DOI:
10.1039/D5TA01130B
(Paper)
J. Mater. Chem. A, 2025,
13, 11475-11485
Coordination-driven safer and sustainable energetic materials†
Received
12th February 2025
, Accepted 17th March 2025
First published on 17th March 2025
Abstract
The convergence of performance optimization and environmental stewardship has positioned coordination-driven approaches at the forefront of energetic material innovation. Now, using 3,6-bis(3,5-dimethyl-1H-pyrazol-1-yl)-1,2,4,5-tetrazine and N,N′-(1,2,4,5-tetrazine-3,6-diyl)dinitramide as precursors (P1 and P4), we have synthesized various coordination-driven polymeric energetic frameworks with potential applications as energetic materials, pyrotechnics, and solid propellants. Unique temperature-controlled reactivity of N,N′-(1,2,4,5-tetrazine-3,6-diyl)dinitramide with bases such as ammonia and alkali metal hydroxides are now reported, which results in the synthesis of new materials in a straightforward manner. These frameworks exhibit high thermal stability, controllable sensitivity, and tunable energy densities, making them versatile candidates for modern energetic applications. Furthermore, coordination-driven syntheses allow for precise structural control, enabling the fine-tuning of properties to meet specific requirements. The environmentally conscious design of these materials emphasizes the reduction of toxic byproducts which align with global sustainability goals.
1 Introduction
To promote the sustainability of energetic materials, azole-based salts with performance-advantaged properties have emerged as a focal point of research, offering potential breakthroughs in efficiency and environmental compatibility.1–5 However, challenges remain in achieving precise control over structural properties, scaling up production processes, and ensuring cost-effectiveness, all of which are critical for the practical implementation of these advanced materials. Coordination-driven polymeric energetic frameworks (CPEs) offer a promising solution to address these challenges.6–10 By utilizing coordination chemistry principles, molecules or metal ions are strategically organized into well-defined, stable structures. Researchers are now exploring innovative approaches to fine-tune these arrangements, aiming to optimize their performance and expand their applications in sustainable technologies.11–15 This approach allows for precise control over the properties of the energetic materials, such as sensitivity, thermal stability, and energy output.21–25 Furthermore, coordination complexes can incorporate eco-friendly ligands and renewable materials, reducing the environmental footprint of energetic compounds. As research continues to explore the potential of CPEs, these materials are poised to redefine the field of energetics.16–20
Recent advances in the design of potassium-azole salts have demonstrated the potential to bridge the gap between high-performance and sustainable development. These materials not only exhibit enhanced thermal stability and controlled sensitivity but also enable the integration of green chemistry principles, such as the use of biodegradable ligands and light metals. To date, several representative potassium derivatives with environmentally friendly characteristics, such as K2DNABT, DTAT-K, K2DNAT, and K2DNAAzT, offer a promising platform for primary initiators (Fig. 1A).21–23 Notably, the four-nitrogen-containing tetrazole backbone has gained prominence in energetics, pyrotechnics, and propellants due to the high heats of formation and ease of syntheses (Fig. 1B).
 |
| | Fig. 1 (A) Azole-based salts with performance-advantaged properties. (B) Application of metal-azole salts. | |
Similar to the tetrazole ring, the tetrazine ring, distinguished by its high nitrogen content and unique reactivity, serves as an environmentally friendly, and versatile ligand for the design of advanced energetic salts.24–30 The ability of the ring nitrogens to form coordination bonds with various metal ions makes it an ideal candidate for creating new coordination materials. By designing tetrazine-based CPEs, researchers can enhance the energy density, thermal stability, and sensitivity of energetic salts while simultaneously reducing their environmental impact. Moreover, the ease of synthesis of tetrazine enables the development of less hazardous energetic materials, aligning with the broader goals of sustainability in the field of energetics. This dual functionality of the tetrazine ring as both a performance-enhancing and eco-conscious component highlights its critical role in the next generation of energetic materials. In this study, we have synthesized new CPEs using easily accessible precursors, 3,6-bis(3,5-dimethyl-1H-pyrazol-1-yl)-1,2,4,5-tetrazine (P1) and N,N′-(1,2,4,5-tetrazine-3,6-diyl)dinitramide (P4). Precursor P4 exhibits distinct reactivity with bases such as ammonia and metal hydroxides. At room temperature, it reacts to form typical salts. However, at elevated temperatures, one of the nitramine groups acts as a leaving group, resulting in the formation of two asymmetrically substituted anions (Fig. 2A).
 |
| | Fig. 2 (A) Reactions of N,N′-(1,2,4,5-tetrazine-3,6-diyl)dinitramide. (B) Effect of salt formation and various interactions on thermal stability. | |
2 Results and discussion
2.1 Synthesis
Precursors P1, P2 and P3 were prepared following literature procedures.31 The small-scale synthesis of P3 from P2 was observed to proceed with relative ease. Reaction of diamino tetrazine (P3) with freshly distilled nitric acid (red fuming) results in the formation of N,N′-(1,2,4,5-tetrazine-3,6-diyl)dinitramide (P4) as a yellow solid (Scheme 1). Precursor P4 upon reaction with metal carbonates in ethanol (95%) at room temperature resulted in formation of CPEs 1–5 in quantitative yields.
 |
| | Scheme 1 Reactions of 3,6-bis(3,5-dimethyl-1H-pyrazol-1-yl)-1,2,4,5-tetrazine (P1) and temperature-based reactivity of N,N′-(1,2,4,5-tetrazine-3,6-diyl)dinitramide (P4). | |
Interestingly, the reaction of P4 with aqueous ammonia in ethanol at 60 °C yields diammonium (3,5-dinitropyrazine-2,6-diyl)bis(nitroamide) (P5) through substitution with an amine, accompanied by the formation of the ammonium salt (Scheme 1). CPEs 6–10 were prepared by neutralizing P5 with 10% H2SO4, followed by reaction with metal carbonates at room temperature. Then P4 was reacted with potassium and sodium carbonates at 60 °C, which produced potassium nitro(6-oxido-1,2,4,5-tetrazin-3-yl)amide (11) and sodium nitro(6-oxido-1,2,4,5-tetrazin-3-yl)amide (12), respectively. Compound 13 was obtained by the neutralization of 12 with 10% H2SO4. Both reactions serve as notable examples of a base substituting one nitramine while facilitating salt formation with another nitramine in the same molecule. Compound 14 was synthesized from precursors P1, P6, or P7. Reaction of P7 with two equivalents of hydrazine resulted in the formation of compound 14.
2.2 Characterization
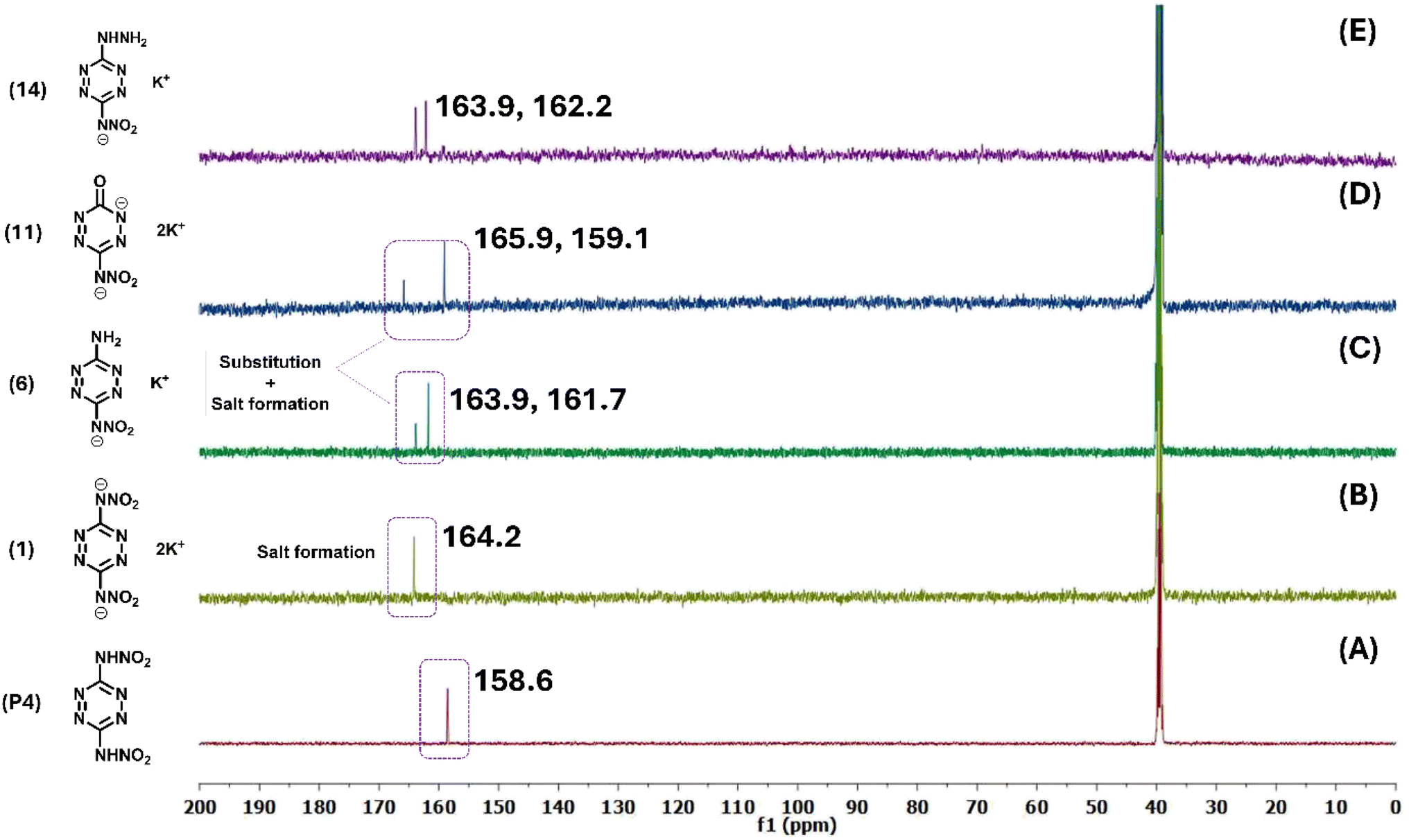
2.2.1 NMR spectroscopy.
All newly synthesized compounds were analyzed and characterized through 1H NMR, 13C NMR, 14N NMR, and 15N NMR spectroscopy. In the 13C NMR spectrum of P4, a carbon signal was observed at 158.6 ppm (Fig. 3A). For CPEs 1–5, this peak shifted downfield to fall in a range of 163.9–164.2 ppm (Fig. 3B). After substitution with ammonia and hydroxide, the carbon signal split into two peaks (Fig. 3C and D). In CPEs 6–10, the carbon signal (C1) attached to the nitramine group appeared in the range of 161.7–161.8 ppm, while the carbon signal (C2) attached to the amine group appeared in the range of 163.4–163.7 ppm. Similarly, in CPEs 11–12, the nitraamine-attached carbon signal (C1) was observed at 159.1 ppm, and the oxygen-attached carbon signal (C2) was found at 166.1 ppm. In the 14N NMR spectrum of P4, the nitro group resonance appeared at −42.3 ppm. In CPEs 1–5, this signal shifted to a range of −11.8 to −12.3 ppm, which is a clear indication of the salt formation.
 |
| | Fig. 3
13C NMR spectra of (A) P4. (B) 1. (C) 6. (D) 11. (E) 14. | |
2.2.2 Crystal structure.
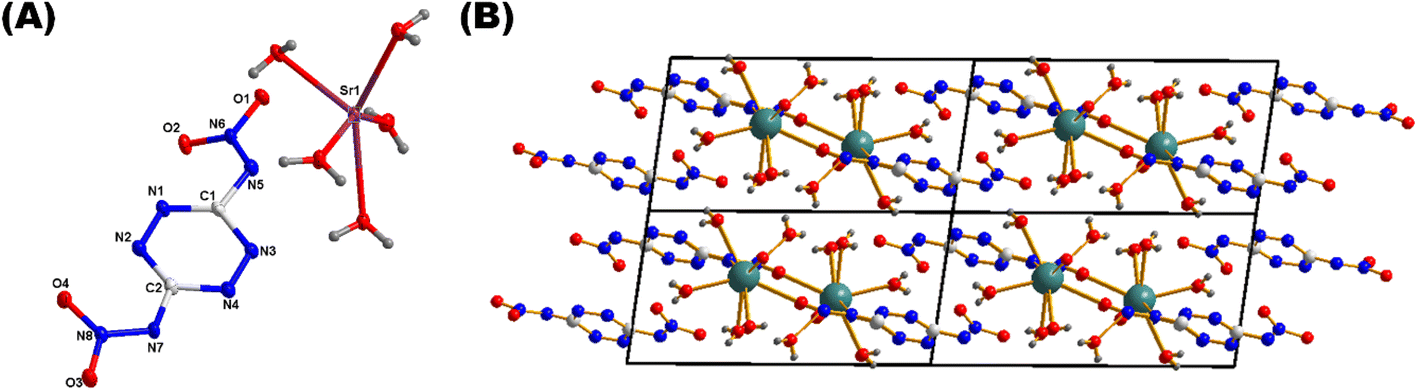
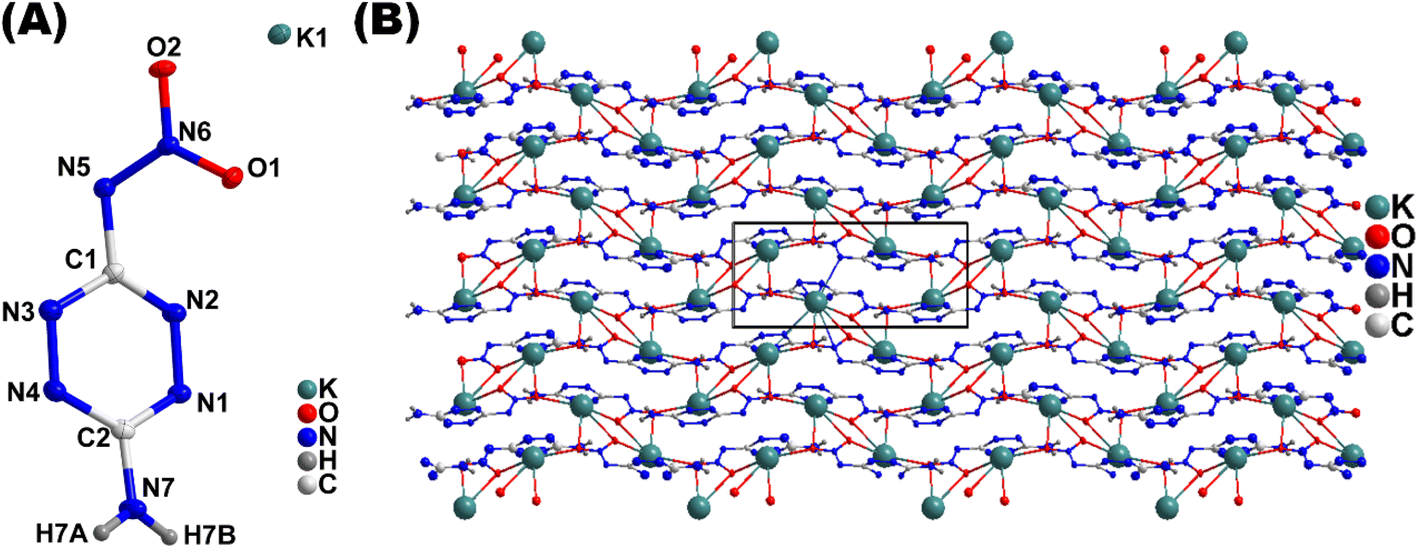
Compound 1·H2O adopts a 3D MOF structure and crystallizes in orthorhombic space group Pnna (Fig. 4a). Each unit cell contains eight chemical formula units. The calculated density for compound 1·H2O is 2.143 g cm−3 at 100 K. In the crystal structure, the potassium atoms are coordinated to O-atoms of the nitraamine groups along with the nitrogen atoms of the tetrazine ring (Fig. 4b). As a result, extended coordination in three-dimensional space is observed, which increases the density and thermal stability of the material (Fig. 4c). The coordination bond lengths between O-atoms (O1, O2, and O1W) and the K+ ions (K1 and K2) range from 2.698 Å to 3.104 Å. The nitrogen atoms of the tetrazine ring and the nitramine group are coordinated to the potassium atoms with bond distances ranging between 2.861 Å and 3.369 Å. The various coordination modes of the dianion and potassium atoms are shown in Fig. 4C and D. Compound 4·4H2O crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Each unit cell contains two chemical formula units (Fig. 5A). The calculated density for compound 4·4H2O is 2.191 g cm−3 at 100 K. The Sr2+ ion is coordinated to four water molecules and O and N atoms of nitramine groups, along with the nitrogen atoms of the tetrazine ring (Fig. 5B). Packing diagram of the compound 4·4H2O shows polymeric nature through extended coordination in three-dimensional space (Fig. 5B). The coordination bond between an O-atom and the Sr2+ ranges from 2.558 Å to 2.921 Å. Compound 6 crystallizes in monoclinic space group P21/c (Fig. 6A). Each unit cell contains four chemical formula units. The calculated density for compound 6 is 1.990 g cm−3 at 100 K. In the crystal packing of 6, the potassium ion (K1) is coordinated to O-atoms of the nitramine groups along with the nitrogen atoms of the tetrazine ring (Fig. 6B). Similarly to compound 1, compound 6 also shows extended coordination in three-dimensional space, which increases the overall density of the material (Fig. 6C).
. Each unit cell contains two chemical formula units (Fig. 5A). The calculated density for compound 4·4H2O is 2.191 g cm−3 at 100 K. The Sr2+ ion is coordinated to four water molecules and O and N atoms of nitramine groups, along with the nitrogen atoms of the tetrazine ring (Fig. 5B). Packing diagram of the compound 4·4H2O shows polymeric nature through extended coordination in three-dimensional space (Fig. 5B). The coordination bond between an O-atom and the Sr2+ ranges from 2.558 Å to 2.921 Å. Compound 6 crystallizes in monoclinic space group P21/c (Fig. 6A). Each unit cell contains four chemical formula units. The calculated density for compound 6 is 1.990 g cm−3 at 100 K. In the crystal packing of 6, the potassium ion (K1) is coordinated to O-atoms of the nitramine groups along with the nitrogen atoms of the tetrazine ring (Fig. 6B). Similarly to compound 1, compound 6 also shows extended coordination in three-dimensional space, which increases the overall density of the material (Fig. 6C).
 |
| | Fig. 4 (A) Labeling scheme for 1·H2O (thermal ellipsoid at 50%). (B) Crystal packing of 1. (C and D) Coordination modes of K+. | |
 |
| | Fig. 5 (A) Labeling scheme for 4·4H2O (thermal ellipsoid at 50%). (B) Crystal packing of 4. | |
 |
| | Fig. 6 (A) Labeling scheme for 6 (thermal ellipsoid at 50%). (B) Crystal packing of 6. | |
The coordination bond between an O-atoms (O1, O2) and K1 ranges from 2.652 Å to 2.858 Å. The nitrogen atoms of the tetrazine ring (N1–N4) and the nitramine group (N5, N6) are coordinated to the potassium atoms with bond distances ranging between 2.934 Å to 3.287 Å. Compound 9·3H2O crystallizes in the monoclinic space group P2/c (Fig. 7A). Each unit cell contains two chemical formula units. The calculated density for compound 9·3H2O is 1.993 g cm−3 at 100 K. In the crystal packing of 9·3H2O, the Sr2+ atoms are coordinated to O and N atoms of the nitramine groups and two water molecules (Fig. 7B).
 |
| | Fig. 7 (A) Labeling scheme for 9·3H2O (thermal ellipsoid at 50%). (B and C) Crystal packing of 9. | |
As a result, compound 9·3H2O shows extended coordination in one dimensional space (Fig. 7C). The coordination bond between an O-atom and the Sr atom ranges from 2.458 Å to 2.666 Å. The nitrogen atoms (N5 and N6) of the nitramine group are coordinated to the Sr atoms with bond distances ranging between 2.847 Å to 3.201 Å. Compound 13 crystallizes in orthorhombic space group Pbca (Fig. 8A). Each unit cell contains eight chemical formula units. The calculated density for compound 13 is 1.976 g cm−3 at 100 K. The unusual high density of compound 13 is attributed to the presence of strong hydrogen bond networks with distances ranging from 1.81 Å to 2.26 Å (Fig. 8B). The crystal packing shows the presence of wave-like packing and π-stacking interactions.
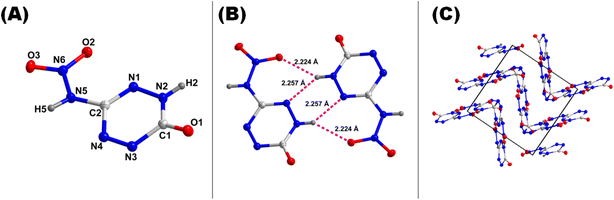 |
| | Fig. 8 (A) Labeling scheme for 13 (thermal ellipsoid at 50%). (B) H-bond interactions. (C) Crystal packing of 13. | |
2.3 Physicochemical properties
2.3.1 Thermal stability.
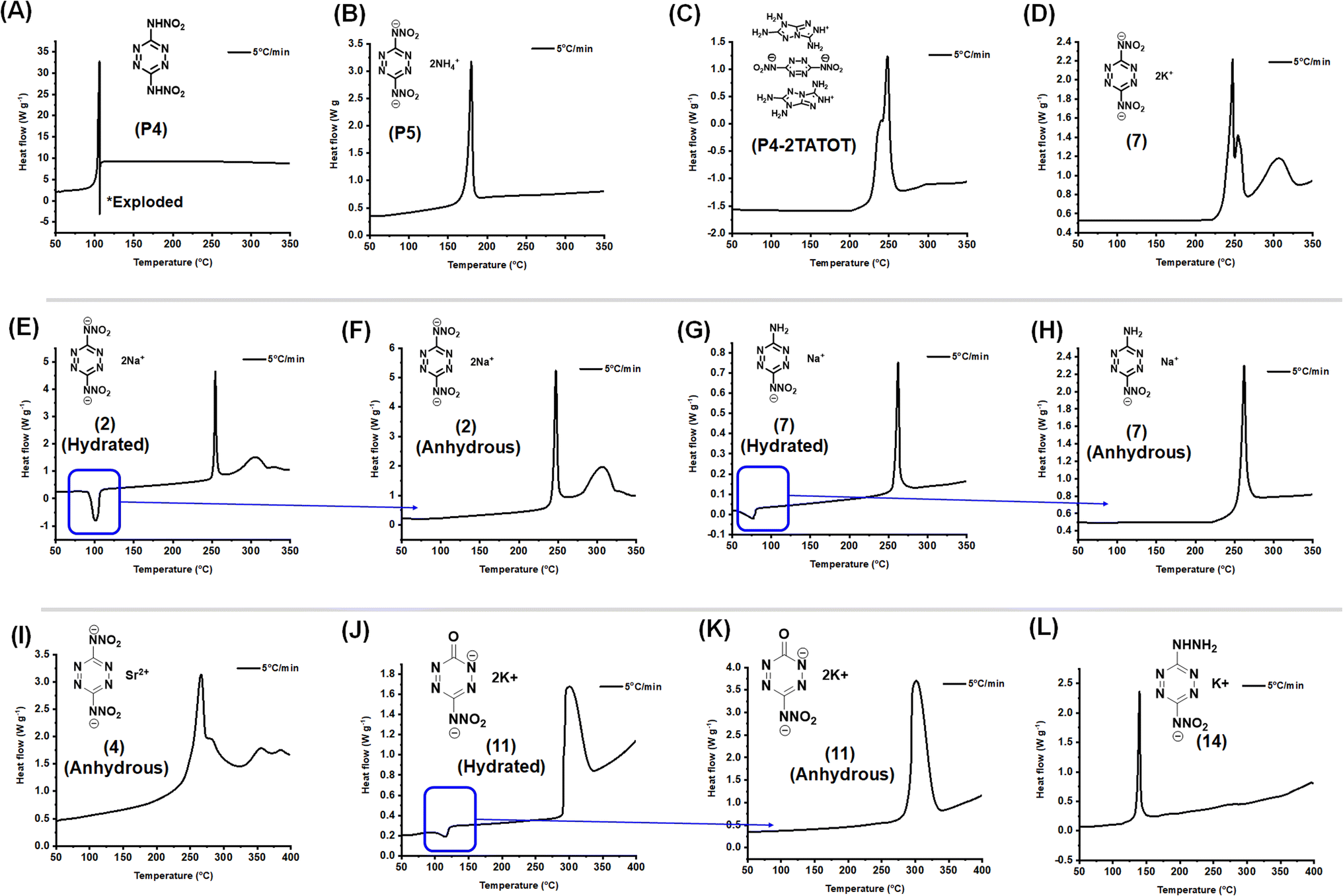
Energetic materials are fundamentally unstable due to their substantial energy load, making them vulnerable to ignition or detonation when subjected to external heat. The ability of these materials to withstand thermal stress is a key factor in evaluating their safety, as it indicates how resistant they are to unintentional combustion or explosion when exposed to heat. The thermal behavior of the new compounds was investigated by differential scanning calorimetry (DSC). In Fig. 9A–D, the comparison of the thermal stabilities of P4 and its energetic salts are given. The dipotassium derivative exhibits highest decomposition owing to the presence of strong coordination packing and rigid structure. The DSC scans show that thermal decomposition initiation temperatures (onset) of potassium derivatives 1, 6, 11 and 14 are 242, 229, 293 and 136 °C, respectively, followed by sharp exothermic peaks at 247, 235, 301 and 140 °C, respectively, indicating that these compounds undergo rapid decomposition upon heating. Notably, all compounds exhibit higher decomposition temperatures than their neutral precursors, highlighting the importance of coordination networks. The water of crystallization or hydration for sodium derivatives was removed by drying the compounds in an oven (120 °C). The onset decomposition temperatures of the sodium derivatives 2, 7 and 12 are 244, 257, and 279 °C, respectively, followed by sharp exothermic peaks at 247, 263, and 305 °C, respectively.
 |
| | Fig. 9 Differential scanning calorimetry (DSC) analysis (A) P4. (B) P5. (C) P4-2TATOT. (D) 7. (E) 2-Hydrated. (F) 2-Anhydrous. (G) 7-Hydrated. (H) 7-Anhydrous. (I) 4-Anhydrous. (J) 11-Hydrated. (K) 11-Anhydrous. (L) 14. | |
2.3.2 Impact and friction sensitivities.
The sensitivities to impact and friction for all new compounds were measured using standard BAM methods. Compounds 1–5, 13, and 14, which are denser, exhibit increased sensitivity to impact and friction. Compounds 6–12 are less sensitive toward impact and friction. The sensitivities of compounds 1·H2O, 6 and 13 are further rationalized from both crystal structure and at the molecular level. At the molecular level, sensitivities of materials toward impact are very closely related to their electrostatic potentials (ESPs). Significant separation of positive and negative charges in different regions of the molecule can indicate instability. Conversely, a well-distributed charge across the molecule suggests lower sensitivity. The electrostatic potentials (ESP) of compounds 1·H2O, 6 and 13 were calculated based on the B3LYP/6-311G(d,p) method with optimized structures.32,33 ESP minima and maxima for compounds 1·H2O, 6 and 13 are −84, −66, −32 kcal mol−1 and +106, +105, +61 kcal mol−1, respectively (Fig. 10). In compound 1·H2O, ESP maps reveal charge separation in different parts of the molecule, with positive regions (red) around potassium atoms and negative regions (blue) on the anion.
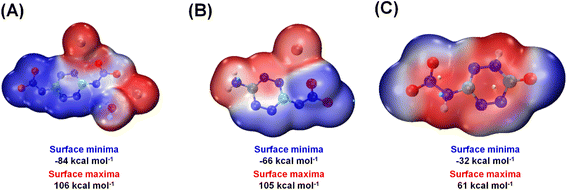 |
| | Fig. 10 Electrostatic potential (ESP) surfaces of compounds (A) 1·H2O, (B) 6, and (C) 13. | |
2.3.3 Detonation properties.
The densities of compounds 1–14 which range between 1.81 g cm−3 to 2.55 g cm−3 were measured using a gas pycnometer at ambient temperature under a helium (He) atmosphere (Table 1). The enthalpies of formation of the new compounds were calculated using an isodesmic method with the Gaussian 03 suite of programs34 giving values for compounds 1–14 in the range from −230.3 to 2281 kJ mol−1 (Table 1). With experimental densities and calculated enthalpies of formation, detonation properties of compounds 1–14 were calculated using EXPLO5 (v7.01.01). The values of detonation velocities and detonation pressures are given in Table 1 and fall between 6945–9214 m s−1 and 18.8–44.8 GPa, respectively, with 13 showing the highest detonation velocity and pressure. The detonation properties of compounds 2, 4, 7, 8 and 13 are comparable to the benchmark explosive RDX.35–38
Table 1 Physicochemical properties of compounds 1–14
|
|
T
d
(°C) |
ISb (J) |
FSc (N) |
ρ
(g cm−3) |
ΔHfe kJ mol−1 |
D
v
(m s−1) |
P
(GPa) |
|
Temperature (onset at 5 °C min−1) of decomposition.
Sensitivity to impact (IS).
Sensitivity to friction (FS).
Density at 25 °C using gas pycnometer.
Molar enthalpy of formation calculated using isodesmic reactions with the Gaussian 03 suite of programs (revision D.01).
Velocity.
Pressure (calculated using EXPLO5 version 7.01.01).
h
50 = 24 cm/2.5 kg hammer (ref. 39)
|
|
1
|
242 |
3 |
40 |
2.06 |
161.8 |
7703 |
25.4 |
|
2
|
253 |
4 |
40 |
2.01 |
275.1 |
8833 |
28.8 |
|
3
|
223 |
5 |
80 |
1.85 |
422.7 |
8878 |
33.6 |
|
4
|
251 |
12 |
80 |
2.22 |
2281.0 |
9017 |
44.8 |
|
5
|
251 |
12 |
80 |
2.53 |
1888.0 |
8496 |
43.3 |
|
6
|
229 |
20 |
240 |
1.86 |
372.3 |
7644 |
23.5 |
|
7
|
257 |
20 |
240 |
1.98 |
434.9 |
9114 |
33.7 |
|
8
|
272 |
20 |
240 |
1.81 |
524.2 |
8947 |
32.2 |
|
9
|
232 |
>20 |
360 |
2.19 |
1395.2 |
8145 |
33.6 |
|
10
|
212 |
>20 |
360 |
2.55 |
985.6 |
8636 |
40.8 |
|
11
|
293 |
5 |
240 |
2.14 |
−230.3 |
6945 |
18.8 |
|
12
|
279 |
>20 |
240 |
1.98 |
−104.0 |
7387 |
18.8 |
|
13
|
98 |
1 |
5 |
1.92 |
255.3 |
9214 |
36.8 |
|
14
|
136 |
4 |
40 |
1.83 |
472.7 |
7900 |
25.0 |
| RDX |
204 |
7.5/5.6h |
120 |
1.80 |
92.6 |
8795 |
34.9 |
2.3.4 Primary initiators.
Potassium-based primary initiators are highly sensitive compounds that utilize potassium as a key component in their chemical structure. While lead azide (LA) has traditionally been the most widely used primary initiator due to its excellent priming ability, concerns over serious heavy-metal pollution have driven the need for greener alternatives. This shift has led to increased interest in potassium-based initiators, which offer similar sensitivity and effectiveness without the environmental risks associated with heavy metals. Compounds 1 and 2 are more thermally stable than some representative potassium-based primary initiators and can be obtained as anhydrous materials as confirmed by CHN and DSC analyses.
Based upon peak decomposition temperatures measured by differential scanning calorimetry (DSC) at different heating rates of 5, 10, 15, and 20 °C min−1, the activation energies (Ek and E0), pre-exponential constant A, and linear correlation coefficients (Rk and R0) were calculated using the methods of Kissinger40 and Ozawa41 (See ESI†). The results show that the activation energy calculated by either method is essentially the same for compound 1 (Ek = 196.11 kJ mol−1; E0 = 194.82 kJ mol−1). Compound 1 exhibits a low activation energy, comparable to that of certain reported primary initiators.42 Similarly, the activation energy for compound 2 was calculated (Ek = 168.97 kJ mol−1; E0 = 168.97 kJ mol−1). For both compounds, impact and friction sensitivities, measured using the standard BAM fall hammer and BAM friction tester, respectively, further indicate high sensitivity, suggesting that they can be readily triggered to undergo intense chemical reactions.
Experiments such as the “red-hot needle” and “heated plate” tests were carried out to determine the energetic behavior of compounds 1 and 2. Generally, achieving detonation in any of the tests is essential, indicating a rapid deflagration to detonation transition (DDT), which is an important characteristic of a primary initiators. For both compounds, detonation was observed in the red-hot needle test (Fig. 11A). In the heated plate test of compounds 1 and 2, two frames within the 10 millisecond differences have been captured, where intense deflagration (Fig. 11B) to detonation was observed which indicates that both of these materials behave as primary initiators.
 |
| | Fig. 11 (A) Hot needle test of 1 and 2. (B) Hot plate test of 1 and 2. | |
2.3.5 Pyrotechnics.
In combustion tests, the coordination-driven polymeric energetic frameworks (CPEs) were evaluated for their pyrotechnic performance. The materials exhibited rapid ignition and sustained combustion, producing intense heat and bright luminescence, indicative of their high energy release and efficient oxidation processes (Fig. 12). Notably, the combustion tests demonstrated that these CPEs have controlled burn rates and produce minimal smoke, aligning with environmentally friendly pyrotechnic standards. These results highlight the potential of the new frameworks as safer and more sustainable alternatives for traditional pyrotechnic compositions, combining superior performance with reduced environmental impact.
 |
| | Fig. 12 (A) 1. (B) 2. (C) 3. (D) 4. (E) 5. (F) 6. (G) 7. (H) 8. (I) 9. (J) 10. (K) 11. (L) 12. (M) 14. | |
2.3.6 Solid propellants.
To evaluate the applicability of 13 and 14 as solid propellants, their performance was evaluated as monopropellants (neat) and composite propellants. The specific impulse (Isp) values were calculated using EXPLO5v7.01.01 software at chamber pressure 7 MPa, expansion pressure ratio (Pc/Pe) of 70, initial T of 3500 K, ambient pressure of 0.1 MPa at equilibrium, and expansion through the nozzle. The Isp values of 13 and 14 as monopropellants (neat) are 266 s and 248 s, respectively, which is higher than AP (ammonium perchlorate) (157 s) and ADN (ammonium dinitramide) (206 s) (Table 2). Additionally, the Isp values of four composite propellant formulations using 13, 14 and RDX (i) 88% of compound and 12% Al, (ii) 78% of compound, 12% Al, and 10% HTPB. (iii) 80% compound, 20% AP (iv) 42% compound, 20% AP, 20% HTPB and 18% Al, were calculated and the results are given in Table 2. The specific impulse values (Isp) of 13 and 14 are comparable to the benchmark explosive RDX.35–38
Table 2 Specific impulse (propulsive properties) for different high energy composite propellant formulations
|
|
I
sp [s]a,x |
I
sp [s]b,x |
I
sp [s]c,x |
I
sp [s]d,x |
I
sp [s]e,x |
|
I
sp – specific impulse of neat compound (monopropellant).
I
sp – specific impulse at 88% compound and 12% Al as fuel additive.
I
sp – specific impulse at 78% compound, 12% Al, and 10% HTPB as a binder.
I
sp – specific impulse at 80% compound, 20% AP.
I
sp – specific impulse at 42% compound, 20% AP, 20% HTPB and 18% Al. xSpecific impulse calculated at an isobaric pressure of 70 bar and initial temperature of 3300 K using EXPLO5 V7.01.
|
|
13
|
266 |
270 |
252 |
266 |
235 |
|
14
|
248 |
246 |
233 |
259 |
233 |
| RDX |
267 |
278 |
259 |
268 |
242 |
3 Conclusion
In this study, we have demonstrated the potential of coordination-driven polymeric energetic frameworks (CPEs) synthesized from 3,6-bis(3,5-dimethyl-1H-pyrazol-1-yl)-1,2,4,5-tetrazine and N,N′-(1,2,4,5-tetrazine-3,6-diyl)dinitramide (P4) as precursors. These frameworks exhibit enhanced thermal stability, tunable sensitivity, and high energy densities, making them promising candidates for various energetic applications, including primary initiators, pyrotechnics and solid propellants. The unique reactivity of precursor P4 with bases underscores the versatility and ease of synthesis of these materials. Our findings highlight the advantages of using coordination-driven approaches to achieve precise structural control, enabling the fine-tuning of material properties to meet specific application requirements. Importantly, the incorporation of eco-friendly ligands and the reduction of toxic byproducts align these materials with global sustainability goals.
Data availability
All data relevant to the work described here are available in the ESI† or from the CCDC.
Conflicts of interest
The authors declare that they have no competing financial interests or personal relationships that could have influenced the work reported in this paper.
Acknowledgements
The diffractometer (Rigaku Synergy S) for SC-XRD was purchased with support from the National Science Foundation (MRI program) under grant no. 1919565. We are grateful to the Fluorine-19 fund for support.
References
- N. Fischer, D. Fischer, T. M. Klapötke, D. G. Piercey and J. Stierstorfer, J. Mater. Chem., 2012, 22, 20418–20422 RSC.
- H. Gao and J. M. Shreeve, Chem. Rev., 2011, 111, 7377–7436 CrossRef CAS PubMed.
- A. J. Bennett, L. M. Foroughi and A. J. Matzger, J. Am. Chem. Soc., 2024, 146, 1771–1775 CrossRef CAS PubMed.
- X. Yu, J. Tang, C. Lei, C. Xue, H. Yang, C. Xiao and G. Cheng, J. Mater. Chem. A, 2024, 12, 29638–29644 RSC.
- P. Bhatia, P. Das and D. Kumar, ACS Appl. Mater. Interfaces, 2024, 16, 64846–64857 CrossRef CAS PubMed.
- M. Jujam, R. Rajak and S. Dharavath, Adv. Funct. Mater., 2025, 35, 2412638 CrossRef CAS.
- J. Singh, A. K. Chinnam, R. J. Staples and J. M. Shreeve, Inorg. Chem., 2022, 61, 16493–16500 CrossRef CAS PubMed.
- S. Li, Y. Wang, C. Qi, X. Zhao, J. Zhang, S. Zhang, S. Pang, S. Li, Y. Wang, C. Qi, X. Zhao, J. Zhang, S. Pang and S. Zhang, Angew. Chem., Int. Ed., 2013, 52, 14031–14035 CrossRef CAS PubMed.
- D. Fischer, T. M. Klapötke and J. Stierstorfer, Angew. Chem., Int. Ed, 2014, 53, 8172–8175 CrossRef CAS PubMed.
- J. Zhang, Y. Du, K. Dong, H. Su, S. Zhang, S. Li and S. Pang, Chem. Mater., 2016, 28, 1472–1480 CrossRef CAS.
- K. Pandey, A. Tiwari, J. Singh, P. Bhatia, P. Das, D. Kumar and J. M. Shreeve, Org. Lett., 2024, 26, 1952–1958 CrossRef CAS PubMed.
- L. Liang, Y. Zhong, J. Chen, J. Zhang, T. Zhang and Z. Li, Inorg. Chem., 2022, 61, 14864–14870 CrossRef CAS PubMed.
- S. Zhang, Q. Yang, X. Liu, X. Qu, Q. Wei, G. Xie, S. Chen and S. Gao, Coord. Chem. Rev., 2016, 307, 292–312 CrossRef CAS.
- R. Rajak, N. Kumar, V. D. Ghule and S. Dharavath, ACS Appl. Mater. Interfaces, 2024, 16, 20670–20680 CAS.
- Y. Liu, P. Yi, L. Gong, X. Yi, P. He, T. Wang and J. Zhang, Inorg. Chem., 2023, 62, 3186–3194 CrossRef CAS PubMed.
- Q. Sun, X. Li, Q. Lin and M. Lu, J. Mater. Chem. A, 2019, 7, 4611–4618 RSC.
- M. Benz, M. S. Gruhne, T. M. Klapötke, N. Krüger, T. Lenz, M. Lommel and J. Stierstorfer, Eur. J. Org. Chem., 2021, 2021, 4388–4392 CAS.
- J. Singh, R. J. Staples and J. M. Shreeve, ACS Appl. Mater. Interfaces, 2021, 13, 61357–61364 CrossRef CAS PubMed.
- W. Hu, J. Tang, X. Ju, Z. Yi, H. Yang, C. Xiao and G. Cheng, ACS Cent. Sci., 2023, 9, 742–747 CrossRef CAS PubMed.
- C. Li, S. Wang, S. Li, H. Yin, Q. Ma and F. X. Chen, ACS Appl. Mater. Interfaces, 2024, 16, 35232–35244 CAS.
- D. Fischer, T. M. Klapötke and J. Stierstorfer, Angew. Chem., Int. Ed., 2014, 53, 8172–8175 CAS.
- Y. Feng, J. Zhang, W. Cao, J. Zhang and J. M. Shreeve, Nat. Commun., 2023, 141, 1–8 Search PubMed.
- D. Kumar and A. J. Elias, Resonance, 2019, 24, 1253–1271 Search PubMed.
- D. E. Chavez, M. A. Hiskey and R. D. Gilardi, Org. Lett., 2004, 6, 2889–2891 CAS.
- M. H. V. Huynh, M. A. Hiskey, D. E. Chavez, D. L. Naud and R. D. Gilardi, J. Am. Chem. Soc., 2005, 127, 12537–12543 CAS.
- D. E. Chavez and M. A. Hiskey, J. Heterocycl. Chem., 1998, 35, 1329–1332 CAS.
- X. Zhang, Y. Wang, Y. Liu, Q. Zhang, L. Hu, C. He and S. Pang, ACS Appl. Mater. Interfaces, 2022, 14, 37975–37981 CAS.
- Y. Liu, G. Zhao, Y. Tang, J. Zhang, L. Hu, G. H. Imler, D. A. Parrish and J. M. Shreeve, J. Mater. Chem. A, 2019, 7, 7875–7884 RSC.
- D. M. Bystrov, A. N. Pivkina and L. L. Fershtat, Molecules, 2022, 27, 5891 CrossRef CAS PubMed.
- M. D. Coburn, G. A. Buntain, B. W. Harris, M. A. Hiskey, K. -Y Lee and D. G. Ott, J. Heterocycl. Chem., 1991, 28, 2049–2050 CrossRef CAS.
- M. D. Coburn, G. A. Buntain, B. W. Harris, M. A. Hiskey, K. -Y Lee and D. G. Ott, J. Heterocycl. Chem., 1991, 28, 2049 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. L. Caricato, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Revision D.01, Gaussian, Inc., Wallingford, CT, 2003 Search PubMed.
-
R. Mayer, J. Köhler and A. Homburg, Explosives, Wiley-VCH, 2007 Search PubMed.
- M. Tang, X. Wang, X. Xu, Z. Zeng, C. Chen, Y. Liu, W. Huang and Y. Tang, J. Mater. Chem. A, 2024, 12, 13081–13085 RSC.
-
T. M. Klapötke, Chemistry of High-Energy Materials, De Gruyter, 6th edn, 2022 Search PubMed.
-
D. Luca, Chemical Rocket Propulsion, Springer Aerospace Technology, 2017 Search PubMed.
- C. B. Storm, J. R. Stine and J. F. Kramer, Chem. Phys. Energ. Mater., 1990, 605–639 CAS.
- K. Yang, F. Bi, Q. Xue, H. Huo, C. Bai, J. Zhang and B. Wang, Dalt. Trans., 2021, 50, 8338–8348 RSC.
- T. Ozawa, Bull. Chem. Soc. Jpn., 1965, 38, 1881–1886 CrossRef CAS.
- D. J. Whelan, R. J. Spear and R. W. Read, Thermochim. Acta, 1984, 80, 149–163 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Richard J.
Staples
a,
Richard J.
Staples