
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContemporary DFT: learning from traditional and recent trends for the development and assessment of accurate exchange–correlation functionals
E.
Brémond
 a,
A. J.
Pérez-Jiménez
b,
C.
Adamo
c and
J. C.
Sancho-García
*b
a,
A. J.
Pérez-Jiménez
b,
C.
Adamo
c and
J. C.
Sancho-García
*b
aUniversité Paris Cité, ITODYS, CNRS, F-75006 Paris, France
bDepartment of Physical Chemistry, University of Alicante, E-03080 Alicante, Spain. E-mail: jc.sancho@ua.es
cChimie ParisTech, PSL Research University, CNRS, Institute of Chemistry for Life and Health Sciences (i-CLeHS), FRE 2027, F-75005 Paris, France
First published on 30th December 2025
Abstract
Density functional theory (DFT) is the most widely and accepted model for calculating the electronic structure of physical systems, but practical applications rely on approximations to the (still) unknown exact exchange–correlation functional. We revisit here the main advances, families, and tipping points for the development of accurate expressions for the exchange–correlation functional, focusing on the historical evolution followed by DFT but also on the underlying reasons for that, while emphasizing both the theoretical foundations and the last methodological and technical developments (exemplified by deep-learned models). The latter are built by training a neural network on a very extended collection of molecular data, with the DM21, the aPBE0-ML, and the Skala functionals now available using such a strategy. This degree of development could have not been possible without the knowledge achieved so far, after more than half a century of investigations and applications of DFT to all kind of systems, and it is rooted on the traditional local (LDA), semi-local (GGA or meta-GGA) and non-local (hybrid and double-hybrid) functionals, which are still important pillars of DFT and are expected to coexist with deep-learned models, as well as on the creation of large and diverse datasets of nearly-exact reference results, which are also needed to train any of the deep-learned models envisioned.
1 Introduction
It is no exaggeration to say that density functional theory (DFT) forms part now of the core of quantum and computational chemistry, electronic structure theories, atomic and molecular physics, theoretical spectroscopy and light-matter interactions, finite-size or periodic materials science, and condensed matter physics, to name just a few of scientific fields more proned to use this theory widely and routinely. Evidently, given the historical and current importance of these fields for the whole scientific knowledge achieved so far in Chemistry, Physics, and Materials Science, this statement can be scaled up to imagine applications and developments comprising systems and properties from all the elements of the periodic table and from all states of matter. Since the main ingredient of any DFT calculation is the exchange–correlation functional, and actually the key for its impactful success and universal adoption by the scientific community, we will focus on its evolution in this perspective, which is summarized in Table 1: the first models were proposed in the late 50s, while the most modern machine-learned models date from the early 2020s, thus covering more than half a century (seven decades in fact) of continuous improvements, studies, and validations.| Jacob's ladder rung | Scaling | Examples | Availability |
|---|---|---|---|
| LDA | O(N3) | SVWN | >1950s |
| GGA | O(N3) | BLYP (semiempirical), PBE (non-empirical), | >1980s |
| meta-GGA | O(N3) | M06-L (semiempirical), r2SCAN (non-empirical) | >1990s |
| Hybrid | O(N4) | B3LYP (semiempirical), PBE0 (non-empirical), ωB97X (semiempirical), RSX-PBE0 (non-empirical) | >1990s |
| Double-hybrid | O(N5) | B2-PLYP (semiempirical), PBE-QIDH (non-empirical), ωB97X-2 (semiempirical), RSX-PBE-QIDH (non-empirical) | >2000s |
| Deep-learned | DM21, CF22D, aPBE0-ML, Skala, etc. | >2020s |
Therefore, considering a context focused on the development and assessment of exchange–correlation functionals, DFT could be now experiencing a “new age”, with the developers of modern functionals based on Machine Learning (ML) advances invoking a “game change”, after a set of deep-learned exchange–correlation functionals have recently appeared with the hope of reaching a competitive accuracy, and at a computational cost similar or even lower, compared with the best among the more traditional models. To better put these new models into the due perspective, we will first summarize here the main theoretical frameworks successfully employed so far for the development of exchange–correlation functionals, with the most modern expression being sufficiently accurate and of general applicability to drive such an immense success of DFT in recent times. We will restrict ourselves to ground-state formulations, not only for simplicity but also because deep-learned expressions are not extensively applied to excited states yet. We refer the readers to literature reviews1–7 on excited-state methods such as TD-DFT, GW, or DFT+DMFT for a deeper discussion. We will also follow a chronological order to also inform the readers about the timeline followed by DFT to reach its current state-of-the-art.
2 A route to DFT through its bibliographic and historical importance
We will first illustrate the importance of DFT along the history by resorting first to bibliographic arguments. In 2014, a literature survey was conducted to disclose the top-10 most cited scientific publications for modern Science,8 among a larger collection of top-100 studies which was also recently updated.9 Entering into that top-100 list required more than 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 citations in 2014. However, that threshold was increased to 30000 in 2025, which also evidences this increase as a byproduct of the large volume of bibliographic records occurred in the last decade. The top-10 list from 2014 included two of the expressions most used for the exchange–correlation functionals: the correlation functional developed10 by Lee–Yang–Parr (LYP) in 1988 and the first hybrid functional11 (named later as B3LYP) developed by Becke in 1993. It is important to remark that about half of the papers in the former (2014) top-100 list have changed due to the irruption of newer papers in the updated (2025) list. However, the update made in 2025 of the top-10 still contains two expressions for exchange–correlation functionals: the LYP correlation functional again, together with the Perdew–Burke–Erzenhof (PBE) exchange and correlation functionals developed12 in 1996 (the previous B3LYP expression ranks now 13). Interestingly, comparing both LYP and PBE models one can already observe another difference which has always permeated the field: while both expressions respond to a solid and deep mathematical formulation, the LYP expression (or the B3LYP one) includes a very reduced set of to-be-fitted parameters, while the PBE expression does not, with the values of the constants of the latter adjusted to reproduce known conditions and numerical limits. Actually, those DFT-based publications are gaining citations at an accelerating rate, and are accompanied into that top-10 by another manuscript dealing with the extension of DFT calculations from molecular to solid-state situations.13 Furthermore, the foundational manuscripts for DFT are also included into both (2014 and 2025) top-100 lists: the Hohenberg–Kohn theorems14 and the Kohn–Sham (KS) orbitalic (or one-electron) model15 in positions 39 and 34 (2014) and 57 and 41 (2025), which also shows the importance of the theoretical framework general to any DFT modern calculation (and not only the importance of the particular model chosen for practical calculations). To conclude this bibliographic analysis, we also notice that the exchange functional developed16 by Becke in 1988 is also listed in positions 25 (2014) and 48 (2025) and that an extension of DFT to mimic non-covalent interactions17 is also part of the last top-100 list at position 92. Those numbers are certainly an impressive achievement for DFT from the bibliometric point of view.
000 citations in 2014. However, that threshold was increased to 30000 in 2025, which also evidences this increase as a byproduct of the large volume of bibliographic records occurred in the last decade. The top-10 list from 2014 included two of the expressions most used for the exchange–correlation functionals: the correlation functional developed10 by Lee–Yang–Parr (LYP) in 1988 and the first hybrid functional11 (named later as B3LYP) developed by Becke in 1993. It is important to remark that about half of the papers in the former (2014) top-100 list have changed due to the irruption of newer papers in the updated (2025) list. However, the update made in 2025 of the top-10 still contains two expressions for exchange–correlation functionals: the LYP correlation functional again, together with the Perdew–Burke–Erzenhof (PBE) exchange and correlation functionals developed12 in 1996 (the previous B3LYP expression ranks now 13). Interestingly, comparing both LYP and PBE models one can already observe another difference which has always permeated the field: while both expressions respond to a solid and deep mathematical formulation, the LYP expression (or the B3LYP one) includes a very reduced set of to-be-fitted parameters, while the PBE expression does not, with the values of the constants of the latter adjusted to reproduce known conditions and numerical limits. Actually, those DFT-based publications are gaining citations at an accelerating rate, and are accompanied into that top-10 by another manuscript dealing with the extension of DFT calculations from molecular to solid-state situations.13 Furthermore, the foundational manuscripts for DFT are also included into both (2014 and 2025) top-100 lists: the Hohenberg–Kohn theorems14 and the Kohn–Sham (KS) orbitalic (or one-electron) model15 in positions 39 and 34 (2014) and 57 and 41 (2025), which also shows the importance of the theoretical framework general to any DFT modern calculation (and not only the importance of the particular model chosen for practical calculations). To conclude this bibliographic analysis, we also notice that the exchange functional developed16 by Becke in 1988 is also listed in positions 25 (2014) and 48 (2025) and that an extension of DFT to mimic non-covalent interactions17 is also part of the last top-100 list at position 92. Those numbers are certainly an impressive achievement for DFT from the bibliometric point of view.
Another milestone for DFT was the awarding in 1998 of the Nobel Prize in Chemistry to Walter Kohn18 for “his development of DFT”, shared with John A. Pople “for his development of computational methods in quantum chemistry”, recognizing in such a way how DFT was a perfect companion for wavefunction-based theories. To bracket the importance of DFT from a different point of view, it is pertinent to comment that the libxc library19–21 of density functional approximations, which is also an example of the state-of-the-art of the field, includes over 600 expressions for the exchange–correlation functional and is being used or interfaced with more than 30 codes. Note that this holds independently of the type of software (proprietary or commercial) or the basis set nature (Slater-type, Gaussian-type, plane waves, or grid-free) used, to name just a pair of the existing differences between the set of massively adopted codes nowadays. Indeed, this proliferation of expressions, implementations, and codes also explains the rise in scientific publications during the last decades:22 the yearly publications citing DFT in the 90s was approximately proportional to O(103) but increased an order of magnitude the next decade, to O(104), and is now doubling its number every 5 years.23 This slope is only comparable to the rate of growth of publications for e.g. climate change, which is somehow surprising for a theory, with the due approximations, having the goal of understanding the electronic structure of matter after calculating the electronic energy and its changes according to the microscopic environment. The growing number of DFT expressions is also accompanied by a higher (formal) computational cost, but this higher scaling with the system size has not precluded the large number of applications leading to this impressive situation. The reasons for that can be roughly traced back to better algorithms and techniques24–27 together with the exponential improvement in scientific computing along the last decades.
3 The form of the exchange–correlation functional as a timeline of DFT advances
A general, but unfortunately unknown, expression for any exchange–correlation functional, Exc[ρ(r)], arises from the contribution of the following terms:| Exc[ρ(r)] = (Vee[ρ(r)] − J[ρ(r)]) + (T[ρ(r)] − Ts[ρ(r)]), | (1) |
 | (2) |
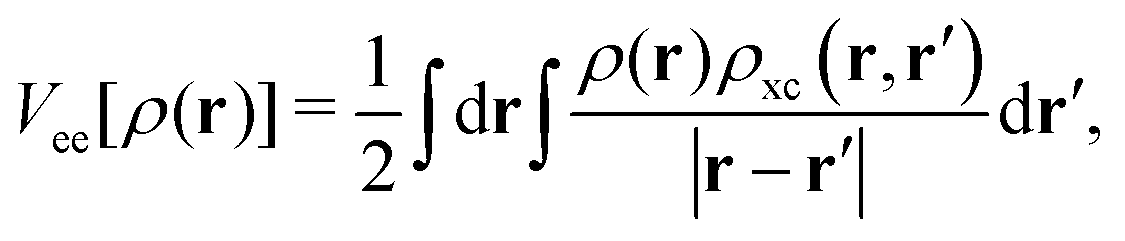 | (3) |
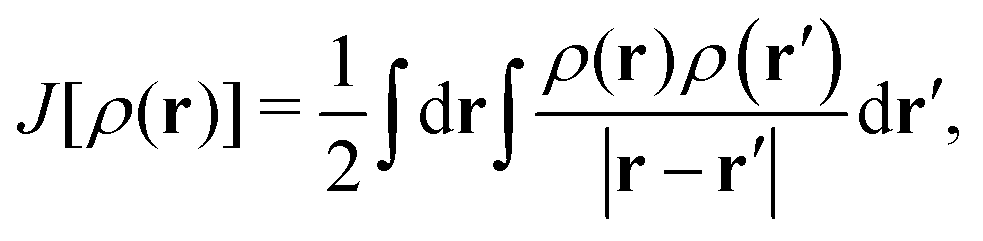 | (4) |
 . On the other hand, ρxc(r,r′) is the exchange–correlation hole of an electron at r, or the reduction in probability around each of the electrons to find another electron at r′. One can thus assume that the possible (general) ingredients of the Exc[ρ(r)] expression can be the density at the reference (or local) point, ρ(r), the density at another (non-local) spatially separated point, ρ(r′), the mathematical form adopted or simplified for the exchange–correlation hole, ρxc(r, r′), and the occupied (or even virtual) orbitals, {ϕi}.
. On the other hand, ρxc(r,r′) is the exchange–correlation hole of an electron at r, or the reduction in probability around each of the electrons to find another electron at r′. One can thus assume that the possible (general) ingredients of the Exc[ρ(r)] expression can be the density at the reference (or local) point, ρ(r), the density at another (non-local) spatially separated point, ρ(r′), the mathematical form adopted or simplified for the exchange–correlation hole, ρxc(r, r′), and the occupied (or even virtual) orbitals, {ϕi}.
We should also comment at this point some of the specific errors made in practical DFT calculations, arising from approximating this formal (and exact up to now) theoretical framework. First of all, it should be noticed that for the exact Exc[ρ(r)] expression, the Vee[ρ(r)] term is not affected by the so-called Self-Interaction Error (SIE). However, when approximated expressions are introduced for the exchange–correlation functional, SIE is not zero anymore, thus leading to one of the intrinsic problems of DFT28,29 and its multiple analysis and approaches to understand and then reduce the impact on practical calculations.30–35 On the other hand, a density-driven error has been recently identified and examined, providing a simple procedure for improving the results of DFT calculations in many situations upon using the Hartree–Fock density in place of the exact density.36,37
Of course, an outcome such as eqn (1) obeys to a solid theoretical framework, rooted first on the Hohenberg–Kohn theorems, proving the existence of an energy density functional for all the electronic systems which is also variationally well-defined, and on the Kohn–Sham approximation, simplifying the treatment after introducing a non-interacting particle systems whose associated density matches that of the exact system formed by interacting particles.15 These KS orbitals, {ϕi}, and their self-energies, 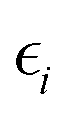 , are self-consistently obtained as part of any DFT calculation after solving iteratively the corresponding one-particle equations:
, are self-consistently obtained as part of any DFT calculation after solving iteratively the corresponding one-particle equations:
 | (5) |
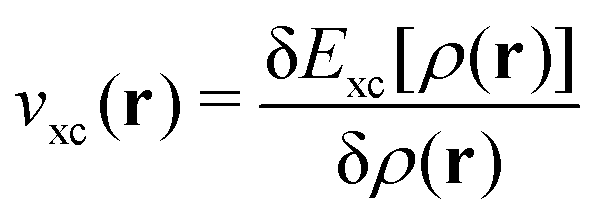 . However, the exchange–correlation functional also enters into this equation through the corresponding potential, explaining why its initial choice is so critical to the performance and accuracy of any DFT calculation, still deserving theoretical and computational efforts after more than half a century of developments. Finally, within this formalism, the exact electronic energy is therefore written as:
. However, the exchange–correlation functional also enters into this equation through the corresponding potential, explaining why its initial choice is so critical to the performance and accuracy of any DFT calculation, still deserving theoretical and computational efforts after more than half a century of developments. Finally, within this formalism, the exact electronic energy is therefore written as: | (6) |
3.1 Former but pedagogical models
Arguably, the first time (1927) in which an electronic energy term could be written as an explicit functional of the density was part of the Thomas-Fermi (TF) model,38,39 a semiclassical theory developed for the electronic structure of many-body systems arising shortly after the Schrödinger equation, circumventing for the first time in history the use of the wavefunction as the primary variable to obtain the energy and associated properties of an N-electron system. The total kinetic energy of the electrons in this model is given by with CTF a constant, which can be thus added to the potential energy of the electrons. However, that model neglected any difference for electron–electron interactions with respect to spin, which was later added thanks to the work40 of Dirac (1930) and again in the simple form
with CTF a constant, which can be thus added to the potential energy of the electrons. However, that model neglected any difference for electron–electron interactions with respect to spin, which was later added thanks to the work40 of Dirac (1930) and again in the simple form  with Cx a constant, which also connects with the Xα method developed later (1951) by Slater. Despite the limitations of these early theories, it might bring the tipping point needed to understand the origin of all the later efforts. Simply speaking, Exc[ρ(r)] is thought to be modelled as
with Cx a constant, which also connects with the Xα method developed later (1951) by Slater. Despite the limitations of these early theories, it might bring the tipping point needed to understand the origin of all the later efforts. Simply speaking, Exc[ρ(r)] is thought to be modelled as  with exc the exchange–correlation energy per unit volume, with the possible separability of terms exc = ex + ec for convenience. Later on, the successful treatment of the system41 known as the “Uniform Electron Gas” corroborated this framework, leading to the same functional form for T[ρ(r)] and Ex[ρ(r)], but with
with exc the exchange–correlation energy per unit volume, with the possible separability of terms exc = ex + ec for convenience. Later on, the successful treatment of the system41 known as the “Uniform Electron Gas” corroborated this framework, leading to the same functional form for T[ρ(r)] and Ex[ρ(r)], but with  and
and  . The correlation energy of this model was also numerically modelled42 by Vosko–Wilk–Nusair (VWN), giving rise to a functional form for Ec[ρ(r)] too. Since both exchange and correlation functionals only depend on ρ(r), the approach was consequently called Local Density Approximation (LDA) as it is known today.
. The correlation energy of this model was also numerically modelled42 by Vosko–Wilk–Nusair (VWN), giving rise to a functional form for Ec[ρ(r)] too. Since both exchange and correlation functionals only depend on ρ(r), the approach was consequently called Local Density Approximation (LDA) as it is known today.
3.2 GGA and meta-GGA models
The form of the LDA exchange and kinetic energies can also be derived from the coordinate scaling of the density, ρλ(r) = λ3ρ(λr), which is one of the known conditions to be satisfied by any model, and where the λ3 factor ensures that the scaled density still integrates to N electrons as it should be. However, the poor predictions of LDA in molecular systems motivated to go a step further considering the inhomogeneity of matter around any spatial reference point. This was done by introducing the dimensionless variable into the integrand of the e.g. exchange functional, which also guarantees the correct coordinate scaling for this kind of expressions:
into the integrand of the e.g. exchange functional, which also guarantees the correct coordinate scaling for this kind of expressions: | (7) |
 which also fits to the long-range behaviour of the energy density,
which also fits to the long-range behaviour of the energy density,  another known condition to be fulfilled by approximate expressions. The introduction of β also allows for some numerical flexibility of the model, and it is actually fitted to reproduce a small set of atomic exchange energies as close as possible. Overall, GGA is not only numerically more accurate than LDA but it also permits to introduce some of the mathematical conditions and exact constraints known for the exact functional. Considering higher-order derivatives of the density, as ∇2ρ(r) by itself or through the kinetic energy density
another known condition to be fulfilled by approximate expressions. The introduction of β also allows for some numerical flexibility of the model, and it is actually fitted to reproduce a small set of atomic exchange energies as close as possible. Overall, GGA is not only numerically more accurate than LDA but it also permits to introduce some of the mathematical conditions and exact constraints known for the exact functional. Considering higher-order derivatives of the density, as ∇2ρ(r) by itself or through the kinetic energy density  has also been explored successfully47,48 leading to the next level of functionals known as meta-GGA. Additionally, this set of elements (i.e., the density, its gradient, and higher-order derivatives) introduced step-by-step into the exchange–correlation integrand motivated their hierarchization, with LDA, GGA, and meta-GGA forming the three (lowest) ladders of a hierarchy known as Jacob's ladder49 (not only symbolically but also considering the timeline of DFT).
has also been explored successfully47,48 leading to the next level of functionals known as meta-GGA. Additionally, this set of elements (i.e., the density, its gradient, and higher-order derivatives) introduced step-by-step into the exchange–correlation integrand motivated their hierarchization, with LDA, GGA, and meta-GGA forming the three (lowest) ladders of a hierarchy known as Jacob's ladder49 (not only symbolically but also considering the timeline of DFT).
On the other hand, there are also examples of GGA exchange–correlation functionals free of adjustable parameters, such as the prominent case of PBE, which introduced in 1996 an enhancement factor for the exchange functional of the form  with s = x/x0 and x0 = 2(3π2)1/3 able to satisfy the correct uniform gas limit, Fx(0) = 1, the spin-scaling relationship of the exact exchange energy,
with s = x/x0 and x0 = 2(3π2)1/3 able to satisfy the correct uniform gas limit, Fx(0) = 1, the spin-scaling relationship of the exact exchange energy,  the expected behaviour at small density variations, Fx(x(r)) = 1 + μx2 if x → 0, and the Lieb-Oxford bound,
the expected behaviour at small density variations, Fx(x(r)) = 1 + μx2 if x → 0, and the Lieb-Oxford bound,  . Note that the values of κ and μ are not longer fitted to any external or reference data but obtained during the own derivation of the model, thus motivating the non-empirical adjective added to these models. The extension of this philosophy to the meta-GGA case is also possible, with the Tao–Perdew–Staroverov–Scuseria (TPSS) functional one of the step further in the hierarchy of models50 thanks to the progressive introduction of more exact constraints without empirical parameters. The most recent example of meta-GGA models is probably the SCAN (strongly constrained and appropriately normed) family of functionals,51,52 with the latest regularized-restored SCAN (or r2SCAN) functional53 including most of the exact constraints known for (meta-)GGAs. It is important to remark that the division between semiempirical and non-empirical functionals still impregnates the field.
. Note that the values of κ and μ are not longer fitted to any external or reference data but obtained during the own derivation of the model, thus motivating the non-empirical adjective added to these models. The extension of this philosophy to the meta-GGA case is also possible, with the Tao–Perdew–Staroverov–Scuseria (TPSS) functional one of the step further in the hierarchy of models50 thanks to the progressive introduction of more exact constraints without empirical parameters. The most recent example of meta-GGA models is probably the SCAN (strongly constrained and appropriately normed) family of functionals,51,52 with the latest regularized-restored SCAN (or r2SCAN) functional53 including most of the exact constraints known for (meta-)GGAs. It is important to remark that the division between semiempirical and non-empirical functionals still impregnates the field.
