
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective ammonia sensing through reversible vapochromism and luminescence ON–OFF switching of a chalcone-based Co(II) complex
Mai
Mukoyama
a,
Masashi
Hashimoto
 ab,
Yuta
Shudo
c,
Hajime
Yagi
ab,
Hayato
Kikuchi
a and
Manabu
Nakaya
*ab
ab,
Yuta
Shudo
c,
Hajime
Yagi
ab,
Hayato
Kikuchi
a and
Manabu
Nakaya
*ab
aDepartment of Material Science, Graduate School of Science, Josai University, 1-1 Keyakidai, Sakado, Saitama 350-0295, Japan. E-mail: nakaya@josai.ac.jp
bDepartment of Chemistry and Biological Science, Faculty of Science, Josai University, 1-1 Keyakidai, Sakado, Saitama 350-0295, Japan
cNanomaterials Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), 1-1-1 Higashi, Tsukuba 305-8565, Japan
First published on 13th April 2026
Abstract
In this work, a chalcone-based cobalt(II) complex exhibits ammonia (NH3)-induced vapochromism with reversible luminescence ON–OFF switching, triggered by partial dissociation and NH3 insertion at the cobalt(II) center.
Stimuli-responsive metal complexes that exhibit optical changes have emerged as a key class of materials for applications in chemosensing, bioimaging, and smart optical devices.1–5 Reversible modulation of the coordination environment and metal oxidation state at the metal center—as well as subtle changes in molecular arrangement within the crystal lattice—provides a versatile platform for constructing systems capable of physically- or chemically-induced electronic or spin switching.6–10 Optically active compounds based on such mechanisms have been widely investigated for sensing industrially relevant gases (O2, CO2, N2, etc.) as well as environmentally hazardous gases and pollutants (NOx, SOx, NH3) and other volatile organic compounds (VOCs).11–14
Polymeric solid materials such as metal–organic frameworks (MOFs), inorganic perovskites and so on have long been explored as highly effective guest adsorbents, owing to their stable porosity and high surface areas.15–19 However, translating adsorption events into clear and readily observable physical responses remains challenging. As a result, systems that enable direct and instantaneous optical readouts for in situ sensing remain relatively limited.20,21 In recent years, however, discrete metal complexes have attracted increasing attention as promising candidates for sensing, particularly industrially relevant gases and pollutants.22–26 These studies underscore the broad applicability of coordination-driven switching mechanisms for the selective and sensitive detection of diverse chemical analytes. On the other hand, molecular compounds can exhibit much faster and more discrete structural or optical changes, yet they typically suffer from limited chemical selectivity toward specific analytes.
Herein, we report a mononuclear cobalt(II) complex bearing a chalcone-based luminescent ligand (L1·Co) that exhibits highly selective and reversible vapochromism and luminescence ON–OFF switching in response to ammonia (NH3) vapor. The switching mechanism involves partial dissociation of the ligand upon NH3 coordination to the Co(II) center, restoring the ligand's intrinsic emission. Subsequent removal of NH3 leads to reformation of the original L1·Co and quenching of the luminescence. These reversible optical changes are supported by reflectance spectroscopy, powder X-ray diffraction (PXRD), gas adsorption analysis, elemental analysis and density functional theory (DFT) calculations.
The luminescent ligand L1, a chalcone derivative bearing a dimethylamine group, was synthesized according to a reported procedure with minor modifications (see Experimental section in the SI).27 Single crystals of L1·Co suitable for single crystal X-ray diffraction (SC-XRD) were obtained by slow evaporation of an acetone solution of L1 and CoCl2·4H2O (Fig. 1), enabling detailed elucidation of the coordination environment at the metal center. Crystallographic parameters are summarized in Table S1. Elemental analysis indicated the presence of a small amount of adsorbed solvent in the powder sample (Table S2); however, the PXRD pattern was consistent with that simulated from the SC-XRD data. This result suggests that the detected solvent molecules are surface-adsorbed, as no lattice solvent molecules were observed in the crystal structure. Notably, the powder sample of L1·Co is structurally identical to the crystalline sample; therefore, it was used in most subsequent experiments.
 | ||
| Fig. 1 Crystal structure of L1·Co. Color code: C, grey; N, blue; H, light blue; O, red; Cl, green; Co, magenta. | ||
Solid-state reflectance spectra of L1 (green dashed line) and L1·Co (red solid line) are shown in Fig. 2a. The reddish-brown L1 ligand exhibited a decrease in reflectance below 600 nm, whereas its cobalt(II) complex L1·Co, with a darker brown color, showed reduced reflectance extending to below 800 nm. This reflectance change upon metal complexation, i.e., red-shift in absorption, is attributed to metal-to-ligand charge transfer (MLCT). As shown in Fig. 2b, the luminescence spectrum of L1 (green dashed line) displays a clear emission maximum at 585 nm (λex = 405 nm), whereas L1·Co exhibits no detectable emission (red solid line).
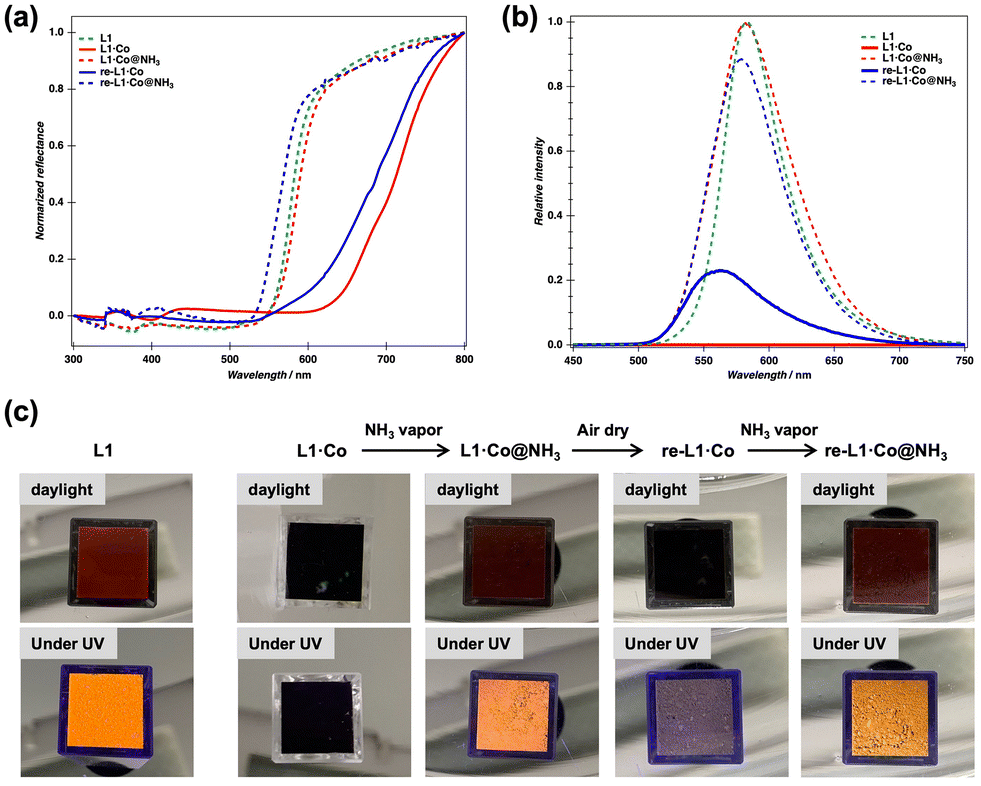 | ||
| Fig. 2 (a) Normalized reflectance spectra and (b) luminescence spectra of L1 (green dashed line), L1·Co (red solid line), L1·Co@NH3 (red dashed line), re-L1·Co (blue solid line) and re-L1·Co@NH3 (blue dashed line). (c) Photographs of powder samples of L1, L1·Co, L1·Co, L1·Co@NH3, re-L1·Co, and re-L1·Co@NH3 (left to right) in a cuvette under daylight (top) and UV light (bottom). | ||
The optical response of L1·Co to NH3 vapor (generated from a 28% aqueous NH3 solution) was investigated. Upon exposure of a powder sample of L1·Co in a cuvette to NH3 vapor, the dark brown color immediately changed to reddish-brown, and the resulting state is denoted as L1·Co@NH3 (Fig. 2c). Notably, L1·Co@NH3 exhibited orange luminescence under UV irradiation, whereas pristine L1·Co showed no detectable emission (middle photographs in Fig. 2c). After air-drying L1·Co@NH3 for a few minutes, the color reverted to the original dark brown state (re-L1·Co in Fig. 2c). Re-exposure to NH3 vapor reproducibly induced the same optical changes, yielding re-L1·Co@NH3. Although the emission intensity of re-L1·Co does not completely return to the original baseline after the recovery step, the ON–OFF switching remains clearly distinguishable. The residual emission is attributed to incomplete removal of NH3 from the solid state, consistent with elemental analysis indicating trace NH3 remaining after drying (see below).
