
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of ketones and mono-fluoro ketones via boron enolates formed by substitution of esters with benzylboronic esters
Pankaj
Kumar†
 *a,
Leah
Mistry†
a,
Josephine M.
Stewart
b and
Graham
Pattison
*a
*a,
Leah
Mistry†
a,
Josephine M.
Stewart
b and
Graham
Pattison
*a
aDepartment of Chemistry, School of Natural Sciences, University of Lincoln, Joseph Banks Laboratories, Green Lane, Lincoln, LN6 7DL, UK. E-mail: pkumar@lincoln.ac.uk; gpattison@lincoln.ac.uk
bSchool of Chemistry, University of St Andrews, North Haugh, St Andrews, KY16 9ST, UK
First published on 25th November 2025
Abstract
Benzylboronic esters can be deprotonated using LiTMP and engaged in useful C–C bond-forming processes. Here we report that they undergo acylation with esters. We also report a fluorinative coupling reaction performed through nucleophilic substitution of a lithiated benzylboronic ester with an ester, and trapping of the resultant boron enolate with the fluorinating agent NFSI.
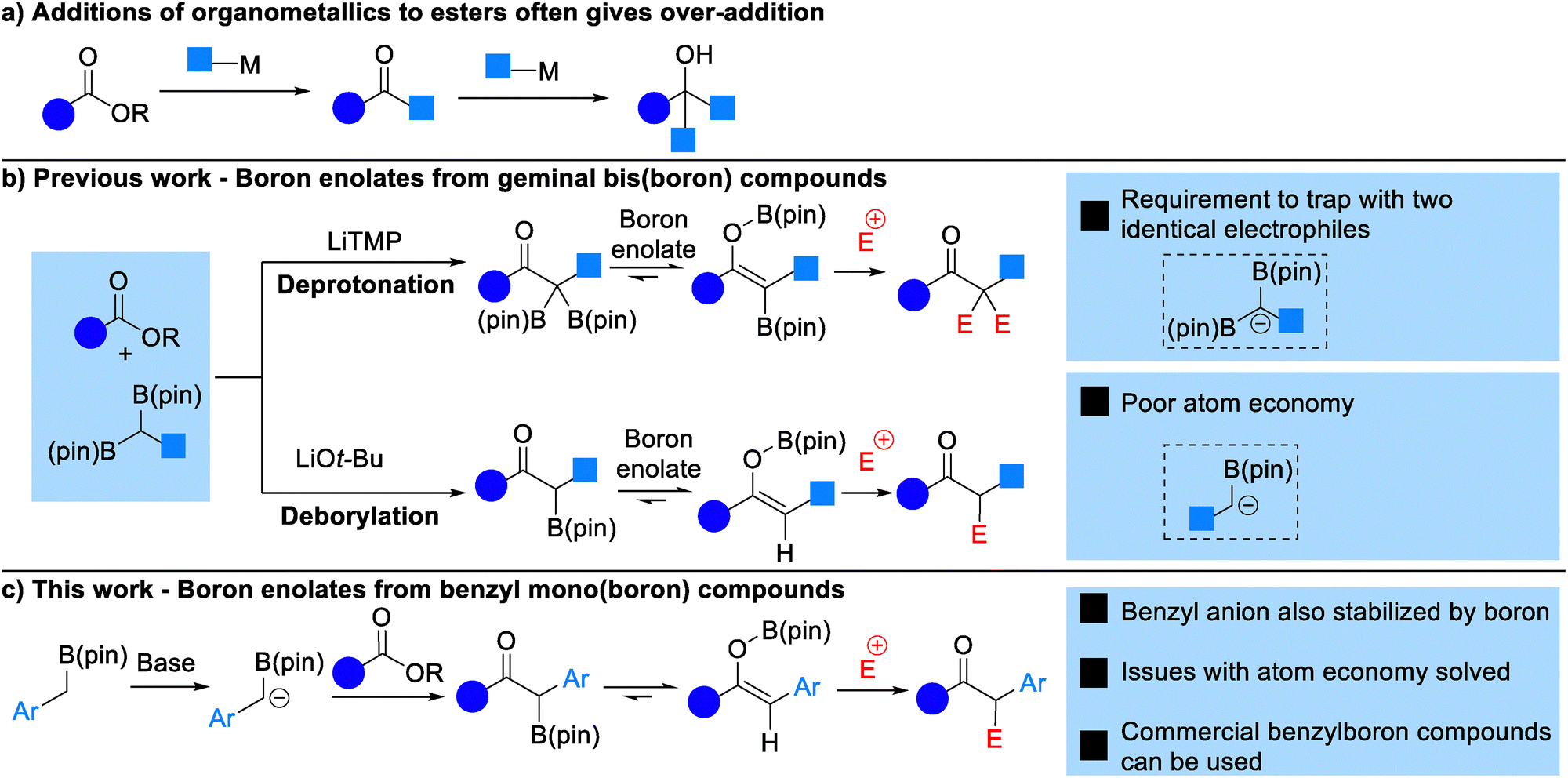
The addition of an organometallic nucleophile, such as a Grignard or organolithium reagent, to an ester to form a ketone is not a reliable transformation. As a ketone is more reactive than an ester the organometallic reagent will typically undergo some degree of addition to the ketone product, forming an alcohol and reducing the yield of the reaction (Scheme 1a). However, the ubiquity of ketones in a range of pharmaceuticals, natural products and materials makes the direct conversion of an ester to a ketone very desirable.
 | ||
| Scheme 1 Concept of boron enolate formation by substitution. | ||
There have been solutions to this problem developed previously, including the Weinreb amide, in which intermolecular coordination of the metal ion to a hydroxylamine stabilizes the tetrahedral intermediate formed on addition of an organometallic.1 However, most hydroxylamines are toxic and N,O-dimethylhydroxylamine specifically inhibits enzymes that protect erythrocytes against oxidative stress.2 Strategies that use reagents of lower toxicity are always valuable. Other methods of achieving the additions of organometallic carbon nucleophiles to carbonyl compounds at the ester oxidation level include addition of organometallic reagents to carboxylic acids (via the carboxylate salt)3 or additions to acid chlorides or anhydrides.4 Metal-catalyzed cross coupling reactions of esters yielding ketones are also known.5 Despite this work, additions to esters to give ketones remains unusual and unpredictable.
We felt that the addition of organoboron reagents to esters presents a potential solution to this problem. Our research group6 along with the groups of Liu7 and Chirik8 (Scheme 1b) have demonstrated that addition of lithitated geminal bis(boron) compounds9 to esters yields boron enolate intermediates that can be trapped by an electrophile. It is the formation of the boron enolate that prevents any over-addition in these reactions, as this intermediate effectively masks the carbonyl group. Geminal bis(boron) compounds are acidic and can be deprotonated at the carbon between the boron atoms by moderately strong sterically-hindered bases such as LiTMP.10 In our previous work we have shown that both boron atoms can be trapped by two equivalents of an electrophile to yield α,α-difunctionalized ketones.6b Alternatively, nucleophilic reagents such as LiOtBu lead to anion formation by deborylation.7a,11a,11b This allows trapping with a single equivalent of electrophile yielding mono-functionalized ketones.
Of course, the need to use a bis(boron) reagent in these reactions limits their atom economy, and in many cases double trapping with two equivalents of electrophile is not desired. We wondered whether equivalent chemistry could be performed using mono-boron esters. We felt that an additional acidifying group may facilitate deprotonation of the boronic ester, so thought that benzyl boronic esters would be a good starting point (Scheme 1c).12 Chirik has reported a single example of the addition of deprotonated benzyl boronic ester to methyl butanoate,8 whilst Zhan demonstrated more broadly that benzylboronic pinacol ester can be deprotonated using LiTMP and the anion trapped with alkyl halides, silyl halides and trifluoromethyl alkenes.13 Furthermore, whilst a range of methods for the synthesis of alkyl-substituted geminal bis(boron) compounds has emerged in recent years, methods that allow the synthesis of benzyl bis(boron) compounds are more limited and include C–H borylation of alkylarenes, and cross-coupling of a diborylzinc reagent.14 The majority of these methods are sensitive and rely on the use of a glovebox and their demonstration on a gram-scale is limited. In contrast a much wider range of methods exist for the synthesis of benzyl mono-boron compounds, including particularly borylation of benzyl halides and decarboxylative borylation.15 A range of benzyl mono-boron compounds are commercially available, in contrast to their geminal bis(boron) counterparts.
Our initial focus was on finding suitable conditions for formation of the boron enolate intermediate using benzyl boronic pinacol ester, and protonation of this intermediate using water. Optimization of the reaction (see SI) showed LiTMP to be a more effective base than NaHMDS, and that leaving the initial substitution step for longer to form the boron enolate could also lead to higher yields. The benzylboronic ester was first deprotonated at 0 °C using LiTMP which is a stronger base than NaHMDS. Bulky bases are required to give deprotonation and prevent deborylation. After deprotonation a solution of the ester was added and heated to 50 °C for 2 hours to achieve boron enolate formation by substitution. Finally, water was added after cooling to room temperature to protonate the boron enolate and yield the desired ketone product.
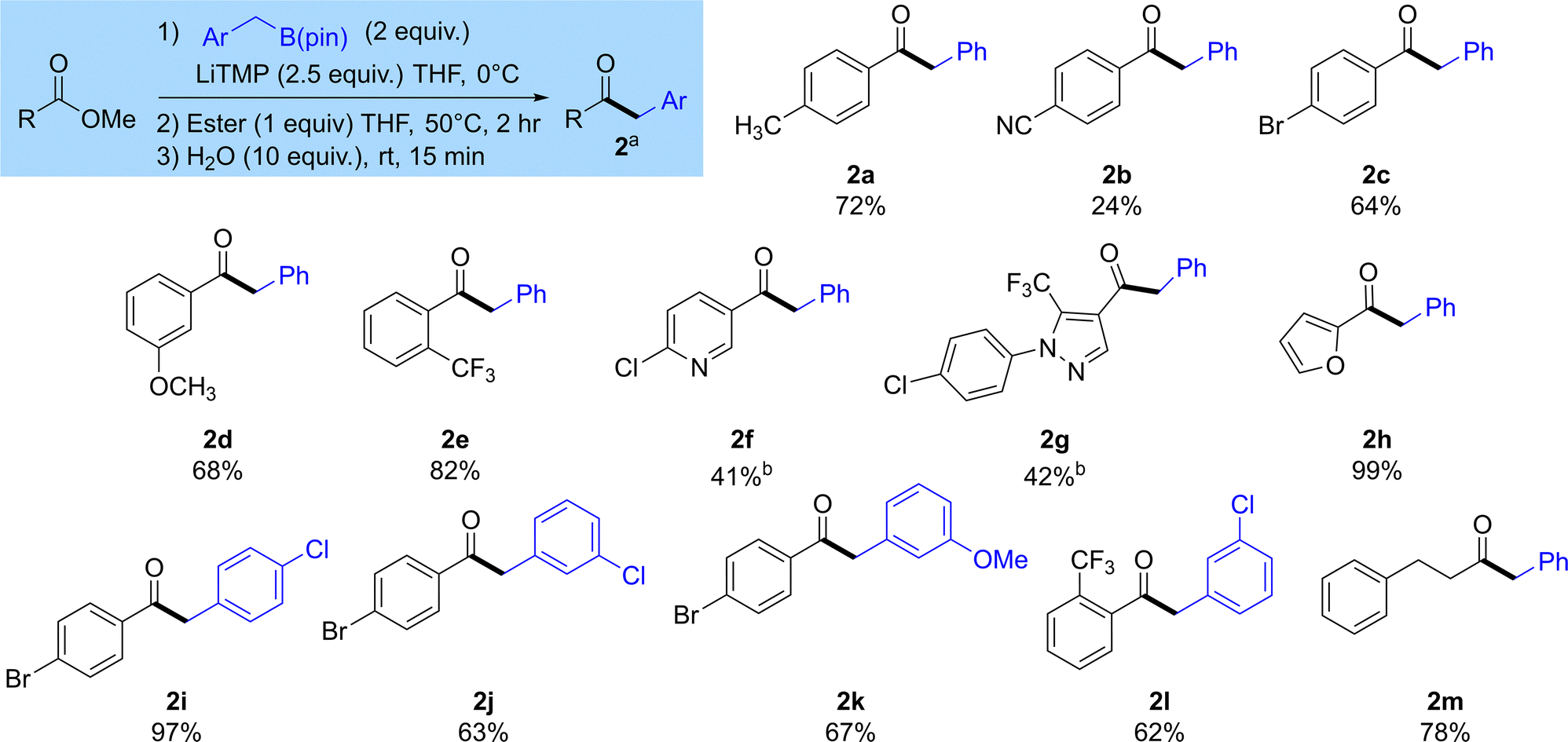
Once we understood more about the optimal conditions of this reaction, we wanted to understand its scope (Scheme 2). Pleasingly, as expected these reactions gave the ketones 2a–m by substitution of the ester with a benzyl group. Aromatic esters are best tolerated in this reaction 2a–l. A range of aromatic esters bearing substituents including alkyl groups, halogens, nitriles and alkoxy substituents at the ortho-, meta- and para-positions successfully gave the desired ketone products 2a–e in good yield. Heterocyclic esters including pyridines, furans and pyrazoles were also good substrates for this reaction, yielding products 2f–h. We also showed that substituents could be tolerated on the benzylboronic ester 2i–l. An alkyl ester could also be used, providing ketone 2m in good yield. We also performed a deuteration experiment by trapping with D2O. Compound 2aD was formed in good yield, and although the mono-deuterated compound was obtained as the major product, incorporation of deuterium was only moderate at 57% (47% mono-deuteration) (see SI).
 | ||
| Scheme 2 Scope of ketone synthesis; aisolated yield; bstarting material was ethyl ester. | ||
We then wanted to show that the boron enolates could be trapped by electrophiles other than a proton. In our previous work we showed that N-fluorobenzenesulfonimide (NFSI) was an excellent electrophilic source of fluorine for trapping boron enolates derived from substitution of esters with geminal bis(boron) compounds,6b providing α,α-difluorinated ketones.16 The fluorinated ketones produced are highly electrophilic and have potential application in medicinal chemistry17 as enzyme inhibitors.18 We wanted to see whether this approach could be extended to the synthesis of mono-fluorinated ketones using benzyl-boronic esters. Mono-fluorination would be expected as in this case only a single boron atom is available for reaction.
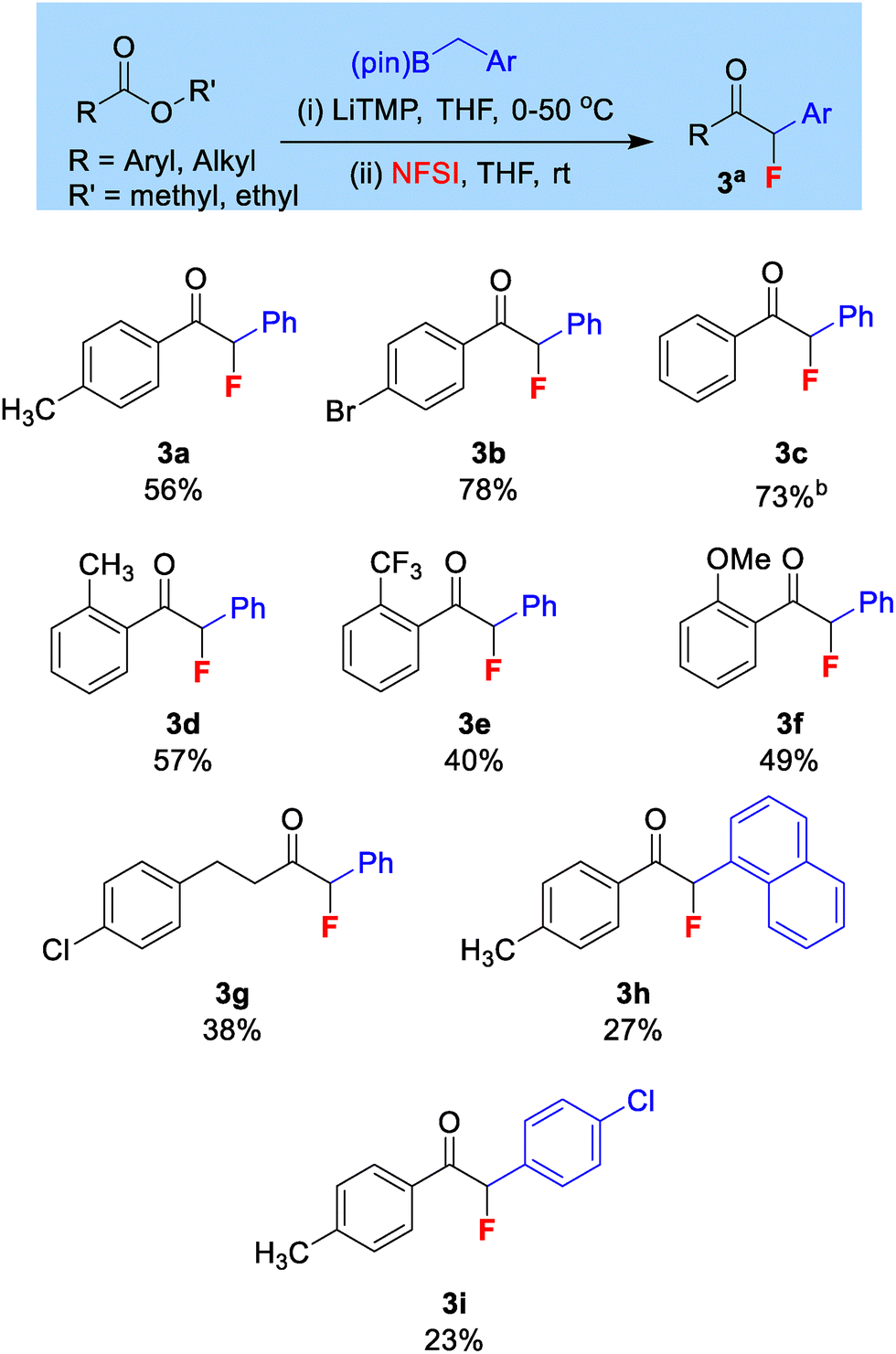
During our optimization (see SI) we observed a tendency to form a mixture of mono-fluorinated and di-fluorinated products. After varying reaction conditions, we could optimize this transformation to afford 62% (NMR yield, isolated to 56%) of the mono-fluorinated ketone 3a with negligible di-fluorinated product. The optimized reaction conditions used 1 equiv. LiTMP, 1.5 equiv. benzylboronic acid pinacol ester and 2 equiv. NFSI.
The tendency for over-fluorination was not observed in our previous geminal-bis(boron) work. We believe that it is observed here due to the acidifying effect of the aryl group introduced from benzylboronic esters. The di-fluorinated side product is believed to form via a second fluorination of the mono-fluorinated product in the presence of residual base and excess NFSI. However, the use of our optimized conditions means minimal di-fluorinated product is produced.
Various ester derivatives were subjected to the reaction conditions (Scheme 3), and unsubstituted and para-substituted benzoate derivatives produced moderate to good yields of the desired mono-fluoro ketones (3a–c). Isolated yields for ortho substituted benzoates were moderate and largely not affected by the electronics of the substituted groups (3d–f). It was also important to demonstrate the mono-fluorinative coupling on alkyl esters, as these have in theory two enolizable sites. Methyl 4-methoxypropanoate gave a 33% NMR conversion to the monofluorinated ketone but could not be isolated to acceptable purity. Methyl 4-chloropropanoate on the other hand afforded 3g in 38% isolated yield, showing fluorination only at the site boron was introduced. Changes to the aryl ring of the benzylboronic ester were also tolerated (3h–i).
 | ||
| Scheme 3 Substrate scope for mono-fluorinative coupling; aisolated yield; bstarting material was ethyl ester. | ||
In some cases, isolated yields were reduced due to challenges in purification. The products are, in general, non-polar in nature and elute with similar Rf values to the starting ester and organoboron by-products. It is also important to ensure that the benzylboronic pinacol ester starting materials are of high purity, as these reagents do show some degree of instability and decomposition. For successful reaction a strong colour change should be observed on addition of LiTMP to the benzylboronic ester, which can be hindered in the presence of decomposition impurities and prevents effective reaction.
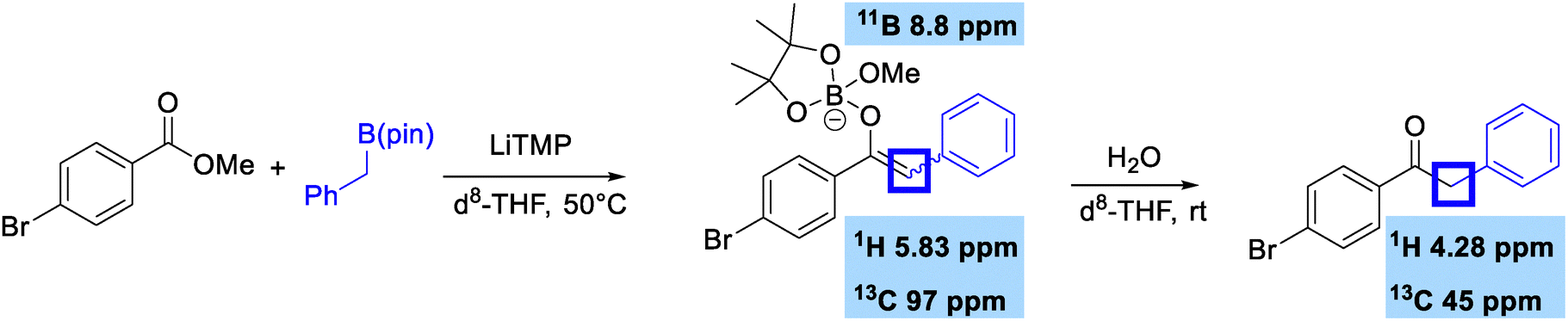
We finally wanted to examine the mechanism of this reaction by attempting to observe the boron enolate intermediate formed on nucleophilic substitution (Scheme 4). Mixing methyl 4-bromobenzoate with benzylboronic pinacol ester in the presence of LiTMP in d8-THF gave a species that had a 1H signal at 5.83 ppm with a HSQC correlation to a 13C signal at 97 ppm. This is very indicative of an electron-rich enol-like alkene. This signal disappeared on addition of water, and formation of product 2c was confirmed. The O-bound nature of the boron enolate was further confirmed by 11B NMR, which showed a peak at 8.8 ppm which is indicative of quaternary species bound to oxygen and suggestive that the methoxide leaving group was involved in coordination to boron, which was also observed in our previous geminal bis(boron) study.6b
 | ||
| Scheme 4 Mechanism study. | ||
In conclusion, we have developed a new method that allows the synthesis of ketones from esters and benzylboronic pinacol esters. This forms a boron enolate that prevents any over-addition, and can be trapped either by water, providing ketones, or by the electrophilic fluorinating agent NFSI to provide an efficient route to fluorinated ketones in a convergent fashion. We have demonstrated that mono-boron enolates can be prepared by nucleophilic substitution with deprotonated benzylic boronate esters, improving atom economy in terms of boron and removing the requirement for a bis(boron) compound in these cases.
We thank EPSRC (Grant Ref: EP/W012626/1) for funding this work. This work was carried out using JBL (Joseph Banks Laboratories) Science facilities at the University of Lincoln.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental methods and data; copies of NMR spectra. See DOI: https://doi.org/10.1039/d5cc05350a.Notes and references
- (a) S. Nahm and S. M. Weinreb, Tetrahedron Lett., 1981, 22, 3815–3818 CAS; (b) R. Senatore, L. Ielo, S. Monticelli, L. Castoldi and V. Pace, Synthesis, 2019, 2792–2808 CAS.
- C. T. A. Evelo, A. A. M. G. Spooren, R. A. G. Bisschops, L. G. M. Baars and J. M. Neis, Blood Cells, Mol., Dis., 1998, 24, 280–295 CAS.
- (a) R. Levine, M. J. Karten and W. M. Kadunce, J. Org. Chem., 1975, 40, 1770–1773 CrossRef CAS; (b) J. Wu, X. Yang, Z. He, X. Mao, T. A. Hatton and T. F. Jamison, Angew. Chem., Int. Ed., 2014, 53, 8416–8420 CrossRef CAS; (c) K. Colas, A. C. V. D. dos Santos and A. Mendoza, Org. Lett., 2019, 21, 7908–7913 CrossRef CAS PubMed; (d) K. Colas, A. C. V. D. dos Santos, S. V. Kohlhepp and A. Mendoza, Chem. – Eur. J., 2022, 28, e202104053 CrossRef CAS PubMed.
- (a) R. K. Dieter, Tetrahedron, 1999, 55, 4177–4236 CAS; (b) X.-j Wang, L. Zhang, X. Sun, Y. Xu, D. Krishnamurthy and C. H. Senanayake, Org. Lett., 2005, 7, 5593–5595 CAS.
- (a) H. Tatamidani, F. Kakiuchi and N. Chatani, Org. Lett., 2004, 6, 3597–3599 CrossRef CAS PubMed; (b) P. Lei, G. Meng, Y. Ling, J. An, S. P. Nolan and M. Szostak, Org. Lett., 2017, 19, 6510–6513 CrossRef CAS; (c) E. E. Stache, T. Rovis and A. G. Doyle, Angew. Chem., Int. Ed., 2017, 56, 3679–3683 CrossRef CAS PubMed; (d) B. Roh, A. O. Farah, B. Kim, T. Feoktistova, F. Moeller, K. D. Kim, P. H.-Y. Cheong and H. G. Lee, J. Am. Chem. Soc., 2023, 145, 7075–7083 CrossRef CAS; (e) W. Wang, C. Shen, L. Zhang and K. Dong, Org. Lett., 2024, 26, 850–854 CrossRef CAS.
- (a) T. C. Stephens and G. Pattison, Org. Lett., 2017, 19, 3498–3501 CrossRef CAS; (b) C. E. Iacono, T. C. Stephens, T. S. Rajan and G. Pattison, J. Am. Chem. Soc., 2018, 140, 2036–2040 CrossRef CAS; (c) G. R. A. Garratt and G. Pattison, Synlett, 2020, 1656–1662 CAS; (d) P. Kumar and G. Pattison, Chem. Commun., 2024, 60, 13887–13890 RSC.
- (a) W. Sun, L. Wang, C. Xia and C. Liu, Angew. Chem., Int. Ed., 2018, 57, 5501–5505 CrossRef CAS; (b) W. Sun, L. Wang, Y. Hu, X. Wu, C. Xia and C. Liu, Nat. Commun., 2020, 11, 3113 CrossRef CAS PubMed; (c) W. Sun, L. Xu, Y. Qin and C. Liu, Nat. Synth., 2023, 2, 413–422 CrossRef CAS; (d) L. Xu, D. Chen, P. Zhang, C. Xia and C. Liu, Chem, 2024, 10, 3474–3487 CrossRef CAS.
- B. Lee and P. J. Chirik, J. Am. Chem. Soc., 2020, 142, 2429–2437 CrossRef CAS PubMed.
- (a) R. Nallagonda, K. Padala and A. Masarwa, Org. Biomol. Chem., 2018, 16, 1050–1064 RSC; (b) W. Jo, J. H. Lee and S. H. Cho, Chem. Commun., 2021, 57, 4346–4353 RSC; (c) C. Zhang, W. Hu and J. P. Morken, ACS Catal., 2021, 11, 10660–10680 CrossRef CAS PubMed.
- (a) D. S. Matteson, R. J. Moody and P. K. Jesthi, J. Am. Chem. Soc., 1975, 97, 5608–5609 CrossRef CAS; (b) D. S. Matteson and R. J. Moody, J. Am. Chem. Soc., 1977, 99, 3196–3197 CrossRef CAS; (c) D. S. Matteson and R. J. Moody, Organometallics, 1982, 1, 20–28 CrossRef CAS.
- (a) K. Hong, X. Liu and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 10581–10584 CrossRef CAS PubMed; (b) T. Fang, B. Ren and C. Liu, Acc. Chem. Res., 2025, 58, 2511–2525 CrossRef CAS.
- (a) M. R. Hollerbach and T. J. Barker, Organometallics, 2018, 37, 1425–1427 CrossRef CAS PubMed; (b) J. C. Hayes, M. R. Hollerbach and T. J. Barker, Tetrahedron Lett., 2020, 61, 151505 CrossRef CAS; (c) S. G. Gierszal and T. J. Barker, Tetrahedron Lett., 2021, 82, 153369 CrossRef CAS; (d) T. J. Barker, A. Bogatkevich, D. W. Crowder, S. G. Gierszal, J. C. Hayes, M. R. Hollerbach and R. W. Russell, Synthesis, 2023, 2639–2647 CrossRef CAS.
- H. Jin, J. Han, X. Liu, C. Feng and M. Zhan, Org. Lett., 2023, 25, 4168–4172 CrossRef CAS.
- (a) S. H. Cho and J. F. Hartwig, Chem. Sci., 2014, 5, 694–698 RSC; (b) W. N. Palmer, J. V. Obligacion, I. Pappas and P. J. Chirik, J. Am. Chem. Soc., 2016, 138, 766–769 CrossRef CAS; (c) H. Lee, Y. Lee and S. H. Cho, Org. Lett., 2019, 21, 5912–5916 CrossRef CAS PubMed; (d) D. Yoshii, T. Yatabe, T. Yabe and K. Yamaguchi, ACS Catal., 2021, 11, 2150–2155 CrossRef CAS; (e) W. Dong, Z. Liu, M. Zhang, Z. Zhang, Y. Wei, G. Liu and W. Zhao, Org. Lett., 2025, 27, 3101–3106 CrossRef CAS; (f) T. Yao, S. Wang, Y. Liu, G. Yin and Y. Li, Org. Lett., 2025, 27, 3691–3696 CAS.
- (a) A. Giroux, Tetrahedron Lett., 2003, 44, 233–235 CrossRef CAS; (b) A. S. Dudnik and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 10693–10697 CrossRef CAS; (c) H. Li, L. Wang, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2012, 51, 2943–2946 CrossRef CAS PubMed; (d) C.-T. Yang, Z.-Q. Zhang, H. Tajuddin, C.-C. Wu, J. Liang, J.-H. Liu, Y. Fu, M. Czyzewska, P. G. Steel, T. B. Marder and L. Liu, Angew. Chem., Int. Ed., 2012, 51, 528–532 CrossRef CAS; (e) T. C. Atack, R. M. Lecker and S. P. Cook, J. Am. Chem. Soc., 2014, 136, 9521–9523 CrossRef CAS; (f) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS; (g) L. Wang, W. Sun and C. Liu, Chin. J. Catal., 2018, 39, 1725–1729 CrossRef CAS; (h) Y. Qin, L. Xu, J. Xu and C. Liu, Chin. J. Org. Chem., 2023, 43, 1868–1874 CrossRef CAS; (i) X. He, E. Shi and J. Xiao, Org. Lett., 2024, 26, 9728–9734 CrossRef CAS PubMed.
- (a) L. S. Dobson and G. Pattison, Chem. Commun., 2016, 52, 11116–11119 RSC; (b) G. Pattison, Beilstein J. Org. Chem., 2017, 13, 2915–2921 CrossRef CAS PubMed; (c) G. Pattison, Eur. J. Org. Chem., 2018, 3520–3540 CAS; (d) G. Pattison, Org. Biomol. Chem., 2019, 17, 5651–5660 Search PubMed.
- (a) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (b) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS; (c) M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS; (d) R. J. Glyn and G. Pattison, J. Med. Chem., 2021, 64, 10246–10259 CrossRef CAS.
- (a) M. H. Gelb, J. P. Svaren and R. H. Abeles, Biochemistry, 1985, 24, 1813–1817 CrossRef CAS PubMed; (b) K. Fearon, A. Spaltenstein, P. B. Hopkins and M. H. Gelb, J. Med. Chem., 1987, 30, 1617–1622 CrossRef CAS PubMed; (c) C. Han, A. E. Salyer, E. H. Kim, X. Jiang, R. E. Jarrard, M. S. Powers, A. M. Kirchhoff, T. K. Salvador, J. A. Chester, G. H. Hockerman and D. A. Colby, J. Med. Chem., 2013, 56, 2456–2465 CrossRef CAS.
Footnote |
| † P. K. and L. M. contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |