microwave field and heterogeneous catalyst can effectively promote the deep deconstruction of polyolefins. These research advances indicate that, by precisely designing the catalyst structure and optimising the reaction conditions, the novel catalytic
technology is able to break through the limitations of traditional heat treatment and realise the efficient conversion of plastics under mild conditions, which provides an important technological support for the construction of a sustainable plastics recycling economy.
Table 4 Summary of the rare earth-based catalysts for polyolefin recycling
| Catalysts |
Plastic types |
Time/h |
P/MPa |
T/°C |
Conv. (%) |
Yield (%) |
Ref. |
| Gas (C1–4) |
Liquid fuel (C5–21) |
Wax (C22–45) |
| β + CeO2 |
LDPE |
2 |
1 |
240 |
97.0 |
3.0 |
90.1 |
6.9 |
267
|
| CeO2 |
LDPE |
2 |
1 |
240 |
4.8 |
95.2 |
2.2 |
2.6 |
267
|
| Ru/CeO2 |
LDPE |
5 |
6 |
240 |
76 |
7.5 |
54 |
15 |
33
|
| Ru/CeO2 |
LDPE |
8 |
6 |
240 |
>99 |
9.7 |
83 |
6.5 |
33
|
| Ru/CeO2 |
HDPE |
10 |
6 |
240 |
>99 |
13 |
83 |
4.1 |
33
|
| Ru/CeO2 |
PP |
72 |
6 |
240 |
>99 |
17 |
72 |
10 |
33
|
| Ru5Pt1/CeO2 |
LDPE |
12 |
0.5 |
200 |
99 |
∼ |
∼ |
∼ |
268
|
| 2 wt% Ru/CeO2 |
LDPE |
18 |
3 |
180 |
∼ |
45.7 |
31.0 |
3.6 |
34
|
| Ru/CeO2 |
LDPE |
4 |
6 |
240 |
93 |
9.2 |
72 |
12 |
269
|
| Ru/CeO2–ZrO2 |
LDPE |
4 |
6 |
240 |
66 |
11 |
45 |
9.3 |
269
|
| 0.2Ru/CeO2 |
LDPE |
6 |
2 |
250 |
97.5 |
3.05 |
94.5 |
∼ |
270
|
| 0.5Ru/CeO2 |
LDPE |
6 |
2 |
250 |
∼ |
16.8 |
76.63 |
∼ |
270
|
| 2Ru/CeO2 |
LDPE |
6 |
2 |
250 |
∼ |
23.06 |
69.45 |
∼ |
270
|
| 5Ru/CeO2 |
LDPE |
6 |
2 |
250 |
67.0 |
62.5 |
4.46 |
∼ |
270
|
| Ru(SA)/CeO2 |
LDPE |
4 |
2 |
240 |
>99 |
15.9 |
27.3 |
56.8 |
271
|
| Ru(NC)/CeO2 |
LDPE |
4 |
2 |
240 |
>99 |
29.4 |
56.8 |
13.8 |
271
|
| Ru(NP)/CeO2 |
LDPE |
4 |
2 |
240 |
>99 |
22.2 |
23.1 |
54.8 |
271
|
| PtSn–0.5Ce/SiAl |
HDPE |
2 |
3 |
270 |
∼ |
12.3 |
85.1 |
2.6 |
272
|
| PtSn–Ce/SiAl |
HDPE |
2 |
3 |
270 |
∼ |
17.6 |
75 |
7.5 |
272
|
| Ru/CeO2-C–HT |
PE |
2 |
3 |
250 |
∼ |
13.2 |
60.6 |
20 |
273
|
| Ru/CeO2-R-WI |
PE |
2 |
3 |
250 |
∼ |
5.7 |
35.8 |
25.9 |
273
|
| Ce/b-ZSM-5 |
LDPE |
5 |
3 |
280 |
96.3 |
85.28 |
11.05 |
∼ |
26
|
| b-ZSM-5 |
LDPE |
5 |
3 |
280 |
86.8 |
78.96 |
7.88 |
∼ |
26
|
| Pt/5Ce-HY |
LDPE |
2 |
3 |
300 |
100 |
19.1 |
80.9 |
0 |
25
|
| Pt/5Ce-HY |
LDPE |
2 |
1 |
300 |
100 |
22.5 |
77.5 |
0 |
25
|
| Ru/CeO2 |
LDPE |
8 |
1 |
250 |
100 |
71.1 |
18.8 |
10.1 |
36
|
| RuMn/CeO2 |
LDPE |
6 |
1 |
250 |
100 |
>99.9 |
0 |
0 |
36
|
| RuMn/CeO2 |
LDPE |
4 |
1 |
250 |
100 |
97.8 |
0.9 |
1.3 |
36
|
| Pt-3Ce/HY |
LDPE |
2 |
2 |
280 |
∼ |
2.9 |
86.5 |
5.1 |
274
|
| Ce_meso_Y |
PE |
4 |
0 |
300 |
100 |
∼ |
∼ |
∼ |
275
|
| 1%Ru-7%Ce/SBA-15 |
LDPE |
18 |
3 |
300 |
100 |
2.9 |
29.9 |
67.3 |
276
|
| 5%Ru-7%Ce/SBA-15 |
LDPE |
18 |
3 |
300 |
100 |
18.3 |
81.5 |
0.2 |
276
|
| Ru–Ce/SiO2 |
LDPE |
18 |
3.5 |
280 |
∼ |
2.6 |
25.7 |
71.7 |
276
|
| FeRu/CeO2 |
LDPE |
1 |
2 |
250 |
100 |
9.2 |
86.4 |
0 |
37
|
| Ru/CeO2 |
LDPE |
1 |
2 |
250 |
100 |
40 |
59.8 |
0 |
37
|
| Rh–CeO2/CeHY |
LDPE |
1 |
2 |
300 |
>99 |
∼ |
78.3 |
∼ |
277
|
| Ni/CeO2 |
LDPE |
1 |
2 |
300 |
∼ |
∼ |
45 |
∼ |
278
|
| 0.125 wt% Ru/CeO2 |
PP |
18 |
3 |
260 |
∼ |
13.3 |
32.5 |
2.1 |
34
|
| 0.253 wt% Rh/CeO2 |
PP |
18 |
3 |
260 |
∼ |
1.2 |
2.8 |
87.2 |
34
|
| 2Ru/CeO2-SA |
PP |
16 |
3 |
250 |
91 |
57 |
34 |
9 |
279
|
| 2Ru/CeO2-NR |
PP |
16 |
3 |
250 |
90 |
63 |
27 |
10 |
279
|
| 2Ru/CeO2-NC |
PP |
16 |
3 |
250 |
94 |
72 |
22 |
6 |
279
|
| PtSn-0.5Ce/SiAl |
PP |
2 |
3 |
270 |
∼ |
14 |
84.4 |
1.6 |
272
|
| PtSn-0.5Ce/SiAl |
LDPE |
2 |
3 |
270 |
∼ |
20.3 |
76.4 |
3.2 |
272
|
| 0.4 wt% Ni/CeO2-SA |
PP |
16 |
3 |
280 |
95 |
18 |
78 |
5 |
280
|
| 0.7 wt% Ni/CeO2-NC |
PP |
16 |
3 |
280 |
96 |
24 |
73 |
4 |
280
|
| 1.2 wt% Ni/CeO2-SA |
PP |
16 |
3 |
280 |
89 |
12 |
77 |
11 |
280
|
| CeO2-SA |
PP |
24 |
3 |
280 |
22 |
12 |
10 |
78 |
280
|
 |
| | Fig. 19 (a) Time-programmed LDPE hydrogenolysis performance of RuMn/CeO2. (b) In situ diffuse reflectance infrared Fourier transform (DRIFT) spectroscopy of PE hydrogenolysis reaction and (c) H2-TPD profile over RuMn/CeO2 and Ru/CeO2 catalysts, (d) C3H8 TPD profile of the used RuMn/CeO2, Ru10Mn/CeO2 and Ru/CeO2 catalysts. (e) Adsorption configuration of H2 (left) and C3H8 (right) over the Ru/RuOx model structure. (f) Mechanism of PE hydrogenolysis over RuMn/CeO2 catalysts. Reprinted with permission from ref. 36. Copyright (2024) American Chemical Society. | |
5.1.3. Hydrocracking.
Hydrocracking reactions rely on a bifunctional catalyst system with both metal and acidic sites. This process begins with the dehydrogenation reaction catalyzed by the metal sites, generating olefin intermediates. Subsequently, these olefin intermediates diffuse to the Brønsted acid sites, where they undergo protonation to form carbocation intermediates. These carbocations then undergo C–C bond cracking and isomerization reactions. The cracked and isomeric intermediates ultimately diffuse back to the metal sites for hydrogenation, yielding short-chain products.266 The catalytic system usually consists of two types of active centres: (1) metal active sites, which mainly include metals such as Pt, Pd, Fe and Ni; and (2) acidic supports such as microporous zeolites (e.g., ZSM-5, MOR, and HY), mesoporous materials (e.g., Al-SBA-15 and MCM-41), and solid acidic metal oxides (e.g., WO3/ZrO2).266,298–302 Among them, rare earth components are often used as promoters to enhance catalytic activity (Table 4). The unique electronic structure and surface properties of rare earth materials can optimise the distribution of active sites and enhance the adsorption of reactants, thus effectively promoting the chemical recycling of PE. Although not a typical metal–acid bifunctional hydrocracking catalyst, Wang et al. successfully prepared Ce-modified ZSM-5 nanosheet catalysts by a simple ion-exchange method, which exhibited excellent catalytic performance due to shorter diffusion paths and higher acidic site concentrations.26 Upon further introduction of Ce species, the acidic site concentration of the catalyst was significantly increased, which not only enhanced the adsorption of substrate molecules, but also promoted the formation of reaction intermediates, and ultimately achieved 96.3% conversion, 80.9% selectivity, and 78.0% yield of C3-C5 alkanes in the hydrocracking reaction of LDPE. Similarly, Wu et al. demonstrated a new strategy to significantly enhance the hydrocracking efficiency of LDPE by introducing a Ce promoter into the Pt/HY catalyst (Fig. 20a).25 The performance of the catalyst was significantly improved by the 5% Ce-modified Pt/HY catalyst, which achieved 100% LDPE conversion and 80.9% liquid fuel selectivity at 300 °C, 30 bar H2, and 2 h of reaction, which were much better than that of the unmodified Pt/HY catalyst (38.8% conversion and 21.3% selectivity) (Fig. 20b and c). Further investigation revealed that even a small amount of Ce (1%) could enhance the LDPE conversion rate, while increasing the Ce content to 5% led to a leap in performance, confirming the highly active nature of the supercage-occupied Ce species. However, excessive Ce loading (10%) induced over-cracking, thereby reducing the liquid fuel selectivity (Fig. 20d). NH3-TPD and Py-IR characterization further confirmed that the introduction of Ce enhanced both the acid strength and the Brønsted acidity, which is favorable for the hydrocracking reaction (Fig. 20e and f). Nevertheless, the variation in Brønsted acidity did not fully align with the catalytic performance. To gain deeper insights into the role of Ce, researchers used n-C16 instead of LDPE as a model substrate and found a similar catalytic trend (Fig. 20g), with n-C16-TPD results showing that 5Ce-HY exhibited the strongest adsorption capacity for intermediates (Fig. 20h). Comparative experiments between Pt/5Ce-HY and Pt@5Ce-HY further revealed a two-step reaction pathway, where the former, possessing external active sites, was more effective in cleaving long-chain LDPE, while the latter suffered from diffusion limitations, leading to lower activity (Fig. 20i). Collectively, these results demonstrate that Ce species not only enhance Brønsted acidity but also promote intermediate adsorption, thereby accelerating the secondary C–C bond cleavage and significantly improving liquid fuel selectivity. Moreover, the researchers synthesized a series of Pt/HY catalysts incorporated with non-cerium rare earth elements (La, Sm, Tb, and Er) through a similar synthesis route and systematically evaluated their catalytic performance. The results showed that the overall catalytic performance of Pt/5La-HY was comparable to that of Pt/5Ce-HY, although its selectivity toward liquid fuels was relatively lower. In contrast, catalysts incorporated with other rare earth elements (such as Sm, Tb, and Er) exhibited significantly reduced catalytic activity and selectivity. Hou et al. prepared a Sm-modified USY zeolite catalyst (5Sm/USY) via a deposition–precipitation method and conducted LDPE recycling experiments under three different reaction modes: pure plasma, thermal catalysis, and plasma catalysis.303 The results showed that under thermal catalytic conditions at 550 °C, the 5Sm/USY catalyst achieved a liquid product yield of 73%, which was slightly lower than that obtained under plasma-catalytic conditions (83%), but significantly higher than that under pure plasma conditions (50%).
 |
| | Fig. 20 (a) The schematic structural model of Pt/5Ce-HY. (b) Catalytic performance of Pt/5Ce-HY. (c) Catalytic performance comparison over prepared catalysts, including Pt/5Ce-HY, Pt/HY, HY, 5Ce-HY, Pt/CeO2, physically-mixed Pt/SiO2 and 5Ce-HY, Pt/HY deposited by CeO2, physically-mixed HY and Pt/CeO2 (Conditions: 300 °C, 2 h, and 3 MPa H2). (d) Selectivity of liquid fuels, (e) NH3-TPD profiles, (f) Py-IR patterns, (g) the hydrocracking of n-C16, (h) n-C16-TPD of samples incorporated with different contents of Ce. (i) Catalytic performance of LDPE (left) and n-C16 (right) over Pt/5Ce-HY and Pt@5Ce-HY. Reprinted with permission from ref. 25. Copyright (2024) Wiley-VCH. | |
5.2. PP recycling
PP is a widely used thermoplastic polymer with excellent chemical resistance,304 mechanical properties and thermal stability, and is one of the most consumed polymers globally with a wide range of applications in the packaging, automotive, textile and food industries.304,305 Depending on the tacticity, polypropylene can be classified into three types:306 isotactic polypropylene (iPP), syndiotactic polypropylene (sPP) and atactic polypropylene (aPP), of which iPP is the most widely used variety due to its high crystallinity and excellent mechanical properties. As global plastics consumption continues to grow, polypropylene consumption is also increasing significantly with polypropylene accounting for around 20% of total global plastics production in 2022, its use is second only to polyethylene.307 However, this rapid growth in polypropylene consumption has also led to a growing waste disposal problem, which has led to chemical recycling becoming more widely recognised as a promising waste treatment option. This section will provide a detailed examination of three representative technologies for polypropylene chemical recycling: catalytic pyrolysis, hydrogenolysis, and hydrocracking.
5.2.1. Catalytic pyrolysis.
Similar to PE, rare earth-based catalysts also play a significant role in the catalytic pyrolysis process of PP. Aboul-Enein et al. efficiently synthesised carbon nanomaterials (CNMs) over La2O3-loaded Ni and Ni–Cu catalysts (total metal loading of 50 wt%) via a two-stage thermocatalytic process (pyrolysis at 500 °C and decomposition at 700 °C) using PP waste plastics as a carbon source.308 It was shown that the La2O3 support effectively inhibited the aggregation of Ni particles, while the formation of Ni–Cu alloys promoted the hybrid growth of large-diameter carbon nanofibres (CNFs) and multi-walled carbon nanotubes (MWCNTs). Compared with the monometallic 50% Ni/La2O3 catalyst, the carbon yield of the bimetallic 40%Ni–10%Cu/La2O3 catalyst was significantly increased from 944% to 1458%. In addition, researchers have used pyrolysis of PP plastics in combination with steam reforming technology to produce hydrogen. La2O3 and CeO2 promoted Ni/Al2O3 catalysts were developed and applied to PP pyrolysis-vapour reforming reaction by Arregi et al. (Fig. 21a).309 The Ni/La2O3–Al2O3 catalyst exhibited excellent catalytic performance. At a higher space time (16.7 gcat min gplastic−1), it enabled complete conversion of PP with a hydrogen yield of 34.9%, whereas at a lower space time (4.1 gcat min gplastic−1), incomplete conversion led to a reduced hydrogen yield of 24.9%. Nevertheless, its overall performance remained significantly superior to that of Ni/Al2O3 and Ni/CeO2–Al2O3. However, the aforementioned catalytic pyrolysis process for PP faces a number of challenges, such as high energy consumption, catalyst sintering, and deactivation of the catalyst, which are drawbacks that limit its further application and optimisation.
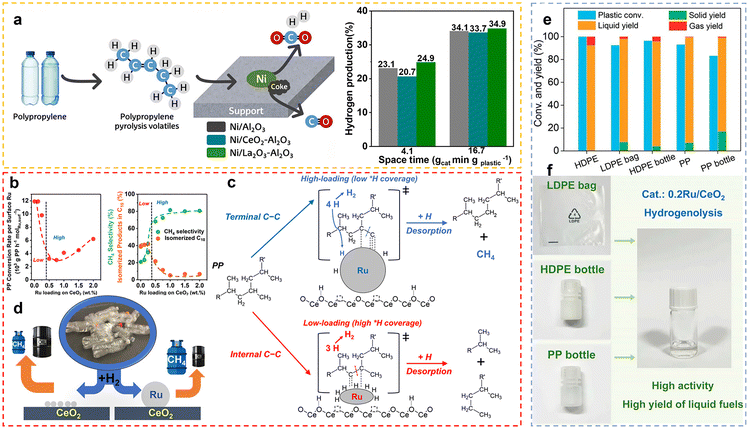 |
| | Fig. 21 (a) Thermal cracking and reforming mechanisms and performance comparison of Ni/Al2O3, Ni/CeO2–Al2O3, and Ni/La2O3–Al2O3 catalysts. Reprinted with permission from ref. 309. Copyright (2020) American Chemical Society. (b) Variations in the performance of Ru/CeO2 in the hydrogenolysis of PP with Ru loading at 260 °C. (c) Mechanism of PP hydrogenolysis on high-loading and low-loading Ru/CeO2. (d) Schematic diagram of the results of the PP hydrogenolysis reaction using high-loading and low-loading Ru/CeO2 catalysts. Reprinted with permission from ref. 34. Copyright (2022) American Chemical Society. (e) Catalytic performance of hydrogenolysis of various polyolefins on Ru SAC. (f) Schematic diagram of hydrogenolysis of various polyolefins on Ru SAC. Reaction conditions: T = 250 °C, PH2 = 2 MPa, stirring rate = 400 rpm, m(Ru)/m(polyolefin) = 1 mg/2000 mg, and reaction times for HDPE, LDPE bag, HDPE bottle, PP, and PP bottle are 6, 8, 8, 18, and 18 h, respectively. Reprinted with permission from ref. 270. Copyright (2023) American Association for the Advancement of Science. | |
5.2.2. Hydrogenolysis.
Hydrogenolysis technologies offer the following advantages over catalytic pyrolysis: lower reaction temperatures, fewer pollutant emissions and higher selectivity for higher-value products. To overcome the challenges of catalytic pyrolysis of PP, researchers have in recent years applied hydrogenolysis to the chemical recycling of PP, which not only helps to improve the recycling efficiency, but also effectively reduces the environmental pollution and generates higher-value chemicals and fuels. Studies indicate that the size and morphology of metal sites critically influence product selectivity in PP hydrogenolysis. Chen et al. evaluated the effect of active site Ru species structure on PP hydrogenolysis reaction (Fig. 21d).34 By systematically modulating the Ru loading, it was found that when the loading was lower than 0.25 wt%, the catalysts exhibited significantly higher PP conversion efficiency, lower CH4 selectivity, and excellent isomerisation ability. However, at loadings ≥0.5 wt%, the catalysts showed reduced intrinsic activity in PP hydrogenolysis and increased methane selectivity (Fig. 21b). Further studies showed that this abrupt change in performance stems from a structural shift in Ru species: at low loadings, the catalysts form highly disordered and sub-nanometre-scale Ru active sites with high surface H coverage that preferentially break the inner C–C bonds rather than the terminal C–C bonds, resulting in a decrease in methane selectivity; whereas at high loadings (≥0.5 wt%), the Ru particles have lower surface H coverage and higher methane selectivity (Fig. 21c). Chu et al. achieved the reduction of methane yield in PP hydrogenolysis reaction to near 0 by reducing the size of Ru in the active centre to a single atom (Fig. 21e).270 In addition, the catalyst is capable of efficiently and selectively degrading a wide range of polyolefins (e.g., HDPE and LDPE) as well as commercial polyolefin plastics (Fig. 21f), which has great potential for industrial applications. It is worth noting that catalytic hydrogenolysis activity depends not only on metal active sites, but also closely on the support material. Tomer et al. conducted a systematic study on the effects of different morphologies of CeO2 supports (nanorods, nanotubes, and non-shaped) loaded with Ru catalysts on the hydrogenolysis reaction of PP.279 Under optimized reaction conditions (220 °C, 30 bar H2, 16 h), the morphology-controlled 2 wt% Ru/CeO2 catalyst (particularly the nanocube configuration) exhibited significantly higher liquid alkane yields compared to the non-shaped support catalyst (58–81% vs. 34–58%). Among these, the 2 wt% Ru/CeO2 nanocube catalyst exhibited the highest activity. Jaydev et al. investigated the effect of Ru nanoparticles supported on different supports (such as TiO2, Al2O3, SiO2, MgO, and CeO2) on the hydrogenolysis of PP.310 The study showed that the catalytic activity of Ru/CeO2 was higher than that of Ru/Al2O3, Ru/SiO2, and Ru/MgO, but comparable to that of Ru/TiO2. Although precious metals (such as Ru, Rh, and Pt) are the most active catalysts for polypropylene hydrocracking, researchers are working to develop non-precious transition metal alternatives due to cost and supply constraints. Inns et al. investigated the performance of Ni/CeO2 catalysts in the hydrogenolysis of polypropylene and studied the effect of CeO2 support morphology on the catalytic performance.280 By standardizing the Ni structure (particle size < 1.3 nm), Ni density, and CeO2 grain size, they compared the performance differences between the nanocube and mixed morphology Ni/CeO2 catalysts. The results showed that both catalysts exhibited excellent liquid alkane yields (65.9–70.9 gliquid gNi−1 h−1) and low methane yields (10%). However, liquid product analysis revealed that the C–C bond cleavage rate for the nanocube catalyst reached 838.1 mmol gNi−1 h−1, which was 75% higher than that of the mixed morphology catalyst. In addition, the liquid products from the nanocube catalyst had a lower molecular weight (Mw = 2786 g mol−1vs. 4599 g mol−1) and a narrower molecular weight distribution (Mn = 1442 g mol−1vs. 2530 g mol−1). These improvements were attributed to improved H-storage and favourable basic properties on the nanocubes due to the exposed (100) facets.
5.2.3. Hydrocracking.
In recent years, hydrocracking technology, as an important complement to hydrogenolysis technology, has demonstrated excellent catalytic efficiency in the chemical recycling of PP. Similar to PE hydrocracking, rare earth-based catalysts also play a key role in this process (Table 4). Zhao et al. developed a cerium-promoted the Pt/HY catalyst that demonstrated exceptional performance in polyethylene catalytic cracking.274 This catalytic system proved equally effective for PP degradation. The tertiary carbon structures in PP molecular chains are more prone to generate carbocation intermediates, which subsequently undergo cracking over zeolites, ultimately yielding products enriched in light hydrocarbons (C1–C4 and C5–C7 fractions). Moreover, Wang et al. used PtSn-based hydrocracking catalysts at different cerium promoter contents for polyolefin conversion experiments.272 The results showed that cerium has a key role in regulating the performance of PtSn catalysts, with the best performance at 0.5 wt% cerium loading. The optimised catalyst can effectively degrade a wide range of plastics, including LDPE, PP and PS, to obtain high-yield fuel range products.
5.3. PET recycling
PET, as one of the most widely consumed synthetic polyesters globally, is extensively used in water bottles,311 textiles312,313 and the food industry due to its high strength, transparency, and thermal stability. However, the massive use of PET has led to severe environmental issues,314 as its waste is difficult to degrade, contributing to a global plastic pollution crisis. To cope with this problem, chemical recycling methods for converting PET into reusable monomers or chemical products have gradually become a focus of research. Currently, chemical recycling of polyester-based plastics mainly includes catalytic pyrolysis, hydrogenolysis and chemical depolymerisation techniques.313,314 Among them, catalytic hydrogenolysis and catalytic pyrolysis can effectively decompose PET into aromatic compounds and other oxygen-containing organic chemicals, but their products cannot be directly used to synthesise new polyesters. In contrast, chemical depolymerisation breaks down PET into monomers such as terephthalic acid (TPA), ethylene glycol (EG), dimethyl terephthalate (DMT) and bis(hydroxyethyl) terephthalate (BHET) by means of hydrolysis, glycolysis or methanolysis.282 Moreover, compared with conventional mechanical recycling, chemical depolymerization has the advantage of being able to process PET waste to obtain monomers that can be repolymerized into materials with properties comparable to virgin PET, thereby overcoming the problem of material degradation in mechanical recycling and making it more suitable for applications requiring high-performance materials, such as food packaging. In this section, we focus on an overview of catalytic pyrolysis and chemical depolymerisation of PET.
5.3.1. Catalytic pyrolysis.
Compared with conventional thermal pyrolysis, catalytic pyrolysis technology offers the advantages of low energy consumption, high economic efficiency, and minimal environmental impact, making it an important method for converting PET waste into fuel and high-value-added chemicals. The commonly used catalysts include zeolites (such as HZSM-5, Hβeta, and HY),315 transition metal oxides, and carbonates (such as CaCO3 and MgCO3).314 Among these catalysts, rare earth-based catalysts have garnered significant attention due to their unique properties. Chattopadhyay et al. studied the catalytic co-pyrolysis of plastics (HDPE, PP, and PET) and biomass in a fixed-bed reactor.316 The results showed that as the plastic content decreased, the liquid product gradually decreased while the gas product increased. During the pyrolysis of a mixture with a biomass-to-plastic ratio of 5:1, the hydrogen yield reached a peak of 47 vol% when using a 40% Co/30% CeO2/30% Al2O3 catalyst. Further, Hsu et al. coupled plastic pyrolysis with steam reforming reactions and developed a two-stage pyrolysis-catalysis process based on UiO-66(Ce)-derived NiO@CeO2 composites for efficiently converting post-consumer plastic waste into hydrogen.317 In this study, nickel oxide was used as the active site. The researchers systematically compared the coke resistance of different supports, including γ-Al2O3, m-ZrO2, and CeO2, and found that CeO2, with its excellent oxygen storage capacity, significantly suppressed coke formation on the catalyst surface. It also promoted the oxidation of deposited carbon by releasing lattice oxygen. The study further validated the applicability of this technology for various post-consumer plastics, including HDPE, PP, PS, and PET. Nabgan et al. developed Ni–Co/CeO2 bimetallic catalysts using impregnation and hydrothermal methods for steam reforming and cracking reactions to generate hydrogen and valuable liquid fuels from PET waste.311 Among them, the hydrothermally synthesized NC–Ce-Hyd catalyst exhibited smaller metal particle sizes and stronger basicity and acidity compared to the impregnation method catalyst, NC–Ce-imp, which significantly improved the catalytic performance. Under reaction conditions at 700 °C, PET conversion increased from 72.8% to 83.8%, and hydrogen yield increased from 56% to 76%. The PET cracking and phenol steam reforming reactions produced several valuable liquid products, such as dibenzofuran, 2-methylphenol, and benzene. Additionally, Nabgan et al. synthesized a Ni–Pd/Al2O3–La2O3 catalyst via the co-impregnation method and systematically evaluated its catalytic behavior in the steam reforming of phenol–PET for hydrogen production.318 The results demonstrated that the catalyst exhibited excellent activity at 700 °C, achieving a feed conversion of 93.87%, which was significantly higher than those of the comparative catalysts Ni/Al (87%) and Ni/Al–La (92%). Under the same conditions, the hydrogen selectivity reached 60.4%, indicating a high efficiency for hydrogen generation. Further investigations revealed that the incorporation of La2O3 not only enhanced the catalyst's basicity but also synergized with Pd to markedly improve the stability of the Ni–Pd/Al2O3–La2O3 catalyst. Even after 36 h of continuous operation, the catalyst maintained excellent activity, with only a slight decline in hydrogen selectivity and feed conversion—significantly outperforming the Ni/Al and Ni/Al–La catalysts in long-term stability. Overall, compared with Ni/Al and Ni/Al–La catalysts, the Ni–Pd/Al2O3–La2O3 system exhibited markedly enhanced performance in terms of activity, selectivity, and stability, offering a promising route for efficient hydrogen production from plastic waste.
5.3.2. Chemical depolymerisation.
In recent years, rare earth-based catalysts have made significant breakthroughs in PET depolymerization reactions. Through catalyst design and process optimization, PET conversion efficiency and product selectivity have been significantly improved. Swapna et al. studied the effects of metal oxide catalysts (MnOx, CeO2, Nb2O5, and TiO2) on the glycolysis of PET.319 The reactions were conducted under mild conditions (180 °C for 3 h), successfully converting waste PET bottles into BHET monomers. Among the catalysts, CeO2-500 exhibited good catalytic performance, with a PET conversion rate of 45% and a BHET monomer yield of 29%. Based on the confirmation of the catalytic potential of CeO2, researchers have begun exploring the enhancement of catalytic performance through nanostructure regulation. Yun et al. synthesized CeO2 nanoparticles (NPs) with tunable sizes (2.7–4.8 nm) and abundant defects using a simple precipitation method combined with KH550 modification and applied them in the glycolysis of PET.320 The oxygen defect-rich CeO2 nanoparticles accelerated the depolymerization reaction by inducing the formation of Ce3+ and providing active sites. The obtained CeO2 nanoparticles with a size of 2.7 nm rapidly depolymerized PET into BHET within 15 minutes at 196 °C, with a PET conversion rate exceeding 98% and a yield exceeding 90%. This demonstrates the potential application value of defect-rich CeO2 nanoparticles in PET depolymerization. Moreover, Pham et al. further investigated the use of CeO2 doped γ-Al2O3 as a catalyst for the glycolysis of waste PET plastics (Fig. 22a).321 The reaction was carried out at 250 °C for 1 h, achieving complete PET conversion. Notably, the 10% Ce/Al2O3 catalyst exhibited high selectivity during glycolysis, with a BHET monomer yield exceeding 70% and minimal by-products, showing significantly better catalytic performance than unmodified γ-Al2O3 (Fig. 22b). The reaction process can be divided into two stages: in the first stage, PET is depolymerized into shorter-chain polymers or oligomers; in the second stage, these oligomers undergo further cleavage to produce dimers and BHET monomers (Fig. 22c). Importantly, the incorporation of CeO2 imparts moderate basicity to the γ-Al2O3 catalyst (Fig. 22d), thereby effectively promoting the overall reaction. Moreover, the catalyst maintained stable catalytic activity over five consecutive reactions without significant deactivation (Fig. 22e). On the basis of achieving efficient depolymerization, researchers have also explored the conversion routes of PET into higher value-added chemicals. Mo et al. developed an innovative two-step catalytic conversion strategy to efficiently convert waste PET plastic into high-value 1,4-cyclohexanedimethanol (CHDM).322 This catalytic strategy consists of two steps: first, using a Pd-modified Ni/CeO2 catalyst for methanolysis and benzene ring hydrogenation reactions to convert PET into dimethyl 1,4-cyclohexanedicarboxylate (DMCD) and then, using a CuMgAl catalyst to hydrogenolyze DMCD to produce CHDM (Fig. 23a). The experimental results demonstrate that the 0.5Pd5Ni/CeO2 catalyst achieves a DMCD yield of 86.5% with a selectivity of 97.4%, surpassing the performance of 5Ni/CeO2 and representing the highest yield reported to date (Fig. 23b). This superior performance is attributed to the dual role of Pd: it not only enhances the hydrogen dissociation capability but also (Fig. 23c and d), by increasing the metallicity of Ni species, suppresses the adsorption and activation of C–O bonds (Fig. 23e). These combined effects collectively drive the significant improvement in DMCD selectivity and yield. Subsequently, DMCD undergoes further hydrogenolysis over the CuMgAl catalyst, yielding 83.8% CHDM.
 |
| | Fig. 22 (a) XRD patterns of γ-Al2O3, 10%Co/Al2O3, 10%Ca/Al2O3 and 10%Ce/Al2O3. (b) Depolymerization efficiency of metal oxide-supported on alumina support catalysts. (c) Catalytic mechanism of PET glycolysis with metal oxide supported–alumina catalyst. (d) CO2-TPD profiles of 10%M/Al2O3 (M = Ce, Ca or Co). (e) Recyclability of 10%Ce/Al2O3 in the glycolysis of PET. Reprinted with permission from ref. 321. Copyright (2023) Elsevier. | |
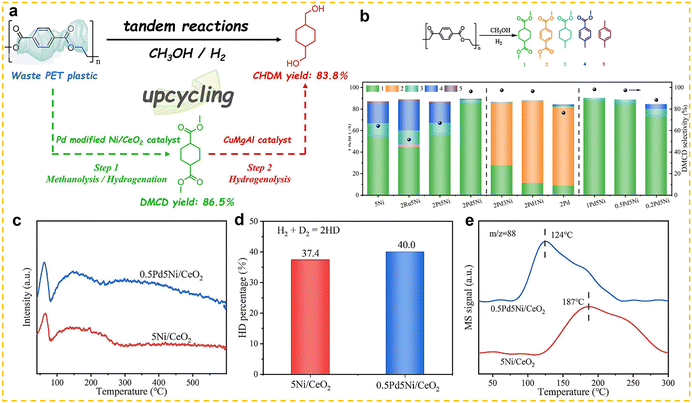 |
| | Fig. 23 (a) Catalytic conversion of PET into CHDM. (b) Catalytic performance of various investigated catalysts. (c) H2-TPD profiles of the 5Ni/CeO2 and 0.5Pd5Ni/CeO2 catalyst. (d) H–D exchange experiments measured at 180 °C, over 5Ni/CeO2 and 0.5Pd5Ni/CeO2 catalysts. (e) Ethyl acetate TPD-MS results of the 5Ni/CeO2 and 0.5Pd5Ni/CeO2 catalysts. Reprinted with permission from ref. 322. Copyright (2024) Elsevier. | |
5.4. PLA recycling
PLA, as a biodegradable bio-based polymer, has gained extensive applications in recent years across fields such as packaging, textiles, biomedical engineering, and agriculture.323 Its industrial production mainly relies on the ring-opening polymerization (ROP) of L-lactide.324,325 With its excellent properties, including biocompatibility, processability, and biodegradability,325,326 PLA has become a promising alternative to traditional petroleum-based plastics. In 2023, global production of biodegradable plastics reached 1.14 million tons, with PLA accounting for 59.4% of that total. Industry forecasts indicate that by 2028, total production of biodegradable plastics will increase significantly to 4.61 million tons, with PLA expected to contribute 70.2% of that growth.327 However, the production costs of PLA remain high, primarily due to the energy-intensive fermentation and purification processes of lactic acid. Additionally, its degradation rate varies significantly across different environments, particularly being slow in marine and ordinary soil conditions. To address the challenges of PLA waste management, chemical recycling has emerged as a crucial solution. This section focuses on two chemical recycling pathways for PLA: catalytic pyrolysis and chemical depolymerisation.
5.4.1. Catalytic pyrolysis.
Catalytic pyrolysis is an important part of chemical recycling and has become a common solution for converting waste plastics into valuable products. However, single catalytic pyrolysis is usually not able to convert all components of plastics into gases, as some intermediate organic compounds have large molecules that are difficult to break down efficiently, resulting in the formation of tar or coke.328 To address this, Wu et al. combined pyrolysis with reforming technology and employed CeMgFeCoNi high-entropy aluminate catalysts (HEOs) for the chemical recycling of waste PLA plastics.328 The HEO catalysts enhanced the structural stability and the number of oxygen vacancies of the high-entropy system through the synergistic effect of multiple elements, which in turn improved its catalytic ability in C–C and C–H bond breaking. It was shown that the highest hydrogen yields up to 124.1 mmol gPLA−1 with a gas yield of 259.8 wt% were achieved during the catalytic reforming process.
5.4.2. Chemical depolymerisation.
Similar to the chemical recycling of PET, chemical depolymerization is also applied to the chemical recycling of PLA. Currently, the main chemical depolymerization methods for PLA include hydrolysis, alcoholysis, and amination. Through chemical depolymerization, PLA plastics can be effectively broken down into valuable low-molecular compounds, providing a sustainable recycling method that helps reduce plastic waste and improve resource circulation efficiency. Rare earth-based catalysts also play an important role in this process. Lehnertz et al. applied the Ru/CeO2 catalyst in the depolymerization of PLA. After just 20 minutes of hydrolysis, the yield of lactic acid (LA) was increased to 94%, while in the absence of the catalyst, the total yield was only 3% (Fig. 24a).329 This result highlights the key role of the Ru/CeO2 catalyst in promoting the conversion of PLA to lactic acid, significantly accelerating the reaction efficiency. In addition to converting PLA into lactic acid monomers through hydrolysis, PLA can also be converted into alanine molecules through ammonolysis. This transformation provides an alternative route for the resource utilization of PLA. Zhao et al. developed an Fe-doped Ru/CeFeOx catalyst, which successfully achieved the efficient conversion of PLA waste into alanine without the addition of H2.330 The optimized Ru/CeFeOx-2 catalyst yielded an alanine production of 70.5% after 18 h of reaction (Fig. 24d), significantly outperforming Ru/CeO2. EPR analysis indicated that the introduction of Fe modulated the electronic structure and promoted the formation of oxygen vacancies (Fig. 24b), while CO2-TPD results confirmed that Fe doping enhanced the surface basicity of the catalyst (Fig. 24c). Collectively, these findings reveal that Fe incorporation not only facilitates the formation of oxygen vacancies but also provides abundant basic sites, thereby promoting the adsorption and activation of ammonium lactate, accelerating the amination reaction, and significantly improving catalytic efficiency (Fig. 24f). Moreover, Ru/CeFeOx-2 maintained moderate cyclic stability over three consecutive runs, with no significant decrease in catalytic activity (Fig. 24e). Wang et al. developed an indium-modified Ru/Ce–In mixed metal oxide catalyst (Ru/Ce3In-MMO) to further enhance the alanine yield (Fig. 25a and b).331 The optimized catalyst achieved an alanine yield of 66.7% under conditions of 180 °C, 0.1 MPa nitrogen pressure, and 10 h of reaction, significantly outperforming single-support catalysts (Ru/CeO2 and Ru/In2O3) (Fig. 25c). Detailed studies revealed that the introduction of indium increased the Ce3+/(Ce3+ + Ce4+) ratio from 16.2% to 28.6% (Fig. 25e), increased the oxygen vacancy concentration from 12.7% to 48.5% (Fig. 25f), expanded the specific surface area, and enhanced surface basicity (Fig. 25d). Density functional theory (DFT) calculations further showed that the adsorption energy of ammonium lactate on Ru/Ce3In-MMO (Ead = −3.95 eV) was much lower than that on Ru/CeO2 (Ead = −2.26 eV), indicating stronger adsorption on the former (Fig. 25g). Thus, the incorporation of In not only promoted the generation of oxygen vacancies but also provided abundant basic sites, thereby enhancing the adsorption and activation of ammonium lactate, which effectively improved catalytic activity and alanine yield. In addition, the catalyst maintained stable activity after three reaction cycles and demonstrated excellent catalytic performance for various real PLA waste substrates, including commercial PLA drinking straws, CPLA straws, and optically pure PLA commonly found in daily life (Fig. 25h and i). This indicates the broad potential of the catalyst for processing different types of PLA waste. The process provides an atom-economic solution for the valorization of PLA waste without the need for external H2, complementing traditional mechanical recycling and biodegradation methods.
 |
| | Fig. 24 (a) Catalytic PLA depolymerisation without a catalyst and Ru/CeO2. Reprinted with permission from ref. 329. Copyright (2022) The Royal Society of Chemistry. (b) The EPR profiles, (c) CO2-TPD curves of Ru/CeO2 and Ru/CeFeOx-2 catalysts. (d) The effect of reaction time on the yield of product over Ru/CeFeOx catalyst. (e) The cycle performance for catalytic amination of PLA to alanine over Ru/CeFeOx catalyst. (f) Reaction pathway for catalytic amination of PLA into alanine over Ru/CeFeOx catalyst. Reprinted with permission from ref. 330. Copyright (2025) Wiley. | |
 |
| | Fig. 25 (a) EDX elemental maps of the Ru/Ce3In-MMO catalyst. (b) Reaction mechanism of catalytic amination of PLA to alanine over Ru/Ce3In-MMO catalysts. (c) Catalytic performance of the as-prepared catalysts for the catalytic amination of PLA to alanine. (d) CO2-TPD profiles of CeO2, In2O3, and Ce3In-MMO supports. XPS spectra of (e) Ce 3d, (f) O 1s of the as-prepared catalysts. (g) Adsorption energy of ammonium lactate over Ru/CeO2 and Ru/Ce3In-MMO. (h) Cycle performance of PLA catalytic amination over the Ru/Ce3In-MMO catalyst. (i) Effect of real PLA substrates on the mass of product over the Ru/Ce3In-MMO catalyst. Reprinted with permission from ref. 331. Copyright (2024) American Chemical Society. | |
5.5. PA recycling
PA, commonly known as nylon, is a class of high-molecular-weight materials characterized by amide groups (–CO–NH–) as their core structure.332 Based on their molecular chain structures, PAs can be categorized into aliphatic (e.g., PA6 and PA66), semi-aromatic (e.g., PA6T), and fully aromatic (aramid) types.332 Aliphatic polyamides dominate the market (accounting for around 85% of global polyamide) due to their high crystallinity and excellent processability, making them widely used in textiles, automotive components, and packaging.333 Semi-aromatic polyamides combine the high-temperature resistance of aromatic rings with the processability of aliphatic chains, making them suitable for electronics and corrosion-resistant equipment.334 Fully aromatic polyamide exhibits exceptional mechanical strength and high thermal resistance, making it ideal for high-demand applications such as the military and aerospace industries.335 In recent years, growing attention has been paid to bio-based polyamides (e.g., PA11 and PA56) owing to the depletion of petrochemical resources and the demand for sustainable materials. However, their commercialization remains slow,336 and most are not biodegradable, which limits their environmental benefits. Meanwhile, polyamide plastic pollution has become an increasingly serious issue, as approximately 10% of marine plastic waste originates from PA fishing nets.337 In this context, chemical recycling technologies have emerged as a promising approach to address environmental pollution. These technologies aim to cleave the polymer backbone, converting polyamides into monomers or chemical feedstocks to achieve resource circularity and reuse. Currently reported chemical recycling methods for polyamides mainly include hydrolysis, ammonolysis, hydrogenolysis, and glycolysis.337–339 For example, Stuyck et al. reported an ammonolysis process for PA-66, in which efficient degradation of PA-66 was achieved using a series of acid catalysts (e.g., CeO2, TiO2, and ortho-Nb2O5) in ethylene glycol solvent under 1 bar NH3 at 200 °C.340 Moreover, Wu et al. investigated the catalytic hydrogenolysis of PA waste using platinum group metal nanoparticles (Ru, Rh, Pd, Ir and Pt) supported on CeO2 (Fig. 26a).35 Among these catalysts, Ru/CeO2 exhibited the highest catalytic activity, achieving complete conversion of N-hexylhexanamide with a methane yield of 76%. The conversion rates for Rh/CeO2, Ir/CeO2, and Pt/CeO2 were >99%, 99%, and 98%, respectively, but they exhibited differences in product selectivity (Fig. 26b). Although n-hexane was the major product, differences in the C–C bond cleavage activity of the catalysts resulted in n-hexane yields of 52%, 57%, and 82%, respectively. In contrast, Pd/CeO2 showed the lowest catalytic activity. The Pt catalyst had a lower tendency to cleave C–C bonds but effectively cleaved C–O and C–N bonds, resulting in higher selectivity for hexane (Fig. 26d). Further density functional theory (DFT) calculations indicated that Pt nanoparticles required higher activation energy to break C–C bonds compared to Ru nanoparticles, which explained the difference in product selectivity. Both Ru/CeO2 and Pt/CeO2 catalysts demonstrated good reusability in hydrogenolysis cycles, maintaining complete conversion with minimal changes in product selectivity after five reaction cycles (Fig. 26c and e). For the hydrogenolysis of PA-12 over Pt/CeO2, the selectivity for n-dodecane improved after each cycle, reaching a maximum yield of 79% in the fifth cycle. The study also showed that the metal nanoparticles in the used catalysts underwent little to no agglomeration during hydrogenolysis, indicating their robustness and potential for long-term use. Although rare earth-based catalysts exhibit high catalytic activity in the chemical recycling of PA, effectively promoting its degradation and reuse, research in this field is still relatively limited. To promote the widespread application of this technology, researchers need to invest more effort in in-depth studies, particularly focusing on selectivity, stability, and regeneration capacity.
 |
| | Fig. 26 (a) STEM-HAADF and EDX analysis of M/CeO2 (M = Ru, Rh, Pd, Ir or Pt) catalysts. (b) Product distribution from the hydrogenolysis of N-hexylhexanamide with M/CeO2 (M = Ru, Rh, Pd, Ir or Pt) and commercial carbon-based catalysts. (c) Product distribution of PA-6 conversion with Ru/CeO2 over five cycles. Reaction conditions: N-hexylhexanamide (250 mg), catalyst (25 mg, 0.5 wt% of metal) and H2 (50 bar) at 325 °C for 2 h. (d) Reaction pathway of alkane formation from the consecutive C–O, C–N and C–C hydrogenolysis of N-hexylhexanamide. (e) Product distribution of PA-12 conversion with Pt/CeO2 over five cycles. Reaction conditions: polymer (250 mg), catalyst (25 mg, 0.5 wt% of metal) and H2 (50 bar) at 325 °C for 24 h (Pt/CeO2). Reprinted with permission from ref. 35. Copyright (2023) Springer Nature. | |
5.6. PC recycling
PCs are a class of transparent thermoplastic polymers that have found widespread applications in electronics, automotive, aerospace industries, optical data storage devices, construction, and medical fields owing to their excellent heat resistance, durability, mechanical strength, and high optical transparency.327,341,342 Among the various PC families, Bisphenol-A-based polycarbonate (BPA–PC), synthesized through the polycondensation of bisphenol A (BPA) with carbonic acid derivatives, is the most widely used.307 Although PCs account for only about 1% of the total plastic production, global demand reached 4.51 million tons in 2022 and continues to grow rapidly, making the development of effective end-of-life treatment strategies increasingly critical.327,343 Traditionally, most PC wastes have not been recycled and are instead landfilled or incinerated. A small fraction can be mechanically recycled, but once the material properties degrade, it is ultimately disposed of through landfilling or energy recovery.342 As a result, chemical recycling of PC has attracted significant attention, being recognized as a practical, economically viable, and environmentally friendlier approach to PC waste management. Current chemical recycling strategies primarily include pyrolysis,342 hydrolysis,344 alcoholysis,345 ammonolysis,346 and hydrogenolysis.264 In the following section, we highlight chemical recycling approaches involving rare earth-based catalytic materials and their recent advances. Taguchi et al. reported that CeO2 exhibits significantly higher catalytic activity for the hydrolysis of polycarbonate compared with Al2O3, MnO2, and ZrO2.344 Notably, when CeO2 nanocrystals were employed, the yield of bisphenol A (BPA) monomers reached approximately 90%, with almost no further decomposition of BPA observed under these conditions. In addition to hydrolysis, alcoholysis is also a common and effective chemical recycling route for polycarbonates. Similar to hydrolysis, alcoholysis can depolymerize polycarbonate into valuable monomers such as bisphenol A or other low-molecular-weight compounds. Recent studies have demonstrated that rare earth-based catalysts also exhibit excellent catalytic performance in alcoholysis. Yang et al. synthesized a CaO/Ce-SBA-15 catalytic material via plasma surface engineering.347 Owing to its abundant basic sites, this catalyst functions as an efficient solid base catalyst for the alcoholysis of polycarbonate (PC). Under the reaction conditions of 130 °C, 3 h, m(Cat)/m(PC) = 0.3:1, n(MeOH)/n(PC) = 8:1, and m(THF)/m(PC) = 1.5:1, the catalyst enabled efficient depolymerization of PC with a bisphenol A (BPA) yield of approximately 95%. Based on this, Liu et al. used a template impregnation method to prepare a CeO2–CaO–ZrO2 catalyst,345 which exhibited superior catalytic performance. Compared with the results reported by Yang et al., this catalyst achieved nearly 100% PC conversion and 96% BPA yield under milder conditions (100 °C, m(catalyst)/m(PC) = 0.05:1, n(MeOH)/n(PC) = 6:1, 2 h). Overall, these studies indicate that CeO2 significantly promotes the alcoholysis reaction of PC in different composite systems by enhancing alkalinity.
5.7. PU recycling
PU is one of the most widely used polymers worldwide, accounting for nearly 8% of total plastic production, with an annual output of approximately 18–24 million tons.307,348,349 It is extensively employed in automotive seats, footwear, furniture, coatings, adhesives, and electronic products.350–352 The key structural unit of PU is the urethane group (–NH–COO–), formed through the reaction between isocyanate groups (–NCO) and hydroxyl groups of polyols.348,353 Because landfill disposal is relatively inexpensive and end-of-life processing can be challenging, landfilling remains the most common disposal method for PU waste, accounting for about 30.8% of the total.307 Consequently, researchers have long been committed to exploring efficient and feasible recycling routes for PU waste. Compared with energy recovery and mechanical recycling, chemical recycling has attracted increasing attention due to its ability to convert waste polymers into reusable chemical feedstocks or monomers, thereby offering a higher potential for resource recovery. Various chemical recycling processes have been developed, including pyrolysis,354 hydrolysis,355 glycolysis,356,357 aminolysis,348 and acidolysis,358–360 among which alcoholysis has demonstrated promising potential for industrial-scale application. Notably, rare earth-based catalysts, owing to their unique redox properties, have been introduced into certain PU chemical recycling processes, where they play an important role in enhancing reaction rates, product selectivity, and overall resource efficiency. Wu et al. evaluated a range of commercially available metal oxides and zeolites for the conversion of model urethane compounds to assess their ability to cleave urethane linkages (Fig. 27a).361 Three simple PU analogues were selected as model compounds, namely phenylurethane (M1), ethane-1,2-diylbis((4-benzylphenyl)carbamate) (M2), and dibutyl(methylenebis(4,1-phenylene))dicarbamate (M3). The reactions were conducted under solvent-free conditions at 200 °C and 10 bar H2. Among the tested catalysts, CeO2 achieved nearly quantitative conversion and exhibited excellent selectivity toward amine products: M1 showed 100% conversion with a 92% yield of aniline, M2 showed 100% conversion with a 90% yield of 4-benzylaniline (4-BA), and M3 showed 94% conversion with a 92% yield of 4,4′-methylene diphenyl diamine (MDA) (Fig. 27b–d). These catalytic performances were markedly superior to those of the other metal oxides and zeolites (SiO2·TiO2·ZrO2·NbO2 ZSM-5 and La2O3). Further investigation revealed that the surface acidity and oxygen vacancies of the CeO2 played a crucial role in this process.
 |
| | Fig. 27 (a) Scheme of the proposed circular life cycle of polyurethane (PU). PU waste can be converted using a metal-oxide catalyst into aniline, which is the precursor for synthesizing new polymers. (b)–(d) Catalyst screening for the conversion of model carbamate compounds using various MOCs. Yield of aniline and alcohol (only for M2 and M3) products from the conversion of (b) M1, (c) M2 and (d) M3, yield of ethanol from the conversion of M1 was not quantified. Reprinted with permission from ref. 361. Copyright (2025) Springer Nature. (e) Schematics of solvent-assisted heterogenous catalysts-driven polyester glycolysis routes. (f) High-resolution Raman spectra of GB-rich and GB-poor CeO2 nanoparticles. (g) High-resolution Ce 3d XPS spectra of GB-rich and GB-poor CeO2 nanoparticles. (h) Photothermal catalytic performance of PC and PET glycolysis over GB-rich CeO2 catalysts with varying reaction temperature. (i) Adsorption energy of EG, EGD and DPC on perfect CeO2 and GB-rich CeO2 nanoparticles. Reprinted with permission from ref. 368. Copyright (2025) Wiley-VCH. | |
5.8. Mixed plastic recycling
The preceding section systematically explored chemical recycling pathways for specific types of plastics (such as PET, PE, and PP), revealing progress in designing efficient catalytic systems tailored to single polymers. However, post-consumer plastic waste in the real world is far from such ‘purity.’ It typically consists of a physical mixture of multiple plastics, including incompatible polymers and even multilayer packaging. This intrinsic heterogeneity renders traditional recycling methods that rely on physical separation both inefficient and costly, severely limiting improvements in overall recycling rates.362,363 Therefore, developing chemical recycling strategies capable of directly processing unseparated mixed plastic waste and selectively converting it into high-value chemicals or fuels has become a key research direction to overcome current plastic recycling bottlenecks. Currently, various catalyst systems have been developed for the chemical recycling of mixed plastics, including Ni/SiO2, Ru/C, Pt/WO3/ZrO2, Ni-Fe/ZSM5 and Ni-Fe/MCM-41 catalysts.364–367 Rare earth-based catalysts are also used in the chemical recycling of mixed plastics. For example, Liu et al. innovatively developed a heterogeneous photothermal catalytic strategy based on grain boundary-enriched CeO2 (GB-rich CeO2) for the selective depolymerization and recycling of mixed polyester (PET/PC) waste.368 By introducing a co-solvent, this study effectively overcame the mass transfer limitations at multiphase interfaces encountered by heterogeneous catalysts in mixed plastics, significantly enhancing the recycling efficiency (Fig. 27e). The obtained GB-rich CeO2 photothermal catalyst exhibited a high concentration of Ce3+ ions and oxygen vacancies (Ov) (Fig. 27f and g). The catalyst achieved sequential depolymerization of PC (97.8% BPA yield) and PET (93.4% BHET yield) under mild conditions (140–190 °C) (Fig. 27h), demonstrating excellent selective recycling performance. The researchers used ethylene glycol diphenyl ester (EGD) and diphenyl carbonate (DPC) as model molecules to replace PET and PC, respectively, and calculated their adsorption energies (Eads) on CeO2 surfaces. The results show that on the perfect CeO2 surface, the Eads values for EG, EGD, and DPC are −0.49, −0.28, and −0.18 eV, respectively. In contrast, on CeO2 surfaces with rich grain boundaries, the Eads values significantly increase to −0.81, −0.68, and −0.47 eV, respectively (Fig. 27i). This suggests that oxygen vacancies preferentially adsorb EGD and DPC. This preference stems from the modulation of the electronic states of the surrounding atoms by the incorporation of the grain boundary. During the catalytic degradation of PC and PET by metal oxides, oxygen vacancies interact with ester group oxygen atoms, promoting the nucleophilic attack of ethylene glycol (EG). In contrast, on the perfect CeO2 surface, the saturated coordination of Ce atoms limits electron transfer, thereby affecting the catalytic performance. However, research on rare earth-based catalysts specifically designed for the chemical recycling of mixed plastics is relatively limited, and their application potential in this complex system remains to be thoroughly explored.
6. Summary and outlook
In this review, we introduce typical synthesis methods for rare earth-based catalysts and systematically summarise the advantages and disadvantages of different synthesis methods. Subsequently, we summarise the in situ characterization of rare earth-based catalysts and their applications in CO2 hydrogenation and plastic waste recycling, respectively, and elaborate in detail on the mechanisms of the rare earth components in the reactions. By modulating the structure and surface properties of rare earth-based catalysts, the activity and selectivity of the reactions can be effectively improved, and the development of sustainable chemical reactions can be promoted, which is especially promising for the realisation of green energy conversion and waste recycling.
Although many significant breakthroughs have been made in recent years in rare earth-based catalysts for waste carbon recycling, there are still many problems that need to be solved: (1) in recent years, rare earth-based catalysts have made remarkable progress in the precise control of shape, size and composition, demonstrating excellent performance in areas such as catalysis and energy conversion. Despite progress in nanostructure modulation, most research is still at the laboratory stage. Efficient, low-cost, and scalable production methods have yet to be realized. Therefore, translating these laboratory results into industrial-scale production methods remains a key challenge. (2) Studies on rare earth-based catalysts for CO2 hydrogenation and plastic recycling have primarily focused on cerium-based materials. The catalytic properties of other rare earth elements, such as La, Y, and Sm, have not been fully explored. Future research should focus on extending the study to these non-cerium-based catalysts to discover new catalytic mechanisms and more efficient catalyst systems. (3) CO2 hydrogenation and plastic hydrogenolysis reactions currently rely on high hydrogen partial pressures and can only proceed under hydrogen excess conditions. However, the high cost of hydrogen makes these processes economically unfeasible. Developing catalysts that can operate efficiently at low hydrogen partial pressures is therefore of significant scientific and practical importance. (4) Currently, most in situ characterization techniques can only be performed under relatively mild or simplified catalytic conditions and cannot fully reproduce harsh reaction environments. This may result in characterization of catalyst structural evolution that does not match the actual behaviour. Therefore, there is an urgent need to develop more advanced in situ/operando characterization techniques for monitoring catalyst structural evolution under realistic, harsh reaction conditions. (5) At present, the chemical recycling of mixed plastics is still mainly limited to small-scale laboratory studies, and most research has focused on only a few types of plastics (PET, PE, PVC, and PP). Research on rare earth-based catalysts in this field is even more scarce, with most existing studies targeting single plastics and lacking systematic investigation of their adaptability and stability in complex mixed systems. It is therefore necessary to develop rare earth-based catalytic systems with high activity and resistance to impurities and to promote their application under scaled-up reaction conditions to improve the efficiency and feasibility of chemical recycling of mixed plastics. (6) Real waste plastics often contain additives such as plasticizers, stabilizers, and dyes, as well as impurities like moisture and food residues, which pose critical challenges for plastic recycling. However, most current studies are still based on high-purity single-polymer model feedstocks, lacking systematic evaluation of complex real-world systems. Future research should establish model systems that better reflect real plastic compositions, systematically investigate the effects of impurities and additives on the performance and stability of rare earth-based catalysts, and on this basis optimize catalyst structures to enhance their stability and applicability under practical conditions. (7) Long-term stability remains a key bottleneck for the industrial application of rare earth-based catalysts in CO2 hydrogenation. Compared with the use of high-purity feed gases, relatively mild conditions, and short-term evaluations in laboratory studies, practical operations typically involve high temperatures, steam-rich environments, and impurities such as CO and H2O, which can easily cause sintering, poisoning, and performance degradation. However, most current studies focus on activity tests under laboratory conditions and lack a systematic evaluation of the durability, deactivation mechanisms, and regenerability of rare earth-based catalysts under such harsh environments. Future work should aim to develop stable rare earth-based catalytic systems resistant to hydrothermal sintering and impurity poisoning, and establish long-term continuous-flow evaluation platforms to promote their application in industrial CO2 hydrogenation. (8) Recently, the coupled conversion of plastics and CO2 into high-value-added products has received much attention. This process not only improves the conversion rate of these wastes but also upgrades plastic waste products and reduces CO2 emissions. However, there are few studies on the use of rare earth-based catalysts in this coupled process, which urgently needs further exploration. In the future, these challenges are expected to be effectively addressed as materials science and experimental techniques continue to advance. We believe that rare earth-based catalysts will play an even more critical role in CO2 hydrogenation and plastics recycling applications.
Author contributions
Xiao Wang, Shuyan Song and Hongjie Zhang proposed the research direction of the review and guided the project. Liangliang Zhang researched the literature and drafted the manuscript. Xiao Wang guided and revised the manuscript.
Conflicts of interest
There are no conflicts to declare.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Acknowledgements
This work was supported by the financial aid from the National Science and Technology Major Project of China (2023YFB3508000), the National Natural Science Foundation of China (22020102003, 22025506, 22271274 and U23A20140) and the Jilin Province Science and Technology Development Plan Project (20240402056GH). X. W. acknowledges funding from the National Natural Science Foundation of China Outstanding Youth Science Foundation of China (Overseas).
Notes and references
-
C. Le Quéré, M. Jones, T. Jarnikova and I. Harris, Global carbon Budget 2024, Earth System Science Data, 2024 DOI:10.5194/essd-2024-519.
- Plastics – the fast Facts 2024. https://plasticseurope.org/knowledge-hub/plastics-the-fast-facts-2024/.
- A. Chamas, H. Moon, J. Zheng, Y. Qiu, T. Tabassum, J. H. Jang, M. Abu-Omar, S. L. Scott and S. Suh, ACS Sustainable Chem. Eng., 2020, 8, 3494–3511 CrossRef CAS.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- K. Zheng, Y. Wu, Z. Hu, S. Wang, X. Jiao, J. Zhu, Y. Sun and Y. Xie, Chem. Soc. Rev., 2023, 52, 8–29 RSC.
- J. Ye, N. Dimitratos, L. M. Rossi, N. Thonemann, A. M. Beale and R. Wojcieszak, Science, 2025, 387, eadn9388 CrossRef CAS PubMed.
- A. Stubbins, K. L. Law, S. E. Muñoz, T. S. Bianchi and L. Zhu, Science, 2021, 373, 51–55 CrossRef CAS PubMed.
- X. Ding, J. Fu, Y. Lyu, L. Ma, Y. Xu and X. Liu, Chem. Eng. J., 2024, 494, 152923 CrossRef CAS.
- Z. Xu, K. Sanchez-Rivera, C. Granger, P. Zhou, A. D. C. Munguia-Lopez, U. M. Ikegwu, S. Avraamidou, V. M. Zavala, R. C. Van Lehn, E. Bar-Ziv, S. De Meester and G. W. Huber, Nat. Chem. Eng., 2025, 2, 407–423 CrossRef.
- CCS projects grow 54% globally but get ‘mixed signals’ in US. https://illuminem.com/illuminemvoices/ccs-projects-grow-54-globally-but-get-mixed-signals-in-us.
- A. S. Pottinger, R. Geyer, N. Biyani, C. C. Martinez, N. Nathan, M. R. Morse, C. Liu, S. Hu, M. de Bruyn and C. Boettiger, Science, 2024, 386, 1168–1173 CrossRef CAS PubMed.
- A. Gulzar, A. Gulzar, M. B. Ansari, F. He, S. Gai and P. Yang, Chem. Eng. J. Adv., 2020, 3, 100013 CrossRef CAS.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef CAS.
- R. Ye, J. Ding, T. R. Reina, M. S. Duyar, H. Li, W. Luo, R. Zhang, M. Fan, G. Feng, J. Sun and J. Liu, Nat. Synth., 2025, 4, 288–302 CrossRef CAS.
- Y. Liu, B. Ma, J. Tian and C. Zhao, Sci. Adv., 2024, 10, eadn0252 CrossRef CAS PubMed.
- Y. Ding, S. Zhang, C. Liu, Y. Shao, X. Pan and X. Bao, Nat. Sci. Rev., 2024, 11, nwae097 CrossRef CAS PubMed.
- Y. Li, M. Wang, X. Liu, C. Hu, D. Xiao and D. Ma, Angew. Chem., Int. Ed., 2022, 61, e202117205 CrossRef CAS PubMed.
- H. Fu, Y. Jiang, M. Zhang, Z. Zhong, Z. Liang, S. Wang, Y. Du and C. Yan, Chem. Soc. Rev., 2024, 53, 2211–2247 RSC.
- W. Yang, X. Wang, S. Song and H. Zhang, Chem, 2019, 5, 1743–1774 CAS.
- H. Yan, N. Zhang and D. Wang, Chem Catal., 2022, 2, 1594–1623 CAS.
- Y. Zhu, C. Chen, P. Cheng, J. Ma, W. Yang, W. Yang, Y. Peng, Y. Huang, S. Zhang and G. Seong, Dalton Trans., 2022, 51, 6506–6518 RSC.
- F. Wang, M. Wei, D. G. Evans and X. Duan, J. Mater. Chem. A, 2016, 4, 5773–5783 RSC.
- X. Guan, X. Wang, X. Zhang, C. Zhang, S. S. C. Chuang and Z. Li, Angew. Chem., Int. Ed., 2025, 64, e202423958 CrossRef CAS PubMed.
- S. Gao, D. Yu, S. Zhou, C. Zhang, L. Wang, X. Fan, X. Yu and Z. Zhao, J. Mater. Chem. A, 2023, 11, 19210–19243 RSC.
- X. Wu, X. Wang, L. Zhang, X. Wang, S. Song and H. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202317594 CrossRef CAS PubMed.
- X. Wang, X. Wu, M. Zhao, R. Zhang, Z. Wang, Y. Li, L. Zhang, X. Wang, S. Song and H. Zhang, Nano Res., 2024, 17, 5645–5650 CrossRef CAS.
- J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek and J. F. Sanz, Science, 2014, 345, 546–550 CrossRef CAS PubMed.
- X. Yang, S. Kattel, S. D. Senanayake, J. A. Boscoboinik, X. Nie, J. Graciani, J. A. Rodriguez, P. Liu, D. J. Stacchiola and J. G. Chen, J. Am. Chem. Soc., 2015, 137, 10104–10107 CrossRef CAS PubMed.
- A. Parastaev, V. Muravev, E. Huertas Osta, A. J. F. van Hoof, T. F. Kimpel, N. Kosinov and E. J. M. Hensen, Nat. Catal., 2020, 3, 526–533 CrossRef CAS.
- H. X. Liu, S. Q. Li, W. W. Wang, W. Z. Yu, W. J. Zhang, C. Ma and C. J. Jia, Nat. Commun., 2022, 13, 867 CrossRef CAS PubMed.
- H. Wang, M. S. Bootharaju, J. H. Kim, Y. Wang, K. Wang, M. Zhao, R. Zhang, J. Xu, T. Hyeon, X. Wang, S. Song and H. Zhang, J. Am. Chem. Soc., 2023, 145, 2264–2270 CrossRef CAS PubMed.
- R. Zhang, X. Wang, K. Wang, H. Wang, X. Sun, W. Shi, S. Song and H. Zhang, Sci. Adv., 2024, 10, eadr3332 CrossRef CAS PubMed.
- Y. Nakaji, M. Tamura, S. Miyaoka, S. Kumagai, M. Tanji, Y. Nakagawa, T. Yoshioka and K. Tomishige, Appl. Catal., B, 2021, 285, 119805 CrossRef CAS.
- L. Chen, L. C. Meyer, L. Kovarik, D. Meira, X. I. Pereira-Hernandez, H. Shi, K. Khivantsev, O. Y. Gutiérrez and J. Szanyi, ACS Catal., 2022, 12, 4618–4627 CrossRef CAS.
- X. Wu, W. T. Lee, R. C. Turnell-Ritson, P. C. L. Delannoi, K. H. Lin and P. J. Dyson, Nat. Commun., 2023, 14, 6524 CrossRef CAS PubMed.
- M. Zhao, X. Chu, F. Wang, Y. Fang, L. Sun, Q. Xie, L. L. Zhang, S. Song, H. Zhang and X. Wang, J. Am. Chem. Soc., 2024, 146, 33104–33111 CrossRef CAS PubMed.
- X. Wang, R. Zhang, X. Wu, Y. Li, Z. Wang, M. Zhao, S. Song, H. Zhang and X. Wang, Angew. Chem., Int. Ed., 2025, 64, e202506035 CrossRef CAS PubMed.
- Y. Jiang, H. Fu, Z. Liang, Q. Zhang and Y. Du, Chem. Soc. Rev., 2024, 53, 714–763 RSC.
- D. Zhang, X. Du, L. Shi and R. Gao, Dalton Trans., 2012, 41, 14455–14475 RSC.
- G. Yan, Y. Tang, Y. Li, Y. Li, L. Nguyen, T. Sakata, K. Higashi, F. F. Tao and P. Sautet, Nat. Catal., 2022, 5, 119–127 CrossRef CAS.
- J. Xu, Y. Wang, K. Wang, M. Zhao, R. Zhang, W. Cui, L. Liu, M. S. Bootharaju, J. H. Kim, T. Hyeon, H. Zhang, Y. Wang, S. Song and X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202302877 CrossRef CAS PubMed.
- L.-L. Zhou, S.-Q. Li, C. Ma, X.-P. Fu, Y.-S. Xu, W.-W. Wang, H. Dong, C.-J. Jia, F. R. Wang and C.-H. Yan, J. Am. Chem. Soc., 2023, 145, 2252–2263 CrossRef CAS PubMed.
- Y. W. Hartati, S. N. Topkaya, S. Gaffar, H. H. Bahti and A. E. Cetin, RSC Adv., 2021, 11, 16216–16235 RSC.
- H.-X. Mai, L.-D. Sun, Y.-W. Zhang, R. Si, W. Feng, H.-P. Zhang, H.-C. Liu and C.-H. Yan, J. Phys. Chem. B, 2005, 109, 24380–24385 CrossRef CAS PubMed.
- J. Ke, J. W. Xiao, W. Zhu, H. Liu, R. Si, Y. W. Zhang and C. H. Yan, J. Am. Chem. Soc., 2013, 135, 15191–15200 CrossRef CAS PubMed.
- R. Zhang, X. Wang, K. Wang, H. Wang, L. Liu, X. Wu, B. Geng, X. Chu, S. Song and H. Zhang, Adv. Energy Mater., 2023, 13, 2203806 CrossRef CAS.
- X. Tian, Z. Wang, M. Ding, S. Zhou, R. Ouyang and Y. Miao, Int. J. Electrochem. Sci., 2020, 15, 10330–10349 CrossRef.
- J. Liu, Z. Zhao, C. Xu and J. Liu, Chin. J. Catal., 2019, 40, 1438–1487 CrossRef CAS.
- Y. Q. Yan, Y. Z. Wu, Y. H. Wu, Z. L. Weng, S. J. Liu, Z. G. Liu, K. Q. Lu and B. Han, ChemSusChem, 2024, 17, e202301778 CrossRef CAS PubMed.
- L. Xue, C. Zhang, H. He and Y. Teraoka, Appl. Catal., B, 2007, 75, 167–174 CrossRef CAS.
- C. Xu, S. Zhong, L. Yuan, M. Yu, Y. Chen, L. Dai and X. Wang, Chem. Eng. J., 2024, 481, 148767 CrossRef CAS.
- Z. Wang and R. Yu, Adv. Mater., 2019, 31, e1800592 CrossRef PubMed.
- I. Y. Kaplin, E. S. Lokteva, E. V. Golubina and V. V. Lunin, Molecules, 2020, 25, 4242 CrossRef CAS PubMed.
- S. Abdollahzadeh-Ghom, C. Zamani, T. Andreu, M. Epifani and J. R. Morante, Appl. Catal., B, 2011, 108–109, 32–38 CrossRef CAS.
- Y. Ke and S.-Y. Lai, Microporous Mesoporous Mater., 2014, 198, 256–262 CrossRef CAS.
- J. Zhang, Y. Cao, C. A. Wang and R. Ran, ACS Appl. Mater. Interfaces, 2016, 8, 8670–8677 CrossRef CAS PubMed.
- H. Xiong, H. Zhou, C. Qi, Z. Liu, L. Zhang, L. Zhang and Z.-A. Qiao, Chem. Eng. J., 2020, 398, 125527 CrossRef CAS.
- L. Liu, X. Yang, Y. Xie, H. Liu, X. Zhou, X. Xiao, Y. Ren, Z. Ma, X. Cheng, Y. Deng and D. Zhao, Adv. Mater., 2020, 32, e1906653 CrossRef PubMed.
- L. Li, H. Peng, Y. Kang, Y. Hao, F. Liu, H. Xin, H. Kang, W. Wang and Z. Lei, ACS Appl. Nano Mater., 2023, 6, 4349–4359 CrossRef CAS.
- J. Zheng, Z. Wang, Z. Chen and S. Zuo, J. Rare earths, 2021, 39, 790–796 CrossRef CAS.
- Q. Yang, D. Gao, C. Li, S. Wang, X. Hu, G. Zheng and G. Chen, Appl. Catal., B, 2023, 328, 122458 CrossRef CAS.
- J. Wei, X. Mu, Y. Hu, L. Liu, X. Wu, Q. Liu, T. Zhang, Y. Peng, J. Cao, C. H. Yan and Y. Tang, Angew. Chem., Int. Ed., 2023, 62, e202302986 CrossRef CAS PubMed.
- K. Ralphs, C. Hardacre and S. L. James, Chem. Soc. Rev., 2013, 42, 7701–7718 RSC.
- M. Leonardi, M. Villacampa and J. C. Menendez, Chem. Sci., 2018, 9, 2042–2064 RSC.
- C. Xu, S. De, A. M. Balu, M. Ojeda and R. Luque, Chem. Commun., 2015, 51, 6698–6713 RSC.
- B. Liu, Y. Li, S. Qing, K. Wang, J. Xie and Y. Cao, CrystEngComm, 2020, 22, 4005–4013 RSC.
- T. Gan, Q. He, H. Zhang, H. Xiao, Y. Liu, Y. Zhang, X. He and H. Ji, Chem. Eng. J., 2020, 389, 124490 CrossRef CAS.
- L. D. Sonawane, A. S. Mandawade, L. N. Bhoye, H. I. Ahemad, S. S. Tayade, Y. B. Aher, A. B. Gite, L. K. Nikam, S. D. Shinde, G. H. Jain, G. E. Patil and M. S. Shinde, Inorg. Chem. Commun., 2024, 164, 112313 CrossRef CAS.
- K. Polychronopoulou, A. F. Zedan, M. S. Katsiotis, M. A. Baker, A. A. AlKhoori, S. Y. AlQaradawi, S. J. Hinder and S. AlHassan, Mol. Catal., 2017, 428, 41–55 CAS.
- X.-Y. Guo, J.-H. Wang, Q. Zhang, T.-Z. Li, H. Dong, C.-J. Jia, C. Li and Y.-W. Zhang, Appl. Catal., B, 2024, 348, 123844 CrossRef CAS.
- A. I. Tsiotsias, N. D. Charisiou, E. Harkou, S. Hafeez, G. Manos, A. Constantinou, A. G. S. Hussien, A. A. Dabbawala, V. Sebastian, S. J. Hinder, M. A. Baker, K. Polychronopoulou and M. A. Goula, Appl. Catal., B, 2022, 318, 121836 CrossRef CAS.
- H. Tang, H. Sun, D. Chen and X. Jiao, Mater. Lett., 2012, 77, 7–9 CrossRef CAS.
- K. Wu, L. D. Sun and C. H. Yan, Adv. Energy Mater., 2016, 6, 1600501 CrossRef.
- M. A. Małecka, L. Kępiński and W. Miśta, J. Alloys Compd., 2008, 451, 567–570 CrossRef.
- J. Zhang, X. Ju, Z. Wu, T. Liu, T. Hu, Y. Xie and Z. Zhang, Chem. Mater., 2001, 13, 4192–4197 CrossRef CAS.
- A. Hadi and I. I. Yaacob, Mater. Lett., 2007, 61, 93–96 CrossRef CAS.
- Y. Li, X. Li, H. Zhang, J. He, K. Su, T. Chen, R. Zhang, H. Xu, Y. Wu, W. Yang and L. Liu, ACS Catal., 2024, 15, 623–638 CrossRef.
- L. Lin, J. He, X. Yan, H. Qi, T. Chen, J. He, Y. Liu, Z. Wu, J. Liu and W. Luo, Angew. Chem., Int. Ed., 2025, 64, e202505871 CrossRef CAS PubMed.
- Y. He, S. Guo, S. Li, L. Zhang and S. Yin, Applied Physics A, 2022, 128, 100 CrossRef CAS.
- N. Rahimi and R. Karimzadeh, Appl. Catal., A, 2011, 398, 1–17 CrossRef CAS.
- T. Zhu, T. Zhang, L. Xiao, C. Zhang and Y. Li, Catalysts, 2025, 15, 836 CrossRef CAS.
- B. Tan, Y. Luo, X. Bi, X. Liang, S. Wang, X. Gao, Z. Zhang and Y. Fang, Heat Mass Transfer, 2019, 55, 3179–3187 CrossRef CAS.
- Q. Hu, P. Zhang, Z. Shi, Y. Huang, J. Li, W. Zhang, X. Chen, X. Luo and Y. Yang, Integr. Ferroelectr., 2022, 228, 230–237 CrossRef CAS.
- L. Zhong, D. Chen and S. Zafeiratos, Catal. Sci. Technol., 2019, 9, 3851–3867 RSC.
- E. Korin, N. Froumin and S. Cohen, ACS Biomater. Sci. Eng., 2017, 3, 882–889 CrossRef CAS PubMed.
- H. Wang, S. Yang, W. Fan, Y. Cui, G. Gong, L. Jiao, S. Chen and J. Qi, ACS Appl. Mater. Interfaces, 2025, 17, 14749–14772 CrossRef CAS PubMed.
- X. Lian, J. Gao, Y. Ding, Y. Liu and W. Chen, J. Phys. Chem. Lett., 2022, 13, 8264–8277 CrossRef CAS PubMed.
- F. Jiang, S. Wang, B. Liu, J. Liu, L. Wang, Y. Xiao, Y. Xu and X. Liu, ACS Catal., 2020, 10, 11493–11509 CrossRef CAS.
- S. Hwang, X. Chen, G. Zhou and D. Su, Adv. Energy Mater., 2019, 10, 1902105 CrossRef.
- H. Y. Chao, K. Venkatraman, S. Moniri, Y. Jiang, X. Tang, S. Dai, W. Gao, J. Miao and M. Chi, Chem. Rev., 2023, 123, 8347–8394 CrossRef CAS PubMed.
- S. Zhang, C. Chen, M. Cargnello, P. Fornasiero, R. J. Gorte, G. W. Graham and X. Pan, Nat. Commun., 2015, 6, 7778 CrossRef PubMed.
- S. Yoon, J. Kim and K. An, Chem Catal, 2025, 5, 101423 CAS.
- J. Moncada, X. Chen, K. Deng, Y. Wang, W. Xu, N. Marinkovic, G. Zhou, A. Martínez-Arias and J. A. Rodriguez, ACS Catal., 2023, 13, 15248–15258 CrossRef CAS.
- J. King, C. Liu and S. S. C. Chuang, Res. Chem. Intermed., 2019, 45, 5831–5847 CrossRef CAS.
- G. Lin, G. Chen and J. Lu, J. Alloys Compd., 2025, 1010, 177552 CrossRef CAS.
- F. Zaera, J. Catal., 2021, 404, 900–910 CrossRef CAS.
- Y. Xie, J. Chen, J. Wen, Z. Li, F. Cao, S. Zhang, Q. Sun, P. Ning, Q. Zhang and J. Hao, ACS Catal., 2024, 14, 12214–12224 CrossRef CAS.
- H. Lee, D. Shin, D. Oh, B. Jeong, K. Y. Kim, C. Hur, J. W. Han and K. An, Chem. Eng. J., 2024, 496, 153758 CrossRef CAS.
- L. Pandit, M. A. Serrer, E. Saraci, A. Boubnov and J. D. Grunwaldt, Chemistry–Methods, 2021, 2, e202100078 CrossRef.
- B. Wu, T. Sun, Y. You, H. Meng, D. M. Morales, M. Lounasvuori, A. Beheshti Askari, L. Jiang, F. Zeng, B. Hu, X. Zhang, R. Tai, Z. J. Xu, T. Petit and L. Mai, Angew. Chem., Int. Ed., 2023, 62, e202219188 CrossRef CAS PubMed.
- S. K. Beaumont, Phys. Chem. Chem. Phys., 2020, 22, 18747–18756 RSC.
- X. Kong, R. Ren, H. Zhu, R. Zhang, Y. Gu, J. Gao, T. Ou, Y. Zhao and J. Zhang, J. Phys. Chem. C, 2025, 129, 3993–4009 CrossRef CAS.
- L. Fang, S. Seifert, R. E. Winans and T. Li, Small, 2022, 18, 2106017 CrossRef CAS PubMed.
- Y. Yan, R. J. Wong, Z. Ma, F. Donat, S. Xi, S. Saqline, Q. Fan, Y. Du, A. Borgna, Q. He, C. R. Müller, W. Chen, A. A. Lapkin and W. Liu, Appl. Catal., B, 2022, 306, 121098 CrossRef CAS.
- L. P. Matte, A. S. Thill, F. O. Lobato, M. T. Novoa, A. R. Muniz, F. Poletto and F. Bernardi, Small, 2022, 18, 2106583 CrossRef CAS PubMed.
- A. M. Bahmanpour, M. Signorile and O. Kröcher, Appl. Catal., B, 2021, 295, 120319 CrossRef CAS.
- C. Zhang, Z. Xiao, X. Guo, X. Tan, J. Gu, J. Li, G. Li, J.-J. Zou and D. Wang, Chem. Eng. J., 2025, 519, 165414 CrossRef CAS.
- W. Zhang, J. Sun, H. Wang and X. Cui, Chem. – Asian J., 2024, 19, e202300971 CrossRef CAS PubMed.
- D. L. Jurković, A. Pohar, V. D. B. C. Dasireddy and B. Likozar, Chem. Eng. Technol., 2017, 40, 973–980 CrossRef.
- Y. Yu, W. Xia, A. Yu, D. S. A. Simakov and L. Ricardez-Sandoval, ChemSusChem, 2025, 18, e202400681 CrossRef CAS PubMed.
- F. Cao, Y. Xiao, Z. Zhang, J. Li, Z. Xia, X. Hu, Y. Ma and Y. Qu, J. Catal., 2022, 414, 25–32 CrossRef CAS.
- Z. Zhao, M. Wang, P. Ma, Y. Zheng, J. Chen, H. Li, X. Zhang, K. Zheng, Q. Kuang and Z.-X. Xie, Appl. Catal., B, 2021, 291, 120101 CrossRef CAS.
- W. Li, J. Gan, Y. Liu, Y. Zou, S. Zhang and Y. Qu, Angew. Chem., Int. Ed., 2023, 62, e202305661 CrossRef CAS PubMed.
- J. Xu, L. Li, J. Pan, W. Cui, X. Liang, Y. Yu, B. Liu, X. Wang, S. Song and H. Zhang, Adv. Sustainable Syst., 2022, 6, 2100439 CrossRef CAS.
- B. Lu, F. Quan, Z. Sun, F. Jia and L. Zhang, Catal. Commun., 2019, 129, 105724 CrossRef CAS.
- J. Zhao, Q. Yang, R. Shi, G. I. N. Waterhouse, X. Zhang, L.-Z. Wu, C.-H. Tung and T. Zhang, NPG Asia Mater., 2020, 12, 5 CrossRef CAS.
- H. Li, B. Ma, J. Tian and C. Zhao, Chem. Eng. J., 2023, 464, 142572 CrossRef CAS.
- Z.-X. Li, K. Xu, W.-W. Wang, X.-P. Fu and C.-J. Jia, Catal. Sci. Technol., 2024, 14, 3483–3492 RSC.
- J. Zhao, X. Liu, Z. Li, K. Feng, Y. Pan, P. Ji, K. Zhao, B. Yan, D. Zhou and D. Su, ACS Catal., 2024, 14, 3158–3168 CrossRef CAS.
- A. Ranjbar, A. Irankhah and S. F. Aghamiri, Res. Chem. Intermed., 2019, 45, 5125–5141 CrossRef CAS.
- S. Wang, N. Cheng, Z. Jiang, S. Tian and L. Han, Energy Fuels, 2025, 39, 18537–18546 CrossRef CAS.
- J. Yu, A. Muhetaer, X. Gao, Z. Zhang, Y. Yang, Q. Li, L. Chen, H. Liu and D. Xu, Angew. Chem.,
Int. Ed., 2023, 62, e202303135 CrossRef CAS PubMed.
- X. Tang, C. Song, H. Li, W. Liu, X. Hu, Q. Chen, H. Lu, S. Yao, X. N. Li and L. Lin, Nat. Commun., 2024, 15, 3115 CrossRef CAS PubMed.
- M. Barreau, D. Salusso, J. Li, J. Zhang, E. Borfecchia, K. Sobczak, L. Braglia, J. J. Gallet, P. Torelli, H. Guo, S. Lin and S. Zafeiratos, Angew. Chem., Int. Ed., 2023, 62, e202302087 CrossRef CAS PubMed.
- W. K. Fan and M. Tahir, J. Environ. Chem. Eng., 2021, 9, 105460 CrossRef CAS.
- Y. Xie, J. Wen, Z. Li, J. Chen, Q. Zhang, P. Ning, Y. Chen and J. Hao, Green Chem., 2023, 25, 130–152 RSC.
- N. M. Martin, P. Velin, M. Skoglundh, M. Bauer and P.-A. Carlsson, Catal. Sci. Technol., 2017, 7, 1086–1094 RSC.
- J. A. H. Dreyer, P. Li, L. Zhang, G. K. Beh, R. Zhang, P. H. L. Sit and W. Y. Teoh, Appl. Catal., B, 2017, 219, 715–726 CrossRef CAS.
- X. Jia, X. Zhang, N. Rui, X. Hu and C.-J. Liu, Appl. Catal., B, 2019, 244, 159–169 CrossRef CAS.
- J. Kirchner, J. K. Anolleck, H. Lösch and S. Kureti, Appl. Catal., B, 2018, 223, 47–59 CrossRef CAS.
- W. Li, X. Nie, X. Jiang, A. Zhang, F. Ding, M. Liu, Z. Liu, X. Guo and C. Song, Appl. Catal., B, 2018, 220, 397–408 CrossRef CAS.
- S. Navarro-Jaén, J. C. Navarro, L. F. Bobadilla, M. A. Centeno, O. H. Laguna and J. A. Odriozola, Appl. Surf. Sci., 2019, 483, 750–761 CrossRef.
- G. Garbarino, D. Bellotti, E. Finocchio, L. Magistri and G. Busca, Catal. Today, 2016, 277, 21–28 CrossRef CAS.
- F. Wang, S. He, H. Chen, B. Wang, L. Zheng, M. Wei, D. G. Evans and X. Duan, J. Am. Chem. Soc., 2016, 138, 6298–6305 CrossRef CAS PubMed.
- T. Zhang, P. Zheng, J. Gao, Z. Han, F. Gu, W. Xu, F. Wang, T. Zhu, G. Xu, Z. Zhong and F. Su, Chem. Eng. J., 2024, 481, 148548 CrossRef CAS.
- Y. Dai, M. Xu, Q. Wang, R. Huang, Y. Jin, B. Bian, C. Tumurbaatar, B. Ishtsog, T. Bold and Y. Yang, Appl. Catal., B, 2020, 277, 119271 CrossRef CAS.
- C. Song, J. Liu, R. Wang, X. Tang, K. Wang, Z. Gao, M. Peng, H. Li, S. Yao, F. Yang, H. Lu, Z. Liao, X.-D. Wen, D. Ma, X. Li and L. Lin, Nat. Chem. Eng., 2024, 1, 638–649 CrossRef.
- H. Song, J. Yang, J. Zhao and L. Chou, Chin. J. Catal., 2010, 31, 21–23 CrossRef CAS.
- G. Tang, D. Gong, H. Liu and L. Wang, Catalysts, 2019, 9, 442 CrossRef CAS.
- J. Ilsemann, A. Sonström, T. M. Gesing, R. Anwander and M. Bäumer, ChemCatChem, 2019, 11, 1732–1741 CrossRef CAS.
- W. Liao, M. Yue, J. Chen, Z. Wang, J. Ding, Y. Xu, Y. Bai, X. Liu, A. Jia, W. Huang and Z. Zhang, ACS Catal., 2023, 13, 5767–5779 CrossRef CAS.
- R. Daroughegi, F. Meshkani and M. Rezaei, Chem. Eng. Sci., 2021, 230, 116194 CrossRef CAS.
- S. Hao, L. Xinzeng, L. Yuling, J. Yang, D. Chuanchuan and L. Bo, Fuel, 2025, 397, 135415 CrossRef CAS.
- D. Pan, Y. Wang, H. Li, Y. Zhang, Q. Liang, M. Zhou, Z. Li and S. Xu, Sep. Purif. Technol., 2024, 332, 125756 CrossRef CAS.
- X. Yue, K. Wang, Z. Yang, W. Ni, Z. Zhang, W. Dai and X. Fu, Chem. Eng. J., 2023, 458, 141491 CrossRef CAS.
- Z.-Y. Zhang, T. Li, X.-L. Sun, D.-C. Luo, J.-L. Yao, G.-D. Yang and T. Xie, J. Catal., 2024, 430, 115303 CrossRef CAS.
- V. Golovanova, M. C. Spadaro, J. Arbiol, V. Golovanov, T. T. Rantala, T. Andreu and J. R. Morante, Appl. Catal., B, 2021, 291, 120038 CrossRef CAS.
- K. Zhang, C. Xu, X. Zhang, Z. Huang, Q. Pian, K. Che, X. Cui, Y. Hu and Y. Xuan, Small, 2024, 20, 2308823 CrossRef CAS PubMed.
- Z. Bian, Y. M. Chan, Y. Yu and S. Kawi, Catal. Today, 2020, 347, 31–38 CrossRef CAS.
- Y. Zhou, P. Zheng, J. Gao, W. Xu, Y. Yang, L. Zhang, T. Zhu, G. Xu, Z. Zhong and F. Su, ACS Catal., 2024, 14, 14285–14296 CrossRef CAS.
- Y. Li, Y. Men, S. Liu, J. Wang, K. Wang, Y. Tang, W. An, X. Pan and L. Li, Appl. Catal., B, 2021, 293, 120206 CrossRef CAS.
- V. Alcalde-Santiago, A. Davó-Quiñonero, D. Lozano-Castelló, A. Quindimil, U. De-La-Torre, B. Pereda-Ayo, J. A. González-Marcos, J. R. González-Velasco and A. Bueno-López, ChemCatChem, 2018, 11, 810–819 CrossRef.
- G. Garbarino, C. Wang, T. Cavattoni, E. Finocchio, P. Riani, M. Flytzani-Stephanopoulos and G. Busca, Appl. Catal., B, 2019, 248, 286–297 CrossRef CAS.
- W. Gac, W. Zawadzki, M. Kuśmierz, G. Słowik and W. Grudziński, Appl. Surf. Sci., 2023, 631, 157542 CrossRef CAS.
- F. Namvar, M. Salavati-Niasari and F. Meshkani, Int. J. Hydrogen Energy, 2023, 48, 1877–1891 CrossRef CAS.
- L. Wang, Y. Qi, Z. Yang, H. Wu, J. Liu, Y. Tang and F. Wang, Green Energy and Resources, 2023, 1, 100036 CrossRef.
- L. Xu, X. Chen, C. Deng, K. Hu, R. Gao, L. Zhang, L. Wang and C. Zhang, Atmosphere, 2023, 14, 1208 CrossRef CAS.
- M. Ren, Y. Zhang, X. Wang and H. Qiu, Catalysts, 2022, 12, 403 CrossRef CAS.
- F. Sha, Z. Han, S. Tang, J. Wang and C. Li, ChemSusChem, 2020, 13, 6160–6181 CrossRef CAS PubMed.
- B. Gao, Z. Wen, Y. Wang, D. Chen, B. Yang, T. Ishihara and L. Guo, ChemCatChem, 2024, 16, e202400814 CrossRef CAS.
- M. L. Schulte, V. Truttmann, D. E. Doronkin, L. Baumgarten, A. Nicolai, D. A. Montalvo Beltran, F. J. Summ, C. Kiener, L. Warmuth, S. Pitter, E. Saraci and J. D. Grunwaldt, Angew. Chem., Int. Ed., 2025, 64, e202423281 CrossRef CAS PubMed.
- H. Zhang, J. Chen, X. Han, Y. Pan, Z. Hao, S. Tang, X. Zi, Z. Zhang, P. Gao, M. Li, J. Lv and X. Ma, Ind. Eng. Chem. Res., 2024, 63, 6210–6221 CrossRef CAS.
- K. W. Ting, T. Toyao, S. M. A. H. Siddiki and K.-I. Shimizu, ACS Catal., 2019, 9, 3685–3693 CrossRef CAS.
- S. K. Sharma, A. Banerjee, B. Paul, M. K. Poddar, T. Sasaki, C. Samanta and R. Bal, J. CO2 Util., 2021, 50, 101576 CrossRef CAS.
- X. Li, Q. Cheng, Y. Zhang, Y. Liu, Y. Pan, D. Zhao, S. Xiong, W. Liu, X. Jiang, J. Yan, X. Duan, Y. Tian and X. Li, Angew. Chem., Int. Ed., 2025, 64, e202424435 CrossRef CAS PubMed.
- W. Wang, Z. Qu, L. Song and Q. Fu, J. Energy Chem., 2020, 40, 22–30 CrossRef.
- J. Yu, M. Yang, J. Zhang, Q. Ge, A. Zimina, T. Pruessmann, L. Zheng, J.-D. Grunwaldt and J. Sun, ACS Catal., 2020, 10, 14694–14706 CrossRef CAS.
- E. M. Fiordaliso, I. Sharafutdinov, H. W. P. Carvalho, J.-D. Grunwaldt, T. W. Hansen, I. Chorkendorff, J. B. Wagner and C. D. Damsgaard, ACS Catal., 2015, 5, 5827–5836 CrossRef CAS.
- Y. Yang, M. Guo and F. Zhao, Chemphyschem, 2024, 25, e202300530 CrossRef CAS PubMed.
- F. Li, Q. Huo, X. Zhou, S. Fang, F. Han, J. Yang, Q. Guan and W. Li, Appl. Catal., B, 2025, 363, 124819 CrossRef CAS.
- C. Huang, S. Zhang, W. Wang, H. Zhou, Z. Shao, L. Xia, H. Wang and Y. Sun, ACS Catal., 2024, 14, 1324–1335 CrossRef CAS.
- A. S. Malik, S. F. Zaman, A. A. Al-Zahrani, M. A. Daous, H. Driss and L. A. Petrov, Appl. Catal., A, 2018, 560, 42–53 CrossRef CAS.
- S. Li, Y. Wang, B. Yang and L. Guo, Appl. Catal., A, 2019, 571, 51–60 CrossRef CAS.
- Z. Shi, Q. Tan and D. Wu, Appl. Catal., A, 2019, 581, 58–66 CrossRef CAS.
- O. A. Ojelade, S. F. Zaman, M. A. Daous, A. A. Al-Zahrani, A. S. Malik, H. Driss, G. Shterk and J. Gascon, Appl. Catal., A, 2019, 584, 117185 CrossRef CAS.
- R. Khobragade, M. Roškarič, G. Žerjav, M. Košiček, J. Zavašnik, N. Van de Velde, I. Jerman, N. N. Tušar and A. Pintar, Appl. Catal., A, 2021, 627, 118394 CrossRef CAS.
- W. L. Ng, P. Sripada, S. Biswas and S. Bhattacharya, Appl. Catal., A, 2022, 646, 118885 CrossRef CAS.
- L. Proaño and C. W. Jones, Appl. Catal., A, 2024, 669, 119485 CrossRef.
- R. Singh, K. Tripathi and K. K. Pant, Fuel, 2021, 303, 121289 CrossRef CAS.
- J. Han, L. Wang, J. Yu, M. Fan and D. Mao, Fuel, 2025, 381, 133262 CrossRef CAS.
- Y. Chen, C. Wang, Y. Liu, Q. Zhang, L. Zhou and Y. Zhang, J. Catal., 2024, 434, 115527 CrossRef CAS.
- X. Lu, X. Garcia, I. Serrano, M. Biset-Peiró, J. Yu, J. Li, J. Arbiol, A. Cabot and J. Llorca, J. Catal., 2025, 450, 116295 CrossRef CAS.
- L. F. Rasteiro, R. A. De Sousa, L. H. Vieira, V. K. Ocampo-Restrepo, L. G. Verga, J. M. Assaf, J. L. F. Da Silva and E. M. Assaf, Appl. Catal., B, 2022, 302, 120842 CrossRef CAS.
- H.-T. Vu, M. Finšgar, J. Zavašnik, N. N. Tušar and A. Pintar, Appl. Surf. Sci., 2023, 619, 156737 CrossRef CAS.
- R. Pothu, H. Mitta, P. Banerjee, R. Boddula, R. K. Srivastava, P. K. Kalambate, R. Naik, A. Bahgat Radwan and N. Al-Qahtani, Mater. Sci. Energy Technol., 2023, 6, 484–492 CAS.
- S. Li, L. Guo and T. Ishihara, Catal. Today, 2020, 339, 352–361 CrossRef CAS.
- F. Lin, X. Jiang, N. Boreriboon, C. Song, Z. Wang and K. Cen, Catal. Today, 2021, 371, 150–161 CrossRef CAS.
- L. Zhang, X. Hu, N. Wang and B. Chen, Catal. Today, 2024, 436, 114773 CrossRef CAS.
- D. Damma, B. Meena and P. G. Smirniotis, Catal. Today, 2025, 458, 115382 CrossRef CAS.
- J. Zuo, W. Na, P. Zhang, X. Yang, J. Wen, M. Zheng and H. Wang, Mol. Catal., 2022, 526, 112357 CAS.
- K. Chen, H. Fang, S. Wu, X. Liu, J. Zheng, S. Zhou, X. Duan, Y. Zhuang, S. Chi Edman Tsang and Y. Yuan, Appl. Catal., B, 2019, 251, 119–129 CrossRef CAS.
- C. Y. Regalado Vera, N. Manavi, Z. Zhou, L.-C. Wang, W. Diao, S. Karakalos, B. Liu, K. J. Stowers, M. Zhou, H. Luo and D. Ding, Chem. Eng. J., 2021, 426, 131767 CrossRef CAS.
- R. Singh, K. Kundu and K. K. Pant, Chem. Eng. J., 2024, 479, 147783 CrossRef CAS.
- W. Wang, X. Zhang, B. Lei and C. Peng, Chem. Eng. J., 2024, 498, 155636 CrossRef CAS.
- T. Ke, L. Wang, X. Guo, J. Yu, J. Lang, Y. H. Hu, M. Fan and D. Mao, Chem. Eng. J., 2025, 503, 158236 CrossRef CAS.
- Z. He, X. Zhang, S. Sun, Y. Zhang, W. Liu, W. Zhan, L. Wang, Q. Dai, Y. Guo, Y. Guo and A. Wang, Chem. Eng. J., 2025, 519, 165240 CrossRef CAS.
- B. Ouyang, W. Tan and B. Liu, Catal. Commun., 2017, 95, 36–39 CrossRef CAS.
- Z. Cai, X. Yu, P. Wang, H. Wu, R. Chong, L. Ren, T. Hu and X. Wang, Chin. J. Catal., 2025, 70, 410–419 CrossRef.
- J. Zhang, X. Sun, C. Wu, W. Hang, X. Hu, D. Qiao and B. Yan, J. Energy Chem., 2023, 77, 45–53 CrossRef CAS.
- A. J. Poerjoto, J. Ashok, N. Dewangan and S. Kawi, J. CO2 Util., 2021, 47, 101498 CrossRef CAS.
- E. J. Choi, Y. H. Lee, D.-W. Lee, D.-J. Moon and K.-Y. Lee, Mol. Catal., 2017, 434, 146–153 CAS.
- Y. Shao, B. Wu, B. Qiu, R. Cai, C. Quan, N. Gao, F. Zeng, X. Fan and H. Chen, AIChE J., 2024, 71, e18617 CrossRef.
- J. Xu, K. Wang, M. Zhao, R. Zhang, W. Cui, L. Liu, X. Wang, J. Wang, S. Song and H. Zhang, Angew. Chem., Int. Ed., 2025, 64, e202423438 CrossRef CAS PubMed.
- R. Singh, V. Pandey and K. K. Pant, ChemCatChem, 2022, 14, e202201053 CrossRef CAS.
- S. Chang, W. Na, J. Zhang, L. Lin and W. Gao, New J. Chem., 2021, 45, 22814–22823 RSC.
- M. A. Rossi, L. H. Vieira, L. F. Rasteiro, M. A. Fraga, J. M. Assaf and E. M. Assaf, React. Chem. Eng., 2022, 7, 1589–1602 RSC.
- J. Guo, Z. Luo, G. Hu and Z. Wang, Greenhouse Gases:Sci. Technol, 2021, 11, 1171–1179 CrossRef CAS.
- L. Kong, Y. Shi, J. Wang, P. Lv, G. Yu and W. Su, Catal. Lett., 2022, 153, 477–492 CrossRef.
- Y. Ji, S. Lin, G. Xu, T. Chen, J. Gong, F. Meng and Y. Wang, Catal. Lett., 2023, 154, 2809–2817 CrossRef.
- R. Ye, L. Ma, J. Mao, X. Wang, X. Hong, A. Gallo, Y. Ma, W. Luo, B. Wang, R. Zhang, M. S. Duyar, Z. Jiang and J. Liu, Nat. Commun., 2024, 15, 2159 CrossRef CAS PubMed.
- S. K. Sharma, B. Paul, R. S. Pal, P. Bhanja, A. Banerjee, C. Samanta and R. Bal, ACS Appl. Mater. Interfaces, 2021, 13, 28201–28213 CrossRef CAS PubMed.
- J. He, C. Yu, Z. Zhao, B. Guan, B. Zhang, Y. Zhang, L. Zhang, Y. Wang, Y. Wang, Y. Wu, J. Guo, Y. Li, T. Wu, Q. Qian, H. Wang and B. Han, Nano Res., 2025, 18, 94907130 CrossRef.
- Q. Tan, Z. Shi and D. Wu, Ind. Eng. Chem. Res., 2018, 57, 10148–10158 CrossRef CAS.
- L. H. Vieira, M. A. Rossi, L. F. Rasteiro, J. M. Assaf and E. M. Assaf, ACS Nanosci. Au, 2024, 4, 235–242 CrossRef CAS PubMed.
- Q. Tan, Z. Shi and D. Wu, Int. J. Energy Res., 2019, 43, 5392–5404 CrossRef CAS.
- H. Zhan, F. Li, P. Gao, N. Zhao, F. Xiao, W. Wei, L. Zhong and Y. Sun, J. Power Sources, 2014, 251, 113–121 CrossRef CAS.
- K. An, S. Zhang, J. Wang, Q. Liu, Z. Zhang and Y. Liu, J. Energy Chem., 2021, 56, 486–495 CrossRef CAS.
- K. Zheng, B. Liu, Y. Xu and X. Liu, J. Fuel Chem. Technol., 2024, 52, 1214–1223 CrossRef CAS.
- S. Ji, F. Hong, D. Mao, Q. Guo and J. Yu, Chem. Eng. J., 2024, 495, 153633 CrossRef CAS.
- F. Hong, D. Mao, T. Meng, G. Wu, H. Mao and J. Yu, J. Rare earths, 2025 DOI:10.1016/j.jre.2025.03.017.
- Y. Lou, F. jiang, W. Zhu, L. Wang, T. Yao, S. Wang, B. Yang, B. Yang, Y. Zhu and X. Liu, Appl. Catal., B, 2021, 291, 120122 CrossRef CAS.
- B. Xie, T. H. Tan, K. Kalantar-Zadeh, J. Zheng, P. Kumar, J. Jiang, S. Zhou, J. Scott and R. Amal, Appl. Catal., B, 2022, 315, 121599 CrossRef CAS.
- Y. Dong, R. Song, Z. Zhang, X. Han, B. Wang, S. Tao, J. Zhao, A. N. Alodhayb, Z. Chen, X. Yi and N. Zhang, Cell Rep. Phys. Sci., 2024, 5, 102227 CrossRef CAS.
- G. Zhang, M. Liu, G. Fan, L. Zheng and F. Li, ACS Appl. Mater. Interfaces, 2022, 14, 2768–2781 CrossRef CAS PubMed.
- T. Yamamura, S. Tada, R. Kikuchi, K. Fujiwara and T. Honma, J. Phys. Chem. C, 2021, 125, 15899–15909 CrossRef CAS.
- L. Wang, L. Wang, J. Zhang, X. Liu, H. Wang, W. Zhang, Q. Yang, J. Ma, X. Dong, S. J. Yoo, J. G. Kim, X. Meng and F. S. Xiao, Angew. Chem., Int. Ed., 2018, 57, 6104–6108 CrossRef CAS PubMed.
- S. Bai, Q. Shao, P. Wang, Q. Dai, X. Wang and X. Huang, J. Am. Chem. Soc., 2017, 139, 6827–6830 CrossRef CAS PubMed.
- X. Li, J. Ke, R. Li, P. Li, Q. Ma and T.-S. Zhao, Chem. Eng. Sci., 2023, 282, 119226 CrossRef CAS.
- R. Salami, Y. Zeng, X. Han, S. Rohani and Y. Zheng, J. Energy Chem., 2025, 101, 345–384 CrossRef CAS.
- Q. Zhang, S. Wang, M. Dong, J. Wang and W. Fan, Sci. China:Chem., 2025, 68, 2310–2321 CrossRef CAS.
- L. Ding, T. Shi, J. Gu, Y. Cui, Z. Zhang, C. Yang, T. Chen, M. Lin, P. Wang, N. Xue, L. Peng, X. Guo, Y. Zhu, Z. Chen and W. Ding, Chem, 2020, 6, 2673–2689 CAS.
- A. H. M. da Silva, L. H. Vieira, C. S. Santanta, M. T. M. Koper, E. M. Assaf, J. M. Assaf and J. F. Gomes, Appl. Catal., B, 2023, 324, 122221 CrossRef CAS.
- X. Wang, P. J. Ramirez, W. Liao, J. A. Rodriguez and P. Liu, J. Am. Chem. Soc., 2021, 143, 13103–13112 CrossRef CAS PubMed.
- X. Ding, J. Fu, Y. Lyu, L. Ma, Y. Xu and X. Liu, Chem. Eng. J., 2024, 494, 152923 CrossRef CAS.
- K. He, Y. Li, Z. Li, J. Chen, L. Tan, T. Qiu, X. Li, X. Ma and C. Dai, Chem. Eng. J., 2024, 501, 157608 CrossRef CAS.
- D. Xu, M. Ding, X. Hong and G. Liu, ACS Catal., 2020, 10, 14516–14526 CrossRef CAS.
- F. Zeng, C. Mebrahtu, X. Xi, L. Liao, J. Ren, J. Xie, H. J. Heeres and R. Palkovits, Appl. Catal., B, 2021, 291, 120073 CrossRef CAS.
- J. Graciani, D. C. Grinter, P. J. Ramírez, R. M. Palomino, F. Xu, I. Waluyo, D. Stacchiola, J. Fdez Sanz, S. D. Senanayake and J. A. Rodriguez, ACS Catal., 2022, 12, 15097–15109 CrossRef CAS.
- Y. Wang, W. Wang, R. He, M. Li, J. Zhang, F. Cao, J. Liu, S. Lin, X. Gao, G. Yang, M. Wang, T. Xing, T. Liu, Q. Liu, H. Hu, N. Tsubaki and M. Wu, Angew. Chem., Int. Ed., 2023, 62, e202311786 CrossRef CAS PubMed.
- K. Zheng, Y. Li, B. Liu, F. Jiang, Y. Xu and X. Liu, Angew. Chem., Int. Ed., 2022, 61, e202210991 CrossRef CAS.
- J. Chen, Y. Zha, B. Liu, Y. Li, Y. Xu and X. Liu, ACS Catal., 2023, 13, 7110–7121 CrossRef CAS.
- J.-n Zheng, K. An, J.-m Wang, J. Li and Y. Liu, J. Fuel Chem. Technol., 2019, 47, 697–708 CrossRef CAS.
- H. Kusama, K. Okabe, K. Sayama and H. Arakawa, J. Jpn. Pet. Inst., 1999, 42, 178–179 CrossRef CAS.
- X. Ding, J. Duan, M. Jia, H. Fan, Y. Lyu, J. Fu and X. Liu, Chem Asian J, 2025, 20, e202401703 CrossRef CAS PubMed.
- K. Liu, M. A. Nawaz and G. Liao, Coord. Chem. Rev., 2025, 535, 216611 CrossRef CAS.
- X. Cui, P. Gao, S. Li, C. Yang, Z. Liu, H. Wang, L. Zhong and Y. Sun, ACS Catal., 2019, 9, 3866–3876 CrossRef CAS.
- K. Cheng, Y. Li, J. Kang, Q. Zhang and Y. Wang, Acc. Chem. Res., 2024, 57, 714–725 CrossRef CAS.
- D. Wang, Z. Xie, M. D. Porosoff and J. G. Chen, Chem, 2021, 7, 2277–2311 CAS.
- F. Mahnaz, V. Dunlap, R. Helmer, S. S. Borkar, R. Navar, X. Yang and M. Shetty, ChemCatChem, 2023, 15, e202300402 CrossRef CAS.
- X. Lu, K. B. Tan, J. Zhao and G. Zhan, Chem Catal, 2025, 5, 101264 CAS.
- K. B. Tan, G. Zhan, D. Sun, J. Huang and Q. Li, J. Mater. Chem. A, 2021, 9, 5197–5231 RSC.
- L. Torrente-Murciano, R. S. Chapman, A. Narvaez-Dinamarca, D. Mattia and M. D. Jones, Phys. Chem. Chem. Phys., 2016, 18, 15496–15500 RSC.
- M. Sedighi and M. Mohammadi, J. CO2 Util., 2020, 35, 236–244 CrossRef CAS.
- M. Ghasemi, M. Mohammadi and M. Sedighi, Microporous Mesoporous Mater., 2020, 297, 110029 CrossRef.
- S. Wang, L. Zhang, W. Zhang, P. Wang, Z. Qin, W. Yan, M. Dong, J. Li, J. Wang, L. He, U. Olsbye and W. Fan, Chem, 2020, 6, 3344–3363 CAS.
- C. Xie, C. Chen, Y. Yu, J. Su, Y. Li, G. A. Somorjai and P. Yang, Nano Lett., 2017, 17, 3798–3802 CrossRef CAS.
- J. Li, T. Yu, D. Miao, X. Pan and X. Bao, Catal. Commun., 2019, 129, 105711 CrossRef.
- C. P. Ferraz, M. Tavares, L. F. Bordini, M. A. S. Garcia, J. M. A. Ribeiro de Almeida, E. F. Sousa-Aguiar and P. N. Romano, Fuel, 2024, 358, 130234 CrossRef CAS.
- W. Zhang, S. Wang, S. Guo, Z. Qin, M. Dong, J. Wang and W. Fan, Catal. Sci. Technol., 2022, 12, 2566–2577 RSC.
- M. Baur, N. K. Mast, J. P. Brahm, R. Habe, T. O. Morgen and S. Mecking, Angew. Chem., Int. Ed., 2023, 62, e202310990 CrossRef CAS PubMed.
- A. Bulati, L. Zhan, Z. Xu and K. Yang, Waste Manag., 2025, 197, 35–49 CrossRef CAS PubMed.
- M. Chu, W. Tu, S. Yang, C. Zhang, Q. Li, Q. Zhang and J. Chen, SusMat, 2022, 2, 161–185 CrossRef CAS.
- M. Selvin, S. Shah, H. J. Maria, S. Thomas, R. Tuladhar and M. Jacob, Ind. Eng. Chem. Res., 2024, 63, 1200–1214 CrossRef CAS.
- M. T. Chin and T. Diao, ACS Catal., 2024, 14, 12437–12453 CrossRef CAS PubMed.
- Y. Ma, X. Jiang, X. Xiang, P. Qu and M. Zhu, Green Chem., 2025, 27, 4040–4093 RSC.
- L. Gan, Z. Dong, H. Xu, H. Lv, G. Liu, F. Zhang and Z. Huang, CCS Chem., 2024, 6, 313–333 CrossRef CAS.
- S. Wang, R. Wang, Y. Jiang, H. Lian, Y. Lv, B. Wang, H. Zhao, G. Zheng, G. Chen and D. Gao, J. Catal., 2025, 451, 116383 CrossRef CAS.
- Y. Yamazaki, X. Jin, W. Sun, K. Nomoto, K. Takahashi, H. Miura, T. Shishido, A. Nakayama and K. Nozaki, ChemRxiv, 2024 DOI:10.26434/chemrxiv-2024-sm1lp.
- M. Tamura, S. Miyaoka, Y. Nakaji, M. Tanji, S. Kumagai, Y. Nakagawa, T. Yoshioka and K. Tomishige, Appl. Catal., B, 2022, 318, 121870 CrossRef CAS.
- M. Chu, X. Wang, X. Wang, X. Lou, C. Zhang, M. Cao, L. Wang, Y. Li, S. Liu, T. K. Sham, Q. Zhang and J. Chen, Research, 2023, 6, 0032 CrossRef CAS PubMed.
- H. Ji, X. Wang, X. Wei, Y. Peng, S. Zhang, S. Song and H. Zhang, Small, 2023, 19, 2300903 CrossRef CAS PubMed.
- H. Wang, T. Yoskamtorn, J. Zheng, P.-L. Ho, B. Ng and S. C. E. Tsang, ACS Catal., 2023, 13, 15886–15898 CrossRef CAS.
- A. Buhori, J.-W. Choi, H. Lee, C. S. Kim, K. H. Kim, K. Kim, W. Yang, J.-M. Ha and C.-J. Yoo, Chem. Eng. J., 2024, 499, 156097 CrossRef CAS.
- P. Zhao, W. Guo, Z. Gui, J. Jiang, Z. Zhu, J.-J. Li, L. Zhao, J. Zhou and Z. Xi, ACS Sustainable Chem. Eng., 2024, 12, 5738–5752 CrossRef CAS.
- T. Kwon, J. Park, K. H. Kang, D. S. Jung, W. Won and I. Ro, Green Chem., 2025, 27, 11769–11781 RSC.
- Z. Ren, Z. Luo and H. Zhang, Catal. Sci. Technol., 2025, 15, 4727–4740 RSC.
- X. Wu, X. Liu, Y. Song, W. Liu, R. Deng, X. Chu, S. Song, H. Zhang and X. Wang, J. Am. Chem. Soc., 2025, 147, 21907–21915 CrossRef CAS PubMed.
- H. Xu, C. Dong, B. Sun, K. Wang, B. Wang and G. Luo, J. Energy Inst., 2025, 121, 102185 CrossRef CAS.
- A. Tomer, M. M. Islam, M. Bahri, D. R. Inns, T. D. Manning, J. B. Claridge, N. D. Browning, C. R. A. Catlow, A. Roldan, A. P. Katsoulidis and M. J. Rosseinsky, Appl. Catal., A, 2023, 666, 119431 CrossRef CAS.
- D. R. Inns, M. Carr, M. Bahri, A. Tomer, T. D. Manning, N. Browning, S. A. Kondrat, J. B. Claridge, A. P. Katsoulidis and M. J. Rosseinsky, J. Mater. Chem. A, 2025, 13, 2032–2046 RSC.
- L. Dai, N. Zhou, Y. Lv, Y. Cheng, Y. Wang, Y. Liu, K. Cobb, P. Chen, H. Lei and R. Ruan, Prog. Energy Combust. Sci., 2022, 93, 101021 CrossRef.
- M. Chu, Y. Liu, X. Lou, Q. Zhang and J. Chen, ACS Catal., 2022, 12, 4659–4679 CrossRef CAS.
- J. Sun, J. Dong, L. Gao, Y. Q. Zhao, H. Moon and S. L. Scott, Chem. Rev., 2024, 124, 9457–9579 CrossRef CAS PubMed.
- B. Luo, H. Liu, R. Shan, J. Zhang, H. Yuan and Y. Chen, Waste Biomass Valor., 2024, 16, 1645–1657 CrossRef.
- J. Huang, A. Veksha, W. P. Chan and G. Lisak, Appl. Catal., A, 2021, 622, 118222 CrossRef CAS.
- M. A. Suarez, L. Santamaria, G. Lopez, E. Fernandez, M. Olazar, M. Amutio and M. Artetxe, Chin. J. Catal., 2025, 69, 149–162 CrossRef.
- J. E. Rorrer, G. T. Beckham and Y. Roman-Leshkov, JACS Au, 2021, 1, 8–12 CrossRef CAS PubMed.
- C. Sun, J. Wang, J. Wang, M. Shakouri, B. Shi, X. Liu, Y. Guo, Y. Hu, X.-P. Wu and Y. Wang, Appl. Catal., B, 2024, 353, 124046 CrossRef CAS.
- Q. Hu, S. Qian, Y. Wang, J. Zhao, M. Jiang, M. Sun, H. Huang, T. Gan, J. Ma, J. Zhang, Y. Cheng and Z. Niu, Nat. Commun., 2024, 15, 10573 CrossRef CAS PubMed.
- B. C. Vance, P. A. Kots, C. Wang, J. E. Granite and D. G. Vlachos, Appl. Catal., B, 2023, 322, 122138 CrossRef CAS.
- S. Kim, B. Yang, O. Y. Gutierrez, W. Zhang, C. Lizandara-Pueyo, P. Ingale, I. Jevtovikj, R. Grauke, J. Szanyi, H. Wang, S. A. Schunk and J. A. Lercher, JACS Au, 2025, 5, 1760–1770 CrossRef CAS PubMed.
- T. Kim, H. Nguyen-Phu, T. Kwon, K. H. Kang and I. Ro, Environ. Pollut., 2023, 331, 121876 CrossRef CAS PubMed.
- S. S. Borkar, R. Helmer, S. Panicker and M. Shetty, ACS Sustainable Chem. Eng., 2023, 11, 10142–10157 CrossRef CAS.
- G. Celik, R. M. Kennedy, R. A. Hackler, M. Ferrandon, A. Tennakoon, S. Patnaik, A. M. LaPointe, S. C. Ammal, A. Heyden, F. A. Perras, M. Pruski, S. L. Scott, K. R. Poeppelmeier, A. D. Sadow and M. Delferro, ACS Cent Sci., 2019, 5, 1795–1803 CrossRef CAS PubMed.
- Y. Zhang, L. Gao, Y. Zhang, X. Zhong, Z. Zhi and B. Zhang, ACS Sustainable Chem. Eng., 2025, 13, 4946–4954 CrossRef CAS.
- C. Wang, K. Yu, B. Sheludko, T. Xie, P. A. Kots, B. C. Vance, P. Kumar, E. A. Stach, W. Zheng and D. G. Vlachos, Appl. Catal., B, 2022, 319, 121899 CrossRef CAS.
- Y. Wang, T. Biddle, C. Jiang, T. Luong, R. Chen, S. Brown, X. Jie and J. Hu, Chem. Eng. J., 2023, 465, 142918 CrossRef CAS.
- P. A. Kots, P. A. Doika, B. C. Vance, S. Najmi and D. G. Vlachos, ACS Sustainable Chem. Eng., 2023, 11, 9000–9009 CrossRef CAS.
- X. Zhong, J. Liu, L. Gao, J. Chen, X. Wang, Y. Zhang, Y. A. Wu, M. Shakeri, X. Zhang and B. Zhang, Nano Res., 2024, 17, 10088–10098 CrossRef CAS.
- N. D. Hesse and R. L. White, J. Appl. Polym. Sci., 2004, 92, 1293–1301 CrossRef CAS.
- J. M. Escola, D. P. Serrano, M. Arroyo and A. Alba, J. Mater. Cycles Waste Manag., 2014, 16, 435–441 CrossRef CAS.
- J. Shang, Y. Li, Y. Hu, T. Zhang, T. Wang, J. Zhang, H. Yan, Y. Liu, X. Chen, X. Feng, X. Zhang, C. Yang and D. Chen, J. Catal., 2024, 430, 115302 CrossRef CAS.
- C. Hou, Y. Diao, T. Yun, J. Han, T. Shi, B. Chen and C. Shi, Appl. Catal., B, 2026, 382, 125944 CrossRef CAS.
- A. Alsabri, F. Tahir and S. G. Al-Ghamdi, Mater. Today: Proc., 2022, 56, 2245–2251 CAS.
- M. D. Samper, D. Bertomeu, M. P. Arrieta, J. M. Ferri and J. Lopez-Martinez, Materials, 2018, 11, 1886 CrossRef PubMed.
- D. Jubinville, E. Esmizadeh, S. Saikrishnan, C. Tzoganakis and T. Mekonnen, Sustain. Mater. Technol., 2020, 25, e00188 CAS.
- O. Guselnikova, O. Semyonov, E. Sviridova, R. Gulyaev, A. Gorbunova, D. Kogolev, A. Trelin, Y. Yamauchi, R. Boukherroub and P. Postnikov, Chem. Soc. Rev., 2023, 52, 4755–4832 RSC.
- A. A. Aboul-Enein and A. E. Awadallah, Polym. Degrad. Stab., 2019, 167, 157–169 CrossRef CAS.
- A. Arregi, M. Seifali Abbas-Abadi, G. Lopez, L. Santamaria, M. Artetxe, J. Bilbao and M. Olazar, ACS Sustainable Chem. Eng, 2020, 8, 17307–17321 CrossRef CAS.
- S. D. Jaydev, M.-E. Usteri, A. J. Martín and J. Pérez-Ramírez, Chem Catal, 2023, 3, 100564 CAS.
- W. Nabgan, B. Nabgan, T. A. Tuan Abdullah, A. A. Jalil, A. Ul-Hamid, M. Ikram, A. H. Nordin and A. Coelho, J. Anal. Appl. Pyrolysis, 2021, 154, 105018 CrossRef CAS.
- T. Muringayil Joseph, S. Azat, Z. Ahmadi, O. Moini Jazani, A. Esmaeili, E. Kianfar, J. Haponiuk and S. Thomas, Case Stud. Chem. Environ. Eng., 2024, 9, 100673 CrossRef CAS.
- M. Babaei, M. Jalilian and K. Shahbaz, J. Environ. Chem. Eng., 2024, 12, 112507 CrossRef CAS.
- H. Jia, H. Ben, Y. Luo and R. Wang, Polymers, 2020, 12, 705 CrossRef CAS PubMed.
- L. Li, C. Wang, A. Zheng and Y. Liao, ACS Sustainable Chem. Eng., 2025, 13, 4599–4610 CrossRef CAS.
- J. Chattopadhyay, T. S. Pathak, R. Srivastava and A. C. Singh, Energy, 2016, 103, 513–521 CrossRef CAS.
- C.-Y. Hsu, W.-T. Chung, T.-M. Lin, R.-X. Yang, S. S. Chen and K. C. W. Wu, Int. J. Hydrogen Energy, 2024, 49, 873–883 CrossRef CAS.
- B. Nabgan, M. Tahir, T. A. T. Abdullah, W. Nabgan, Y. Gambo, R. Mat and I. Saeh, Int. J. Hydrogen Energy, 2017, 42, 10708–10721 CrossRef CAS.
- B. Swapna, N. Singh, S. Patowary, P. Bharali, G. Madras and P. Sudarsanam, Catal. Sci. Technol., 2024, 14, 5574–5587 RSC.
- L.-X. Yun, H. Wu, Z.-G. Shen, J.-W. Fu and J.-X. Wang, ACS Sustainable Chem. Eng., 2022, 10, 5278–5287 CrossRef CAS.
- D. D. Pham, A. N. T. Cao, P. Senthil Kumar, T. B. Nguyen, H. Tran Nguyen, P. T. T. Phuong, D. L. T. Nguyen, W. Nabgan, T. H. Trinh, D.-V. N. Vo and T. M. Nguyen, Chem. Eng. Sci., 2023, 282, 119356 CrossRef CAS.
- S. Mo, J. Kou, J. Zeng, K. Song, Y. Zhang, S. He, Y. Hu, Y. Guo, X. Liu, X. Chen and Y. Wang, Chem. Eng. J., 2024, 500, 157249 CrossRef CAS.
- N. F. Zaaba and M. Jaafar, Polym. Eng. Sci., 2020, 60, 2061–2075 CrossRef CAS.
- C. L. Reichert, E. Bugnicourt, M. B. Coltelli, P. Cinelli, A. Lazzeri, I. Canesi, F. Braca, B. M. Martinez, R. Alonso, L. Agostinis, S. Verstichel, L. Six, S. Mets, E. C. Gomez, C. Issbrucker, R. Geerinck, D. F. Nettleton, I. Campos, E. Sauter, P. Pieczyk and M. Schmid, Polymers, 2020, 12, 1558 CrossRef CAS PubMed.
- J. Payne and M. D. Jones, ChemSusChem, 2021, 14, 4041–4070 CrossRef CAS PubMed.
- Y. Miao, Y. Zhao, J. Gao, J. Wang and T. Zhang, J. Am. Chem. Soc., 2024, 146, 4842–4850 CrossRef CAS PubMed.
- S. Kumar, D. Sajwan, D. Sharma and V. Krishnan, Adv. Sustainable Syst., 2025, 9, 2500003 CrossRef CAS.
- J. Wu, X. He, L. Xu, Q. Song, Z. Tian, C. Wang, M. Zhao, G. Zhou and Y. Gao, Int. J. Hydrogen Energy, 2025, 142, 1102–1112 CrossRef CAS.
- M. S. Lehnertz, J. B. Mensah and R. Palkovits, Green Chem., 2022, 24, 3957–3963 RSC.
- Q. Zhao, J. Hu, Z. Gui, Z. Chang, C. Zhang, Y. Chen, Y. Huang, P. Zhang and F. Wang, ChemSusChem, 2025, 18, e202401727 CrossRef CAS PubMed.
- S. Wang, J. Hu, Q. Zhao, G. Liu, Z. Gui, X. Liu, Y. Huang, P. Zhang, Y. Chen and F. Wang, ACS Sustainable Chem. Eng., 2024, 12, 11754–11766 CrossRef CAS.
- L. Zheng, M. Wang, Y. Li, Y. Xiong and C. Wu, Molecules, 2024, 29, 1742 CrossRef CAS PubMed.
- Y. Jiang and K. Loos, Polymers, 2016, 8, 243 CrossRef PubMed.
- J. Morales and D. Rodrigue, Macromol. Mater. Eng., 2024, 310, 2400304 CrossRef.
- J. A. Reglero Ruiz, M. Trigo-Lopez, F. C. Garcia and J. M. Garcia, Polymers, 2017, 9, 414 CrossRef PubMed.
- J. A. Lee, J. Y. Kim, J. H. Ahn, Y.-J. Ahn and S. Y. Lee, Trends Chem., 2023, 5, 873–891 CrossRef CAS.
- V. Hirschberg and D. Rodrigue, J. Polym. Sci., 2023, 61, 1937–1958 CrossRef CAS.
- Z. Liu and Y. Ma, ACS Eng. Au, 2024, 4, 432–449 CrossRef CAS.
- A.-J. Minor, R. Goldhahn, L. Rihko-Struckmann and K. Sundmacher, Chem. Eng. J., 2023, 474, 145333 CrossRef CAS.
- W. Stuyck, K. Janssens, M. Denayer, F. De Schouwer, R. Coeck, K. V. Bernaerts, J. Vekeman, F. De Proft and D. E. De Vos, Green Chem., 2022, 24, 6923–6930 RSC.
- Y. Liu and X. B. Lu, J. Polym. Sci., 2022, 60, 3256–3268 CrossRef CAS.
- J. G. Kim, Polym. Chem., 2020, 11, 4830–4849 RSC.
- B. J. Seewoo, E. V. S. Wong, Y. R. Mulders, L. M. Goodes, E. Eroglu, M. Brunner, A. Gozt, P. Toshniwal, C. Symeonides and S. A. Dunlop, Heliyon, 2024, 10, e32912 CrossRef CAS PubMed.
- M. Taguchi, Y. Ishikawa, S. Kataoka, T. Naka and T. Funazukuri, Catal. Commun., 2016, 84, 93–97 CrossRef CAS.
- F. Liu, Y. Xiao, X. Sun, G. Qin, X. Song and Y. Liu, Chem. Eng. J., 2019, 369, 205–214 CrossRef CAS.
- K. Hatakeyama, T. Kojima and T. Funazukuri, J. Mater. Cycles Waste Manage., 2013, 16, 124–130 CrossRef.
- Y. Yang, C. Wang, F. Liu, X. Sun, G. Qin, Y. Liu and J. Gao, J. Mater. Sci., 2019, 54, 9442–9455 CrossRef CAS.
- A. Kemona and M. Piotrowska, Polymers, 2020, 12, 1752 CrossRef CAS PubMed.
- B. Liu, Z. Westman, K. Richardson, D. Lim, A. L. Stottlemyer, T. Farmer, P. Gillis, V. Vlcek, P. Christopher and M. M. Abu-Omar, ACS Sustainable Chem. Eng., 2023, 11, 6114–6128 CrossRef CAS.
- J. Li, H. Zhu, D. Fang, X. Huang, C. Zhang and Y. Luo, J. Environ. Chem. Eng., 2023, 11, 110269 CrossRef CAS.
- J. Banik, D. Chakraborty, M. Rizwan, A. H. Shaik and M. R. Chandan, Waste Manag. Res., 2023, 41, 1063–1080 CrossRef CAS PubMed.
- A. Magnin, L. Entzmann, A. Bazin, E. Pollet and L. Averous, ChemSusChem, 2021, 14, 4234–4241 CrossRef CAS PubMed.
- J. Zhang, G. H. Yeoh and I. I. Kabir, Fire, 2025, 8, 64 CrossRef.
- R. Lattimer, M. Polce and C. Wesdemiotis, J. Anal. Appl. Pyrolysis, 1998, 48, 1–15 CrossRef CAS.
- W. Yang, Q. Dong, S. Liu, H. Xie, L. Liu and J. Li, Procedia Environ. Sci., 2012, 16, 167–175 CrossRef CAS.
- L. Zhao and V. Semetey, ACS Omega, 2021, 6, 4175–4183 CrossRef CAS PubMed.
- T. Calvo-Correas, L. Ugarte, P. J. Trzebiatowska, R. Sanzberro, J. Datta, M. Á. Corcuera and A. Eceiza, Polym. Degrad. Stab., 2017, 144, 411–419 CrossRef CAS.
- B. Liu, Z. Westman, K. Richardson, D. Lim, A. L. Stottlemyer, T. Farmer, P. Gillis, N. Hooshyar, V. Vlcek, P. Christopher and M. M. Abu-Omar, ACS Sustain Chem. Eng., 2024, 12, 4435–4443 CrossRef CAS PubMed.
- H. He, H. Su, H. Yu, K. Du, F. Yang, Y. Zhu, M. Ma, Y. Shi, X. Zhang, S. Chen and X. Wang, ACS Sustainable Chem. Eng., 2023, 11, 5515–5523 CrossRef CAS.
- M. Grdadolnik, A. Drincic, A. Oreski, O. C. Onder, P. Utrosa, D. Pahovnik and E. Zagar, ACS Sustain Chem. Eng., 2022, 10, 1323–1332 CrossRef CAS PubMed.
- X. Wu, R. C. Turnell-Ritson, P. Han, J. C. Schmidt, L. Piveteau, N. Yan and P. J. Dyson, Nat. Commun., 2025, 16, 4322 CrossRef CAS PubMed.
- Y. Shi, X. Diao, N. Ji, H. Ding, Z. Ya, D. Xu, R. Wei, K. Cao and S. Zhang, ACS Catal., 2024, 15, 841–868 CrossRef.
- K. P. Sullivan, A. Z. Werner, K. J. Ramirez, L. D. Ellis, J. R. Bussard, B. A. Black, D. G. Brandner, F. Bratti, B. L. Buss and X. Dong, Science, 2022, 378, 207–211 CrossRef CAS PubMed.
- D. Yao, H. Yang, Q. Hu, Y. Chen, H. Chen and P. T. Williams, Appl. Catal., B, 2021, 280, 119413 CrossRef CAS.
- J. E. Rorrer, C. Troyano-Valls, G. T. Beckham and Y. Román-Leshkov, ACS Sustain Chem. Eng., 2021, 9, 11661–11666 CrossRef CAS.
- M. Wang, Y. Gao, S. Yuan, J. Deng, J. Yang, J. Yan, S. Yu, B. Xu and D. Ma, Nat. Chem. Eng., 2024, 1, 376–384 CrossRef.
- S. Liu, P. A. Kots, B. C. Vance, A. Danielson and D. G. Vlachos, Sci. Adv., 2021, 7, eabf8283 CrossRef CAS PubMed.
- Y. Liu, P. Yan, X. Li, Q. Li, S. Li, H. Han, M. Chu, J. Fu, M. Cao, P. Xu, Q. Zhang, L. He and J. Chen, Adv. Mater., 2025, 37, e2412740 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Hongjie
Zhang
*ab,
Hongjie
Zhang