The reproducibility of the switching behavior over repeated NH3 exposure and drying cycles is shown in Fig. S1. Although the ON/OFF ratio was slightly decreased, reversibility was confirmed for at least three cycles. The reversible response can be attributed to NH3 molecules, as no color or luminescence change was observed when L1·Co was exposed to water vapor under identical experimental conditions (Fig. S2). Furthermore, L1·Co exhibited high selectivity toward NH3 over other volatile organic solvents and amines, which can be rationalized by the strong coordination affinity of ammonia and its small molecular size. A visually detectable luminescence response was maintained down to 1 wt% aqueous ammonia. This corresponds to an estimated NH3 vapor concentration of ca. 8600 ppm under ambient conditions (based on Henry's law28). While this detection limit does not reach the sub-ppm levels reported for some porous or device-based sensors, the present system offers distinct advantages as a structurally defined discrete molecular metal complex, enabling reversible coordination-driven switching with a direct optical readout clearly observable to the naked eye.
The changes in color and luminescence feature of L1·Co before and after exposure to NH3 vapor were further examined by optical spectroscopic measurements, as summarized in Fig. 2a and b. The reflectance spectrum of L1·Co@NH3 (red dashed line) is nearly identical to that of L1 (green dashed line). This observation implies that the coordination environment formed between the cobalt(II) center and the L1 ligand is disrupted upon NH3 insertion,29 resulting in the generation of free L1. Consistent with this interpretation, the luminescence feature of L1·Co@NH3 (red dashed line) is recovered upon exposure to NH3 vapor. Upon air-drying L1·Co@NH3, the reflectance spectrum of re-L1·Co (blue solid line) again exhibits a red-shift, indicating partial removal of coordinated NH3 molecules and the reformation of the original metal–ligand coordination environment. Associated with this process, the luminescence intensity decreases again (blue solid line); however, it does not completely vanish and exhibits a slight spectral shift. This behavior is attributed to residual NH3 molecules remaining in the solid, which leads to incomplete dissociation of the original coordination environment. As a result, reversible luminescence ON–OFF switching is clearly observed upon alternating exposure to NH3 vapor and air-drying (blue dashed line).
To reactivate the luminescence feature, partial dissociation of the L1 ligand from the Co(II) center is required. Metal complexes containing unpaired spins, such as cobalt(II) complex L1·Co, are generally prone to luminescence quenching due to efficient non-radiative relaxation pathways. Even if only the chloride (Cl) atoms coordinated to the Co(II) center are replaced by NH3 molecules, nonradiative relaxation is still expected to remain operative. However, if L1 ligands were to be completely removed from the Co(II) center, i.e., decomposition of the metal complex formation, re-complexation in the solid-state would be difficult to achieve. Thus, NH3 molecules are likely to be partially inserted through dissociation of the coordination bonds involving the pyridine N atoms, whose coordination donor ability is reduced by the electron-withdrawing effect of the adjacent carbonyl group.30
To clarify this hypothesis, density functional theory (DFT) structural optimizations and time-dependent DFT (TD-DFT) calculations at the B3LYP31 theoretical level were performed using the Gaussian 16 program32 with the 6-311G(d,p) basis sets for all other atoms. The absorption of the L1 ligand is attributed to an intra-ligand charge transfer (ILCT) from the dimethylaminophenyl moiety to the carbonyl-pyridine site (Fig. S3). In contrast, both L1·Co and L1·Co@NH3 have three excitations with oscillator strength greater than 0.1 in their main absorption. To assign the characteristics of these excitations, natural transition orbital (NTO) analyses33 were performed. In L1·Co, although the ILCT transition is still present, MLCT dominates one of these excitations (ca. 38%), thereby suppressing the ILCT contribution and quenching the luminescence (Fig. S4). Upon exposure to NH3 vapor, the electronic structure of L1·Co@NH3 shifts back toward an ILCT-dominant excited state (Fig. S5). This result indicates that NH3 coordination likely weakens the competing MLCT pathway, thereby enabling the re-emergence of ILCT-driven luminescence. Besides, although the emission of L1·Co@NH3 originates predominantly from a ligand-centered excited state, additional nonradiative relaxation pathways associated with the cobalt(II) center are still operative. It should be noted that the present TD-DFT calculations primarily describe ligand- or metal-centered charge-transfer, whereas the d–d states are not explicitly described within this computational operation.
Photoluminescence lifetime measurements were performed to further elucidate the optical response (Fig. S6 and Table S3). The decay profile of the free ligand L1 (green plots) displays lifetimes of τ1 = 0.34 ns and τ2 = 1.06 ns, whereas that of the cobalt(II) complex after NH3 exposure, L1·Co@NH3 (red plots), exhibits longer lifetimes of τ1 = 0.56 ns and τ2 = 2.51 ns. In contrast, pristine L1·Co without NH3 exposure does not show a detectable lifetime owing to the absence of observable luminescence. Considering that the ligand is only partially dissociated from the central Co(II) ion upon NH3 coordination, rather than being fully released, the observed lifetimes—distinct from those of free L1—are reasonable.
In addition to the spectroscopic observations and DFT results, the response mechanism toward NH3 vapor was investigated by elemental analysis, PXRD, gas adsorption isotherms, and thermogravimetric analysis (TGA). After exposure of L1·Co to NH3 vapor, elemental analysis revealed a marked increase in nitrogen content for L1·Co@NH3, indicating the uptake of NH3 molecules (+H2O and +5.5NH3, Table S2). The NH3 adsorption isotherm measured at 25 °C shows a gradual uptake at lower pressures, followed by a steep increase beginning at PNH3 = 21 kPa, indicative of gate-opening (GO) adsorption behavior, to reach an adsorption amount of 211 mL (stp) g−1 (5.4 mol per formula unit) at 100 kPa (Fig. S7). This is in good agreement with the results of elemental analysis for L1·Co@NH3.
On the other hand, the PXRD pattern of L1·Co@NH3 differs from that of pristine L1·Co after exposure to NH3 vapor (Fig. 3). That is, the optical change caused by NH3 vapor is accompanied by a structural change due to the GO adsorption behavior. Notably, the PXRD pattern of re-L1·Co is nearly identical to that of L1·Co@NH3, even though its optical properties revert toward those of pristine L1·Co. Elemental analysis of re-L1·Co dried for 3 h indicates the presence of a small portion of residual NH3 molecules (+1.8H2O and +2.8NH3, Table S2). After further air-drying for 1 and 3 days, CHN analysis showed no significant change compared to the 3 h dried sample, remaining within experimental uncertainty. This indicates that NH3 desorption predominantly occurs within the initial 3 h, with approximately 3 mol released before reaching a steady composition. Nevertheless, the reflectance spectra clearly indicate re-coordination of L1 to the Co(II) center, demonstrating that the residual NH3 molecules remaining in re-L1·Co are adsorbed in a coordination-free manner or replace chloride ions, rather than being directly bound to the metal center. Furthermore, TGA of L1·Co@NH3 shows a weight loss of ca.13% upon heating to 450 K (Fig. S8), which is consistent with the release of 0.5 H2O and 5 NH3 molecules. Elemental analysis of L1·Co@NH3 after thermal treatment at 450 K indicates the presence of residual 0.5H2O and 0.5NH3 molecules (bottom in Table S2). The PXRD pattern of the thermally treated L1·Co@NH3 is identical to that of pristine L1·Co, indicating recovery of the original crystal structure. However, a slight shift of the main diffraction peak toward higher 2θ (7.72° for L1·Co and 7.84° for thermally treated L1·Co@NH3) suggests a minor lattice contraction. Although the overall diffraction pattern closely resembles that of pristine L1·Co, such subtle densification may reduce the local free volume and/or diffusion pathways required for guest uptake, potentially leading to reduced responsiveness.34,35 This effect is therefore more likely associated with bulk structural contraction rather than solely with residual guest molecules adsorbed on the surface.
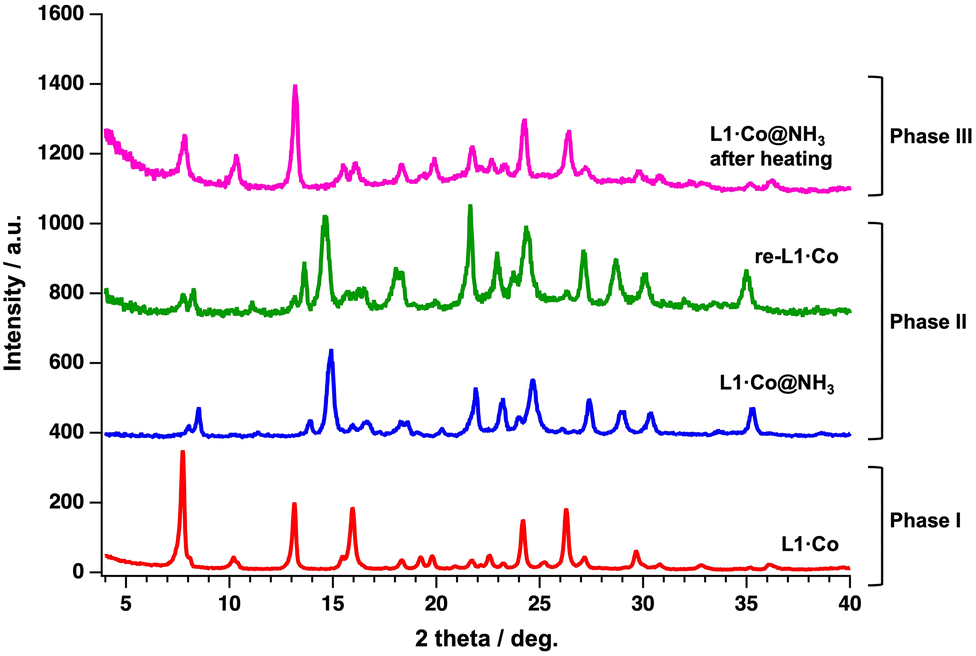 | ||
| Fig. 3 PXRD patterns of L1·Co (red), L1·Co@NH3 (blue), re-L1·Co (green) and L1·Co@NH3 after thermal treatment (magenta), λ = 1.54 Å. | ||
Fig. 4 presents a schematic illustration of the NH3 vapor responsivity of L1·Co, in which each state is categorized as Phases I–III. The corresponding phases are also reflected in the PXRD patterns shown in Fig. 3. As described above, pristine L1·Co exhibits no luminescence and is defined as Phase I. In Phase II, L1·Co@NH3 and re-L1·Co reversibly interconvert, accompanied by distinct color changes and luminescence ON–OFF switching. The exact coordination environment at the Co(II) center in Phase II cannot be unambiguously determined. Therefore, we propose a plausible coordination model (X = NH3 or Cl−) consistent with elemental analysis, spectroscopic data, and PXRD results. Elemental analysis indicates the presence of two chloride ions, suggesting that Cl− remains in the lattice as counter anions even when NH3 coordinates to the metal center. A small amount of NH3 also persists after drying L1·Co@NH3 without significant structural change, as supported by PXRD. These residual NH3 molecules are likely involved in maintaining the reversible optical response of Phase II. In contrast, thermal treatment of L1·Co@NH3 generates Phase III, in which the original PXRD pattern of L1·Co is restored, while approximately 0.5 mol of NH3 and H2O remain in the material. Unlike Phase II, the presence of these residual guest molecules in Phase III likely hinders further NH3 uptake, thereby suppressing the coordination-driven optical response.
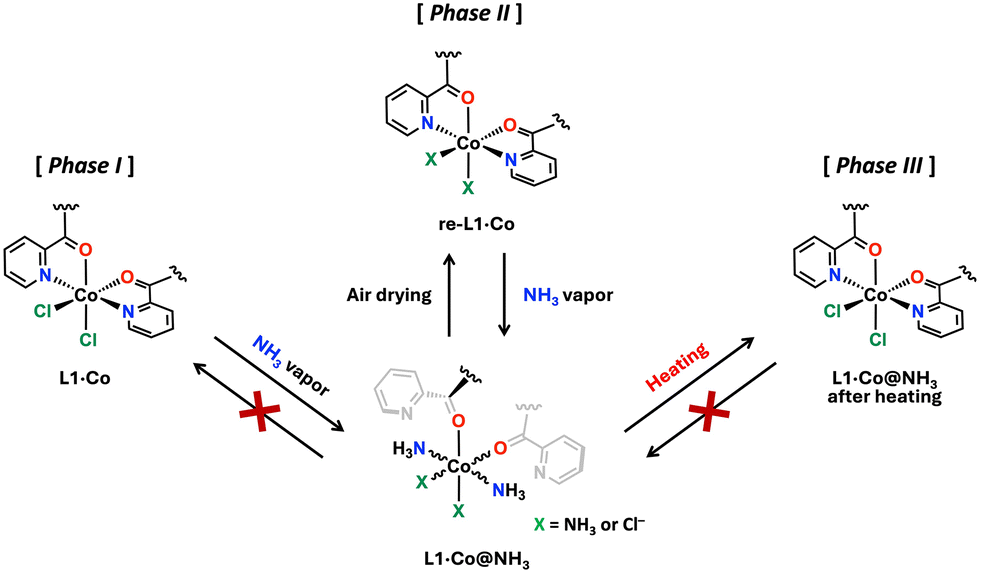 | ||
| Fig. 4 Schematic diagram of NH3 vapor responsivity of L1·Co focusing on the coordination geometry. Phase I: The pristine L1·Co with no luminescent activity. Phase II: L1·Co@NH3 and re-L1·Co with reversible luminescent ON–OFF behavior. Phase III: Thermally treated L1·Co@NH3 with no luminescent activity. | ||
In conclusion, we have demonstrated that a structurally defined chalcone-based Co(II) complex exhibits highly selective and reversible vapochromism together with luminescence ON–OFF switching in response to NH3 vapor. A combination of spectroscopic measurements, PXRD, gas adsorption analysis, and DFT calculations strongly suggests that this behavior most likely originates from a coordination-driven switching process involving partial ligand dissociation and reversible NH3 binding. The present study highlights a molecular design for fast and selective vapor sensing based on the reversible metal–ligand reconfiguration in discrete coordination compounds.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting the results of this study have been included in the supplementary information (SI). See DOI: https://doi.org/10.1039/d5cc07385e.CCDC 2512162 contains the supplementary crystallographic data for this paper.36
Acknowledgements
This work was supported by a JSPS KAKENHI Grant-in-Aid for Early-Career Scientists (No. JP22K14695). M. N. also acknowledges financial support from the Iketani Science and Technology Foundation (ISTF, No. 0351080-A) and the Kurita Water and Environment Foundation (No. 25H015).References
- A. Fernández-Acebes and J.-M. Lehn, Chem. – Eur. J., 1999, 5, 3285 CrossRef.
- Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007 RSC.
- P. Hajivand, J. C. Jansen, E. Pardo, D. Armentano, T. F. Mastropietro and A. Azadmehr, Coord. Chem. Rev., 2024, 501, 215558 CrossRef CAS.
- N. Dey, D. Biswakarma and S. Bhattacharya, ACS Sustainable Chem. Eng., 2019, 7, 569 CrossRef CAS.
- S. Fujii, H. Yagi, T. Kawaguchi, M. Ishikawa, N. Izumiyama and M. Nakaya, Dalton Trans., 2023, 52, 10206 RSC.
- S. Khanra, M. Sahad E, S. Das, S. Chakraborty, P. Brandao, B. de Bruin, B. C. Das and N. D. Paul, Adv. Funct. Mater., 2025, 35, 2502728 CrossRef CAS.
- P. Sarkar, L. T. Manamel, P. Saha, C. Jana, A. Sarmah, K. U. Mohanan, B. C. Das and C. Mukherjee, Mater. Horiz., 2025, 12, 246 RSC.
- S. Sinha, M. Sahad E, R. Mondal, S. Das, L. T. Manamel, P. Brandão, B. de Bruin, B. C. Das and N. D. Paul, J. Am. Chem. Soc., 2022, 144, 20442 CrossRef CAS PubMed.
- R. Mondal, A. K. Guin, S. Chakraborty and N. D. Paul, J. Org. Chem., 2022, 87, 2921 CrossRef CAS PubMed.
- D. Sengupta, S. Goswami, R. Banerjee, M. J. Guberman-Pfeffer, A. Patra, A. Dutta, R. Pramanick, S. Narasimhan, N. Pradhan, V. Batista, T. Venkatesan and S. Goswami, Chem. Sci., 2020, 11, 9226 RSC.
- S. A. Hilderbrand, M. H. Lim and S. J. Lippard, J. Am. Chem. Soc., 2004, 126, 4972 CrossRef CAS PubMed.
- M. H. Lim and S. J. Lippard, J. Am. Chem. Soc., 2005, 127, 12170 CrossRef CAS PubMed.
- X. Zhang, B. Li, Z.-H. Chen and Z.-N. Chen, J. Mater. Chem., 2012, 22, 11427 RSC.
- D. Saito, T. Galica, E. Nishibori, M. Yoshida, A. Kobayashi and M. Kato, Chem. – Eur. J., 2022, 28, e202200703 CrossRef CAS PubMed.
- Y. Shen, A. Tissot and C. Serre, Chem. Sci., 2022, 13, 13978 RSC.
- H. Yuan, N. Li, W. Fan, H. Cai and D. Zhao, Adv. Sci., 2022, 9, 2104374 CrossRef CAS PubMed.
- Z. Gao, Y. Lai, Y. Tao, L. Xiao, L. Zhang and F. Luo, ACS Cent. Sci., 2021, 7, 1066 CrossRef CAS PubMed.
- P. Qin, B. A. Day, S. Okur, C. Li, A. Chandresh, C. E. Wilmer and L. Heinke, ACS Sens., 2022, 7, 1666 CrossRef CAS PubMed.
- S. Wang, Y. Fu, T. Wang, W. Liu, J. Wang, P. Zhao, H. Ma, Y. Chen, P. Cheng and Z. Zhang, Nat. Commun., 2023, 14, 7261 CrossRef CAS PubMed.
- H. Yoshino, M. Saigo, T. Ehara, K. Miyata, K. Onda, J. Pirillo, Y. Hijikata, S. Takaishi, W. Kosaka, K. Otake, S. Kitagawa and H. Miyasaka, Angew. Chem., Int. Ed., 2025, 64, e202413830 CrossRef CAS PubMed.
- A. Tiwari, M. Arjumanda and A. Yella, Nanoscale, 2024, 16, 22152 RSC.
- O. S. Wenger, Chem. Rev., 2013, 113, 3686 CrossRef CAS PubMed.
- P. Kar, M. Yoshida, Y. Shigeta, A. Usui, A. Kobayashi, T. Minamidate, N. Matsunaga and M. Kato, Angew. Chem., Int. Ed., 2017, 56, 2345 CrossRef CAS PubMed.
- S. Kusumoto, K. Inaba, H. Suda, M. Nakaya, R. Tokunaga, P. Thuéry, R. Haruki, T. Kanazawa, S. Nozawa, Y. Kim, S. Hayami and Y. Koide, Inorg. Chem., 2023, 62, 16222 CrossRef CAS PubMed.
- T. Murakami, A. Masuno, M. Okazaki and S. Ohta, Inorg. Chem., 2025, 64, 15332 CrossRef CAS PubMed.
- O. Baumeyer, A. Wu, A. Pandya, P. Nelson, P. C. Hillesheim, M. Zeller, G. M. Carignan, J. Li and D. W. Ki, Chem. Commun., 2025, 61, 10170 RSC.
- J. M. Len, N. Hussein, S. Malla, K. Mcintosh, R. Patidar, M. Elangovan, K. Chandrabose, N. S. H. N. Moorthy, M. Pandey, D. Raman, P. Trivedi and A. K. Tiwari, Molecules, 2021, 26, 4214 CrossRef CAS PubMed.
- R. Sander, Atmos. Chem. Phys., 2023, 23, 10901 CrossRef CAS.
- B. E. R. Snyder, A. B. Turkiewicz, H. Furukawa, M. V. Paley, E. O. Velasquez, M. N. Dods and J. R. Long, Nature, 2023, 613, 287 CrossRef CAS PubMed.
- E. D. Raczyńska, J.-F. Gal and P.-C. Maria, Int. J. Mass Spectrom., 2017, 418, 130 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775 CrossRef CAS.
- J. E. Mann, R. Gao, S. S. London and J. A. Swift, Mol. Pharmaceutics, 2023, 20, 5554 CrossRef CAS PubMed.
- A. Yao, H. Xu, K. Shao, C. Sun, C. Qin, X. Wang and Z. Su, Nat. Commun., 2025, 16, 1385 CrossRef CAS PubMed.
- CCDC 2512162: Experimental Crystal Structure Determination, 2026 DOI:10.5517/ccdc.csd.cc2qb3hp.
| This journal is © The Royal Society of Chemistry 2026 |