
Ultrafast Zn2+ solvation dynamics unmask a hopping mechanism in eutectic battery electrolytes
Aruna K.
Mora
 *ab and
Prabhat K.
Singh
*ab
*ab and
Prabhat K.
Singh
*ab
aRadiation & Photochemistry Division, Bhabha Atomic Research Centre, Mumbai 400 085, India. E-mail: arunm@barc.gov.in; prabhatk@barc.gov.in
bHomi Bhabha National Institute, Anushaktinagar, Mumbai-400085, India
First published on 27th November 2025
Abstract
Understanding and controlling ultrafast Zn2+ solvation in deep-eutectic electrolytes is pivotal for advancing safe, high-efficiency Zn batteries yet has remained experimentally unexplored. Two-dimensional IR spectroscopy of tunable Zn(TFSI)2–acetamide media, with thiocyanate anion (SCN−) as a vibrational probe, reveals composition-driven slowing of picosecond solvation dynamics, however, paradoxically, the slowest solvation accompanies the highest conductivity, implying a hopping-like Zn2+ migration mechanism. 2DIR measurements provide a direct molecular link between electrolyte composition and macroscopic charge transport, offering a powerful platform for rational design of high-performance Zn-ion batteries.
Rechargeable zinc-ion batteries (ZIBs) are attractive for grid-scale storage because Zn metal is earth-abundant, intrinsically safe, and offers one of the highest volumetric capacities among practical anodes.1,2 Yet their coulombic efficiency is still compromised by dendritic Zn growth and parasitic hydrogen evolution at the Zn|electrolyte interface.3 Unlike Li metal, Zn seldom develops a robust solid–electrolyte interphase (SEI), so recent effort has shifted toward electrolyte engineering to regulate the Zn2+ solvation sheath itself.4 Highly concentrated “water-in-salt” formulations and, more recently, deep-eutectic solvents (DESs) have emerged as effective routes: by coordinating anions or donor molecules directly to Zn2+.5 DESs were selected in this study because they combine wide electrochemical stability (≈ 2.4 V vs. Zn/Zn2+), non-flammability, negligible vapour pressure, and high ionic tunability with low cost and environmental compatibility. Their high salt solubility and tunable coordination environment enhance coulombic efficiency (≈98%) and suppress dendritic Zn growth compared to aqueous Zn(TFSI)2 (≈70%). These media suppress water activity, limit side reactions, and deliver nearly quantitative plating/stripping efficiencies. For example, Zhang et al. demonstrated an in situ ZnF2-rich organic/inorganic hybrid SEI from an acetamide–Zn(TFSI)2 eutectic electrolyte (ZES), achieving nearly 100% CE and dendrite-free cycling even at high areal capacity.6 This work highlighted the critical role of electrolyte solvation structure (e.g. anion–Zn complexes) in directing interphase chemistry. In fact, highly concentrated and eutectic electrolytes have been shown to improve Zn reversibility by altering the Zn2+ solvation shell.1,7–9 Chen et al. recently designed a hydrated deep eutectic Zn electrolyte with a water-deficient [Zn(H2O)2(eg)2(OTf)2] solvation structure, dramatically suppressing water side reactions while boosting Zn2+ transport.10 Together, these studies demonstrate that solvation-sheath modulation—rather than classical SEI formation—has become the master variable for achieving stable performance in Zn metal batteries, although the formation of a stable SEI remains indispensable.
Despite these structural insights, the dynamics of Zn2+ solvation—how fast the coordination cage reorganises and whether that timescale depends on electrolyte composition—remain essentially unknown. Such ultrafast motions are non-trivial, because they set the molecular friction that governs ion mobility and, by extension, plating efficiency. We therefore hypothesise that composition-dependent changes in the picosecond solvation dynamics will correlate directly with macroscopic properties such as ionic conductivity and viscosity. Ultrafast two-dimensional infrared (2DIR) spectroscopy offers a uniquely sensitive route to test this hypothesis. By monitoring the frequency–frequency correlation function (FFCF) of a vibrational reporter, 2DIR measures spectral diffusion—the hallmark of solvent reorganisation—on femto- to picosecond timescales. A well-known approach is to dissolve a trace amount of vibrational probe (e.g. SCN−) into the electrolyte. The CN stretch of thiocyanate is a sensitive reporter of its local environment, as shown in ionic-liquid and salt solutions where SCN− binds weakly or strongly to cations. For instance, in Li+–SCN− solutions, contact ion pairs (CIPs) give a CN frequency blue-shifted by ∼16 cm−1 relative to free SCN−, producing a resolvable IR doublet.11 In 2DIR the nodal-line slope (NLS)—the slope of the nodal line connecting the 0–1 bleach and 1–2 excited-state absorption—decays from ∼1 (fully correlated) toward 0 with waiting time, thereby reporting the solvation (spectral-diffusion) timescales.12 Here we apply 2DIR with the SCN− probe to Zn2+ eutectic electrolytes. We focus on a ZES system (ratios 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 to 1:9) known for its wide electrochemical stability and ability to dissolve multivalent salts.10 Unlike dilute aqueous Zn electrolytes, these nonaqueous ZES electrolytes can be tuned by composition. We systematically vary the Zn:acetamide ratio and measure how the SCN− CN stretching band behaves in both linear IR and ultrafast 2DIR spectroscopy. The goal is to correlate molecular solvation dynamics with macroscopic transport properties. We show that the Frequency-frequency correlation function (FFCF) decay of the 2DIR spectra resolves two distinct dynamical components which lengthen systematically with acetamide content. Finally, we relate these molecular timescales to bulk transport properties, revealing an unexpected inverse relationship that hints at a possible hopping-type Zn2+ migration mechanism (Scheme 1). Thus, 2DIR not only fills the current knowledge gap in solvation dynamics but also provides a predictive link between electrolyte composition and battery performance.
:4 to 1:9) known for its wide electrochemical stability and ability to dissolve multivalent salts.10 Unlike dilute aqueous Zn electrolytes, these nonaqueous ZES electrolytes can be tuned by composition. We systematically vary the Zn:acetamide ratio and measure how the SCN− CN stretching band behaves in both linear IR and ultrafast 2DIR spectroscopy. The goal is to correlate molecular solvation dynamics with macroscopic transport properties. We show that the Frequency-frequency correlation function (FFCF) decay of the 2DIR spectra resolves two distinct dynamical components which lengthen systematically with acetamide content. Finally, we relate these molecular timescales to bulk transport properties, revealing an unexpected inverse relationship that hints at a possible hopping-type Zn2+ migration mechanism (Scheme 1). Thus, 2DIR not only fills the current knowledge gap in solvation dynamics but also provides a predictive link between electrolyte composition and battery performance.
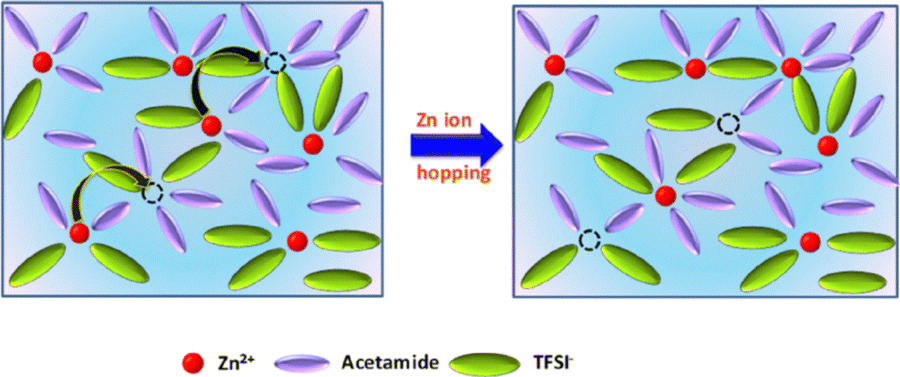 | ||
| Scheme 1 Schematic illustration of Zn2+ hopping mechanism in Zn(TFSI)2—acetamide deep-eutectic solvent (ZES) electrolytes. | ||
The steady-state FTIR spectra of SCN− in ZES show a prominent CN stretching band around 2090 cm−1 (Fig. 1). Across the series the ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) peak red-shifts only marginally—from 2093.1 cm−1 in the 1:4 mixture to 2087.8 cm−1 in the 1:9 mixture—while the full width at half maximum (FWHM) contracts slightly, from 43.2 cm−1 to 40.7 cm−1 (Table S1, SI). These ≤ 5% changes confirm that the average solvation polarity sensed by SCN− remains nearly constant despite varying composition. In other words, replacing some acetamide by Zn(TFSI)2 does not dramatically alter the dielectric environment of the CN probe. We also note that the band remains broad (FWHM ≈ 41–43 cm−1) and slightly asymmetric, consistent with a distribution of environments. By analogy to Li–SCN systems, such line broadening often arises from a superposition of “free” SCN− ions and those bound in contact ion pairs (CIPs).11 In Li+–SCN− solutions, the CN stretch of a Li+–SCN− contact ion pair appears ∼16 cm−1 higher than that of free SCN−.11 In the present Zn-based electrolytes, we similarly expect that some SCN− may form CIPs with Zn2+ (or possibly ion aggregates), while other SCN− remains more solvent-separated. However, the linear spectra alone do not fully resolve multiple peaks; the dominant feature remains a single broad line. The lack of large shifts suggests that neither population dominates overwhelmingly and that the overall polarity (solvent vs. ion content) is stable. In summary, the linear IR data serve mainly to identify the probe band and confirm that its baseline frequency is only weakly dependent on composition. Any subtle peak splitting or shoulder could hint at CIP formation, but the key finding is that FTIR alone cannot capture kinetic information of such states. This underscores the need for time-resolved measurements to uncover kinetic information.
N) peak red-shifts only marginally—from 2093.1 cm−1 in the 1:4 mixture to 2087.8 cm−1 in the 1:9 mixture—while the full width at half maximum (FWHM) contracts slightly, from 43.2 cm−1 to 40.7 cm−1 (Table S1, SI). These ≤ 5% changes confirm that the average solvation polarity sensed by SCN− remains nearly constant despite varying composition. In other words, replacing some acetamide by Zn(TFSI)2 does not dramatically alter the dielectric environment of the CN probe. We also note that the band remains broad (FWHM ≈ 41–43 cm−1) and slightly asymmetric, consistent with a distribution of environments. By analogy to Li–SCN systems, such line broadening often arises from a superposition of “free” SCN− ions and those bound in contact ion pairs (CIPs).11 In Li+–SCN− solutions, the CN stretch of a Li+–SCN− contact ion pair appears ∼16 cm−1 higher than that of free SCN−.11 In the present Zn-based electrolytes, we similarly expect that some SCN− may form CIPs with Zn2+ (or possibly ion aggregates), while other SCN− remains more solvent-separated. However, the linear spectra alone do not fully resolve multiple peaks; the dominant feature remains a single broad line. The lack of large shifts suggests that neither population dominates overwhelmingly and that the overall polarity (solvent vs. ion content) is stable. In summary, the linear IR data serve mainly to identify the probe band and confirm that its baseline frequency is only weakly dependent on composition. Any subtle peak splitting or shoulder could hint at CIP formation, but the key finding is that FTIR alone cannot capture kinetic information of such states. This underscores the need for time-resolved measurements to uncover kinetic information.