3.3 Hybrid and double-hybrid models
All the LDA, GGA, and meta-GGA functionals share the formal (asymptotic) scaling of O(N3) with respect to the system size N, although differing in their numerical quadrature and possible algorithmic speedups, but also share some of the limitations imposed by the functional form chosen as starting point, see eqn (7). GGA and meta-GGA are also considered as semi-local functionals, in the sense that some degree of non-locality is introduced thanks to that dependence on ∇ρ(r) and ∇2ρ(r), but the importance of non-locality has prompted other developments. For instance, it is well-known that local or semi-local functionals fail to deliver accurate band gaps of extended systems and key materials (i.e., insulators or semiconductors) due to the self-interaction error,54 and that the incorporation of a fraction of non-local exchange reduces the systematic underestimation of band gaps.55,56 On the other hand, the introduction of non-local exchange is also known to benefit thermochemistry and thermochemical kinetics calculations.57,58 Finally, we also mention their importance for accurate predictions for electron transport, photocurrent generation, and photovoltaics applications.59–61 Overall, global hybrid functionals add a relatively low-to-medium fraction of exact-exchange energy to a GGA or meta-GGA exchange functional, improving considerably their accuracy, which have therefore prompted its wide use in Chemistry, Materials Science, or Condensed Matter Physics.62Actually, the adiabatic connection method (ACM) allows to go a step further with respect to GGA or meta-GGA models, representing the majority of the calculations performed today at least at the molecular and supramolecular scales, thus briefly revisiting here the foundations and formal framework. The motivation is to find a mathematically admissible connection63–65 between the non-interacting (fictitious) and interacting (real) particle system, thanks to a λ-dependent electron–electron interaction term written as  . If λ ∈ [0,1], both systems are defined at the limits λ = 0 (fictitious) and λ = 1 (real). The exchange–correlation functional therefore represents the interpolation path between them, expressed as:
. If λ ∈ [0,1], both systems are defined at the limits λ = 0 (fictitious) and λ = 1 (real). The exchange–correlation functional therefore represents the interpolation path between them, expressed as:
 | (8) |
 , with Ψλ the wavefunction minimizing 〈
, with Ψλ the wavefunction minimizing 〈![[T with combining circumflex]](https://www.rsc.org/images/entities/i_char_0054_0302.gif) + λ
+ λ![[V with combining circumflex]](https://www.rsc.org/images/entities/i_char_0056_0302.gif) ee〉 whilst giving the exact density. The pending task is thus to guess the form of
ee〉 whilst giving the exact density. The pending task is thus to guess the form of  with the simplest (linear66 and quadratic67) expressions being
with the simplest (linear66 and quadratic67) expressions being  and
and  respectively. The information needed to infer the final functional form of Exc[ρ(r)] is again based on a set of known conditions, such as:
respectively. The information needed to infer the final functional form of Exc[ρ(r)] is again based on a set of known conditions, such as: | (9) |
 | (10) |
 | (11) |
Interestingly, the combination of EEXXx[{ϕi}], EPT2c[{ϕi,ϕa}], and Exc[ρ(r)] energy terms can be easily controlled and tuned step-by-step by introducing a linear combination of them:
| Exc[ρ(r)] = λxEEXXx[{ϕi}] + (1 − λx)Ex[ρ(r)] + Ec[ρ(r)], | (12) |
| Exc[ρ(r)] = λxEEXXx[{ϕi}] + (1 − λx)Ex[ρ(r)] + λcEPT2c[{ϕi,ϕa}] + (1 − λc)Ec[ρ(r)], | (13) |
The preference for the fitting procedure of those values has led to the proliferation of expressions such as the original B3PW9111 or the later popularized B3LYP,70 for which the particular combination of energy terms is:
 | (14) |
 | ||
| Fig. 1 Illustration of the Jacob's ladder in DFT, where any upper rung of the hierarchy adds more physical information and complexity, thus allowing to reach a more accurate representation of chemical systems. | ||
The advantages of developing and working with non-empirical expressions (i.e., PBE) can also be extended to these (double-)hybrid expressions too,83 as it is exemplified by the parameter-free PBE0-n models:
 | (15) |
Actually, the large number of exchange–correlation functionals available, belonging to the categories briefly discussed above, motivated the compilation of the libxc library,19 encompassing all the developments along the last 50 years and rapidly being interfaced to main codes.20
3.4 Other variants and corrections
Complementarily to these LDA, GGA, meta-GGA, hybrid or double-hybrid approximations, there have also been transversal developments transferable to all the models, such as the range-separation of the two-electron operator, or the use of a spatial-dependent EXX weight, in the hope of correcting some of the identified deficiencies of DFT. As a matter of illustration, hybrid and double-hybrid functionals still formally miss the correct long-range behaviour of the exchange potential, which should asymptotically go as . A possible solution to the problem, since λx → 1 is known to deteriorate the accuracy of the corresponding expressions for e.g. thermochemical calculations, is to force that the amount of exact exchange increases as the interaction becomes more long-ranged, by splitting the electron–electron interaction term:
. A possible solution to the problem, since λx → 1 is known to deteriorate the accuracy of the corresponding expressions for e.g. thermochemical calculations, is to force that the amount of exact exchange increases as the interaction becomes more long-ranged, by splitting the electron–electron interaction term: | (16) |
 | (17) |
Note that any range-separated hybrid functional can also be made non-empirical thanks to a recent development disclosing101,102 the relationship between λx and ω, and that range-separated semiempirical (e.g. ωB97X-2103) and non-empirical (e.g. RSX-PBE-QIDH104) double-hybrid functionals are also available. Another degree of empiricism is introduced if the value of ω is adapted to any of the system and/or geometries considered, which defines the optimally-tuned range-separated functionals.105 On the other hand, local hybrid functionals106 involve position-dependent ratio of EXX and an exchange (GGA or meta-GGA) functional, with the latter models107 employing τw(r) and the conventional KS kinetic-energy density τ(r) in the form  .
.
Particular to the case of double-hybrid functionals, there is also the possibility of allowing the splitting of the PT2 term in Eq. (13) into the same-spin and opposite-spin correlation contributions,108 scaled by the corresponding coefficients css and cos:
| EPT2c = cssEss-PT2c + cosEos-PT2c, | (18) |
Finally, it is also known that non-covalent (long-range) electronic effects are not completely introduced by any rung of the Jacob's ladder, although this omission is attenuated by the correlation energy introduced by the PT2 term of double-hybrid functionals but it is also largely dependent on the λc weight. However, the flexibility (and additivity) of DFT energy terms also allows to add a posteriori this energy, once the densities ρ(r) and ρ(r′) are available, thanks to the e.g. VV10111 correlation energy functional, taken here as example of a modern and widely used non-local correlation functional:
 | (19) |
 . The parameter b can be optimized to merge this correction to any of the available GGA, meta-GGA, hybrid, and double-hybrid functionals.112,113 The VV10 expression is part of a broader family of density functional methods for van der Waals interactions (vdW-DF), which can be found as part of the libvdwxc library114 (vdW-DF1, vdW-DF2, vdW-DF-C09, vdW-DF-cx, vdW-DF-optb86, rev-vdW-DF2, vdW-DF-optPBE, vdW-DF-optB88, BEEF-vdW, and mBEEF-vdW) where the non-local correlation forces are captured through a formal analysis of screened response in the electron gas.
. The parameter b can be optimized to merge this correction to any of the available GGA, meta-GGA, hybrid, and double-hybrid functionals.112,113 The VV10 expression is part of a broader family of density functional methods for van der Waals interactions (vdW-DF), which can be found as part of the libvdwxc library114 (vdW-DF1, vdW-DF2, vdW-DF-C09, vdW-DF-cx, vdW-DF-optb86, rev-vdW-DF2, vdW-DF-optPBE, vdW-DF-optB88, BEEF-vdW, and mBEEF-vdW) where the non-local correlation forces are captured through a formal analysis of screened response in the electron gas.
Other cost-efficient alternatives also exist, such as the widely used Dn methods17,115 (D2, D3, and D4) of S. Grimme et al., the eXchange-hole Dipole Moment (XDM) formalism,116 the density-dependent energy correction117 (dDsC), or the tailored DH-SVPD basis set118 for double-hybrid functionals such as PBE-QIDH. Due to its widespread use to efficiently (but more empirically) incorporate the non-covalent interactions at a negligible computational cost, the expression for the modern versions of those Dn methods is:
 | (20) |
 | (21) |
4 Optimal fitting and datasets in DFT
All the advances in DFT over the last decades have been fostered by the extensive benchmarking done in parallel for all the models, either by the own developers or as a part of larger and comparative studies, using extended datasets for that and providing guidance to any user about what functionals are recommended in general or for some specific or tailored application. Computational protocols,119 tutorial reviews,120 and best practices guidelines121,122 for a wise use of DFT are rooted on the robust and general performance of the models on these datasets after the adequate and step-by-step calibration studies done. On the other hand, in the case of semiempirical functionals, these datasets are also very often used for the fitting of the parameters entering into the model. Therefore, the importance of designing and using datasets for past, current, and future studies and developments cannot be underrated. The design of the reference datasets (not specifically for DFT) has reached a high degree of maturity,123 which also means to include chemically relevant systems of varied sizes and composition together with a wide variety of properties. We here mention some of the most general-purpose employed datasets, such as the GMTKN55,124 the W4,125 the MGCDB84,121 or the ACCDB,126 together with some other more suited to a particular electronic structure problem, such as the treatment of non-covalent interactions (e.g. S66 × 8127,128), the treatment of very large systems (e.g. CiM13129), the extension to excited-states (e.g. QUEST130), etc. to name just a few of the most known examples nowadays. Regarding the Materials Science field, and thus the large-scale datasets available, we can mention Materials Project,131,132 JARVIS,133,134 and Materials Cloud.135Of particular importance for DFT was also the derivation of the Minnesota datasets,136 created along several years of efforts and refinements and used to develop some of the most used exchange–correlation functionals, and the generalized use of the GMTKN55 dataset in comparison studies, basically covering basic properties and reaction energies for small systems, reaction energies for large systems and isomerization reactions, reaction barrier heights, and intra- and intermolecular non-covalent interactions. Another effort to create meta-data is the NCIAtlas collection137–141 of non-covalent interactions of several types. Finally, we also emphasize the large-scale dataset Open Molecules 2025 (OMol25),142 recently launched as an example of the culmination of the work around compilation of datasets. OMol25 is composed of small molecules, biomolecules, metal complexes, and electrolytes, covering 83 elements of the periodic table and systems up to a size of 350 atoms, and includes more than 100 million of DFT calculations at the ωB97M-V/def2-TZVPD level of theory. The motivation behind that huge dataset was to create a comprehensive data for training of machine learning models performing accurately for molecular chemistry.
Note also that these larger and modern datasets are rooted on historical developments mostly done for wavefunction-based theories, such as W4,143 HEAT,144 FPA,145 FPD,146 Gn theories,147 ccCA,148 to name just a few of them, also learning about the minimum level of theory needed to deliver sub-kcal mol−1 or even sub-kJ mol−1 accuracy for e.g. thermochemical calculations. That means that the reference (or nearly-exact) results needed for modern datasets are usually obtained at the (DLPNO-)CCSD(T)/CBS level of theory; i.e., using CCSD(T) (Coupled-Cluster Singles, Doubles, and perturbatively estimated Triples) possibly assisted by DLPNO (Domain Local Pair Natural Orbitals149), a technique applied in recent years to extend the size of the system tackled,150 and with basis sets large enough to reach the Complete Basis Set (CBS) limit. This model chemistry represents the best trade-off between accuracy and computational cost, although higher levels of CC theory can also be employed if those results are believed to be not sufficiently converged.151
Despite the emergence of lower cost methods able to deliver reference results, such as those provided by DLPNO, databases of reactions remain poorly diverse and are mostly focused on small molecular systems, not often representative of systems in use for chemical (real-world) applications. Following this line of research, some efforts were recently done to develop chemical databases gathering reactions more and more representative of current trend in chemistry, and containing new systems not yet used to parameterized semi empirical approaches, opening thus the road to unbiased benchmarking.152 As as example of this, we mention the BH9 database153 gathering a total of 449 real-life organic chemistry reactions developed with the purpose of filling this gap. It is also worth noting that machine-learning techniques are an open road to improve the diversity of these databases,154 with some new datasets (e.g. CYCLO70) already available built under this principle, even if yet marginally applied for that purpose.
Delving into the use of datasets for the optimization of the parameters entering into widely used models, also recognizing that the following division is necessarily arbitrary, users have qualitatively divided the functionals into marginally parameterized (if the exchange and correlation functionals contains very few parameters, between 5–10, and the reference data were limited but chemically meaningful, as e.g. BLYP10,16 or B3LYP11), moderately parameterized (if more parameters are introduced into the models, but not exceeding 10–20, using extended datasets for that, as e.g. B97M-V155), and heavily or highly parameterized (if a relatively large number of parameters is used, in the range of 30–50, and intensively trained, as e.g. M06-L and M06-2X74). Note that this rough classification also basically obeys to the temporal timeline followed for the development of DFT models, since the number and quality of the reference data and datasets in the 80s and 90s were necessarily more modest and smaller than in the next decades. Note also that the final number of parameters of an exchange–correlation functional, specially true for those heavily parameterized, is not critically determined by the range-separation (i.e., only ω is added), or the hybrid (i.e., only λx is needed) or double-hybrid (i.e., only λc is required, and possibly but not always css and cos) schemes, although their ultimate performance it is. On the other hand, non-empirical (or minimally empirical) functionals are obviously not parameterized, neither being proned to properties or systems covered as part of the training sets, so that a genuine use of the datasets for assessment purposes is done. A different consideration deserves the coupling of any correction for non-covalent interactions with the exchange–correlation functional, independently of semiempirical or non-empirical models, since the corresponding parameterization cannot be neglected.
Motivated by the difficulties to express the exchange–correlation functional in a compact and mathematical meaningful analytical form, some authors156–158 searched in the late 90s and early 00s for a more systematic yet optimal functional form based on extensive parameterization of polynomial or other continuum expansions. We show just an example of this approach, which additionally avoided the splitting of the exchange and correlation expressions:
 | (22) |
 , and where ωabcd are parameters, with the remaining functions being Aa = ρa↑ + ρa↓,
, and where ωabcd are parameters, with the remaining functions being Aa = ρa↑ + ρa↓,  ,
,  , and
, and 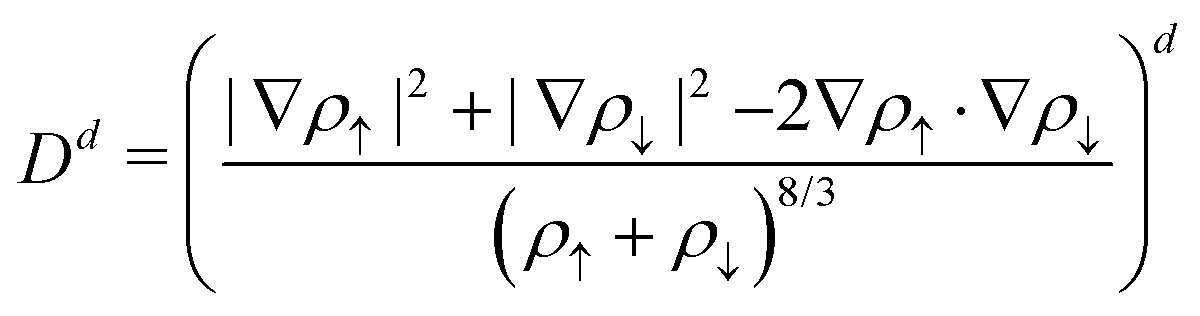 , with in principle no limits imposed for the set of (a, b, c, d) exponents and corresponding parameters. However, this line of research was more an exploration of the limit of accuracy (and redundancy of parameters and/or training data) that is ultimately possible with GGA or meta-GGA inspired expressions, but it constitutes a first step towards more systematic and modern approaches. Overall, the intense parameterization done in recent times has also disclosed some bottlenecks of semiempirical approaches, such as the bias-variance tradeoff,159 the numerical instabilities possibly caused by an overfitting,160 and the importance of the quality of the data used for the fitting.161
, with in principle no limits imposed for the set of (a, b, c, d) exponents and corresponding parameters. However, this line of research was more an exploration of the limit of accuracy (and redundancy of parameters and/or training data) that is ultimately possible with GGA or meta-GGA inspired expressions, but it constitutes a first step towards more systematic and modern approaches. Overall, the intense parameterization done in recent times has also disclosed some bottlenecks of semiempirical approaches, such as the bias-variance tradeoff,159 the numerical instabilities possibly caused by an overfitting,160 and the importance of the quality of the data used for the fitting.161
Another recent approach195 circumvents the need to develop new exchange–correlation functionals, and their optimization, and simply weights the total (ground-state) energy by functional-specific weights, in the form  with
with ![[small omega, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e0b4.gif) i the normalized weights and Ei the total energies calculated with the X density functionals entering the ensemble. This procedure guarantees the size-consistency of the results and the easy calculation of forces and potentials, and it could be also extended to densities with
i the normalized weights and Ei the total energies calculated with the X density functionals entering the ensemble. This procedure guarantees the size-consistency of the results and the easy calculation of forces and potentials, and it could be also extended to densities with  . The GMTKN55 data are used to train the ensemble with up to 77 functionals belonging to 2nd–5th rungs of Jacob's ladder included into the regression procedure, to reach an optimal functional (DENS24) achieving an error as small as 1.6 kcal mol−1 for that GMTKN55 dataset. The transferability of the ensemble and the basis set dependence of the model was also tested, as well as ways to reduce the associated computational cost to get each of the Ei energies.
. The GMTKN55 data are used to train the ensemble with up to 77 functionals belonging to 2nd–5th rungs of Jacob's ladder included into the regression procedure, to reach an optimal functional (DENS24) achieving an error as small as 1.6 kcal mol−1 for that GMTKN55 dataset. The transferability of the ensemble and the basis set dependence of the model was also tested, as well as ways to reduce the associated computational cost to get each of the Ei energies.
5 Deep-learned DFT models
We have summarized in previous sections how the conscious and meticulous work for many decades is behind the current success of DFT, regardless some generalized flaws of the theory162 such as the (many-electron) self-interaction error, or some limits imposed by the search and optimization of the parameter space. On the other hand, the recent rise of machine learning for the design of exchange–correlation functionals163 has consequently led to a new paradigm of work, which we will also try to summarize here with three examples of functionals fully machine-learned, but rooted on the prescriptions and knowledge so far achieved and belonging to the 3rd–4th rungs of the Jacob's ladder: the DeepMind (DM21),164 the “adaptative” PBE0 (aPBE0-ML)165 and the recently presented Skala model.166 Interestingly, two of these models (DM21 and Skala) are developed by private companies, Google Deepmind and Microsoft (Microsoft Research – AI for Science, and Microsoft Quantum), respectively. A common point of all these ML-based approaches is the interest to replace traditional, empirically or physically motivated approximations, with a neural-network mapping from electron density (and related magnitudes) to exchange–correlation energies. Note also that prior to DM21, there was some attempts to develop machine-learned exchange–correlation functionals (e.g. ML-ωPBE167) but not for such a general purpose as those developed later. Additionally, the use of neural network had not demonstrated any comparable or superior performance with respect to traditional (or analytical, from a mathematical point of view) functionals, as those summarized before, before the level of accuracy achieved by DM21, aPBE0-ML, or Skala functionals. For other pioneering approaches, we also mention OrbNet,168 NeuralXC,169 DeePHF,170 or CF22D.1715.1 The deepmind functional
In the DM21 model172 the proportion of exact-exchange varies locally in space, thus belonging to the family of local hybrid functionals, and also incorporates their range-separated values, which has the advantage of (formally) removing the delocalization error.173 The explicit form of the DM21 functional integrates local energies calculated by a MultiLayer Perceptron (MLP) which took as input both local and non-local features of the occupied KS orbitals: | (23) |
 the square norm of the gradient of the spin densities and of the total density, |∇ρσ(r)|2 and |∇ρ(r)|2, and the kinetic energy density,
the square norm of the gradient of the spin densities and of the total density, |∇ρσ(r)|2 and |∇ρ(r)|2, and the kinetic energy density,  . The local energies are:
. The local energies are:| eLDAx(r) = −Cx[ρ↑(r) + ρ↓(r)]4/3 | (24) |
 | (25) |
DM21 has recently been applied to transition metal chemistry,176 which can be seen as a proof-of-concept for its transferability since DM21 was exclusively trained on main-group chemistry as said before; more particularly, it was applied to the TMC151 dataset, featuring transition metal dimer dissociation energies, metal–organic reaction energies, and barriers of complexes of second- and third-row transition metals. The authors concluded that despite a similar performance to B3LYP, the model faced convergence issues most of the times, thus complicating its self-consistent implementation; however, the use of B3LYP densities instead (DM21@B3LYP) improved the results, although at the price of a higher computational cost comparable even to i.e. B2-PLYP, a double-hybrid exchange–correlation functional. One possible solution for this, already pointed out by the authors of the study, would be to incorporate transition metal reactions into the machine-learned functional, with the due care to not deteriorate the performance of main-group chemistry, borrowing again one of the strategies followed historically, that is, extending and extending the size and diversity of the datasets used for the development and assessment of the models.
DM21 has also been applied to neutral and charged water clusters,177 with a varying degree of accuracy as a function of the water cluster size: (H2O)n (n = 2–10), H3O+·(H2O)n and OH−·(H2O)n clusters with n = 2–6. The functional performed similarly to one the best range-separated meta-GGA functionals (ωB97M-V) but the errors for the normalized interaction energies (i.e., interaction energies divided by n) increased with the cluster size, thus being size-dependent. The authors also introduced a many-body potential (MB-DM21) to correct for missing many-body non-covalent interactions, and applied the corrected model to calculate some properties of liquid water with partial success, recommending to improve the functional form and physical content of machine-learned density functionals. Besides energy calculations, DM21 has also been extended to geometry optimizations and potential energy surfaces (PES). As another example of application of DM21, we mention the DM21-based calculations178 of some PES upon variation of bond angles (e.g., H–O–H in water) and bond lengths (e.g., C–H or C–C bond length in common organic molecules such as C4H8). The authors found a very good agreement between CCSD(T) and DM21-based results for all the PES tackled, including the stretched H2 and H2+ PES at long H–H distances, comparable or better than results obtained at the hybrid PW6B95/cc-pVnZ level.
5.2 The Skala functional
The first step of the developers of the Skala functional166 was the compilation of an unprecedented dataset of physically meaningful values (149079) to train the model, including a set of Total Atomization Energies (TAE) as the largest subset, which is a property known to be difficult to calculate and has always been part of both early and modern datasets. The TAE dataset is composed by 78650 values with an expected accuracy of ±1 kcal mol−1 relative to experiments, and achieved by a suited wavefunction-based protocol such as CCSD(T)/CBS. The TAE were calculated for molecules containing up to five non-H atoms (Li–F, Na–Cl) and the total number of points was complemented by other datasets. The latter included atomic (total energies, electron affinities, and ionization potentials), molecular (conformational energies, proton affinities, and ionization potentials), reaction kinetics, and both intra- and intermolecular non-covalent interactions from the NCIAtlas collection (D442x10, SH250x10, R739x5, and HB300SPXx10).
The chosen form of the Skala functional was also motivated by satisfying energetically relevant constraints such as the high-density uniform coordinate scaling, the Lieb-Oxford lower bound, and the LDA limit if the enhancement factor is made fθ(x(r)) = 1, which can be achieved by the form:
 | (26) |
 | (27) |
Looking more in detail at the performance of Skala for the different subsets of the families of the GMTKN55 dataset, the largest error was found for the SIE4x4 (self-interaction-error related systems, with 13.6 kcal mol−1) subset, the DIE60 (relative energies between C60 isomers, 8.54 kcal mol−1), BHPERI (barrier heights of pericyclic reactions, 3.13 kcal mol−1), WATER27 (Binding energies in (H2O)n, H+·(H2O)n and OH−·(H2O)n, 2.47 kcal mol−1), and IDISP (intramolecular dispersion interactions, 3.46 kcal mol−1). On the other hand, the errors given by DM21 for those subsets are 4.92, 11.35, 0.77, 1.96, and 1.37 kcal mol−1, respectively, evidencing the large discrepancies between both models when looking at their performances for the individual subsets. Actually, the largest error dropped by DM21 were found for the SIE4x4 (4.92 kcal mol−1) and DIE60 (11.35 kcal mol−1) for the families of basic properties and reaction energies for small systems, and reaction energies for large systems and isomerization reactions, respectively. For the family of reaction barrier heights, the DM21 largest error was found for the BH76 dataset (barrier heights of various reaction types, 2.08 kcal mol−1). Considering the intra- and intermolecular families of non-covalent interactions, the DM21 largest errors are again found for the WATER27 (1.96 kcal mol−1) and IDISP (1.37 kcal mol−1) datasets.
Additionally, Skala was also tested166 for equilibrium geometries with a deviation of 0.012 Å for the bond lengths of the CCse21 dataset,185 a typical deviation by semi-local functionals186 and larger than the values (mostly around 0.04–0.05 Å) provided by the rest of the functionals (r2SCAN, B97M-V, B3LYP, M06-2X, ωB97X-V, and ωB97M-V) chosen with the exception of revPBE which also leads to a deviation of 0.012 Å. By perusing now187 the LMGB35 dataset17 of light atoms main group bond lengths, Skala gave a mean absolute error of 0.014 Å, compared with 0.006 Å by DM21 and thus competitive with PBE0 results. However, except BO, F2+, N2+, NF, NH+, O2+, and OH+, those molecules belonging to LMGB35 are also included into the W4–17 used for training the DM21. Skala was also applied to the HMGB11 dataset of heavy atoms main group bond length, providing larger deviations (of 0.03 Å) than for light atoms, but comparable to those achieved by revPBE or ωB97X-V functionals.
Finally, taken from the original study,166 the authors also compared Skala and DM21 cost. We remind that the former deep-learned form (Skala) avoids a position-dependent fraction of exact-exchange (DM21) with a clear benefit for the (CPU-based) computational cost which is substantially lower by two orders of magnitude. For this comparison, the authors used a sample of molecules of varying sizes, from 50 to 900 atoms and from 1000 to 20000 orbitals, roughly speaking, taken also from established datasets such as S30L188 HS13L189 and NCI16L.190
5.3 The adaptative PBE0 functional
The “adaptative” PBE0 functional (aPBE0) was also recently developed165 by von Lilienfeld et al., based on the hypothesis that a system-dependent optimal λoptx value exists, which is actually a system-specific scalar label leading to vanishing error for the model if compared with reference of high-level electronic structures methods, in the form:| Exc[ρ(r)] = λoptx{(ZI, RI)I, S}EEXXx[{ϕi}] + (1 − λoptx{(ZI, RI)I, S})Ex[ρ(r)] + Ec[ρ(r)], | (28) |
After having confirmed the hypothesis that a significant improvement of the results can be obtained by a system-dependent mixing of semi-local and exact-exchange, the authors explored the possibility of deep-learned λoptx values: using the generated λoptx values as training data, they use a predictive model to infer values for any other system of interest, naming this variant as aPBE0-ML consequently. They predicted on-the-fly the λoptx values for the revised QM9 dataset193 (revQM9) consisting on 130000 molecules, roughly speaking. The deep-learned λoptx values followed again a Gaussian-like distribution, peaking at λx = 0.44, with a standard deviation of 0.037, and thus very close to the λx = 0.42 value disclosed before. The TAE of the reduced set of 50 molecules showed now an error of 1.32 kcal mol−1 by aPBE0-ML, and thus slightly larger than that by aPBE0 itself. HOMO–LUMO gaps errors, with respect to nearly-exact GW values for 100 organic molecules from the QM7b dataset,194 are also reduced from 3.52 (PBE0) to 0.86 (aPBE0-ML) eV, with aPBE0-ML again on top-tier of the models tested (PBE, BLYP, r2SCAN, PBE0, B3LYP, M06-2X, LC-ωPBE, and ωB97XD) with the exception of M06-2X for which a slightly lower error (0.67 eV) was found.
6 Conclusions
The field of DFT has been living a golden age for decades, as it is clearly evidenced by the large volume of scientific publications, advances, and exchange–correlation functionals expressions implemented in codes and exploited in endless applications. Generally speaking, the main advances and developments of DFT has gone in parallel with those of modern Science. For instance, the deployment of large computing facilities and hardware/software improvements has allowed the application of DFT to systems of all sizes and chemical composition, thus going a step further with respect to earlier studies. However, other issues have also arised. Among them, we mention some of the most critical ones, such as the increasing cost of the calculations with rungs of Jacob's ladder and how to alleviate it, the dependence and nature of the errors with respect to the system size and composition, the different performance (and the reasons for that) between the class of exchange–correlation functionals employed, etc. That continuous validation and assessment of models was also possible by preserving the accuracy of wavefunction-based results, which are very often taken as reference, at all size scales, thanks to the introduction of extended and chemically diverse datasets, from both the composition of the systems and the properties included in them.This rich and very fruitful context is now also experiencing another step forward due to deep-learned expressions for the exchange–correlation functional. However, the underlying hypothesis and expressions are deeply rooted on the lessons achieved so far, as well as on the key role played by the ingredients defining the exchange–correlation integrand. Furthermore, in a kind of virtuous circle, the size and availability of datasets has also been invigorated by the need to train those deep-learned expressions, which can also be seen as a source of opportunities for assessing and comparing more traditional expressions. With the due efforts in both directions, the field of DFT is expected to still grow strongly in the next decades, full of opportunities and success.
Conflicts of interest
There are no conflicts to declare.Data availability
The data that supports the findings of this study are available within the article or are available from the corresponding authors upon reasonable request.Acknowledgements
The work in Alicante is supported by grant PID2023-152372NB-I00 funded by MICIU/AEI/10.13039/501100011033. E. B. thanks ANR (Agence Nationale de la Recherche) and CGI (Commissariat à l'Investissement d'Avenir) for their financial support to this work through Labex SEAM (Science and Engineering for Advanced Materials and devices), Grant no. ANR-10-LABX-096, ANR-18-IDEX-0001, and ANR-21-CE29-0003. Funded by the European Union (ERC, MaMa, no. 101097351). Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or the European Research Council Executive Agency. Neither the European Union nor the granting authority can be held responsible for them.References
- K. Giesbertz, K. Pernal, O. Gritsenko and E. Baerends, Excitation energies with time-dependent density matrix functional theory: Singlet two-electron systems, J. Chem. Phys., 2009, 130, 114104 CrossRef CAS PubMed.
- M. E. Casida and M. Huix-Rotllant, Progress in time-dependent density-functional theory, Annu. Rev. Phys. Chem., 2012, 63, 287–323 Search PubMed.
- A. D. Laurent and D. Jacquemin, TD-DFT benchmarks: a review, Int. J. Quantum Chem., 2013, 113, 2019–2039 Search PubMed.
- C. Adamo and D. Jacquemin, The calculations of excited-state properties with Time-Dependent Density Functional Theory, Chem. Soc. Rev., 2013, 42, 845–856 Search PubMed.
- X. Blase, I. Duchemin and D. Jacquemin, The Bethe-Salpeter equation in chemistry: relations with TD-DFT, applications and challenges, Chem. Soc. Rev., 2018, 47, 1022–1043 RSC.
- E. Bremond, M. Savarese, C. Adamo and D. Jacquemin, Accuracy of TD-DFT geometries: a fresh look, J. Chem. Theory Comput., 2018, 14, 3715–3727 Search PubMed.
- S. Ghosh, P. Verma, C. J. Cramer, L. Gagliardi and D. G. Truhlar, Combining wave function methods with density functional theory for excited states, Chem. Rev., 2018, 118, 7249–7292 CrossRef CAS PubMed.
- R. Van Noorden, B. Maher and R. Nuzzo, The top 100 papers, Nat. News, 2014, 514, 550 CrossRef CAS PubMed.
- R. Van Noorden, These are the most-cited research papers of all time, Nature, 2025, 640, 591 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- A. D. Becke, Density-Functional Thermochemistry. III. The Role of Exact Exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- P. Hohenberg and W. Kohn, Inhomogeneous electron gas, Phys. Rev., 1964, 136, B864 Search PubMed.
- W. Kohn and L. J. Sham, Self-consistent equations including exchange and correlation effects, Phys. Rev., 1965, 140, A1133 Search PubMed.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 Search PubMed.
- W. Kohn, Nobel Lecture: Electronic structure of matter—wave functions and density functionals, Rev. Mod. Phys., 1999, 71, 1253 Search PubMed.
- M. A. Marques, M. J. Oliveira and T. Burnus, Libxc: A library of exchange and correlation functionals for density functional theory, Comput. Phys. Commun., 2012, 183, 2272–2281 CrossRef CAS.
- S. Lehtola, C. Steigemann, M. J. Oliveira and M. A. Marques, Recent developments in libxc – A comprehensive library of functionals for density functional theory, SoftwareX, 2018, 7, 1–5 Search PubMed.
- S. Lehtola and A. J. Karttunen, Free and open source software for computational chemistry education, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1610 CAS.
- M. Dumaz, R. Boucher, M. A. Marques and A. H. Romero, Authorship and citation cultural nature in Density Functional Theory from solid state computational packages, Scientometrics, 2021, 126, 6681–6695 Search PubMed.
- R. Haunschild, A. Barth and W. Marx, Evolution of DFT studies in view of a scientometric perspective, J. Cheminf., 2016, 8, 1–12 Search PubMed.
- T.-S. Lee, J. P. Lewis and W. Yang, Linear-scaling quantum mechanical calculations of biological molecules: The divide-and-conquer approach, Comput. Mater. Sci., 1998, 12, 259–277 Search PubMed.
- E. Artacho, D. Sánchez-Portal, P. Ordejón, A. Garcia and J. M. Soler, Linear-scaling ab initio calculations for large and complex systems, Phys. Status Solidi B, 1999, 215, 809–817 CrossRef CAS.
- F. Neese, An improvement of the resolution of the identity approximation for the formation of the Coulomb matrix, J. Comput. Chem., 2003, 24, 1740–1747 Search PubMed.
- H. Neugebauer, P. Pinski, S. Grimme, F. Neese and M. Bursch, Assessment of DLPNO-MP2 approximations in double-hybrid DFT, J. Chem. Theory Comput., 2023, 19, 7695–7703 Search PubMed.
- Y. Zhang and W. Yang, A challenge for density functionals: Self-interaction error increases for systems with a noninteger number of electrons, J. Chem. Phys., 1998, 109, 2604–2608 CrossRef CAS.
- P. Mori-Sánchez, A. J. Cohen and W. Yang, Many-electron self-interaction error in approximate density functionals, J. Chem. Phys., 2006, 125, 201102 CrossRef PubMed.
- J. P. Perdew and A. Zunger, Self-interaction correction to density-functional approximations for many-electron systems, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 23, 5048 CrossRef CAS.
- V. Polo, E. Kraka and D. Cremer, Electron correlation and the self-interaction error of density functional theory, Mol. Phys., 2002, 100, 1771–1790 CrossRef CAS.
- M. Lundberg and P. E. Siegbahn, Quantifying the effects of the self-interaction error in DFT: When do the delocalized states appear?, J. Chem. Phys., 2005, 122, 224103 CrossRef PubMed.
- J. Gräfenstein and D. Cremer, The self-interaction error and the description of non-dynamic electron correlation in density functional theory, Theor. Chem. Acc., 2009, 123, 171–182 Search PubMed.
- T. Schmidt and S. Kümmel, One-and many-electron self-interaction error in local and global hybrid functionals, Phys. Rev. B, 2016, 93, 165120 CrossRef.
- D. R. Lonsdale and L. Goerigk, The one-electron self-interaction error in 74 density functional approximations: a case study on hydrogenic mono-and dinuclear systems, Phys. Chem. Chem. Phys., 2020, 22, 15805–15830 RSC.
- S. Vuckovic, S. Song, J. Kozlowski, E. Sim and K. Burke, Density functional analysis: The theory of density-corrected DFT, J. Chem. Theory Comput., 2019, 15, 6636–6646 CrossRef CAS PubMed.
- E. Sim, S. Song, S. Vuckovic and K. Burke, Improving results by improving densities: Density-corrected density functional theory, J. Am. Chem. Soc., 2022, 144, 6625–6639 Search PubMed.
- L. H. Thomas, The calculation of atomic fields, Math. Proc. Cambridge Philos. Soc., 1927, 23, 542–548 Search PubMed.
- E. Fermi, Un metodo statistico per la determinazione di alcune priorieta dell’atome, Rend. Accad. Naz. Lincei, 1927, 6, 32 Search PubMed.
- P. A. Dirac, Note on exchange phenomena in the Thomas atom, Math. Proc. Cambridge Philos. Soc., 1930, 26, 376–385 Search PubMed.
- E. Wigner, Effects of the electron interaction on the energy levels of electrons in metals, Trans. Faraday Soc., 1938, 34, 678–685 Search PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis, Can. J. Phys., 1980, 58, 1200–1211 Search PubMed.
- J. P. Perdew, Generalized gradient approximations for exchange and correlation: A look backward and forward, Phys. B, 1991, 172, 1–6 Search PubMed.
- Y. Zhao and D. G. Truhlar, Construction of a generalized gradient approximation by restoring the density-gradient expansion and enforcing a tight Lieb-Oxford bound, J. Chem. Phys., 2008, 128, 184109 Search PubMed.
- A. Ruzsinszky, G. I. Csonka and G. E. Scuseria, Regularized gradient expansion for atoms, molecules, and solids, J. Chem. Theory Comput., 2009, 5, 763–769 Search PubMed.
- R. Peverati, Y. Zhao and D. G. Truhlar, Generalized gradient approximation that recovers the second-order density-gradient expansion with optimized across-the-board performance, J. Phys. Chem. Lett., 2011, 2, 1991–1997 CrossRef CAS.
- E. Engel and S. Vosko, Fourth-order gradient corrections to the exchange-only energy functional: Importance of ∇2ρ contributions, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 10498 CrossRef CAS PubMed.
- J. P. Perdew, S. Kurth, A. Zupan and P. Blaha, Accurate density functional with correct formal properties: A step beyond the generalized gradient approximation, Phys. Rev. Lett., 1999, 82, 2544 Search PubMed.
- J. P. Perdew and K. Schmidt, Jacob's ladder of density functional approximations for the exchange-correlation energy, AIP Conf. Proc., 2001, 577, 1–20 Search PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Climbing the density functional ladder: Nonempirical meta-generalized gradient approximation designed for molecules and solids, Phys. Rev. Lett., 2003, 91, 146401 Search PubMed.
- J. Sun, A. Ruzsinszky and J. P. Perdew, Strongly constrained and appropriately normed semilocal density functional, Phys. Rev. Lett., 2015, 115, 036402 CrossRef PubMed.
- A. P. Bartók and J. R. Yates, Regularized SCAN functional, J. Chem. Phys., 2019, 150, 161101 CrossRef PubMed.
- J. W. Furness, A. D. Kaplan, J. Ning, J. P. Perdew and J. Sun, Accurate and numerically efficient r2SCAN meta-generalized gradient approximation, J. Phys. Chem. Lett., 2020, 11, 8208–8215 CrossRef CAS PubMed.
- A. J. Garza and G. E. Scuseria, Predicting band gaps with hybrid density functionals, J. Phys. Chem. Lett., 2016, 7, 4165–4170 CrossRef CAS PubMed.
- J. Muscat, A. Wander and N. Harrison, On the prediction of band gaps from hybrid functional theory, Chem. Phys. Lett., 2001, 342, 397–401 Search PubMed.
- H. Xiao, J. Tahir-Kheli and W. A. Goddard III, Accurate band gaps for semiconductors from density functional theory, J. Phys. Chem. Lett., 2011, 2, 212–217 Search PubMed.
- Y. Zhao and D. G. Truhlar, Hybrid meta density functional theory methods for thermochemistry, thermochemical kinetics, and noncovalent interactions: the MPW1B95 and MPWB1K models and comparative assessments for hydrogen bonding and van der Waals interactions, J. Phys. Chem. A, 2004, 108, 6908–6918 Search PubMed.
- L. Goerigk and S. Grimme, A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 Search PubMed.
- S. Mandal and R. Pati, Mechanism behind the switching of current induced by a gate field in a semiconducting nanowire junction, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 115306 Search PubMed.
- A. Tal, P. Liu, G. Kresse and A. Pasquarello, Accurate optical spectra through time-dependent density functional theory based on screening-dependent hybrid functionals, Phys. Rev. Res., 2020, 2, 032019 Search PubMed.
- W. Sun, T. van der Heide, V.-Q. Vuong, T. Frauenheim, M. A. Sentef, B. Aradi and C. R. Lien-Medrano, Hybrid Functional DFTB Parametrizations for Modeling Organic Photovoltaic Systems, J. Chem. Theory Comput., 2025, 21, 5103–5117 CrossRef CAS PubMed.
- G. I. Csonka, J. P. Perdew and A. Ruzsinszky, Global hybrid functionals: A look at the engine under the hood, J. Chem. Theory Comput., 2010, 6, 3688–3703 CrossRef CAS.
- W. Yang, Generalized adiabatic connection in density functional theory, J. Chem. Phys., 1998, 109, 10107–10110 CrossRef CAS.
- M. Levy, Density-functional exchange correlation through coordinate scaling in adiabatic connection and correlation hole, Phys. Rev. A: At., Mol., Opt. Phys., 1991, 43, 4637 Search PubMed.
- M. Ernzerhof, Construction of the adiabatic connection, Chem. Phys. Lett., 1996, 263, 499–506 Search PubMed.
- A. D. Becke, A new mixing of Hartree-Fock and local density-functional theories, J. Chem. Phys., 1993, 98, 1372–1377 Search PubMed.
- É. Brémond, J. C. Sancho-Garca, Á. J. Pérez-Jiménez and C. Adamo, Communication: double-hybrid functionals from adiabatic-connection: the QIDH model, J. Chem. Phys., 2014, 141, 031101 CrossRef PubMed.
- A. J. Cohen, P. Mori-Sánchez and W. Yang, Assessment and formal properties of exchange-correlation functionals constructed from the adiabatic connection, J. Chem. Phys., 2007, 127, 034101 CrossRef PubMed.
- D. Khan, SCAN based non-linear double hybrid density functional, J. Chem. Phys., 2025, 163, 144115 Search PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- V. Barone and C. Adamo, Theoretical study of direct and water-assisted isomerization of formaldehyde radical cation. A comparison between density functional and post-Hartree-Fock approaches, Chem. Phys. Lett., 1994, 224, 432–438 CrossRef CAS.
- C. Adamo and V. Barone, Toward reliable adiabatic connection models free from adjustable parameters, Chem. Phys. Lett., 1997, 274, 242–250 Search PubMed.
- N. C. Handy and A. J. Cohen, Left-right correlation energy, Mol. Phys., 2001, 99, 403–412 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- S. Grimme, Semiempirical hybrid density functional with perturbative second-order correlation, J. Chem. Phys., 2006, 124, 034108 CrossRef PubMed.
- F. Tran, J. Stelzl and P. Blaha, Rungs 1 to 4 of DFT Jacob's ladder: Extensive test on the lattice constant, bulk modulus, and cohesive energy of solids, J. Chem. Phys., 2016, 144, 204120 Search PubMed.
- A. Karton, How reliable is DFT in predicting relative energies of polycyclic aromatic hydrocarbon isomers? comparison of functionals from different rungs of jacob's ladder, J. Comput. Chem., 2017, 38, 370–382 CrossRef CAS PubMed.
- I. Y. Zhang and X. Xu, On the top rung of Jacob's ladder of density functional theory: Toward resolving the dilemma of SIE and NCE, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11, e1490 Search PubMed.
- A. Karton, Big data benchmarking: how do DFT methods across the rungs of Jacob's ladder perform for a dataset of 122k CCSD(T) total atomization energies?, Phys. Chem. Chem. Phys., 2024, 26, 14594–14606 Search PubMed.
- J. Simmie and K. Somers, Snakes on the rungs of Jacob's ladder: anomalous vibrational spectra from double-hybrid DFT methods, J. Phys. Chem. A, 2020, 124, 6899–6902 Search PubMed.
- A. Karton, S. L. Waite and A. J. Page, Performance of DFT for C60 isomerization energies: a noticeable exception to Jacob's ladder, J. Phys. Chem. A, 2018, 123, 257–266 Search PubMed.
- S. Mandal, K. Haule, K. M. Rabe and D. Vanderbilt, Electronic correlation in nearly free electron metals with beyond-DFT methods, npj Comput. Mater., 2022, 8, 181 Search PubMed.
- J. P. Perdew, M. Ernzerhof and K. Burke, Rationale for mixing exact exchange with density functional approximations, J. Chem. Phys., 1996, 105, 9982–9985 Search PubMed.
- M. Ernzerhof and G. E. Scuseria, Assessment of the Perdew-Burke-Ernzerhof exchange-correlation functional, J. Chem. Phys., 1999, 110, 5029–5036 Search PubMed.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- C. A. Guido, E. Brémond, C. Adamo and P. Cortona, Communication: One third: A new recipe for the PBE0 paradigm, J. Chem. Phys., 2013, 138, 021104 CrossRef PubMed.
- J. Toulouse, K. Sharkas, E. Brémond and C. Adamo, Communication: Rationale for a new class of double-hybrid approximations in density-functional theory, J. Chem. Phys., 2011, 135, 101102 Search PubMed.
- K. Sharkas, J. Toulouse and A. Savin, Double-hybrid density-functional theory made rigorous, J. Chem. Phys., 2011, 134, 064113 CrossRef PubMed.
- E. Brémond and C. Adamo, Seeking for parameter-free double-hybrid functionals: the PBE0-DH model, J. Chem. Phys., 2011, 135, 024106 Search PubMed.
- E. Brémond, M. Savarese, Á. J. Pérez-Jiménez, J. C. Sancho-Garca and C. Adamo, Systematic improvement of density functionals through parameter-free hybridization schemes, J. Phys. Chem. Lett., 2015, 6, 3540–3545 CrossRef PubMed.
- T. Tsuneda and K. Hirao, Long-range correction for density functional theory, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 375–390 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- H. Iikura, T. Tsuneda, T. Yanai and K. Hirao, A long-range correction scheme for generalized-gradient-approximation exchange functionals, J. Chem. Phys., 2001, 115, 3540–3544 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Systematic optimization of long-range corrected hybrid density functionals, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed.
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 Search PubMed.
- D. Tozer and N. Handy, On the determination of excitation energies using density functional theory, Phys. Chem. Chem. Phys., 2000, 2, 2117–2121 RSC.
- D. Tozer, Relationship between long-range charge-transfer excitation energy error and integer discontinuity in Kohn–Sham theory, J. Chem. Phys., 2003, 119, 12697–12699 CrossRef CAS.
- T. M. Henderson, B. G. Janesko and G. E. Scuseria, Range separation and local hybridization in density functional theory, J. Phys. Chem. A, 2008, 112, 12530–12542 CrossRef CAS PubMed.
- T. Stein, L. Kronik and R. Baer, Reliable prediction of charge transfer excitations in molecular complexes using time-dependent density functional theory, J. Am. Chem. Soc., 2009, 131, 2818–2820 CrossRef CAS PubMed.
- T. Körzdörfer and J.-L. Brédas, Organic electronic materials: recent advances in the DFT description of the ground and excited states using tuned range-separated hybrid functionals, Acc. Chem. Res., 2014, 47, 3284–3291 CrossRef PubMed.
- É. Brémond, Á. J. Pérez-Jiménez, J. C. Sancho-Garca and C. Adamo, Range-separated hybrid density functionals made simple, J. Chem. Phys., 2019, 150, 201102 CrossRef PubMed.
- É. Brémond, Á. J. Pérez-Jiménez, J. C. Sancho-Garca and C. Adamo, Range-separated hybrid and double-hybrid density functionals: A quest for the determination of the range-separation parameter, J. Chem. Phys., 2020, 152, 244124 CrossRef PubMed.
- N. Mardirossian and M. Head-Gordon, Survival of the most transferable at the top of Jacob's ladder: Defining and testing the ωB97M(2) double hybrid density functional, J. Chem. Phys., 2018, 148, 241736 CrossRef PubMed.
- E. Brémond, M. Savarese, Á. J. Pérez-Jiménez, J. C. Sancho-Garca and C. Adamo, Range-Separated Double-Hybrid Functional from Nonempirical Constraints, J. Chem. Theory Comput., 2018, 14, 4052–4062 Search PubMed.
- A. Karolewski, L. Kronik and S. Kümmel, Using optimally tuned range separated hybrid functionals in ground-state calculations: Consequences and caveats, J. Chem. Phys., 2013, 138, 204115 Search PubMed.
- J. Jaramillo, G. E. Scuseria and M. Ernzerhof, Local hybrid functionals, J. Chem. Phys., 2003, 118, 1068–1073 CrossRef CAS.
- T. M. Maier, A. V. Arbuznikov and M. Kaupp, Local hybrid functionals: Theory, implementation, and performance of an emerging new tool in quantum chemistry and beyond, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, 9, e1378 Search PubMed.
- S. Grimme, Improved second-order Møller-Plesset perturbation theory by separate scaling of parallel-and antiparallel-spin pair correlation energies, J. Chem. Phys., 2003, 118, 9095–9102 CrossRef CAS.
- S. Kozuch and J. M. Martin, Spin-component-scaled double hybrids: an extensive search for the best fifth-rung functionals blending DFT and perturbation theory, J. Comput. Chem., 2013, 34, 2327–2344 Search PubMed.
- É. Brémond, M. Savarese, J. C. Sancho-Garca, Á. J. Pérez-Jiménez and C. Adamo, Quadratic integrand double-hybrid made spin-component-scaled, J. Chem. Phys., 2016, 144, 124104 CrossRef PubMed.
- O. A. Vydrov and T. Van Voorhis, Nonlocal van der Waals density functional: The simpler the better, J. Chem. Phys., 2010, 133, 244103 CrossRef PubMed.
- W. Hujo and S. Grimme, Performance of the van der Waals Density Functional VV10 and (hybrid) GGA Variants for Thermochemistry and Noncovalent Interactions, J. Chem. Theory Comput., 2011, 7, 3866–3871 CrossRef CAS PubMed.
- J. Aragó, E. Ort and J. C. Sancho-Garca, Nonlocal van der Waals approach merged with double-hybrid density functionals: Toward the accurate treatment of noncovalent interactions, J. Chem. Theory Comput., 2013, 9, 3437–3443 CrossRef PubMed.
- A. H. Larsen, M. Kuisma, J. Löfgren, Y. Pouillon, P. Erhart and P. Hyldgaard, libvdwxc: A library for exchange-correlation functionals in the vdW-DF family, Modell. Simul. Mater. Sci. Eng., 2017, 25, 065004 CrossRef.
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, A generally applicable atomic-charge dependent London dispersion correction, J. Chem. Phys., 2019, 150, 154122 CrossRef PubMed.
- A. Otero-De-La-Roza and E. R. Johnson, Non-covalent interactions and thermochemistry using XDM-corrected hybrid and range-separated hybrid density functionals, J. Chem. Phys., 2013, 138, 204109 CrossRef CAS PubMed.
- S. N. Steinmann and C. Corminboeuf, Comprehensive benchmarking of a density-dependent dispersion correction, J. Chem. Theory Comput., 2011, 7, 3567–3577 CrossRef CAS PubMed.
- J. S. Garcia, E. Brémond, M. Campetella, I. Ciofini and C. Adamo, Small basis set allowing the recovery of dispersion interactions with double-hybrid functionals, J. Chem. Theory Comput., 2019, 15, 2944–2953 CrossRef CAS PubMed.
- A. Karton, A computational chemist's guide to accurate thermochemistry for organic molecules, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2016, 6, 292–310 CAS.
- P. Morgante and R. Peverati, The devil in the details: a tutorial review on some undervalued aspects of density functional theory calculations, Int. J. Quantum Chem., 2020, 120, e26332 Search PubMed.
- N. Mardirossian and M. Head-Gordon, Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals, Mol. Phys., 2017, 115, 2315–2372 CrossRef CAS.
- M. Bursch, J.-M. Mewes, A. Hansen, S. Grimme and D. F. T. Best-practice, protocols for basic molecular computational chemistry, Angew. Chem., Int. Ed., 2022, 61, e202205735 Search PubMed.
- A. Karton and M. T. De Oliveira, Good Practices in Database Generation for Benchmarking Density Functional Theory, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2025, 15, e1737 Search PubMed.
- L. Goerigk, A. Hansen, C. Bauer, S. Ehrlich, A. Najibi and S. Grimme, A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions, Phys. Chem. Chem. Phys., 2017, 19, 32184–32215 RSC.
- A. Karton, N. Sylvetsky and J. M. Martin, W4-17: a diverse and high-confidence dataset of atomization energies for benchmarking high-level electronic structure methods, J. Comput. Chem., 2017, 38, 2063–2075 CrossRef CAS PubMed.
- P. Morgante and R. Peverati, ACCDB: a collection of chemistry databases for broad computational purposes, J. Comput. Chem., 2019, 40, 839–848 Search PubMed.
- J. Rezác, K. E. Riley and P. Hobza, S66: A well-balanced database of benchmark interaction energies relevant to biomolecular structures, J. Chem. Theory Comput., 2011, 7, 2427–2438 CrossRef PubMed.
- B. Brauer, M. K. Kesharwani, S. Kozuch and J. M. L. Martin, The S66x8 benchmark for noncovalent interactions revisited: explicitly correlated ab initio methods and density functional theory, Phys. Chem. Chem. Phys., 2016, 18, 20905–20925 RSC.
- Z. Ni, Y. Guo, F. Neese, W. Li and S. Li, Cluster-in-molecule local correlation method with an accurate distant pair correction for large systems, J. Chem. Theory Comput., 2021, 17, 756–766 Search PubMed.
- M. Veril, A. Scemama, M. Caffarel, F. Lipparini, M. Boggio-Pasqua, D. Jacquemin and P.-F. Loos, QUESTDB: A database of highly accurate excitation energies for the electronic structure community, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11, e1517 Search PubMed.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner and G. Ceder, et al., Commentary: The Materials Project: A materials genome approach to accelerating materials innovation, APL Mater., 2013, 1, 011002 CrossRef.
- M. K. Horton, P. Huck, R. X. Yang, J. M. Munro, S. Dwaraknath, A. M. Ganose, R. S. Kingsbury, M. Wen, J. X. Shen and T. S. Mathis, et al., Accelerated data-driven materials science with the Materials Project, Nat. Mater., 2025, 24, 1522–1532 CrossRef CAS PubMed.
- K. Choudhary, K. F. Garrity, A. C. Reid, B. DeCost, A. J. Biacchi, A. R. Hight Walker, Z. Trautt, J. Hattrick-Simpers, A. G. Kusne and A. Centrone, et al., The joint automated repository for various integrated simulations (JARVIS) for data-driven materials design, npj Comput. Mater., 2020, 6, 173 CrossRef.
- K. Choudhary, D. Wines, K. Li, K. F. Garrity, V. Gupta, A. H. Romero, J. T. Krogel, K. Saritas, A. Fuhr and P. Ganesh, et al., JARVIS-Leaderboard: a large scale benchmark of materials design methods, npj Comput. Mater., 2024, 10, 93 CrossRef.
- L. Talirz, S. Kumbhar, E. Passaro, A. V. Yakutovich, V. Granata, F. Gargiulo, M. Borelli, M. Uhrin, S. P. Huber and S. Zoupanos, et al., Materials Cloud, a platform for open computational science, Sci. Data, 2020, 7, 299 CrossRef PubMed.
- Y. Shu, Z. Zhu, S. Kanchanakungwankul and D. G. Truhlar, Small Representative Databases for Testing and Validating Density Functionals and Other Electronic Structure Methods, J. Phys. Chem. A, 2024, 128, 6412–6422 Search PubMed.
- J. Rezac, Non-covalent interactions atlas benchmark data sets: Hydrogen bonding, J. Chem. Theory Comput., 2020, 16, 2355–2368 Search PubMed.
- J. Řezáč, Non-covalent interactions atlas benchmark data sets 2: Hydrogen bonding in an extended chemical space, J. Chem. Theory Comput., 2020, 16, 6305–6316 CrossRef PubMed.
- K. Kříž, M. Novacek and J. Rezac, Non-covalent interactions atlas benchmark data sets 3: Repulsive contacts, J. Chem. Theory Comput., 2021, 17, 1548–1561 CrossRef PubMed.
- K. Kříž and J. Řezáč, Non-covalent interactions atlas benchmark data sets 4: σ-hole interactions, Phys. Chem. Chem. Phys., 2022, 24, 14794–14804 Search PubMed.
- J. Řezáč, Non-Covalent Interactions Atlas benchmark data sets 5: London dispersion in an extended chemical space, Phys. Chem. Chem. Phys., 2022, 24, 14780–14793 RSC.
- D. S. Levine, M. Shuaibi, E. W. C. Spotte-Smith, M. G. Taylor, M. R. Hasyim, K. Michel, I. Batatia, G. Csanyi, M. Dzamba and P. Eastman, et al., The open molecules 2025 (omol25) dataset, evaluations, and models, arXiv, 2025, preprint, arXiv:2505.08762 DOI:10.48550/arXiv2505.08762.
- A. Karton, E. Rabinovich, J. M. Martin and B. Ruscic, W4 theory for computational thermochemistry: In pursuit of confident sub-kJ mol−1 predictions, J. Chem. Phys., 2006, 125, 144108 Search PubMed.
- A. Tajti, P. G. Szalay, A. G. Császár, M. Kállay, J. Gauss, E. F. Valeev, B. A. Flowers, J. Vázquez and J. F. Stanton, HEAT: High accuracy extrapolated ab initio thermochemistry, J. Chem. Phys., 2004, 121, 11599–11613 CrossRef CAS PubMed.
- P. M. Nelson, Z. L. Glick and C. D. Sherrill, Approximating large-basis coupled-cluster theory vibrational frequencies using focal-point approximations, J. Chem. Phys., 2023, 159, 094104 CrossRef CAS PubMed.
- D. Feller, K. A. Peterson and D. A. Dixon, Further benchmarks of a composite, convergent, statistically calibrated coupled-cluster-based approach for thermochemical and spectroscopic studies, Mol. Phys., 2012, 110, 2381–2399 CrossRef CAS.
- L. A. Curtiss, P. C. Redfern and K. Raghavachari, Gn theory, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 810–825 CAS.
- N. J. DeYonker, T. R. Cundari and A. K. Wilson, The correlation consistent composite approach (ccCA): An alternative to the Gaussian-n methods, J. Chem. Phys., 2006, 124, 114104 Search PubMed.
- D. G. Liakos and F. Neese, Is it possible to obtain coupled cluster quality energies at near density functional theory cost? Domain-based local pair natural orbital coupled cluster vs modern density functional theory, J. Chem. Theory Comput., 2015, 11, 4054–4063 Search PubMed.
- I. Sandler, J. Chen, M. Taylor, S. Sharma and J. Ho, Accuracy of DLPNO-CCSD(T): Effect of basis set and system size, J. Phys. Chem. A, 2021, 125, 1553–1563 Search PubMed.
- A. Karton, Highly accurate CCSDT(Q)/CBS reaction barrier heights for a diverse set of transition structures: Basis set convergence and cost-effective approaches for estimating post-CCSD(T) contributions, J. Phys. Chem. A, 2019, 123, 6720–6732 CrossRef CAS PubMed.
- É. Brémond, H. Li, Á. J. Pérez-Jiménez, J. C. Sancho-Garca and C. Adamo, Tackling an accurate description of molecular reactivity with double-hybrid density functionals, J. Chem. Phys., 2022, 156, 161101 Search PubMed.
- V. K. Prasad, Z. Pei, S. Edelmann, A. Otero-de-la Roza and G. A. DiLabio, BH9, a New Comprehensive Benchmark Data Set for Barrier Heights and Reaction Energies: Assessment of Density Functional Approximations and Basis Set Incompleteness Potentials, J. Chem. Theory Comput., 2022, 18, 151–166 CrossRef CAS PubMed.
- J. E. Alfonso-Ramos, C. Adamo, É. Brémond and T. Stuyver, Improving the reliability of, and confidence in, DFT functional benchmarking through active learning, J. Chem. Theory Comput., 2025, 21, 1752–1761 CrossRef CAS PubMed.
- N. Mardirossian and M. Head-Gordon, Mapping the genome of meta-generalized gradient approximation density functionals: The search for B97M-V, J. Chem. Phys., 2015, 142, 074111 CrossRef PubMed.
- F. A. Hamprecht, A. J. Cohen, D. J. Tozer and N. C. Handy, Development and assessment of new exchange-correlation functionals, J. Chem. Phys., 1998, 109, 6264–6271 CrossRef CAS.
- D. J. Tozer and N. C. Handy, Development of new exchange-correlation functionals. 2, J. Phys. Chem. A, 1998, 102, 3162–3168 CrossRef CAS.
- A. D. Becke, Exploring the limits of gradient corrections in density functional theory, J. Comput. Chem., 1999, 20, 63–69 Search PubMed.
- R. Peverati, Fitting elephants in the density functionals zoo: Statistical criteria for the evaluation of density functional theory methods as a suitable replacement for counting parameters, Int. J. Quantum Chem., 2021, 121, e26379 Search PubMed.
- S. E. Wheeler and K. Houk, Integration grid errors for meta-GGA-predicted reaction energies: Origin of grid errors for the M06 suite of functionals, J. Chem. Theory Comput., 2010, 6, 395–404 Search PubMed.
- B. Chan, W. Dawson and T. Nakajima, Data Quality in the Fitting of Approximate Models: A Computational Chemistry Perspective, J. Chem. Theory Comput., 2024, 20, 10468–10476 Search PubMed.
- A. J. Cohen, P. Mori-Sánchez and W. Yang, Challenges for density functional theory, Chem. Rev., 2012, 112, 289–320 CrossRef CAS PubMed.
- R. Pederson, B. Kalita and K. Burke, Machine learning and density functional theory, Nat. Rev. Phys., 2022, 4, 357–358 Search PubMed.
- J. Kirkpatrick, B. McMorrow, D. H. Turban, A. L. Gaunt, J. S. Spencer, A. G. Matthews, A. Obika, L. Thiry, M. Fortunato and D. Pfau, et al., Pushing the frontiers of density functionals by solving the fractional electron problem, Science, 2021, 374, 1385–1389 Search PubMed.
- D. Khan, A. J. Price, B. Huang, M. L. Ach and O. A. von Lilienfeld, Adapting hybrid density functionals with machine learning, Sci. Adv., 2025, 11, eadt7769 Search PubMed.
- G. Luise, C.-W. Huang, T. Vogels, D. P. Kooi, S. Ehlert, S. Lanius, K. J. Giesbertz, A. Karton, D. Gunceler and M. Stanley, et al., Accurate and scalable exchange-correlation with deep learning, arXiv, 2025, preprint, arXiv:2506.14665 DOI:10.48550/arXiv.2506.14665.
- C.-W. Ju, E. J. French, N. Geva, A. W. Kohn and Z. Lin, Stacked ensemble machine learning for range-separation parameters, J. Phys. Chem. Lett., 2021, 12, 9516–9524 CrossRef CAS PubMed.
- Z. Qiao, M. Welborn, A. Anandkumar, F. R. Manby and T. F. Miller, OrbNet: Deep learning for quantum chemistry using symmetry-adapted atomic-orbital features, J. Chem. Phys., 2020, 153, 124111 Search PubMed.
- S. Dick and M. Fernandez-Serra, Machine learning accurate exchange and correlation functionals of the electronic density, Nat. Commun., 2020, 11, 3509 Search PubMed.
- Y. Chen, L. Zhang, H. Wang and W. E, Ground state energy functional with Hartree-Fock efficiency and chemical accuracy, J. Phys. Chem. A, 2020, 124, 7155–7165 Search PubMed.
- Y. Liu, C. Zhang, Z. Liu, D. G. Truhlar, Y. Wang and X. He, Supervised learning of a chemistry functional with damped dispersion, Nat. Comput. Sci., 2023, 3, 48–58 Search PubMed.
- J. P. Perdew, Artificial intelligence “sees” split electrons, Science, 2021, 374, 1322–1323 Search PubMed.
- K. R. Bryenton, A. A. Adeleke, S. G. Dale and E. R. Johnson, Delocalization error: The greatest outstanding challenge in density-functional theory, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2023, 13, e1631 Search PubMed.
- I. S. Gerasimov, T. V. Losev, E. Y. Epifanov, I. Rudenko, I. S. Bushmarinov, A. A. Ryabov, P. A. Zhilyaev and M. G. Medvedev, Comment on “Pushing the frontiers of density functionals by solving the fractional electron problem”, Science, 2022, 377, eabq3385 Search PubMed.
- J. Kirkpatrick, B. McMorrow, D. H. Turban, A. L. Gaunt, J. S. Spencer, A. G. Matthews, A. Obika, L. Thiry, M. Fortunato and D. Pfau, et al., Response to Comment on “Pushing the frontiers of density functionals by solving the fractional electron problem”, Science, 2022, 377, eabq4282 Search PubMed.
- H. Zhao, T. Gould and S. Vuckovic, Deep Mind 21 functional does not extrapolate to transition metal chemistry, Phys. Chem. Chem. Phys., 2024, 26, 12289–12298 Search PubMed.
- E. Palos, E. Lambros, S. Dasgupta and F. Paesani, Density functional theory of water with the machine-learned DM21 functional, J. Chem. Phys., 2022, 156, 161103 Search PubMed.
- B. Jijila, V. Nirmala, P. Selvarengan, D. Kavitha, V. Arun Muthuraj and A. Rajagopal, Employing neural density functionals to generate potential energy surfaces, J. Mol. Model., 2024, 30, 65 Search PubMed.
- F. Ju, X. Wei, L. Huang, A. J. Jenkins, L. Xia, J. Zhang, J. Zhu, H. Yang, B. Shao and P. Dai, et al., Acceleration without disruption: DFT software as a service, J. Chem. Theory Comput., 2024, 20, 10838–10851 Search PubMed.
- Q. Sun, T. C. Berkelbach, N. S. Blunt, G. H. Booth, S. Guo, Z. Li, J. Liu, J. D. McClain, E. R. Sayfutyarova and S. Sharma, et al., PySCF: the Python-based simulations of chemistry framework, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1340 Search PubMed.
- A. D. Becke, G. Santra and J. M. Martin, A double-hybrid density functional based on good local physics with outstanding performance on the GMTKN55 database, J. Chem. Phys., 2023, 158, 151103 Search PubMed.
- A. D. Becke, A remarkably simple dispersion damping scheme and the DH24 double hybrid density functional, J. Chem. Phys., 2024, 160, 204118 CrossRef CAS PubMed.
- E. Brémond, M. Rodríguez-Mayorga, A.-J. Pérez-Jiménez, C. Adamo and J.-C. Sancho-García, Assessment of the nonempirical r2SCAN-QIDH double-hybrid density functional against large and diverse datasets, J. Chem. Phys., 2023, 159, 141101 Search PubMed.
- É. Brémond, Á. J. Pérez-Jiménez, J. C. Sancho-Garca and C. Adamo, SOS1-RSX-QIDH: A spin-opposite-scaled range-separated-exchange quadratic-integrand double-hybrid density functional, J. Chem. Phys., 2023, 159, 234104 CrossRef PubMed.
- M. Piccardo, E. Penocchio, C. Puzzarini, M. Biczysko and V. Barone, Semi-experimental equilibrium structure determinations by employing B3LYP/SNSD anharmonic force fields: Validation and application to semirigid organic molecules, J. Phys. Chem. A, 2015, 119, 2058–2082 Search PubMed.
- É. Brémond, M. Savarese, N. Q. Su, Á. J. Pérez-Jiménez, X. Xu, J. C. Sancho-Garca and C. Adamo, Benchmarking density functionals on structural parameters of small-/medium-sized organic molecules, J. Chem. Theory Comput., 2016, 12, 459–465 CrossRef PubMed.
- K. Kulaev, A. Ryabov, M. Medvedev, E. Burnaev and V. Vanovskiy, On the practical applicability of modern DFT functionals for chemical computations. Case study of DM21 applicability for geometry optimization, arXiv, 2025, preprint, arXiv:2501.12149 DOI:10.48550/arXiv.2501.12149.
- R. Sure and S. Grimme, Comprehensive benchmark of association (free) energies of realistic host-guest complexes, J. Chem. Theory Comput., 2015, 11, 3785–3801 Search PubMed.
- J. Gorges, S. Grimme and A. Hansen, Reliable prediction of association (free) energies of supramolecular complexes with heavy main group elements-the HS13L benchmark set, Phys. Chem. Chem. Phys., 2022, 24, 28831–28843 RSC.
- J. Gorges, B. Baedorf, S. Grimme and A. Hansen, Efficient computation of the interaction energies of very large non-covalently bound complexes, Synlett, 2023, 1135–1146 CrossRef CAS.
- B. Huang, O. A. von Lilienfeld, J. T. Krogel, A. Benali and D. M. C. Toward, accuracy across chemical space with scalable Δ-QML, J. Chem. Theory Comput., 2023, 19, 1711–1721 CrossRef CAS PubMed.
- M. Schwilk, D. N. Tahchieva and O. A. von Lilienfeld, Large yet bounded: Spin gap ranges in carbenes, arXiv, 2020, preprint, arXiv:2004.10600 DOI:10.48550/arXiv.2004.10600.
- R. Ramakrishnan, P. O. Dral, M. Rupp and O. A. Von Lilienfeld, Quantum chemistry structures and properties of 134 kilo molecules, Sci. Data, 2014, 1, 1–7 Search PubMed.
- B. Huang and O. A. Von Lilienfeld, Ab initio machine learning in chemical compound space, Chem. Rev., 2021, 121, 10001–10036 Search PubMed.
- Y. Rui, Y. Chen, E. Ivanova, V. B. Kumar, S. Smiga, I. Grabowski and P. O. Dral, The best DFT functional is the ensemble of functionals, Adv. Sci., 2024, 11, 2408239 Search PubMed.
| This journal is © the Owner Societies 2026 |