
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of aryl sulfides via visible light-induced solventylation in diarylazo sulfides
Hawraz Ibrahim M.
Amin
 abc,
Stefano
Protti
a and
Maurizio
Fagnoni
*a
abc,
Stefano
Protti
a and
Maurizio
Fagnoni
*a
aPhotoGreen Lab, Department of Chemistry, University of Pavia, Viale Taramelli 12, 27100, Pavia, Italy. E-mail: maurizio.fagnoni@unipv.it
bDepartment of Chemistry, College of Science, Salahaddin University-Erbil, Erbil 44001, Iraq
cNational Interuniversity Consortium of Materials Science and Technology, Via G. Giusti, 9, 50121, Firenze, Italy
First published on 19th January 2026
Abstract
A metal-free C(sp3)–H bond thiolation of common organic solvents to give valuable aryl sulfides was carried out under very mild conditions upon visible light irradiation of colored diarylazo sulfides.
Aryl sulfides are key scaffolds in several drugs or biologically active compounds that exhibit anti-tumour, anticonvulsant, anti-inflammatory and antioxidant properties.1–3 Some examples are the azole antifungal medication Butoconazole,4 the anorectic drug Tiflorex5,6 and the coccidiostat medicine Toltrazuril.7 These derivatives are mainly accessed starting from aryl electrophiles and thiols by metal-catalyzed C–S bond formation8–12 or under photocatalytic conditions.13 Metal-free, uncatalyzed preparation of aryl sulfides is obviously desirable.
Recently, we discovered that diarylazo sulfides (I) when irradiated in the solid state in a water suspension underwent the cleavage of the S–N bond and the resulting intimate radical pair led to diaryl sulfides II upon nitrogen extrusion (Scheme 1, paths a and b).14 We wondered whether by switching to homogeneous conditions we could divert the fate of the reaction, since the initial photohomolytic cleavage could release a free radical pair composed of a thiyl radical III and an aryldiazenyl radical IV (path c).
 | ||
| Scheme 1 Reaction design for the synthesis of solventylated derivatives V. | ||
The latter intermediate easily loses nitrogen, and the resulting reactive aryl radical can trigger a hydrogen atom abstraction (HAT) reaction with the solvent (Solv-H, path d), ultimately producing valuable aryl sulfides V by radical coupling between the resulting Solv˙ and the persistent ArS˙ in what is considered a solventylation reaction (path e).
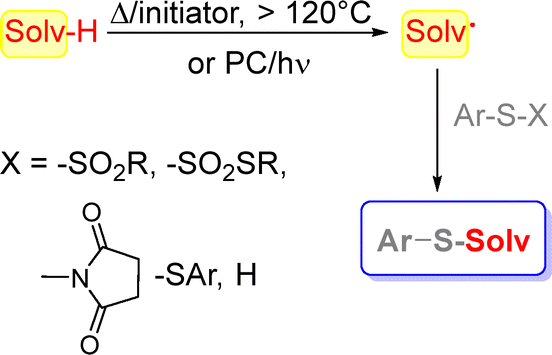
The preparation of compounds V under metal-free conditions is only sparsely reported and is resumed in Scheme 2. The strategy makes use of a solution of Ar-S-X derivatives in a chosen solvent (Solv-H) by heating (>120 °C) in the presence of a radical initiator (e.g. tert-butylperoxide) or by irradiation in the presence of a photocatalyst via generation of a Solv˙ intermediate.
 | ||
| Scheme 2 Metal-free routes to Ar-S-Solv derivatives. | ||
To this aim, alkyl arylsulfinates,15 sodium sulfinates,16 thiosulfonates17,18 and 1-(arylthio)pyrrolidine-2,5-diones19 were used as sulfenylation agents. Alternatively, the thiolation of a C(sp3)–H bond may be obtained by using diaryldisulfides20–27 or, more rarely, a thiophenol28 as sulfur sources. In this way, the thiolation of ethers, amides, cycloalkanes and so on was pursued.
In view of these premises, the aim of the present work is to achieve the hydrogen atom transfer-assisted C(sp3)–H thioarylation of common organic solvents to form adducts V (Scheme 1).
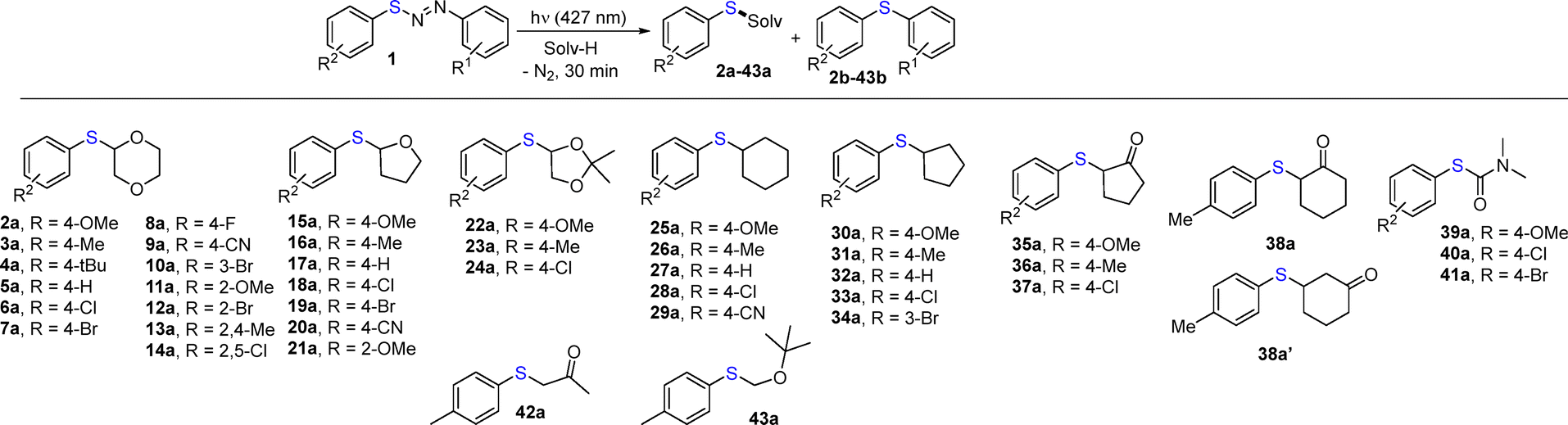
Diarylazo sulfides 1a–q are bench-stable and coloured derivatives14 and have been prepared starting from the corresponding anilines and aryl sulfides in up to 89% yield (Chart S1, SI). We initially tested the reaction of 1-(4-chlorophenyl)-2-(p-tolylthio)diazene 1b upon visible light irradiation (λ = 427 nm), by using 1,4-dioxane as the coupling partner (Table S1).
Thus, a 0.05 M solution of 1b in DMC in the presence of 1,4-dioxane (10 equiv.) was irradiated for 16 h and low amounts of the adduct 3a (11% yield) were accompanied by the undesired photoextrusion product 3b as the main product (entry 1). The same reaction was then performed in neat 1,4-dioxane again, leading to a mixture in which 3a is now by far the major product (64%, entry 2). On shortening the irradiation time to 30 min, a total consumption of 1b was likewise observed and the process (despite not clean) led to 82% of 3a (entries 3 and 4). Increasing the concentration of 1b up to 0.2 M and testing diarylazo sulfide 1p in place of 1b did not improve the yield (Table S1, entries 5–7). The reaction was completely suppressed when the vessel was covered by aluminium foil (entry 8).
With the reaction conditions in hand (Table S1, entry 4), we investigated the synthesis of various 2-(arylthio)-1,4-dioxanes by retaining the 4-chlorophenyl group as the hydrogen abstractor (Table 1). The reaction led to the desired compounds 2a–14a in good to excellent yields, independent of the nature and the position of the substituents on the aromatic ring. Variable amounts of byproducts 2b–14b derived from photoextrusion were likewise observed. In the preparation of compound 6a, we tested different diarylazo sulfides as the aryl radical precursors (1n, 1o) with lower efficiency compared to the same reaction carried out starting from 1e. Substituents such as methyl and cyano in the aromatic ring (compounds 1m and 1q) likewise led to a satisfying yield (>70%) of 7a. The reaction was extended to incorporate THF in the aryl sulfide structure. To this aim, sulfides 15a–21a were isolated in yields ranging from 40 to 66%. By shifting to a different oxygen heterocycle (2,2-dimethyl-1,3-dioxolane), the yields increased up to 83% in the preparation of 22a–24a again accompanied by diaryl sulfides 22b–24b. We then moved to cycloalkane solutions and cyclohexyl (25a–29a) and cyclopentyl adducts (30a–34a) were formed in variable yields (mostly around 60%). Cycloalkanones were tested, and while cyclopentanone regioselectively afforded α-thioaryl adducts 35a–37a with a yield in the range of 59–69%, cyclohexanone yielded a mixture of α- and β-adducts (compounds 38a and 38a′ with the former slightly preferred). In the reaction with DMF, carbamothioates 39a–41a, valuable building blocks for the synthesis of potentially bioactive derivatives,29,30 were regioselectively formed but only in moderate yields (Table 1). In acetone and tert-butyl methyl ether the solventylation of the starting diarylazo sulfides gave the desired sulfides 42a and 43a in less than 43% yield, while diaryl sulfides 39b–43b were formed in comparable amounts.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Diarylazo sulfides 1 | Medium (Solv-H) | % Yield | % Yield | Diarylazo sulfides 1 | Medium (Solv-H) | % Yield | % Yield |
| a Reaction conditions: An N2 saturated solution of diarylazo sulfides 1a–1q (0.05 M), irradiated for 30 min at 427 nm (a 32 W Kessil lamp) in the chosen medium. | |||||||
| 1a, R2 = 4-OMe, R1 = 4-Cl | 1,4-Dioxane | 2a, 86% | 2b, 12% | 1a, R2 = 4-OMe, R1 = 4-Cl | 2,2-Dimethyl-1,3-dioxolane | 22a, 83% | 22b, 11% |
| 1b, R2 = 4-Me, R1 = 4-Cl | 1,4-Dioxane | 3a, 82% | 3b, 8% | 1b, R2 = 4-Me, R1 = 4-Cl | 2,2-Dimethyl-1,3-dioxolane | 23a, 75% | 23b, 10% |
| 1c, R2 = 4-tBu, R1 = 4-Cl | 1,4-Dioxane | 4a, 77% | 4b, 19% | 1e, R2 = 4-Cl, R1 = 4-Cl | 2,2-Dimethyl-1,3-dioxolane | 24a, 73% | 24b, 10% |
| 1d, R2 = 4-H, R1 = 4-Cl | 1,4-Dioxane | 5a, 80% | 5b, 11% | 1a, R2 = 4-OMe, R1 = 4-Cl | Cyclohexane | 25a, 64% | 25b, 17% |
| 1e, R2 = 4-Cl, R1 = 4-Cl | 1,4-Dioxane | 6a, 83% | 6b, 12% | 1b, R2 = 4-Me, R1 = 4-Cl | Cyclohexane | 26a, 47% | 26b, 9% |
| 1n, R2 = 4-Cl, R1 = 4-CO2Me | 1,4-Dioxane | 6a, 65% | 6b, 13% | 1d, R2 = 4-H, R1 = 4-Cl | Cyclohexane | 27a, 68% | 27b, 11% |
| 1o, R2 = 4-Cl, R1 = 4-NO2 | 1,4-Dioxane | 6a, 27% | 6b, 12% | 1n, R2 = 4-Cl, R1 = 4-CO2Me | Cyclohexane | 28a, 63% | 28b, 10% |
| 1m, R2 = 4-Br, R1 = 4-Me | 1,4-Dioxane | 7a, 77% | 7b, 13% | 1g, R2 = 4-CN, R1 = 4-Cl | Cyclohexane | 29a, 41% | 29b, 12% |
| 1q, R2 = 4-Br, R1 = 4-CN | 1,4-Dioxane | 7a, 70% | 7b, 9% | 1a, R2 = 4-OMe, R1 = 4-Cl | Cyclopentane | 30a, 72% | 30b, 12% |
| 1f, R2 = 4-F, R1 = 4-Cl | 1,4-Dioxane | 8a, 78% | 8b, 8% | 1b, R2 = 4-Me, R1 = 4-Cl | Cyclopentane | 31a, 61% | 31b, 9% |
| 1g, R2 = 4-CN, R1 = 4-Cl | 1,4-Dioxane | 9a, 61% | 9b, 6% | 1d, R2 = 4-H, R1 = 4-Cl | Cyclopentane | 32a, 61% | 32b, 7% |
| 1h, R2 = 3-Br, R1 = 4-Cl | 1,4-Dioxane | 10a, 84% | 10b, 13% | 1e, R2 = 4-Cl, R1 = 4-Cl | Cyclopentane | 33a, 65% | 33b, 12% |
| 1i, R2 = 2-OMe, R1 = 4-Cl | 1,4-Dioxane | 11a, 71% | 11b, 5% | 1h, R2 = 3-Br, R1 = 4-Cl | Cyclopentane | 34a, 63% | 34b, 10% |
| 1j, R2 = 2-Br, R1 = 4-Cl | 1,4-Dioxane | 12a, 75% | 12b, 13% | 1a, R2 = 4-OMe, R1 = 4-Cl | Cyclopentanone | 35a, 68% | 35b, 13% |
| 1k, R2 = 2,4-Me, R1 = 4-Cl | 1,4-Dioxane | 13a, 80% | 13b, 14% | 1b, R2 = 4-Me, R1 = 4-Cl | Cyclopentanone | 36a, 67% | 36b, 14% |
| 1l, R2 = 2,5-Cl, R1 = 4-Cl | 1,4-Dioxane | 14a, 77% | 14b, 13% | 1e, R2 = 4-Cl, R1 = 4-Cl | Cyclopentanone | 37a, 59% | 37b, 9% |
| 1a, R2 = 4-OMe, R1 = 4-Cl | THF | 15a, 61% | 15b, 11% | 1b, R2 = 4-Me, R1 = 4-Cl | Cyclohexanone | 38a, 34%; | 38b, 10% |
| 38a′, 23% | |||||||
| 1b, R2 = 4-Me, R1 = 4-Cl | THF | 16a, 51% | 16b, 9% | 1a, R2 = 4-OMe, R1 = 4-Cl | DMF | 39a, 35% | 39b, 14% |
| 1d, R2 = 4-H, R1 = 4-Cl | THF | 17a, 59% | 17b, 8% | 1e, R2 = 4-Cl, R1 = 4-Cl | DMF | 40a, 44% | 40b, 17% |
| 1e, R2 = 4-Cl, R1 = 4-Cl | THF | 18a, 56% | 18b, 9% | 1m, R2 = 4-Br, R1 = 4-Me | DMF | 41a, 31% | 41b, 13% |
| 1m, R2 = 4-Br, R1 = 4-Me | THF | 19a, 52% | 19b, 9% | 1b, R2 = 4-Me, R1 = 4-Cl | Acetone | 42a, 46% | 42b, 43% |
| 1g, R2 = 4-CN, R1 = 4-Cl | THF | 20a, 40% | 20b, 11% | 1b, R2 = 4-Me, R1 = 4-Cl | tert-Butyl methyl ether | 43a, 32% | 43b, 36% |
| 1i, R2 = 2-OMe, R1 = 4-Cl | THF | 21a, 66% | 21b, 8% | ||||
The preparation of 9a was repeated on a larger scale (1 mmol) under simulated solar light conditions (Scheme 3a and SI, Section S1.4) without any appreciable decrease in yield (60%). To assess the radical nature of the process, we irradiated (427 nm, 30 min) a cyclohexane/DMC 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solution of diarylazo sulfide 1q (0.05 mmol) in the presence of (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO, 10 equiv.). GC-MS analysis of the crude mixture showed the presence of 1-(cyclohexyloxy)-2,2,6,6-tetramethylpiperidine 44 as the major product together with a minor amount of diaryl sulfide 7b (Scheme 3b and SI). Finally, a value >1 of the consumption quantum yield (Φ−1 = 9.4) of 1b in 1,4-dioxane was determined (Section S1.2, SI).
:1 solution of diarylazo sulfide 1q (0.05 mmol) in the presence of (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO, 10 equiv.). GC-MS analysis of the crude mixture showed the presence of 1-(cyclohexyloxy)-2,2,6,6-tetramethylpiperidine 44 as the major product together with a minor amount of diaryl sulfide 7b (Scheme 3b and SI). Finally, a value >1 of the consumption quantum yield (Φ−1 = 9.4) of 1b in 1,4-dioxane was determined (Section S1.2, SI).
 | ||
| Scheme 3 (a) Large-scale synthesis of 9a under solar simulated conditions. (b) TEMPO trapping experiment. | ||
Based on these results, we proposed a mechanism illustrated in Scheme 4. Visible light photolysis of compounds 1a–q induced photohomolysis of the N–S bond, releasing, upon nitrogen loss, an aryl radical and a thiyl radical. Radical coupling led to the formation of a small amount of adducts 2b–43b resulting from nitrogen photoextrusion as previously observed in the solid state14 but still present in solution. However, the photogenerated aryl radical may readily act as a hydrogen atom transfer (HAT) agent from the medium to form Solv˙.31–36
 | ||
| Scheme 4 The proposed mechanism for the formation of compounds 2a–43a and 2b–43b. | ||
The radical mechanism of the reaction was confirmed by the intermediacy of Solv˙ (see the isolation of TEMPO adduct 44 in the reaction with 1q, Scheme 3b). Interestingly, a better electrophilic aryl radical is no guarantee of reaction success (see the case of 6a where the strong 4-nitrophenyl radical37 gave unsatisfactory performance).
Moreover, in the synthesis of 7a, incorporation of a 4-methylphenyl or a 4-cyanophenyl residue in the starting diarylazo sulfides did not change the overall yield appreciably (Table 1). Quantum yield measurement pointed to a radical chain reaction.
The most likely pathway might be the addition of Solv˙ to the starting diarylazo sulfides with the concomitant formation of adducts 2a–43a together with an aryl radical capable of restarting the HAT process. A possible competitive HAT reaction by a thiyl radical on the solvent may not be ruled out but this process is not so efficient when compared to the hydrogen abstracting capabilities of aryl radicals.26,27,38,39 Minor amounts of aryl sulfides may arise from the radical coupling between Solv˙ and the thiyl radical.40
In the case of cycloalkanones and DMF regioselectivity in hydrogen abstraction (and the subsequent reaction onto 1) is an issue. In cyclopentanone, only the more labile α-hydrogen41 is removed (Table 1). This is in contrast with the C–H cleavage induced by other HAT agents where the β-hydrogens are selectively removed.42,43 In the case of cyclohexanone radicals the α-isomer is more stable (resonantly stabilized) than the β- and γ-isomers.44 In our case a mixture of α- and β-adducts was isolated in a comparable yield, in contrast to that observed in the hydrogen abstraction from a sulfate radical anion.43 The selectivity in HAT reactions of aryl radicals is still an open issue.34 The product distribution observed in the latter cases reflects the lability of C–H bonds in cycloalkanones indicating thermodynamic rather than kinetic control of the abstraction step usually observed in electrophilic hydrogen abstractors.42 Moreover, the feasibility of the addition of Solv˙ to 1 may strongly affect the final product distribution.
In DMF, the abstraction of the H-atom of the –CHO group is claimed to be preferred over the methyl group H-atom.45 This was effectively observed in the abstraction by an aryl radical46 but not by the decatungstate anion photocatalyst where an exclusive formation of an α-amido radical resulted.47
In conclusion, a metal-free C(sp3)–H bond thiolation of common organic solvents has been proposed herein. The process is based on the visible light activation in diarylazo sulfides, which causes the release of a thiyl radical (a persistent radical) and a reactive aryl radical, which abstracts a hydrogen atom from the reaction medium to form a solvent derived carbon radical prone to give the desired solventylation adduct by reaction with the starting diarylazo sulfide 1. The reaction is particularly efficient with cyclic ethers (e.g. 1,4-dioxane) but modest results arose from open chain solvents (e.g. acetone, DMF or t-butyl methyl ether). The current protocol does not make use of additives (e.g., TBHP and zinc salts) as in related photocatalyzed preparations.25,26 The main disadvantage of the method is the poor atom economy (the aryl moiety is lost in the reaction). Nevertheless, this is a nice example where a visible light-induced metal-free forging of an ArS-C bond was devised under very mild conditions.
This work was conceptualized by M. F. and S. P., and experimentation was performed by H. I. M. A. The first draft of the manuscript was prepared by M. F., and the final version was edited and revised by S. P. and M. F.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental details and NMR spectra. See DOI: https://doi.org/10.1039/d5cc06764b.Acknowledgements
The authors acknowledge support from the Ministero dell’Università e della Ricerca (MUR) and the University of Pavia through the program “Dipartimenti di Eccellenza 2023–2027”. The authors gratefully acknowledge financial support from the European Union's Life Programme under grant agreement no. 101074164 (CROSS-LIFE—CROtonic Acid from Sewage Sludge).References
- K. L. Dunbar, D. H. Scharf, A. Litomska and C. Hertweck, Chem. Rev., 2017, 117, 5521–5577 CrossRef CAS PubMed.
- E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832–2842 CrossRef CAS PubMed.
- C. Zhao, K. P. Rakesh, L. Ravidar, W.-Y. Fang and H.-L. Qin, Eur. J. Med. Chem., 2019, 162, 679–734 CrossRef CAS PubMed.
- F. Qin, Q. Wang, C. Zhang, C. Fang, L. Zhang, H. Chen, M. Zhang and F. Cheng, Infect. Drug Resist., 2018, 11, 1893–1901 Search PubMed.
- T. Silverstone, J. Fincham and J. Plumley, Br. J. Clin. Pharmacol., 1979, 7, 353–356 Search PubMed.
- J. F. Giudicelli, C. Richer and A. Berdeaux, Br. J. Clin. Pharmacol., 1976, 3, 113–121 CrossRef CAS PubMed.
- M. Diaferia, F. Veronesi, G. Morganti, L. Nisoli and D. P. Fioretti, Parasitol. Res., 2013, 112, 163–168 Search PubMed.
- T. Kondo and T. Mitsudo, Chem. Rev., 2000, 100, 3205–3220 CrossRef CAS PubMed.
- S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400–5449 CrossRef CAS PubMed.
- I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596–1636 CrossRef CAS PubMed.
- C. C. Eichman and J. P. Stambuli, Molecules, 2011, 16, 590–608 CrossRef CAS PubMed.
- M. Kazemi, L. Shiri and H. Kohzadi, Phosphorus, Sulfur Silicon Relat. Elem., 2015, 190, 978–1003 CrossRef CAS.
- J. Feng, Y. Zhang, X. Wang, J. Liu, V. Benazzi, K. Lu, X. Zhao and S. Protti, Adv. Synth. Catal., 2023, 365, 3413–3431 CrossRef CAS.
- H. I. M. Amin, L. D. Terlizzi, S. Protti and M. Fagnoni, ChemistryEurope, 2025, 3, e202500182 CrossRef.
- Y. Li, F. Zhu, Z. Wang and X.-F. Wu, Chem. – Asian J., 2016, 11, 3503–3507 Search PubMed.
- F. Zhao, Q. Tan, D. Wang, J. Chen and G.-J. Deng, Adv. Synth. Catal., 2019, 361, 4075–4081 CrossRef CAS.
- W.-Z. Bi, W.-J. Zhang, Z.-J. Li, Y.-H. He, S.-X. Feng, Y. Geng, X.-L. Chen and L.-B. Que, Org. Biomol. Chem., 2021, 19, 8701–8705 RSC.
- W.-Z. Bi, Q.-P. Liu, C.-Y. Li, W.-J. Zhang, S.-X. Feng, Y. Geng, X.-L. Chen and L.-B. Qu, New J. Chem., 2023, 47, 9035–9039 RSC.
- J. Grover, G. Prakash, C. Teja, G. K. Lahiri and D. Maiti, Green Chem., 2023, 25, 3431–3436 RSC.
- R.-Y. Tang, Y.-X. Xie, Y.-L. Xie, J.-N. Xiang and J.-H. Li, Chem. Commun., 2011, 47, 12867–12869 RSC.
- S.-R. Guo, Y.-Q. Yuan and J.-N. Xiang, Org. Lett., 2013, 15, 4654–4657 CrossRef CAS PubMed.
- J. Zhao, H. Fang, J. Han, Y. Pan and G. Li, Adv. Synth. Catal., 2014, 356, 2719–2724 CrossRef CAS PubMed.
- B. Du, B. Jin and P. Sun, Org. Lett., 2014, 16, 3032–3035 CrossRef CAS PubMed.
- S. K. Sahoo, J. Chem. Sci., 2015, 127, 2151–2157 CrossRef CAS.
- X. Zhu, X. Xie, P. Li, J. Guo and L. Wang, Org. Lett., 2016, 18, 1546–1549 CrossRef CAS PubMed.
- L. I. Panferova, M. O. Zubkov, V. A. Kokorekin, V. V. Levin and A. D. Dilman, Angew. Chem., Int. Ed., 2021, 60, 2849–2854 CrossRef CAS PubMed.
- S. W. Lardy, V. L. Lerda and V. A. Schmidt, J. Org. Chem., 2024, 89, 15062–15067 CrossRef CAS PubMed.
- J. Feng, G. Lu, M. Lv and C. Cai, Synlett, 2015, 915–920 CAS.
- A. Serhouni, F. Calzaferri, Y. Bessina, A. van der Lee, P. B. Arimondo, M. Louet, J.-Y. Winum and M. Lopez, Bioorg. Med. Chem., 2025, 118, 117988 Search PubMed.
- R. Romagnoli, P. G. Baraldi, M. D. Carrion, O. Cruz-Lopez, M. Tolomeo, S. Grimaudo, A. Di Cristina, M. R. Pipitone, J. Balzarini, A. Brancale and E. Hamel, Bioorg. Med. Chem., 2010, 18, 5114–5122 CrossRef CAS PubMed.
- M. Anselmo, A. Basso, S. Protti and D. Ravelli, ACS Catal., 2019, 9, 2493–2500 Search PubMed.
- J. Kang, H. S. Hwang, V. K. Soni and E. J. Cho, Org. Lett., 2020, 22, 6112–6116 CrossRef CAS PubMed.
- P. Capurro, V. Ricciardiello, P. Lova, C. Lambruschini, S. Protti and A. Basso, ACS Omega, 2022, 7, 48564–48571 CrossRef CAS PubMed.
- J.-L. Tu and B. Huang, Chem. Commun., 2024, 60, 11450–11465 RSC.
- W. A. Pryor, J. V. Echols Jr. and K. Smith, J. Am. Chem. Soc., 1966, 88, 1189–1199 CrossRef CAS.
- F.-D. Kopinke, G. Zimmermann and K. Anders, J. Org. Chem., 1989, 54, 3571–3576 CrossRef CAS.
- J. J. A. Garwood, A. D. Chen and D. A. Nagib, J. Am. Chem. Soc., 2024, 146, 28034–28059 CAS.
- W. A. Pryor, G. Gojon and D. F. Church, J. Org. Chem., 1978, 43, 793–800 CrossRef CAS.
- A. K. Z. Zachmann, J. A. Drappeau, S. Liu and E. J. Alexanian, Angew. Chem., Int. Ed., 2024, 63, e202404879 CrossRef CAS PubMed.
- T. Li, K. Liang, J. Tang, Y. Ding, X. Tong and C. Xia, Chem. Sci., 2021, 12, 15655–15661 RSC.
- W. Li, L. Ye, Q. Fang, J. Zou, J. Yang and Y. Li, Energy Fuels, 2021, 35, 14023–14034 CrossRef CAS.
- M. Okada, T. Fukuyama, K. Yamada, I. Ryu, D. Ravelli and M. Fagnoni, Chem. Sci., 2014, 5, 2893–2898 RSC.
- M. Ueda, A. Kitano and H. Matsubara, Org. Biomol. Chem., 2021, 19, 4775–4782 RSC.
- S. Thiona, Z. S. G. Dayma and P. Dagauta, Fuel, 2018, 220, 908–915 CrossRef.
- X. Bai, Y. Li, S. Liu, R. He, J. Liang, G. Yin, S. Zheng, Y. Qian and X. Wang, Fuel, 2023, 353, 129164 CrossRef CAS.
- Q. Zhou, Y. Zhang, J. Shang, R. Qi, X. Ren, Z. Xu and C. Wang, ACS Catal., 2025, 15, 17504–17512 CrossRef CAS.
- S. Angioni, D. Ravelli, D. Emma, D. Dondi, M. Fagnoni and A. Albini, Adv. Synth. Catal., 2008, 350, 2209–2214 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |