
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactivity of ketenyl anions towards ammonia
Prakash
Duari
 ,
Mike
Jörges
,
Sunita
Mondal
,
Kai-Stephan
Feichtner
and
Viktoria H.
Gessner
*
,
Mike
Jörges
,
Sunita
Mondal
,
Kai-Stephan
Feichtner
and
Viktoria H.
Gessner
*
Faculty of Chemistry and Biochemistry, Ruhr-University Bochum, 44801, Bochum, Germany. E-mail: viktoria.gessner@rub.de
First published on 5th February 2026
Abstract
Alkali metal ketenyls, [M(RCCO)], were found to exhibit diverging reactivities towards ammonia depending on the substitution pattern. Ketenyl anions with strong electron-withdrawing groups (R = CN or tosyl) react with NH3 to form β-ketoamides, while the phosphinoyl substituted systems (R = Ph2P(E), E = S, Se) activate all three N−H bonds, resulting in a trianionic triamide. This triamide exhibits a dimeric structure with the six potassium cations forming a unique planar triangular {K6}6+ cluster.
Ammonia (NH3) is a fundamental building block in the chemical industry and serves as a key precursor for the synthesis of a wide range of nitrogen-containing compounds.1–4 The development of systems capable of efficiently activating ammonia and facilitating nitrogen transfer to other substrates offers promising opportunities for atom-economical pathways to valuable N-containing molecules.5 Despite significant advances in transition-metal-mediated N−H functionalization of various amines, the activation of ammonia remains a formidable challenge owing to its high bond dissociation energy (homolytic BDE ≈ 107 kcal mol−1) and strong tendency to form thermodynamically stable Lewis acid-base adducts.6,7 Usually, coordination of ammonia to a metal centre decreases the N−H bond dissociation energy, thereby facilitating subsequent hydrogen atom abstraction and deprotonation reactions.8,9 Nonetheless, oxidative addition of ammonia to metal centres has been accomplished only in a few cases.10,11
In recent years, main-group systems have emerged as attractive alternatives to transition-metal-based catalysts due to the generally higher natural abundance and the lower toxicity of p-block elements, as well as their reduced tendency to form unreactive Werner-type complexes with ammonia.12 In 2007, Bertrand and Schoeller et al. reported the first example of a transition metal-free ammonia splitting at the carbene carbon centre in alkyl(amino)carbenes (A, Fig. 1a).13 Since then, numerous ambiphilic main-group compounds incorporating group 13 to 15 elements have demonstrated similar reactivity, such as various carbene-like species,14–18 geometrically constrained phosphorus systems (e.g.B),19 and Lewis acidic species that enable NH3 activation through cooperative addition across an element-ligand bond (C, Fig. 1a).20,21
 | ||
| Fig. 1 (a) Selected examples for the activation of ammonia by main group compounds; (b) reaction of neutral ketenes with ammonia and (c) diverging reactivity of ketenyl anions with ammonia reported in this work. | ||
Except for singlet carbenes, carbon compounds usually do not readily undergo N−H activation through simultaneous formation of new C−N and C−H bonds. Strong carbon bases such as Grignard or organolithium reagents usually lead to ammonia deprotonation to form the corresponding metal amides MNH2, whereas electrophiles – if active – result in ammonium salt formation or – under basic conditions – in alkyl/aryl amine formation. Addition reactions of ammonia to C−C multiple bonds are typically not facile without a potent catalyst.2 Like carbenes, ketenes are ambiphilic carbon species (Fig. 1b). They possess a polarized C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, which facilitates the direct addition of N−H bonds from various amines. Moreover, the direct addition of ammonia to ketenes is feasible, typically resulting in the formation of the corresponding amides, which provide access to a variety of follow-up products, including nitriles or carboxylic acids.22–24 Computational studies by Tidwell and Nguyen showed that the amination takes place via initial attack of ammonia at the CO bond of the ketene to give an enol amide. This step was shown to require two molecules of NH3 to stabilize a six-membered cyclic transition state.25,26
C bond, which facilitates the direct addition of N−H bonds from various amines. Moreover, the direct addition of ammonia to ketenes is feasible, typically resulting in the formation of the corresponding amides, which provide access to a variety of follow-up products, including nitriles or carboxylic acids.22–24 Computational studies by Tidwell and Nguyen showed that the amination takes place via initial attack of ammonia at the CO bond of the ketene to give an enol amide. This step was shown to require two molecules of NH3 to stabilize a six-membered cyclic transition state.25,26
Recently, our group established a novel synthetic route to ketenyl anions, [RCCO]− through an exchange of PPh3 at the ylidic carbon centre of α-metalated ylides with carbon monoxide.27 Using this approach, phosphinoyl,28,29 tosyl,30 and cyano-substituted31–33 ketenyl anions have been prepared.34,35 Structural and computational analyses revealed that these anions exhibit an intermediate electronic structure between the ketene form, R–CΘCO and the ynolate form, R–C≡C–OΘ. The isolated ketenyl anions served as versatile precursors to neutral ketenes and a broad range of other carbonyl containing compounds, including heterocycles.31,32 The reactivity of ketenyl anions towards ammonia was so far only investigated for the cyano-substituted ketenyl anion 1CN, which resulted in the formation of β-ketoamide 2CN through the reaction of 2 equiv. of the ketenyl anion with NH3 (Scheme 1).31 This transformation presumably proceeds via initial generation of cyanoketene through deprotonation of ammonia, followed by nucleophilic attack of a second equivalent of 1CN and subsequent reaction with another equivalent of ammonia.
 | ||
| Scheme 1 Synthesis of 2CN and 2Tos from the corresponding ketenyl anions 1CN and 1Tos, respectively (Ts = tosyl, 18-c-6 = 18-crown-6, 12-c-4 = 12-crown-4). | ||
To probe the generality of this reaction, we extended our studies to other ketenyl anions. Treatment of the tosyl-substituted ketenyl anion 1Tos with ammonia resulted in the formation of the analogous β-ketoamide 2Tos in 44% isolated yield. NMR spectroscopy revealed that 2Tos exists as an enol tautomer, as evidenced by two distinct sets of tosyl resonances along with a characteristic CH signal at 4.47 ppm in the 1H NMR spectrum. Additionally, the 13C NMR spectrum shows a peak at 85.2 ppm for the enolic carbon atom. This similarity aligns with our earlier observations that cyano- and tosyl-substituted derivatives often display comparable behavior due to their related electronic properties.36
We next turned our attention to the reactivity of the thiophosphinoyl-substituted potassium ketenyl 1PS. Exposure of a THF solution of 1PS to NH3 gas in a J. Young NMR tube resulted in the selective formation of a new species as indicated by the appearance of a new peak at δ = 22.21 ppm in the 31P{1H} NMR spectrum. The reaction progressed slowly at ambient temperature, reaching complete conversion only after 48 h. Crystallization by vapor diffusion of hexane into the THF solution afforded off-white crystals in 85% isolated yield. Single-crystal X-ray diffraction analysis revealed the new compound to be potassium salt 3PS with a trianionic triamide. Analogously, the reaction of the selenium compound 1PSe with NH3 afforded 3PSe as an off-white crystalline solid in 83% yield.
Compounds 3PS and 3PSe are formed by activation of all three N−H bonds of ammonia through formal addition across the C−C bond of three ketenyl anions. In the solid-state, both compounds form a dimeric structure with the potassium cations being sandwiched in between two triamide molecules (Fig. 2a). The six cations arrange to a planar, triangular {K6}6+ cluster, with three cations forming the edges, while the remaining ones are positioned along each side. Thus, the triangular array can formally be viewed as subdivided into four smaller triangles. Overall, the complex possesses pseudo-C3 symmetry with the symmetry axis passing through the two central nitrogen atoms of the triamide and the centre of the K6 triangle.
 | ||
| Fig. 2 Representation of 3PS (a) from the front, (b) from the side (THF molecules are omitted), and (c) as a single unit (THF molecules and potassium ions are omitted). Ellipsoids are drawn at the 50% probability level. All H atoms except H1, H3, and H5 are omitted for clarity. Important bond lengths [Å] and angles [°]: C1–C2 1.369(6), C3–C4 1.380(6), C5–C6 1.378(6), C2–O1 1.270(5), C4–O2 1.277(6), C6–O3 1.277(5), N1–C2 1.438(6), N1–C4 1.431(6), N1–C6 1.437(6) P1–C1 1.748(5), P2–C3 1.742(5), P3–C5 1.750(5), P1–C1–C2 121.0(3), P2–C3–C4 123.2(4), P3–C5–C6 122.9(4), C2–N1–C4 119.1(3), C4–N1–C6 119.3(3), C6–N1–C2 119.6(4) (for a single unit). | ||
It is interesting to note that both neutral and ionic potassium clusters have been extensively studied through computational methods.37,38 However, the isolation of small potassium clusters such as K2, K3, and K4 has previously only been accomplished in krypton matrices at 15 K and characterized by Raman spectroscopy.39 For neutral K6 clusters, two energetically feasible geometries have been theoretically predicted, a triangular planar, D3h-symmetric structure similar to the planar triangle in dimeric 3 and a pentagonal pyramidal (C5v) structure. For the planar K6 structure, K⋯K distances of 4.29 and 4.60 Å were calculated, which are considerably longer than those observed in 3PS. The average K⋯K distance in the outer triangle of the {K6}6+ cluster amounts to approx. 3.675 Å, and those in the inner triangle to approx. 4.384 Å. While covalent bonding has been discussed in neutral clusters, we assume that the six cationic potassium ions in 3 are assembled into a unique triangular structure due to the symmetry of the triamide. The potassium ions in 3PS and 3PSe are coordinated by the oxygen and sulfur or selenium atoms of the triamide as well as by six additional THF solvent molecules. The C1−C2 bond length in 3PS of 1.369(6) Å indicates an elongated CC double bond, whereas the C2−O1 distance of 1.270(5) Å lies between that typical CO double and single bonds,40 consistent with predominant enolate character as depicted in Scheme 2.28 The average C–N–C angle (∼119.0°) is consistent with trigonal planar geometry around the nitrogen centres.
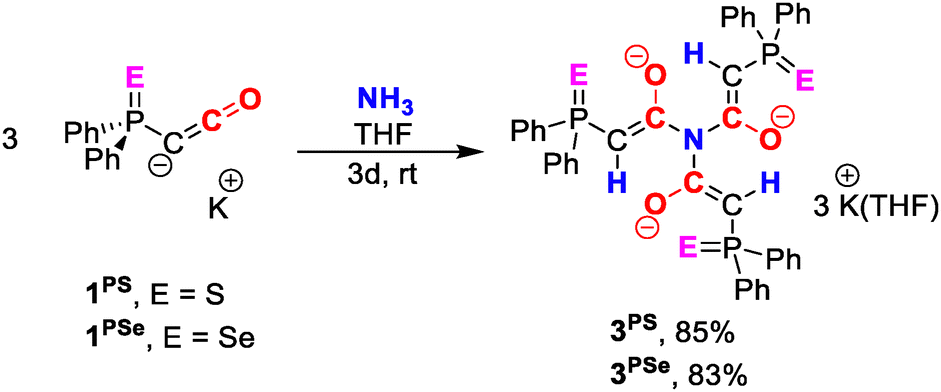 | ||
| Scheme 2 Synthesis of 3PS and 3PSe from the corresponding ketenyl anions, 1PS and 1PSe, respectively. | ||
In THF solution, both trianions exhibit highly symmetric 1H and 13C{1H} NMR patterns, with a characteristic doublet at approx. 4.00 ppm with a large 2JHP coupling constant (e.g. 25.7 Hz for 3PS) in the 1H NMR spectrum, corresponding to the enolate proton. The 13C{1H} NMR spectrum shows two doublets at approx. 63 ppm for the C1 carbon atom and at 173 ppm for the carbonyl carbon. The coupling constants for the C1 and C2 carbon atoms are considerably smaller than those of the parent ketenyl anions 1 due to the reduced bond orders in the PCC linkage (see Table 1). Interestingly, while both compounds feature singlets in the 31P{1H} NMR spectrum, the selenium compound 3PSe exhibits two slightly different 1JPSe coupling constants and two different signals at −236.3 and −236.6 ppm in the 77Se NMR spectrum. Since this observation cannot be explained by a monomeric structure of 3PSe in solution, we hypothesized that the two different 77Se signals arise from different coordination modes in the dimers. Indeed, diffusion-ordered NMR spectroscopy (DOSY) experiments confirmed the preservation of the dimeric structure of 3 in solution (see the SI, Fig. S15). Additionally, at elevated temperatures, the two selenium resonances coalesce into a single signal, indicating rapid interconversion between the two isomers in solution. Upon cooling back to room temperature, the signal splitting is restored, demonstrating that this process is reversible. We propose that the two isomers originate from different arrangements of the two triamides relative to the {K6} triangle. These arrangements result in a C3h symmetric isomer, as observed in the solid-state, and another isomer with D3-symmetry (see Fig. S14 for visual representations).
| δ(P) | δ(C1) | 1 J PC | δ(C2) | 2 J PC | C1−C2 | |
|---|---|---|---|---|---|---|
| 1PS | 22.5 | 2.4 | 175.0 | 142.7 | 40.7 | 1.240(8) |
| 3PS | 32.4 | 63.8 | 110.9 | 173.2 | 9.1 | 1.369(6) |
| 1PSe | 6.2 | 2.5 | 164.0 | 143.9 | 39.0 | 1.220(1) |
| 3PSe | 19.1 | 63.1 | 102.4 | 173.3 | 9.2 | 1.379(6) |
Mechanistically, the formation of 3 is proposed to proceed through an initial proton transfer from ammonia to the ketenyl anion, resulting in the generation of the corresponding protonated ketene and potassium amide (KNH2). The potassium amide subsequently attacks at the carbonyl carbon of the protonated ketene, affording a potassium enolate intermediate. Owing to its enhanced nucleophilicity relative to free ammonia, this intermediate undergoes further reactions with two additional equivalents of the ketenyl anion to yield the final trianionic products 3. The second and third activation steps proceed significantly faster than the initial proton transfer. This conclusion is supported by the observation of a single resonance corresponding to the final product in the 31P{1H} NMR spectrum, with no intermediates detectable under the reaction conditions.
Notably, ketenyl anions 1PS and 1PSe exhibit no reactivity towards ammonia in the presence of 18-crown-6 or [2,2,2]-cryptand, even upon heating to 70 °C. This highlights the crucial role of cation–anion interactions in facilitating ammonia activation. Encapsulation of potassium by these macrocyclic ligands presumably suppresses beneficial cooperative effects (e.g. through coordination of NH3 to potassium) and impedes the formation of the {K6}6+ framework. To further probe the influence of the counter-cation, the corresponding lithium salts of 1PS and 1PSe (without additional co-ligands) were examined. Analogously, these species exhibited no reactivity toward ammonia, likely due to their pronounced ynolate character, which disfavours initial proton abstraction from ammonia.28
Initially we hypothesized that the divergent reactivity of ketenyl anions 1CN and 1Tos, leading to the β-ketoamides 2 instead of 3, arises from their lower basicity due to the stronger electron-withdrawing ability of the tosyl and cyano group. We assumed that, in contrast to the triamide formation, the reaction to 2 is initiated by nucleophilic attack of ammonia at the ketenyl C2 carbon atom. However, attempts to optimize the structure of the putative NH3 adduct resulted in spontaneous ammonia elimination suggesting that the ketenyl anion cannot act as an electrophile and that the selectivity is instead governed by steric effects. We therefore propose that both reactions are initiated by deprotonation of NH3. While the resulting ketene readily reacts with another equivalent of the ketenyl anions 1CN or 1Tos, it preferentially reacts with the less nucleophilic NH2− rather than with the sterically more demanding ketenyl anions 1PS and 1PSe, leading to triamide 3 instead of 2 (Fig. S1 and Scheme 3).
 | ||
| Scheme 3 Reaction of 3PS with an excess amount of H2O to form diamide. | ||
To investigate whether the neutral triamides can be generated from 3, the thiophosphinoyl compound 3PS was reacted with various electrophiles. Due to its high negative charge, 3PS demonstrates pronounced reactivity. However, reactions with MeI and TMSCl proceeded unselectively, resulting in complex reaction mixtures. In contrast, treatment of 3PS with excess water afforded diamide 4PS, which precipitated directly from the reaction mixture as a colourless crystalline solid in 74% yield (Scheme 3). 4PS is characterized by a doublet at 3.83 ppm with a 2JHP coupling constant of 25.0 Hz for the CH2 protons and a broad singlet at 9.79 ppm for the N−H proton in the 1H NMR spectrum. The molecular structure of 4PS was unambiguously confirmed by single-crystal XRD analysis, which revealed a C1−C2 bond length of 1.509(2) Å, significantly longer than that in 3PS, and a C2−O1 bond length of 1.216(2) Å, notably shorter than that in 3PS (Fig. 3).
 | ||
| Fig. 3 Crystal structure of 4PS. Ellipsoids are drawn at the 50% probability level. All H atoms except at C1 and C3 are omitted for clarity. For crystallographic details, see the SI. Important bond lengths [Å] and angles [°]: P1–C1 1.826(2), C1–C2 1.509(2), C2–O1 1.216(2), P1–S1 1.961(1), P2–C3 1.828(2), C3–C4 1.512(2), C4–O2 1.211(2), P2–S2 1.950(1), P1–C1–C2 112.8(1), P2–C3–C4 115.4(1). | ||
In conclusion, we reported that ketenyl anions exhibit diverging reactivity toward ammonia depending on the nature of the ketenyl substituent. Ketenyl anions bearing a strongly anion-stabilizing substituent, such as a cyano or tosyl group, undergo cleavage of a single N−H bond of ammonia through reaction with two equivalents of the ketenyl anion to yield β-ketoamides. In contrast, the more basic phosphinoyl-substituted ketenyl anions react with ammonia via cleavage of all three N−H bonds, affording the trianionic triamide complexes 3 through reaction with 3 equivalents of the anion. The triamides form dimers both in solution and in the solid-state, with six potassium cations assembling to a planar triangle. Besides the substitution pattern, the choice of alkali metal and co-ligands proved crucial for the selective reaction of ketenyl anions with ammonia, suggesting a broader potential to access structurally diverse amides from these anions.
P. D. prepared and characterized 3PSe and 4PS, conducted the XRD analyses, and wrote the first draft of the manuscript. M. J. and S. M. prepared and characterized 3PS and 2Tos, respectively. K.-S. F. helped with the XRD refinement of 3PS. V. H. G. supervised the project and finalized the manuscript.
This work was supported by RESOLV, funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC-2033 – project number 390677874, and the European Union (ERC, CarbFunction, 101086951). Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or the European Research Council. Neither the European Union nor the granting authority can be held responsible for them. P. D. acknowledges the German Academic Exchange Service for a PhD scholarship. We thank Dr Dvoyashkin for helping with the VT NMR experiments.
Conflicts of interest
There are no conflicts of interest.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental details and characterization data for new compounds, including NMR spectra and a link to a repository with the original data files (PDF).CCDC 2505652–2505654 contain the supplementary crystallographic data for this paper.41a–c
References
- S. A. Lawrence, Amines: Synthesis, Properties and Applications, Cambridge University Press, New York, 2004 Search PubMed.
- J. L. Klinkenberg and J. F. Hartwig, Angew. Chem., Int. Ed., 2010, 50, 86–95 CrossRef PubMed.
- J. I. van der Vlugt, Chem. Soc. Rev., 2010, 39, 2302 RSC.
- D. M. Roundhill, Chem. Rev., 1992, 92, 1 CrossRef CAS.
- H. Kim and S. Chang, Acc. Chem. Res., 2017, 50(3), 482–486 Search PubMed.
- D. H. Mordaunt, M. N. R. Ashfold and R. N. Dixon, J. Chem. Phys., 1996, 104, 6460–6471 Search PubMed.
- A. Werner, Z. Anorg. Chem., 1893, 3, 267–330 Search PubMed.
- P. L. Dunn, B. J. Cook, S. I. Johnson, A. M. Appel and R. M. Bullock, J. Am. Chem. Soc., 2020, 142(42), 17845–17858 Search PubMed.
- H.-Y. Liu, H. M. C. Lant, C. C. Cody, J. Jelušić, R. H. Crabtree and G. W. Brudvig, ACS Catal., 2023, 13(7), 4675–4682 Search PubMed.
- J. Zhao, A. S. Goldman and J. F. Hartwig, Science, 2005, 307, 1080–1082 CrossRef CAS PubMed.
- E. Morgan, D. F. MacLean, R. McDonald and L. Turculet, J. Am. Chem. Soc., 2009, 131, 14234–14236 Search PubMed.
- P. P. Power, Nature, 2010, 463, 171–177 Search PubMed.
- G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439 CrossRef CAS PubMed.
- Z. Zhu, X. Wang, Y. Peng, H. Lei, J. C. Fettinger, E. Rivard and P. P. Power, Angew. Chem., Int. Ed., 2009, 48, 2031–2034 CrossRef CAS PubMed.
- Y. Peng, B. D. Ellis, X. Wang and P. P. Power, J. Am. Chem. Soc., 2008, 130, 12268–12269 Search PubMed.
- A. Jana, C. Schulzke and H. W. Roesky, J. Am. Chem. Soc., 2009, 131, 4600–4601 CrossRef CAS PubMed.
- U. Siemeling, C. Färber, C. Bruhn, M. Leibold, D. Selent, W. Baumann, M. von Hopffgarten, C. Goedecke and G. Frenking, Chem. Sci., 2010, 1, 697 RSC.
- A. V. Protchenko, J. I. Bates, L. M. A. Saleh, M. P. Blake, A. D. Schwarz, E. L. Kolychev, A. L. Thompson, C. Jones, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2016, 138, 4555–4564 Search PubMed.
- S. M. McCarthy, Y.-C. Lin, D. Devarajan, J. W. Chang, H. P. Yennawar, R. M. Rioux, D. H. Ess and A. T. Radosevich, J. Am. Chem. Soc., 2014, 136, 4640–4650 Search PubMed.
- J. Cui, Y. Li, R. Ganguly, A. Inthirarajah, H. Hirao and R. Kinjo, J. Am. Chem. Soc., 2014, 136, 16764–16767 CrossRef CAS PubMed.
- F. Krämer, J. Paradies, I. Fernández and F. Breher, Nat. Chem., 2024, 16, 63–69 Search PubMed.
- A. F. Hegarty and P. O’Neill, Tetrahedron Lett., 1987, 28, 901–904 CrossRef CAS.
- L. F. Clarke, A. F. Hegarty and P. O'Neill, J. Org. Chem., 1992, 57, 362–366 Search PubMed.
- Y.-L. Zhong, D. R. Gauthier, Jr., Y.-J. Shi, M. McLaughlin, J. Y. L. Chung, P. Dagneau, B. Marcune, S. W. Krska, R. G. Ball, R. A. Reamer and N. Yasuda, J. Org. Chem., 2012, 77, 3297–3310 CrossRef CAS PubMed.
- K. Sung and T. T. Tidwell, J. Am. Chem. Soc., 1998, 120, 3043–3048 CrossRef CAS.
- G. Raspoet and M. T. Nguyen, J. Org. Chem., 1998, 63, 9669–9677 CrossRef CAS.
- M. Jörges, F. Krischer and V. H. Gessner, Science, 2022, 378, 1331–1336 CrossRef PubMed.
- P. Duari, S. Mondal, M. Jörges and V. H. Gessner, Chem. Commun., 2024, 60, 9372–9375 Search PubMed.
- M. Jörges, S. Mondal, M. Kumar, P. Duari, F. Krischer, J. Löffler and V. H. Gessner, Organometallics, 2024, 43, 585–593 CrossRef PubMed.
- F. Krischer, M. Jörges, T.-F. Leung, H. Darmandeh and V. H. Gessner, Angew. Chem., Int. Ed., 2023, 62, e202309629 Search PubMed.
- F. Krischer, V. S. S. N. Swamy, K.-S. Feichtner, R. J. Ward and V. H. Gessner, Angew. Chem., Int. Ed., 2024, 63, e202403766 Search PubMed.
- T. Wang, Z. Guo, L. E. English, D. W. Stephan, A. R. Jupp and M. Xu, Angew. Chem., Int. Ed., 2024, 63, e202402728 Search PubMed.
- R. Wei, X.-F. Wang, D. A. Ruiz and L. L. Liu, Angew. Chem., Int. Ed., 2023, 62, e202219211 CrossRef CAS PubMed.
- F. Krischer and V. H. Gessner, JACS Au, 2024, 4, 1709–1722 Search PubMed.
- A. Das, Q. Le Dé and V. H. Gessner, Nat. Rev. Chem., 2025, 9, 523–536 CrossRef PubMed.
- P. Duari, A. Linke, M. Shishkova, Q. L. Dé, A. Das and V. H. Gessner, Angew. Chem., Int. Ed., 2025, 64, e202516374 CrossRef CAS PubMed.
- A. Kornath, R. Ludwig and A. Zoermer, Angew. Chem., Int. Ed., 1998, 37, 1575–1577 CrossRef CAS PubMed.
- A. Banerjee, T. K. Ghanty and A. Chakrabarti, J. Phys. Chem. A, 2008, 112, 12303–12311 CrossRef CAS PubMed.
- L. Padilla-Campos and E. Chávez, J. Mol. Struct. THEOCHEM, 2010, 958, 92–100 CrossRef CAS.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- (a) CCDC 2505652: Experimental Crystal Structure Determination, 2026 DOI:10.5517/ccdc.csd.cc2q3bhp; (b) CCDC 2505653: Experimental Crystal Structure Determination, 2026 DOI:10.5517/ccdc.csd.cc2q3bjq; (c) CCDC 2505654: Experimental Crystal Structure Determination, 2026 DOI:10.5517/ccdc.csd.cc2q3bkr.
| This journal is © The Royal Society of Chemistry 2026 |