
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElucidating the effect of Fe substitution on structural and redox stability of Na2Mn3O7†
Hugh B.
Smith
 a,
Gi-Hyeok
Lee
b,
Bachu Sravan
Kumar
a,
Aubrey N.
Penn
c,
Victor
Venturi
a,
Yifan
Gao
a,
Ryan C.
Davis
d,
Kevin Hunter
Stone
d,
Adrian
Hunt
e,
Iradwikanari
Waluyo
e,
Eli
Stavitski
e,
Wanli
Yang
b and
Iwnetim I.
Abate
*a
a,
Gi-Hyeok
Lee
b,
Bachu Sravan
Kumar
a,
Aubrey N.
Penn
c,
Victor
Venturi
a,
Yifan
Gao
a,
Ryan C.
Davis
d,
Kevin Hunter
Stone
d,
Adrian
Hunt
e,
Iradwikanari
Waluyo
e,
Eli
Stavitski
e,
Wanli
Yang
b and
Iwnetim I.
Abate
*a
aDepartment of Materials Science and Engineering, Massachusetts Institute of Technology, 77 Massachusetts Ave., Cambridge, MA 02139, USA. E-mail: iabate@mit.edu
bAdvanced Light Source, Lawrence Berkeley National Laboratory, 1 Cyclotron Rd, Berkeley, CA 94720, USA
cMIT.nano, Massachusetts Institute of Technology, 77 Massachusetts Ave., Cambridge, MA 02139, USA
dStanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, 2575 Sand Hill Rd, Menlo Park, CA 94025, USA
eNational Synchrotron Light Source II, Brookhaven National Laboratory, Upton, NY 11973, USA
First published on 11th March 2025
Abstract
Sodium-ion batteries have the potential to meet the growing demand for energy storage due to their low costs stemming from natural resource abundances, but their cathode energy densities must be improved to be comparable to those of lithium-ion batteries. One strategy is accessing high voltage capacity through high-valent redox reactions. Such reactions usually cause instability in cathode materials, but Na2Mn3O7 (NMO) has demonstrated excellent performance and reversibility in the high-valent regime due to its unique lattice structure with ordered Mn vacancies. This work expands the universality of the ordered vacancy as a design principle and increases the material candidates with such exceptional electrochemical behavior. Our approach involves synergizing cationic ordered vacancies with tunable metal–ligand hybridization through partial metal substitution. In particular, we successfully incorporated Fe3+ for Mn4+ in NMO to make Na2.25Mn2.75Fe0.25O7 and achieved improved high-valent redox behavior. Fe substitution leads to larger specific capacities (171 vs. 159 mA h g−1 first cycle), enhanced cycle stability (97 vs. 60 mA h g−1 after 50 cycles), and superior rate performance. This study lays the foundation for developing new cathode materials with stable high-valent redox through substitution of redox-active transition metals by employing cationic ordered vacancies and partial transition metal substitution as design principles in tandem.
1. Introduction
The scalable adoption of batteries for decarbonization and energy accessibility requires addressing current challenges including (1) high cost, (2) unethical and environmentally unfriendly mining practices, (3) dependence on critical minerals and their supply chains, and (4) unsafe battery chemistries. These issues can be addressed in part through the careful design of electrode materials. Sodium-ion batteries (NIBs) offer a promising alternative to lithium-ion batteries (LIBs) for grid storage or electric vehicle applications due to their superior raw material abundance, significantly lower cost (up to half in terms of $ kW−1 h−1), and ease of scalable production using existing Li-ion battery manufacturing methods and infrastructure.1–6 Even with these excellent attributes, to be a competitive technology, the energy densities of NIB cathode materials must be comparable with those of LIBs. Layered transition metal oxides (TMOs), NaxTMO2 where TM is a transition metal, have emerged as the most energy dense class of NIB cathode materials compared to Prussian blue/white analogs and polyanions.7 The path to achieving the highest energy density in TMOs should involve high-valent redox reactions (i.e., the over oxidation of TMs or oxygen). Such redox reactions typically lead to a high driving force to form covalent bonds, leading to structural rearrangements, resulting in voltage hysteresis, voltage fading, and capacity losses.8 One material exhibits highly reversible high-valent redox with negligible hysteresis and specific capacities exceeding 200 mA h g−1 in large voltage windows: Na2Mn3O7 (NMO) where one out of every seven Mn sites is vacant (Na4/7[Mn6/7□1/7]O2 where □ represents an ordered vacancy).9–15Yamada's group was the first to experimentally verify that the high voltage capacity stemmed from anionic redox reactions.13 Recent work from Abate et al. used advanced X-ray characterization and computational techniques to develop a deeper mechanistic understanding of this reaction, proving that the high-valent redox reaction was O2−/O− redox stabilized by an electron hole polaron on the oxygen as opposed to structural rearrangement mediated by the formation of an O–O dimer.9 The formation of O− in NMO was further confirmed using electron paramagnetic resonance (EPR).16 Moreover, the O2− ions bordering the ordered vacancy are the ones that undergo this redox reaction because their non-bonding 2p orbitals are at the Fermi level and are thus first to oxidize.9,12–14,17 The ordered vacancy is central (a design principle) to the reversible and low-hysteresis high-valent redox behavior in NMO. In this work, we seek to test the universality of this design principle and expand the material candidates with such exceptional high-valent redox behavior. Does the design principle only work with Mn, or does it work with other TMs too? Does the TM have to be in a +4 valence state as in NMO to stabilize the ordered vacancy? Is there competition between the strength of hybridization between TM–O (chemical) and the ordered vacancy (structural) in terms of driving forces that determine the stability of the structure upon cycling? How do redox-active substitutes impact the high-valent redox reactions and overall electrochemical performance? These are some of the questions that need to be asked to test the universality of the design principle. In this work, by substituting Fe3+ for Mn4+ in NMO, we aim to answer these questions.
Similar studies have substituted Al,18 Mg,19 and Ti20,21 for Mn in NMO, demonstrating that the ordered vacancy structure can be maintained with isovalent or aliovalent cationic substitution including TMs. While these substitutions were found to increase stability in the material during cycling, they are not redox active, so they decrease the contributions to capacity from TM redox reactions. Further, the impact of redox-active substitutions on high-valent redox in NMO is still unknown. Fe, however, is redox active and has long been known to demonstrate excellent electrochemical behavior with a specific capacity of 190 mA h g−1 in P2–Na2/3[Mn1/2Fe1/2]O2.22 Since Fe is adjacent to Mn on the periodic table and has a similar mass, its substitution will not meaningfully decrease specific capacity in NMO. Further, in one formula unit of NMO, three O2− ions are bordering the ordered vacancy and are thus stably redox active, but only a maximum of two of them can contribute to capacity since there are only two Na+ ions per formula unit. By substituting Fe3+ for Mn4+, the charge can be balanced through excess Na+, unlocking additional contributions to capacity from O2−/O− redox reactions. Since Fe3+ has two more electrons than Mn4+, more coulombic repulsion between TMO layers is expected, resulting in increased interlayer spacing. This would lead to improved Na+ diffusion kinetics and better rate capability. Fe substitution in NMO has been unsuccessfully attempted before, leading to a common P2 structure with no ordered vacancies,23 but we demonstrate that such substitution into the ordered vacancy lattice is possible.
We seek to further experimentally investigate the predicted high-valent electrochemical behavior of an Fe oxide system. During high-valent redox, it is the O2− ions in sodiated Mn oxides that are directly oxidized because the density of states (DOS) of these materials close to the Fermi level is dominated by contributions from O2− ions, and this has been computationally predicted and experimentally verified for NMO.9,12–15 Using NMO as a model oxide system for stable high-valent redox, we will investigate the behavior of Fe oxide overoxidation through comparing an Fe-substituted NMO system with the pristine system. This may lead to enhanced electrochemical performance and a better understanding of the impact of tuning hybridization strength in TM–O bonds on high-valent redox behavior.
2. Results and discussion
We investigated Na2+xMn3−xFexO7 (NMFO) by substituting a fraction of Mn in Na2Mn3O7 (NMO) with Fe (x ≈ 0.25, which will be discussed later). Assuming Fe3+ is substituting for Mn4+ in NMO, the charge difference is compensated with extra Na+. This has been successfully demonstrated as a charge compensation technique when synthesizing an NMO derivative with aliovalent doping.182.1 Structural, chemical, and morphological characterization
Fig. 1a shows the Rietveld refinement of the as-synthesized Na2.25Mn2.75Fe0.25O7 (NMFO), which was performed using the structural model for Na2Mn3O7 (NMO) (ICSD #5665).14 Refinement reveals a triclinic structure and P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group (Table S1†) except the small peak at 2θ ≈ 19°, which likely corresponds to a minor α-NaFeO2 impurity phase (Fig. S1†). In addition, we found that the interlayer spacing in NMFO is 5.6035 Å, corresponding to the (10) planes, compared to 5.5669 Å for NMO according to our Rietveld refinement (Fig. 1a and S2†). The small [001] peak at 2θ ≈ 6.5° is formed from the ordering of the vacancies and TM ions in the TMO slab, so its presence confirms the existence of ordered vacancies globally.13 The lattice with ordered vacancies is illustrated in Fig. 1b. This model uses the structure of NMO and partially substitutes Fe for Mn to the NMFO stoichiometry. There are two unique Na+ sites in the lattice: Na1 is directly above the ordered vacancy and is prismatically coordinated while Na2 is in an octahedral site not directly adjacent to an ordered vacancy (Fig. 1b).9 The morphology of NMFO is dominated by platelet particles that are approximately 1–2 μm in diameter and about 100 nm in thickness (Fig. 1c). STEM imaging also confirmed the layered structure (Fig. 1d) and the presence of ordered vacancies along the [02] zone axis (Fig. 1e). The ordered vacancy columns (Fig. 1e) are represented by the ordered larger, darker circles, which indicate the absence of atomic columns. The identification of this zone axis is shown in Fig. S3.† Fig. S4† shows STEM imaging displaying the same ordered vacancy structure in pristine NMO, confirming this unique structure in NMO was achieved for NMFO. Fig. S5† shows the effect of precursor stoichiometry on phase purity of the synthesized material.
space group (Table S1†) except the small peak at 2θ ≈ 19°, which likely corresponds to a minor α-NaFeO2 impurity phase (Fig. S1†). In addition, we found that the interlayer spacing in NMFO is 5.6035 Å, corresponding to the (10) planes, compared to 5.5669 Å for NMO according to our Rietveld refinement (Fig. 1a and S2†). The small [001] peak at 2θ ≈ 6.5° is formed from the ordering of the vacancies and TM ions in the TMO slab, so its presence confirms the existence of ordered vacancies globally.13 The lattice with ordered vacancies is illustrated in Fig. 1b. This model uses the structure of NMO and partially substitutes Fe for Mn to the NMFO stoichiometry. There are two unique Na+ sites in the lattice: Na1 is directly above the ordered vacancy and is prismatically coordinated while Na2 is in an octahedral site not directly adjacent to an ordered vacancy (Fig. 1b).9 The morphology of NMFO is dominated by platelet particles that are approximately 1–2 μm in diameter and about 100 nm in thickness (Fig. 1c). STEM imaging also confirmed the layered structure (Fig. 1d) and the presence of ordered vacancies along the [02] zone axis (Fig. 1e). The ordered vacancy columns (Fig. 1e) are represented by the ordered larger, darker circles, which indicate the absence of atomic columns. The identification of this zone axis is shown in Fig. S3.† Fig. S4† shows STEM imaging displaying the same ordered vacancy structure in pristine NMO, confirming this unique structure in NMO was achieved for NMFO. Fig. S5† shows the effect of precursor stoichiometry on phase purity of the synthesized material.
 | ||
Fig. 1 Structural, morphological, and chemical characterization of Na2.25Mn2.75Fe0.25O7 (NMFO). (a) Synchrotron powder X-ray diffraction of NMFO with Rietveld refined pattern. Majority pure phase NMFO with minor α-NaFeO2 impurity. (b) Lattice structure of NMFO viewing the [10] and 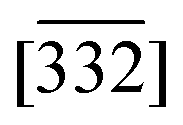 directions, top-down and side view of TMO layers, respectively. The two unique Na sites are labeled. The seven TM sites for one unit cell are also labeled, including the vacant site. (c) SEM image of NMFO particles. (d) HAADF STEM image of NMFO displaying the layered structure with visible atomic columns coming from the TMs. (e) HAADF STEM image of NMFO viewing the [02] direction, showing the ordered vacancies. The overlaid Mn lattice (magenta color) in (d) and (e) is from the known NMO lattice. The yellow boxes in (e) show the repeat unit of seven TM sites along this zone axis where one is vacant. (f) EDX elemental mapping of NMFO. The pink arrow indicates the particle from which the image in (e) was captured. The red semicircles indicate an impurity α-NaFeO2 particle. directions, top-down and side view of TMO layers, respectively. The two unique Na sites are labeled. The seven TM sites for one unit cell are also labeled, including the vacant site. (c) SEM image of NMFO particles. (d) HAADF STEM image of NMFO displaying the layered structure with visible atomic columns coming from the TMs. (e) HAADF STEM image of NMFO viewing the [02] direction, showing the ordered vacancies. The overlaid Mn lattice (magenta color) in (d) and (e) is from the known NMO lattice. The yellow boxes in (e) show the repeat unit of seven TM sites along this zone axis where one is vacant. (f) EDX elemental mapping of NMFO. The pink arrow indicates the particle from which the image in (e) was captured. The red semicircles indicate an impurity α-NaFeO2 particle. | ||
The particle where the ordered vacancies were captured (Fig. 1e) is indicated by the pink arrow in Fig. 1f along with the EDX chemical mapping. The homogeneous distributions of Mn and Fe on this particle suggest Fe's integration into the NMO lattice. More EDX mapping is shown in Fig. S6.† Electron energy loss spectroscopy (EELS) analysis on the same particle corroborates the EDX results (Fig. 2). The Fe L-edge peak (both L3 and L2) is present at multiple locations moving from the edge to the bulk, confirming the presence of Fe throughout the particle. Moreover, the Mn L3-peak shifts to higher energies moving from the particle edge (A in Fig. 2) towards the bulk (D in Fig. 2), which is indicative of oxidation from edge to bulk. This suggests that the surface has lower valent Mn species than the bulk, which could be from surface reduction.9 The surface is also coated with a few unit cells without the ordered vacancy present, requiring the Mn to reduce to preserve charge neutrality (Fig. S4†). Furthermore, the negligible presence of Mn in the particle near the bottom right of Fig. 1f, circled in red, may indicate the presence of α-NaFeO2 impurity observed in XRD.
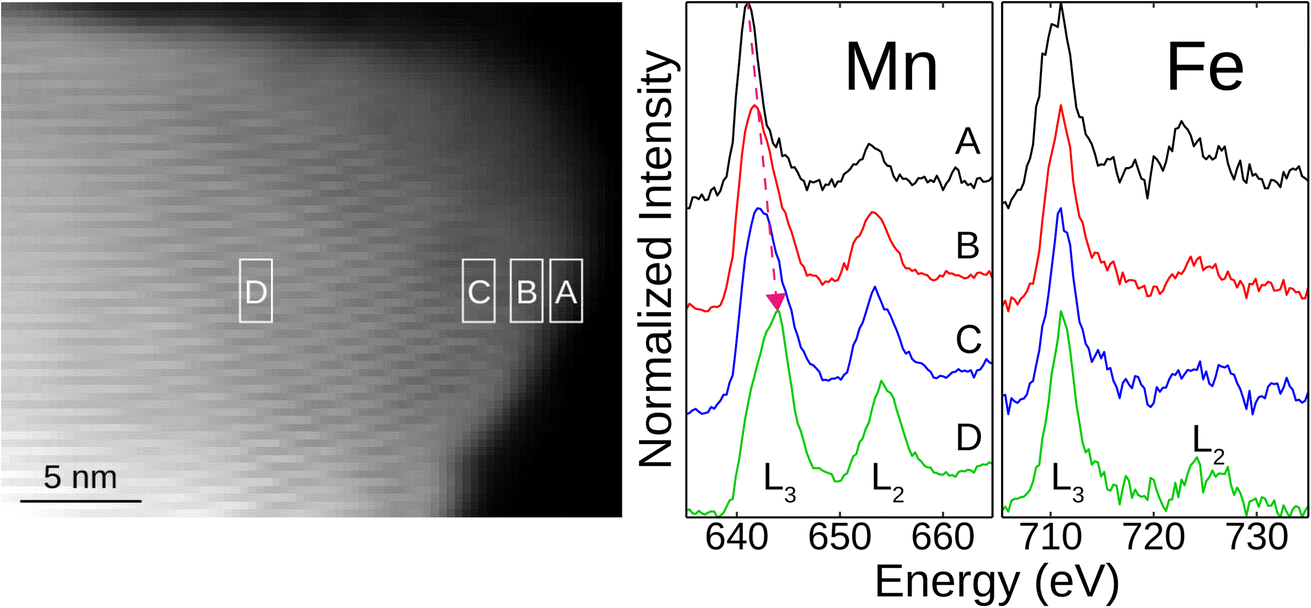 | ||
| Fig. 2 EELS measurements for Na2.25Mn2.75Fe0.25O7. The particle on which these measurements are taken is shown on the left with the white boxes indicating the areas of the particle on which the measurements were taken moving from the edge of the particle (A) to the bulk (D). The associated EELS spectra are shown on the right, and the Mn and Fe L-edges are labeled. The pink arrow indicates the shift in Mn L3-edge. | ||
2.2 Computational investigation
To predict the impact of Fe substitution on redox mechanisms and capacity, we calculated the density of states (DOS) and projected DOS (pDOS) of pristine NMO and NMFO using density functional theory (DFT) (Fig. 3a and b). For this computational investigation, NMFO was modeled using the composition Na2.5Mn2.5Fe0.5O7 instead of the synthesized Na2.25Mn2.75Fe0.25O7. The supercell for the former requires only half as many atoms as the latter, leading to analysis of Na2.5Mn2.5Fe0.5O7 being much more practical in terms of computational resources. The impact of Fe3+ substitution in Na2.5Mn2.5Fe0.5O7 and Na2.25Mn2.75Fe0.25O7 is theoretically the same in terms of mechanism and would only differ in magnitude. | ||
| Fig. 3 Projected density of states (pDOS) for (a) Na2Mn3O7 (NMO) and (b) Na2.5Mn2.5Fe0.5O7 (NMFO). The yellow box highlights the area corresponding to the removal of one electron per formula unit, representing the amount of electrons estimated to be removed in the electrochemical voltage window shown later. (c) Fractional contributions of each element for pristine NMO and NMFO from the highlighted region in (a and b). | ||
The bands near the Fermi level will participate in the redox reactions upon desodiation/charging. We adapted a method we previously developed24 to determine the energy region of the band near the Fermi level (highlighted in yellow in Fig. 3a and b) which corresponds to one electron per formula unit (e.g., Na2Mn3O7 → Na1Mn3O7 during charging). This energy region was determined by the total DOS. We then obtained the relative contribution of each element in this energy region (capacity) by integrating the pDOS and determining the ratios of integrated results for each element (Fig. 3c). The results of our analysis show that oxygen has the highest contribution to the capacity in NMO and NMFO upon charging (Fig. 3c). However, this contribution is relatively suppressed by the high-valent redox activity of the cations in NMFO due to the enhanced TM–O hybridization strength (i.e., stronger orbital overlap between Fe and O in Fig. 3b). This is consistent with other predictions for charging in the high-valent regime for Mn and Fe oxides.8 Since anionic redox is generally related to irreversible structural distortion, the suppression of anionic redox activity in NMFO could enable reversible and low-hysteresis cycling.25 Additionally, the pDOS suggest that if we were to desodiate (charge) beyond one Na+ (1 e−), there would be more capacity that can be obtained from the combined anionic and cationic redox, suggesting NMFO could have a higher energy density than NMO.
2.3 Redox mechanisms
The second electrochemical cycle was chosen to explore the redox mechanisms in this material to capture redox mechanisms starting from the fully discharged state, which is more sodiated than the pristine state. The second cycle electrochemical profiles of Na2.25Mn2.75Fe0.25O7 (NMFO) and Na2Mn3O7 (NMO) at C/20 over the voltage range 1.5–4.7 V are shown in Fig. 4a. The first electrochemical cycle was in the voltage range 1.5–4.3 V, which fully sodiates the samples without causing irreversible structural rearrangement or major electrolyte decomposition that may happen above 4.3 V. The two voltage plateaus at ∼4.2 V and ∼4.5 V are characteristic of O2−/O− redox and correspond to phase changes where first the Na2 site (Na+ site not adjacent to the ordered TM vacancy, shown in Fig. 1b) is removed in the lower plateau, and the Na1 site (Na+ site above the ordered TM vacancy, shown in Fig. 1b) is removed in the higher plateau. This is because the Na2 site has a higher cation–cation repulsion compared to the Na1 site, which borders the TM vacancy, so the Na2 ion will more readily be extracted.10,12–15,18–20,24,26 In the lower voltage regions below ∼3 V, there is a voltage plateau during charging, which corresponds to the Mn3+/Mn4+ redox couple. Upon discharge, this lower voltage plateau shifts to be more sloping, corresponding to solid–solution behavior. The voltage profiles of the first cycle are additionally explored in Fig. S8.†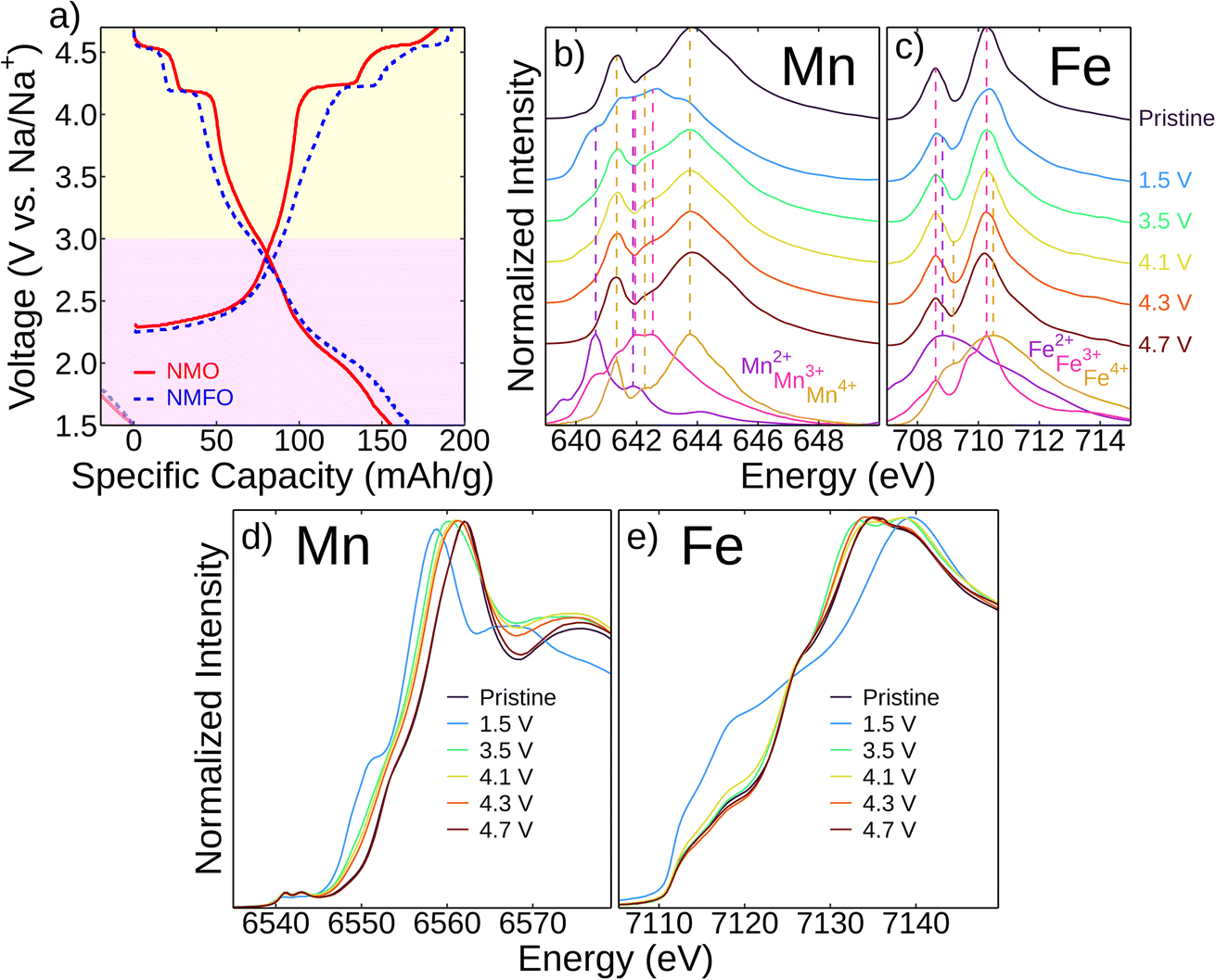 | ||
| Fig. 4 Redox mechanisms investigated with XAS measurements of Na2.25Mn2.75Fe0.25O7 (NMFO) at different states of charge. (a) Second cycle voltage profiles of Na2Mn3O7 (NMO) and NMFO at C/20 over the voltage range 1.5–4.7 V. The high- and low-valent regime are colored yellow and purple, respectively. The end of the first cycle discharge, which was 1.5–4.3 V, is shown as negative capacity with faded lines. Mn (b) and Fe (c) L3-edge iPFY measurements at different states of charge during the second cycle charge (first cycle 1.5–4.3 V) with reference measurements. The vertical dotted lines align with the resonances of the reference spectra. Mn (d) and Fe (e) K-edge XANES spectra at different states of charge during the second cycle charge (first cycle 1.5–4.3 V). | ||
We employed ex situ X-ray absorption spectroscopy (XAS) to study the redox mechanisms in NMFO during the second cycle and determine the contributions of the cations and anions during cycling (Fig. 4). While EELS is capable of spatially resolving the spectra with relatively rough excitation energy resolution (∼1.5 eV in this work), as shown in Fig. 2, XAS reveals the detailed electronic configuration with higher resolution (in this work, ∼0.06–0.07 eV for soft XAS and ∼0.7 eV for hard XAS). As shown in Fig. 4b and c, more splittings in the Mn and Fe L3-edges are observed in the given excitation energy range compared to EELS in Fig. 2. Using soft XAS, we probed the Mn and Fe L3-edges of NMFO at different states of charge (Fig. 4b and c). Here, inverse partial fluorescence yield (iPFY) mode was used to collect undistorted XAS of the Mn and Fe L3-edges from the subsurface of the materials (<200 nm from the surface).
Fig. 4b shows that the Mn L3-edge of the pristine sample reflects the pattern of the Mn4+ reference. When fully discharged (1.5 V), this edge maintains a pattern close to that of the Mn3+ reference, and charging to 3.5 V results in features of the Mn4+ reference. This larger change in oxidation state between 1.5–3.5 V (clearly shown in the difference plot in Fig. S9a†) is expected as this corresponds to the low voltage plateau on the electrochemical profile (Fig. 4a), which contributes significant capacity. Linear combination fitting from the reference scans also shows that the most Mn redox occurs in this voltage region (Fig. S9c and e†). There is slight oxidation between 3.5–4.7 V but not exceeding an Mn4+ oxidation state. Fig. 4c shows that Fe is also redox active. Its pristine spectrum maintains a pattern close to that of the Fe3+ reference.27 When fully discharged (1.5 V), the spectrum shows features of the Fe2+ reference pattern with subsequent charging to 3.5 V resulting in oxidation of Fe to Fe3+. Between 3.5–4.7 V, there is oxidation towards Fe4+ (also shown in the difference plot in Fig. S9b† and the linear combination fitting in Fig. S9d and e†), which is a higher oxidation state than the pristine sample. Soft XAS measurements during first cycle charging are shown in Fig. S7† and are in agreement with the second cycle measurements.
Similarly, we used hard XAS to probe the Mn and Fe K-edges (Fig. 4d and c, respectively), which shows a progression of redox reactions throughout cycling consistent with the Mn and Fe L3-edge results. Both the Mn L3- and K-edge results are consistent with spectra found in the literature for NMO.9,12 The very high voltage (i.e., >4.3 V) charging behavior is also consistent with other studies of oxide cathodes where Mn can continue to oxidize but not past the Mn4+ state and Fe is oxidized to a state between Fe3+ and Fe4+.28–31 Ultimately, the low voltage capacity is dominated by Mn and Fe redox, but lower levels of TM oxidation at high voltage are insufficient to explain the observed capacity in this regime. Thus, oxygen redox must be contributing significantly to the reversible high-voltage capacity.
Oxygen redox was probed with resonant inelastic X-ray scattering (RIXS), which is known to be sensitive to the different oxidation states of oxygen in TMO battery cathodes. Fig. 5 shows O K-edge RIXS results. Previous RIXS measurements of NMO9 reported a negligible signature for the O–O dimer formation, which typically appears at the excitation energy of ∼531 eV through an emission at ∼524 eV and low-energy excitation intensities close to the elastic scattering.32 Instead, dedicated O− is formed at a characteristic excitation energy of ∼527.5 eV,9,33 and the presence of this specie in NMO was further confirmed by electron paramagnetic resonance (EPR).16 NMFO exhibits a similar redox mechanism where, in the high-valent regime, O2−/O− is the dominant redox mechanism until 4.3 V. When charging to higher voltages (4.7 V), this mechanism is then accompanied by a slight contribution from O–O dimer formation. The O–O dimer signatures here are relatively weak compared with typical O–O dimer forming systems in both Li-rich and Na layered compounds (Fig. 5a),9,33–36 but integrating over the excitation (Fig. 5b) and emission (Fig. 5c) energies provides a clear contrast between the pristine and charged states, indicating formation of both O–O bonding and dedicated O− states at 4.7 V, which is also observed in NMO (Fig. 5d) after O− is given enough time to overcome the kinetic barrier to form an O–O dimer.9 Like in NMO, the low voltage capacity in NMFO is derived from TM redox. In the high voltage regime, the capacity in NMO comes solely from oxygen redox, but in NMFO, our results show that while oxygen redox still dominates, some of it is shifted to TM redox, which is in agreement with our computationally derived results (Fig. 3).
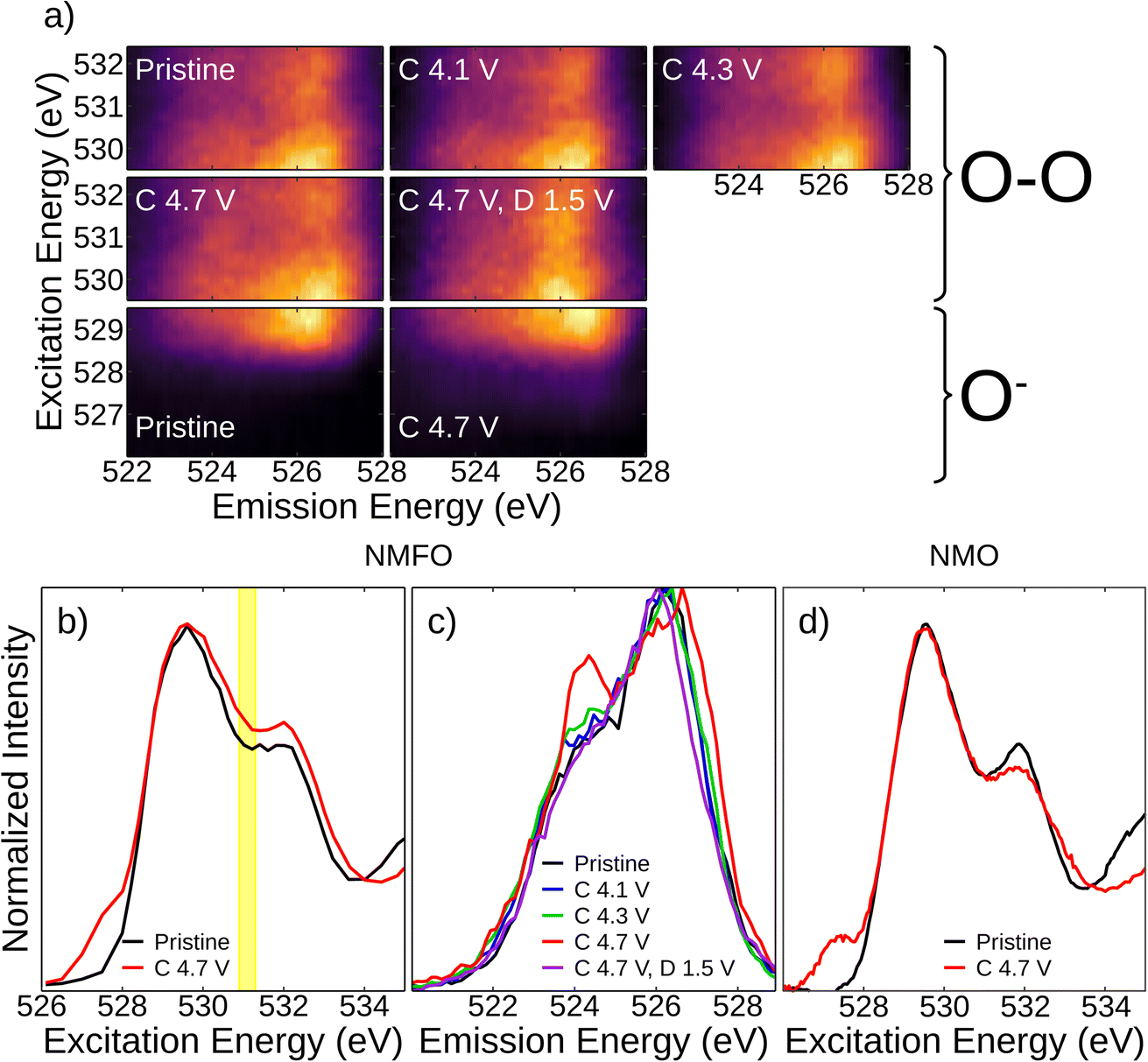 | ||
| Fig. 5 Oxygen redox in Na2.25Mn2.75Fe0.25O7 (NMFO) at different states of charge. (a) O K-edge RIXS maps of NMFO at different states of charge. (b) O K-edge XAS (via integration of RIXS maps) for NMFO pristine and fully charged. (c) Emission spectra for the region corresponding to excitation energies of 530.9–531.3 eV, highlighted in yellow in (b). (d) O K-edge XAS of Na2Mn3O7 at the same voltages as (b). | ||
Furthermore, we investigated the global and local structural stability of NMFO during cycling. XRD of NMFO at different states of charge (Fig. S10†) shows that the majority NMFO phase remains stable throughout cycling with no phase changes observed. Investigating the Mn and Fe K-edge extended X-ray absorption fine structure (EXAFS) revealed the evolution of the local structure (Fig. S11†). The peaks corresponding to TM–TM bonds and TM-O bonds remain stable upon desodiation.
2.4 Electrochemical evaluation
The electrochemical performance of NMFO compared to NMO was evaluated over the voltage window of 1.5–4.3 V vs. Na/Na+ (Fig. 6) where the electrolyte is stable (Fig. S13†). Above 4.3 V, the electrolyte decomposes, and a significant amount of O2− ions are oxidized, which could lead to a very strong driving force to form covalent bonds coupled with in-plane Mn migration, leading to material instability.8,9,33 The electrochemical cycling was carried out with a current density corresponding to C/20 (based on 1C = 155 mA g−1 (ref. 18) then recalculated after the first cycle based on measured discharge capacity). Based on the first charge capacity of NMO and NMFO, the amount of Fe3+ incorporated into the Na2+xMn3−xFexO7 lattice was estimated to be x ≈ 0.25 (Fig. S14†). During the first discharge, both NMO and NMFO show clear plateaus at ∼4.2 V, which corresponds to O2−/O− redox, and at ∼2.0 V, which corresponds to Mn3+/Mn4+ redox.9,12–15 During the first discharge, in the high-valent regime (>3.0 V), the capacity contribution in NMO is dominated by the O2−/O− redox plateau, which contributes a discharge capacity of ∼30 mA h g−1. Further discharge leads to a steep drop in voltage then a Mn3+/Mn4+ redox plateau, leading to TM redox contributing ∼130 mA h g−1. For NMFO, the O2−/O− redox plateau contributes ∼20 mA h g−1 and is followed by a sloping profile. This could be characteristic of O2−/O− redox shifting to TM redox as was predicted by computation (Fig. 3) and has been observed in Li-ion chemistries.35 The total discharge capacity that TM redox contributes for NMFO is ∼150 mA h g−1. Similar observations are made for cycling in a larger voltage window (1.5–4.7 V vs. Na/Na+) and at higher C-rate (i.e., C/10) cycling in the same voltage range (Fig. S16†). A detailed explanation of this is presented in the ESI.† | ||
| Fig. 6 Electrochemical testing of Na2Mn3O7 (NMO) and Na2.25Mn2.75Fe0.25O7 (NMFO) over the voltage range 1.5–4.3 V vs. Na/Na+. Select voltage profiles of (a) NMO and (b) NMFO cycled at C/20. (c) Cycling performance at C/20 with specific capacity (left axis, filled points) and coulombic efficiency (right axis, empty points) as a function of cycle. (d) Rate performance evaluation. | ||
Over the first three cycles, the discharge capacity of NMO drops significantly (from 159 mA h g−1 to 88 mA h g−1). A closer look at the cycling profiles hints that the capacity loss is severe in the lower voltage region (i.e., during Mn3+/Mn4+ redox) as seen from Fig. 6a. This could primarily be due to TM dissolution, which is common in lithium- and sodium-containing TMOs that derive capacity from Mn redox reactions.37–41 An ICP-OES investigation of the TM content in the electrolyte after cycling confirms that TM dissolution is an issue (Table S4†). The rapid capacity loss in the lower voltage regime coupled with relative stability in the high voltage regime of NMO (Fig. 6a) causes the average discharge voltage to increase from ∼2.4 V to a maximum of ∼3.0 V (Fig. S17†). A similar phenomenon occurs when cycling at C/10 (Fig. S16f†). With progressive cycling (>10 cycles), the low voltage regime of NMO remains relatively stable while the high voltage O2−/O− redox plateau fades, causing the average discharge voltage to decrease (Fig. 6c).
The initial discharge capacity is higher for NMFO (171 mA h g−1), likely stemming from the fact that NMFO can accommodate more Na in its structure. Most of the initial capacity fade from cycling comes from the low voltage cycling (again due to Mn-dissolution) but degrades at a much slower rate than in NMO (Fig. 6b), causing the overall specific capacity to degrade much slower (Fig. 6c). An ICP-OES investigation of the TM content in the electrolyte after cycling confirms that TM dissolution is substantially less significant for NMFO than NMO (Table S4†). In NMFO, both the high and low voltage regimes degrade slowly (Fig. 6b), leaving the average discharge voltage relatively constant (Fig. S17†). This leads to the proportions of capacity from oxygen and transition metal redox relatively stable compared to NMO (Fig. S18†). At C/10, the low-valent capacity degrades quickly, which is similar to NMO, but the high-valent capacity degrades much slower than in NMO (Fig. S16e†). This suggests that Fe introduces an increased energetic penalty for in-plane TM migration, leading to less structural deformation. NMFO maintains superior performance over NMO in terms of specific capacity (Fig. 6c) and specific energy (Fig. S19†) at higher cycles (97 to 60 mA h g−1 after 50 cycles, 49.5 to 19.6 mA h g−1 after 100 cycles). Future efforts to improve cycle stability will focus on issues related to TM dissolution and will include coating with carbon, alumina, or other materials as possible solutions.
NMFO also outperforms NMO in terms of rate capability (Fig. 6d). To study the kinetics further, electrochemical impedance spectroscopy (EIS) was performed on pristine cells of NMO and NMFO (Fig. S21†). NMFO had a superior Na diffusivity (DNa+) of 3.78 × 10−16 cm2 s−1 compared to 2.56 × 10−16 cm2 s−1 for NMO. Galvanostatic intermittent titration technique (GITT) was also used to study the kinetics throughout charge and discharge of NMO and NMFO (Fig. S22†). The DNa+ of NMO varies between ∼9.3 × 10−15 and 3.9 × 10−10 cm2 s−1 during cycling while that of NMFO varies between ∼2.7 × 10−12 and ∼1.3 × 10−9 cm2 s−1. For a given voltage and half cycle (i.e., charge or discharge), NMFO has a superior DNa+. The methodology of calculating the DNa+ is explained in the ESI.† The primary limiting factor for Na+ kinetics in NMFO is the fact that the Na+ ion must transition through different coordination environments as it moves through the material.
Compared to other cationic substitutions studied for NMO, Fe is very competitive. Substitution of Mg for Mn in NMO at low amounts (x = 0–0.06 in Na2Mn3−xMgxO7) results in decreasing specific capacity contributions from O redox while increasing and stabilizing low voltage Mn redox, leading to enhanced cycle stability and rate performance.19 Isovalent substitution of Ti4+ in NMO at greater quantity (x = 0.5 in Na2Mn3−xTixO7) leads to structural stability and enhanced cycle stability without increasing specific capacity since a significant amount of redox active Mn is replaced by redox inactive Ti.20,21 Aliovalent substitution of Al (x = 0.4 in Na2+xMn3−xAlxO7) leads to improved cycle stability and specific capacity from higher Na content but a loss of low-hysteresis O redox.18 Our findings show that substitution of aliovalent and redox active Fe3+ for Mn4+ in NMO shifts some O redox to TM redox, which improves the stability of O redox while maintaining low-hysteresis redox. This is similar to the impact of Mg substitution on high-valent redox mechanisms.19 However, in NMFO, the specific capacity contributions from TM redox are significantly increased from all cations being redox active. Like with Al substitution, the increased Na content of the pristine material contributes to a larger overall specific capacity as well.18 Further, TM redox is more reversible in NMFO compared to NMO, and rate performance is also improved (Fig. 6).
3. Conclusions
Although high-valent redox is a crucial mechanism for enhancing the energy density of battery cathodes, a limited number of materials and design rules exist to harness this enhanced capacity reversibly with low voltage hysteresis. The ordered cationic vacancy in Na2Mn3O7 (NMO) is a unique case where this is possible. By coupling two design principles, ordered vacancies and partial metal substitution, we discovered that Fe substitution for Mn in NMO can enhance TM–O hybridization, enabling reversible and stable high-valent cationic and anionic capacity while maintaining the robust ordered vacancy structure motif. Through combined computational and experimental investigation, we discovered that although Na2.25Mn2.75Fe0.25O7 (NMFO) exhibits a similar high-valent redox mechanism as NMO, its ability to accommodate more Na in its structure and its increased interlayer spacing (due to the extra electrons on Fe3+versus Mn4+) result in superior electrochemical performance during cycling in terms of specific capacity, energy density, cycle stability, and rate capability.Our findings suggest that ordered cationic vacancies can be achieved in redox active TMs beyond Mn, and by combining this with other design rules, material candidates with exceptional high-valent redox behavior can be expanded. In addition, the partial substitution of TMs not only tunes TM–O hybridization strength and governs the redox mechanism/redox-active species, but it can also tune structures (such as interlayer distance or phases), having a significant impact on material stability, rate capability, and overall electrochemical performance in the high-valent regime. This paves the way for more studies to thoroughly establish the relationships between TM–O hybridization strength, stable lattice structures, and high-valent electrochemical performance. We aim to establish such relationships with future studies that incorporate various metals.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Author contributions
IIA conceived the project; IAA and WY supervised the experiments; IAA supervised the theoretical calculations; HBS synthesized the materials and performed electrochemical measurements; RCD, ES, AH, IW, and GHL performed XAS measurements; GHL performed RIXS measurements; KHS performed XRD measurements; ANP and HBS performed STEM and EDX measurements; HBS and VV performed the theoretical calculations; YG performed ICP-OES measurements. HBS analyzed electrochemical, XAS, RIXS, XRD, STEM, EDX and DFT data. In addition, the analysis was performed by GHL (RIXS), BKS, ANP (STEM/EELS), YG (ICP-OES), AH (sXAS), IW (sXAS), and ES (XANES). All authors contributed in interpreting the data; VV assisted in interpreting the simulation results; HBS wrote the manuscript with inputs from all authors; IIA directed the overall research.Conflicts of interest
Massachusetts Institute of Technology has filed for patents on subject matter related to this manuscript in which HBS and IIA are listed inventors.Acknowledgements
The authors would like to thank Anirudh Adavi for SEM measurements and Corson Chao for the initial Na2Mn3O7 synthesis. This work was supported by the MIT Startup Fund. This research used BL 8.0.1 of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. This research used BLs 8-ID and 23-ID-2 of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. This work was carried out in part through the use of MIT.nano's facilities, which is supported by the National Science Foundation under award ECCS-1542152. The computational work used Expanse at San Diego Supercomputer Center through allocation MAT230005 from the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program, which is supported by National Science Foundation grants #2138259, #2138286, #2138307, #2137603, and #2138296.References
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 1–16 CrossRef.
- M. Ferraro and G. Tumminia, Techno-economics Analysis on Sodium-Ion Batteries, https://www.springerprofessional.de/en/techno-economics-analysis-on-sodium-ion-batteries-overview-and-p/26703648, accessed March 12, 2024.
- H. S. Hirsh, Y. Li, D. H. S. Tan, M. Zhang, E. Zhao and Y. S. Meng, Adv. Energy Mater., 2020, 10, 2001274 CrossRef CAS.
- M. Munjal, T. Prein, M. M. Ramadan, H. B. Smith, V. Venugopal, J. L. M. Rupp, I. I. Abate, E. A. Olivetti and K. J. Huang, Joule, 2025 DOI:10.1016/j.joule.2025.101871.
- M. Walter, M. V. Kovalenko and K. V. Kravchyk, New J. Chem., 2020, 44, 1677–1683 RSC.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- B. Sayahpour, H. Hirsh, S. Parab, L. H. B. Nguyen, M. Zhang and Y. S. Meng, MRS Energy Sustain., 2022, 9, 183–197 CrossRef.
- W. E. Gent, I. I. Abate, W. Yang, L. F. Nazar and W. C. Chueh, Joule, 2020, 4, 1369–1397 CrossRef CAS.
- I. I. Abate, C. D. Pemmaraju, S. Y. Kim, K. H. Hsu, S. Sainio, B. Moritz, J. Vinson, M. F. Toney, W. Yang, W. E. Gent, T. P. Devereaux, L. F. Nazar and W. C. Chueh, Energy Environ. Sci., 2021, 14, 4858–4867 RSC.
- E. Adamczyk and V. Pralong, Chem. Mater., 2017, 29, 4645–4648 CrossRef CAS.
- F. M. Chang and M. Jansen, Z. Anorg. Allg. Chem., 1985, 531, 177–182 CAS.
- Y. Li, X. Wang, Y. Gao, Q. Zhang, G. Tan, Q. Kong, S. Bak, G. Lu, X.-Q. Yang, L. Gu, J. Lu, K. Amine, Z. Wang and L. Chen, Adv. Energy Mater., 2019, 9, 1803087 Search PubMed.
- B. Mortemard de Boisse, S. Nishimura, E. Watanabe, L. Lander, A. Tsuchimoto, J. Kikkawa, E. Kobayashi, D. Asakura, M. Okubo and A. Yamada, Adv. Energy Mater., 2018, 8, 1800409 CrossRef.
- B. Song, M. Tang, E. Hu, O. J. Borkiewicz, K. M. Wiaderek, Y. Zhang, N. D. Phillip, X. Liu, Z. Shadike, C. Li, L. Song, Y.-Y. Hu, M. Chi, G. M. Veith, X.-Q. Yang, J. Liu, J. Nanda, K. Page and A. Huq, Chem. Mater., 2019, 31, 3756–3765 CrossRef CAS.
- A. Tsuchimoto, X.-M. Shi, K. Kawai, B. Mortemard de Boisse, J. Kikkawa, D. Asakura, M. Okubo and A. Yamada, Nat. Commun., 2021, 12, 631 CrossRef CAS PubMed.
- C. Zhao, H. Liu, F. Geng, B. Hu and C. Li, Energy Storage Mater., 2022, 48, 290–296 CrossRef.
- Y. Lai, H. Xie, P. Li, B. Li, A. Zhao, L. Luo, Z. Jiang, Y. Fang, S. Chen, X. Ai, D. Xia and Y. Cao, Adv. Mater., 2022, 34, 2206039 CrossRef CAS PubMed.
- C. Soares, B. Silván, Y.-S. Choi, V. Celorrio, V. R. Seymour, G. Cibin, J. M. Griffin, D. O. Scanlon and N. Tapia-Ruiz, J. Mater. Chem. A, 2022, 10, 7341–7356 RSC.
- D. P. Siriwardena, J. F. S. Fernando, T. Wang, K. L. Firestein, C. Zhang, H. E. A. Brand, M. W. M. Jones, C. M. Kewish, P. Berntsen, T. Jenkins, C.-E. M. Lewis, J. E. von Treifeldt, D. P. Dubal and D. V. Golberg, Electrochim. Acta, 2021, 394, 139139 CrossRef CAS.
- Y. Liu, C. Wang, M. Ren, H. Fang, Z. Jiang and F. Li, J. Energy Chem., 2022, 63, 351 Search PubMed.
- S. F. Linnell, E. J. Kim, Y.-S. Choi, M. Hirsbrunner, S. Imada, A. Pramanik, A. F. Cuesta, D. N. Miller, E. Fusco, B. E. Bode, J. T. S. Irvine, L. C. Duda, D. O. Scanlon and A. R. Armstrong, J. Mater. Chem. A, 2022, 10, 9941–9953 RSC.
- N. Yabuuchi, M. Kajiyama, J. Iwatate, H. Nishikawa, S. Hitomi, R. Okuyama, R. Usui, Y. Yamada and S. Komaba, Nat. Mater., 2012, 11, 512–517 CrossRef CAS PubMed.
- L. Rakhymbay, A. Namazbay, M. Karlykan, M. Abilkairova, A. Konarov and Z. Bakenov, Int. J. Electrochem. Sci., 2022, 17, 221234 CrossRef CAS.
- I. Abate, S. Y. Kim, C. D. Pemmaraju, M. F. Toney, W. Yang, T. P. Devereaux, W. C. Chueh and L. F. Nazar, Angew. Chem., 2021, 133, 10975–10982 CrossRef.
- W. E. Gent, I. I. Abate, W. Yang, L. F. Nazar and W. C. Chueh, Joule, 2020, 4, 1369–1397 CrossRef CAS.
- J. Wan, Y. Qiu, X. Sun, M. Ou, J. Xu, X. Zhang, Y. Liu, S. Sun, Y. Xu, C. Fang, L. Huang, P. K. Chu and J. Han, ACS Appl. Mater. Interfaces, 2022, 14, 38769–38777 CrossRef CAS PubMed.
- X. Liu, J. Liu, R. Qiao, Y. Yu, H. Li, L. Suo, Y. Hu, Y.-D. Chuang, G. Shu, F. Chou, T.-C. Weng, D. Nordlund, D. Sokaras, Y. J. Wang, H. Lin, B. Barbiellini, A. Bansil, X. Song, Z. Liu, S. Yan, G. Liu, S. Qiao, T. J. Richardson, D. Prendergast, Z. Hussain, F. M. F. de Groot and W. Yang, J. Am. Chem. Soc., 2012, 134, 13708–13715 CrossRef CAS PubMed.
- X.-L. Li, T. Wang, Y. Yuan, X.-Y. Yue, Q.-C. Wang, J.-Y. Wang, J. Zhong, R.-Q. Lin, Y. Yao, X.-J. Wu, X.-Q. Yu, Z.-W. Fu, Y.-Y. Xia, X.-Q. Yang, T. Liu, K. Amine, Z. Shadike, Y.-N. Zhou and J. Lu, Adv. Mater., 2021, 33, 2008194 CAS.
- M.-H. Cao, R.-Y. Li, S.-Y. Lin, S.-D. Zheng, L. Ma, S. Tan, E. Hu, Z. Shadike, X.-Q. Yang and Z.-W. Fu, J. Mater. Chem. A, 2021, 9, 27651–27659 RSC.
- C. Dräger, F. Sigel, R. Witte, R. Kruk, L. Pfaffmann, S. Mangold, V. Mereacre, M. Knapp, H. Ehrenberg and S. Indris, Phys. Chem. Chem. Phys., 2018, 21, 89–95 RSC.
- E. McCalla, M. T. Sougrati, G. Rousse, E. J. Berg, A. Abakumov, N. Recham, K. Ramesha, M. Sathiya, R. Dominko, G. Van Tendeloo, P. Novák and J.-M. Tarascon, J. Am. Chem. Soc., 2015, 137, 4804–4814 CrossRef CAS PubMed.
- Z. Zhuo, Y. Liu, J. Guo, Y. Chuang, F. Pan and W. Yang, J. Phys. Chem. Lett., 2020, 11, 2618–2623 CrossRef CAS PubMed.
- R. A. House, U. Maitra, M. A. Pérez-Osorio, J. G. Lozano, L. Jin, J. W. Somerville, L. C. Duda, A. Nag, A. Walters, K.-J. Zhou, M. R. Roberts and P. G. Bruce, Nature, 2020, 577, 502–508 CrossRef CAS PubMed.
- K. Dai, J. Wu, Z. Zhuo, Q. Li, S. Sallis, J. Mao, G. Ai, C. Sun, Z. Li, W. E. Gent, W. C. Chueh, Y. Chuang, R. Zeng, Z. Shen, F. Pan, S. Yan, L. F. J. Piper, Z. Hussain, G. Liu and W. Yang, Joule, 2019, 3, 518–541 CrossRef CAS.
- W. E. Gent, K. Lim, Y. Liang, Q. Li, T. Barnes, S.-J. Ahn, K. H. Stone, M. McIntire, J. Hong, J. H. Song, Y. Li, A. Mehta, S. Ermon, T. Tyliszczak, D. Kilcoyne, D. Vine, J.-H. Park, S.-K. Doo, M. F. Toney, W. Yang, D. Prendergast and W. C. Chueh, Nat. Commun., 2017, 8, 2091 CrossRef PubMed.
- J. Hong, W. E. Gent, P. Xiao, K. Lim, D.-H. Seo, J. Wu, P. M. Csernica, C. J. Takacs, D. Nordlund, C.-J. Sun, K. H. Stone, D. Passarello, W. Yang, D. Prendergast, G. Ceder, M. F. Toney and W. C. Chueh, Nat. Mater., 2019, 18, 256–265 CrossRef CAS PubMed.
- M. Lin, L. Ben, Y. Sun, H. Wang, Z. Yang, L. Gu, X. Yu, X.-Q. Yang, H. Zhao, R. Yu, M. Armand and X. Huang, Chem. Mater., 2015, 27, 292–303 CrossRef CAS.
- N. P. W. Pieczonka, Z. Liu, P. Lu, K. L. Olson, J. Moote, B. R. Powell and J.-H. Kim, J. Phys. Chem. C, 2013, 117, 15947–15957 CAS.
- R. Qiao, K. Dai, J. Mao, T.-C. Weng, D. Sokaras, D. Nordlund, X. Song, V. S. Battaglia, Z. Hussain, G. Liu and W. Yang, Nano Energy, 2015, 16, 186–195 CAS.
- R. Qiao, Y. Wang, P. Olalde-Velasco, H. Li, Y.-S. Hu and W. Yang, J. Power Sources, 2015, 273, 1120–1126 CAS.
- D. Tang, Y. Sun, Z. Yang, L. Ben, L. Gu and X. Huang, Chem. Mater., 2014, 26, 3535–3543 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta08203f |
| This journal is © The Royal Society of Chemistry 2025 |