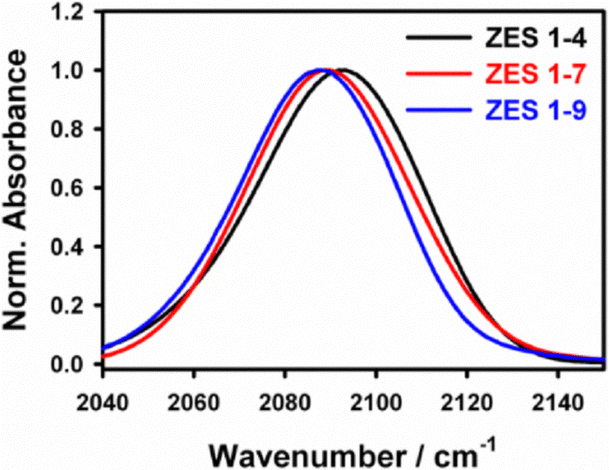 | ||
| Fig. 1 Linear FTIR spectra of the SCN− CN stretch in Zn(TFSI)2:acetamide electrolytes at different molar ratios. The band is broad and shifts only slightly with composition, indicating a nearly constant solvation polarity. | ||
Fig. 2 plots the population-relaxation traces of the SCN−ν(CN) band for the three ZES compositions. The bi-exponential fits yield average lifetimes T1 = 14.0 ± 0.5 ps (1:4), 11.2 ± 0.4 ps (1:7) and 9.2 ± 0.4 ps (1:9); the values decrease as the viscosity drops from 2.13 to 0.53 Pa s (Table S2, SI). The ∼1.5-fold change in T1 mirrors the four-fold change in fluidity, indicating that vibrational energy transfer is promoted in the more mobile, acetamide-rich liquid—most likely because the lower-frequency bath modes that accept energy from the CN stretch become more densely populated as the liquid loosens. Nevertheless, the decrease in vibrational lifetime trend is modest and contrasting compared with the three-fold increase observed in the slow spectral-diffusion time (τslow, vide infra), showing that T1 reflects only bulk dissipation pathways and provides limited insight into the structural reorganisation that governs spectral diffusion and, ultimately, ion transport.
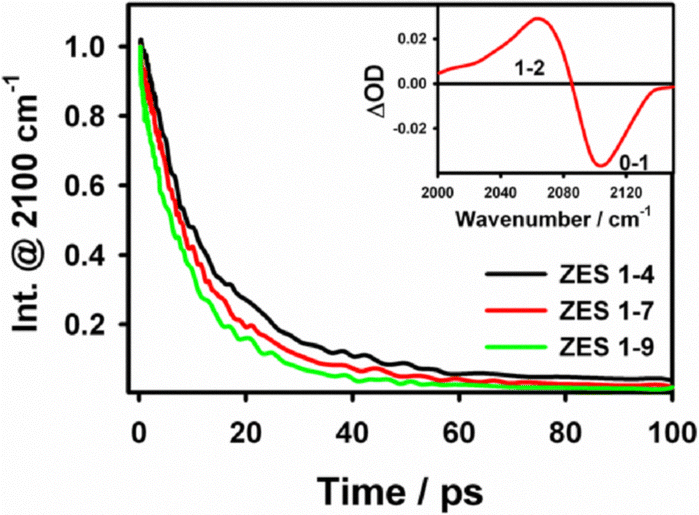 | ||
| Fig. 2 Normalized vibrational population decays at the bleach wavelength (2100 cm−1) of SCN− band in three electrolyte compositions (Zn:Ace = 1:4, 1:7, 1:9). The inset shows a pump–probe spectra displaying a ground-state bleach (0–1) and excited-state absorption (1–2) band. | ||
To access those collective solvent fluctuations, we turn to two-dimensional IR spectroscopy. By analysing the nodal-line-slope decay of the 2DIR spectra, we extract the frequency–frequency correlation function (FFCF) and resolve sub- and multi-picosecond components that are invisible to pump–probe measurements alone. Fig. 3A–D shows 2D IR spectra of the SCN− ν(CN) band in the 1:4 and 1:9 ZES electrolyte at waiting times Tw = 0.5 ps and 20 ps. At early time, the peak is highly elongated along the diagonal (ωpump = ωprobe), reflecting inhomogeneous broadening in which each oscillator “remembers’’ its initial frequency. By 20 ps, the contour is nearly round for 1:4 composition, indicating that solvent motions have randomized the frequencies—a hallmark of spectral diffusion. In contrast, for 1:9 composition, at Tw = 20 ps, the contours still maintain a significant elongation, suggesting a slower rate of spectral diffusion. The decay of this elongation is quantified by the nodal-line-slope method, which provides a direct measure of the frequency–frequency correlation function (FFCF).12
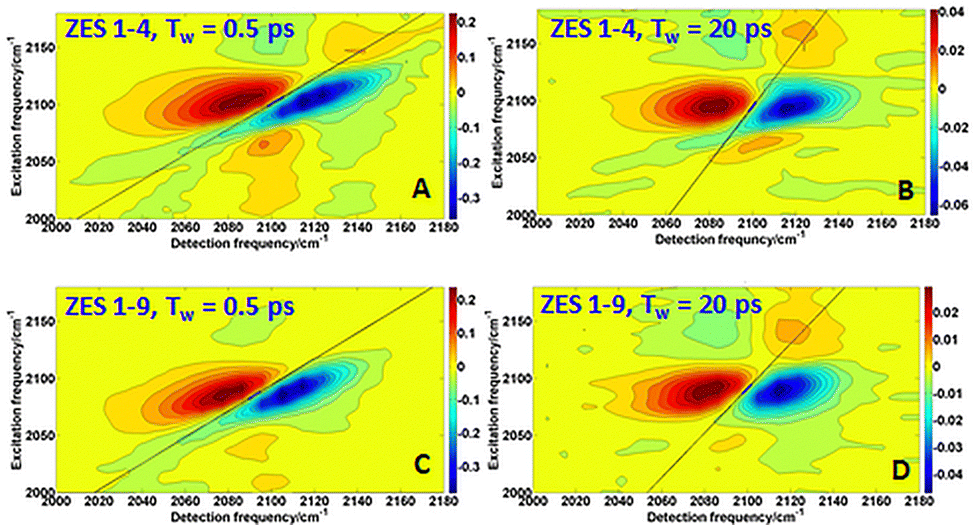 | ||
| Fig. 3 2DIR spectra of the SCN− CN stretch at waiting times Tw = 0.5 ps and Tw = 20 ps, for the Zn:acetamide 1:4 and 1:9 electrolyte. Contour lines (in red/blue for positive/negative absorptive signal) show that the peak elongation along the diagonal (ωpump = ωprobe) at early time relaxes to a more circular shape by 20 ps. | ||
Fig. 4 depicts the frequency–frequency correlation functions (FFCFs) extracted from nodal-line-slope analysis for the three ZES compositions. Each decay is accurately described by a bi-exponential expression, C(t) = Aslowexp(−t/τslow) + Afastexp(−t/τfast) and the resulting fit parameters are presented in Table 1. A clear compositional trend appears in the FFCF analysis (Table 1). In the Zn-rich 1:4 electrolyte the slow component accounts for ∼60% of the spectral variance with τslow = 7.4 ± 0.3 ps, while the remaining ∼40% relaxes in 0.38 ± 0.05 ps. Moving to the intermediate 1:7 mixture lengthens the slow decay to 15.0 ± 0.5 ps (amplitude = 0.63) and stretches the fast process to 0.78 ± 0.10 ps (amplitude = 0.37). The acetamide-rich 1:9 sample displays a slow component (20.25 ps) of amplitude 0.67 while the accompanying fast component is 0.92 ± 0.08 ps with amplitude of 0.33. Accordingly, the population-weighted average correlation time τavg rises from 4.7 ps to 13.9 ps across the series, even though the steady-state CN frequency shifts by <3 cm−1.
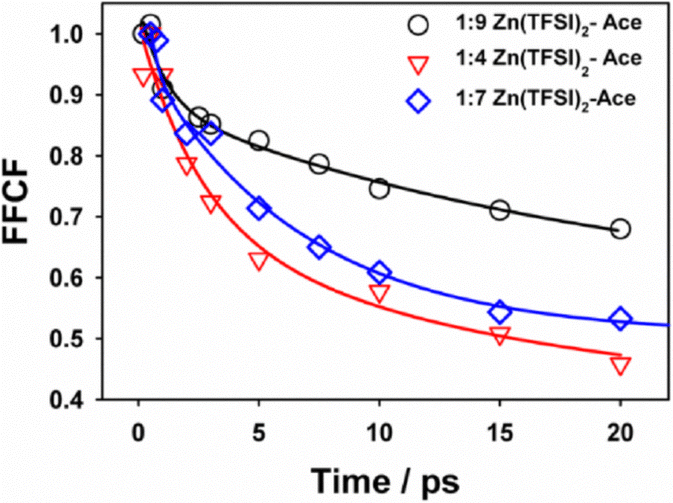 | ||
| Fig. 4 FFCF decay at different Zn-Acetamide composition. | ||
| Zn:Ace | τ slow/ps | A slow | τ fast/ps | A fast | τ avg/ps |
|---|---|---|---|---|---|
| 1–4 | 7.43 ± 0.3 | 0.61 | 0.38 ± 0.05 | 0.39 | 4.68 |
| 1–7 | 15.02 ± 0.5 | 0.63 | 0.78 ± 0.10 | 0.37 | 9.75 |
| 1–9 | 20.25 ± 1.02 | 0.67 | 0.92 ± 0.08 | 0.33 | 13.87 |
The presence of two well-separated timescales indicates two distinct solvation regimes for the SCN− probe. We attribute the sub-picosecond component to rapid frequency modulation within a tightly bound Zn2+–SCN− contact ion pair: even though the ion pair is strongly coordinated, librational motions of the surrounding ligands and partial ligand exchanges can scramble the electric field at the CN bond on this ultrafast timescale. The longer component is assigned to SCN− situated in a more weakly bound, outer-sphere environment; significant frequency changes in this case require complete solvent-shell reorganisation or binding events and therefore occur on the order of tens of picoseconds. Such frequency fluctuations specifically track changes in the local Zn2+ coordination configuration rather than bulk solvent reorganization, since the timescales of spectral diffusion correspond to the exchange dynamics of Zn2+ around the probe site.
The seeming paradox—that the “bound’’ species relaxes more rapidly than the “free’’ species—is resolved by recognising that the inner-sphere complex explores a restricted set of configurations that interchange quickly, whereas an outer-sphere SCN− must wait for a complete cage rearrangement before its frequency is perturbed. The systematic increase of both τslow and its fractional amplitude with rising acetamide content reveals a progressive shift from inner-sphere to outer-sphere solvation as the Zn2+ concentration decreases. Classical-molecular-dynamics simulations on the same ZES system10 indeed predict co-existing intimate and loose Zn2+ coordination motifs, lending independent support to this two-population picture. Thus, the 2DIR data show that the picosecond solvation dynamics slow substantially as the electrolyte becomes more acetamide-rich; the implications of this dynamical slowdown for macroscopic charge transport are examined in the next section. It is worth emphasising that the equilibrium solvation structure, judged by the FTIR band maximum and the modest (≈ 40%) drop in vibrational lifetime T1, changes only slightly, whereas the kinetic aspect of solvation varies by almost 200%. In other words, composition leaves static polarity nearly intact but dramatically tunes the reorganisation rate of the solvation shell. Such a decoupling of static and dynamic properties is invisible to conventional IR spectroscopy yet becomes obvious in 2DIR, underscoring the power of ultrafast methods for electrolyte design. Importantly, this compositional control over solvation timescales has not been reported previously by any other method in Zn-battery electrolytes. It reveals that simply tuning the salt:solvent ratio in a deep eutectic system can modulate ultrafast solvent reorganization.
Table 2 lists the slow FFCF time constant (τslow), ionic conductivity (σ), and shear viscosity (η) for the three electrolytes. An unexpected inverse correlation emerges: the acetamide-rich 1:9 mixture, which displays the longest solvation time (τslow ≈ 20 ps), also exhibits the highest conductivity (0.51 mS cm−1) and the lowest viscosity (0.53 Pa s).10 Conversely, the Zn-rich 1:4 liquid possesses a τslow of only 7.4 ps yet conducts Zn2+ three-times more slowly while being roughly four-times more viscous. Initially, this seems at odds with a Stokes–Einstein picture, in which faster local solvent fluctuations (short τ) would be expected to accompany enhanced ion mobility; instead, the data imply that rapid spectral diffusion does not guarantee efficient charge transport in these deep-eutectic solvents.
We interpret this decoupling in terms of an activated hopping mechanism. In the low-viscosity, acetamide-rich electrolyte, Zn2+ ions reside predominantly in outer-sphere complexes separated by extended solvent cages. Long intervals of relatively slow cage relaxation (τslow) are punctuated by infrequent but sizeable hops between coordination sites, yielding a high net conductivity despite slow local dynamics. In contrast, the viscous, Zn-rich liquid hosts a percolating network of contact ion pairs; here the solvation shell fluctuates quickly, yet each Zn2+ is topologically caged by neighbouring anions, so long-range motion is hindered. Such behaviour parallels ion transport in super-concentrated “water-in-salt” electrolytes and glassy ionic conductors, where hopping between discrete sites rather than continuous viscous flow dominates diffusion.13,14 The present DES system therefore illustrates that local solvation kinetics and global ion mobility can be anticorrelated when composition simultaneously alters viscosity and the topology of ionic domains.
Our conclusions resonate with earlier static-structure studies. Chang et al. showed that reducing free water in a hydrated glycol-DES reshapes the Zn2+ solvation shell and boosts Zn2+ transference, achieving 99.6% plating efficiency.15 Qiu et al. demonstrated that in the same acetamide DES studied here, anion-complexed Zn species foster an in-situ ZnF2-rich interphase and nearly 100% coulombic efficiency.4 Both reports underscore the leverage gained by tailoring the cation's coordination environment; our 2DIR results add the missing dynamical dimension, proving that composition not only fixes which ligands bind Zn2+ but also how fast the coordination cage reorganises—information essential for rational electrolyte design. Future temperature-dependent conductivity and molecular-dynamics studies should test the hopping hypothesis and relate activation energies to the τslow values reported here, thereby providing a basis for the hopping model proposed here.
In summary, we have shown that ultrafast two-dimensional IR spectroscopy can directly resolve Zn2+ solvation dynamics in battery electrolytes. In Zn(TFSI)2–acetamide deep-eutectic solvents, the SCN− probe reveals two dynamical regimes: a fast decay component assigned to contact ion pairs and a composition-dependent slow component. Raising the acetamide-to-Zn ratio from 4:1 to 9:1 nearly triples the slow correlation time—from ∼7 ps to ∼20 ps—while the static CN frequency shifts by <3 cm−1. Surprisingly, the electrolyte with the slowest solvation (1:9) displays the highest ionic conductivity and the lowest viscosity, inverting the expectation that rapid solvent fluctuations promote ion motion. This decoupling points to an activated, hopping-like migration of Zn2+, where long waiting periods between coordination hops are offset by low viscous drag in the acetamide-rich medium. These results establish a link between picosecond solvation kinetics and macroscopic charge transport. Electrolyte designers must therefore optimise not only which ligands bind Zn2+ but also how quickly those ligands exchange. Tuning deep-eutectic composition offers a practical handle to decouple fast conductivity from rapid local relaxation, potentially enabling electrolytes that combine wide electrochemical windows with high Zn plating efficiency. By bridging molecular dynamics with device-level performance, ultrafast vibrational probes provide a powerful roadmap for the rational engineering of Zn-ion and other multivalent electrolytes.
AKM conceived the idea, performed the experiments, analysed the data, made the figures. PKS conceived the idea, analysed the data, supervised the project, wrote the first and final draft.
The authors gratefully acknowledge the support of Department of Atomic Energy for generous funding.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental procedures for electrolyte preparation, FTIR and ultrafast 2D IR methods; details of the nodal-line-slope (NLS) analysis; full FFCF fitting parameters with uncertainties (Table S3); vibrational lifetime and viscosity data (Table S2); FTIR peak positions (Table S1); and raw 2DIR spectra for all Zn:acetamide compositions at different waiting times (Fig. S1). See DOI: https://doi.org/10.1039/d5cc04087f.Notes and references
- H.-Y. Shi, Y.-J. Ye, K. Liu, Y. Song and X. Sun, Angew. Chem., Int. Ed., 2018, 57, 16359–16363 CrossRef CAS PubMed.
- S. Li, Y. Liu, X. Zhao, Q. Shen, W. Zhao, Q. Tan, N. Zhang, P. Li, L. Jiao and X. Qu, Adv. Mater., 2021, 33, 2007480 CrossRef CAS PubMed.
- Q. Zhang, J. Luan, L. Fu, S. Wu, Y. Tang, X. Ji and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 15841–15847 CrossRef CAS PubMed.
- H. Qiu, X. Du, J. Zhao, Y. Wang, J. Ju, Z. Chen, Z. Hu, D. Yan, X. Zhou and G. Cui, Nat. Commun., 2019, 10, 5374 CrossRef.
- J. Shi, T. Sun, J. Bao, S. Zheng, H. Du, L. Li, X. Yuan, T. Ma and Z. Tao, Adv. Funct. Mater., 2021, 31, 2102035 CrossRef CAS.
- Q. Zhang, K. Xia, Y. Ma, Y. Lu, L. Li, J. Liang, S. Chou and J. Chen, ACS Energy Lett., 2021, 6, 2704–2712 CrossRef CAS.
- S. Huang, F. Wan, S. Bi, J. Zhu, Z. Niu and J. Chen, Angew. Chem., Int. Ed., 2019, 58, 4313–4317 CrossRef CAS.
- Y. Hu, Z. Liu, L. Li, S. Guo, X. Xie, Z. Luo, G. Fang and S. Liang, Natl. Sci. Rev., 2023, 10, nwad220 CrossRef CAS PubMed.
- W. Chen, Y. Wang, F. Wang, Z. Zhang, W. Li, G. Fang and F. Wang, Adv. Mater., 2024, 36, 2411802 CrossRef CAS.
- R. Chen, C. Zhang, J. Li, Z. Du, F. Guo, W. Zhang, Y. Dai, W. Zong, X. Gao, J. Zhu, Y. Zhao, X. Wang and G. He, Energy Environ. Sci., 2023, 16, 2540–2549 RSC.
- K.-K. Lee, K.-H. Park, D. Kwon, J.-H. Choi, H. Son, S. Park and M. Cho, J. Chem. Phys., 2011, 134, 064506 CrossRef.
- K. Kwac and M. Cho, J. Chem. Phys., 2003, 119, 2256–2263 CrossRef CAS.
- S. D. Jones, J. Bamford, G. H. Fredrickson and R. A. Segalman, ACS Polym. Au, 2022, 2, 430–448 CrossRef CAS PubMed.
- Z. Yu, L. A. Curtiss, R. E. Winans, Y. Zhang, T. Li and L. Cheng, J. Phys. Chem. Lett., 2020, 11, 1276–1281 CrossRef CAS.
- C. Li, R. Kingsbury, A. S. Thind, A. Shyamsunder, T. T. Fister, R. F. Klie, K. A. Persson and L. F. Nazar, Nat. Commun., 2023, 14, 3067 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |